

Abstract
Plasma biomarkers studies are based on the differential expression of proteins between different treatment groups or between diseased and control populations. Most mass spectrometry-based methods of protein quantitation, however, are based on the detection and quantitation of peptides, not intact proteins. For peptide-based protein quantitation to be accurate, the digestion protocols used in proteomic analyses must be both efficient and reproducible. There have been very few studies, however, where plasma denaturation/digestion protocols have been compared using absolute quantitation methods. In this paper, 14 combinations of heat, solvent [acetonitrile, methanol, trifluoroethanol], chaotropic agents [guanidine hydrochloride, urea], and surfactants [sodium dodecyl sulfate (SDS) and sodium deoxycholate (DOC)] were compared with respect to their effectiveness in improving subsequent tryptic digestion. These digestion protocols were evaluated by quantitating the production of proteotypic tryptic peptides from 45 moderate- to high-abundance plasma proteins, using tandem mass spectrometry in multiple reaction monitoring mode, with a mixture of stable-isotope labeled analogues of these proteotypic peptides as internal standards. When the digestion efficiencies of these 14 methods were compared, we found that both of the surfactants (SDS and DOC) produced an increase in the overall yield of tryptic peptides from these 45 proteins, when compared to the more commonly used urea protocol. SDS, however, can be a serious interference for subsequent mass spectrometry. DOC, on the other hand, can be easily removed from the samples by acid precipitation. Examining the results of a reproducibility study, done with 5 replicate digestions, DOC and SDS with a 9 h digestion time produced the highest average digestion efficiencies (~80%), with the highest average reproducibility (<5% error, defined as the relative deviation from the mean value). However, because of potential interferences resulting from the use of SDS, we recommend DOC with a 9 h digestion procedure as the optimum protocol.
Keywords: protein digestion, deoxycholate, urea, sodium dodecyl sulfate, heat denaturation, solvent denaturation
Introduction
Plasma is an extremely complex (1010-fold protein concentration range) and highly proteinaceous (60–80 mg/mL) matrix that is frequently used for the determination of disease bio-markers.1 Most methods of mass spectrometry-based protein quantitation rely on the quantitation of peptides as molecular surrogates for their parent proteins. A key assumption of all such methods is that a protein is converted to its component peptides reproducibly and efficiently. If the abundance of a selected peptide is used to represent the abundance of the protein, then this efficiency is assumed to be 100%. This, however, has previously been shown not to be the case.2,3 Any variation in digestion efficiency will affect the amount of peptide available for detection, which can adversely affect the reproducibility of the entire analysis and the accuracy of the results.
For multiple reaction monitoring (MRM) and other quantitative proteomics methods based on comparing the signals of native peptides to the signals of standard peptides, quantitation of a protein is based on the amounts of specific selected peptides. [Note: for a review of protein quantitation methods, see ref 4; peptide-based quantitation methods include AQUA,5,6 SISCAPA,7,8 MRM,9 and iMALDI.10-12] Thus, the completeness of digestion is not really important. In other words, it does not matter whether the entire protein is digested because only the production of these specific target peptides is important. Digestion of the remainder of the protein is not important. Whether the digestion goes to completion (i.e., whether the digestion process is “finished”) is important, but only because it affects the reproducibility of the results. If the digestion consistently and reproducibly only went to 50% completion, for example, and if this fact were known, it could be corrected for. However, the percent completion of digestion is usually not known, and if the percent completion of digestion is not reproducible, this would lead to errors in both absolute and relative quantitation measurements.
Why does the digestion process vary from protein to protein? There are structural reasons for variations in protein digestibilities. These include disulfide bridges, specific folds (as in prions), solubility issues (membrane proteins), glycosylation, and combinations of these features that can keep the enzyme from being able to access all or some of the cleavage sites. There are biological reasons that have led to the evolution of these structural features. Some proteins function as protease inhibitors (e.g., eppin), others are located in an enzyme-rich environment and must themselves be resistant to proteolysis. Proteolytic enzymes in particular need to be resistant to autolysis. This means that not all proteins are alike with respect to ease of proteolytic digestion, and this can lead to variabilities in digestion efficiencies between proteins. However, because enzymes are, in general, more resistant to proteolysis, it also provides a “window of opportunity” for the selection of digestion conditions where the target proteins can be unfolded but the enzyme itself is not denatured and remains active.
Previous studies comparing digestion efficiency could only monitor the “completeness” of the digestion by determining the sequence coverage of the peptides produced from target proteins3,13,14 or by comparing the number of successful protein or peptide identifications.3,14-17 “Completion” of the digestion was determined by monitoring peptide abundances to determine the point at which there was no further change in either peptide abundances or total peptide yield.
The ideal denaturing and digestion protocol would be one where complete digestion of the protein occurred in a short time period, resulting in the maximum observed peptide signal, followed by a “steady state” condition where no change in the observed peptide concentrations were observed. Such a protocol would be robust in that small changes in digestion time would not be an important factor, resulting in optimum digestion reproducibility.
In this current study, by using a set of stable-isotope labeled peptides that were developed for the quantitation of proteins in human plasma, we are now able to determine and compare the absolute amounts of peptides formed from 45 plasma proteins in a single analysis, when different denaturation/digestion procedures are used. Being able to measure the absolute quantities of the peptides produced allows an accurate and quantitative comparison of the digestion efficiencies of the 14 different denaturation/digestion protocols studied.
Materials and Methods
Human Plasma Collection
Plasma was collected in-house from a volunteer who provided written informed consent. Blood from a healthy male donor who had fasted for 16 h was collected by venous puncture using a 21-gauge Becton Dickinson (BD) Sample Needle (Becton Dickinson, Oakville, ON). Sixty milliliters of blood was collected in twenty 3.0 mL lavender Vacutainer K2 EDTA tubes (Becton Dickinson), which contain 5.4 mg EDTA per tube. The samples were immediately centrifuged twice at 1,000 × g for 15 min at room temperature to remove any cells. The plasma was then aliquoted into 1.0 mL sterile cryotubes and immediately frozen. The collection process took less than 1 h, and the plasma samples were kept at −80 °C.
Reagents and Chemicals. Chemicals and Solvents
All reagents were ACS grade or higher; all solvents used, including water, were LC/MS grade. Urea Sigma Ultra, ammonium bicarbonate (ABC), sodium deoxycholate (DOC), guanidine hydrochloride (GnHCl), 2,2,2,-trifluoroethanol (TFE), iodoacetamide (IAA), and formic acid were purchased from Sigma-Aldrich (Oakville, ON). Sodium dodecyl sulfate electrophoresis grade (SDS), Bond Breaker TCEP Solution (TCEP), Optima LC/MS grade acetonitrile (ACN), and Optima LC/MS grade water were purchased from Thermo-Fisher Scientific (Waltham, MA). LC/MS grade methanol was purchased from EMD Chemicals (Gibbstown, NJ). Sequencing-grade modified trypsin Gold was purchased from Promega (Madison, WI).
Stable-Isotope Labeled Internal Standard (SIS) Peptides
Proteotypic tryptic peptides containing isotopically-coded amino acids ([13C6]Arg or [13C6]Lys) representing 45 high- and moderate-abundance plasma proteins (Supporting Information Table 1) were synthesized at a 5 μmol scale using Fmoc chemistry on a Protein Technologies Prelude peptide synthesizer (Tuscon, AZ). An in-house cocktail of these 45 isotopically-labeled peptide standards peptides was created, with the concentration of the internal standard mixture balanced to give a stable-isotope labeled peptide peak area within a factor of 10 of the endogenous peptide peak area when the stable-isotope labeled peptide is added to a standard plasma tryptic digest.9
Denaturation Protocols
Plasma tryptic digestions were carried out using various chemical protocols and solvent protocols, alone and in combination. A common digestion method, using DOC as the denaturant, was included as a control during each experiment so that the methods could be accurately compared. Figure 1 outlines the experimental design for all of these different extraction protocols; the numbers refer to the specific stage at which denaturants were added in each protocol (Table 1).
Figure 1.

Analytical scheme for comparison of digestion methods. The numbers refer to the steps described in Table 1.
Table 1.
Summary of Reagents Used to Denature EDTA-Plasma Prior to or during Tryptic Digestion
| denaturing condition | addition during workflow step no. |
denaturing concentration | concentration during digestion |
heat | |
|---|---|---|---|---|---|
| chaotrope | urea | 1 | 8M | <1 M | N |
| urea | 1 | 8M | <1 M | Y | |
| GnHCl | 1 | 5.6 M | <1 M | N | |
| GnHCl | 1 | 5.6 M | <1 M | Y | |
|
| |||||
| surfactant | DOC | 2 | 5% w/v | 1% w/v | N |
| DOC | 2 | 5% w/v | 1% w/v | Y | |
| SDS | 2 | 0.11% w/v | 0.025% w/v | N | |
| SDS | 2 | 0.11% w/v | 0.025% w/v | Y | |
|
| |||||
| solvent | TFE | 1 | 50% v/v | 5% v/v | N |
| MeOH | 3 | 20% v/v | 20% v/v | N | |
| ACN | 3 | 40% v/v | 40% v/v | N | |
|
| |||||
| combination | TFE + MeOH | 1 | 50% v/v | 5% v/v | N |
| 3 | 20% v/v | 20% v/v | |||
| DOC + MeOH | 2 | 5% w/v | 1% w/v | N | |
| 3 | 20% v/v | 20% v/v | |||
| DOC + TFE | 2 | 5% w/v | 1% w/v | N | |
| 1 | 50% v/v | 5% v/v | |||
Urea Denaturation Protocol
Both raw and heat-denatured (boiled) plasma samples were prepared for urea denaturation as follows. To prevent degradation of the urea into cyanate ions that can carbamylate primary amines when exposed to heat,18 the plasma samples were heat-denatured prior to urea denaturation. A 50 μL aliquot of 10-fold diluted plasma (25 mM ABC) was heat-denatured by boiling for 5 min at 100 °C. This sample was allowed to cool on ice for 5 min, followed by chemical denaturation with 62.5 μL of 8 M urea. After approximately 5 min, the sample was diluted with 257.1 μL of 25 mM ABC, reduced with 41.1 μL of 50 mM TCEP (5 mM final concentration), and alkylated with 45.6 μL of 100 mM iodoacetamide (10 mM final concentration) as indicated below. The urea concentration was reduced to 1 M during digestion by dilution with 25 mM ABC in order to maintain the activity of the trypsin.
For urea denaturation without heat, 5 μL of undiluted human plasma was denatured with 40 μL of 9 M urea. After approximately 5 min, the urea-denatured sample was diluted with 25 mM ABC to give 10-fold diluted plasma sample. The urea concentration during digestion was reduced to 0.72 M by dilution with 25 mM ABC to maintain the activity of the trypsin.
Guanidine HCl Protocol
The GnHCl digestion protocol of Buscher et al.19 in which 1.0 M GnHCl was used was modified in accordance with the instructions in the Promega trypsin product insert.20 The GnHCl concentration was kept below 1.0 M during tryptic digestion in order to maintain the activity of the trypsin in the presence of this strong chaotropic agent. Both raw and heat-denatured (boiled) samples were prepared as in the urea protocol, with the following modifications: a 5 μL aliquot of undiluted plasma was denatured with 75.6 μL of 6 M GnHCl (in 25 mM ABC) at which point the sample was either boiled for 5 min or left on ice. Both samples were then diluted with 289.6 μL of 25 mM ABC, thereby reducing the GnHCl concentration to 0.9 M during digestion.
