

Abstract
Motivation: Clusters of protein–DNA interaction events involving the same transcription factor are known to act as key components of invertebrate and mammalian promoters and enhancers. However, detecting closely spaced homotypic events from ChIP-Seq data is challenging because random variation in the ChIP fragmentation process obscures event locations.
Results: The Genome Positioning System (GPS) can predict protein–DNA interaction events at high spatial resolution from ChIP-Seq data, while retaining the ability to resolve closely spaced events that appear as a single cluster of reads. GPS models observed reads using a complexity penalized mixture model and efficiently predicts event locations with a segmented EM algorithm. An optional mode permits GPS to align common events across distinct experiments. GPS detects more joint events in synthetic and actual ChIP-Seq data and has superior spatial resolution when compared with other methods. In addition, the specificity and sensitivity of GPS are superior to or comparable with other methods.
Availability: http://cgs.csail.mit.edu/gps
Contact: gifford@mit.edu; mahony@mit.edu
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
The precise physical description of where transcription factors, histones, RNA polymerase II and other proteins interact with the genome provides an invaluable mechanistic foundation for understanding gene regulation. ChIP-Seq (Chromatin immunoprecipitation followed by high-throughput sequencing) has become an indispensable tool for genome-wide profiling of protein-DNA interactions (Barski et al., 2007; Johnson et al., 2007; Robertson et al., 2007).
Computational methods are necessary to predict the location of protein–DNA interaction events from ChIP-Seq data because random variation in the ChIP DNA fragmentation process obscures the actual location of interaction events. Thus while ChIP-Seq DNA sequence reads are mapped to precise bases in the genome, these reads do not manifestly indicate the location of the protein–DNA interaction events that caused them. We define spatial resolution to be the difference between the computationally predicted location of a protein–DNA binding event and the midpoint of its actual location. An ideal computational method for analyzing ChIP-Seq data would accurately localize protein-DNA interaction events (high spatial resolution), would include no false events (high specificity), would include all true events (high sensitivity), and would be able to resolve closely spaced DNA-protein interactions (joint event discovery).
Joint event discovery is important because it can capture cooperative biological regulatory mechanisms in proximal genomic locations (Pepke et al., 2009). Homotypic clusters of transcription factor binding sites (TFBS) have been extensively studied in Drosophila (Lifanov et al., 2003). Such regulatory mechanisms may be common in mammalian genomes as 40–60% of certain ChIP-Seq defined protein-DNA interaction regions contain more than one motif within 200 bp (Jothi et al., 2008; Valouev et al., 2008). Furthermore, homotypic clusters of TFBS occupy nearly 2% of the human genome and may act as key components of almost half of the human promoters and enhancers (Gotea et al., 2010). Thus, homotypic event discovery is necessary to fully reveal the transcription factor regulatory interactions present in ChIP-Seq data.
Existing ChIP-Seq computational methods (Park, 2009; Pepke et al., 2009) do not simultaneously consider multiple events as the cause for observed reads in the context of a probabilistic model at mammalian genome scale. To detect binding events, PeakSeq extends the length of mapped reads to create peaks (Rozowsky et al., 2009), MACS shifts the mapped position of reads a fixed distance towards their 3′-ends (Zhang et al., 2008), FindPeaks aggregates overlapping reads (Fejes et al., 2008), SISSRs identifies positive to negative strand transition points at read accumulations (Jothi et al., 2008), cisGenome scans for the center of modes of the 5′ and 3′ peaks (Ji et al., 2008), and QuEST (Valouev et al., 2008) and spp (Kharchenko et al., 2008) use kernel density estimation methods. All of these methods use statistical detection criteria such as overlapping read counts or read distribution strand symmetry to estimate the location of a protein–DNA interaction event. A recent method named CSDeconv deconvolves proximal binding events using a computed spatial read distribution (Lun et al., 2009), although it is at present computationally impractical on entire mammalian genomes. In addition, recent evaluations showed that while all these methods identified binding sites with a highly significant overlap with the corresponding sequence motif (Laajala et al., 2009), and exhibited similar sensitivity and specificity, there are pronounced differences in their spatial resolution (Wilbanks and Facciotti, 2010).
