


Abstract
Although the nucleolar localization of proteins is often believed to be mediated primarily by non-specific retention to core nucleolar components, many examples of short nucleolar targeting sequences have been reported in recent years. In this article, 46 human nucleolar localization sequences (NoLSs) were collated from the literature and subjected to statistical analysis. Of the residues in these NoLSs 48% are basic, whereas 99% of the residues are predicted to be solvent-accessible with 42% in α-helix and 57% in coil. The sequence and predicted protein secondary structure of the 46 NoLSs were used to train an artificial neural network to identify NoLSs. At a true positive rate of 54%, the predictor’s overall false positive rate (FPR) is estimated to be 1.52%, which can be broken down to FPRs of 0.26% for randomly chosen cytoplasmic sequences, 0.80% for randomly chosen nucleoplasmic sequences and 12% for nuclear localization signals. The predictor was used to predict NoLSs in the complete human proteome and 10 of the highest scoring previously unknown NoLSs were experimentally confirmed. NoLSs are a prevalent type of targeting motif that is distinct from nuclear localization signals and that can be computationally predicted.
INTRODUCTION
The nucleolus is a prominent non-membrane-contained nuclear structure known primarily as the site of ribosome biogenesis and assembly (1). In the past two decades however, the nucleolus has been shown to be involved in various other cellular functions including assembly of diverse ribonucleoprotein particles (RNPs), cell-cycle progression and proliferation regulation, as well as the response to numerous forms of cellular stress (2–6). Many of the processes that occur, at least in part, in the nucleolus require the re-location, often cyclical or conditional, of nucleoplasmic and even cytoplasmic proteins to the nucleolus (2–4,7). Consistent with this, the nucleolar proteome is large with currently over 4500 distinct human proteins that have been identified in purified nucleoli (8) and has been shown to respond dynamically to various treatments (9,10). The nucleolus thus accommodates a large and dynamic volume of cellular traffic, which presumably requires tight regulation of its protein targeting mechanisms. However, as highlighted in two recent reviews, widely accepted mechanisms of protein targeting to the nucleolus remain elusive (6,11).
In contrast, protein targeting to membrane-bound cellular compartments is well characterized and a small number of short targeting sequence motifs are predominantly used. These short targeting motifs are generally recognized by the import machinery of the target compartment. Such is the case for nuclear localization signals (NLSs) for targeting across the nuclear envelope (12), signal peptides for co-translational entry into the secretory pathway at the endoplasmic reticulum (13) as well as mitochondrial targeting peptides (14) and peroxisomal targeting signals (15). Protein localization in the nucleolus, on the contrary, is not generally well understood and is widely believed to be the result of interaction by high affinity binding to nucleolar core components such as ribosomal DNA, RNA or major protein components (16). Thus, nucleolar localization would result from retention in the nucleolus rather than targeting to this compartment.
However, in the past 15 years, numerous reports of unrelated human proteins harbouring nucleolar localization sequences (NoLSs) have been published (summarized in Table 1). Not all these motifs have been rigorously tested, but many have been shown to be sufficient for targeting reporter proteins to the nucleolus. While some of these NoLSs have been manually aligned with previously known NoLSs, no systematic study of these motifs has been reported. Here, we investigate the characteristics of these experimentally validated NoLSs and use them as a training set to computationally predict NoLSs in the entire human proteome.
Table 1.
Experimentally Validated NoLSs (EVN) dataset
| Accession | Protein name | NoLS | Targets reporter protein to nucleolusa |
|---|---|---|---|
| NP_001012270 | BIRC5 | MQRKPTIRRKNLRLRRK | GFP |
| NP_006161 | NOP2 | SKRLSSRARKRAAKRRLG | β-Gal (but requires additional NLS) |
| NP_005336 | HSPA1A | FKRKHKKDISQNKRAVRR | GFP |
| NP_937862 | ING1b (NoLS-1) | DKPNSKRSRRQRNNENR | GFP |
| NP_937862 | ING1b (NoLS-2) | TPKEKKAKTSKKKKRSKAKA | GFP |
| NP_005238 | FGF3 | GKGVQPRRRRQKQSPDNLEP | N/A |
| NP_006618 | POP4 | RHKRKEKKKKAKGLSARQRRELR | GFP |
| NP_945316 | PTHLH | GKKKKGKPGKRREQEKKKRRT | β-gal |
| NP_003778 | NOL4 | KEKIQAIIDSCRRQFPEYQERAR | N/A |
| NP_001002 | RPS7 | RRILPKPTRKSRTKNKQKRPR | N/A |
| NP_001034800 | DEDD | LKRRRA | N/A |
| NP_001091059 | RPP38 | KIKKLIPNPNKIRKPPKSKKATPK | GFP |
| NP_478102 | CDKN2A | QLRRPRHSHPTRARRCP | GFP |
| NP_003133 | SSB | QESLNKWKSKGRRFKGKGKGNKAAQPGSGKGK | PTB-GFP |
| NP_005560 | LIMK2 | KKRTLRKNDRKKR | GFP |
| NP_001997 | FGF2 | RSRKYTSWYVALKR | GFP |
| NP_477352 | PI4KA | SKKTNRGSQLHKYYMKRRTL | Soybean trypsin inhibitor |
| NP_002383 | MDM2 | KKLKKRNK | Thioredoxin |
| NP_003945 | MAP3K14 | RKKRKKK | GFP |
| NP_078908 | SAP30L | RRYKRHYK | N/A |
| NP_951038 | MDFIC | GRCRRLANFPGRKRRRRRR | GFP |
| NP_848927 | MTDH (NoLS-1) | KSKKKKKKKKKQGE | GFP |
| NP_848927 | MTDH (NoLS-2) | KQIKKKKKARRET | GFP |
| NP_078805 | CDC73 (NoLS-1) | RRAATENIPVVRRPDRK | GFP |
| NP_078805 | CDC73 (NoLS-2) | KKKQGCQRENETLIQRRK | GFP |
| NP_078905 | MLF1IP | MAPRGRRRPRPHRSEGARRSKNTLERTHS | GFP |
| NP_060239 | G2E3 | RKHDDCPNKYGEKKTKEK | N/A |
| NP_077289 | NOL12 | KRKHPRRAQDSKKPPRAPRTSKAQRRR | GFP fused to rat NOL12-NoLS |
| NP_039252 | NRG1 | MSERKEGRGKGKGKKKERGSGKK | GFP |
| NP_055318 | UTP20 | KKKMKKHKNKSEAKKRK | GFP |
| NP_849193 | STT3B | KQKYLSKKTTKRKRGYIKNKLVFKKGKKISKKTV | GFP |
| NP_068810 | RELA | EQPKQRGMRFRYKCEGRSAGSIPGER | N/A |
| NP_112578 | INO80B | HGHGVHKKKHKKHKKKHKKKHH | N/A |
| AAB60345 | L1 ORF2 | RLKIKGQRKIYQANGKQKK | N/A |
| AAH01024 | GNL3 | KRPKLKKASKRMTCHKRYKIQKKVREHHRKLRLEAKKQGHKKPRK | N/A |
| NP_002511 | NPM1 | QDLWQWRKSL | GFP |
| NP_937983 | TERT | MPRAPRCRAVRSLLR | GFP |
| NP_003277 | TOP1 | NKKKKPKKE | N/A |
| NP_796375 | MIDN | QQKRLRRKARRDARGPYHWSPSRKAGRS | GFP |
| NP_004851 | FXR2 | RPQRRNRSRRRRNR | N/A |
| NP_000347 | TCOF1 | KRKKDKEKKEKKKKAKKASTKDSESPSQKKKKKKKKTAEQTV | GFP |
| NP_004695 | RRP9 | GQEHRLGRWWRIKEARNSVCIIPLRRVPVPPAAGS | N/A |
| NP_150241 | PML | DRPLVFFDLKIDN | GFP |
| NP_061940 | GNL3L | MMKLRHKNKKPGEGSKGHKKISWPYPQPA KQNGKKATSKVPSAPHFVHPN | GFP |
| NP_004251 | RECQL4 | KQAWKQKWRKK | GFP |
| NP_068778 | PPP1R11 | HRKGRRR | N/A |
aIndicates whether this NoLS has been shown to target a reporter protein to the nucleolus when fused to it. The reporter protein chosen is indicated and references are provided in Supplementary File 1.
