

Abstract
BACKGROUND AND PURPOSE
Lipoxin A4 (LXA4) is a lipid mediator involved in the resolution of inflammation. Increased levels of LXA4 in synovial fluid and enhanced expression of the formyl peptide receptor 2/lipoxin A4 receptor (FPR2/ALX) in the synovial tissues of rheumatoid arthritis patients have been reported. Endothelins (ETs) play a pivotal pro-inflammatory role in acute articular inflammatory responses. Here, we evaluated the anti-inflammatory role of LXA4, during the acute phase of zymosan-induced arthritis, focusing on the modulation of ET-1 expression and its effects.
EXPERIMENTAL APPROACH
The anti-inflammatory effects of LXA4, BML-111 (agonist of FPR2/ALX receptors) and acetylsalicylic acid (ASA) pre- and post-treatments were investigated in a murine model of zymosan-induced arthritis. Articular inflammation was assessed by examining knee joint oedema; neutrophil accumulation in synovial cavities; and levels of prepro-ET-1 mRNA, leukotriene (LT)B4, tumour necrosis factor (TNF)-α and the chemokine KC/CXCL1, after stimulation. The direct effect of LXA4 on ET-1-induced neutrophil activation and chemotaxis was evaluated by shape change and Boyden chamber assays respectively.
KEY RESULTS
LXA4, BML-111 and ASA administered as pre- or post-treatment inhibited oedema and neutrophil influx induced by zymosan stimulation. Zymosan-induced preproET-1 mRNA, KC/CXCL1, LTB4 and TNF-α levels were also decreased after LXA4 pretreatment. In vitro, ET-1-induced neutrophil chemotaxis was inhibited by LXA4 pretreatment. LXA4 treatment also inhibited ET-1-induced oedema formation and neutrophil influx into mouse knee joints.
CONCLUSION AND IMPLICATION
LXA4 exerted anti-inflammatory effects on articular inflammation through a mechanism that involved the inhibition of ET-1 expression and its effects.
Keywords: LXA4, BML-111, acetylsalicylic acid, ET-1, BOC-1, oedema formation, neutrophil
Introduction
Rheumatoid arthritis (RA) is an autoimmune disease characterized by synovial hyperplasia, progressive destruction of cartilage and bone, neoangiogenesis and infiltration of the joint synovium by activated inflammatory leucocytes, leading to chronic inflammation of the joints (Harris, 1991). A critical factor that contributes to joint damage is the excessive production of inflammatory mediators by resident and/or infiltrating inflammatory cells. Free radicals, extracellular matrix-degrading enzymes, pro-inflammatory cytokines [including interleukin (IL)-6, IL-1 and tumour necrosis factor (TNF)-α], chemokines (such as CXCL1) and lipid mediators [like leukotriene (LT)B4] are among the main mediators involved in this process (Maini et al., 1999). Our group has recently shown that endogenous endothelins (ETs) are inflammatory mediators that also play a crucial pro-inflammatory role in articular inflammation by regulating oedema formation, leucocyte influx and the production of inflammatory mediators, including TNF-α, chemokines and LTB4 (Conte et al., 2008). Accordingly, ET-1 levels have been shown to be increased in the serum (Haq et al., 1999; Kuryliszyn-Moskal et al., 2006) and synovial fluids (Vaudo et al., 2004) of RA patients when compared to healthy subjects. ET-1 is a 21-amino acid isopeptide that is mainly produced by endothelial cells from a 200-residue prepropeptide (Inoue et al., 1989).
Lipoxin A4 (LXA4) is another mediator involved in inflammation that is endogenously derived from arachidonic acid metabolism, and has marked anti-inflammatory and pro-resolving effects (Yacoubian and Serhan, 2007). The C-15 epimeric form of LXA4, aspirin-triggered LXA4 (15-epi-LXA4 or ATL), has been shown to potently inhibit neutrophil activity (chemotaxis, adhesion and transmigration) (Filep et al., 2005) and leucocyte trafficking in various inflammatory disorders. The anti-inflammatory effects of both LXA4 and ATL have been demonstrated in several experimental models of inflammation, including ischaemia/reperfusion (Souza et al., 2007), paw oedema (Menezes-de-Lima et al., 2006), air pouch (Chiang et al., 2000) and gastric damage (Fiorucci et al., 2002). Furthermore, its pro-resolution actions have been previously described in periodontitis (Serhan et al., 2003) and lung inflammation evoked by carrageenan or Escherichia coli septicemia (El Kebir et al., 2009). LXA4 exerts its anti-inflammatory effects through binding to formyl-peptide receptor-like (FPRL1), also called formyl peptide receptor 2/lipoxin A4 receptor (FPR2/ALX; receptor nomenclature follows Alexander et al., 2009), which is a G protein-coupled receptor that is primarily expressed on neutrophils and monocytes (Chiang et al., 2006). A recent study has demonstrated that synovial fluid and tissues obtained from RA patients had elevated levels of LXA4 and FPR2/ALX receptor expression, respectively (Hashimoto et al., 2007).
In this study, we investigated the effect of LXA4 treatment on zymosan-induced articular inflammation by evaluating oedema formation, leucocyte influx and the production of pro-inflammatory mediators. In addition, because ET-1 plays a pivotal role in articular inflammation by inducing the production of cytokines, chemokines and LTs, we also investigated the ability of LXA4 to impair ET-1-triggered knee joint inflammation and neutrophil activation in vitro.
Methods
Animals
All animal care and experimental procedures performed were approved by the institution's Ethical Committee for Animal Care and Use (CEUA, Fiocruz, Rio de Janeiro, Brazil). Male C57BL/6 mice (20–25 g) were obtained from the Oswaldo Cruz Foundation Breeding Unit (Fiocruz). The mice were kept in plastic cages with free access to food and fresh water in a room with a controlled temperature (22–24°C) and light cycle (12 h light/dark) at the experimental animal facility until use.
Induction of joint inflammation
Joint inflammation was induced by intra-articular (i.art.) injection of zymosan (500 µg per cavity in 25 µL sterile saline) or ET-1 (10 pmol per cavity) by inserting a 27.5 G needle through the suprapatellar ligament into the left knee joint cavity, as previously described (Penido et al., 2006; Conte et al., 2008). Control animals received an i.art. injection of an equal volume of sterile saline.
Pretreatments
LXA4 was administered i.art. in doses ranging from 1 to 20 ng per cavity, in a final volume of 25 µL sterile saline, 60 min before i.art. zymosan stimulation. BML-111, an LXA4 receptor agonist, was dissolved in sterile saline and administered i.art. at 200 ng per cavity 60 min before zymosan stimulation in a final volume of 25 µL. BOC-1, an antagonist of the G protein-coupled LXA4 receptor, was administered i.art. 90 min before articular stimulation at 20 ng per cavity in a final volume of 25 µL. Acetylsalicylic acid (ASA; 300 mg·kg−1) was diluted in 0.5% carboxymethylcellulose, and given orally (p.o.) 30 min before i.art. zymosan stimulation. Indomethacin (3 mg·kg−1) was diluted in a 5% bicarbonate solution, and given orally 30 min before zymosan stimulation. Respective control groups were injected with the same volume of sterile saline.
Post-treatments
In this set of experiments, mice were treated with LXA4, BML-111 and ASA 1 and 6 h after zymosan i.art. injection. LXA4 and BML-111 were administered i.p. at 5 µg·kg−1 in a final volume of 100 µL of sterile saline. ASA (300 mg·kg−1) was diluted in 0.5% carboxymethylcellulose and was given orally (p.o.) in a final volume of 100 µL. Evaluations of oedema formation, total and differential counts were performed 10 h after zymosan stimulation. The control group was injected i.p. with the same volume of sterile saline.
