


Abstract
The quest for a universal and efficient method of identifying species has been a longstanding challenge in biology. Here, we show that accurate identification of species in all domains of life can be accomplished by multiplex analysis of variable-length sequences containing multiple insertion/deletion variants. The new method, called SPInDel, is able to discriminate 93.3% of eukaryotic species from 18 taxonomic groups. We also demonstrate that the identification of prokaryotic and viral species with numeric profiles of fragment lengths is generally straightforward. A computational platform is presented to facilitate the planning of projects and includes a large data set with nearly 1800 numeric profiles for species in all domains of life (1556 for eukaryotes, 105 for prokaryotes and 130 for viruses). Finally, a SPInDel profiling kit for discrimination of 10 mammalian species was successfully validated on highly processed food products with species mixtures and proved to be easily adaptable to multiple screening procedures routinely used in molecular biology laboratories. These results suggest that SPInDel is a reliable and cost-effective method for broad-spectrum species identification that is appropriate for use in suboptimal samples and is amenable to different high-throughput genotyping platforms without the need for DNA sequencing.
INTRODUCTION
The correct identification of entities belonging to the biological category we call ‘species’ is frequently inhibited by both theoretical and practical limitations. The long-running debate on the meaning of this category has produced over 20 different concepts of species that challenge any attempt to identify a species accurately (1). Additionally, the multiplicity of biological properties and methods currently used for identification have resulted in many practical limitations that often lead to contradictory results (2–6).
Developments in the field of molecular biology have revolutionized the way that we classify and identify species. Initial methods based on protein analysis had significant limitations, such as rapid protein degradation in samples under stress conditions, a high risk of cross-reactivity or problems associated with tissue specificity. These limitations were only overcome with the advent of DNA-based systems. Information encoded in nucleic acid sequences can be examined using multiple methods for molecular-recognition purposes: hybridization between a known genomic or synthetic DNA probe and the target DNA, generation of species-unique patterns of amplified products by PCR (coupled with enzymatic restriction or not) and direct sequencing (3,4,6). Nevertheless, many of these methods are limited by: (i) the need for high amounts of quality DNA neither degraded nor chemically modified, (ii) the occurrence of non-specific DNA hybridizations, (iii) the difficulty of interpreting electrophoretic profiles in mixtures and (iv) the high dependence on laboratory conditions, hampering the standardization of results for inter- and intra-laboratory comparisons [for further information see (2–6)].
Direct DNA sequencing of cytosolic and mitochondrial ribosomal RNA (rRNA) (7–10) and protein-coding genes, e.g. cytochrome b (11–13) and cytochrome c oxidase I (14), is presently the most effective analytic and diagnostic approach for species identification. Conventional capillary sequencing methods usually rely on the inspection of a single large amplicon, over 300 bp (8,13,14), to obtain enough information for a confident discrimination, which poses serious problems for the typing of suboptimal DNA samples and increases the likelihood of null results (for instance, due to intraspecific polymorphisms on PCR primer-binding sites). Also, detection of mixtures requires species-specific primer sets or time-consuming and expensive cloning steps. The so called ‘next-generation’ of sequencing technologies have already offered promising results in metagenomic studies (15–17) but are still rarely used in species identification procedures and have not been subjected to validation studies (18,19).
Although the field will inevitably move towards more economical and faster sequencing technologies, it is still imperative to conceive other ways of retrieving information enclosed in nucleic acid sequences. This search can open new avenues in the development of valuable methodologies for genetic screening, and ultimately, the identification of individuals and species without directly determining DNA or RNA sequences. To address this need, we designed an alternative approach for biological identification in a wide range of life forms based on the length of rRNA gene regions. The use of size variation in hypervariable regions or related approaches has already proved to be efficient in the identification of species (20–27).
The SPecies Identification by Insertions/Deletions (SPInDel) method described here makes use of the evolution of rRNA gene sequences across species (Figure 1): hypervariable regions, regions containing multiple indels that potentially allow for differentiation by the determination of sequence length, are found interspersed with highly conserved domains, regions presenting none or low sequence variability used as anchors (10,28–31). Thus, a species can be defined by a unique set of fragment lengths (a ‘SPInDel profile’) that can be interrogated using the present array of high-throughput genotyping platforms, ranging from gel and capillary electrophoresis to mass spectrometry and high performance liquid chromatography. Numeric profiles of fragment lengths yield a considerable power of discrimination: in theory, and for simplicity’s sake assuming no functional constrains and independence of SPInDel loci, a survey of just 6 hypervariable regions with 20 alleles each (or 11 regions with 5 alleles each) is enough to discriminate all eukaryotic species on Earth, which are estimated to be between 5 and 15 million in number (32) (Figure 2a).
Figure 1.

Schematic illustration of the strategy used in the species identification by the insertions/deletions (SPInDel) method. (a) Illustration of the sequence alignment for three hypothetical species. Four conserved regions (green boxes) define three hypervariable domains (dotted brown lines). A section of the alignment is magnified to show the presence of multiple gaps in hypervariable regions. (b) Each species is identified by a numeric profile resulting from the combination of lengths in hypervariable regions.
Figure 2.

Species discrimination achieved with the SPInDel method. (a) Using a logarithmic scale, the four straight lines represent the possible number of different profiles obtained when combining lengths from hypervariable regions, considering 2, 5, 20 or 100 alleles per region. For simplicity’s sake, we assumed that each allele is sampled once and that there is independence among loci. The dotted line represents the estimated maximum number of eukaryotic species on Earth (15 million). (b) Maximum frequency of species-specific profiles observed by combining SPInDel hypervariable regions in 18 eukaryotic groups. The maximum frequency of species-specific SPInDel profiles (y-axis) is plotted for all m-combinations from a set with n hypervariable regions (x-axis), for m from 1 to n. The complete set of values for each taxonomic group can be better visualized on the Supplementary Figure S18.
We present a new method of species identification that includes the following: (i) a large data set comprising nearly 1800 numeric profiles for the identification of eukaryotic (n = 1556), prokaryotic (n = 105) and viral species (n = 130); (ii) a publicly available computational platform for data analysis accompanied by a collaborative online workspace (http://www.portugene.com/SPInDel/SPInDel_web.html and http://groups.google.com/group/spindel); (iii) a multiplex PCR profiling kit for the identification of 10 eutherian species by capillary electrophoresis; and (iv) a validation study performed in highly processed food products, including the identification of species from mixtures.
MATERIALS AND METHODS
Nucleotide sequences
The reference sequences for all complete mitochondrial genomes available on April 2009 were retrieved from the NCBI Entrez Nucleotide database (http://www.ncbi.nlm.nih.gov) with the Geneious 4.6.4 software (33). A total of 1757 complete mitochondrial genomes, mitochondrial chromosomes (e.g. Spizellomyces punctatus) and plasmids (e.g. Oryza sativa and Zea mays) were extracted (Supplementary Table S1). We arranged these sequences into 18 major taxonomic groups comprising at least six sequences each (Table 1) and excluded 96 sequences belonging to underrepresented taxonomic groups. In each group, we excluded the following: (i) duplicated reference sequences for the same species; for instance, Rattus norvegicus AC_000022.2 was removed, but R. norvegicus NC_001665.2 was kept; (ii) sequences lacking clearly annotated rRNA genes; and (iii) sequences in which rRNA genes have several introns. We included sequences identified as belonging to subspecies or hybrids. The adjacent large and small subunit rRNA genes were used in Platyhelminthes, Arthropoda and seven Vertebrate groups (Supplementary Figure S1). In the remaining taxa, only the large subunit rRNA gene was investigated because the large and small subunit rRNA genes were not contiguous.
Table 1.
General description of standard SPInDel profiles on eukaryotic, prokaryotic and viral groups
| Taxonomic group | Number of reference sequences (N) | Number of conserved regions | Number of hypervariable regions (n) | Average number of pairwise differences (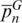 ) ) |
Average number of pairwise differences per hypervariable region | Number of species-specific profiles (Nsp) | Frequency of species-specific profiles (fnG) | Number of species with species-shared profiles | Number of species-shared profiles (Nsh) (number of species with shared profiles) |
||
|---|---|---|---|---|---|---|---|---|---|---|---|
| From different genera | From the same genus | Between subspecies or hybrids | |||||||||
| Eukaryotes | |||||||||||
| Viridiplantae (Vi) | 23 | 9 | 8 | 6.51 | 0.81 | 19 | 0.83 | 4 | 0 | 0 | 2 (4) |
| Stramenopiles (St) | 14 | 11 | 10 | 7.71 | 0.77 | 14 | 1.00 | 0 | 0 | 0 | 0 |
| Fungi (Fu) | 42 | 7 | 6 | 5.90 | 0.98 | 40 | 0.95 | 2 | 0 | 1 (2) | 0 |
| Alveolata (Al) | 6 | 8 | 7 | 4.67 | 0.67 | 6 | 1.00 | 0 | 0 | 0 | 0 |
| Porifera (Po) | 20 | 10 | 9 | 8.65 | 0.96 | 20 | 1.00 | 0 | 0 | 0 | 0 |
| Cnidaria (Cn) | 32 | 8 | 7 | 5.64 | 0.81 | 27 | 0.84 | 5 | 0 | 2 (5) | 0 |
| Platyhelminthes (Pl) | 29 | 10 | 9 | 7.93 | 0.88 | 29 | 1.00 | 0 | 0 | 0 | 0 |
| Echinodermata (Ec) | 20 | 6 | 5 | 3.91 | 0.78 | 18 | 0.90 | 2 | 0 | 1 (2) | 0 |
| Nematoda (Ne) | 29 | 6 | 5 | 4.00 | 0.80 | 27 | 0.93 | 2 | 0 | 1 (2) | 0 |
| Arthropoda (Ar) | 204 | 6 | 5 | 4.72 | 0.94 | 196 | 0.96 | 8 | 0 | 4 (8) | 0 |
| Mollusca (Mo) | 50 | 6 | 5 | 4.17 | 0.83 | 48 | 0.96 | 2 | 0 | 1 (2) | 0 |
| Actinopterygii (Ac) | 539 | 11 | 10 | 7.48 | 0.75 | 492 | 0.91 | 47 | 7 (14) | 10 (31) | 1 (2) |
| Amphibia (Am) | 84 | 7 | 6 | 5.12 | 0.85 | 81 | 0.96 | 3 | 0 | 1 (3) | 0 |
| Testudines (Te) | 29 | 11 | 10 | 7.74 | 0.77 | 29 | 1.00 | 0 | 0 | 0 | 0 |
| Lepidosauria (Le) | 69 | 8 | 7 | 5.36 | 0.77 | 69 | 1.00 | 0 | 0 | 0 | 0 |
| Aves (Av) | 98 | 12 | 11 | 8.90 | 0.81 | 94 | 0.96 | 4 | 0 | 0 | 1 (4) |
| Metatheria (Me) | 31 | 13 | 12 | 8.95 | 0.75 | 31 | 1.00 | 0 | 0 | 0 | 0 |
| Eutheria (Eu) | 237 | 13 | 12 | 9.63 | 0.80 | 211 | 0.89 | 26 | 3 (10) | 3 (7) | 4 (9) |
| Total [percentage of species from total N] | 1556 | 1451 [93.3%] | (Mean: 0.95) | 105 [6.75%] | 10 (24) [1.54%] | 24 (62) [3.98%] | 8 (19) [1.22%] | ||||
| Intra-species data sets | Number of species-shared profiles (Nsh) | ||||||||||
| Caenorhabditis briggsae | 25 | 6 | 5 | 0.08 | 0.02 | 1 | 0.04 | 24 | 1 | ||
| Drosophila simulans | 28 | 6 | 5 | 1.40 | 0.28 | 0 | 0 | 28 | 5 | ||
| Bos taurus | 145 | 13 | 12 | 0.51 | 0.04 | 3 | 0.02 | 142 | 6 | ||
| Homo sapiens | 5664 | 13 | 12 | 0.43 | 0.04 | 10 | 0.002 | 5654 | 18 | ||
| Prokaryotes | |||||||||||
| Crenarchaeota (Archaea) | 48 | 8 | 7 | 3.60 | 0.51 | 30 | 0.62 | 18 | 6 | ||
| Tenericutes (Bacteria) | 57 | 8 | 7 | 3.78 | 0.54 | 25 | 0.44 | 32 | 10 | ||
| Viruses | |||||||||||
| Lentivirus (Retroviridae) | 11 | 6 | 5 | 3.65 | 0.73 | 4 | 0.36 | 7 | 3 | ||
| Papillomaviridae (dsDNA viruses) | 94 | 6 | 5 | 3.68 | 0.74 | 75 | 0.80 | 19 | 9 | ||
| Rhabdoviridae (ssRNA viruses) | 25 | 6 | 5 | 3.38 | 0.68 | 10 | 0.40 | 15 | 6 | ||
A set of complete mtDNA sequences from Caenorhabditis briggsae, Drosophila simulans, Bos taurus and Homo sapiens was retrieved from the NCBI Entrez Nucleotide database for the assessment of intraspecific diversities. These species were selected because they present the largest number of available complete mtDNA sequences returned in an initial Entrez Nucleotide search. We then extracted the largest possible number of sequences for each species using several search statements on the Entrez Search Field (e.g. ‘complete mitochondrion species name’ and ‘complete mtDNA species name’).