MATERIALS AND METHODS
Datasets
Positive examples of NoLSs were manually curated from the literature and are referred to as the experimentally validated NoLSs (EVN, listed in Table 1 and detailed in Supplementary File 1) set.
Three types of negatives were considered:
Non-NoLS NLSs that were manually curated from the literature and the NLSdb (17) and are listed in Supplementary File 2.
Randomly chosen sequences of length 20 from cytoplasmic non-nucleolar proteins as annotated by Uniprot (18).
Randomly chosen sequences of length 20 from nucleoplasmic non-nucleolar proteins as annotated by Uniprot (18).
The training/testing dataset should be a representative set that maximizes coverage while minimizing redundancy (19,20). Redundancy filtering was performed by ensuring that all the corresponding full-length proteins from which the sub-sequences are extracted to generate the datasets are <30% identical over their entire sequence to any other corresponding full-length protein used to generate the dataset. In addition to this, we also verified that our datasets are non-redundant by extending all the sub-sequences considered to a size of 50 (the length of the longest EVN NoLS) and aligning them pairwise using the fasta program (version 35.04) (21). All extended NoLS pairs have at most 13 exact matches in local alignments, representing <30% sequence identity between the pairs.
For the purpose of training the ANN, several different combinations of the datasets were investigated and their performance compared by cross-validation. The one that was settled on consists of unbalanced datasets comprising 20 copies of the positive examples, 5 copies of the non-NoLS NLSs negatives, ∼1000 cytoplasmic negatives and 180 nucleoplasmic negatives. When 3-fold cross-validation was performed, care was taken to ensure that all copies of a given sequence (for NoLSs and non-NoLS NLSs which were used in more than one copy) were placed in the same group.
Encoding
For the sequence encoding, windows of 13 residues in size were sparsely encoded in a binary manner using a reduced alphabet of size 12 with the follow groupings: {K, R, Q, P, H, ED, STY, N, C, W, ILVAMG, F}. For example, the sequence NSAT would be encoded as the binary vector 000000010000000000100000000000000010000000100000. This reduced alphabet was chosen to ensure that frequent residues in NoLSs are represented as singlets while under-represented residues in NoLSs are grouped by chemical similarity. Other sequence encodings were considered but did not outperform the encoding described here as assessed by cross-validation.
For the sequence encoding, a window size of 13 was chosen for several reasons: (i) bipartite NLSs are between 15 and 17 residues in length according to Prosite (22) and thus a window size shorter than this might minimize the number of NLSs wrongly predicted as NoLSs, (ii) larger window sizes lead to larger artificial neural networks (ANNs) and a higher possibility to overfitting, (iii) the accuracy by 3-fold cross-validation is substantially worse when the window size is greater than 16 or smaller than 11, and 4) an odd number for the window size makes it easier to assign a score to the middle residue.
Additional information including protein characteristics and secondary structure were also considered and encoded using nine floating point numbers:
- a representation SL of the length L of the protein
400 was chosen as a threshold as this is the approximate average length of human proteins as defined by IPI version 3.40 (23).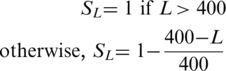
- a representation D of the relative distance between the sub-sequence considered and the middle of the full-length protein
where x is the position of the subsequence considered and m is the position of the middle of the protein.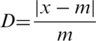
- and 7 measures of protein secondary structure all predicted by Jpred (24) over a region R covering the window of size 13 considered and three flanking residues on either side:
- ○ the proportion of residues in R predicted as belonging to an α-helix
- ○ the proportion of residues in R predicted as belonging to a β-sheet
- ○ the proportion of residues in R predicted as located in a coil
- ○ the average confidence of the three above predictions over region R, as estimated by Jpred (24)
- ○ the proportion of buried residues in R predicted at a relative solvent accessibility threshold of >25%
- ○ the proportion of buried residues in R predicted at a relative solvent accessibility threshold of >5%
- ○ the proportion of buried residues in R predicted at a relative solvent accessibility threshold of >0%
When only the sequence information is used, a binary vector of size 156 is created (window of size 13 × alphabet of size 12). If in addition to sequence, protein characteristics and secondary structure are considered, a vector of size 165 (156 + 9) is created.
ANNs
The Stuttgart Neural Network Simulator (SNNS; http://www.ra.cs.uni-tuebingen.de/SNNS/) was used to train ANNs for the purpose of predicting NoLSs. Many different combinations of neural network architecture and parameters were investigated. Most performed equally well, indicating that the method is relatively insensitive to parameter changes, and many of the default settings were chosen. The combination settled on is described here. ANNs were built with either 156 or 165 input nodes (depending on the encoding used, see ‘Encoding’ section), 9 hidden nodes and 1 output node. The chosen target outputs were 0 for non-NoLSs and 1 for NoLSs. The learning function used was batch backpropopagation, the initialization function was Randomize_Weights and the update function was Topological_Order.
During 3-fold cross-validation, ANNs were trained until the prediction performance on the validation set started decreasing (∼4000 cycles).
For the receiver operating characteristic (ROC) plots, the ANN was trained and validated on all three types of negatives combined and it is just for testing purposes that the three types of negatives were considered separately as well as combined (see Figure 3).
Figure 3.