SDS Protocol
Both raw and heat-denatured (boiled) plasma samples were prepared as in the urea protocol with the following modifications: a 50 μL aliquot of 10-fold diluted plasma (25 mM ABC) was denatured with 62.5 μL of 0.2% w/v SDS at which point the sample was either boiled for 5 min or left on ice. Both samples were then diluted with 257.1 μL of 25 mM ABC, reduced with 41.1 μL of 50 mM TCEP (5 mM final concentration), and alkylated with 45.6 μL of 100 mM iodoacetamide (10 mM final concentration) as indicated below. The final SDS concentration during digestion was 0.025% w/v to maintain the activity of the trypsin.
DOC Protocol
Both raw and heat-denatured (boiled) plasma samples were prepared as in the urea protocol, but with the following modifications: a 50 μL aliquot of 10-fold diluted plasma (25 mM ABC) was denatured with 50 μL of 10% w/v DOC at which point the sample was either boiled for 5 min or left on ice. Both samples were then diluted with 269.6 μL of 25 mM ABC, reduced with 41.1 μL of 50 mM TCEP (5 mM final concentration), and alkylated with 45.6 μL of 100 mM iodoacetamide (10 mM final concentration) as indicated below. The DOC concentration during digestion was reduced to 1% w/v which is compatible with trypsin activity.21
TFE Protocol
A 5 μL aliquot of undiluted plasma was denatured with 45 μL of 50% v/v TFE in 25 mM ABC to give 10-fold diluted plasma. Samples were diluted with 319.6 μL of 25 mM ABC, reduced with 41.1 μL of 50 mM TCEP (5 mM final concentration), and alkylated with 45.6 μL of 100 mM iodoacetamide (10 mM final concentration) as indicated below. The TFE concentration during digestion was reduced to 5% v/v by dilution with 25 mM ABC to maintain the activity of the trypsin.
ACN Protocol
A 50 μL aliquot of 10-fold diluted plasma in 25 mM ABC was diluted with 157.6 μL of 25 mM ABC, reduced with 23.1 μL of 50 mM TCEP (5 mM final concentration), and alkylated with 25.6 μL of 100 mM iodoacetamide (10 mM final concentration) as indicated below. Immediately prior to trypsin addition, ACN was added to give a final concentration of 40% v/v during digestion.13 The solvent addition was done prior to the addition of the trypsin in order to ensure that the plasma samples and the trypsin were only exposed to 40% v/v ACN and to prevent protein precipitation.13,22
MeOH Protocol
A 50-μL aliquot of 10-fold diluted plasma in 25 mM ABC was diluted with 238.6 μL of 25 mM ABC, reduced with 32.1 μL of 50 mM TCEP (5 mM final concentration), and alkylated with 35.6 μL of 100 mM iodoacetamide (10 mM final concentration) as indicated below. Prior to trypsin addition, MeOH was added to give a final concentration of 20% v/v during digestion. Solvent was added prior to the addition of the trypsin in order to ensure that plasma samples and trypsin were only exposed to 20% v/v MeOH and to prevent protein precipitation.23
Reduction, Alkylation, and Digestion
All of the denatured samples were reduced with 5 mM TCEP, which corresponds to a 22.5-fold excess over the ~26 mM concentration of protein cysteine in plasma that we have calculated from the known plasma protein concentration. The reaction was kept at 60 °C for 30 min. Samples were alkylated with 10 mM iodoacetamide (which corresponds to a 50-fold excess over the calculated plasma protein cysteine concentration24) at 37 °C for 30 min in the dark. Denatured samples not compatible with these incubation temperatures (i.e., those containing urea) were reduced and alkylated at room temperature. Modified sequencing-grade trypsin was added to the reduced and alkylated samples (43.8 μL of 0.4 mg/mL trypsin in 25 mM ABC) at a 20:1 protein/enzyme ratio, and samples were digested at 37 °C. Plasma was diluted 100-fold during the sample preparation process, resulting in a 500 μL final volume for all samples. Samples were collected in 20 μL aliquots at 0.5, 1, 2, 4, 6, 9, 12, and 23 h time points. The tryptic digestion was stopped by acidifying the sample with 80 μL of a 0.5% formic acid solution containing the cocktail of 45 SIS peptide standards. The sample was stored at −80 °C until analysis.
LC-MRM/MS Analysis
Prior to MS analysis, the digested samples containing DOC were centrifuged (Eppendorf centrifuge 5415D, Mississauga, ON) at room temperature for 2 min at 16,000 × g to effectively pellet DOC and remove it from the digest. All digests were then desalted and concentrated by solid phase extraction (SPE) using Waters Oasis (10 mg) columns according to the manufacturer's instructions (Waters, Mississauga, ON). Briefly, the SPE columns were conditioned and equilibrated with 1 mL MeOH and 1 mL dH2O, respectively. The sample solution (90 μL sample + 500 μL 0.1% formic acid) was then loaded onto the column, and the columns were washed with 1 mL dH20. The samples were eluted with 200 μL 50% ACN, 0.1% formic acid and lyophilized (Thermo Electron Corporation, ModulYoD Freeze-Dryer). Lyophilized samples were rehydrated with 0.1% formic acid to give 1 μg/μL protein concentrations.
An Eksigent NanoLC-1Dplus HPLC (Dublin, CA) coupled online to an Applied Biosystems/MDS Sciex 4000 QTRAP was used for the analyses. The desalted plasma digest samples (1 μL) were injected onto a reversed-phase capillary column (75 μm i.d. × 15 cm) packed in-house with Magic C18AQ (5 μm i.d. particles, 100 Å pore size, Michrom, Auburn, CA). The flow rate was 300 nL/min. A sample loading time of 6 min at 100% solvent A (2% acetonitrile, 0.1% formic acid) was followed by a 32-min linear gradient from 0 to 23% solvent B (98% acetonitrile, 0.1% formic acid), a 9-min linear gradient from 23% to 43% solvent B, a 2-min linear gradient to 80% solvent B held for 2 min, followed by a 10-min column equilibration step with 100% solvent A.
An Applied Biosystems (AB) Sciex 4000 QTRAP (Applied Biosystems, Streetsville, ON) equipped with a nanoelectrospray ionization source and controlled by Analyst 1.5 software was used for all of the LC-MRM/MS analyses. The following parameters were used: 1900–2000 V ion spray voltage; 25 nitrogen curtain gas; 150 °C interface heater temperature, 3.5 × 10−5 Torr vacuum gauge pressure; unit resolution on Q1 and Q3.
Uncoated fused silica emitter tips (20 μm inner diameter, 10 μm tip, New Objective, Woburn, MA), 1–3 L/min Ion Source Gas (GS1), and postcolumn addition makeup flow were used. Make-up solvent (80% v/v isopropanol, 10% v/v acetonitrile) at a flow rate of 50 nL/min was added using a PicoPlus 11 syringe pump (Harvard Apparatus, Holliston, MA). The 60 min MRM acquisition method was constructed using 90 MRM pairs with the tuned DP and CE voltages, which had previously been developed for these peptides, and a default collision cell exit potential of 23 V, (see Supporting Information Table 2, from ref 9). As was done in this previous study, an 8 min wide scheduled MRM retention time window with a 2 min cycle time was used for all data acquisition.
Data Analysis
MultiQuant 1.1 (Applied Biosystems), with the integration algorithm MQL for peak integration, was used for MRM data analysis. Peak integration parameters were set with a total smoothing width of 1 point, a retention window of 120 s, a peak splitting factor of 2 points, and report largest peak enabled. Other peak integration parameters were used at default settings. Peak area ratios were generated by dividing the integrated peak areas of the extracted ion chromatograms for the natural peptide by the peak areas of the coeluting, equivalent MRM transitions of the isotopically-labeled peptide.
Results and Discussion
Currently there are numerous published protocols for proteolytic digestion, involving various methods of chaotropic, surfactant, and solvent based denaturation.2,5,13,14,16,17,19,21,25-27 It is well-documented that altering trypsin digestion conditions can affect which peptides are observed from proteins in proteolytic digests and in turn the overall sequence coverage of proteins,17 as was clearly demonstrated in the characterization of the urine proteome by Adachi et al.28 Primary and secondary structure of proteins can confer differing levels of resistance to proteolysis, which can result in poor accessibility of trypsin to internal cleavage sites.29 Advantages of in-gel digestion for mass spectrometry-based identification of hydro-phobic membrane proteins can largely be attributed to the denaturing power of SDS during the sample preparation step prior to gel electrophoresis. Recent hybrid methods for sample preparation have focused on combining the denaturing properties of SDS with the ease and improved recovery of in-solution digestion.30
With the intent of adapting sample preparation for MRM analysis to high-throughput liquid-handling robotics platforms, protocols were selected on the basis of their simplicity, cost effectiveness, and previous use in proteomics sample preparation, as well as on compatibility of the chemical or solvent system with tryptic activity and downstream mass spectrometric analysis.
Overnight (16 h) trypsin digestion at 37 °C is routinely used in proteomics, but this is for convenience only. Examining the tryptic digests at defined time points during the digestion process could give insights as to when digestion has actually reached completion. Decreasing the time required for tryptic digestion could dramatically impact sample throughput and time to first result, as well as the reducing the amount of tryptic autolysis products.
A schematic workflow for the digestion of human plasma is shown in Figure 1. Whenever possible, all digests were prepared from a common EDTA-plasma standard sample. This human plasma sample was diluted 1/10 with 25 mM ABC and was used for all digestions to minimize errors that might result from pipetting small volumes of highly proteinaceous material. The volumes of all samples were kept identical during reduction, alkylation, and digestion. To ensure that the amount of trypsin was not a limiting factor in our experiments, all samples were digested using a 1:20 ratio of trypsin to substrate rather than the 1:50 ratio commonly cited in the literature.17,21
Digestion Profiles
The 45 proteins can be divided into 4 groups depending on their digestion profiles (Supporting Information Figure 1). For most proteins, including plasma retinol binding protein and transthyretin, a maximum signal was achieved within 4 h of digestion followed by a “plateau” over the following 20 h. This is the ideal case and allows the development of a robust digestion protocol where small variations in digestion time would not have a significant impact on peptide production. The peptide abundances in a second group of analytes, which included fibrinogen γ chain and apolipoprotein A-II, continued to increase during the 24 h period studied, without ever reaching a maximum. Other proteins, like haptoglobin β and α-2-antiplasmin, also generated a maximum signal within 4 h of digestion, but the signal then decreased as the period of digestion increased. For a fourth group of proteins, which included vitamin D binding protein and complement component C9, the shape of the digestion efficiency curve depended on the digestion method used. For these 3 groups of “non-ideal” proteins, consistent digestion times are required in order to achieve reproducible results.