Prokaryotic 16S rRNA sequences were obtained from the Greengenes web application (34). We extracted all classified sequences from the phyla Crenarchaeota (Archaea) and Tenericutes (Bacteria) with a minimum nucleotide size of 1250 and without nucleotide ambiguities. Redundant sequences belonging to the same species and sequences without a clear species assignment were subsequently removed. Nucleotide sequences of viral reference genomes were retrieved from the NCBI Viral Genomes project (http://www.ncbi.nlm.nih.gov).
All sequences in each group were aligned using the default settings of the MUSCLE 3.6 software (35). In the case of Arthropoda, Actinopterygii, Eutheria (Figure 3a) and intraspecific groups, we only ran the first two iterations of the MUSCLE algorithm (the –maxiters 2 option) as recommended by the software user's guide when dealing with a large number of sequences. The alignment of duplicated rRNA genes on mtDNA was also performed as described earlier.
Figure 3.

Species identification in Eutheria using the SPInDel method. (a) Partial overview of the sequence alignment of mitochondrial small and large subunit ribosomal RNA genes from 237 eutherian species (obtained with the Geneious software). The plot displays the identity across all sequences for every position (high and low identity values represented by green and red bars, respectively). SPInDel conserved regions (dark areas in the alignment) are indicated by lettered arrows. (b) Sliding window analysis of nucleotide diversity (100 bp windows with a 1 bp overlap) along aligned rRNA gene sequences showing the abrupt increase and decrease in diversity values resulting from multiple alignment gaps. (c) Homo sapiens 12S rRNA secondary structure retrieved from the Comparative RNA Web Site showing the location of SPInDel conserved regions A to F. (d) Example of standard SPInDel profiles for 10 species: Homo sapiens (Hs), Mus musculus (Mm), Oryctolagus cuniculus (Oc), Equus caballus (Ec), Felis catus (Fc), Canis familiaris (Cf), Bos taurus (Bt), Ovis aries (Oa), Capra hircus (Ch) and Sus scrofa (Ss). (e) Mismatch distribution of pairwise comparisons between 237 profiles revealed that 98.9% diverge by more than four hypervariable regions. (f) Frequency of species-specific alleles (black bars on the right) and the average number of pairwise differences (blue bars on the left) in each hypervariable region.
Selection of SPInDel conserved and hypervariable regions
We identified SPInDel conserved and hypervariable regions using a sliding window analysis of nucleotide diversity (π) (36) and the number of segregating sites (S) across rRNA gene alignments using the DnaSP ver. 5.10 software (37) (Figure 3b and Supplementary Figure S2). A window of 100 nucleotides was moved along the alignment in 1 nt steps, and the values for each parameter (π and S) were assigned to the nucleotide at the midpoint of each window. Sites with alignment gaps were counted in the window length. Standard diversity measures were also calculated in the DnaSP software (Supplementary Table S2).
SPInDel conserved regions were selected as potential primer-binding sites using, whenever possible, the following criteria: (i) location in highly conserved domains 17–25 nt long; (ii) GC content between 30% and 70%; (iii) corresponding to primers with a predicted melting temperature (Tm) between 54 and 63°C; and (iv) absence of self-complementarity. Salt adjusted Tm values (38) and hairpin and self-dimerization of primers were calculated using the Oligo Calc webserver (39).
The selected conserved regions for Eutheria (labeled A–F) and Amphibia (labeled E–G) were identified on the RNA structural model of the Homo sapiens 12S rRNA and the Xenopus laevis 16S rRNA, respectively (Figure 3c and Supplementary Figure S3). Structural diagrams incorporating secondary and tertiary interactions were retrieved from the Comparative RNA Web (CRW) Site (40) (http://www.rna.ccbb.utexas.edu). We also used the conservation diagram superimposed onto the Zea mays (accession number X00794) mitochondrial small subunit rRNA secondary structure available on the CRW site. This diagram indicates the maximum and minimum numbers of nucleotides observed in some variable domains based on the alignment of 901 sequences (Supplementary Figures S3 and S4).
The DNA secondary structures of the Homo sapiens 12S rRNA (accession number J01415) and Xenopus laevis 16S rRNA genes (accession number M10217) were predicted by free energy minimization methods (41) in the mfold web server (42) as described earlier (43). Folding temperatures of 37 and 20°C were used for H. sapiens and X. laevis rRNA gene sequences, respectively (Supplementary Figure S5).
A total of 6044 profiles from Eutheria, including H. sapiens and B. taurus intra-species profiles, were used to test for correlations among hypervariable regions. A hierarchical clustering analysis (HCA) was performed using the R package ‘pvclust’ (44). The hierarchical clustering was performed using average linkage correlation as a metric and 1000 bootstraps were used to estimate the bootstrap probabilities (BP). The results were presented in a dendrogram (Supplementary Figure S6). The same data set was examined by principal component analysis (PCA) using the programming language R. A 2D diagram of the PCA results was created by exporting the principal component values to Microsoft Excel.
We tested the discriminatory efficiency of the SPInDel approach on the eutherian data set by PCA, considering profiles as variables. A multidimensional scaling (MDS) analysis was also performed with the R package ‘MASS’ (45), using the dissimilarity matrix obtained from the number of differences between profiles generated on the SPInDel workbench (Figure 4a and Supplementary Figure S7). The same matrix was used to construct a UPGMA (Unweighted Pair Group Method with Arithmetic Mean) tree (Figure 4b). An in-house developed algorithm was used to cluster profiles based on a dissimilarity matrix obtained from the number of differences between the profiles from different species. The output was exported in the Newick format from the SPInDel workbench and edited in the FigTree v1.3.1 software (http://tree.bio.ed.ac.uk/).
Figure 4.

Discrimination of eutherian species using standard SPInDel profiles. (a) MDS plot obtained from the matrix of pairwise distances measured by the number of different loci between profiles. Species were labeled according to their taxonomic category. (b) UPGMA polar tree representing the phylogenetic relationship among the most represented eutherian groups. The input matrix and UPGMA tree were generated by the SPInDel workbench.
Calculations on SPInDel profiles
We defined ‘standard SPInDel profile’ as the set of fragment length of all contiguous SPInDel hypervariable regions observed in a sequence (AB length; BC length; CD length; Figure 3d). The fragment length also includes the two flanking conserved regions. The potential use of SPInDel profiles for species identification purposes requires the existence of ‘species-specific SPInDel profiles’: those that are only found in one species within a taxonomic group and allow their unequivocal identification. Thus, the discriminatory capacity of the SPInDel approach in a particular taxonomic group 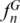 can be easily measured by the frequency of species-specific SPInDel profiles in that group,
can be easily measured by the frequency of species-specific SPInDel profiles in that group,
where G denotes the taxonomic group under investigation according to the two-letter code in Table 1, n is the number of SPInDel hypervariable regions included on the profile, Nsp is the number of species-specific SPInDel profiles and N is the total number of sequences represented on group G. For instance, 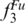 indicates the frequency of species-specific SPInDel profiles with three hypervariable regions observed in the Fungi group. The number of species-shared profiles (Nsh) refers to those that were found in more than one species. Accordingly, the total number of different profiles (Ndp) is Nsp + Nsh.
indicates the frequency of species-specific SPInDel profiles with three hypervariable regions observed in the Fungi group. The number of species-shared profiles (Nsh) refers to those that were found in more than one species. Accordingly, the total number of different profiles (Ndp) is Nsp + Nsh.