ROC plots. The predictor was trained by 3-fold cross-validation using all types of negatives combined. The true positive rates (TPRs) versus false positive rates (FPRs) are plotted for the three different types of negatives tested collectively (allNegativeTypesCombined) and separately: randomly chosen cytoplasmic sequences (referred to as cyto), randomly chosen nucleoplasmic sequences (referred to as nuc) and curated non-NoLS NLSs (labelled nls). The accuracy measures of two encodings are shown: encodings based only on sequence (Seq) and encodings based on both sequence and additional structure elements (Seq-Struct). The diagonal line indicates the performance that would be expected at random.
Characterization of predicted NoLS-containing proteins
For the characterization of predicted NoLS-containing proteins, ‘experimental’ subcellular localization annotations were downloaded from Uniprot (18) for all human proteins. DAVID (25) was used to compare the GO biological process term enrichment between the list of predicted NoLS-containing proteins that exist in RefSeq and the list of all human RefSeq proteins that were considered by our predictor as background.
Cell culture and transfection
The human osteosarcoma cell line U2OS was cultured as adherent cells in Dulbeccos’s modified eagle medium (DMEM) (Invitrogen) supplemented with 10% fetal bovine serum, 100 U/ml penicillin/streptomycin and 2 mM l-glutamine. Transfection was done using Effectene (QIAGEN) as per the manufacturer protocol.
Cloning
The oligonucleotides corresponding to each NoLS considered (see Supplementary File 3 for their nucleotide sequences and Table 4 for their amino acid sequences) were annealed by first heating them at 95°C and then letting them cool down to room temperature. The resulting double-stranded DNA was then cloned into pEGFP-C1 (Clontech) using the restriction enzymes Bgl II and Kpn I.
Table 4.
Sequences of NoLSs chosen for experimental validation
| Protein name | NoLS sequence chosen for experimental validation |
|---|---|
| RBBP6 | SQDSKKKKKKKEKKKHKKHKKHKKHKKH |
| RNF213 | SWTVQESKKKKRKKKKKGNKSASSE |
| C1orf35 | HRKSKKEKKKKKKRKHKKEKKKKDKEHRRP |
| DDX10 | KKHSHRQNKKKQLRKQLKKPEWQVERE |
| SF3B2 | GRSTVSVSKKEKNRKRRNRKKKKKPQRVRGVSSE |
| RBM34 | KAVLLKTKKKGQKKSGRPKKQRKQK |
| CEBPZ | AKSIIKKKKHFKKKRIKTTQKTKKQRK |
| SMARCA2 | QAQAAKEKKKRRRRKKKAEENAEGG |
| AP3D1 | RRHRQKLEKDKRRKKRKEKEERTKGKKKSKK |
| SRP72 | QPKEQGQGDLKKKKKKKKGKLPKNYDPK |
Immunofluorescence
Cells were grown on glass coverslips and fixed with 1% paraformaldehyde in PBS for 10 min. Cells were then permeabilized in PBS containing 0.5% Triton X-100 for 10 min and mounted on slides with Vectashield (Vector Laboratories Inc.) containing DAPI. Fluorescence imaging was performed on a DeltaVision Spectris widefield deconvolution microscope (Applied Precision), using a CoolMax charge-coupled device camera (Roper Scientific). Cells were imaged using a 60 × NA 1.4 Plan-Apochromat objective (Olympus) and the appropriate filter sets (Chroma Technology Corp.), with 20 optical sections of 0.5 μM each acquired. SoftWorX software (Applied Precision) was used for both acquisition and deconvolution.
RESULTS
General NoLS characteristics
A dataset of experimentally validated NoLSs was assembled by extensive manual curation of the literature. Reported NoLSs of length >50 residues were discarded as their critical residues have likely not been precisely defined and/or the NoLS might form a signal patch and exist only in the folded protein. The remaining 46 NoLSs are shown in Table 1. These will be referred to as the experimentally validated NoLS (EVN) set.
Visual inspection of the EVN sequences reveals a high proportion of basic amino acids. In fact, 48% of the residues found in these sequences are lysines or arginines. The average residue frequency for all amino acids in EVN sequences is shown in Supplementary File 4.
The secondary structure predictor Jpred 3 (24) was used to analyze the protein regions that contain NoLSs (Figure 1). EVN sequences are localized in regions predicted to be almost uniquely α-helices or coils (Figure 1A) and found predominantly at the surface of proteins (Figure 1B). An analysis of the position of experimentally validated NoLSs in full-length proteins shows that known NoLSs localize predominantly at the ends of proteins (Figure 1C). In fact, 22 of the 46 NoLSs examined are found in the 25% of residues closest to the protein termini. NoLSs are thus localized in protein regions that are easily accessible.
Figure 1.

NoLS characteristics. (A) NoLSs are predominantly found in regions predicted by Jpred (24) as α-helices or coils and very rarely in regions predicted as extended β-strands. (B) NoLSs localize predominantly at the surface of proteins as predicted by Jnet (24) either at relative solvent accessibility thresholds <25% (JnetSol25), <5% (JnetSol5) or 0% (JnetSol0). (C) NoLSs are found predominantly at the ends of proteins. The error bars represent standard deviation.
NoLS vs NLS
NLSs target proteins to the nucleus. Numerous and diverse NLSs have been reported and mechanisms of recognition of NLSs have been extensively studied (12,26). NoLSs and NLSs have very similar amino acid compositions (a high prevalence of basic residues in both cases) and while there is mounting evidence that these two types of signals are recognized as different by the cell, little attention has been given to distinguishing and systematically characterizing both types of signals. NoLSs and NLSs can be collectively grouped into three classes:
NLS-only signals that target proteins to the nucleus but do not cause significant accumulation in the nucleolus [e.g. PTMA is nucleoplasmic and harbours a bipartite and non-NoLS NLS (27)].
NoLS-only signals that cause proteins to accumulate in the nucleolus but are unable to mediate nuclear envelop translocation. These are usually found in proteins that also contain an NLS-only signal. For example, the proteins NOP2 (28) and PPP1R11 described below.
Joint NoLS-NLS regions which can both target proteins across the nuclear envelope and cause proteins to accumulate in the nucleolus. For example, UTP20 is reported to contain overlapping NLS and NoLS near its C-terminus (29).
To confirm that these signals are necessary and sufficient for this targeting, they are usually fused to reporter proteins and visualized by microscopy (see Table 1 for examples of experimentally confirmed NoLSs).
Several proteins are reported to contain two ‘NLSs’, one of which seems to allow entry into the nucleus (an NLS-only signal) and the other which targets nuclear proteins to the nucleolus (an NoLS-only signal). For example, PPP1R11 (protein phosphatase-1 inhibitor-3) is mainly nucleolar. It has two basic stretches that have different targeting roles. The most N-terminal basic motif (residues 32–37) serves as an NLS and the protein accumulates in the cytoplasm when this signal is mutated. In contrast, a C-terminal motif (residues 94–100) functions as an NoLS and the protein is nuclear but non-nucleolar when this motif is absent (30).