Calculation of Digestion Efficiencies
SIS peptides were added to all time-point samples in equal amounts. To obtain the plots shown in Figure 2, peak area ratios (light:heavy) were generated as integrated peak areas of the endogenous peptide (light) to the peak areas of their coeluting SIS peptide (heavy). To provide a reference method, each set of experiments contained a DOC digest as a common standard condition (green line). A linear data-normalization step using the average response from 4 DOC data sets was used to correct the time points of other treatments and to facilitate comparisons between experiments. To generate these plots, a two point rolling average was applied in order to reduce local variation (note that rolling averaging affects only the line and not the data points in these plots).
Figure 2.


(A) Representative plots showing the production of target proteotypic peptides from 6 plasma proteins as a function of chaotropic or surfactant denaturation and digestion time. (B) Representative plots showing the production of target proteotypic peptides from the same 6 plasma proteins as a function of solvent denaturation and digestion time. (C) Representative plots showing the production of target proteotypic peptides from the same 6 plasma proteins as a function of a combination of solvent denaturation, chaotropic or surfactant denaturation, and digestion time. (D) Representative plots showing the production of target proteotypic peptides from 6 plasma proteins as a function of heat denaturation and digestion time.
Denaturation with Chaotropic Agents and Surfactants
A literature survey of plasma proteomics-related publications reveals that urea is a widely used chaotropic agent for solubilizing protein pellets, IEF sample prep, and general in-solution enzymatic digestion of proteins.31 The mechanism of protein denaturation by urea has been widely studied (ref 32 and references therein) and is thought to start with urea attachment to charged histidine and then to the positively charged amino acids and the amide groups,33 leading to disruption of hydrogen bonds, followed by solvation with water and urea.32,34 Urea, however, can degrade into ammonium cyanate when exposed to heat, which leads to carbamylation of free primary amines.
Stronger chaotropic chemicals that are incapable of modifying proteins, such as GnHCl and SDS, can also be used during tryptic digestion,19,35 but each of these chemicals presents additional challenges. The concentration required for GnHCl denaturation can severely reduce the enzymatic activity of the trypsin.36 SDS is not compatible with direct sample injection as it is not possible to remove SDS from samples with reversed-phase HPLC configurations. Even low levels of SDS (<0.01%) interfere with reversed-phase separation of peptides37,38 and suppress ionization in both MALDI and electrospray techniques, and off-line removal of SDS (for example, with SCX Ziptips) adds additional steps to the protocol and can lead to sample loss.
Sodium deoxycholate (DOC) is a bile salt surfactant with unique properties that make it compatible with easy sample preparation for MS-based proteomics. DOC is inexpensive and is easily removed prior to MS analysis or solid phase extraction, as it is acid-insoluble and precipitates at low pH levels.39 Additionally, trypsin retains its full activity in 1% w/v DOC solutions.17
Analysis of the tryptic peptides from all 45 proteins revealed that the rate of digestion for these analytes vary widely even within a single chemical denaturation method (urea, GnHCl, DOC, or SDS) (Figure 2 and Supporting Information Figure 1). Six representative plots of tryptic peptide production versus time, as a function of the chemical denaturant used, are shown in Figure 2A, and the remainder of these plots are provided as Supporting Information Figure 1.
It proved difficult to maintain the activity of trypsin in digests that contained GnHCl as a denaturant. The concentration of GnHCl was reduced to 0.9 M prior to the addition of trypsin, as recommended by Promega40 and in accordance with values found in the literature.19 However, in this study we found that GnHCl-containing digests consistently showed lower efficiencies of digestion, resulting in lower peak area ratios in the MRM analysis than with the other methods. SDS was found to increase the production of proteotypic peptides from digestion-resistant proteins when digestion was performed for longer than 9 h. SDS, however, is associated with contamination of the HPLC column and signal suppression37,38 and is therefore not the ideal denaturant. Our results indicate that DOC could be used as an alternative to SDS.
Solvent Denaturation
Solvent denaturation protocols are attractive as they alter the solution chemistry of the digestion by causing partial unfolding or some alteration of the protein's secondary structure. Volatile solvents can be easily removed from samples prior to MS analysis. Non-salt-based denaturation protocols have also been shown to provide reproducible and sensitive detection when compared to traditional protocols using urea.16 Russell et al.13 and Simon et al.22 reported that trypsin maintains its proteolytic activity in mixed-solvent systems containing up to 80% organic solvent. The use of organic solvents as denaturants must be optimized to determine the proper concentration suitable for both solubilization of proteins and retention of tryptic activity.
Our solvent digestion protocol using MeOH was adapted from studies indicating that the addition of MeOH enhances the activity of trypsin when compared to aqueous conditions.27,41 The solvent digestion protocol using ACN was adapted from Russell et al.13 In both protocols, solvent is added immediately prior to the addition of trypsin in order to prevent protein precipitation before digestion. A tryptic digestion protocol using 50% v/v TFE as a denaturant, reported by Adachi et al.,28 was modified for use in our experiments. As was the case for methanol and acetonitrile, consideration was given to obtaining concentrations of TFE solvent that both denature plasma proteins and are compatible with tryptic digestion. To ensure that the plasma was exposed to an appropriate concentration of TFE, denaturation was done prior to sample dilution. Sample dilution reduced the concentration of TFE to 5% v/v, which was compatible with efficient tryptic enzyme activity.
Figure 2B shows representative plots showing the production of target proteotypic peptides, from the same 6 plasma proteins as shown in Figure 2A, as a function of solvent denaturation and digestion time. All of the remaining digestion profiles are provided in Supporting Information Figure 1. DOC was included as a control treatment to ensure reproducibility and allow for comparison between solvent and chemical denaturant experiments.
Both MeOH and TFE produced tryptic digestion profiles similar to those obtained with DOC. Although other studies have reported the use 40% acetonitrile, and even up to 80% acetonitrile as a denaturant,42 in our experiments, a significant reduction in digestion efficiency was observed for all 45 analytes in the presence of 40% v/v acetonitrile, even though no protein precipitation was observed. However, moderate digestion of some proteins, such as haptoglobin β (Figure 2B), was observed when using acetonitrile denaturation. These results indicate that it is possible to obtain tryptic digestion efficiencies equivalent to that obtained with DOC by using solvent denaturation.
Combined Chemical and Solvent Denaturation
The data above indicate that tryptic digestion efficiencies equivalent to that obtained with DOC (a chemical denaturant) could be obtained with solvent denaturation, using MeOH or TFE. To determine if the digestion protocols could be further improved, solvent denaturation was combined with sodium deoxycholate denaturation. Digestion protocols were designed to examine the effects of the following combined treatments: DOC/TFE, DOC/MeOH, and TFE/MeOH (Figure 2C).
The combination of chemical and solvent denaturation did not significantly improve digestion efficiencies, compared to the digestion efficiencies obtained with sodium deoxycholate alone. The strongest combined denaturation method was found to be DOC/MeOH, with 29 proteins attaining a maximum signal under these conditions. However, it should be noted that 22 proteins attained their maximum signals with DOC alone in this experiment (Figure 3).
Figure 3.

Ranking of (A) chemical, (B) solvent, and (C) combined denaturant protocols based on the number of proteins (i.e., proteotypic peptides) reaching maximum signal using each digestion protocol. The maximum signal is considered to be the highest peak area ratio for a single analyte that was obtained during the experiment, under any of the experimental conditions used. In each part the maximum average peak area ratio was determined for each of the 45 analytes and sorted by time and the four conditions. For example in part A, none of the 45 analytes achieved maximum signal using GnHCl at any time during digestion when compared to SDS, urea, and DOC.
Although proteotypic peptides for 41 of the 45 proteins studied reached their maximum signals and improvement of overall digestion efficiency could be achieved for certain proteins, complete digestion of other proteins was never attained, and the digestion efficiencies of several proteins were even reduced when these combined systems were used (Figure 2C). For example, both α-1-acid glycoprotein and antithrombin-III peptide production decreased 2-fold when reduced with a combined denaturation method. This suggests that the combination of denaturants may have reduced the efficiency of the trypsin and upset the balance between creating a strongly denaturing environment and maintaining trypsin enzyme activity. Tryptic digestion profiles of all 45 proteins in the presence of all of the chaotropic and solvent denaturation methods are provided in Supporting Information Figure 1.
Heat Denaturation
Heat is often used as a strong denaturing condition in the analysis of proteins, assisting in the unfolding of proteins. Heat denaturation is routinely used in SDS-PAGE sample preparation and is widely regarded as one of the strongest methods of protein denaturation. In this portion of the study, the chemical denaturation protocols using SDS, urea, DOC, and GnHCl were modified for use with heat denaturation. Each of the four chemical denaturant protocols was applied to two aliquots of plasma, all of which were taken from the same stock sample of human EDTA-plasma, one of which was heat-treated. Our results indicated that heat denaturation does not have a dramatic effect on the rate of tryptic digestion when combined with SDS, DOC, or GnHCl denaturation. However, heat denaturation of plasma samples prior to chemical denaturation with urea improved the efficiencies of the urea digestion protocol for 32 out of the 45 proteins examined (Figure 2D and Supporting Information Figure 1).
Comparison of Methods by Frequency of Maximum Signal
To compare the digestion results from these different digestion protocols, we determined not only the maximum signal but the frequency at which this maximum intensity was reached using the 10 protocols that showed significant differences (protocols using heat did not show significant effect and were not included in this portion of the study). As can be seen from Figure 3, the maximum observed peptide production from 38 of the 45 proteins was obtained with either the SDS or the DOC protocols, with 41 of the 45 proteins achieving their maximum signals using these protocols. Thus, for proteins that are hard to solubilize, detergents such as SDS and DOC may be better choices of denaturants than urea or GnHCl. Over the range of denaturing conditions studied, TFE and MeOH were found to outperform DOC for 7 of the 45 proteins studied. However, the use of ACN resulted in no peptide reaching maximum peptide signal compared with the other solvents.
Comparison of Methods, Including Reproducibilities
It is difficult to rank these methods by digestion efficiency alone, especially since the results differ so widely from protein to protein. Although the profiles are very informative, each time point represents only a single technical replicate. Therefore, since the reproducibility of the method is of paramount importance for accurate quantitation, we chose to characterize the complete technical reproducibility of these protocols by generating 5 replicate samples of a standard plasma sample digested for 4, 9, and 16 h. We selected the 4 most promising methods on the basis of the results above (DOC, DOC/MeOH, DOC/TFE, and TFE/MeOH) and compared them to SDS (arguably the strongest denaturant in the study) and urea (representing the most commonly used method), for a total of 6 digestion protocols, 3 digestion times, and 5 replicates, giving 90 unique digests. The results of these replicate analyses are shown in Table 2 and Figures 4-7.
Table 2.