The average number of pairwise differences in a sample of SPInDel profiles was estimated using the equation for nucleotide diversity (π) (36), adapted as follows:
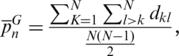 |
where k and l are indices that refer to individual SPInDel profiles, dkl is the number of SPInDel hypervariable regions (from the total set of n) that differ in length between profiles k and l and N is the total number of sequences represented in group G. The average number of pairwise differences per locus is 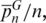 where n is the number of loci (i.e. hypervariable regions). The frequency distribution of the number of SPInDel hypervariable regions that differ between all pairs of SPInDel profiles in a taxonomic group (mismatch distribution; Figure 3e and Supplementary Figure S8) was estimated as described earlier for DNA sequences (46,47).
where n is the number of loci (i.e. hypervariable regions). The frequency distribution of the number of SPInDel hypervariable regions that differ between all pairs of SPInDel profiles in a taxonomic group (mismatch distribution; Figure 3e and Supplementary Figure S8) was estimated as described earlier for DNA sequences (46,47).
The two measures and 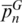 yielded disparate values for many loci as a result of their different sensitivities to the allele distributions. The parameter
yielded disparate values for many loci as a result of their different sensitivities to the allele distributions. The parameter 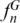 only considers the existence of unique alleles and disregards information from shared alleles. However, parameter has an opposite behavior that focuses on the uneven distribution of alleles among sequences, regardless of whether they are characteristic of a certain species. The former measure is appropriate to ascertain the discriminatory efficiency of the system concerning species identification, while the latter is a better indicator of the overall genetic diversity at each locus.
only considers the existence of unique alleles and disregards information from shared alleles. However, parameter has an opposite behavior that focuses on the uneven distribution of alleles among sequences, regardless of whether they are characteristic of a certain species. The former measure is appropriate to ascertain the discriminatory efficiency of the system concerning species identification, while the latter is a better indicator of the overall genetic diversity at each locus.
SPInDel profiles of unknown origin can be identified by a k-nearest neighbor method using a database of known profiles. The k-nearest neighbor algorithm is a supervised learning approach that finds the k closest matches in a database of known profiles using a distance metric. The SPInDel workbench uses the discrete metric: if x = y then d(x,y) = 0; otherwise, d(x,y) = 1. x is the length of one hypervariable region in the observed profile and y the length of the corresponding region in a profile from the reference database. The similarity values are obtained by summing the score of the discrete metric over all hypervariable regions in the profile. The discrete metric was chosen since it is not clear how distances between fragment lengths can be translated into evolutionary distances. The discrete metric means that length differences gets punished equally hard independent of the degree to which the lengths differ. The k nearest neighbor algorithm was implemented using Biopython and the discrete distance metric was added to the library.
A modified leave-one-out cross validation was used to test the accuracy of the SPInDel k-nearest neighbor classification algorithm. The modification ensures that there is always one profile left for each of the class labels in the reference data set. This is necessary because there are some classes with only one or few species or genera in the data set. The classification performance was calculated as the fraction of correct assignments (Supplementary Table S3).
SPInDel workbench
We developed a computational platform for project management, alignment of nucleotide sequences, visualization and selection of conserved regions, calculation of the properties of PCR primers, prediction of SPInDel profiles and diverse statistical and phylogenetic analyses. The workbench includes a database management system with aligned and curated gene sequences and SPInDel profiles for the identification of nearly 1800 species in all domains of life (1556 for eukaryotes, 105 for prokaryotic and 130 for viral species). Detailed information on the workbench can be found on Supplementary Figure S9 and at http://www.portugene.com/SPInDel/SPInDel_web.html.
Experimental data
We collected 94 samples from humans and 9 domestic mammalian species belonging to unrelated individuals and, when applicable, different breeds (Supplementary Table S4). Eight samples of non-mammalian species were also used (Supplementary Table S5). DNA was extracted from various types of biological materials, such as dried blood on FTA paper (Whatman, Clifton, NJ, USA), buccal swabs, muscle and liver, using standard Chelex (Biorad, Hercules, CA, USA), phenol–chloroform or saline extraction protocols. Processed food samples were obtained from supermarkets and retail stores and included meat mixtures with complex matrices subject to strong thermal and chemical treatments. These samples had to be subjected to mechanical homogenization prior to a standard phenol–chloroform extraction.
Using the SPInDel workbench, we performed the following: (i) aligned rRNA gene sequences from the 10 target species; (ii) selected the eutherian conserved regions A to M; (iii) retrieved degenerate primers; (iv) generated species-specific profiles; and (v) tested the discriminatory efficiency of the procedure. Conserved regions were used for the design of only forward or reverse primers to allow the simultaneous amplification of all loci by multiplex PCR (Figure 5a).
Figure 5.

Experimental application and validation of a SPInDel profiling kit for identification of eutherian species. (a) Graphical representation of the seven ribosomal RNA hypervariable regions amplified by multiplex PCR. PCR primers (arrows) were named using letters A to M, their orientation (F, for forward, R for reverse) and labeling with fluorescent dyes (blue, orange and green arrows). (b) The products of multiplex PCRs of three eutherian and one avian species are shown on a silver stained polyacrylamide gel. Each species has a unique pattern of migration because of differences in the length of amplicons (hypervariable regions). Identification of species using such gels is only possible by comparing the banding pattern of the target sample with those of reference samples analyzed with the same procedure. (c) Electropherogram illustrating a SPInDel profile from a human reference sample obtained by capillary electrophoresis with multidye fluorescence detection. The profile is displayed in a four-color fluorescent system, in which green, blue and yellow channels were used for detection of amplified products and red was used for a size marker. The species identification is achieved by running the SPInDel numeric profile of the target sample against a reference database (SPInDel workbench).
Degenerate primers were used in six conserved regions (A, B, D, F, H and M) to accommodate interspecific base differences and avoid loss of amplification due to mispriming (Supplementary Table S6). All primers retrieved from the workbench had similar melting temperatures (Tm near 60°C) to achieve a good balance among PCR products in the multiplex reaction. The final set of 23 PCR primers was screened for potential cross-reactivity in the multiplex reaction (all possible primer-primer combinations were tested) using the AutoDimer version 1.0 program with default settings (48). PCR primers were purchased from Thermo Electron Corp. (Waltham, MA, USA) with reverse phase HPLC purification and some were 5′-end labeled with 6-Carboxyfluorescein (6-FAM), 6-Carboxy-2′-, 4-, 7-, 7′-tetrachlorofluorescein (TET) and 6-Carboxy-2′-, 4-, 4′-, 5′-, 7-, 7′-hexachlorofluorescein (HEX) fluorescent dyes (Supplementary Table S6).
We tested each pair of primers by singleplex PCR on a Thermocycler GeneAmp PCR System 2700 (Applied Biosystems, Foster City, CA) using 2 μl of extracted DNA in a 12.5 μl reaction volume containing 1.25 μl of 10× PCR buffer, 0.25 μl of the four dNTPs (0.2 mM each), 1.0 μl of MgCl2 (25 mM), 1.25 μl of each primer (2.5 μM) and 0.1 μl of Taq polymerase (Bioron Ludwigshafen, Germany). After a 95°C pre-incubation step of 2 min, PCRs were performed for a total of 35 cycles of denaturation at 95°C for 30 s, annealing at 58°C for 30 s, and extension at 72°C for 1 min, with a final extension step of 10 min at 72°C. PCR products were separated by 12% polyacrylamide gel electrophoresis and visualized by silver staining (Figure 5b). To avoid an overlap between alleles from two hypervariable regions with similar size and the same fluorescent labels, all amplicons had size ranges at least 75 bp apart (Supplementary Figure S10). Although we tested primers labeled with three dyes (6-FAM, TET, HEX) to explore the full potential of the detection system, two dyes are sufficient to combine these seven loci in a single multiplex reaction (locus DE could be labeled with TET dye).
After optimization, multiplex PCRs were performed on all samples, including on those from non-mammalian species and processed food product (Supplementary Table S7), using the SPInDel profiling kit by combining 2 μl of extracted DNA, 1 μl of primer mix (2 μM of each primer) and 5 μl of Multiplex PCR Master Mix (Qiagen GmbH, Germany) in a 10 μl final volume. PCRs were performed as follows: an initial denaturation step at 95°C for 15 min, followed by 30 cycles of 30 s at 94°C, 90 s at 56°C and 1 min at 72°C and a final extension step of 50 min at 72°C. A long final extension step is recommended to promote complete adenylation (the non-template addition) in all PCR products. We produced ladders for each locus by mixing amplified products of each allele in balanced concentrations (Supplementary Figure S11). Each allelic ladder was then re-amplified to increase the final stock volume.
Samples were prepared for fragment-size detection by adding 1 µl of PCR product to 15 µl deionized formamide containing 0.75 µl GeneScanTM 500 TAMRA size standard (Applied Biosystems). The separation and detection of amplified products were accomplished with the ABI PrismTM 310 Genetic Analyzer (Applied Biosystems) using filter set C with 6-FAM, TET and HEX (Figure 5c, Supplementary Figures S12 and S13). Sizing was performed using the GeneScanTM 350 TAMRA size standard and the ABI PrismTM GeneScanTM v3.1.2 software package (Applied Biosystems). Positive and negative controls were used in all reactions.
RESULTS
Gaps in ribosomal RNA gene alignments
We decided to use mitochondrial rRNA genes instead of cytosolic versions because: (i) several reference mitochondrial genomes are available from which complete rRNA genes can be retrieved; (ii) on average, mtDNA has a high mutation rate than nuclear DNA (with the exception of plants) and thus accumulates more indels; and (iii) the high number of mtDNA copies per cell increases the probability of obtaining results from degraded/low-copy DNA samples. Therefore, we aligned 1556 mitochondrial rRNA gene sequences belonging to species from 18 eukaryotic groups (Figure 3a; Supplementary Table S1 and Figure S1). The proportion of sites with gaps in each alignment varies from <15% in Alveolata (10.0 %) and Metatheria (14.1%) to >90% in Fungi (91.3%) (Supplementary Table S2 and Figure S14). The inspection of sequence alignments revealed that most observed gaps result from small insertion/deletion (indel) polymorphisms across species. Although most rRNA genes with large or numerous introns have been excluded to allow feasible alignments, the inclusion of a few rRNA genes with small introns explains the high number of gaps observed in the intron-rich mtDNA of Fungi and Viridiplantae (Supplementary Figure S14). The highest values of nucleotide diversity and number of variable sites, not considering sites with gaps, were observed in invertebrate groups (Supplementary Table S2). It should be noted that the inclusion of more species in a taxonomic group yields fewer conserved and longer hypervariable regions.