Prediction of NoLSs using ANNs
The EVN dataset was used to investigate whether known NoLSs can be identified computationally and predicted at the proteome level. ANNs were chosen as a machine learning method to predict NoLSs because they perform well at pattern recognition tasks and have been used successfully to identify other protein targeting motifs (31,32). For this task, the aim is to differentiate between NoLS and non-NoLS sequences. For training purposes, the ANN thus requires both positive examples of NoLSs (the EVN dataset) and examples of sequences that do not target proteins to the nucleolus (referred to as the negative training set). As described in the ‘Materials and methods’ section, the negative training set was generated by combining three groups of non-NoLS sequences: (i) randomly chosen protein sub-sequences of 20 residues from cytoplasmic proteins not annotated as localizing to the nucleolus, (ii) randomly chosen protein sub-sequences of 20 residues from nucleoplasmic proteins not annotated as localizing to the nucleolus and (iii) reported NLSs for which there is no evidence that they also localize proteins to the nucleolus (NLS-only signals, as described above). As NLSs and NoLSs have similar amino acid compositions, NLSs represent the most difficult group of negatives to predict against. Non-NoLS NLSs used in the negative training set were identified by manual curation of the literature and of NLSdb (17). However, in assembling this dataset, it became obvious that many reported NLSs might also be NoLSs (joint NoLS-NLS regions as described above) or are found in nucleolar proteins and no investigation has been performed to check whether these NLSs are also NoLSs. For example, NLS27 and NLS30 from NLSdb (17) refer to the NLS of the protein LEF1 described in (33). However, while some microscopy pictures in (33) show LEF1 accumulating in structures that resemble nucleoli, and Entrez Gene annotates LEF1 as being nucleolar, no further investigation has been undertaken to clarify the true nature of the LEF1 ‘NLS’. Reported NLSs found in proteins localized to the nucleolus were excluded from the negative training set.
Positive and negative training set sequences were encoded as described in Figure 2 and the ‘Materials and methods’ section. ANNs were built using the SNNS (http://www.ra.cs.uni-tuebingen.de/SNNS/).
Figure 2.

Prediction of NoLSs using an ANN. (A) Sequence windows of size 13 overlapping with an offset of 1 are sparsely encoded into binary vectors of size 165 based on their amino acid sequence, position within the full-length protein sequence and elements of secondary structure. (B) The encoded vectors are fed to the ANN which outputs one score for each input window, attributed to the central residue of the window. (C) Peptides of length 20 are predicted as NoLSs if the average score of the 8 windows of size 13 they contain is >0.8.
Measures of accuracy
Cross-validation
Three-fold cross-validation experiments were performed to measure the accuracy of the predictor. The positive and negative datasets were randomly divided into three non-overlapping sets used respectively for training, validating and testing the ANN. The reported accuracy is the average of the different training, validating and testing combinations. Figure 3 summarizes the performance of the predictor as a ROC plot in which the true positive rate (TPR) is plotted against the false positive rate (FPR) of the predictor. The predictor was trained on the combination of all three types of negative examples as described above and subsequently tested on this combination of negatives (points labelled allNegativeTypesCombined). To investigate how well the predictor performs on the different types of negatives, in Figure 3 we also provide a breakdown of their estimated accuracy separately. This was done by training the predictor in a cross-validation manner on all three types of negatives combined and then considering each of these types of negatives separately for testing. As shown in Figure 3, including secondary structure information as well as sequence (solid lines) consistently results in higher accuracy compared to using only sequence (dashed lines) for all negative types. As expected, the predictor performs better on negatives randomly generated from nucleoplasmic or cytoplasmic non-nucleolar proteins than when tested with reported NLSs. To yield low FPRs while maintaining a reasonably high TPR, the threshold to predict NoLSs was set to an average output score of 0.8 over 8 consecutive windows (as described in the ‘Materials and methods’ section and in Figure 2). At this score, the average TPR is measured to be 54% and the FPRs are measured to be 0.26% for the randomly chosen cytoplasmic sequences, 0.80% for the randomly chosen nucleoplasmic sequences and 12% for the NLSs.
Independent validation on NoLS-containing proteins of human-infecting viruses
Numerous and diverse viral proteins have been shown to localize in the nucleoli of their host’s cells (34). Viral proteins that have an experimentally identified and validated NoLS were used as an independent test of our human-trained predictor. As shown in Table 2, all NoLS-containing viral proteins considered were predicted to harbour at least one NoLS that overlaps with the experimentally validated NoLS.
Table 2.
Positions of experimentally validated and computationally predicted viral NoLSs
| Protein name | Virus | Accession | Predicted NoLS position | Experimentally determined NoLS position | Reference for experimentally determined NoLS position |
|---|---|---|---|---|---|
| tat | HIV | NP_057853 | 43–68 | 48–61 | (43) |
| rev | HIV | NP_057854 | 28–57 | 33–52 | (44) |
| rex | HTLV-1 | NP_057863 | 1–26 | 1–20 | (45) |
| NS1A | Influenza A | P03495 | 208–237 | 216–237 | (46) |
| US11 | HSV1 | NP_044674 | 86–105, 113–160 | 88–125 | (47) |
| RL1 | HSV1 | P08353 | 1–22 | 1–16 | (48) |
| ORF57 | HVS | NP_040259 | 114–139 | 91–94, 119–128 | (11,49) |
Independent experimental validation of human proteins
The entire EVN dataset was encoded by considering both sequence and elements of structure and used to train an ANN which was then applied to the whole human proteome as defined by IPI version 3.40 (23). Supplementary File 5 shows the list of human proteins predicted to harbour a NoLS. The proteome-wide prediction of NoLSs may also be searched and downloaded from http://www.compbio.dundee.ac.uk/www-nod/.
The predicted human NoLSs were ranked by score and ten of the highest scoring human NoLSs were chosen for experimental validation. Amongst the highest scoring NoLSs, care was taken to select diverse proteins including uncharacterized proteins (e.g. RNF213, C1orf35), mainly cytoplasmic proteins (AP3D1, SRP72), nucleoplasmic proteins (SMARCA2, CEBPZ) and a nucleolar protein for which no NoLS has been described (RBM34). These proteins selected for experimental validation are shown in Table 3 and the sequences of their NoLSs are shown in Table 4. Their respective high-scoring NoLSs were cloned downstream of GFP, expressed in U2OS cells and visualized by microscopy. GFP alone as well as a fusion protein of GFP cloned upstream of a region of protein RBM34 that is not predicted to be a NoLS (residues 324–345 of RBM34) were used as negative controls. As shown in Figure 4 and Supplementary File 6, all predicted NoLSs that were successfully cloned are capable of causing the accumulation of the GFP fusion protein in the nucleolus. The negative controls GFP and GFP-RBM34 (324–345) do not accumulate in the nucleolus. Interestingly, while all the predicted NoLS fusion proteins tested display a strong signal in the nucleolus, the extent of nucleoplasmic and cytoplasmic accumulations vary considerably for the different NoLSs. As the number of experimentally validated NoLSs increases in the future, it will become possible to investigate the differences between these signals and to determine whether they are NoLS-only or joint NoLS-NLS signals.