Measurement of knee joint swelling
Knee joint swelling was evaluated by measurement of the transverse diameters of the left knee joints using a digital caliper (Digmatic Caliper, Mitutoyo Corporation, Kanagawa, Japan). Values of knee joint thickness are expressed as the difference (Δ) between the diameter measured before (basal) and after induction of articular inflammation in millimetres (mm).
Collection of synovial fluid and leucocyte counts
Mice were killed by an excess of CO2 at 6 h after i.art. injection of zymosan. Knee synovial cavities were washed with 300 µL of phosphate-buffered saline (PBS) containing EDTA (10 mM) by inserting a 21 G needle into the mouse knee joint, and the synovial washes were recovered by aspiration. Total leucocyte counts were performed in a Neubauer chamber under an optical microscope, after dilution in Turk's fluid (2% acetic acid). Differential neutrophil counts were performed using May–Grunwald–Giemsa-stained cytospins (1000× magnification) (Cytospin 3, Shandon Inc., Pittsburgh, PA, USA), and values are expressed as numbers of cells per cavity (×105). After cellular counts, the synovial washes were centrifuged at 416×g for 10 min at 4°C, and the supernatant was stored at −80°C for further analysis.
Paw oedema
Mice were injected intraplantarly (i.pl.) with ET-1 (30 pmol per paw) into the right hind paw, following a previously described protocol (Henriques et al., 1987). The contralateral paw was injected with the same volume of sterile PBS and was used as a control. All inflammatory agents were dissolved in sterile PBS before injection. Paw oedema was measured at 60 min after stimuli using digital plethysmometer (Ugo Basile, Comerio, Italy). LXA4 (10 ng per paw) was diluted in sterile PBS and concomitantly i.pl. administered with the inflammatory mediators as described by Menezes-de-Lima et al. (2006). The final volume injected into the paw (summing agonists and treatments) was never higher than 50 µL. Results are expressed in microlitres, and the difference (Δ) between the right and left paws was taken as oedema volume.
elisa
Levels of TNF-α and KC/CXCL1 in the knee joint washes were evaluated by sandwich elisa using matched antibody pairs (Quantikine, R&D Systems, Minneapolis, MN, USA) according to the manufacturer's instructions.
Enzymic immuno-assay for LTB4 and LXA4
LTB4 and LXA4 levels were evaluated in cell-free synovial washes recovered from zymosan-stimulated C57BL/6 mice after zymosan (500 µg per cavity) stimulation. LTB4 was assayed by immunosorbent assay (EIA) according to the manufacturer's protocol (Cayman Chemical, Ann Arbor, MI, USA). In the specific case of LXA4, samples were extracted with C18 Sep-Pak cartridges (Waters, Milford, MA, USA) pre-activated with methanol and deionized water. The column was washed with deionized water followed by petroleum ether. Materials in the ethyl formate eluate were dried under a stream of N2, and immediately measured by EIA (Neogen, Lexington, KY, USA) according to the manufacturer's instructions.
Real-time RT–PCR
Quantitative PCR (QPCR) was performed as previously described (Verri et al., 2008). Briefly, mice were killed by excessive CO2 inhalation, 2 h after i.art. injection of zymosan (500 µg per cavity), and knee joint complexes were harvested. Samples were homogenized in Trizol reagent, and total RNA was extracted using the SV Total RNA Isolation System (Promega Biosciences, San Luis Obispo, CA, USA). QPCR was performed in an ABI Prism 7000 Sequence Detection System using SYBR-green fluorescence (Applied Biosystems, Wilmington, NC, USA). The following primers were used: preproET-1, sense: 5′-TGT GTC TAC TTC TGC CAC CT-3′, antisense: 5′-CAC CAG CTG CTG ATA GAT AC-3′; β-actin, sense: 5′-AGC TGC GTT TTA CAC CCT TT-3′, anti-sense: 5′-AAG CCA TGC CAA TGT TGT CT-3′. The expression of β-actin mRNA was used as control for tissue integrity in all samples.
Chemotaxis assay
Neutrophils isolated from C57BL/6 mouse bone marrow were purified by Percoll discontinuous gradients, washed and resuspended in RPMI 1640 supplemented with 10% FBS and then assayed in a 48-well microchemotaxis Boyden chamber (Neuroprobe Inc., Cabin John, MD, USA). The bottom wells of the chamber were filled with 28 µL of a chemo-attractant stimulus, KC (10 nM), ET-1 (1–1000 nM) or RPMI 1640 (control), whereas the upper wells were filled with neutrophils (105 cells; 50 µL) that had been previously incubated with BOC-1 (1 nM) (or medium) for 30 min before LXA4 (1–100 nM, 60 min), followed by ET-1 (100 nM) stimulation for 60 min (37°C, 5%, CO2). The bottom and upper cells were separated with a 3 µm polycarbonate filter (Nuclepore, Pleasanton, CA, USA). The chamber was incubated in humidified air with a 5% CO2 atmosphere at 37°C for 60 min. Cells that migrated completely through the filter were counted under light microscopy (×1000). Neutrophil chemotaxis was calculated and expressed as the mean number of migrated cells in five random high-power fields per well (in quadruplicate).
Shape change assay
Human peripheral polymorphonuclear leucocytes (PMNLs) were obtained by venepuncture, from male adult healthy volunteers (25–30 years; with full consent), who had taken no systemic medication for at least 72 h before donating blood. About 20 mL of venous blood was taken into 2 mL of sodium citrate anticoagulant (3.8% w/v solution), and neutrophils were isolated and purified by dextran sedimentation followed by Percoll discontinuous gradients, as previously described (Penido et al., 2001). Aliquots of 5 × 105 neutrophils (∼90–95% final purity) were incubated in the presence or absence of ET-1 (0.3–10 nM) or KC (0.1 nM) in a 37°C shaking water bath for 6 min, after which the reaction was stopped by the addition of a paraformaldehyde solution (0.25%) at 4°C and placed on ice until analysis. To evaluate the LXA4 (1–100 nM) -induced shape change, samples were pretreated for 15 min (37°C) before stimulation. Samples were immediately analysed on a FACSCalibur flow cytometer (Becton Dickinson, San Jose, CA, USA). Acquisition was set using the FL-2 fluorescence channel, through which human neutrophils can be distinguished from eosinophils by means of their auto-fluorescence characteristics. Forward scatter (FSC-H), side scatter (SSC-H) and FL-2 data were saved. Five hundred neutrophils were acquired for each of the triplicate samples. As measurement of shape change, data are reported as the change in FSC-H compared with buffer-treated cells.
Statistical analysis
Results are reported as the mean ± SEM, and were statistically analysed by means of anova followed by Newman–Keuls–Student test or Student's t-test. Values of P≤ 0.05 were regarded as significant.
Materials
PBS, Tween-20, o-phenylenediamine dihydrochloride, EDTA sodium salt, Hank's balanced salt solution, BSA, ET-1 and ASA were purchased from Sigma Chemical Co. (St Louis, MO, USA). TNF-α and KC/CXCL1 matched antibody pairs were obtained from R&D Systems. LXA4[5(S),6(R),15(S)-trihydroxyeicosa-7-trans-9-trans-11-cis-13-transtetraenoic acid] and BML-111 [5(S),6(R),7-trihydroxyheptanoic acid methyl ester] were purchased from Cayman Chemical Company. BOC-1 (N-tert-butoxy-carbonyl-methionyl-leucyl-phenylalanine) was purchased from MP Biomedicals (Solon, OH, USA).
Results
Pretreatment with LXA4 and the lipoxin receptor agonist BML-111 reduced zymosan-induced articular inflammatory response
Previous reports from our group have demonstrated that i.art. injection of zymosan induced an articular inflammatory response within 6 h, characterized by a significant increase in oedema formation and massive neutrophil influx (approximately 90% of total leucocytes) (Penido et al., 2006; Conte et al., 2008). We examined the anti-inflammatory effects of LXA4 in this murine model of articular inflammation, and observed that the i.art. pretreatment of mice with all doses of LXA4 (1, 10 and 20 ng per cavity) clearly reduced zymosan-induced oedema formation at 6 h, achieving a 74% inhibition at 20 ng per cavity (Figure 1A). Interestingly, LXA4 inhibited zymosan-induced total leucocyte influx (Figure 1B) only at the highest dose, mainly due to the inhibition of neutrophil influx (60% inhibition) as measured by synovial washes at 6 h (Figure 1C).