We present the Genome Positioning System (GPS), a high-resolution genome-wide ChIP-Seq analysis method that can accurately detect closely spaced protein-DNA interaction events (joint events). GPS detects more joint events in synthetic and actual ChIP-Seq data and has superior spatial resolution when compared with other methods. In addition, the specificity and sensitivity of GPS are superior to or comparable with other methods.
2 METHODS
2.1 GPS model overview
GPS has three phases: spatial distribution discovery, event discovery and the determination of event significance. In its first phase, GPS summarizes the observed spatial distribution of reads from protein–DNA interaction events in the input ChIP-Seq data. The farther a mapped read is located from an event, the less likely it is to be caused by the event (Fig. 1b). We assume in GPS that for a given ChIP-Seq experiment, every interaction event will produce the same characteristic distribution of reads. While this assumption will not always be true, we have found that it produces good results in practice. Note that reads from joint events will spatially mix with one another along the genome, presenting a challenge for precisely estimating the multiplicity and exact positions of proximal protein–DNA interaction events (Fig. 1a).
Fig. 1.

GPS probabilistically models ChIP-Seq read spatial distributions. (a) Protein-DNA interaction events at positions 1 and 2 on the genome result in DNA end sequence reads in the ChIP-Seq protocol. (b) The observed spatial read density (blue: ‘+’ strand, red: ‘−’ strand) from ∼4000 CTCF events aligned with respect to the CTCF motif position at each event (c) GPS models ChIP-Seq reads as being generated by a mixture of binding events at every genomic base, with each event producing the characteristic spatial read density. (d) A sparse prior on mixture components causes GPS to assign events to as few bases as possible to explain the observed reads (green and orange reads). In GPS, a given read can be explained by more than one event (yellow reads).
In its second phase, GPS employs a probabilistic mixture model to assign an event probability to every base in the genome. Each potential event's contribution to generating the observed reads is modeled (Fig. 1c and d). A sparse prior on event probabilities provides a complexity penalty that biases events to have their probability mass at a single base position. Event probabilities are selected to maximize the penalized likelihood of observed reads using a multi-resolution EM algorithm that segments the genome into efficiently solvable subproblems. GPS uses the number of reads assigned to a base by the mixture model as a measure of the relative strength of a predicted event at that base.
In its third and final phase, GPS filters discovered events by comparing the number of reads at the predicted events to the corresponding normalized number of reads in the control channel. We compute the statistical significance using the binomial distribution (Rozowsky et al., 2009) and correct for multiple hypothesis testing by applying a Benjamini-Hochberg correction (Benjamini and Hochberg, 1995).
2.2 GPS mixture model
GPS is based on a generative mixture model that describes the likelihood of an observed set of ChIP-Seq reads from a set of protein–DNA interaction events. Each event contributes a distribution of reads surrounding its genomic position to the mixture of reads. We assume that reads are independent conditioned on the locations of their underlying causal events.
GPS performs event discovery by finding the set of protein–DNA interaction events that maximizes the penalized likelihood of the observed ChIP-Seq reads. We consider N ChIP-Seq reads that have been mapped to genome locations R = {r1,…, rN} and M possible protein–DNA interaction events at genome locations B = {b1,…, bM}. We represent the latent assignments of reads to the location of events that caused them as Z = {z1,…, zN}, where zn = j when j is the index of the event located at position bj that caused read n.
The conditional probability of read rn being generated from event j is
where emp(d) is the empirical spatial distribution that models the probability of a read occurring d bases away from its corresponding event position. Strand sense is handled by sn = 0 or sn = 1 if read rn is mapped to the forward strand or reverse strand, respectively. We assume that all the events in one ChIP-Seq experiment have the same empirical spatial distribution. The empirical spatial distribution is obtained from ChIP-Seq data (see below).
The probability of a read is a convex combination of possible binding events
where M is the number of possible events, π denotes the parameter vector of mixing probabilities, and πj is the probability of event j, with ∑j=1M πj = 1.