Table 3.
NoLSs chosen for experimental validation
| Protein name | Accession | NoLS score | Subcellular localization of protein if known | Function/ process of protein if known | Reference for localization/ process annotations | NoLS cloned successfully | Experimentally validated as nucleolar-targeting |
|---|---|---|---|---|---|---|---|
| RBBP6 | Q7Z6E9-4 | 0.981 | N/A | N/A | N/A | Yes | Yes |
| RNF213 | Q9HCF4-3 | 0.977 | N/A | N/A | N/A | Yes | Yes |
| C1orf35 | Q9BU76-1 | 0.976 | N/A | N/A | N/A | Yes | Yes |
| DDX10 | Q13206 | 0.970 | N/A | RNA helicase | (50) | Yes | Yes |
| SF3B2 | Q13435 | 0.966 | Spliceosomal complex | RNA splicing | (51–53) | Yes | Yes |
| RBM34 | P42696 | 0.966 | Nucleolar (inferred from electronic annotation) | RNA binding (inferred from electronic annotation) (18) | (18) | No | N/A |
| CEBPZ | Q03701 | 0.959 | Nucleus | Transcription | (54) | Yes | Yes |
| SMARCA2 | P51531-2 | 0.958 | Nucleoplasm | Regulation of transcription | (55) | Yes | Yes |
| AP3D1 | O14617-4 | 0.957 | Golgi apparatus | Intracellular protein transport | (56) | Yes | Yes |
| SRP72 | O76094 | 0.950 | Mainly cytoplasmic but nucleolar for complex assembly | Signal particle recognition binding | (57) | Yes | Yes |
| USP36 | Q9P275-2 | 0.931 | Nucleolar | Ubiquitin-dependent protein degradation (inferred from electronic annotation) (18) | (18,58) | N/A | Yes [independently validated (35)] |
Figure 4.

Experimental validation by microscopy. (A) Fusion constructs of NoLSs chosen for experimental validation and successfully cloned downstream of GFP (Table 3) were transfected into U2OS cells and the resulting proteins were visualized by microscopy [GFP-NoLS() labelled columns]. The DAPI columns show staining of the DNA in these cells. (B) GFP and GFP-RBM34(324–345) were used as negative controls. The bars represent 15 µm.
In choosing the candidates for experimental validation, we also noticed that USP36 (described in Table 3), a high scoring candidate, has been recently validated by an independent group. Endo and colleagues experimentally identified a functional NoLS between positions 1076 and 1091 of USP36 (35), while we predict an NoLS between residues 1073 and 1102.
Characteristics of NoLS-containing proteins
Analysis of whole-proteome predictions of NoLS reveals that a significantly larger proportion of proteins annotated as nucleolar are predicted to contain a NoLS than proteins annotated as localized in all other major cellular compartments (Figure 5). Of proteins annotated as nucleolar in Uniprot (18), 54% are predicted to harbour a NoLS. Thirty-nine percent of nuclear-annotated human proteins and 43% of nucleoplasmic or nuclear envelope human proteins are predicted to contain a NoLS. Since the nucleolus is contained within the nucleus, it is likely that many nucleolar proteins are still simply annotated as nuclear. As for the nucleoplasmic or nuclear envelope proteins predicted to have a NoLS, further experiments and a higher coverage of the localization annotations will be required to determine whether these proteins can also localize to the nucleolus or represent false-positive predictions. Amongst cytoplasmic proteins, between 25% (cytosolic proteins) and 5% (peroxisomal proteins) are predicted to contain NoLSs. While some of these proteins surely represent false-positive predictions, others are likely to represent true NoLS-containing proteins that might conditionally localize to the nucleolus. Numerous such examples have been reported (36–42).
Figure 5.

Characteristics of predicted NoLS-containing proteins. For all cellular compartments considered, the fraction of proteins predicted to harbour a NoLS is shown. Protein counts for each compartment are indicated in parenthesis beside the compartment name. The compartment groups labelled with an asterisk include proteins annotated as being in this and any other compartment except the nucleolus. The 261 proteins in the nucleolus group represent all proteins annotated as being nucleolar regardless of any other localization annotations they may have (indicated by double asterisks). The error bars were determined by bootstrap.
In addition to the Uniprot localization annotations which are predominantly derived from microscopy experiments reported in the literature, we have also mapped our predictions of NoLSs onto the quantitative proteomic analysis of subcellular proteome localization described recently (10). In this study, the relative abundance of proteins in different cellular compartments was measured by harvesting nucleolar, nucleoplasmic and cytoplasmic cellular extracts each grown in the presence of amino acids labelled with different isotopes and then by pooling together the different fractions and analysing them by mass spectrometry. Table 5 shows the fraction of proteins that harbour at least one NoLS depending on their relative abundance ratios in the nucleolus. Similar to the Uniprot annotations, 48% of proteins that are both more nucleolar than nucleoplasmic and more nucleolar than cytoplasmic are predicted to harbour a NoLS. In contrast, ∼25% of proteins that are more nucleoplasmic or cytoplasmic than nucleolar have a predicted NoLS and only 16% of proteins that are more nucleoplasmic and cytoplasmic than nucleolar harbour a predicted NoLS.
Table 5.
Comparison between NoLS predictions and protein localization ratios from ref. (10)
| Localization abundance ratios | Total protein count | Protein count with predicted NoLSs | Fraction of proteins with NoLS (%) |
|---|---|---|---|
| Nucleolar/Cytoplasmic > 1 Nucleolar/Nucleoplasmic > 1 | 347 | 165 | 47.6 |
| Nucleolar/Cytoplasmic ≤ 1 Nucleolar/Nucleoplasmic ≤ 1 | 1402 | 229 | 16.3 |
| Nucleolar/Cytoplasmic ≤ 1 Nucleolar/Nucleoplasmic > 1 | 406 | 102 | 25.1 |
| Nucleolar/Cytoplasmic > 1 Nucleolar/Nucleoplasmic ≤ 1 | 290 | 75 | 25.9 |
Significantly enriched Gene Ontology (GO) biological process annotations of all predicted NoLS-containing human proteins are shown in Table 6. The most prevalent terms associated with predicted NoLS-containing proteins involve transcription, processing of RNA and regulation of chromatin which agree well with the biological process annotations of many of the proteins that contain the EVN sequences.