Figure 1.

LXA4 inhibits zymosan-induced oedema formation and neutrophil migration through FPR2/ALX receptors. Pretreatment with exogenous LXA4 (1–20 ng per cavity; i.art.) 60 min before zymosan (500 µg per cavity; i.art.) stimulation reduced zymosan-induced oedema formation (A), total leucocyte (B) and neutrophil influx (C) in the knee joint cavity of C57BL/6 mice. In another set of experiments, treatment with BOC-1 (20 ng per cavity; i.art., 90 min prior to stimulation) and exogenous LXA4 (20 ng per cavity; i.art., 60 min prior to stimulation) before i.art. injection of zymosan (500 µg per cavity) prevented the anti-inflammatory actions of LXA4 on zymosan-induced oedema formation (D), total leucocyte (E) and neutrophil influx (F). Respective control groups were injected with the same volume of sterile saline. Knee joint diameter was evaluated with a digital caliper, and knee synovial cells were recovered 6 h after zymosan stimulation. Results are expressed as the mean ± SEM from at least six animals per group. *P≤ 0.05, significant differences between stimulated (zymosan) and non-stimulated groups; +P≤ 0.05, significant differences between treated (BOC-1) and untreated groups.
In order to address the involvement of the FPR2/ALX receptor in the therapeutic effects of exogenous lipoxins in this model, mice received the FPR2/ALX receptor antagonist BOC-1 (20 ng per cavity; i.art.), 30 min prior to LXA4 treatment (20 ng per cavity; i.art.). As expected, BOC-1 reversed the therapeutic effects of LXA4 (20 ng per cavity) on zymosan-induced oedema formation (Figure 1D), total leucocyte (Figure 1E) and neutrophil (Figure 1F) accumulation in synovial washes at 6 h. To confirm the participation of the FPR2/ALX receptor in the anti-inflammatory effects mediated by LXA4 in zymosan-induced articular inflammation, C57BL/6 mice were pretreated with a LXA4 receptor agonist, BML-111 (200 ng per cavity; i.art.). This pretreatment significantly inhibited zymosan-induced oedema formation (Figure 2A), total leucocyte (Figure 2B) and neutrophil influx (Figure 2C) into the knee joint cavity.
Figure 2.
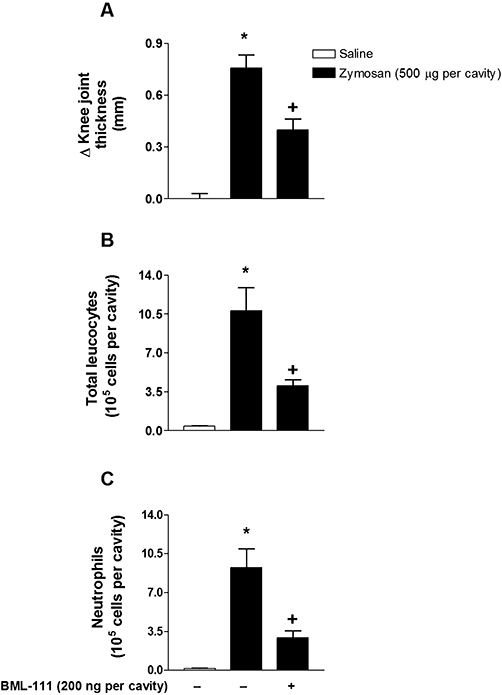
BML-111 inhibits 6 h zymosan-induced oedema formation and neutrophil migration. Pretreatment with BML-111 (200 ng per cavity; i.art.) 60 min before i.art. injection of zymosan (500 µg per cavity) inhibited zymosan-induced oedema formation (A), total leucocyte (B) and neutrophil influx (C) into the knee joint cavity of C57BL/6 mice. The control group was injected with the same volume of sterile saline. Knee joint diameter was evaluated with a digital caliper, and knee synovial cells were recovered 6 h after zymosan stimulation. Results are expressed as the mean ± SEM from at least six animals per group. *P≤ 0.05, significant differences between stimulated (zymosan) and non-stimulated groups; +P≤ 0.05, significant differences between treated (BML-111) and untreated groups.
ASA pretreatment reduced articular zymosan-induced inflammation
In another set of experiments, we addressed the effects of ASA, a non-steroidal anti-inflammatory drug that produces endogenous aspirin-triggered lipoxins (ATLs), during the zymosan-induced knee articular inflammatory response. As demonstrated in Figure 3, ASA (300 mg·kg−1; p.o.) treatment clearly reduced zymosan-induced oedema formation, total leucocyte and neutrophil (Figure 3A–C) influx into the knee joint cavity of C57BL/6 mice within 6 h compared to the zymosan-stimulated group (50, 68 and 68% inhibition respectively). Interestingly, BOC-1 (20 ng per cavity; i.art.) significantly reversed the anti-inflammatory effects of ASA on zymosan-induced total leucocyte (Figure 3B) and neutrophil (Figure 3C) influx, whereas it did not alter zymosan-induced articular oedema formation (Figure 3A). In addition, pretreatment with BOC-1 (20 ng per cavity; i.art.) alone did not alter zymosan induced-oedema formation and cellular influx. Moreover, as demonstrated in Figure 3D, the i.art. injection of zymosan failed to induce endogenous production of lipoxin within the first 6 h, in contrast with the significant increase in lipoxin production observed at later time-points. The increased lipoxin production at a later time-point (72 h) was accompanied by a significant decrease in zymosan-induced neutrophil influx (at 24 h, 10.75 ± 2.26; at 72 h, 1.42 ± 0.67 × 105 neutrophils per cavity; n = 6, P≤ 0.05) and of knee joint swelling (at 24 h, 0.92 ± 0.22; at 72 h, 0.30 ± 0.07 Δ mm; n = 6, anova, P≤ 0.05).
Figure 3.