Some mitochondrial genomes contain duplicated regions. The existence of two or more different copies of a particular gene within the same genome can result in mixed profiles and lead to misclassifications. Here, we analyzed 10 species with duplicated rRNA genes that fulfilled our criteria for inclusion in the SPInDel database: 3 from Viridiplantae, 1 from Stramenopiles, 5 from Alveolata and 1 from Nematoda. The sequence alignment of duplicated rRNA genes showed no indel events in any of these cases and a few base substitutions in Alveolata species (Supplementary Figure S15). These results demonstrate that our identification procedure is not hampered by the existence of duplicated genes at least in this set of species. On the contrary, sequencing-based approaches could be more prone to dubious classifications due to the occurrence of base substitutions, as observed in Alveolata species.
Eukaryotic SPInDel profiles
We performed a sliding window analysis of nucleotide sequence variability across mitochondrial rRNA genes to identify highly conserved regions, i.e. regions with no or minimal variability at the sequence level (‘SPInDel conserved regions’), and regions with high numbers of indels across species (‘SPInDel hypervariable regions’, here also considered as polymorphic loci). The nucleotide diversity (π) and the number of segregating sites (S) vary markedly across rRNA genes, with several peaks of high π and S values on multiple parts of the alignments (Figure 3b; Supplementary Figure S2). The abrupt rise and sudden decline in diversity values result from an imbalanced distribution of gaps along the sequence alignment with indel-rich sections interspersed with highly conserved domains. SPInDel conserved regions were designated alphabetically starting at the 5′-end of the rRNA gene, and SPInDel hypervariable regions were designated by the combination of SPInDel conserved regions that define them (e.g. ‘AB’ designates the hypervariable region defined by the conserved regions ‘A’ and ‘B’; Figure 1). The hypervariable regions presented here are relative to each taxonomic group under study. For instance, hypervariable regions AB in Viridiplantae and Eutheria are not comparable because they represent different sections of the rRNA sequence alignments.
We defined a ‘SPInDel profile’ as the numeric set of fragment lengths measured in nucleotides (also designated here as alleles) in two or more SPInDel hypervariable regions from a DNA sequence (e.g. 176, 101, 119 in species 1 of Figure 1). A ‘standard SPInDel profile’ refers to profiles that include all contiguous hypervariable regions (Figure 3d shows standard profiles observed in some eutherian species). We generated 1556 standard profiles using the sequence alignment of rRNA gene as indicated in Supplementary Figure S1, after defining conserved regions for 18 taxonomic groups of eukaryotes (Supplementary Table S1). The utility of SPInDel as a taxonomic tool requires the existence of ‘species-specific SPInDel profiles’, i.e. profiles unique within a taxonomic group that therefore allow the identification of a species. We observed 1451 species-specific profiles among the 18 eukaryotic groups for mitochondrial rRNA genes, which meant that 93.3% of all SPInDel profiles are unique (Table 1). The frequency of species-specific SPInDel profiles (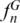 ) was higher than 0.90 in all eukaryotic groups (mean value of 0.95), except in Viridiplantae due to the inclusion of some subspecies profiles (
) was higher than 0.90 in all eukaryotic groups (mean value of 0.95), except in Viridiplantae due to the inclusion of some subspecies profiles (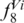 = 0.83) (Table 1; Supplementary Figure S16). In seven groups, all species had a unique profile (
= 0.83) (Table 1; Supplementary Figure S16). In seven groups, all species had a unique profile (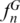 = 1.00). The number of species with shared profiles was 105 (6.75% of the total number of species). When we excluded subspecies and hybrids with equal profiles (19 cases), this value decreased to 5.53%. For instance, the same profile was observed in Zea mays mays and Z. mays parviglumis (but this profile was different from that of Z. luxurians and Z. perennis) and in Anguilla bicolor bicolor and A. bicolor pacifica (Supplementary Table S8). Similarly, the three subspecies of the red junglefowl (Gallus gallus gallus, G. gallus spadiceus and G. gallus bankiva) had the same profile, but this profile was different from other species of the same genus (G. lafayetii, G. sonneratii and G. varius). We also noticed that some domestic species and their wild ancestors had equivalent profiles, in agreement with the well-known difficulty of devising a satisfactory genetic definition for ‘domestic species’ in comparison to its wild ancestor, as in the cases of dog (Canis lupus familiaris) and wolf (C. lupus lupus) or the llama (Lama glama) and guanaco (Lama guanicoe).
= 1.00). The number of species with shared profiles was 105 (6.75% of the total number of species). When we excluded subspecies and hybrids with equal profiles (19 cases), this value decreased to 5.53%. For instance, the same profile was observed in Zea mays mays and Z. mays parviglumis (but this profile was different from that of Z. luxurians and Z. perennis) and in Anguilla bicolor bicolor and A. bicolor pacifica (Supplementary Table S8). Similarly, the three subspecies of the red junglefowl (Gallus gallus gallus, G. gallus spadiceus and G. gallus bankiva) had the same profile, but this profile was different from other species of the same genus (G. lafayetii, G. sonneratii and G. varius). We also noticed that some domestic species and their wild ancestors had equivalent profiles, in agreement with the well-known difficulty of devising a satisfactory genetic definition for ‘domestic species’ in comparison to its wild ancestor, as in the cases of dog (Canis lupus familiaris) and wolf (C. lupus lupus) or the llama (Lama glama) and guanaco (Lama guanicoe).
Shared profiles were mostly observed among species belonging to the same genus (3.98% of the total number of species). In many cases, this apparent limitation of our approach simply reflects the lack of clear boundaries between species, particularly those exposed to recent hybridizations or with short evolutionary histories (sometimes referred to as ‘species complexes’ to indicate their close genetic similarity). For instance, the only shared profile in Fungi was between Aspergillus tubingensis and A. niger from the A. niger species complex, which is one of the most difficult groups to classify and identify (Supplementary Table S8). In Cnidaria, the same profile was shared by the three members of the Montastraea annularis coral species complex (M. annularis, M. franksi and M. faveolata), whose species status has been disputed for many years (49). Similarly, the same profile was observed in two morphospecies of the coral genus Pocillopora (P. eydouxi and P. damicornis). Note that in both cases these species are impossible to differentiate not only by SPInDel profiles but also by sequencing of rRNA genes, a situation which was also observed in some species of Actinopterygii, Aves and Eutheria (the number of haplotypes is lower than the number of reference sequences used; Supplementary Table S2). The presence of shared profiles and rRNA sequences indicates that discrimination of these species would also not be possible by mtDNA sequencing, at least on rRNA genes.
In the largest animal phyla, Arthropoda, only four profiles were found shared among 204 species, with 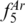 = 0.96. Two profiles included four species of the Bactrocera dorsalis complex of tropical fruit flies, a recently recognized, rapidly evolving species complex with multiple sibling species (50) (Supplementary Table S8). The four pest species with shared profiles are morphologically similar (50), but our approach differentiates B. carambolae/B. papaya from B. dorsalis/B. philippinensis. We only found 24 species from different genera with equal profiles (1.54% of the total number of species), and they were restricted to Actinopterygii (seven profiles shared by 14 species from a total of 539) and Eutheria (three profiles shared by 10 species from a total of 237).
= 0.96. Two profiles included four species of the Bactrocera dorsalis complex of tropical fruit flies, a recently recognized, rapidly evolving species complex with multiple sibling species (50) (Supplementary Table S8). The four pest species with shared profiles are morphologically similar (50), but our approach differentiates B. carambolae/B. papaya from B. dorsalis/B. philippinensis. We only found 24 species from different genera with equal profiles (1.54% of the total number of species), and they were restricted to Actinopterygii (seven profiles shared by 14 species from a total of 539) and Eutheria (three profiles shared by 10 species from a total of 237).
Overall, these results show the great efficiency of the SPInDel approach for species-level assignments of different eukaryotic lineages using mitochondrial rRNA genes, with successful identification in >95% of cases. Only a small number of species are impossible to distinguish with our approach (Table 1; Supplementary Table S8), although most of these can be discriminated using additional hypervariable regions (e.g. the additional analysis of small subunit rRNA gene sequences in certain groups not described here). In other situations, the simple addition of a conserved region to split a hypervariable domain is sufficient for identification. For example, D. melanogaster and D. simulans have two indel polymorphisms in the hypervariable region EF (one insertion in each species), resulting in the same sequence length. The inclusion of a conserved region (e.g. Z) between these two indels creates two hypervariable regions (EZ and ZF) in which each one differentiates between species by a single nucleotide.
We also measured the level of discrimination allowed by the SPInDel approach by considering the number of pairwise differences between profiles, i.e. the higher the number of different hypervariable regions between two profiles, the lower the probability of misclassification. The average number of pairwise differences per hypervariable region (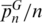 ) varies from 0.67 in Alveolata to 0.98 in Fungi (Table 1). Values higher than 0.75 were found in most groups, indicating that on average, two profiles diverge by more than three quarters of their loci. This pattern is clearly seen in the histograms representing the distribution of all individual pairwise profile comparisons (mismatch distribution) because most groups presented unimodal distributions with high mean values of pairwise mismatches (Figure 3e; Supplementary Figure S8). In several groups, most pairwise comparisons were made between completely different profiles (last column on the histograms; as in Viridiplantae, Fungi, Porifera or Arthropoda).
) varies from 0.67 in Alveolata to 0.98 in Fungi (Table 1). Values higher than 0.75 were found in most groups, indicating that on average, two profiles diverge by more than three quarters of their loci. This pattern is clearly seen in the histograms representing the distribution of all individual pairwise profile comparisons (mismatch distribution) because most groups presented unimodal distributions with high mean values of pairwise mismatches (Figure 3e; Supplementary Figure S8). In several groups, most pairwise comparisons were made between completely different profiles (last column on the histograms; as in Viridiplantae, Fungi, Porifera or Arthropoda).
Although we focus our attention on performing identification at the species level, higher-level taxonomic categories can also be discriminated using our approach. The phylogenetic, principal component and MDS analyses show that the assignment of individuals to higher taxonomic levels is achievable in Eutheria. In some cases, taxonomic divisions such as families and orders appeared as nested monophyletic lineages or clusters in concordance with current taxonomy (Figure 4; Supplementary Figure S7). However, some discrepancies are clear in a few categories, such as Chiroptera and Insectivora. These clustering approaches can still be very useful to retrieve some taxonomic information from casework samples belonging to species not present in the database.