Table 6.
Most significantly enriched GO annotations of predicted NoLS-containing proteins
| Biological process GO term | Protein counta | Benjamini-adjusted P-valueb | Fold enrichmentc |
|---|---|---|---|
| GO:0006351∼transcription, DNA-dependent | 1008 | 5.42E−100 | 1.73 |
| GO:0032774∼RNA biosynthetic process | 1008 | 1.23E−99 | 1.73 |
| GO:0006355∼regulation of transcription, DNA-dependent | 988 | 1.03E−98 | 1.74 |
| GO:0045449∼regulation of transcription | 1036 | 1.02E−98 | 1.71 |
| GO:0051276∼chromosome organization and biogenesis | 221 | 2.10E−39 | 2.26 |
| GO:0006323∼DNA packaging | 185 | 1.54E−35 | 2.34 |
| GO:0006325∼establishment and/or maintenance of chromatin architecture | 181 | 1.37E−34 | 2.34 |
| GO:0006259∼DNA metabolic process | 338 | 2.05E−30 | 1.76 |
| GO:0016568∼chromatin modification | 120 | 1.23E−22 | 2.35 |
| GO:0045934∼negative regulation of nucleobase, nucleoside, nucleotide and nucleic acid metabolic process | 165 | 5.54E−20 | 1.97 |
| GO:0016481∼negative regulation of transcription | 151 | 2.65E−18 | 1.97 |
| GO:0031324∼negative regulation of cellular metabolic process | 174 | 7.51E−15 | 1.76 |
| GO:0045892∼negative regulation of transcription, DNA-dependent | 111 | 5.30E−14 | 2.02 |
| GO:0006333∼chromatin assembly or disassembly | 79 | 2.23E−13 | 2.28 |
| GO:0008380∼RNA splicing | 113 | 1.27E−12 | 1.94 |
| GO:0016071∼mRNA metabolic process | 140 | 1.51E−12 | 1.80 |
aProtein count of all predicted NoLS-containing proteins that are annotated with this GO term.
bThe Benjamini-adjusted P-value was calculated by DAVID (25).
cEnrichment of this GO term in predicted NoLS-containing proteins compared to all human refseq proteins. Only GO terms with fold enrichment >1.7 are shown here.
DISCUSSION
NoLSs are emerging as a predominant mechanism in the targeting of proteins to the nucleolus. Through careful curation of the literature, we have identified 46 NoLSs, most of which are required for nucleolar targeting of the proteins that encode them and can target non-nucleolar reporter proteins to the nucleolus. As a group, these NoLSs contain a high proportion of basic amino acids making them similar to NLSs. Because of this similarity, NLSs and NoLSs are often perceived as analogous and interchangeably used to annotate proteins. In particular, short basic stretches in proteins are often assumed to be NLSs and even when experimental validation is performed, often no attention is given to the particular intra-nuclear localization of the protein even though this provides valuable clues about its function in the cell. Because of this, numerous NoLSs are annotated as NLSs.
Given the very different nature of their target compartments, the similarity between NLSs and NoLSs is somewhat surprising: NLSs specify translocation across the nuclear envelope, a double membrane surrounding the nucleus, whereas NoLSs ensure accumulation in the nucleolus, a membrane-less subcompartment within the nucleus. The similarity between NLSs and NoLSs has likely delayed the systematic characterization of NoLSs because of the extra difficulty of identifying clear and meaningful examples of both true NoLSs and true non-NoLSs. To overcome this problem, we have performed extensive curation of the literature making possible the accurate prediction of these motifs on a proteome-wide level. In future experiments, it will be important to consistently recognize and annotate NLSs and NoLSs as distinct, which will undoubtedly lead to improved predictions. A larger number of examples of true NLS-only signals, NoLS-only signals and joint NLS-NoLSs will help in better defining these signals and differentiating them. In addition to this, studies such as this one should help in the construction of precisely targeted fusion proteins, ensuring that proteins are not highly enriched in the nucleolus when the aim is to locate them in the nucleoplasm.
A small number of proteins have been proposed to act as transporters to the nucleolus [e.g. B23/NPM1 which shuttles between the cytoplasm and nucleolus and binds several NoLS-containing proteins (28)]. Alternatively, NoLSs might instead bind to nucleolar RNA thus causing the targeting of the proteins that contain them to the nucleolus. Further investigations will be required to clarify whether protein transporters are widely used for the nucleolar targeting of NoLS-containing proteins or whether other mechanisms are predominantly employed for this purpose. The NoLS predictions should serve as a good starting point to experimentally address these questions.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
M.S.S. is a recipient of a post-doctoral fellowship from the Caledonian Research Foundation. A.I.L. is a Wellcome Trust Principal Research Fellow. A.I.L. and F.M.B. are funded in part by the European Commission’s FP7 (GA HEALTH-F4-2008-201648/PROSPECTS) (www.prospects-fp7.eu/) and by a Wellcome Trust programme grant (073980/Z/03/Z). G.J.B. acknowledges funding from the Wellcome Trust (WT083481). Funding for open access charge: Wellcome Trust grant WT083481.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank Drs Tom Walsh and Peter Troshin for technical expertise.