Reproducibilities of Six Digestion Protocols and 3 Digestion Times, 5 Replicate Digestions; bold = Highest Digestion Efficiency Observed for Each Protein
| protein | time (h) | DOC | SDS | urea | TFE/MeOH | DOC/TFE | DOC/MeOH |
|---|---|---|---|---|---|---|---|
| heparin cofactor II | 4 | 0.92 ± 4.1% | 0.96 ± 2.6% | 1.00 ± 3.1% | 0.93 ± 3.4% | 0.95 ± 7.0% | 0.94 ± 6.3% |
| 9 | 0.88 ± 5.0% | 0.91 ± 3.9% | 0.96 ± 4.9% | 0.90 ± 7.3% | 0.92 ± 4.5% | 0.92 ± 5.4% | |
| 16 | 0.86 ± 2.8% | 0.92 ± 3.9% | 0.92 ± 4.7% | 0.93 ± 5.8% | 0.91 ± 3.4% | 0.94 ± 2.5% | |
| complement C3 | 4 | 1.00 ± 4.2% | 1.00 ± 2.0% | 0.99 ± 3.1% | 0.84 ± 3.1% | 0.91 ± 2.6% | 0.81 ± 10.1% |
| 9 | 0.95 ± 3.9% | 0.94 ± 1.5% | 0.95 ± 6.4% | 0.88 ± 7.2% | 0.93 ± 2.2% | 0.83 ± 5.2% | |
| 16 | 0.89 ± 5.4% | 0.94 ± 3.3% | 0.93 ± 3.7% | 0.91 ± 4.3% | 0.90 ± 2.4% | 0.87 ± 6.9% | |
| apolipoprotein E | 4 | 0.97 ± 4.5% | 0.96 ± 2.5% | 0.98 ± 1.8% | 1.00 ± 5.6% | 0.95 ± 3.3% | 1.00 ± 6.7% |
| 9 | 0.91 ± 2.9% | 0.88 ± 4.2% | 0.91 ± 5.0% | 0.97 ± 3.2% | 0.96 ± 3.7% | 0.96 ± 4.5% | |
| 16 | 0.83 ± 2.3% | 0.85 ± 2.7% | 0.80 ± 3.2% | 0.99 ± 3.1% | 0.86 ± 4.6% | 0.92 ± 1.9% | |
| apolipoprotein A-I | 4 | 0.96 ± 3.4% | 0.85 ± 2.8% | 0.79 ± 1.9% | 0.99 ± 2.7% | 1.00 ± 1.9% | 0.92 ± 5.7% |
| 9 | 0.94 ± 1.0% | 0.84 ± 2.1% | 0.85 ± 4.9% | 0.97 ± 2.7% | 0.97 ± 2.2% | 0.95 ± 3.5% | |
| 16 | 0.90 ± 1.8% | 0.85 ± 2.2% | 0.86 ± 3.0% | 0.99 ± 1.4% | 0.95 ± 1.6% | 0.96 ± 4.1% | |
| haptoglobin β chain | 4 | 0.99 ± 4.6% | 1.00 ± 1.2% | 0.92 ± 2.3% | 0.86 ± 3.8% | 0.95 ± 3.3% | 0.92 ± 8.4% |
| 9 | 0.90 ± 1.1% | 0.88 ± 1.7% | 0.86 ± 5.0% | 0.89 ± 7.7% | 0.90 ± 2.3% | 0.90 ± 4.4% | |
| 16 | 0.79 ± 4.2% | 0.84 ± 2.2% | 0.78 ± 2.2% | 0.92 ± 5.8% | 0.83 ± 2.2% | 0.88 ± 5.0% | |
| fibrinogen α chain | 4 | 0.93 ± 4.7% | 0.97 ± 0.8% | 0.92 ± 2.0% | 0.92 ± 1.7% | 1.00 ± 3.1% | 0.95 ± 5.9% |
| 9 | 0.88 ± 2.0% | 0.86 ± 1.2% | 0.84 ± 4.0% | 0.89 ± 3.7% | 0.96 ± 3.8% | 0.89 ± 3.3% | |
| 16 | 0.80 ± 1.8% | 0.83 ± 3.8% | 0.75 ± 2.8% | 0.88 ± 3.0% | 0.91 ± 0.5% | 0.89 ± 3.5% | |
| albumin, serum | 4 | 0.95 ± 4.0% | 1.00 ± 2.1% | 0.81 ± 2.6% | 0.87 ± 3.2% | 0.99 ± 3.0% | 0.93 ± 7.2% |
| 9 | 0.89 ± 1.2% | 0.90 ± 2.5% | 0.76 ± 3.5% | 0.87 ± 3.9% | 0.95 ± 2.1% | 0.92 ± 4.0% | |
| 16 | 0.84 ± 1.9% | 0.90 ± 2.9% | 0.73 ± 2.4% | 0.89 ± 2.2% | 0.92 ± 2.5% | 0.92 ± 4.5% | |
| L-selectin | 4 | 0.85 ± 10.4% | 0.98 ± 10.9% | 0.99 ± 14.7% | 0.72 ± 12.7% | 0.79 ± 16.5% | 0.78 ± 8.6% |
| 9 | 0.87 ± 9.8% | 0.92 ± 9.0% | 0.89 ± 15.7% | 0.79 ± 12.6% | 1.00 ± 11.5% | 0.86 ± 8.3% | |
| 16 | 0.86 ± 7.5% | 0.97 ± 13.7% | 0.85 ± 16.4% | 0.89 ± 7.7% | 0.86 ± 20.3% | 0.83 ± 11.6% | |
| α-2-antiplasmin | 4 | 0.94 ± 3.9% | 0.97 ± 2.4% | 0.91 ± 1.9% | 1.00 ± 2.7% | 0.94 ± 4.1% | 0.98 ± 4.0% |
| 9 | 0.81 ± 3.2% | 0.82 ± 2.0% | 0.84 ± 4.1% | 0.93 ± 2.3% | 0.86 ± 1.5% | 0.93 ± 3.2% | |
| 16 | 0.71 ± 2.1% | 0.73 ± 1.5% | 0.73 ± 3.1% | 0.88 ± 3.1% | 0.73 ± 2.6% | 0.87 ± 4.5% | |
| complement C4 β chain | 4 | 0.89 ± 8.6% | 0.97 ± 3.9% | 0.63 ± 10.5% | 0.88 ± 4.6% | 0.96 ± 6.3% | 0.90 ± 6.6% |
| 9 | 0.82 ± 3.8% | 0.91 ± 5.9% | 0.74 ± 11.8% | 0.98 ± 6.9% | 0.87 ± 4.1% | 0.87 ± 2.9% | |
| 16 | 0.77 ± 5.5% | 0.80 ± 10.4% | 0.79 ± 8.0% | 1.00 ± 5.0% | 0.81 ± 8.0% | 0.81 ± 7.5% | |
| angiotensinogen | 4 | 0.92 ± 7.3% | 1.00 ± 3.2% | 0.64 ± 6.2% | 0.91 ± 4.4% | 0.80 ± 5.8% | 0.82 ± 5.1% |
| 9 | 0.85 ± 2.5% | 0.85 ± 6.4% | 0.63 ± 12.0% | 0.87 ± 4.4% | 0.79 ± 6.4% | 0.79 ± 7.2% | |
| 16 | 0.82 ± 4.2% | 0.83 ± 9.5% | 0.65 ± 11.3% | 0.94 ± 4.4% | 0.84 ± 5.6% | 0.81 ± 2.9% | |
| afamin | 4 | 0.98 ± 3.9% | 0.98 ± 1.6% | 0.63 ± 5.0% | 0.83 ± 2.5% | 0.93 ± 7.0% | 1.00 ± 2.4% |
| 9 | 0.93 ± 3.7% | 0.92 ± 4.8% | 0.64 ± 5.7% | 0.88 ± 4.0% | 0.91 ± 6.2% | 0.98 ± 4.4% | |
| 16 | 0.87 ± 2.1% | 0.87 ± 5.1% | 0.62 ± 4.8% | 0.88 ± 2.4% | 0.92 ± 3.7% | 0.95 ± 3.2% | |
| apolipoprotein A-IV | 4 | 0.92 ± 2.4% | 0.95 ± 3.0% | 1.00 ± 2.9% | 0.62 ± 4.8% | 0.72 ± 5.5% | 0.76 ± 10.4% |
| 9 | 0.90 ± 4.7% | 0.91 ± 4.0% | 0.97 ± 4.7% | 0.78 ± 2.8% | 0.84 ± 2.2% | 0.89 ± 4.3% | |
| 16 | 0.87 ± 3.0% | 0.88 ± 2.5% | 0.92 ± 4.2% | 0.91 ± 5.4% | 0.87 ± 3.0% | 0.93 ± 4.7% | |
| vitamin D-binding protein | 4 | 0.79 ± 4.8% | 0.61 ± 20.5% | 0.67 ± 29.1% | 0.69 ± 8.2% | 0.84 ± 30.2% | 0.90 ± 11.0% |
| 9 | 0.87 ± 12.8% | 0.60 ± 4.5% | 0.84 ± 5.3% | 0.71 ± 6.8% | 0.92 ± 14.9% | 0.95 ± 7.4% | |
| 16 | 0.95 ± 6.6% | 0.61 ± 16.0% | 0.75 ± 17.1% | 0.81 ± 10.6% | 1.00 ± 22.3% | 0.96 ± 15.8% | |
| β-2-glycoprotein I | 4 | 0.92 ± 6.0% | 0.95 ± 2.4% | 1.00 ± 1.8% | 0.60 ± 3.0% | 0.94 ± 3.3% | 0.90 ± 9.0% |
| 9 | 0.92 ± 3.1% | 0.88 ± 2.2% | 0.90 ± 4.8% | 0.66 ± 13.4% | 0.95 ± 2.6% | 0.90 ± 4.4% | |
| 16 | 0.88 ± 1.9% | 0.87 ± 1.2% | 0.81 ± 4.1% | 0.70 ± 7.6% | 0.92 ± 2.2% | 0.90 ± 2.3% | |
| α-1B-glycoprotein | 4 | 0.87 ± 7.6% | 0.91 ± 5.5% | 0.59 ± 6.9% | 0.93 ± 3.4% | 0.87 ± 6.3% | 0.85 ± 6.3% |
| 9 | 0.95 ± 4.6% | 0.88 ± 3.1% | 0.76 ± 5.3% | 0.97 ± 4.5% | 1.00 ± 3.8% | 1.00 ± 5.7% | |
| 16 | 0.92 ± 3.8% | 0.93 ± 6.9% | 0.75 ± 9.4% | 0.97 ± 4.0% | 0.95 ± 6.4% | 0.99 ± 7.3% | |
| hemopexin | 4 | 0.93 ± 4.7% | 1.00 ± 1.7% | 0.62 ± 4.9% | 0.85 ± 1.5% | 0.89 ± 3.1% | 0.92 ± 6.0% |
| 9 | 0.89 ± 1.6% | 0.85 ± 3.2% | 0.58 ± 3.7% | 0.81 ± 3.2% | 0.89 ± 2.2% | 0.90 ± 3.3% | |
| 16 | 0.92 ± 2.5% | 0.93 ± 3.2% | 0.58 ± 4.6% | 0.91 ± 3.0% | 0.95 ± 1.9% | 0.96 ± 3.4% | |
| kininogen-1 | 4 | 0.93 ± 3.5% | 1.00 ± 1.9% | 0.60 ± 5.7% | 0.59 ± 2.1% | 0.86 ± 2.9% | 0.65 ± 9.1% |
| 9 | 0.91 ± 2.4% | 0.91 ± 2.4% | 0.57 ± 4.1% | 0.67 ± 4.9% | 0.87 ± 1.8% | 0.67 ± 7.1% | |
| 16 | 0.90 ± 3.2% | 0.92 ± 3.2% | 0.58 ± 3.6% | 0.74 ± 6.9% | 0.87 ± 3.3% | 0.73 ± 6.0% | |
| clusterin | 4 | 0.57 ± 7.0% | 0.69 ± 6.3% | 0.66 ± 4.9% | 0.69 ± 10.3% | 0.61 ± 2.1% | 0.63 ± 6.7% |
| 9 | 0.60 ± 10.7% | 0.75 ± 5.6% | 1.00 ± 8.7% | 0.91 ± 6.7% | 0.63 ± 7.2% | 0.60 ± 4.5% | |
| 16 | 0.58 ± 6.9% | 0.80 ± 6.5% | 0.90 ± 3.8% | 0.98 ± 7.8% | 0.55 ± 11.3% | 0.59 ± 5.7% | |