Region by region and combinatory analyses
In order to assess the individual contribution of each hypervariable region to the discriminatory efficiency of standard profiles, we estimated 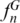 and
and 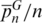 using the SPInDel workbench (Figure 3f; Supplementary Figure S17). We next asked how many hypervariable regions would be necessary to identify species with high accuracy within each taxonomic group. All profiles with n hypervariable regions were generated and compared in the SPInDel workbench by obtaining all n-combinations. When only one locus was considered (n = 1), the maximum observed
using the SPInDel workbench (Figure 3f; Supplementary Figure S17). We next asked how many hypervariable regions would be necessary to identify species with high accuracy within each taxonomic group. All profiles with n hypervariable regions were generated and compared in the SPInDel workbench by obtaining all n-combinations. When only one locus was considered (n = 1), the maximum observed 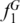 varied from 0.03 in Actinopterygii and Eutheria to more than 0.80 in Viridiplantae, Alveolata, Porifera and Echinodermata (Figure 2b; Supplementary Figure S18). These values drastically increased when profiles with two loci were considered (n = 2); in two-combined profiles, the maximum
varied from 0.03 in Actinopterygii and Eutheria to more than 0.80 in Viridiplantae, Alveolata, Porifera and Echinodermata (Figure 2b; Supplementary Figure S18). These values drastically increased when profiles with two loci were considered (n = 2); in two-combined profiles, the maximum 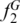 was higher than 0.80 in 15 groups out of 18. Actinopterygii and Eutheria reached a maximum
was higher than 0.80 in 15 groups out of 18. Actinopterygii and Eutheria reached a maximum 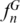 higher than 0.80 with four and five combined regions, respectively. This result clearly shows that just a few loci are sufficient to identify most species within a group. Interestingly, joining regions with the highest individual discriminative values did not necessarily lead to higher discriminatory efficiencies for combined regions. For instance, in Arthropoda, the
higher than 0.80 with four and five combined regions, respectively. This result clearly shows that just a few loci are sufficient to identify most species within a group. Interestingly, joining regions with the highest individual discriminative values did not necessarily lead to higher discriminatory efficiencies for combined regions. For instance, in Arthropoda, the 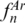 of separated loci were 0.37 (DE), 0.13 (EF), 0.07 (AB), 0.04 (BC) and 0.03 (CD), but when combined, ‘DE; EF’ had a lower
of separated loci were 0.37 (DE), 0.13 (EF), 0.07 (AB), 0.04 (BC) and 0.03 (CD), but when combined, ‘DE; EF’ had a lower 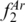 value (0.85) than ‘BC; DE’ (0.89) and ‘AB; DE’ (0.88). The highest
value (0.85) than ‘BC; DE’ (0.89) and ‘AB; DE’ (0.88). The highest 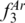 value for a three-locus profile was obtained with ‘AB; BC; DE’ (0.96), which did not include the region (EF) with the second highest value of
value for a three-locus profile was obtained with ‘AB; BC; DE’ (0.96), which did not include the region (EF) with the second highest value of 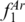 .
.
These observations suggest that variations in some hypervariable regions could be correlated. In order to test this hypothesis, we implemented hierarchical clustering and principal component analyses on the complete eutherian data set. In fact, adjacent regions of eutherian rRNAs were highly correlated, particularly the pairs BC/DE, EF/GH and JK/LM (Supplementary Figure S6). One possible explanation for this phenomenon, assuming non-recombination of mtDNA in eutherian species (51), is the occurrence of an insertion (or deletion) of a nucleotide into a stem region followed by a compensatory insertion (or deletion) on the complementary strand in order to maintain the structural stability. The length of adjacent regions would be under co-evolution because paired bases are usually close in the sequence. Epistatic interactions between alleles to preserve the structural properties of DNA or proteins have already been detected in coding (52) and non-coding (43) regions of mtDNA. Further work is necessary to explain these striking results.
Intra-species data sets
The effectiveness of SPInDel depends upon the existence of low intraspecific variation, so we analyzed four large intra-species data sets from Nematoda (Caenorhabditis briggsae), Arthropoda (D. simulans) and Eutheria (Bos taurus, Homo sapiens). Values of 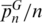 lower than 0.05 were observed in C. briggsae, B. taurus and H. sapiens (although reaching 0.28 in D. simulans), which indicates that all profiles from the same species diverged by a small number of hypervariable regions (Table 1 and Supplementary Figure S16). The mismatch distribution of intra-species data sets clearly shows that most profiles are equal, with the exception of D. simulans (Supplementary Figure S19). From 5664 H. sapiens rRNA gene sequences, only 10 individuals (0.2%) had unique profiles (Table 1), whereas most sequences were equal or divergent by only one out of 12 hypervariable regions. Based on these results, we concluded that high levels of intraspecific divergence are uncommon, although detailed sampling of SPInDel profiles in more species is needed to verify this observation.
lower than 0.05 were observed in C. briggsae, B. taurus and H. sapiens (although reaching 0.28 in D. simulans), which indicates that all profiles from the same species diverged by a small number of hypervariable regions (Table 1 and Supplementary Figure S16). The mismatch distribution of intra-species data sets clearly shows that most profiles are equal, with the exception of D. simulans (Supplementary Figure S19). From 5664 H. sapiens rRNA gene sequences, only 10 individuals (0.2%) had unique profiles (Table 1), whereas most sequences were equal or divergent by only one out of 12 hypervariable regions. Based on these results, we concluded that high levels of intraspecific divergence are uncommon, although detailed sampling of SPInDel profiles in more species is needed to verify this observation.
SPInDel profiles of prokaryotes and viruses
We tested the SPInDel concept on the cytosolic 16S rRNA genes of two prokaryotic groups: Crenarchaeota (Archaea) and Tenericutes (Bacteria). The alignment of rRNA genes revealed that 10.4 and 21.6% of sites had gaps in Crenarchaeota and Tenericutes, respectively (Supplementary Table S2). These values were lower than those found in most eukaryotic groups for mitochondrial rRNAs. Even so, several species had unique SPInDel profiles, with 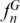 reaching 0.62 in Crenarchaeota and 0.44 in Tenericutes. The mismatch distribution of pairwise differences showed that 80% of profile comparisons diverged by three or more loci in Crenarchaeota, while this value reached 88% in Tenericutes (Supplementary Figure S20). The
reaching 0.62 in Crenarchaeota and 0.44 in Tenericutes. The mismatch distribution of pairwise differences showed that 80% of profile comparisons diverged by three or more loci in Crenarchaeota, while this value reached 88% in Tenericutes (Supplementary Figure S20). The 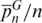 was 0.51 and 0.54 in Crenarchaeota and Tenericutes, respectively. Identical profiles were almost always derived from species of the same genus (Supplementary Table S8).
was 0.51 and 0.54 in Crenarchaeota and Tenericutes, respectively. Identical profiles were almost always derived from species of the same genus (Supplementary Table S8).
To verify whether the SPInDel concept could be generalized to taxa without rRNA genes, we tested all available reference genomic sequences from five taxonomic groups of viruses: Lentivirus (Retroviridae), Papillomaviridae (dsDNA viruses), Rhabdoviridae (ssRNA negative-strand viruses), Flaviviridae and Picornaviridae (ssRNA positive-strand viruses). Mosaics of conserved and hypervariable regions suitable for SPInDel implementation were found on the pol gene of Lentivirus, L1 ORF of Papillomavirida and the L gene of Rhabdoviridae. No such pattern was found on the sequence alignment of ssRNA positive-strand viruses. Values of 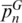 were similar to those of some eukaryotic groups (0.68 to 0.74), while
were similar to those of some eukaryotic groups (0.68 to 0.74), while 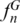 varied from 0.36 in Lentivirus to 0.80 in Papillomaviridae (Table 1). This result clearly demonstrates that our methodology can be used in different genomic regions wherever conserved domains are interspersed with variable-length sequences.
varied from 0.36 in Lentivirus to 0.80 in Papillomaviridae (Table 1). This result clearly demonstrates that our methodology can be used in different genomic regions wherever conserved domains are interspersed with variable-length sequences.
SPInDel profiling kit
We proved the efficacy of the SPInDel concept by discriminating 10 eutherian species (Supplementary Table S4) using the length of seven hypervariable regions determined by gel and capillary electrophoresis (Figure 5). The experiment was designed using the SPInDel workbench (Supplementary Figure S9 and S21).
As a first step toward a multiplex assay, we tested seven primer pairs using singleplex PCR on two to five samples from each target species (Figure 5a). Amplification and correct sizes of PCR products were confirmed on polyacrylamide gels (data not shown) for six loci with PCR primer melting temperatures (Tm) of 58° C. Locus FG yielded no PCR product in several attempts; therefore, a new forward primer was designed between positions 1569 and 1588 (instead of 1549 and 1570) according to the H. sapiens mtDNA reference sequence numbering (Supplementary Table S6). Then, the seven primer pairs were combined at similar concentrations (2 μM) and tested in a multiplex reaction under annealing-temperature gradient PCR from 48 to 64°C. A Tm = 56°C resulted in the amplification of all desired loci with similar band intensities and no relevant non-specific amplicons. Some species are clearly discriminated by conventional polyacrylamide gels as long as reference samples are analyzed simultaneously (Figure 5b).
Subsequently, amplified products were analyzed by capillary electrophoresis with multicolor fluorescence detection, which resolves similar sized PCR products labeled with spectrally distinguishable dyes; in this case, three dyes were used (Supplementary Figure S10). Hypervariable regions AC and FG yielded peaks with lower intensities that were, nevertheless, within optimal detection limits (Figure 5c). We observed some differences between the expected and observed length of amplified products, most likely because of differences in purine and pyrimidine contents, which are known to influence the mobility of DNA molecules. To compensate for this difference, we used internal size standards and the establishment of a home-made allelic ladder to accurately identify alleles (Supplementary Figure S11).
The three dye multiplex PCR assay was used to analyze 94 reference samples of human and domestic species (Supplementary Table S4). In some cases, traces of non-specific amplification (e.g. peaks resulting from cross-amplifications between primers designed for different loci) were detected but did not interfere with the expected profile (Figure 5c). For instance, a peak close to locus JK recurrently appeared on most Felis catus samples, but it did not prevent the correct interpretation of the profile. We obtained successful amplifications for the seven loci in all samples with the exception of seven Capra hircus individuals for which no traces of amplified products were observed in locus FG, possibly due to polymorphisms on primer-binding sites. Because the profiles of these 10 species diverge by at least six hypervariable regions (Supplementary Figure S21), incomplete profiles missing a few hypervariable regions are informative enough for unambiguous identification.