REFERENCES
- 1.Scheer U, Hock R. Structure and function of the nucleolus. Curr. Opin. Cell Biol. 1999;11:385–390. doi: 10.1016/S0955-0674(99)80054-4. [DOI] [PubMed] [Google Scholar]
- 2.Boisvert FM, van Koningsbruggen S, Navascues J, Lamond AI. The multifunctional nucleolus. Nat. Rev. Mol. Cell Biol. 2007;8:574–585. doi: 10.1038/nrm2184. [DOI] [PubMed] [Google Scholar]
- 3.Olson MO, Dundr M, Szebeni A. The nucleolus: an old factory with unexpected capabilities. Trends Cell Biol. 2000;10:189–196. doi: 10.1016/s0962-8924(00)01738-4. [DOI] [PubMed] [Google Scholar]
- 4.Olson MO, Hingorani K, Szebeni A. Conventional and nonconventional roles of the nucleolus. Int. Rev. Cytol. 2002;219:199–266. doi: 10.1016/S0074-7696(02)19014-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pederson T. The plurifunctional nucleolus. Nucleic Acids Res. 1998;26:3871–3876. doi: 10.1093/nar/26.17.3871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pederson T, Tsai RY. In search of nonribosomal nucleolar protein function and regulation. J. Cell Biol. 2009;184:771–776. doi: 10.1083/jcb.200812014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pederson T. Growth factors in the nucleolus? J. Cell Biol. 1998;143:279–281. doi: 10.1083/jcb.143.2.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ahmad Y, Boisvert FM, Gregor P, Cobley A, Lamond AI. NOPdb: Nucleolar Proteome Database–2008 update. Nucleic Acids Res. 2009;37:D181–184. doi: 10.1093/nar/gkn804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Andersen JS, Lam YW, Leung AK, Ong SE, Lyon CE, Lamond AI, Mann M. Nucleolar proteome dynamics. Nature. 2005;433:77–83. doi: 10.1038/nature03207. [DOI] [PubMed] [Google Scholar]
- 10.Boisvert FM, Lam YW, Lamont D, Lamont AI. A quantitative proteomic analysis of subcellular proteome localization and changes induced by DNA damage. Mol. Cell Proteomics. 2010;9:457–470. doi: 10.1074/mcp.M900429-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Emmott E, Hiscox JA. Nucleolar targeting: the hub of the matter. EMBO Rep. 2009;10:231–238. doi: 10.1038/embor.2009.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boulikas T. Nuclear localization signals (NLS) Crit. Rev. Eukaryot. Gene Expr. 1993;3:193–227. [PubMed] [Google Scholar]
- 13.von Heijne G. The signal peptide. J. Membr. Biol. 1990;115:195–201. doi: 10.1007/BF01868635. [DOI] [PubMed] [Google Scholar]
- 14.Gavel Y, Nilsson L, von Heijne G. Mitochondrial targeting sequences. Why ‘non-amphiphilic' peptides may still be amphiphilic. FEBS Lett. 1988;235:173–177. doi: 10.1016/0014-5793(88)81257-2. [DOI] [PubMed] [Google Scholar]
- 15.Gould SJ, Keller GA, Hosken N, Wilkinson J, Subramani S. A conserved tripeptide sorts proteins to peroxisomes. J. Cell Biol. 1989;108:1657–1664. doi: 10.1083/jcb.108.5.1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Carmo-Fonseca M, Mendes-Soares L, Campos I. To be or not to be in the nucleolus. Nat. Cell Biol. 2000;2:E107–E112. doi: 10.1038/35014078. [DOI] [PubMed] [Google Scholar]
- 17.Nair R, Carter P, Rost B. NLSdb: database of nuclear localization signals. Nucleic Acids Res. 2003;31:397–399. doi: 10.1093/nar/gkg001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.The Universal Protein Resource. (UniProt) in 2010. Nucleic Acids Res. 2010;38:D142–D148. doi: 10.1093/nar/gkp846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hobohm U, Scharf M, Schneider R, Sander C. Selection of representative protein data sets. Protein Sci. 1992;1:409–417. doi: 10.1002/pro.5560010313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nielsen H, Engelbrecht J, von Heijne G, Brunak S. Defining a similarity threshold for a functional protein sequence pattern: the signal peptide cleavage site. Proteins. 1996;24:165–177. doi: 10.1002/(SICI)1097-0134(199602)24:2<165::AID-PROT4>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 21.Pearson WR, Lipman DJ. Improved tools for biological sequence comparison. Proc. Natl Acad. Sci. USA. 1988;85:2444–2448. doi: 10.1073/pnas.85.8.2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sigrist CJ, Cerutti L, de Castro E, Langendijk-Genevaux PS, Bulliard V, Bairoch A, Hulo N. PROSITE, a protein domain database for functional characterization and annotation. Nucleic Acids Res. 2010;38:D161–D166. doi: 10.1093/nar/gkp885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kersey PJ, Duarte J, Williams A, Karavidopoulou Y, Birney E, Apweiler R. The International Protein Index: an integrated database for proteomics experiments. Proteomics. 2004;4:1985–1988. doi: 10.1002/pmic.200300721. [DOI] [PubMed] [Google Scholar]
- 24.Cole C, Barber JD, Barton GJ. The Jpred 3 secondary structure prediction server. Nucleic Acids Res. 2008;36:W197–W201. doi: 10.1093/nar/gkn238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 26.Cokol M, Nair R, Rost B. Finding nuclear localization signals. EMBO Rep. 2000;1:411–415. doi: 10.1093/embo-reports/kvd092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rubtsov YP, Zolotukhin AS, Vorobjev IA, Chichkova NV, Pavlov NA, Karger EM, Evstafieva AG, Felber BK, Vartapetian AB. Mutational analysis of human prothymosin alpha reveals a bipartite nuclear localization signal. FEBS Lett. 1997;413:135–141. doi: 10.1016/s0014-5793(97)00824-7. [DOI] [PubMed] [Google Scholar]
- 28.Valdez BC, Perlaky L, Henning D, Saijo Y, Chan PK, Busch H. Identification of the nuclear and nucleolar localization signals of the protein p120. Interaction with translocation protein B23. J. Biol. Chem. 1994;269:23776–23783. [PubMed] [Google Scholar]
- 29.Liu J, Du X, Ke Y. Mapping nucleolar localization sequences of 1A6/DRIM. FEBS Lett. 2006;580:1405–1410. doi: 10.1016/j.febslet.2006.01.064. [DOI] [PubMed] [Google Scholar]
- 30.Huang HS, Pozarowski P, Gao Y, Darzynkiewicz Z, Lee EY. Protein phosphatase-1 inhibitor-3 is co-localized to the nucleoli and centrosomes with PP1gamma1 and PP1alpha, respectively. Arch. Biochem. Biophys. 2005;443:33–44. doi: 10.1016/j.abb.2005.08.021. [DOI] [PubMed] [Google Scholar]
- 31.Baldi P, Brunak S. Bioinformatics: The Machine Learning Approach. 2nd edn. Cambridge, MA: MIT Press; 2001. [Google Scholar]
- 32.Nielsen H, Engelbrecht J, Brunak S, von Heijne G. Identification of prokaryotic and eukaryotic signal peptides and prediction of their cleavage sites. Protein Eng. 1997;10:1–6. doi: 10.1093/protein/10.1.1. [DOI] [PubMed] [Google Scholar]