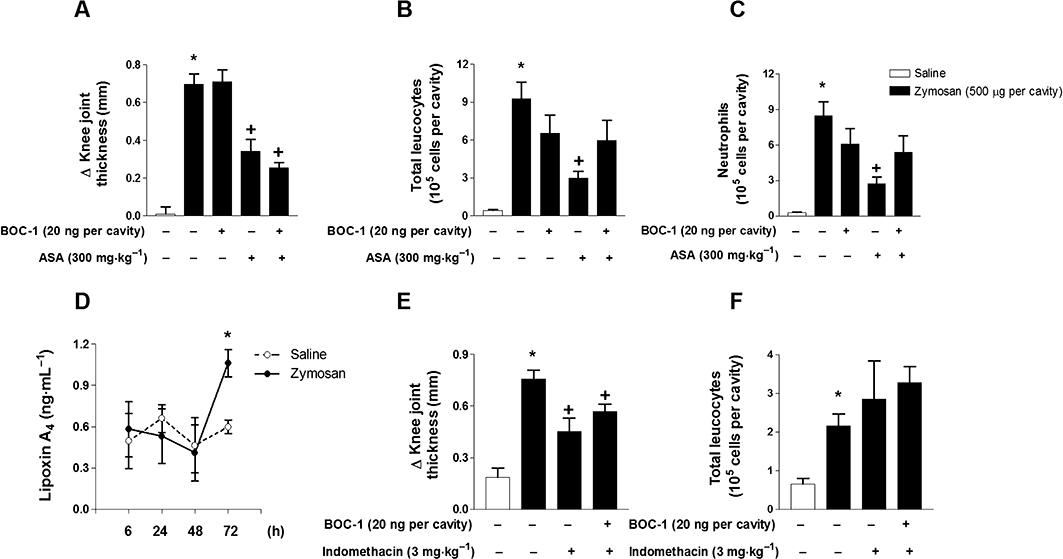
ASA significantly reduces articular neutrophil influx and oedema formation induced by zymosan. Pretreatments with BOC-1 (20 ng per cavity; i.art.) and ASA (300 mg per cavity; p.o.) were performed 60 and 30 min, respectively, prior to zymosan (500 µg per cavity; i.art.) stimulation. BOC-1 pretreatment blocked the inhibitory actions of ASA on zymosan-induced oedema formation (A), total leucocyte (B) and neutrophil influx (C) into the knee joint cavity of C57BL/6 mice. Zymosan-induced LXA4 levels in knee synovial washes (D) were determined by EIA 6, 24, 48 and 72 h after stimulation. In another set of experiments, C57BL/6 mice were pretreated with BOC-1 (20 ng per cavity; i.art.) and indomethacin (3 mg·kg−1; p.o.) 60 and 30 min, respectively, prior to zymosan (500 µg per cavity; i.art.) stimulation. BOC-1 did not reverse the effects of indomethacin pretreatment on zymosan-induced oedema formation (E) and total leucocyte influx (F) within 6 h after stimulation. The control group was injected with the same volume of sterile saline. Knee joint diameter was evaluated with a digital caliper, and knee synovial cells were recovered 6 h after zymosan stimulation. Results are expressed as the mean ± SEM from at least six animals per group. *P≤ 0.05, significant differences between stimulated (zymosan) and non-stimulated groups; +P≤ 0.05, significant differences between treated (BOC and ASA) and untreated groups.
Confirming the participation of ATLs acting through FPR2/ALX receptors on zymosan-induced arthritis, treatment of mice with indomethacin (3 mg·kg−1), a non-steroidal anti-inflammatory drug that does not trigger ATL formation, led to a significant reduction of zymosan-induced oedema formation (Figure 3E), whereas no alteration was observed on the total leucocyte influx (Figure 3F). Furthermore, no significant change was observed when BOC-1 (200 ng per cavity) was co-administered to mice that received zymosan or indomethacin (3 mg·kg−1, i.p.).
LXA4 pretreatment reduced the levels of pro-inflammatory mediators in articular zymosan-injected knee joints
To investigate if LXA4 exerts its anti-inflammatory effects by modulating the production of inflammatory mediators, we analysed its effects on the expression levels of the ET-1 peptide precursor, preproET-1, mRNA, and the production of KC/CXCL1, LTB4 and TNF-α. As demonstrated in Figure 4, preproET-1 mRNA expression in articular tissue, as well as TNF-α, KC/CXCL1 and LTB4 production in synovial washes, was increased after i.art. zymosan injection. In situ pretreatment with LXA4 markedly reduced zymosan-induced preproET-1 mRNA expression, as well as TNF-α, KC/CXCL1 and LTB4 levels (Figure 4A–D).
Figure 4.
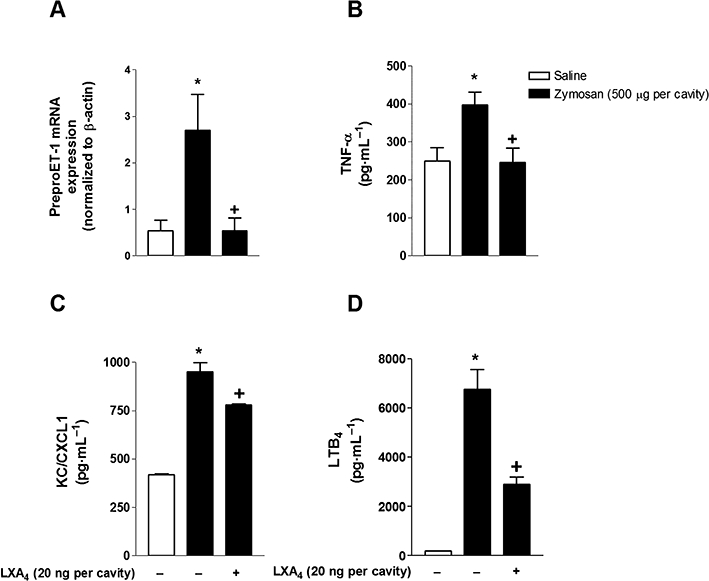
LXA4 inhibits the zymosan-induced increase of preproET-1 mRNA expression and KC/CXCL1, LTB4 and TNF-α production. Mice were treated with LXA4 (20 ng per cavity) 1 h before the i.art. injection of zymosan (500 µg per cavity). After 2 h of zymosan stimulation, samples of the knee joints were collected for real-time PCR analysis (A) (n = 5 mice per group, each sample was analysed in triplicate). TNF-α (B), KC/CXCL1 (C) and LTB4 (D) levels in knee synovial washes were determined 6 h after i.art. injection of zymosan (500 µg per cavity). Results are expressed as the mean ± SEM from at least six animals per group. *P≤ 0.05, significant differences between stimulated (zymosan) and non-stimulated groups; +P≤ 0.05, significant differences between treated (LXA4) and untreated groups.
LXA4 impaired human neutrophil activation and chemotaxis in vitro
Our previous results showed that ET-1 triggered neutrophil accumulation in vivo (Conte et al., 2008), and data from another group demonstrated ETA/B receptor expression by human neutrophils (Mencarelli et al., 2009). Together, these data led us to assess the effects of LXA4 on ET-1-induced neutrophil activation and migration in vitro. As shown in Figure 5, ET-1 (100 nM) induced a significant increase in neutrophil shape change that was blocked by pretreatment with LXA4, as demonstrated in the representative dot plots of side scatter (SSC-H) versus forward scatter (FSC-H). As a control for the assays, possible cytotoxic effects were evaluated. ET-1 (1 and 100 nM) or LXA4 (10 and 100 nM) pretreatment did not induce neutrophil cell death (data not shown). As shown in Figure 6A, ET-1 (1–1000 nM) increased the number of migrating neutrophils compared to basal migration in control wells. In addition, it is noteworthy that ET-1 (100 and 1000 nM) induced higher levels of neutrophil transmigration than KC (10 µM), a rodent chemokine used as positive control. LXA4 (1–100 nM) in vitro pretreatment, 60 min prior to ET-1 (100 nM) stimulation, blocked neutrophil migration in all doses used (Figure 6B). Confirming the in vitro participation of FPR2/ALX receptor in LXA4 anti-inflammatory effects on ET-1-induced neutrophil migration, BOC-1 (30 min before LXA4 pretreatment) inhibited neutrophil migration induced by ET-1 (Figure 6C).
Figure 5.

LXA4 blocks ET-1-induced human neutrophil shape change in vitro. Human polymorphonuclear leucocytes purified from peripheral venous blood were pretreated in vitro with LXA4 (1–100 nM) 15 min before ET-1(100 nM)-induced neutrophil shape change. Representative dot plots of FSC-H versus SSC-H of human neutrophils exposed to LXA4 pretreatment 15 min before the ET-1-induced increase in forward scatter characteristics. Control groups were treated with RPMI under the same conditions. Results are expressed as mean of FSC-H, and data represent the mean ± S.E.M. from at least quadruplicate wells per group of two separate experiments. *P≤ 0.05, significant differences between stimulated (ET-1) and non-stimulated groups; +P≤ 0.05, significant differences between treated (LXA4) and untreated groups.
Figure 6.