Testing multiple members of the same species showed that peak sizes were consistently identical among individuals. However, we did detect intra-specific polymorphisms in two species. A shorter allele was observed in a F. catus individual for locus DE, although species identification was unequivocal. Interestingly, wild and domestic rabbits (Oryctolagus cuniculus) had different profiles, as all wild individuals presented a shorter allele at locus HI and a longer allele at locus JK. Additional samples are required to demonstrate that wild and domestic variants of rabbits can be distinguished by our method. In any case, identification of O. cuniculus samples (wild or domestic) was unequivocal among these 10 species because species-specific profiles diverge by at least six loci (Supplementary Figure S21).
The design of PCR primers in highly conserved regions significantly increases the probability of successful amplifications in highly divergent species. We tested the SPInDel profiling kit using standard PCR conditions in representative species from Arthropoda, Mollusca, Actinopterygii and Aves (Supplementary Table S5). In all cases, amplified products were obtained that defined very different profiles among classes that can be used for preliminary identifications, at least at the class level (Supplementary Figure S12). The different length of amplicons observed for different classes suggests that cross contaminations are also easily detected. Overall, these results confirmed that broad-range SPInDel systems can be easily devised because primers designed for eutherian species yield amplified products for very divergent taxonomic groups.
The co-amplification of seven informative rRNA regions in a single multiplex PCR and the length of the targeted amplicons (five out of seven loci had alleles <350 bp in length) enables this method to be used for suboptimal DNA samples (Supplementary Figure S10). This was demonstrated by the typing of nine processed food samples that had been subjected to severe physical and chemical alterations (Supplementary Table S7). We obtained complete profiles in six cases, while three samples yielded incomplete Sus scrofa profiles. As expected, longer regions (FG and AC) were more difficult to amplify in suboptimal samples. Despite this limitation, we were able to unequivocally identify all species reported on the food products’ labels, including products that contained mixtures of species. The identification was done by comparing the numeric profiles obtained by capillary electrophoresis with those of the reference samples. For instance, we easily detected porcine and bovine biological material in a mixed meat food product (Supplementary Figure S13).
DISCUSSION
We developed a new resource for the taxonomical identification of eukaryotic species based on the particular mode of sequence evolution of mitochondrial ribosomal RNA (rRNA) genes. The core element of the protein synthesis machinery is the ribosome, a complex catalytic machine composed of rRNA molecules and several different proteins (the ribosomal proteins) (53). All mitochondrial genomes harbor genes coding for the small and large subunit rRNAs, whereas genes for 5S rRNA only occur in a few species (54).
The rRNA molecules are folded into precise 3D structures that form the compact core of the ribosome and determine its overall shape (28,31,53) (Figure 3c). The structural features of rRNA molecules include regular unknotted helical elements composed of canonical base pairs (the so-called secondary structure) along with more complex longer-range intramolecular tertiary interactions, such as pseudoknots (i.e. the interaction between a loop of one stem and residues outside that stem). Although biologically active RNAs most likely undergo functional conformational transitions, similar rRNA architecture is evident in higher-order rRNA structural models obtained by comparative sequence analysis and experimental methods (28,31). Interestingly, homology in secondary and tertiary interactions in rRNA structural motifs is achieved with very different primary sequences, which means that different sequences can correspond to the same 3D structure and molecular function. In general, alignments of primary rRNA gene sequences from different species show alternating regions of nucleotide conservation and variation (Figure 3b; Supplementary Figure S2), both in terms of nucleotide substitutions (commonly called ‘SNPs’) and insertion/deletion (indel) events (10,30). The presence of indels results in sequences of different lengths and introduces gaps in the alignment, typically denoted by a dash ‘–’ (Figure 3a). Thus, the overall length of these hypervariable regions is poorly conserved and these regions are often referred to as expansion segments or D (divergent) domains (29,30).
The DNA sequence analysis of both cytosolic and mitochondrial rRNA genes has been used as a primary tool for phylogenetic reconstructions, mainly because they occur in all organisms, are functionally conserved and have a mosaic structure of conserved and variable regions that allows for inferences on a wide range of phylogenetic time scales (7,10,55). As pointed out previously (14), the broad taxonomic use of rRNA genes is constrained by the prevalence of indels that greatly complicate sequence alignments. However, our concept for biological identification circumvents this apparent limitation by using rRNA gene sequences in a different manner: conserved regions are used to define variable segments in which a combination of sequence lengths is characteristic of each species (Figure 1). Because the conserved core of nucleotide sequences is retained in all rRNA sequences [some were postulated to date back to the RNA world (56)], our approach has the potential to work for very general taxonomic identifications. Moreover, as suggested by our investigation of different groups of viruses (Table 1), other genomic regions may present similar patterns of sequence evolution and thus may also be suitable for species identification using the SPInDel concept.
The mosaicism seen in the distribution of gaps throughout rRNA genes is most likely the result of purifying selection acting to maintain the rRNA architecture with differences in the base-to-base mutability of rRNA gene sequences. It has been demonstrated that there is considerable heterogeneity in the relative rates of evolution of different structural categories (stems, loops, bulges and junctions) within cytosolic rRNA and that these rates vary across phylogenetic domains (31). Although evolutionary rates for mitochondrial rRNA genes are still poorly defined, indel proliferation is likely most constrained in regions corresponding to functionally-relevant stems and in elements with unpaired bases of strong structural and functional relevance. For instance, most SPInDel conserved regions selected from the eutherian 12S rRNAs include unpaired bases at multi-helix junctions (e.g. regions A and B) or near pseudoknots (region E) (Figure 3c and Supplementary Figure S3). Figure 3c clearly shows that one of the most divergent regions with multiple indels found in Eutheria belongs to a large loop between conserved regions E and F. The conservation diagram for eukaryotic mitochondrial small subunit rRNA also shows a great variability in the number of nucleotides within hypervariable regions (Supplementary Figure S4). For instance, different eutherian species can have between 3 and 400 nt in hypervariable region EF. Therefore, the proliferation of indels is most likely associated with unconstrained regions such as large loops.
The patchy structure observed in rRNA gene alignments could also be influenced by heterogeneity in the mutation rate across mitochondrial DNA positions, as a result of differences in replication errors and/or DNA damage. For instance, the transient formation of secondary structures in DNA during replication and transcription may protect paired bases from chemical alterations, while rendering others vulnerable, such as unpaired and mispaired bases of stem-loop structures (57–59). This is particularly relevant in mtDNA genes because of the highly mutagenic environment of mitochondria, which is rich in oxygen-free radicals. In fact, this could help to explain why mitochondrial rRNAs have higher rates of indels (resulting in a higher power of discrimination for SPInDel) than do their cytosolic counterparts (Table 1). Moreover, rRNAs are synthesized at a higher rate than mRNAs in human mitochondria, with a 15- to 60-fold excess of truncated transcripts containing the two rRNAs (and no mRNAs) relative to full length transcripts encoding all mRNAs, the two rRNAs, and most of the tRNAs (60–62). This high rate of synthesis significantly increases the propensity for transcription-driven mutagenesis in rRNA genes. In agreement with these observations, we observed long stretches of paired bases intermingled with large loops in the DNA sequences encoding the H. sapiens 12S rRNA and X. laevis 16S rRNA molecules (Supplementary Figure S5). This sequence structure is in agreement with the high folding potential observed for the genomic region encoding rRNAs and the resemblance between DNA and complementary RNA structures that was previously detected in human mtDNA (43,63). These results suggest that heterogeneity in the rate of nucleotide changes in DNA sequences (and its mosaic structure) could indirectly reflect the folding potential of derived RNA molecules.
One great benefit of using our molecular approach for the identification of species is its use of indel polymorphisms. Overall, this class of polymorphisms has an intra-species frequency that is significantly lower than that of SNPs. For example, it was estimated that ∼20% of all human polymorphisms in the nuclear genome are indels, consistent with the values found in D. melanogaster (16%), C. elegans (25%) and A. thaliana (37%) (64). A survey of 60 published intraspecific data sets revealed that only 30 studies reported indels, which comprised on average 12% of all variable sites in the non-coding mtDNA control region (65). Similarly, our method is also less prone to misclassifications due to the occurrence of mitochondrial heteroplasmy, i.e. when there are two or more mtDNA types present within the cells of a single individual. This is because small indels are less likely to occur in tissues than base substitutions, particularly in protein-coding regions (66).
The complex mutational mechanism underlying indel events also helps to explain the low probability of having an excess of polymorphic variants of this class within a species or individual. Indels are less prone to recurrent mutations (i.e. identical insertions or deletions occurring in independent lineages), which means that there is a low probability that similar sequences originated by convergence (homoplasy). In this regard, an investigation of 38 published interspecific data sets (including mitochondrial rRNAs) proved that indels have significantly less homoplasy than base alterations, while representing a considerable portion of the potential phylogenetic informative content (67). The insertion of a nucleotide that restores a previous deletion at the same position or vice versa (a phenomenon known as ‘back mutation’) is also very unlikely in this class of polymorphisms. In fact, the low diversity observed in hypervariable regions in the intraspecific data sets (Table 1) and in the 94 tested eutherian samples corroborates these theoretical expectations.
Despite the low intra-species diversity in rRNA genes, indel polymorphisms have a sufficiently rapid evolutionary rate of accumulation that allows for discrimination between closely related taxa. The frequency of species-specific SPInDel profiles, even when subspecies were included in taxonomic groups, was higher than 0.90 in most cases (mean value of 0.95), which indicates that 93.3% of all SPInDel profiles are unique (1451 species-specific profiles from a total of 1556 species). In any case, well-sampled data sets are still necessary to prove that intraspecific variation and interspecific divergence in rRNA variable-length regions do not overlap for various taxonomic groups.
The occurrence of intraspecific or intraindividual variability in hypervariable regions does not pose serious problems for the SPInDel approach because it relies on the analysis of multiple loci, which presents a clear advantage over methods targeting a single locus (11,13,14). In cases where one (or more) SPInDel hypervariable region(s) have an unexpected length (i.e. different from the reference length), a correct identification is still possible based on the information from the remaining loci. In the most difficult cases, SPInDel should at least serve as a guide for further research and/or should exclude some possibilities. The same is true in the case of the occurrence of null alleles by possible limitations of the screening technique. As shown in Figure 2b, combining only two or three hypervariable regions results in high frequency values of species-specific SPInDel profiles. The guarantee against misclassifications resulting from missing data or unexpected allelic variants relies on the fact that most profiles diverge by several loci, which was clearly shown by mismatch distributions (Figure 3e and Supplementary Figure S8).