The overall likelihood of the observed set of reads is then,
Our assumption is that binding events are relatively sparse throughout the genome. To model this assumption, we place a negative Dirichlet prior distribution (Bicego et al., 2007; Figueiredo and Jain, 2002) p(π) on π:
where α is a tuning parameter to adjust the degree of sparseness. If for event j, the value of πj becomes zero (see component elimination below), the model is restructured to eliminate it.
2.3 EM algorithm
We solve for the MAP (maximum a posteriori) solution for π using the Expectation–Maximization (EM) algorithm (Dempster et al., 1977). The complete data log penalized likelihood is
where 1(zn = j) is the indicator function.
We initialize mixing probabilities π with uniform probabilities, πj = 1/M, where j = 1,…, M.
At the E step, we use the current parameter estimates π to evaluate the expectation of Z given R,
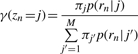 |
We can interpret γ(zn = j) as the fraction of read n that is assigned to event j. This is referred to as a ‘soft assignment’ because read n can be assigned partially to multiple events.
At the M step, on iteration i we find parameter 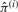 to maximize the expected complete-data log penalized likelihood,
to maximize the expected complete-data log penalized likelihood,
under the constraint ∑j=1M πj = 1. By simplifying, we find
where Nj is the expected number of reads assigned to event j.
As we iteratively estimate  , we use a component elimination method (Figueiredo and Jain, 2002). If Nj ≤ α , we set πj = 0 to eliminate event j. Our final estimate of
, we use a component elimination method (Figueiredo and Jain, 2002). If Nj ≤ α , we set πj = 0 to eliminate event j. Our final estimate of 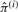 is
is
The sparseness parameter α can be interpreted as the minimum number of reads that an event needs to survive the EM iterations. The EM algorithm is deemed to have converged when the change in likelihood falls below a specified threshold.
Our implementation of component elimination includes two special cases. To avoid premature elimination of components during EM iterations, we start with α = 0 for a number of iterations to allow nascent components to gain support from the data. We then set α to our desired value. This is because when the number of components M is large, no component may have enough initial support to prevent π from being immediately forced to zero. Furthermore, in a single iteration we do not eliminate all the components that meet the criteria Nj ≤ α. Instead, we only eliminate the components with the lowest value of Nj at each iteration. This allows the data points that supported the eliminated components to be re-distributed immediately to support the other components.
At the convergence of the EM algorithm, the GPS mixture model produces a list of non-zero-probability events πj ≠ 0, and the ‘soft’ read assignments to these events γ(zn = j). We do not use the mixing probabilities π in subsequent analysis because we segment the genome into regions for analysis, and π values are dependent on the region analyzed.
We define event strength as the expected number of reads associated with the event. Thus the event strength of event j is calculated as
2.4 Empirical spatial distribution of reads
GPS iteratively estimates the empirical spatial distribution of reads directly from ChIP-Seq data. Given a set of events, we count all the reads at each position relative to the corresponding event positions. Only the base positions within 250 bp of the event are counted because typical ChIP-Seq protocols performs a size selection in the range of ∼150–300 bp (Park, 2009) and we have empirically found that the probability of generating reads at positions further than 250 bp is not significant. The initial set of events for estimating the empirical spatial distribution can be defined by using known motifs or by finding the center of the forward and reverse read profiles (Zhang et al., 2008). With the predicted event positions from GPS, we can re-estimate the empirical spatial distribution and use it for more accurate prediction. This process can be repeated until convergence (Supplementary Fig. 1).
2.5 Statistical significance of predicted events
To evaluate the statistical significance of predicted events when we have a control dataset, we compare the number of reads of the IP event to the number of reads in the corresponding region in the control sample.
For non-overlapping events, we count the number of control reads in the range of the empirical spatial distribution (±250 bp). For joint events, we need to assign control reads to the corresponding events. We run the EM algorithm without the sparse prior (no component elimination, equivalent to α = 0) on the control data, initializing the events j at the same positions as predicted IP events. The M step of EM algorithm is modified as
where Nj = ∑n=1N γ(zn = j).