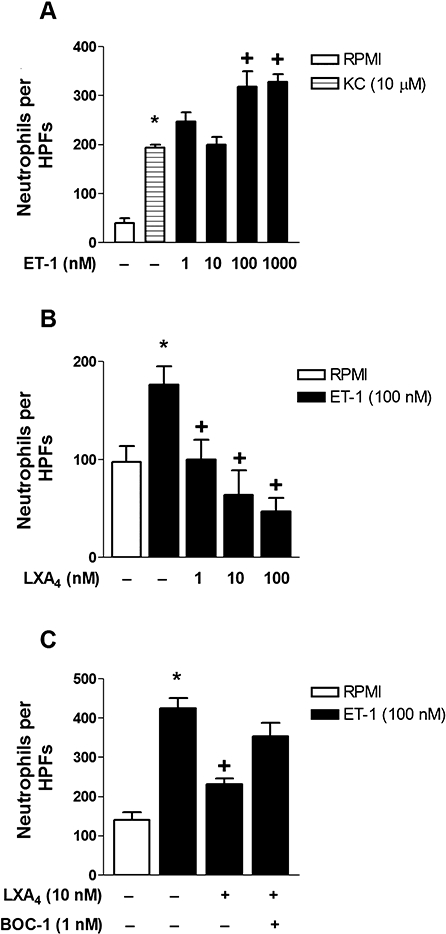
LXA4 inhibits ET-1-induced murine neutrophil chemotaxis. In vitro stimulation with KC (10 µM) or ET-1 (1–1000 nM), for 60 min at 37°C, induced murine bone marrow-derived neutrophil chemotaxis (A). In vitro pretreatment with LXA4 (1–100 nM; 60 min prior to stimulation) inhibited ET-1(100 nM)-induced chemotaxis at all doses used (B). BOC-1 (1 nM; 90 min prior to stimulation) pretreatment reversed LXA4 inhibition of neutrophil chemotaxis induced by ET-1 (C). Control groups were treated with RPMI under the same conditions. Results are expressed as numbers of neutrophils per high-power fields (HPFs) per well. Data represent the mean ± SEM from at least quadruplicate wells per group of one out of two separate experiments. *P≤ 0.05, significant differences between stimulated (ET-1, KC) and non-stimulated groups; +P≤ 0.05, significant differences between treated (LXA4, BOC-1) and untreated groups; anova followed by a Newman–Keuls–Student test.
LXA4 inhibited ET-1-induced articular and paw inflammation
According to our previous results (Conte et al., 2008), ET-1 (10 pmol per cavity; i.art.) injection induced a significant articular inflammatory response that was characterized by marked articular oedema formation and neutrophil accumulation in synovial washes. LXA4 (20 ng per cavity) pretreatment significantly reduced ET-1-induced oedema formation (Figure 7A), and corroborating previous published data (Kassuya et al., 2008), we also observed that i.pl. injection of ET-1 (30 pmol per paw) induced marked increase in paw oedema 1 h after stimulation that was significantly inhibited by LXA4 (20 ng per paw; i.pl.) co-administration (inset). Moreover, LXA4 (20 ng per cavity) pretreatment significantly decreased ET-1-induced total leucocyte accumulation (Figure 7B), which was mainly due to reduced neutrophil influx (Figure 7C) and TNF-α production (Figure 7D).
Figure 7.
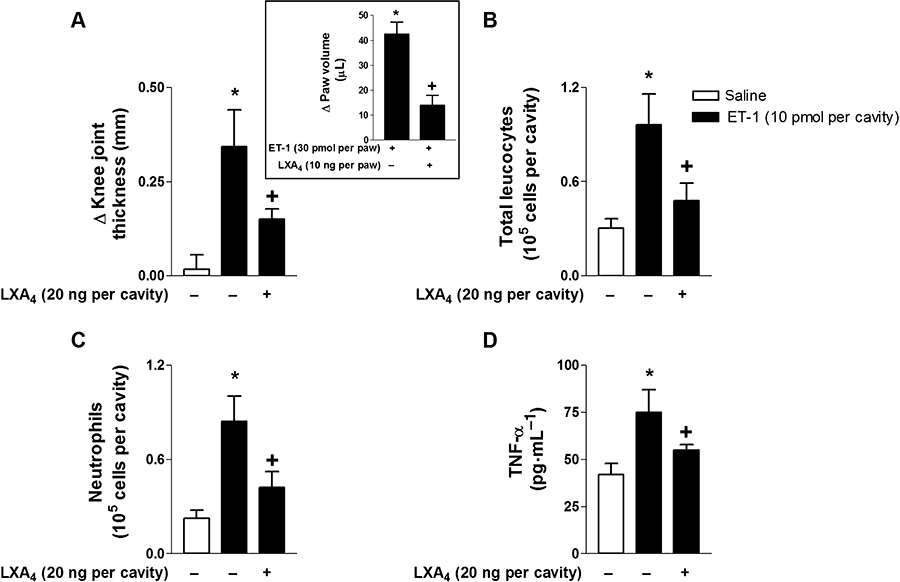
ET-1-induced articular and paw inflammation are impaired by LXA4 pretreatment. LXA4 (20 ng per cavity; i.art.) pretreatment 60 min prior to ET-1 (10 pmol per cavity; i.art.) stimulation reduced ET-1-induced oedema formation (A), total leucocyte (B) and neutrophil influx (C) into the knee joint cavity of C57BL/6 mice. Respective control groups were injected with the same volume of sterile saline. Knee joint diameter was evaluated with a digital caliper, and knee synovial cells were recovered 6 h after ET-1 stimulation. The inset shows that paw oedema induced by ET-1 (30 pmol per paw; i.pl.) was inhibited by LXA4 (20 ng per paw; i.pl.) pretreatment. Values represent the differences of volume (in microlitres) between ET-1- and vehicle-injected paws 60 min after stimulation. Results are expressed as the mean ± SEM from at least five animals per group. *P≤ 0.05, significant differences between stimulated (ET-1) and non-stimulated groups; +P≤ 0.05, significant differences between treated (LXA4) and untreated groups; anova followed by Newman–Keuls–Student test (A–D) or Student's t-test (inset).
LXA4 post-treatment reduced zymosan-induced articular inflammatory response
Corroborating the results obtained with experiments using pretreated mice, post-treatments with LXA4 (5 µg·kg−1; i.p), BML-111 (5 µg·kg−1; i.p) and ASA (300 mg·kg−1; p.o.) also reduced oedema formation (Figure 8A), total leucocyte (Figure 8B) and neutrophil influx (Figure 8C) to zymosan-stimulated joints at 10 h.
Figure 8.

LXA4, BML-111 and ASA post-treatments inhibited articular inflammation induced by zymosan stimulation. In vivo administration of LXA4 (5 µg·kg−1; i.p.), BML-111 (5 µg·kg−1; i.p.) or ASA (300 mg·kg−1; p.o.) 1 and 6 h following i.art. injection of zymosan (500 µg per cavity; i.art.) reduced oedema formation (A), total leucocyte (B) and neutrophil influx (C) into the knee joint cavity of C57BL/6 mice. The control group received the same volume of sterile saline. Knee joint diameter and knee synovial cell accumulation were analysed 10 h after zymosan stimulation. Results are expressed as the mean ± SEM from at least six animals per group. *P≤ 0.05, significant differences between stimulated (zymosan) and non-stimulated groups; +P≤ 0.05, significant differences between treated (LXA4, BML-111, ASA) and untreated groups; anova followed by Newman–Keuls–Student test.
Discussion
In the present study, we demonstrated that LXA4 exerted an anti-inflammatory effect on zymosan-induced arthritis through a mechanism that involved activation of FPR2/ALX receptors. We have also shown for the first time that the LXA4 anti-inflammatory effect on articular inflammation involves the blockade of ET-1 production and action.