The identification of a species using our method is achieved by running the SPInDel numeric profile of the target sample against a reference database. This can be easily done by using the ‘search profile’ option available on the SPInDel workbench (Supplementary Figure S9). The profile can be generated by capillary electrophoresis with the use of allelic ladders (Figure 5c), or by any other related technique. However, if the exact determination of the fragment lengths is not possible, as in some agarose and polyacrylamide gels, the identification is still achievable by comparing the banding pattern of the target sample with those of reference samples analyzed with the same procedure. Equal patterns suggest that the samples might belong to the same species (Figure 5b).
Here, we demonstrated the applicability and robustness of our approach using conventional multiplex PCR coupled with capillary electrophoresis in highly processed food samples (Supplementary Table S7). Complete profiles were obtained in almost all cases, even in samples that were subjected to strong physical and chemical alterations (Supplementary Figure S13). This system is appropriate for low-quantity and/or degraded DNA samples due to (i) the use of mtDNA as a target for PCR, which is usually present in many copies per cell, provides a clear advantage over nuclear genome-based methods; (ii) the targeting of short amplicons and (iii) amenability to multiplexing. In fact, correct identifications were possible on partial profiles including only short amplicons (Supplementary Table S7). Therefore, we have selected numerous SPInDel hypervariable regions with short alleles in all taxonomic groups for inclusion in the workbench to facilitate the analysis of suboptimal samples. One advantage over existing approaches is the possibility of identifying species that co-exist in a sample using conventional laboratory equipment and without the need for time-consuming cloning steps or expensive microarray devices. For instance, a mixture of pork and bovine materials was easily identified in meat products (Supplementary Figure S13).
The utility of the SPInDel approach is not restricted to the boundaries imposed by the taxonomic groups we have initially selected (Table 1). As presented here, our method is limited by the need of knowing the broad taxonomic group where the identification is aimed. However, the structural and functional conservation of some sections of rRNAs suggests that broader screening tests can be easily designed. In fact, the alignment of a representative species from each eukaryotic group disclosed a handful of well-conserved regions of potential utility (Supplementary Figure S22), as previously reported by others for cytosolic rRNA genes (7,10,55). Also, we were able to amplify DNA from avian and fish samples using PCR primers intended for mammalian species (Figure 5b and Supplementary Figure S12). These results suggest that this method can be used for taxonomic classification of a wide range of species as long as appropriate conserved regions are selected.
The simplicity of the SPInDel concept makes it easily adaptable to diverse high-throughput genotyping platforms with a reduced cost per sample (Supplementary Table S9). A myriad of research fields can benefit from this methodology: biomedical sciences (e.g. characterization of clinical pathogens), forensics (e.g. identification of human remains in mixtures of biological materials), food quality control (e.g. detection of fraudulent description of food contents), ecology (e.g. identification of bones, feces, hair or seeds found in the field), ancient DNA studies and paleogenetics, among others. Further work is now necessary to explore the potential applicability of sequence length determination in species identification procedures using, for instance, nanotechnology and bioelectronic systems (68–70). In theory, a lab-on-a-chip platform can be developed to determine the length of hypervariable regions with high-throughput multiplexing capability. Hopefully, this development will aid in the long-standing quest for a portable system that would allow rapid species identification in the field.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Funding for open access charge: Research grants SFRH/BPD/44637/2008 (to F.P.); SFRH/BDE/15581/2006 (to B.vA.); SFRH/BD/37261/2007 (to N.P.); Research project PTDC/CVT/100881/2008; Portuguese Foundation for Science and Technology (FCT). IPATIMUP is an Associate Laboratory of the Portuguese Ministry of Science, Technology and Higher Education and is partially supported by FCT.
Conflict of interest statement. The subject matter of this paper is covered by a patent application in Portugal (number 103599), filed 26 November 2006. Inventors: F.P., B.A. and L.G.
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful to Alexandra Lopes, Ana Goios, Cíntia Alves, Mafalda Rocha and Marta Novais for helping with sample collection. We also thank the Portuguese sheep and goat breeders associations for providing samples.
REFERENCES
- 1.Hey J. The mind of the species problem. Trends Ecol. Evol. 2001;16:326–329. doi: 10.1016/s0169-5347(01)02145-0. [DOI] [PubMed] [Google Scholar]
- 2.Monis P, Giglio S, Keegan A, Thompson R. Emerging technologies for the detection and genetic characterization of protozoan parasites. Trends Parasitol. 2005;21:340–346. doi: 10.1016/j.pt.2005.05.012. [DOI] [PubMed] [Google Scholar]
- 3.Mothershed EA, Whitney AM. Nucleic acid-based methods for the detection of bacterial pathogens: present and future considerations for the clinical laboratory. Clin. Chim. Acta. 2006;363:206–220. doi: 10.1016/j.cccn.2005.05.050. [DOI] [PubMed] [Google Scholar]
- 4.Pereira F, Carneiro J, Amorim A. Identification of species with DNA-based technology: current progress and challenges. Recent Pat. DNA Gene Seq. 2008;2:187–200. doi: 10.2174/187221508786241738. [DOI] [PubMed] [Google Scholar]
- 5.Sites JW, Marshall JC. Delimiting species: a Renaissance issue in systematic biology. Trends Ecol. Evol. 2003;18:462–470. [Google Scholar]
- 6.Wells JD, Stevens JR. Application of DNA-based methods in forensic entomology. Annu. Rev. Entomol. 2008;53:103–120. doi: 10.1146/annurev.ento.52.110405.091423. [DOI] [PubMed] [Google Scholar]
- 7.Clarridge JE. Impact of 16S rRNA gene sequence analysis for identification of bacteria on clinical microbiology and infectious diseases. Clin. Microbiol. Rev. 2004;17:840–862. doi: 10.1128/CMR.17.4.840-862.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Imaizumi K, Akutsu T, Miyasaka S, Yoshino M. Development of species identification tests targeting the 16S ribosomal RNA coding region in mitochondrial DNA. Int. J. Legal Med. 2007;121:184–191. doi: 10.1007/s00414-006-0127-5. [DOI] [PubMed] [Google Scholar]
- 9.Kolbert CP, Persing DH. Ribosomal DNA sequencing as a tool for identification of bacterial pathogens. Curr. Opin. Microbiol. 1999;2:299–305. doi: 10.1016/S1369-5274(99)80052-6. [DOI] [PubMed] [Google Scholar]
- 10.Sonnenberg R, Nolte A, Tautz D. An evaluation of LSU rDNA D1-D2 sequences for their use in species identification. Front Zool. 2007;4:6. doi: 10.1186/1742-9994-4-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hsieh HM, Chiang HL, Tsai LC, Lai SY, Huang NE, Linacre A, Lee JC. Cytochrome b gene for species identification of the conservation animals. Forensic Sci. Int. 2001;122:7–18. doi: 10.1016/s0379-0738(01)00403-0. [DOI] [PubMed] [Google Scholar]
- 12.Kocher TD, Thomas WK, Meyer A, Edwards SV, Paabo S, Villablanca FX, Wilson AC. Dynamics of mitochondrial DNA evolution in animals: amplification and sequencing with conserved primers. Proc. Natl Acad. Sci. USA. 1989;86:6196–6200. doi: 10.1073/pnas.86.16.6196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Parson W, Pegoraro K, Niederstatter H, Foger M, Steinlechner M. Species identification by means of the cytochrome b gene. Int. J. Legal Med. 2000;114:23–28. doi: 10.1007/s004140000134. [DOI] [PubMed] [Google Scholar]
- 14.Hebert PDN, Cywinska A, Ball SL, Dewaard JR. Biological identifications through DNA barcodes. P. Roy. Soc. B-Biol. Sci. 2003;270:313–321. doi: 10.1098/rspb.2002.2218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fierer N, Breitbart M, Nulton J, Salamon P, Lozupone C, Jones R, Robeson M, Edwards RA, Felts B, Rayhawk S, et al. Metagenomic and small-subunit rRNA analyses reveal the genetic diversity of bacteria, archaea, fungi, and viruses in soil. Appl. Environ. Microb. 2007;73:7059–7066. doi: 10.1128/AEM.00358-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gill SR, Pop M, Deboy RT, Eckburg PB, Turnbaugh PJ, Samuel BS, Gordon JI, Relman DA, Fraser-Liggett CM, Nelson KE. Metagenomic analysis of the human distal gut microbiome. Science. 2006;312:1355–1359. doi: 10.1126/science.1124234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huber JA, Mark Welch D, Morrison HG, Huse SM, Neal PR, Butterfield DA, Sogin ML. Microbial population structures in the deep marine biosphere. Science. 2007;318:97–100. doi: 10.1126/science.1146689. [DOI] [PubMed] [Google Scholar]
- 18.Mardis ER. Next-generation DNA sequencing methods. Annu. Rev. Genom. Hum. Genet. 2008;9:387–402. doi: 10.1146/annurev.genom.9.081307.164359. [DOI] [PubMed] [Google Scholar]
- 19.Metzker ML. Sequencing technologies: the next generation. Nat. Rev. Genet. 2010;11:31–46. doi: 10.1038/nrg2626. [DOI] [PubMed] [Google Scholar]
- 20.Buntjer JB, Lenstra JA. Mammalian species identification by interspersed repeat PCR fingerprinting. J. Ind. Microbiol. Biot. 1998;21:121–127. [Google Scholar]
- 21.Ferri G, Alu M, Corradini B, Licata M, Beduschi G. Species Identification Through DNA “Barcodes”. Genet. Test. Mol. Bioma. 2009;13:421–426. doi: 10.1089/gtmb.2008.0144. [DOI] [PubMed] [Google Scholar]
- 22.Kitano T, Umetsu K, Tian W, Osawa M. Two universal primer sets for species identification among vertebrates. Int. J. Legal Med. 2007;121:423–427. doi: 10.1007/s00414-006-0113-y. [DOI] [PubMed] [Google Scholar]