To account for differences between IP and control dataset sizes, we multiply the control reads by a scaling factor. We divide long non-specific-binding regions (defined by excluding the ‘enriched regions’) into short segments (length 10 Kb) and perform least-squares linear regression using all the read count pairs of IP and control segments that have at least one mapped fragment. The slope of the regression is then the scaling factor, FIP/C, between the read counts from the IP and control (Kharchenko et al., 2008; Rozowsky et al., 2009).
Using a statistical testing method proposed by Rozowsky et al. (2009), we calculate the P-value from the cumulative distribution function for the binomial distribution using the corresponding IP and scaled control read counts,
where k is the scaled control read count, n is ceiling of total count of IP and scaled control reads, P = 0.5, which is the probability under the null hypothesis that reads should occur with equal likelihood from the IP as from the control data.
To correct for multiple hypothesis testing, we apply a Benjamini–Hochberg correction to adjust the P-value (Benjamini and Hochberg, 1995). All the predicted events that are tested for significance are ranked by P-value from most significant to least significant. For each event, the Q-value is given by
where count is the total number of events tested. Significant events are then selected using a Q-value threshold.
If control data is not available, we apply a statistical test proposed by Zhang et al. (2008) that uses a dynamic Poisson distribution to account for local biases. The dynamic parameter of a local Poisson model for the candidate event is defined as
where the λBG, λ5 kb, λ10 kb are λ estimated from corresponding chromosome (background), 5 kb or 10 kb window centered at the event location, to capture the background variability at both global and local scales. The P-value is calculated to be the upper tail of the Poisson distribution,
where nevent is the read count of the candidate event. To correct for multiple hypothesis testing, we apply a Benjamini–Hochberg correction as above.
2.6 Artifact filtering
GPS filters the predicted events by computing the Kullback–Leibler divergence (Kullback and Leibler, 1951) from the empirical read distribution to the read distribution of each predicted event,
where event() is the distribution of non-zero read count of the event computed from the EM algorithm, and emp() is the empirical read distribution with the corresponding positions of the non-zero reads, i is the index of the non-zero read positions.
Events with a Kullback–Leibler divergence value higher than a user defined threshold are discarded.
2.7 Event alignment across independent experiments
To align events across experiments, we first apply the GPS model to the combined data from all experiments to discover a global set of events, and then use this global set of events to condition a modified version of GPS for experiment specific event discovery (Fig. 2). Events from the global set represent a superset of potential events for each experiment. Thus, we allow only the π components corresponding to these global events to be non-zero for the discovery of experiment specific events. A uniform initial weighting is assigned to these possible events. Since the global set of events is sparse, we do not use a sparse prior on π for each condition. The complete alignment algorithm is described in the Supplementary Notes.
Fig. 2.

Probabilistic model for GPS event alignment.
2.8 GPS has an efficient implementation
We have implemented GPS in Java, and our software is available for download from our website (http://cgs.csail.mit.edu/gps).
For computational efficiency GPS independently processes separable genomic regions. We identify separable regions with a conservative method that spatially segments the genome at read gaps that are larger than the width of empirical spatial distribution (500 bp) and further excludes regions that contain fewer than six reads. The segmented protein binding regions are typically a few thousand base pairs long.
To further reduce memory requirements and run time, GPS estimates events in two stages for each region. In the first stage, events are spaced at 5 bp intervals to make a rough estimate of event locations. In the second stage, events are spaced at 1 bp near locations predicted in the first stage.
For the CTCF ChIP-Seq experiment in this study (∼4.2 million IP reads and ∼7.9 million control reads), GPS requires 750 MB of main memory, and runs for 21 min on an AMD 64-bit 2.3 GHz computer.
3 RESULTS
3.1 GPS predictions have higher spatial resolution
We analyzed the performance of GPS on ChIP-seq data profiling the insulator binding factor CTCF (CCCTC-binding factor) (Chen et al., 2008), as the strong CTCF motif allows us to reliably measure spatial resolution. We used GFP ChIP-Seq data (Chen et al., 2008) in the third phase of GPS to control for non-specific binding.