Several human arthropathies, including RA, are characterized by significant joint oedema, movement-induced joint hyperalgesia and the infiltration of inflammatory cells (Kraan et al., 2000). In this regard, the role of neutrophils in the pathogenesis of tissue lesions in arthritis has long been recognized (Jonsson et al., 2005). Previous work from our group has demonstrated that i.art. administration of zymosan into mouse knee joints induced marked acute oedema formation, hypernociception and significant leucocyte influx, mainly due to neutrophil accumulation in knee synovial washes (Penido et al., 2006; Conte et al., 2008; Guerrero et al., 2008), which correspond to features of human RA articular inflammation. Moreover, in accordance with RA clinical data, this murine model was also characterized by a significant increase of important pro-inflammatory mediators, such as ETs, TNF-α and LTB4, that have pivotal roles in articular oedema formation and leucocyte accumulation (Conte et al., 2008).
Among the mediators chronically produced in the joints of RA patients, LXA4 and ATL have been shown to be increased when compared to osteoarthritis, a phenomenon that occurs in parallel to increased expression of FPR2/ALX receptors in the RA synovium (Hashimoto et al., 2007). Interestingly, LXA4 and ATLs are described as endogenous ‘stop signals’ that regulate chronic leucocyte trafficking and neutrophil activity, and promote resolution of the inflammatory process (Takano et al., 1997). Such effects have been demonstrated to be mediated by FPR2/ALX receptors (Takano et al., 1997; Devchand et al., 2003; Chiang et al., 2006) in several inflammatory models (McMahon and Godson, 2004), and, more recently, experimental evidence suggests a protective role for 12/15-lipoxygenase products (including LXA4) in experimental models of arthritis (Kronke et al., 2009). It is noteworthy that we observed, in an experimental model of zymosan-induced arthritis, an increase in LXA4 production at later time-points, which was accompanied by a significant amelioration of articular inflammation. This is additional evidence of the anti-inflammatory and pro-resolution actions of LXA4. Moreover, Zhang et al. (2008) have reported the therapeutic effects of BML-111 (FPR2/ALX receptor agonist) in collagen-induced arthritis. In line with these reports, the present study clearly demonstrates that LXA4, as well as BML-111, markedly reduced articular zymosan-induced neutrophil accumulation through an FPR2/ALX receptor-operated mechanism, because it is reversed by BOC-1 treatment. It is noteworthy that, in mice, BOC-1 antagonizes both FPR2/ALX and FPR1 receptors; however, in this species, LXA4 has also been shown to bind both of these receptors (Takano et al., 1997; Vaughn et al., 2002; Ye et al., 2009).
The finding that ASA treatment markedly reduced zymosan-induced total leucocyte influx, mainly due to decreased neutrophil trafficking into the knee synovium, suggests that ASA treatment leads to endogenous ATL production in vivo that triggers FPR2/ALX receptor-orchestrated mechanisms, culminating in reduced leucocyte trafficking. This conclusion is supported by the reversal of ASA anti-inflammatory effects on zymosan-induced leucocyte influx induced by BOC-1 (FPR2/ALX receptor antagonist), as well as by the lack of effects of indomethacin on zymosan-induced total leucocyte accumulation in knee synovial washes, in the presence or absence of BOC-1. Together, these results support the proposition that ASA treatment induced ATL production, a phenomenon that is not triggered by other NSAIDs.
Previous reports demonstrated that LXA4 and its analogues inhibited plasma leakage in parallel to the impairment of granulocyte migration (Bandeira-Melo et al., 2000; Jin et al., 2007). The effects of lipoxin on plasma leakage have commonly been considered an indirect consequence of the reduction of neutrophil adhesion and transmigration induced by such lipid mediators (Bandeira-Melo et al., 2000). However, subsequent investigation has revealed that the potent anti-oedematogenic effect of LXA4 occurred independently of leucocyte accumulation, which suggested that LXA4 might directly affect endothelial cells (Menezes-de-Lima et al., 2006). Interestingly, our results suggest that LXA4 inhibits articular plasma leakage independently of neutrophil migration, because LXA4 significantly inhibited zymosan-induced oedema formation in all doses used, but did not reduce articular neutrophil influx at 1 or 10 ng per cavity. In addition, supporting this notion, we demonstrated that acute paw oedema induced by ET-1 was significantly reduced by LXA4. Moreover, our data show that the anti-oedematogenic effects of ASA seem to be related to the blockade of cyclooxygenase and arachidonic acid derivatives, rather than by ATL binding to FPR2/ALX receptors. This possibility was supported by the fact that the anti-oedematogenic effects of ASA and indomethacin were not altered by BOC-1 pretreatment. In line with this hypothesis, LXA4 and ATL also directly interacted with vascular CysLT1 receptors, functioning as antagonists (Gronert et al., 2001), in addition to activating FPR2/ALX receptors on leucocytes. This finding could partially explain why BOC-1 did not affect the inhibition by LXA4 of vascular leakage. In addition, other LXA4-independent enzymic pathways have been described to be involved in the anti-oedematogenic effects of ASA. These effects can be triggered by other lipid mediators capable of controlling the resolution of inflammatory response, such as resolvins and protectins, and remain to be investigated (Yacoubian and Serhan, 2007).
Overall, the mechanisms involved in LXA4 anti-inflammatory effects seem to depend on the blockade of articular pro-inflammatory mediators, including ET-1, TNF-α, CXCL1 and LTB4. Indeed, we have previously demonstrated that ETs play a crucial pro-inflammatory role in articular inflammation, modulating these mediators (Conte et al., 2008). We additionally demonstrated for the first time that ET-1-induced articular inflammation was prevented by LXA4 through a mechanism that involves the impairment of TNF-α production. In accordance with this, TNF-α-based therapies are currently clinically employed to treat RA patients. However, some RA patients do not respond successfully to these treatments. New therapeutic targets for the treatment of RA include LTB4 and its receptors (Mathis et al., 2007), supporting our data that the beneficial effects of LXA4 also involve the impairment of LTB4 production in parallel with reduced CXCL1 levels and acute neutrophil accumulation. Moreover, in contrast to current TNF-targeting therapies used in RA (soluble receptors and anti-TNF antibodies), LXA4 inhibits the production of TNF-α and other mediators. These data therefore suggest that LXA4 treatment might present additive effects to current anti-TNF-α therapies or at least allow for the use of lower doses of anti-TNF therapies, if used in association with LXA4, to achieve better clinical outcomes. LXA4 decreased in vitro neutrophil chemotaxis, adherence and transmigration across the vascular endothelium and epithelium (McMahon and Godson, 2004; Filep et al., 2005), CD11b/CD18 expression (Filep et al., 1999) and blocked superoxide generation (Levy et al., 1999) and TNF-α production and actions (Hachicha et al., 1999). Previous in vitro data demonstrate that ETs cause direct neutrophil activation (Toffoli et al., 2007), adhesion (Zouki et al., 1999) and migration in vitro (Elferink and de Koster, 1994). Interestingly, in the present study, we showed that LXA4 seemed to directly impair ET-1-induced neutrophil activation and migration through a mechanism that needs further elucidation. Fierro et al. (2003) reported that the exposure of PMNLs to LXA4 and 15-epi-LXA4 markedly decreased LTB4-induced transmigration across both human microvessel endothelial and epithelial cells, reinforcing the notion that lipoxins act directly on neutrophils, impairing their activation and transmigration. It is interesting to note that post-treatment with LXA4 also reduced the established articular inflammation, suggesting that this lipid mediator might be useful as a putative new anti-inflammatory drug.
Taken together, these results demonstrated a significant anti-inflammatory effect of LXA4, acting via FPR2/ALX receptors, in a murine model of articular inflammation. Additionally, our data contribute to the understanding of the anti-inflammatory mechanisms mediated by LXA4 that involve the blockade of ET-1 pro-inflammatory production and effects, and highlight the importance of ETs in the context of articular inflammation. Moreover, our data indicate that LXA4, ATLs and other FPR2/ALX receptor agonists can represent promising therapeutic tools for the treatment of RA and other articular inflammatory diseases.