| α-1-antichymotrypsin | 4 | 0.89 ± 4.7% | 0.94 ± 3.7% | 0.53 ± 4.4% | 0.93 ± 4.5% | 1.00 ± 4.0% | 0.93 ± 5.2% |
| 9 | 0.86 ± 1.7% | 0.81 ± 5.9% | 0.54 ± 5.0% | 0.88 ± 3.5% | 0.93 ± 2.5% | 0.89 ± 4.8% | |
| 16 | 0.79 ± 2.9% | 0.85 ± 4.5% | 0.53 ± 4.4% | 0.91 ± 4.2% | 0.93 ± 5.4% | 0.90 ± 4.3% | |
| fibrinogen β chain | 4 | 0.88 ± 3.5% | 1.00 ± 4.2% | 0.53 ± 2.8% | 0.96 ± 2.4% | 0.82 ± 3.5% | 0.92 ± 7.1% |
| 9 | 0.85 ± 2.3% | 0.86 ± 2.8% | 0.79 ± 3.3% | 0.98 ± 1.7% | 0.86 ± 3.6% | 0.93 ± 5.7% | |
| 16 | 0.70 ± 3.5% | 0.74 ± 1.9% | 0.80 ± 2.2% | 0.94 ± 3.6% | 0.76 ± 2.7% | 0.85 ± 4.9% | |
| complement C4 γ chain | 4 | 1.00 ± 7.5% | 0.93 ± 4.8% | 0.60 ± 8.4% | 0.72 ± 6.2% | 0.89 ± 4.0% | 0.64 ± 12.6% |
| 9 | 0.95 ± 9.2% | 0.87 ± 4.8% | 0.56 ± 5.6% | 0.74 ± 13.4% | 0.90 ± 5.9% | 0.65 ± 7.8% | |
| 16 | 0.86 ± 2.8% | 0.85 ± 4.2% | 0.53 ± 7.5% | 0.78 ± 12.4% | 0.84 ± 7.0% | 0.70 ± 8.6% | |
| gelsolin, isoform 1 | 4 | 0.95 ± 4.4% | 0.95 ± 9.0% | 0.55 ± 5.5% | 0.97 ± 3.1% | 0.93 ± 3.9% | 0.93 ± 6.1% |
| 9 | 0.94 ± 6.3% | 0.83 ± 2.5% | 0.57 ± 9.7% | 0.96 ± 3.1% | 0.91 ± 2.8% | 0.94 ± 7.5% | |
| 16 | 0.87 ± 2.6% | 0.85 ± 4.9% | 0.52 ± 6.3% | 1.00 ± 5.8% | 0.91 ± 3.0% | 0.96 ± 3.4% | |
| apolipoprotein B-100 | 4 | 0.94 ± 5.4% | 1.00 ± 1.5% | 0.63 ± 3.7% | 0.73 ± 2.3% | 0.70 ± 6.7% | 0.78 ± 10.3% |
| 9 | 0.92 ± 3.8% | 0.78 ± 3.0% | 0.60 ± 6.1% | 0.82 ± 1.3% | 0.78 ± 4.5% | 0.88 ± 8.5% | |
| 16 | 0.87 ± 6.7% | 0.69 ± 2.8% | 0.49 ± 5.8% | 0.91 ± 2.1% | 0.80 ± 0.6% | 0.91 ± 5.4% | |
| α-2-macroglobulin | 4 | 0.46 ± 9.5% | 0.49 ± 3.2% | 0.60 ± 3.2% | 0.64 ± 5.0% | 0.47 ± 15.1% | 0.46 ± 10.3% |
| 9 | 0.55 ± 3.9% | 0.56 ± 3.5% | 1.00 ± 8.0% | 0.92 ± 17.3% | 0.61 ± 6.8% | 0.62 ± 8.5% | |
| 16 | 0.49 ± 6.0% | 0.50 ± 8.5% | 0.69 ± 9.7% | 0.76 ± 8.3% | 0.54 ± 10.8% | 0.56 ± 4.2% | |
| coagulation factor XIIa HC | 4 | 0.98 ± 5.9% | 0.80 ± 2.6% | 0.45 ± 4.2% | 0.79 ± 4.0% | 0.95 ± 3.8% | 0.88 ± 7.9% |
| 9 | 0.94 ± 3.7% | 0.86 ± 5.7% | 0.58 ± 10.1% | 0.84 ± 8.8% | 0.96 ± 4.2% | 0.89 ± 6.5% | |
| 16 | 0.95 ± 1.0% | 0.89 ± 6.7% | 0.59 ± 1.9% | 0.89 ± 5.8% | 1.00 ± 5.9% | 0.91 ± 5.7% | |
| inter-R-trypsin inhibitor HC | 4 | 0.92 ± 2.9% | 0.96 ± 2.6% | 0.72 ± 5.4% | 0.77 ± 5.5% | 0.67 ± 2.9% | 0.40 ± 12.7% |
| 9 | 0.92 ± 4.2% | 0.99 ± 1.8% | 0.67 ± 4.5% | 0.84 ± 6.7% | 0.74 ± 3.0% | 0.44 ± 9.1% | |
| 16 | 0.93 ± 1.1% | 1.00 ± 4.7% | 0.62 ± 5.9% | 0.92 ± 4.6% | 0.80 ± 4.0% | 0.49 ± 7.7% | |
| complement factor H | 4 | 0.97 ± 4.4% | 0.95 ± 1.6% | 0.40 ± 15.6% | 0.96 ± 5.6% | 1.00 ± 3.7% | 0.87 ± 6.3% |
| 9 | 0.76 ± 7.8% | 0.73 ± 5.4% | 0.72 ± 11.4% | 0.82 ± 16.4% | 0.82 ± 5.0% | 0.81 ± 4.4% | |
| 16 | 0.51 ± 18.1% | 0.44 ± 9.7% | 0.62 ± 13.0% | 0.78 ± 10.7% | 0.52 ± 43.7% | 0.70 ± 4.0% | |
| plasma retinol binding protein | 4 | 0.76 ± 9.1% | 0.72 ± 13.1% | 0.34 ± 5.7% | 0.68 ± 2.4% | 0.54 ± 3.4% | 0.78 ± 4.6% |
| 9 | 0.88 ± 3.4% | 0.73 ± 7.3% | 0.36 ± 6.8% | 0.82 ± 5.6% | 0.65 ± 2.5% | 0.94 ± 4.4% | |
| 16 | 0.83 ± 5.0% | 0.86 ± 26.7% | 0.35 ± 7.8% | 0.88 ± 5.3% | 0.69 ± 5.0% | 1.00 ± 7.2% | |
| vitronectin | 4 | 0.69 ± 2.8% | 0.95 ± 2.6% | 0.88 ± 2.3% | 0.38 ± 3.1% | 0.34 ± 9.8% | 0.51 ± 7.8% |
| 9 | 0.79 ± 1.8% | 0.84 ± 4.2% | 0.97 ± 4.7% | 0.51 ± 12.0% | 0.51 ± 2.1% | 0.66 ± 6.7% | |
| 16 | 0.85 ± 7.0% | 0.96 ± 7.8% | 1.00 ± 5.3% | 0.68 ± 9.3% | 0.65 ± 4.2% | 0.80 ± 4.2% | |
| α-1-acid glycoprotein 1 | 4 | 0.13 ± 2.3% | 0.42 ± 5.9% | 0.07 ± 11.1% | 0.03 ± 13.6% | 0.02 ± 9.3% | 0.05 ± 15.5% |
| 9 | 0.29 ± 4.8% | 0.70 ± 9.1% | 0.34 ± 7.7% | 0.08 ± 7.1% | 0.04 ± 7.3% | 0.13 ± 13.9% | |
| 16 | 0.45 ± 3.0% | 1.00 ± 8.8% | 0.69 ± 8.4% | 0.16 ± 13.2% | 0.06 ± 7.3% | 0.23 ± 9.1% | |
| antithrombin-III | 4 | 0.35 ± 4.5% | 0.61 ± 9.4% | 0.39 ± 6.4% | 0.08 ± 9.5% | 0.12 ± 15.4% | 0.14 ± 23.6% |
| 9 | 0.57 ± 4.8% | 0.82 ± 7.1% | 0.68 ± 11.2% | 0.14 ± 5.7% | 0.20 ± 8.0% | 0.25 ± 11.5% | |
| 16 | 0.75 ± 3.2% | 1.00 ± 12.4% | 0.77 ± 9.1% | 0.22 ± 16.5% | 0.24 ± 2.0% | 0.32 ± 5.2% | |
| apolipoprotein A-II | 4 | 0.47 ± 4.4% | 0.34 ± 3.1% | 0.30 ± 3.0% | 0.70 ± 2.1% | 0.38 ± 2.3% | 0.64 ± 5.4% |
| 9 | 0.65 ± 1.7% | 0.49 ± 2.3% | 0.47 ± 4.1% | 0.85 ± 0.8% | 0.47 ± 1.7% | 0.84 ± 3.3% | |
| 16 | 0.76 ± 2.1% | 0.60 ± 2.6% | 0.58 ± 2.9% | 1.00 ± 2.1% | 0.53 ± 3.0% | 0.98 ± 3.0% | |
| apolipoprotein C-I | 4 | 0.91 ± 4.4% | 0.01 ± 10.0% | 0.68 ± 4.1% | 0.33 ± 4.8% | 0.30 ± 6.5% | 0.22 ± 17.0% |
| 9 | 1.00 ± 1.9% | 0.02 ± 15.8% | 0.76 ± 4.9% | 0.53 ± 5.2% | 0.47 ± 4.1% | 0.46 ± 8.2% | |
| 16 | 0.96 ± 2.8% | 0.02 ± 4.9% | 0.75 ± 3.1% | 0.71 ± 4.7% | 0.56 ± 4.1% | 0.68 ± 5.8% | |
| apolipoprotein C-III | 4 | 0.26 ± 8.2% | 0.27 ± 16.1% | 0.99 ± 2.1% | 0.62 ± 3.5% | 0.07 ± 5.8% | 0.46 ± 9.4% |
| 9 | 0.48 ± 6.8% | 0.40 ± 6.4% | 0.94 ± 4.6% | 0.83 ± 2.4% | 0.13 ± 13.6% | 0.72 ± 4.0% | |
| 16 | 0.69 ± 3.5% | 0.62 ± 5.1% | 0.88 ± 3.3% | 1.00 ± 1.9% | 0.19 ± 6.9% | 0.95 ± 5.6% | |
| ceruloplasmin | 4 | 0.75 ± 8.1% | 0.88 ± 3.6% | 0.73 ± 2.0% | 0.29 ± 8.6% | 0.35 ± 12.3% | 0.28 ± 15.5% |
| 9 | 0.90 ± 4.5% | 0.96 ± 3.4% | 0.93 ± 10.4% | 0.42 ± 11.4% | 0.46 ± 4.2% | 0.43 ± 10.1% | |
| 16 | 0.94 ± 3.9% | 0.95 ± 5.6% | 1.00 ± 4.5% | 0.52 ± 8.4% | 0.54 ± 4.1% | 0.55 ± 10.7% | |
| complement component C9 | 4 | 0.76 ± 7.9% | 1.00 ± 4.9% | 0.88 ± 7.2% | 0.62 ± 5.0% | 0.55 ± 18.4% | 0.17 ± 12.3% |
| 9 | 0.81 ± 12.2% | 0.89 ± 12.3% | 0.81 ± 13.4% | 0.62 ± 22.1% | 0.63 ± 6.5% | 0.24 ± 11.3% | |
| 16 | 0.84 ± 9.7% | 0.97 ± 8.4% | 0.81 ± 14.9% | 0.73 ± 13.2% | 0.67 ± 20.5% | 0.31 ± 16.4% | |
| complement factor B | 4 | 0.66 ± 4.6% | 0.85 ± 1.6% | 0.72 ± 3.5% | 0.34 ± 1.5% | 0.31 ± 4.4% | 0.35 ± 9.9% |
| 9 | 0.82 ± 2.3% | 0.91 ± 3.1% | 0.96 ± 4.5% | 0.48 ± 7.7% | 0.41 ± 4.8% | 0.51 ± 10.4% | |
| 16 | 0.89 ± 4.0% | 0.94 ± 2.5% | 1.00 ± 3.3% | 0.58 ± 8.9% | 0.48 ± 3.8% | 0.62 ± 7.3% | |
| fibrinogen γ chain | 4 | 0.32 ± 24.4% | 0.50 ± 8.0% | 0.24 ± 14.6% | 0.15 ± 5.0% | 0.18 ± 18.7% | 0.16 ± 19.9% |
| 9 | 0.39 ± 21.8% | 0.77 ± 3.9% | 0.23 ± 11.4% | 0.31 ± 7.7% | 0.23 ± 7.1% | 0.34 ± 14.9% | |