We found that the spatial resolution of GPS on the CTCF data is superior to the spatial resolution produced by seven published ChIP-Seq analysis methods (Fig. 3a, Supplementary Fig. 2): MACS (Zhang et al., 2008), SISSRs (Jothi et al., 2008), cisGenome (Ji et al., 2008), QuEST (Valouev et al., 2008), FindPeaks (Fejes et al., 2008), spp-wtd and spp-mtc (Kharchenko et al., 2008). Because different methods predict different sets of binding events (Supplementary Table 1), we limit our comparison to a matched set of events. From the 34 019 top ranking predictions by each method, 7653 events are predicted by all eight methods and correspond to the same high-scoring CTCF binding motif. Of these matched events, 86.5% of the predictions by GPS are within 20 bp of the CTCF binding motif, while between 65.2% and 75.9% of predictions by other methods are within 20 bp. GPS has an average spatial resolution of 11.08 ± 10.27 bp, compared to 14.50 ± 12.61 bp for SISSRs, 16.07 ± 12.29 bp for MACS, 16.66 ± 14.20 bp for cisGenome, 17.52 ± 13.59 bp for QuEST, 15.22 ± 11.47 for FindPeaks, 16.46 ± 12.95 for spp-wtd and 16.08 ± 14.88 for spp-mtc. SISSRs, MACS and two spp methods were shown to have better spatial resolution than seven other methods in a recent performance evaluation (Wilbanks and Facciotti, 2010), and thus our analysis of CTCF data shows that GPS has superior spatial resolution to these seven methods.
Fig. 3.

GPS improves the effective spatial resolution and accuracy in resolving proximal binding events. (a) Fraction of predicted CTCF binding events with a motif within the given distance with event discovery by GPS, SISSRs, MACS, cisGenome, QuEST, FindPeaks, spp-wtd and spp-mtc. Events shown were predicted by all eight methods and had a CTCF motif within 100 bp. (b) Fraction of binary events recovered vs. the distance between the generated synthetic events for GPS, SISSRs, MACS and QuEST. (c) Example of a predicted binary CTCF event that contains coordinately located CTCF motifs. (d) Number of GABP events discovered by GPS, SISSRs, MACS, cisGenome, and QuEST in regions that contain clustered GABP motifs within 500 bp.
By evaluating the fraction of identified binding sites that contain a CTCF binding motif, we found that GPS, MACS and FindPeaks achieve higher specificity overall than the other methods and GPS performs better for the high-ranking predictions (Supplementary Fig. 3). GPS also achieves marginally better sensitivity in discovering binding events that are supported by CTCF binding motifs (Supplementary Fig. 4).
3.2 GPS discovers more joint events
Using synthetic data we found that GPS is able to detect more joint events than other methods. We generated synthetic events by placing ChIP-seq binding events from actual CTCF data at pre-defined intervals (Supplementary Notes). GPS detects 99.7% of joint events that are 200 bp apart, while SISSRs only detects 54.5–78.0% of joint events that are 200–750 bp apart, respectively. MACS and QuEST detect joint events only when they are more than 280 bp apart. MACS detects 97.9% and QuEST detects 88.4% of joint events when they are separated by 750 bp (Fig. 3b).
GPS is also able to predict more joint events than the other methods we tested on actual ChIP-Seq data. For example, GPS uniquely detects two CTCF events in mouse ES cells over proximal CTCF motifs that are 99 bp apart on chromosome 8 (Fig. 3c). However, the CTCF dataset does not contain a sufficient number of joint events to effectively evaluate the methods on a whole genome scale (Supplementary Table 2). We selected a human Growth Associated Binding Protein (GABP) ChIP-Seq dataset for our evaluation because GABP ChIP-Seq data were previously reported to contain joint events (Lun et al., 2009; Valouev et al., 2008). We identified 581 candidate sites of joint events that all had at least one event detected by all five methods and where each site contains two or more GABP motifs separated by <500 bp. GPS identified joint events in 122 candidate sites, while SISSRs and QuEST detected joint events at fewer than 83 of the candidate sites, and MACS and cisGenome only identified 3 and 5 of the candidate sites as containing joint events respectively (Fig. 3d). In addition, we compared GPS with CSDeconv on a 2 Mb region of GABP ChIP-Seq data that CSDeconv could process (Lun et al., 2009). GPS found four joint events with clustered GABP motifs while CSDeconv found two joint events (Supplementary Table 3 and Supplementary Fig. 5).