Acknowledgments
This work was supported by grants from Conselho Nacional de Pesquisa (CNPq), Brazil and Coordenação de Aperfeicoamento de Pessoal de Nível Superior (CAPES), Brazil. F.P.C. is a student in the Post-Graduation Program in Cellular and Molecular Biology of Oswaldo Cruz Institute and a fellow of CNPq.
Glossary
Abbreviations
- ASA
acetylsalicylic acid
- ATL
aspirin-triggered lipoxin
- ET
endothelin
- FPR2/ALX receptor
formyl peptide receptor 2/lipoxin A4 receptor
- i.art.
intra-articular
- i.pl.
intraplantar
- LT
leukotriene
- LXA4
lipoxin A4
- RA
rheumatoid arthritis
Conflict of interest
There is no conflict of interest to declare.
Supplemental material
References
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 4th edn. Br J Pharmacol. 2009;158(Suppl 1):S1–S254. doi: 10.1111/j.1476-5381.2009.00499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandeira-Melo C, Serra MF, Diaz BL, Cordeiro RS, Silva PM, Lenzi HL, et al. Cyclooxygenase-2-derived prostaglandin E2 and lipoxin A4 accelerate resolution of allergic edema in Angiostrongylus costaricensis-infected rats: relationship with concurrent eosinophilia. J Immunol. 2000;164:1029–1036. doi: 10.4049/jimmunol.164.2.1029. [DOI] [PubMed] [Google Scholar]
- Chiang N, Fierro IM, Gronert K, Serhan CN. Activation of lipoxin A(4) receptors by aspirin-triggered lipoxins and select peptides evokes ligand-specific responses in inflammation. J Exp Med. 2000;191:1197–1208. doi: 10.1084/jem.191.7.1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang N, Serhan CN, Dahlen SE, Drazen JM, Hay DW, Rovati GE, et al. The lipoxin receptor ALX: potent ligand-specific and stereoselective actions in vivo. Pharmacol Rev. 2006;58:463–487. doi: 10.1124/pr.58.3.4. [DOI] [PubMed] [Google Scholar]
- Conte FP, Barja-Fidalgo C, Verri WA, Jr, Cunha FQ, Rae GA, Penido C, et al. Endothelins modulate inflammatory reaction in zymosan-induced arthritis: participation of LTB4, TNF-α, and CXCL-1. J Leukoc Biol. 2008;84:652–660. doi: 10.1189/jlb.1207827. [DOI] [PubMed] [Google Scholar]
- Devchand PR, Arita M, Hong S, Bannenberg G, Moussignac RL, Gronert K, et al. Human ALX receptor regulates neutrophil recruitment in transgenic mice: roles in inflammation and host defense. FASEB J. 2003;17:652–659. doi: 10.1096/fj.02-0770com. [DOI] [PubMed] [Google Scholar]
- Elferink JG, de Koster BM. Endothelin-induced activation of neutrophil migration. Biochem Pharmacol. 1994;48:865–871. doi: 10.1016/0006-2952(94)90356-5. [DOI] [PubMed] [Google Scholar]
- El Kebir D, József L, Pan W, Wang L, Petasis NA, Serhan CN, et al. 15-Epi-lipoxin A4 inhibits myeloperoxidase signaling and enhances resolution of acute lung injury. Am J Respir Crit Care Med. 2009;180:311–319. doi: 10.1164/rccm.200810-1601OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fierro IM, Colgan SP, Bernasconi G, Petasis NA, Clish CB, Arita M, et al. Lipoxin A4 and aspirin-triggered 15-epi-lipoxin A4 inhibit human neutrophil migration: comparisons between synthetic 15 epimers in chemotaxis and transmigration with microvessel endothelial cells and epithelial cells. J Immunol. 2003;170:2688–2694. doi: 10.4049/jimmunol.170.5.2688. [DOI] [PubMed] [Google Scholar]
- Filep JG, Zouki C, Petasis NA, Hachicha M, Serhan CN. Anti-inflammatory actions of lipoxin A(4) stable analogs are demonstrable in human whole blood: modulation of leukocyte adhesion molecules and inhibition of neutrophil–endothelial interactions. Blood. 1999;94:4132–4142. [PubMed] [Google Scholar]
- Filep JG, Khreiss T, Jozsef L. Lipoxins and aspirin-triggered lipoxins in neutrophil adhesion and signal transduction. Prostaglandins Leukot Essent Fatty Acids. 2005;73:257–262. doi: 10.1016/j.plefa.2005.05.014. [DOI] [PubMed] [Google Scholar]
- Fiorucci S, de Lima OM, Jr, Mencarelli A, Palazzetti B, Distrutti E, McKnight W, et al. Cyclooxygenase-2-derived lipoxin A4 increases gastric resistance to aspirin-induced damage. Gastroenterology. 2002;23:1598–1606. doi: 10.1053/gast.2002.36558. [DOI] [PubMed] [Google Scholar]
- Gronert K, Martinsson-Niskanen T, Ravasi S, Chiang N, Serhan CN. Selectivity of recombinant human leukotriene D(4), leukotriene B(4), and lipoxin A(4) receptors with aspirin-triggered 15-epi-LXA(4) and regulation of vascular and inflammatory responses. Am J Pathol. 2001;158:3–9. doi: 10.1016/S0002-9440(10)63937-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrero AT, Verri WA, Jr, Cunha TM, Silva TA, Schivo IR, Dal-Secco D, et al. Involvement of LTB4 in zymosan-induced joint nociception in mice: participation of neutrophils and PGE2. J Leukoc Biol. 2008;83:122–130. doi: 10.1189/jlb.0207123. [DOI] [PubMed] [Google Scholar]
- Hachicha M, Pouliot M, Petasis NA, Serhan CN. Lipoxin (LX)A4 and aspirin-triggered 15-epi-LXA4 inhibit tumor necrosis factor 1alpha-initiated neutrophil responses and trafficking: regulators of a cytokine–chemokine axis. J Exp Med. 1999;189:1923–1930. doi: 10.1084/jem.189.12.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haq A, El-Ramahi K, Al-Dalaan A, Al-Sedairy ST. Serum and synovial fluid concentrations of endothelin-1 in patients with rheumatoid arthritis. J Med. 1999;30:51–60. [PubMed] [Google Scholar]
- Harris ED., Jr Pathogenesis of rheumatoid arthritis: its relevance to therapy in the ‘90s. Trans Am Clin Climatol Assoc. 1991;102:260–268. discussion 268–270. [PMC free article] [PubMed] [Google Scholar]
- Hashimoto A, Hayashi I, Murakami Y, Sato Y, Kitasato H, Matsushita R, et al. Antiinflammatory mediator lipoxin A4 and its receptor in synovitis of patients with rheumatoid arthritis. J Rheumatol. 2007;34:2144–2153. [PubMed] [Google Scholar]
- Henriques MG, Silva PM, Martins MA, Flores CA, Cunha FQ, Assreuy-Filho J, et al. Mouse paw oedema. A new model for inflammation. Braz J Med Biol Res. 1987;20:243–249. [PubMed] [Google Scholar]
- Inoue A, Yanagisawa M, Kimura S, Kasuya Y, Miyauchi T, Goto K, et al. The human endothelin family: three structurally and pharmacologically distinct isopeptides predicted by three separate genes. Proc Natl Acad Sci U S A. 1989;86:2863–2867. doi: 10.1073/pnas.86.8.2863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin SW, Zhang L, Lian QQ, Liu D, Wu P, Yao SL, et al. Posttreatment with aspirin-triggered lipoxin A4 analog attenuates lipopolysaccharide-induced acute lung injury in mice: the role of heme oxygenase-1. Anesth Analg. 2007;104:369–377. doi: 10.1213/01.ane.0000252414.00363.c4. [DOI] [PubMed] [Google Scholar]