- 23.Nakamura H, Muro T, Imamura S, Yuasa I. Forensic species identification based on size variation of mitochondrial DNA hypervariable regions. Int. J. Legal Med. 2009;123:177–184. doi: 10.1007/s00414-008-0306-7. [DOI] [PubMed] [Google Scholar]
- 24.Pinhal D, Gadig OBF, Wasko AP, Oliveira C, Ron E, Foresti F, Martins C. Discrimination of Shark species by simple PCR of 5S rDNA repeats. Genet. Mol. Biol. 2008;31:361–365. [Google Scholar]
- 25.Pun KM, Albrecht C, Castella V, Fumagalli L. Species identification in mammals from mixed biological samples based on mitochondrial DNA control region length polymorphism. Electrophoresis. 2009;30:1008–1014. doi: 10.1002/elps.200800365. [DOI] [PubMed] [Google Scholar]
- 26.Stamoulis P, Stamatis C, Sarafidou T, Mamuris Z. Development and application of molecular markers for poultry meat identification in food chain. Food Control. 2010;21:1061–1065. [Google Scholar]
- 27.Tobe SS, Linacre AMT. A multiplex assay to identify 18 European mammal species from mixtures using the mitochondrial cytochrome b gene. Electrophoresis. 2008;29:340–347. doi: 10.1002/elps.200700706. [DOI] [PubMed] [Google Scholar]
- 28.Gutell RR, Larsen N, Woese CR. Lessons from an evolving ribosomal RNA – 16S and 23S ribosomal-RNA structures from a comparative perspective. Microbiol. Rev. 1994;58:10–26. doi: 10.1128/mr.58.1.10-26.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hassouna N, Michot B, Bachellerie JP. The complete nucleotide sequence of mouse 28S rRNA gene. Implications for the process of size increase of the large subunit rRNA in higher eukaryotes. Nucleic Acids Res. 1984;12:3563–3583. doi: 10.1093/nar/12.8.3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Larson A. Evolutionary analysis of length-variable sequences: divergent domains of ribosomal RNA. In: Miyamoto MM, Cracraft J, editors. Phylogenetic Analysis of DNA Sequences. Oxford University Press, Oxford; 1991. pp. 221–248. [Google Scholar]
- 31.Smit S, Widmann J, Knight R. Evolutionary rates vary among rRNA structural elements. Nucleic Acids Res. 2007;35:3339–3354. doi: 10.1093/nar/gkm101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dirzo R, Raven PH. Global state of biodiversity and loss. Annu. Rev. Environ. Resour. 2003;28:137–167. [Google Scholar]
- 33.Drummond AJ, Ashton B, Burton S, Cheung M, Cooper A, Heled J, Moir R, Stones-Havas S, Sturrock S, Thierer T, et al. Geneious v5.1. 2010 http://www.geneious.com/ (24 September 2010, date last accessed) [Google Scholar]
- 34.DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, Huber T, Dalevi D, Hu P, Andersen GL. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microb. 2006;72:5069–5072. doi: 10.1128/AEM.03006-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nei M, Li WH. Mathematical model for studying genetic variation in terms of restriction endonucleases. Proc. Natl Acad. Sci. USA. 1979;76:5269–5273. doi: 10.1073/pnas.76.10.5269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25:1451–1452. doi: 10.1093/bioinformatics/btp187. [DOI] [PubMed] [Google Scholar]
- 38.Howley PM, Israel MA, Law MF, Martin MA. Rapid method for detecting and mapping homology between heterologous DNAs - evaluation of polyomavirus genomes. J. Biol. Chem. 1979;254:4876–4883. [PubMed] [Google Scholar]
- 39.Kibbe WA. OligoCalc: an online oligonucleotide properties calculator. Nucleic Acids Res. 2007;35:W43–W46. doi: 10.1093/nar/gkm234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cannone JJ, Subramanian S, Schnare MN, Collett JR, D’Souza LM, Du YS, Feng B, Lin N, Madabusi LV, Muller KM, et al. The Comparative RNA Web (CRW) Site: an online database of comparative sequence and structure information for ribosomal, intron, and other RNAs. BMC Bioinformatics. 2002;3:2. doi: 10.1186/1471-2105-3-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.SantaLucia J. A unified view of polymer, dumbbell, and oligonucleotide DNA nearest-neighbor thermodynamics. Proc. Natl Acad. Sci. USA. 1998;95:1460–1465. doi: 10.1073/pnas.95.4.1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pereira F, Soares P, Carneiro J, Pereira L, Richards MB, Samuels DC, Amorim A. Evidence for variable selective pressures at a large secondary structure of the human mitochondrial DNA control region. Mol. Biol. Evol. 2008;25:2759–2770. doi: 10.1093/molbev/msn225. [DOI] [PubMed] [Google Scholar]
- 44.Shimodaira H. Approximately unbiased tests of regions using multistep-multiscale bootstrap resampling. Ann. Stat. 2004;32:2616–2641. [Google Scholar]
- 45.Torgerson W. Theory and Methods of Scaling. New York: Wiley; 1958. [Google Scholar]
- 46.Rogers AR, Harpending H. Population growth makes waves in the distribution of pairwise genetic differences. Mol. Biol. Evol. 1992;9:552–569. doi: 10.1093/oxfordjournals.molbev.a040727. [DOI] [PubMed] [Google Scholar]
- 47.Slatkin M, Hudson RR. Pairwise comparisons of mitochondrial DNA sequences in stable and exponentially growing populations. Genetics. 1991;129:555–562. doi: 10.1093/genetics/129.2.555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vallone PM, Butler JM. AutoDimer: a screening tool for primer-dimer and hairpin structures. Biotechniques. 2004;37:226–231. doi: 10.2144/04372ST03. [DOI] [PubMed] [Google Scholar]
- 49.Fukami H, Budd AF, Levitan DR, Jara J, Kersanach R, Knowlton N. Geographic differences in species boundaries among members of the Montastraea annularis complex based on molecular and morphological markers. Evolution. 2004;58:324–337. [PubMed] [Google Scholar]
- 50.Clarke AR, Armstrong KF, Carmichael AE, Milne JR, Raghu S, Roderick GK, Yeates DK. Invasive phytophagous pests arising through a recent tropical evolutionary radiation: the Bactrocera dorsalis complex of fruit flies. Annu. Rev. Entomol. 2005;50:293–319. doi: 10.1146/annurev.ento.50.071803.130428. [DOI] [PubMed] [Google Scholar]
- 51.Elson JL, Lightowlers RN. Mitochondrial DNA clonality in the dock: can surveillance swing the case? Trends Genet. 2006;22:603–607. doi: 10.1016/j.tig.2006.09.004. [DOI] [PubMed] [Google Scholar]
- 52.Azevedo L, Carneiro J, van Asch B, Moleirinho A, Pereira F, Amorim A. Epistatic interactions modulate the evolution of mammalian mitochondrial respiratory complex components. BMC Genomics. 2009;10:266. doi: 10.1186/1471-2164-10-266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lodish H, Berk A, Matsudaira P, Kaiser CA, Krieger M, Scott MP, Zipursky L, Darnell J. Molecular Cell Biology. New York: W. H. Freeman; 2003. Basic molecular genetic mechanisms. [Google Scholar]
- 54.Gray MW, Burger G, Lang BF. Mitochondrial evolution. Science. 1999;283:1476–1481. doi: 10.1126/science.283.5407.1476. [DOI] [PubMed] [Google Scholar]
- 55.Woese CR. Bacterial Evolution. Microbiol. Rev. 1987;51:221–271. doi: 10.1128/mr.51.2.221-271.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gilbert W. Origin of Life: the RNA World. Nature. 1986;319:618. [Google Scholar]
- 57.Datta A, Jinksrobertson S. Association of increased spontaneous mutation rates with high levels of transcription in yeast. Science. 1995;268:1616–1619. doi: 10.1126/science.7777859. [DOI] [PubMed] [Google Scholar]
- 58.Hoede C, Denamur E, Tenaillon O. Selection acts on DNA secondary structures to decrease transcriptional mutagenesis. PLoS Genet. 2006;2:1697–1701. doi: 10.1371/journal.pgen.0020176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wright BE. A biochemical mechanism for nonrandom mutations and evolution. J. Bacteriol. 2000;182:2993–3001. doi: 10.1128/jb.182.11.2993-3001.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bonawitz ND, Clayton DA, Shadel GS. Initiation and beyond: Multiple functions of the human mitochondrial transcription machinery. Mol. Cell. 2006;24:813–825. doi: 10.1016/j.molcel.2006.11.024. [DOI] [PubMed] [Google Scholar]
- 61.Martin M, Cho JY, Cesare AJ, Griffith JD, Attardi G. Termination factor-mediated DNA loop between termination and initiation sites drives mitochondrial rRNA synthesis. Cell. 2005;123:1227–1240. doi: 10.1016/j.cell.2005.09.040. [DOI] [PubMed] [Google Scholar]
- 62.Montoya J, Gaines GL, Attardi G. The pattern of transcription of the human mitochondrial ribosomal RNA genes reveals two overlapping transcription units. Cell. 1983;34:151–159. doi: 10.1016/0092-8674(83)90145-9. [DOI] [PubMed] [Google Scholar]
- 63.Saccone C, Attimonelli M, Sbisa E. Structural elements highly preserved during the evolution of the D-Loop-containing region in vertebrate mitochondrial DNA. J. Mol. Evol. 1987;26:205–211. doi: 10.1007/BF02099853. [DOI] [PubMed] [Google Scholar]
- 64.Weber JL, David D, Heil J, Fan Y, Zhao CF, Marth G. Human diallelic insertion/deletion polymorphisms. Am. J. Hum. Genet. 2002;71:854–862. doi: 10.1086/342727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pearce JM. Minding the gap: frequency of indels in mtDNA control region sequence data and influence on population genetic analyses. Mol. Ecol. 2006;15:333–341. doi: 10.1111/j.1365-294X.2005.02781.x. [DOI] [PubMed] [Google Scholar]
- 66.Magnacca KN, Brown MJF. Tissue segregation of mitochondrial haplotypes in heteroplasmic Hawaiian bees: implications for DNA barcoding. Mol. Ecol. Resour. 2010;10:60–68. doi: 10.1111/j.1755-0998.2009.02724.x. [DOI] [PubMed] [Google Scholar]
- 67.Simmons MP, Ochoterena H, Carr TG. Incorporation, relative homoplasy, and effect of gap characters in sequence-based phylogenetic analyses. Syst. Biol. 2001;50:454–462. [PubMed] [Google Scholar]
- 68.Cheng MMC, Cuda G, Bunimovich YL, Gaspari M, Heath JR, Hill HD, Mirkin CA, Nijdam AJ, Terracciano R, Thundat T, et al. Nanotechnologies for biomolecular detection and medical diagnostics. Curr. Opin. Chem. Biol. 2006;10:11–19. doi: 10.1016/j.cbpa.2006.01.006. [DOI] [PubMed] [Google Scholar]
- 69.Fortina P, Kricka LJ, Surrey S, Grodzinski P. Nanobiotechnology: the promise and reality of new approaches to molecular recognition. Trends Biotechnol. 2005;23:168–173. doi: 10.1016/j.tibtech.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 70.Wang J. Nanomaterial-based electrochemical biosensors. Analyst. 2005;130:421–426. doi: 10.1039/b414248a. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.