3.3 GPS aligns events across multiple experiments
Because transcription factors bind at sequence-specific sites, we would expect them to bind at the same genome location in different conditions. We have generalized the GPS model to optionally align events across multiple experiments by considering the data from independent experiments simultaneously. In alignment mode, a global mixture component biases events to be aligned, and also provides improved spatial resolution for events that are shared by a collection of experiments (Supplementary Fig. 6). In addition, event alignment across experimental conditions provides a straightforward way to detect events that are lost and gained between experimental conditions.
We performed GPS in multicondition alignment mode using human CTCF ChIP-Seq data from two different cell types (GM12878, HUVEC) produced by the ENCODE project (Birney et al., 2007) and determined the distribution of the distances between all the events across conditions. We limit our evaluation to a distance range of <500 bp because events outside of this window are likely to involve independent sites. Within this window, GPS aligns 45.5% of the events to be at the same genomic position (Fig. 4a). The presence of closely spaced unaligned events (Fig. 4a) demonstrates that GPS alignment does not force all the proximal events across conditions to be aligned. When GPS was performed on data from both cell types independently, only 13.7% of events are within 10 bp, and about 30–40% of the event distances are in the range of 10–100 bp (Fig. 4b). This suggests that events occurring at the same genomic position in two conditions are likely to be predicted to occur at different locations with independent event discovery. Thus the alignment mode of GPS provides a simple way to recognize events that are gained and lost in a particular condition.
Fig. 4.

GPS in alignment mode. (a) Histogram of distance between predicted human CTCF events across two conditions shows that GPS in alignment mode aligns proximal events while continuing to discover separated discrete events. (b) Histogram of distance between predicted CTCF events across two conditions when GPS is run independently on each condition.
4 DISCUSSION
GPS is a novel computational method that uses a probabilistic mixture model to predict the most likely positions of binding events at single-base resolution based on a characteristic spatial distribution of reads. Our analysis with synthetic and actual ChIP-Seq data demonstrates the value of our approach in resolving closely spaced joint events and improving event spatial resolution.
GPS's ability to resolve homotypic events from ChIP-Seq data will facilitate the genome-wide study of cooperative binding on gene expression under specific biological conditions. Homotypic binding sites have been shown to act as key components of invertebrate and mammalian promoters and enhancers (Gotea et al., 2010; Lifanov et al., 2003). In addition, modeling based approaches have demonstrated that identifying homotypic binding is important for the faithful reproduction of biological behaviors (Segal et al., 2008).
GPS also provides improved spatial resolution, specificity, and sensitivity when compared with contemporary methods. The high spatial resolution of GPS can be used to produce a position-specific prior (Bailey et al., 2010; Narlikar et al., 2006) that can be used by motif discovery methods to limit motif search to tight genomic regions around events (Supplementary Notes, Supplementary Fig. 8), or can exclude event locations for co-factor motif discovery.
In addition, we expect that alternative empirical read distributions can be used for different kinds of events, such as histone location, as the GPS framework is inherently adaptable to other empirical read distributions.
Supplementary Material
ACKNOWLEDGEMENTS
We thank A. Rolfe and C. C. Reeder for many helpful discussions.
Funding: National Institutes of Health (5R01HG002668, P01-NS055923 and 1-UL1-RR024920 to D.K.G.).
Conflict of Interest: none declared.