- Jonsson H, Allen P, Peng SL. Inflammatory arthritis requires Foxo3a to prevent Fas ligand-induced neutrophil apoptosis. Nat Med. 2005;11:666–671. doi: 10.1038/nm1248. [DOI] [PubMed] [Google Scholar]
- Kassuya CAL, Rogerio AP, Calixto JB. The role of ETA and ETB receptor antagonists in acute and allergic inflammation in mice. Peptides. 2008;29:1329–1337. doi: 10.1016/j.peptides.2008.03.008. [DOI] [PubMed] [Google Scholar]
- Kraan MC, Haringman JJ, Ahern MJ, Breedveld FC, Smith MD, Tak PP. Quantification of the cell infiltrate in synovial tissue by digital image analysis. Rheumatology (Oxford) 2000;39:43–49. doi: 10.1093/rheumatology/39.1.43. [DOI] [PubMed] [Google Scholar]
- Kronke G, Katzenbeisser J, Uderhardt S, Zaiss MM, Scholtysek C, Schabbauer G, et al. 12/15-Lipoxygenase counteracts inflammation and tissue damage in arthritis. J Immunol. 2009;183:3383–3389. doi: 10.4049/jimmunol.0900327. [DOI] [PubMed] [Google Scholar]
- Kuryliszyn-Moskal A, Klimiuk PA, Sierakowski S, Ciolkiewicz M. A study on vascular endothelial growth factor and endothelin-1 in patients with extra-articular involvement of rheumatoid arthritis. Clin Rheumatol. 2006;25:314–319. doi: 10.1007/s10067-005-0007-2. [DOI] [PubMed] [Google Scholar]
- Levy BD, Fokin VV, Clark JM, Wakelam MJ, Petasis NA, Serhan CN. Polyisoprenyl phosphate (PIPP) signaling regulates phospholipase D activity: a ‘stop’ signaling switch for aspirin-triggered lipoxin A4. FASEB J. 1999;13:903–911. doi: 10.1096/fasebj.13.8.903. [DOI] [PubMed] [Google Scholar]
- McMahon B, Godson C. Lipoxins: endogenous regulators of inflammation. Am J Physiol Renal Physiol. 2004;286:F189–F201. doi: 10.1152/ajprenal.00224.2003. [DOI] [PubMed] [Google Scholar]
- Maini RN, Taylor PC, Paleolog E, Charles P, Ballara S, Brennan FM, et al. Anti-tumour necrosis factor specific antibody (infliximab) treatment provides insights into the pathophysiology of rheumatoid arthritis. Ann Rheum Dis. 1999;58:I56–I60. doi: 10.1136/ard.58.2008.i56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathis S, Jala VR, Haribabu B. Role of leukotriene B4 receptors in rheumatoid arthritis. Autoimmun Rev. 2007;7:12–17. doi: 10.1016/j.autrev.2007.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mencarelli M, Pecorelli A, Carbotti P, Valacchi G, Grasso G, Muscettola M. Endothelin receptor A expression in human inflammatory cells. Regul Pept. 2009;158:1–5. doi: 10.1016/j.regpep.2009.06.004. [DOI] [PubMed] [Google Scholar]
- Menezes-de-Lima O, Jr, Kassuya CA, Nascimento AF, Henriques MG, Calixto JB. Lipoxin A4 inhibits acute edema in mice: implications for the anti-edematogenic mechanism induced by aspirin. Prostaglandins Other Lipid Mediat. 2006;80:123–135. doi: 10.1016/j.prostaglandins.2006.05.016. [DOI] [PubMed] [Google Scholar]
- Penido C, Castro-Faria-Neto HC, Vieira-de-Abreu A, Figueiredo RT, Pelled A, Martins MA, et al. LPS induces eosinophil migration via CCR3 signaling through a mechanism independent of RANTES and Eotaxin. Am J Respir Cell Mol Biol. 2001;25:707–716. doi: 10.1165/ajrcmb.25.6.4401. [DOI] [PubMed] [Google Scholar]
- Penido C, Conte FP, Chagas MS, Rodrigues CA, Pereira JF, Henriques MG. Antiinflammatory effects of natural tetranortriterpenoids isolated from Carapa guianensis Aublet on zymosan-induced arthritis in mice. Inflamm Res. 2006;55:457–464. doi: 10.1007/s00011-006-5161-8. [DOI] [PubMed] [Google Scholar]
- Serhan CN, Jain A, Marleau S, Clish C, Kantarci A, Behbehani B, et al. Reduced inflammation and tissue damage in transgenic rabbits overexpressing 15-lipoxygenase and endogenous anti-inflammatory lipid mediators. J Immunol. 2003;171:6856–6865. doi: 10.4049/jimmunol.171.12.6856. [DOI] [PubMed] [Google Scholar]
- Souza DG, Fagundes CT, Amaral FA, Cisalpino D, Sousa LP, Vieira AT, et al. The required role of endogenously produced lipoxin A4 and annexin-1 for the production of IL-10 and inflammatory hyporesponsiveness in mice. J Immunol. 2007;179:8533–8543. doi: 10.4049/jimmunol.179.12.8533. [DOI] [PubMed] [Google Scholar]
- Takano T, Fiore S, Maddox JF, Brady HR, Petasis NA, Serhan CN. Aspirin-triggered 15-epi-lipoxin A4 (LXA4) and LXA4 stable analogues are potent inhibitors of acute inflammation: evidence for anti-inflammatory receptors. J Exp Med. 1997;185:1693–1704. doi: 10.1084/jem.185.9.1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toffoli MC, Gabra BH, Teixeira CF, Sirois P, Jancar S. Endothelins mediate neutrophil activation, ProMMP-9 release and endothelial cell detachment. Inflammation. 2007;30:28–37. doi: 10.1007/s10753-006-9018-7. [DOI] [PubMed] [Google Scholar]
- Vaudo G, Marchesi S, Gerli R, Allegrucci R, Giordano A, Siepi D, et al. Endothelial dysfunction in young patients with rheumatoid arthritis and low disease activity. Ann Rheum Dis. 2004;63:31–35. doi: 10.1136/ard.2003.007740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughn MW, Proske RJ, Haviland DL. Identification, cloning, and functional characterization of a murine lipoxin A4 receptor homologue gene. J Immunol. 2002;169:3363–3369. doi: 10.4049/jimmunol.169.6.3363. [DOI] [PubMed] [Google Scholar]
- Verri WA, Jr, Guerrero AT, Fukada SY, Valerio DA, Cunha TM, Xu D, et al. IL-33 mediates antigen-induced cutaneous and articular hypernociception in mice. Proc Natl Acad Sci U S A. 2008;105:2723–2728. doi: 10.1073/pnas.0712116105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yacoubian S, Serhan CN. New endogenous anti-inflammatory and proresolving lipid mediators: implications for rheumatic diseases. Nat Clin Pract Rheumatol. 2007;3:570–579. doi: 10.1038/ncprheum0616. [DOI] [PubMed] [Google Scholar]
- Ye RD, Boulay F, Wang JM, Dahlgren C, Gerard C, Parmentier M, et al. International Union of Basic and Clinical Pharmacology. LXXIII. Nomenclature for the formyl peptide receptor (FPR) family. Pharmacol Rev. 2009;61:119–161. doi: 10.1124/pr.109.001578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Zhang X, Wu P, Li H, Jin S, Zhou X, et al. BML-111, a lipoxin receptor agonist, modulates the immune response and reduces the severity of collagen-induced arthritis. Inflamm Res. 2008;57:157–162. doi: 10.1007/s00011-007-7141-z. [DOI] [PubMed] [Google Scholar]
- Zouki C, Baron C, Fournier A, Filep JG. Endothelin-1 enhances neutrophil adhesion to human coronary artery endothelial cells: role of ET(A) receptors and platelet-activating factor. Br J Pharmacol. 1999;127:969–979. doi: 10.1038/sj.bjp.0702593. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.