| 16 | 0.52 ± 7.3% | 1.00 ± 5.8% | 0.24 ± 9.9% | 0.52 ± 10.1% | 0.24 ± 10.7% | 0.58 ± 10.0% | |
| plasminogen | 4 | 0.51 ± 3.5% | 0.64 ± 1.8% | 0.43 ± 4.4% | 0.37 ± 4.4% | 0.32 ± 4.8% | 0.46 ± 8.5% |
| 9 | 0.75 ± 2.2% | 0.83 ± 2.1% | 0.65 ± 6.0% | 0.51 ± 4.1% | 0.41 ± 6.2% | 0.65 ± 7.5% | |
| 16 | 0.88 ± 5.0% | 1.00 ± 3.9% | 0.77 ± 6.1% | 0.66 ± 3.4% | 0.48 ± 5.9% | 0.86 ± 7.6% | |
| prothrombin | 4 | 0.30 ± 3.8% | 0.63 ± 11.6% | 0.47 ± 14.4% | 0.17 ± 6.1% | 0.13 ± 31.3% | 0.26 ± 12.3% |
| 9 | 0.44 ± 8.4% | 0.78 ± 5.3% | 0.74 ± 11.2% | 0.26 ± 2.8% | 0.19 ± 16.0% | 0.40 ± 3.2% | |
| 16 | 0.58 ± 3.8% | 1.00 ± 4.5% | 0.94 ± 5.1% | 0.36 ± 9.3% | 0.22 ± 17.2% | 0.54 ± 8.0% | |
| serum amyloid P | 4 | 0.07 ± 9.5% | 0.79 ± 2.3% | 0.01 ± 19.5% | 0.63 ± 2.5% | 0.73 ± 4.3% | 0.76 ± 3.4% |
| 9 | 0.28 ± 2.9% | 0.73 ± 12.9% | 0.04 ± 27.2% | 0.73 ± 19.7% | 0.82 ± 9.7% | 1.00 ± 3.3% | |
| 16 | 0.60 ± 7.4% | 0.75 ± 11.2% | 0.32 ± 9.6% | 0.81 ± 10.3% | 0.77 ± 6.4% | 1.00 ± 9.9% | |
| transferrin | 4 | 0.76 ± 5.8% | 0.99 ± 4.1% | 0.76 ± 5.0% | 0.40 ± 4.7% | 0.29 ± 5.5% | 0.46 ± 11.2% |
| 9 | 0.99 ± 2.6% | 0.92 ± 7.1% | 0.94 ± 5.6% | 0.84 ± 10.3% | 0.50 ± 3.3% | 0.76 ± 6.4% | |
| 16 | 0.99 ± 3.3% | 1.00 ± 6.8% | 0.84 ± 3.6% | 0.98 ± 7.2% | 0.63 ± 3.4% | 0.94 ± 6.6% | |
| transthyretin | 4 | 0.71 ± 4.8% | 0.03 ± 3.9% | 0.10 ± 11.4% | 0.39 ± 4.4% | 1.00 ± 2.0% | 0.75 ± 6.0% |
| 9 | 0.71 ± 3.5% | 0.02 ± 7.8% | 0.10 ± 8.4% | 0.41 ± 3.8% | 0.97 ± 2.6% | 0.80 ± 4.8% | |
| 16 | 0.71 ± 1.9% | 0.02 ± 17.9% | 0.12 ± 7.5% | 0.46 ± 3.0% | 0.96 ± 2.1% | 0.83 ± 3.1% | |
| zinc-α-2-glycoprotein | 4 | 0.74 ± 8.0% | 0.58 ± 7.6% | 0.01 ± 66.5% | 0.70 ± 3.5% | 0.63 ± 6.5% | 0.72 ± 6.6% |
| 9 | 0.89 ± 3.7% | 0.75 ± 11.5% | 0.03 ± 27.8% | 1.00 ± 4.4% | 0.89 ± 7.9% | 0.93 ± 2.1% | |
| 16 | 0.77 ± 6.5% | 0.67 ± 8.4% | 0.05 ± 18.6% | 0.85 ± 3.9% | 0.80 ± 3.2% | 0.87 ± 4.9% |
Figure 4.
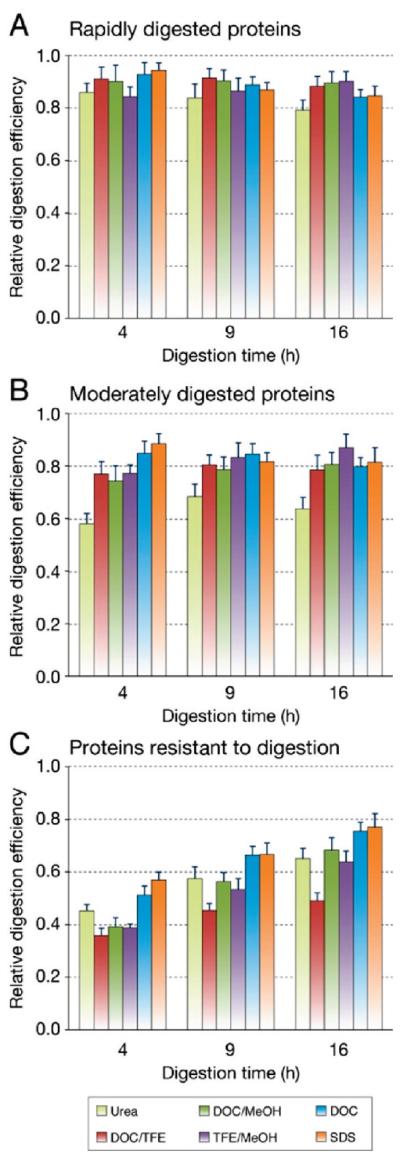
Protein classification by range of digestion efficiency. Proteins were classified into three equal groups based on the range of digestion efficiency observed between methods and digestion times: proteins that were rapidly digested to equivalent levels regardless of the denaturation methods (A), proteins with an intermediate range of digestion efficiencies (B), and proteins either slowly or poorly digested with at least one protocol (C). Error bars represent the average standard deviation of normalized peak area ratios measured in 5 replicate digests.
Figure 7.

(A) Average digestion efficiencies as a function of digestion time and treatment. (B) Average % errors as function of digestion time and treatment. (Note: each % error is a measure of the deviation from the mean efficiency and is actually a ± value. The absolute values were used to create this figure).
Comparison of Digestion Efficiencies
To normalize data for each analyte, the maximum value of these average peak area ratios (light:heavy) for these 5-fold replicate analyses was determined, and the 90 peak area ratios were normalized by dividing each by this maximum value (from this point forward, this will be referred to as normalized data). The minimum average peak area ratio was also identified for each analyte, and all 45 proteins were ranked. The minimum average peak area ratio of an analyte is the lowest response for that analyte under all conditions and digestion lengths. This effectively describes the range of digestion efficiencies observed: the higher the minimum average peak area ratio of an analyte, the more rapid the digestion, whereas the maximum peak area ratio gives information on “completeness” of the digest.
To compensate for the different levels of signal observed for the 45 analytes and to determine if greater differences exist between protocols when looking at different protein classes, proteins were ranked on the basis of minimum average peak area ratios, and the 6 most effective protocols were compared. To better reveal differences between digestion conditions, all 45 proteins were divided into three equal groups based on the range of digestion efficiencies observed: proteins that were rapidly digested (Figure 4A), proteins that were moderately digested (Figure 4B) and proteins resistant to digestion (Figure 4C).
The results show that peptides from 31 of the 45 proteins reach a steady state of tryptic digestion products after 8 h of tryptic digestion and showed no further increase or decrease for up to 16 h. For 25 of these 31 proteins, rapid and complete digestion was observed after only 4 h of tryptic digestion. For the remaining 14 proteins, however, 10 proteins actually show a decrease in signal with prolonged digestion (9 and 16 h). Proteins that are slow to digest show a progressive increase in signal over longer digestion periods, with all 6 denaturation methods. Signals from 4 of the analytes kept increasing even after 16 h of digestion and did not reach a plateau during this time period. Thus, the optimal digestion protocols and reproducibility are analyte-dependent (Figure 5).
Figure 5.

Relative tryptic peptide production efficiencies of the six most effective denaturation protocols on 45 human plasma proteins for either 4 (white), 9 (light gray), or 16 h (dark gray). Digestion protocols are sorted from most effective (right) to least effective (left) when considering all 45 analytes. Plasma proteins are sorted by relative tryptic peptide production efficiencies at 4 h when DOC is used as a denaturant, sorted in order of increasing range of measured relative digestion efficiency (top to bottom) with the most effectively digested proteins at the top and the least effectively digested proteins at the bottom: proteins that were rapidly digested to equivalent levels regardless of the denaturation methods (A), proteins with an intermediate range of digestion efficiencies (B), and proteins either slowly or poorly digested with at least one protocol (C).