REFERENCES
- Bailey TL, et al. The value of position-specific priors in motif discovery using MEME. BMC Bioinformatics. 2010;11:179. doi: 10.1186/1471-2105-11-179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barski A, et al. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. Roy. Statist. Soc. Ser. B. 1995;57:289–300. [Google Scholar]
- Bicego M, et al. Proceedings of the 14th International Conference on Image Analysis and Processing (ICIAP 2007). Modena: IEEE Computer Society; 2007. Sparseness achievement in hidden Markov models; pp. 67–72. [Google Scholar]
- Birney E, et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447:799–816. doi: 10.1038/nature05874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, et al. Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell. 2008;133:1106–1117. doi: 10.1016/j.cell.2008.04.043. [DOI] [PubMed] [Google Scholar]
- Dempster AP, et al. Maximum likelihood from incomplete data via the EM algorithm. J. Roy. Stat. Soc. Ser. B. 1977;39:1. [Google Scholar]
- Fejes AP, et al. FindPeaks 3.1: a tool for identifying areas of enrichment from massively parallel short-read sequencing technology. Bioinformatics. 2008;24:1729–1730. doi: 10.1093/bioinformatics/btn305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figueiredo MAT, Jain AK. Unsupervised learning of finite mixture models. IEEE Trans. Pattern Anal. Mach. Intelligence. 2002;24:381–396. [Google Scholar]
- Gotea V, et al. Homotypic clusters of transcription factor binding sites are a key component of human promoters and enhancers. Genome Res. 2010;20:565–577. doi: 10.1101/gr.104471.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji H, et al. An integrated software system for analyzing ChIP-chip and ChIP-seq data. Nat. Biotechnol. 2008;26:1293–1300. doi: 10.1038/nbt.1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DS, et al. Genome-wide mapping of in vivo protein-DNA interactions. Science. 2007;316:1497–1502. doi: 10.1126/science.1141319. [DOI] [PubMed] [Google Scholar]
- Jothi R, et al. Genome-wide identification of in vivo protein-DNA binding sites from ChIP-Seq data. Nucleic Acids Res. 2008;36:5221–5231. doi: 10.1093/nar/gkn488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharchenko PV, et al. Design and analysis of ChIP-seq experiments for DNA-binding proteins. Nat. Biotechnol. 2008;26:1351–1359. doi: 10.1038/nbt.1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kullback S, Leibler RA. On information and sufficiency. Ann. Math. Stat. 1951;22:79–86. [Google Scholar]
- Laajala TD, et al. A practical comparison of methods for detecting transcription factor binding sites in ChIP-seq experiments. BMC Genomics. 2009;10:618. doi: 10.1186/1471-2164-10-618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lifanov AP, et al. Homotypic regulatory clusters in Drosophila. Genome Res. 2003;13:579–588. doi: 10.1101/gr.668403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lun DS, et al. A blind deconvolution approach to high-resolution mapping of transcription factor binding sites from ChIP-seq data. Genome Biol. 2009;10:R142. doi: 10.1186/gb-2009-10-12-r142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narlikar L, et al. Informative priors based on transcription factor structural class improve de novo motif discovery. Bioinformatics. 2006;22:e384–e392. doi: 10.1093/bioinformatics/btl251. [DOI] [PubMed] [Google Scholar]
- Park PJ. ChIP-seq: advantages and challenges of a maturing technology. Nat. Rev. Genet. 2009;10:669–680. doi: 10.1038/nrg2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pepke S, Wold B, Mortazavi A. Computation for ChIP-seq and RNA-seq studies. Nat. Methods. 2009;6:S22–S32. doi: 10.1038/nmeth.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson G, et al. Genome-wide profiles of STAT1 DNA association using chromatin immunoprecipitation and massively parallel sequencing. Nat. Methods. 2007;4:651–657. doi: 10.1038/nmeth1068. [DOI] [PubMed] [Google Scholar]
- Rozowsky J, et al. PeakSeq enables systematic scoring of ChIP-seq experiments relative to controls. Nat. Biotechnol. 2009;27:66–75. doi: 10.1038/nbt.1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal E, et al. Predicting expression patterns from regulatory sequence in Drosophila segmentation. Nature. 2008;451:535–540. doi: 10.1038/nature06496. [DOI] [PubMed] [Google Scholar]
- Valouev A, et al. Genome-wide analysis of transcription factor binding sites based on ChIP-Seq data. Nat. Methods. 2008;5:829–834. doi: 10.1038/nmeth.1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilbanks EG, Facciotti MT. Evaluation of algorithm performance in ChIP-seq peak detection. PLoS One. 2010;5:e11471. doi: 10.1371/journal.pone.0011471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, et al. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.