- 33.Prieve MG, Guttridge KL, Munguia J, Waterman ML. Differential importin-alpha recognition and nuclear transport by nuclear localization signals within the high-mobility-group DNA binding domains of lymphoid enhancer factor 1 and T-cell factor 1. Mol. Cell Biol. 1998;18:4819–4832. doi: 10.1128/mcb.18.8.4819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hiscox JA. RNA viruses: hijacking the dynamic nucleolus. Nat. Rev. Microbiol. 2007;5:119–127. doi: 10.1038/nrmicro1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Endo A, Kitamura N, Komada M. Nucleophosmin/B23 regulates ubiquitin dynamics in nucleoli by recruiting deubiquitylating enzyme USP36. J. Biol. Chem. 2009;284:27918–27923. doi: 10.1074/jbc.M109.037218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dang CV, Lee WM. Nuclear and nucleolar targeting sequences of c-erb-A, c-myb, N-myc, p53, HSP70, and HIV tat proteins. J. Biol. Chem. 1989;264:18019–18023. [PubMed] [Google Scholar]
- 37.Henderson JE, Amizuka N, Warshawsky H, Biasotto D, Lanske BM, Goltzman D, Karaplis AC. Nucleolar localization of parathyroid hormone-related peptide enhances survival of chondrocytes under conditions that promote apoptotic cell death. Mol. Cell Biol. 1995;15:4064–4075. doi: 10.1128/mcb.15.8.4064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stegh AH, Schickling O, Ehret A, Scaffidi C, Peterhansel C, Hofmann TG, Grummt I, Krammer PH, Peter ME. DEDD, a novel death effector domain-containing protein, targeted to the nucleolus. Embo J. 1998;17:5974–5986. doi: 10.1093/emboj/17.20.5974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Caron E, Cote C, Parisien M, Major F, Perreault C. Identification of two distinct intracellular localization signals in STT3-B. Arch. Biochem. Biophys. 2006;445:108–114. doi: 10.1016/j.abb.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 40.Stark LA, Dunlop MG. Nucleolar sequestration of RelA (p65) regulates NF-kappaB-driven transcription and apoptosis. Mol. Cell Biol. 2005;25:5985–6004. doi: 10.1128/MCB.25.14.5985-6004.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Antoine M, Reimers K, Dickson C, Kiefer P. Fibroblast growth factor 3, a protein with dual subcellular localization, is targeted to the nucleus and nucleolus by the concerted action of two nuclear localization signals and a nucleolar retention signal. J. Biol. Chem. 1997;272:29475–29481. doi: 10.1074/jbc.272.47.29475. [DOI] [PubMed] [Google Scholar]
- 42.Goyal P, Pandey D, Siess W. Phosphorylation-dependent regulation of unique nuclear and nucleolar localization signals of LIM kinase 2 in endothelial cells. J. Biol. Chem. 2006;281:25223–25230. doi: 10.1074/jbc.M603399200. [DOI] [PubMed] [Google Scholar]
- 43.Siomi H, Shida H, Maki M, Hatanaka M. Effects of a highly basic region of human immunodeficiency virus Tat protein on nucleolar localization. J. Virol. 1990;64:1803–1807. doi: 10.1128/jvi.64.4.1803-1807.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bohnlein E, Berger J, Hauber J. Functional mapping of the human immunodeficiency virus type 1 Rev RNA binding domain: new insights into the domain structure of Rev and Rex. J. Virol. 1991;65:7051–7055. doi: 10.1128/jvi.65.12.7051-7055.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nosaka T, Siomi H, Adachi Y, Ishibashi M, Kubota S, Maki M, Hatanaka M. Nucleolar targeting signal of human T-cell leukemia virus type I rex-encoded protein is essential for cytoplasmic accumulation of unspliced viral mRNA. Proc. Natl Acad. Sci. USA. 1989;86:9798–9802. doi: 10.1073/pnas.86.24.9798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Melen K, Kinnunen L, Fagerlund R, Ikonen N, Twu KY, Krug RM, Julkunen I. Nuclear and nucleolar targeting of influenza A virus NS1 protein: striking differences between different virus subtypes. J. Virol. 2007;81:5995–6006. doi: 10.1128/JVI.01714-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Catez F, Erard M, Schaerer-Uthurralt N, Kindbeiter K, Madjar JJ, Diaz JJ. Unique motif for nucleolar retention and nuclear export regulated by phosphorylation. Mol. Cell Biol. 2002;22:1126–1139. doi: 10.1128/MCB.22.4.1126-1139.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cheng G, Brett ME, He B. Signals that dictate nuclear, nucleolar, and cytoplasmic shuttling of the gamma(1)34.5 protein of herpes simplex virus type 1. J. Virol. 2002;76:9434–9445. doi: 10.1128/JVI.76.18.9434-9445.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Boyne JR, Whitehouse A. Nucleolar trafficking is essential for nuclear export of intronless herpesvirus mRNA. Proc. Natl Acad. Sci. USA. 2006;103:15190–15195. doi: 10.1073/pnas.0604890103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Savitsky K, Ziv Y, Bar-Shira A, Gilad S, Tagle DA, Smith S, Uziel T, Sfez S, Nahmias J, Sartiel A, et al. A human gene (DDX10) encoding a putative DEAD-box RNA helicase at 11q22-q23. Genomics. 1996;33:199–206. doi: 10.1006/geno.1996.0184. [DOI] [PubMed] [Google Scholar]
- 51.Gozani O, Feld R, Reed R. Evidence that sequence-independent binding of highly conserved U2 snRNP proteins upstream of the branch site is required for assembly of spliceosomal complex A. Genes Dev. 1996;10:233–243. doi: 10.1101/gad.10.2.233. [DOI] [PubMed] [Google Scholar]
- 52.Neubauer G, King A, Rappsilber J, Calvio C, Watson M, Ajuh P, Sleeman J, Lamond A, Mann M. Mass spectrometry and EST-database searching allows characterization of the multi-protein spliceosome complex. Nat. Genet. 1998;20:46–50. doi: 10.1038/1700. [DOI] [PubMed] [Google Scholar]
- 53.Zhou Z, Licklider LJ, Gygi SP, Reed R. Comprehensive proteomic analysis of the human spliceosome. Nature. 2002;419:182–185. doi: 10.1038/nature01031. [DOI] [PubMed] [Google Scholar]
- 54.Lum LS, Sultzman LA, Kaufman RJ, Linzer DI, Wu BJ. A cloned human CCAAT-box-binding factor stimulates transcription from the human hsp70 promoter. Mol. Cell Biol. 1990;10:6709–6717. doi: 10.1128/mcb.10.12.6709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Muchardt C, Reyes JC, Bourachot B, Leguoy E, Yaniv M. The hbrm and BRG-1 proteins, components of the human SNF/SWI complex, are phosphorylated and excluded from the condensed chromosomes during mitosis. EMBO J. 1996;15:3394–3402. [PMC free article] [PubMed] [Google Scholar]
- 56.Simpson F, Peden AA, Christopoulou L, Robinson MS. Characterization of the adaptor-related protein complex, AP-3. J. Cell Biol. 1997;137:835–845. doi: 10.1083/jcb.137.4.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Politz JC, Yarovoi S, Kilroy SM, Gowda K, Zwieb C, Pederson T. Signal recognition particle components in the nucleolus. Proc. Natl Acad. Sci. USA. 2000;97:55–60. doi: 10.1073/pnas.97.1.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Barbe L, Lundberg E, Oksvold P, Stenius A, Lewin E, Bjorling E, Asplund A, Ponten F, Brismar H, Uhlen M, et al. Toward a confocal subcellular atlas of the human proteome. Mol. Cell Proteomics. 2008;7:499–508. doi: 10.1074/mcp.M700325-MCP200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.