Although no single digestion protocol produced the highest signal for all of the proteins, if the target protein is known, selection of the appropriate digestion protocol can increase analyte signal more than 3-fold. For example, although DOC produced the highest peptide signals for most of the proteins, apolipoprotein C-I showed its highest signal when SDS was used. What we have presented here is the “best” method for general shotgun proteomics. Certainly, if the target protein is known, 3–5 peptides should be selected and examined for digestion efficiencies and for MRM tuning as well, as was previously pointed out.9
Comparison of Methods for Reproducibility
As can be seen in Table 2, there is a large amount of protein to protein variability, but the protein displaying this maximum variability is not the same in every treatment. For example, zinc α-2-glycoprotein showed the overall highest variability (66%) with 4 h of digestion with urea. With other treatments, however, this protein gave % error values (defined as the relative deviation from the mean) close to the overall average. In contrast, almost all of the % errors for L-selectin are above average, while hemopexin showed very low % errors in all treatments.
The average % errors for each treatment and digestion time are given in Figure 6B. For the 9 and 16 h digestion times, the DOC treatment showed the lowest % error (the highest reproducibility) of all 6 treatments, a % error of 4.72% and 4.30%, respectively. Even for the 4 h digestion time, the DOC treatment had a % error of only 5.79%. Urea showed poor reproducibility relative to the other treatments, with the highest (or one of the highest) % errors in all treatments and at all digestion times.
Figure 6.

Digestion efficiency (% completion) for DOC, DOC/MeOH, DOC/TFE, TFE/MeOH, SDS, and urea for three digestion times: (A) 4 h, (B) 9 h, and (C) 16 h. Reproducibility distribution (% error) for DOC, DOC/MeOH, DOC/TFE, TFE/MeOH, SDS, and urea, for three digestion times: (D) 4 h, (E) 9 h, and (F) 16 h.
The results in Table 2 clearly demonstrate the complexity of the protein digestion process and the challenges in obtaining reproducible and effective digestion of a mixture of proteins. Looking at the % digestion of α-1-glycoprotein (classified as a Group C or “resistant” protein, in Figures 4 and 5), we find that the digestion efficiencies increased with digestion time all treatments but reached widely differing values (from 6% with DOC/TFE to 100% with SDS). In contrast, transthyretin (another Group C protein) shows very little effect of digestion time but a large effect of the choice of denaturant (ranging from 2% to 100% efficiency).
The optimum digestion conditions for each protein can be found in Supporting Information Figure 1. However, the target protein may not always be known, as in shotgun proteomics or biomarker discovery, where the optimum method for a large group of proteins must be selected. Figure 6A–C shows the frequency distribution of digestion efficiencies (% completion) of the different treatments, for each digestion time. Looking at Figure 6A and B, the frequency distribution of digestion efficiencies shift to higher values (higher % completion) as one moves from 4 to 9 h digestion times. When one compares the distribution of % completion at 9 and 16 h digestion times (Figure 6B and C), the efficiencies for the combined treatments (DOC/MeOH and DOC/TFE) actually shift to lower % efficiencies. Comparing DOC (dark blue line) and SDS (pink line) at the three digestion times shows an increased % completion from 4 to 9 h but not much change between 9 and 16 h, whereas urea (yellow line) was not as efficient as DOC or SDS at any of the digestion times.
The comparison of the different treatments with respect to reproducibility (% errors), at the three different digestion times, is shown in Figure 6D–F. In general, increasing the digestion time from 4 to 9 h (Figure 6D and E) shifts the frequency distribution of % errors to lower values, reflecting an increase in reproducibility, probably indication that digestion was not complete digestion at the shorter digestion times. The shift from 9 to 16 h digestion time (Figure 6F) does not have as much effect on the frequency distributions as did the change from 4 to 9 h. The longer digestion times did reduce the % errors for the “outliers”; for example, the highest % errors for urea were 68%, 27%, and 18% at 4, 9, and 16 h, respectively.
Examining the average digestion efficiencies (Figure 7A), SDS and DOC show the best overall efficiencies at the 9 and 16 h time points, while the “combinations” of DOC/TFE or DOC/MeOH may show some promise at shorter digestion times. However, in terms of % error, these combined treatments show higher % errors than either SDS or DOC. When both digestion efficiency and % error are considered, DOC is the best overall method (at 9 and 16 h digestion times), where it shows a higher digestion efficiency and a lower % error than SDS. Under the digestion conditions used, urea shows the lowest digestion efficiency and one of the highest average % errors. Therefore, considering both digestion efficiency and reproducibility, DOC appears to be the best method for general protein digestion.
Interestingly, SDS, DOC, and urea appear to reach a “plateau” in terms of the average digestion efficiencies (~80%) and do not show much change between 4 and 16 h. The % errors for SDS and DOC, however, are reduced with increasing digestion time, with DOC showing the lowest % error (and therefore the highest reproducibility) at both the 9 and 16 h digestion times.
The recommendation of DOC as a denaturant is consistent with the investigation of Lin et al.17 regarding the use of DOC, in which they found that DOC aided solubilization and led to improvements in tryptic digestion, particularly for proteolytically resistant proteins. In contrast to the Lin study, however, we did not observe any deleterious effect of longer digestion times on the average DOC digestion efficiency or reproducibility, although we did observe this phenomenon for a few proteins. In our study, comparing the 4, 9, and 16 h DOC digestion times, the longer digestion times produced higher average reproducibilities (±5.8%, ±4.7%, and ±4.3% errors, respectively).
Conclusions
Our goal in studying the different digestion protocols was to determine the conditions under which the highest possible percentage of the protein has been digested in the most reproducible way, in other words, where the production of tryptic peptides has reached its highest plateau. Although this can be achieved for almost every protein, we found that the “optimum achievable” digestion conditions are protein-dependent, and there is no one common procedure that is best for all of the proteins.
Even in this relatively small set of proteins, wide variations in digestion performance were observed. For 31 of the 45 proteins, “ideal” conditions could be achieved. For these proteins, reproducible and stable levels of tryptic digestion products (as determined by MRM peak area ratios) were obtained after 9 h of tryptic digestion, and the peptide MRM ratios showed no further increase or decrease for up to 16 h at 37 °C. For 25 of these 31 proteins, the maximum MRM ratios were observed after only 4 h of tryptic digestion. For the remaining 14 proteins, however, either the signal did not reach a plateau during this 16 h time period, or the peptide signals reached a maximum and then decreased. For these “non-ideal” proteins, consistent digestion times are required in order to achieve reproducible results.
Improvement in digestion efficiencies was achieved with several proteins by altering denaturation conditions. Although there is no single “best” digestion method for all 45 human plasma proteins, one must take into account the analyte signal intensity produced and the digestion time required for maximum digestion, as well as whether consistent digestion performance across all 45 analytes is needed for a particular application. Although one single digestion protocol may never be 100% efficient for all proteins, protocols can be selected and optimized for proteins of interest in order to ensure reproducibility and optimal detection. For clinical applications involving detection of biomarker proteins, digestion protocols should be selected specific to the target proteins of interest.
It is also worth noting that different protocols can yield different steady state conditions, in other words, the digestion reaches “completion”, but the digestion is still not “complete” (Supporting Information Figure 1). Transthyretin and plasma retinol binding protein are good examples of this (Figure 2A–C). Twenty-four proteins reached a steady state with nearly all of the digestion protocols (Supporting Information Figure 1). The proteotypic peptides from 21 proteins do not reach a steady state within the 24 h digestion times with most of the digestion conditions used. The proteotypic peptides from 10 proteins continued to increase with increasing digestion time and never reached a plateau under most of the digestion conditions studied. The proteotypic peptides from 5 proteins decreased with increasing digestion time under most digestion conditions, and there were 6 proteins whose digestion was so treatment-dependent that no generalization could be made. For the 3 non-steady-state groups of proteins, care must be taken to precisely control the digestion times in order to achieve reproducible digestion for comparative proteomics studies. This is another advantage of methods performing absolute quantitation using SIS peptides. On the basis of the relative peak areas of the SIS and native peptides, it would be possible to create calibration curves in order to correct for less than 100% digestion of the target protein.
Ultimately, using absolute quantitation methods, we have found that there are digestion protocols better than urea. Tryptic digestion in solutions containing urea routinely showed lower digestion efficiencies than methods using SDS or DOC, achieving maximum peptide production for only 10 out of 45 proteins (Figure 3). Both SDS and DOC have been shown to give higher tryptic peptide production, which in turn leads to increased signal intensities and therefore a more sensitive MRM assay. However, SDS contaminates MS instrumentation, interferes with chromatographic resolution, and is difficult to completely remove from sample digests. With a 9 h digestion, DOC produced one of the highest average digestion efficiencies (~80%), with the highest average reproducibility (<5% error). We therefore conclude that the best method for untargeted protein denaturation is DOC with a 9 h digestion time. The DOC denaturation protocol is also simple and amenable to automation. However, it should be stressed that, in order to maximize reproducibility between analytical runs, sample preparation must still be carefully defined and rigorous standardized operating protocols must be used.
Supplementary Material
Acknowledgment
We would like to thank Genome Canada and Genome BC for providing funding for the proteomics platform facility. This work was supported in part by a grant from the National Institutes of Health Grants 1U24 CA126476 (PI: Steven A. Carr, Broad Institute of Harvard and MIT) as part of the NCI's Clinical Proteomic Technologies Assessment in Cancer Program.
Abbreviations
- ABC
ammonium bicarbonate
- CE
collision energy
- CID
collision induced dissociation
- DOC
sodium deoxycholate
- CV
coefficient of variation
- Da
Daltons
- GnHCl
guanidine hydrochloride
- LC
liquid chromatography
- MRM
multiple reaction monitoring
- MS
mass spectrometry/ spectrometer
- MS/MS
tandem MS
- Q1
quadrupole 1
- Q3
quadrupole 3
- SDS
sodium dodecyl sulfate
- SIS
stable-isotope labeled internal standard
- TOF
time-of-flight
Footnotes
Supporting Information Available: This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Anderson NL, Anderson NG. The human plasma proteome: history, character, and diagnostic prospects. Mol. Cell. Proteomics. 2002;1(11):845–867. doi: 10.1074/mcp.r200007-mcp200. [DOI] [PubMed] [Google Scholar]
- 2.Barnidge DR, Goodmanson MK, Klee GG, Muddiman DC. Absolute quantification of the model biomarker prostate-specific antigen in serum by LC-MS/MS using protein cleavage and isotope dilution mass spectrometry. J. Proteome Res. 2004;3(3):644–652. doi: 10.1021/pr049963d. [DOI] [PubMed] [Google Scholar]
- 3.Klammer AA, MacCoss MJ. Effects of modified digestion schemes on the identification of proteins from complex mixtures. J. Proteome Res. 2006;5:695–700. doi: 10.1021/pr050315j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Elliott M, Smith D, Kuzyk M, Parker CE, Borchers CH. Recent trends in quantitative proteomics. J. Mass Spectrom. 2009;44(12):1637–1660. doi: 10.1002/jms.1692. [DOI] [PubMed] [Google Scholar]
- 5.Gerber SA, Rush J, Stemman O, Kirschner MW, Gygi SP. Absolute quantification of proteins and phosphoproteins from cell lysates by tandem MS. Proc. Natl. Acad. Sci. U.S.A. 2003;100(12):6940–6945. doi: 10.1073/pnas.0832254100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kirkpatrick DS, Gerber SA, Gygi SP. The absolute quantification strategy: a general procedure for the quantification of proteins and post-translational modifications. Methods (San Diego, CA, U. S.) 2005;35(3):265–273. doi: 10.1016/j.ymeth.2004.08.018. [DOI] [PubMed] [Google Scholar]
- 7.Anderson NL, Anderson NG, Haines LR, Hardie DB, Olafson RW, Pearson TW. Mass spectrometric quantitation of peptides and proteins using stable isotope standards and capture by anti-peptide antibodies (SISCAPA) J. Proteome Res. 2004;3(2):235–244. doi: 10.1021/pr034086h. [DOI] [PubMed] [Google Scholar]
- 8.Anderson NL, Jackson A, Smith D, Hardie D, Borchers C, Pearson TW. SISCAPA peptide enrichment on magnetic beads using an in-line bead trap device. Mol. Cell. Proteomics. 2009;8(5):995–1005. doi: 10.1074/mcp.M800446-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kuzyk M, Smith D, Yang J, Cross T, Jackson A, Hardie D, Anderson L, Borchers C. MRM-based, multiplexed, absolute quantitation of 45 proteins in human plasma. Mol. Cell. Proteomics. 2009;8(8):1860–1877. doi: 10.1074/mcp.M800540-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jiang J, Parker CE, Hoadley KA, Perou CM, Boysen G, Borchers CH. Development of an immuno tandem mass spectrometry (iMALDI) assay for EGFR diagnosis. Proteomics: Clin. Appl. 2007;1:1651–1659. doi: 10.1002/prca.200700009. [DOI] [PubMed] [Google Scholar]
- 11.Jiang J, Parker CE, Fuller JR, Kawula TH, Borchers CH. An immunoaffinity tandem mass spectrometry (iMALDI) assay for detection of Francisella tularensis. Anal. Chim. Acta. 2007;605:70–79. doi: 10.1016/j.aca.2007.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shah B, Borchers CH. iMALDI: A targeted proteomics approach to the differentiation of EGFR and its isoforms; Presented at the 57th ASMS Conference on Mass Spectrometry and Allied Topics; Philadelphia, PA. May 31–June 4, 2009. [Google Scholar]
- 13.Russell WK, Park Z-Y, Russell DH. Proteolysis in mixed organic-aqueous solvent systems: Applications for peptide mass mapping using mass spectrometry. Anal. Chem. 2001;73(11):2682–2685. doi: 10.1021/ac001332p. [DOI] [PubMed] [Google Scholar]
- 14.Hervey WJ, Strader MB, Hurst GB. Comparison of digestion protocols for microgram quantities of enriched protein samples. J. Proteome Res. 2007;6(8):3054–3061. doi: 10.1021/pr070159b. [DOI] [PubMed] [Google Scholar]
- 15.McDonald WH, Ohi R, Miyamoto DT, Mitchison TJ, Yates JR. Comparison of three directly coupled HPLC MS/MS strategies for identification of proteins from complex mixtures: single-dimension LC-MS/MS, 2-phase MudPIT, and 3-phase MudPIT. Int. J. Mass Spectrom. 2002;219(1):245–251. [Google Scholar]
- 16.Ru QC, Zhu LA, Katenhusen RA, Silberman J, Brzeski H, Liebman M, Shriver CD. Exploring human plasma proteome strategies: high efficiency in-solution digestion protocol for multidimensional protein identification technology. J. Chromatogr., A. 2006;1111(2):175–191. doi: 10.1016/j.chroma.2005.06.080. [DOI] [PubMed] [Google Scholar]
- 17.Lin Y, Zhou J, Bi D, Chen P, Wang X, Liang S. Sodium-deoxycholate-assisted tryptic digestion and identification of proteolytically resistant proteins. Anal. Biochem. 2008;377(2):259–266. doi: 10.1016/j.ab.2008.03.009. [DOI] [PubMed] [Google Scholar]
- 18.Kresge N, Simoni RD, Hill RL. Cyanate chemistry, enzyme mechanisms, and gene amplification: The work of George R. Stark. J. Biol. Chem. 2007;282:e23. [Google Scholar]
- 19.Buscher BAP, Gerritsen H, van Schoell I, Cnubben NHP, Bruell LP. Quantitative analysis of Tenecteplase in rat plasma samples using LC-MS/MS as an alternative for ELISA. J. Chromatogr., B: Anal. Technol. Biomed. Life Sci. 2007;852(1–2):631–634. doi: 10.1016/j.jchromb.2006.12.053. [DOI] [PubMed] [Google Scholar]
- 20.Wilkinson JM. Fragmentation of polypeptides by enzymic methods. In: Darbre A, editor. Practical Protein Chemistry: A Handbook. John Wiley and Sons; New York, NY: 1986. [Google Scholar]
- 21.Kuhn E, Wu J, Karl J, Liao H, Zolg W, Guild B. Quantification of C-reactive protein in the serum of patients with rheumatoid arthritis using multiple reaction monitoring mass spectrometry and 13C-labeled peptide standards. Proteomics. 2004;4:1175–1186. doi: 10.1002/pmic.200300670. [DOI] [PubMed] [Google Scholar]
- 22.Simon LM, Laszlo K, Vertesi A, Bagi K, Szajani B. Stability of hydrolytic enzymes in water-organic solvent systems. J. Mol. Catal. B: Enzym. 1998;4(1–2):41–45. [Google Scholar]
- 23.Blonder J, Conrads TP, Yu LR, Terunuma A, Janini GM, Issaq HJ, Vogel J, Veenstra TD. A detergent- and cyanogen bromide-free method for integral membrane proteomics: Application to Halobacterium purple membranes and the human epidermal membrane proteome. Proteomics. 2004;4:31–45. doi: 10.1002/pmic.200300543. [DOI] [PubMed] [Google Scholar]
- 24.Anderson NL. 2009. personal communication.
- 25.MezaJ E, MillerC A, FischerS M. Improved tryptic digestion of proteins using 2,2,2-trifluoroethanol (TFE); Poster presentation at ABRF 2004; Portland, OR. February 28–March 2, 2004. [Google Scholar]
- 26.Blonder J, Hale ML, Chan KC, Yu L-R, Lucas DA, Conrads TP, Zhou M, Popoff MR, Issaq HJ, Stiles BG, Veenstra TD. Quantitative profiling of the detergent-resistant membrane proteome of iota-b toxin induced Vero cells. J. Proteome Res. 2005;4(2):523–531. doi: 10.1021/pr049790s. [DOI] [PubMed] [Google Scholar]
- 27.Blonder J, Chan KC, Issaq HJ, Veenstra TD. Identification of membrane proteins from mammalian cell/tissue using methanol-facilitated solubilization and tryptic digestion coupled with 2D-LC-MS/MS. Nat. Protoc. 2006;1(6):2784–2790. doi: 10.1038/nprot.2006.359. [DOI] [PubMed] [Google Scholar]
- 28.Adachi J, Kumar C, Zhang Y, Olsen J, Mann M. The human urinary proteome contains more than 1500 proteins, including a large proportion of membrane proteins. GenomeBiology. 2006;7:R80. doi: 10.1186/gb-2006-7-9-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Oberg KA, Ruysschaert JM, Azarkan M, Smolders N, Zerhouni S, Wintjens R, Amrani A, Looze Y. Papaya glutamine cyclase, a plant enzyme highly resistant to proteolysis, adopts an all-β conformation. Eur. J. Biochem. 1998;258:214–222. doi: 10.1046/j.1432-1327.1998.2580214.x. [DOI] [PubMed] [Google Scholar]
- 30.Wisniewski JR, Zougman A, Nagaraj N, Mann M. Universal sample preparation method for proteome analysis. Nat. Methods. 2009;6(5):359–632. doi: 10.1038/nmeth.1322. [DOI] [PubMed] [Google Scholar]
- 31.Canas B, Pineiro C, Calvo E, Lopez-Ferrer D, Gallardo JM. Trends in sample preparation for classical and second generation proteomics. J. Chromatogr., A. 2007;1153:235–258. doi: 10.1016/j.chroma.2007.01.045. [DOI] [PubMed] [Google Scholar]
- 32.Monera OD, Kay CM, Hodges RS. Protein denaturation with guanidine hydrochloride or urea provides a different estimate of stability depending on the contributions of electrostatic interactions. Protein Sci. 1994;3:1984–1991. doi: 10.1002/pro.5560031110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Almarza J, Rincon L, Bahsas A, Brito F. Molecular mechanism for the denaturation of proteins by urea. Biochemistry. 2009;48:7608–7613. doi: 10.1021/bi9007116. [DOI] [PubMed] [Google Scholar]
- 34.Bennion BJ, Daggett V. The molecular basis for the chemical denaturation of proteins by urea. Proc. Natl. Acad. Sci. U.S.A. 2003;100:5142–5147. doi: 10.1073/pnas.0930122100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Anderson L, Hunter CL. Quantitative mass spectrometric multiple reaction monitoring assays for major plasma proteins. Mol. Cell. Proteomics. 2006;5(4):573–588. doi: 10.1074/mcp.M500331-MCP200. [DOI] [PubMed] [Google Scholar]
- 36.Trypsin sequencing grade product overview. BoehringerMannheim: 2004. [Google Scholar]
- 37.Stone KL, LoPresti MB, Williams KR. Enzymatic digestion of proteins and HPLC peptide isolation in the subnanomole range. In: Fini C, Floridi A, Finelli VN, Wittman-Liebold B, editors. Laboratory Methodology in Biochemistry. CRC Press; Boca Raton, FL: 1990. pp. 181–205. Chapter 11. [Google Scholar]
- 38.The Handbook of Analysis and Purification of Peptides and Proteins by Reversed-Phase HPLC. 3rd ed. GraceVydac; Deerfield, IL: 2002. Grace/Vydac, Appendix C: The effect of surfactants on reversed-phase separations; pp. 56–57. [Google Scholar]
- 39.Zhou J, Zhou TY, Cao R, Liu Z, Shen JY, Chen P, Wang XC, Liang SP. Evaluation of the application of sodium deoxycholate to proteomic analysis of rat hippocampal plasma membrane.rat hippocampal plasma membrane. J. Proteome Res. 2006;5:2547–2553. doi: 10.1021/pr060112a. [DOI] [PubMed] [Google Scholar]
- 40.Promega_Trypsin Promega Usage Information. 4/2009, Part no. 9PIV5113, www.promega.com.
- 41.Nelson CJ, Hegeman AD, Harms AC, Sussman MR. A quantitative analysis of Arabidopsis plasma membrane using trypsin-catalyzed (18)O labeling. Mol. Cell. Proteomics. 2006;5(8):1382–1395. doi: 10.1074/mcp.M500414-MCP200. [DOI] [PubMed] [Google Scholar]
- 42.Strader MB, Tabb DL, Hervey WJ, Pan CH, Hurst GB. Efficient and specific trypsin digestion of microgram to nanogram quantities of proteins in organic-aqueous solvent systems. Anal. Chem. 2006;78(1):125–134. doi: 10.1021/ac051348l. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.