

Abstract
Mutations at position 187 in secreted gelsolin enable aberrant proteolysis at the 172–173 and 243–244 amide bonds, affording the 71-residue amyloidogenic peptide deposited in Familial Amyloidosis of Finnish Type (FAF). Thermodynamic comparisons of two different domain 2 constructs were carried out to study possible effects of the mutations on proteolytic susceptibility. In the construct we consider to be most representative of domain 2 in the context of the full-length protein (134–266), the D187N FAF variant is slightly destabilized relative to wild type (WT) under the conditions of urea denaturation, but exhibits a Tm identical to WT. The D187Y variant is less stable to intermediate urea concentrations and exhibits a Tm that is estimated to be ≈5°C lower than WT (pH 7.4, Ca2+-free). Although the thermodynamic data indicate that the FAF mutations may slightly destabilize domain 2, these changes are probably not sufficient to shift the native to denatured state equilibrium enough to enable the proteolysis leading to FAF. Biophysical data indicate that these two FAF variants may have different native state structures and possibly different pathways of amyloidosis.
Amyloid diseases are associated with abnormal fibrillar protein deposits (1–7) from the self-assembly of misfolded proteins or peptides (3, 8–11). Both amyloid and intermediates of assembly have been implicated as causative agents in these diseases (12–15). Deposition of a fragment of mutated human plasma gelsolin (173–243) putatively causes Familial Amyloidosis of Finnish Type (FAF), a disease characterized by lattice corneal dystrophy, progressive cranial neuropathy, and skin elasticity complications (16–18).
Gelsolin is an actin-binding protein that nucleates, caps, and severs actin filaments. Phosphatidylinositol 4,5-bisphosphate (PIP2) and Ca2+ ions regulate gelsolin's dissociation from and binding to actin, respectively (19–22). Human gelsolin is expressed as an 81-kDa protein in cytoplasm and 84-kDa protein in plasma (23). The secreted protein differs by a signal sequence required for export and a 25-residue N-terminal extension. The cysteines at positions 188 and 201 form a disulfide bond in the plasma, but remain reduced in the cytoplasmic form (24).
The mutation of Asp-187 in domain 2 to either an Asn or a Tyr allows aberrant proteolysis at the 172–173 amide bond during secretion (25). A subsequent proteolytic cleavage generates the 173–243 amyloidogenic fragment.
Sequence comparisons and limited proteolysis defined gelsolin to be composed of six domains (26–28). The crystal structure of horse plasma gelsolin, highly homologous to human, in the absence of Ca2+ confirmed this architecture and revealed a fold common to each domain (Figs. 1 and 2; ref. 29). The fold consists of a central five- or six-stranded β-sheet sandwiched between a long α-helix parallel to the β-strands and a short α-helix almost perpendicular to the strands (Fig. 2). The definition of domain 2 based on sequence homology and proteolysis sensitivity includes residues 150–266, whereas the structure requires that residues 134–149 be added as they form strand A′ of the β-sheet (27, 29). The crystal structure shows that Asp-187 is within hydrogen bonding distance of Gln-164, Lys-166, and Asn-184 (Fig. 2). Burtnick et al. (29) propose that the mutation of Asp-187 could disrupt this hydrogen bonding network and destabilize the local structure resulting in 172–173 proteolysis.
Figure 1.

Water-accessible surface area representation of horse plasma gelsolin. The color scheme is as follows: S1, yellow; S2, blue; S6, green; the S2-S3 loop (251–266), orange; and S3, S4, and S5, gray.
Figure 2.

Ribbon diagram representation structure of domain 2 from horse plasma gelsolin including the loop that connects S2 to S3. The other five domains are omitted for clarity. S2 is colored blue and the loop connecting S2 to S3 is orange. The strands of the β-sheet are labeled A′ to E in order from the N to C termini of the construct. The side chain of the FAF-associated mutation, residue D187, is illustrated as a ball and stick model, as are the three residues to which it is proposed to hydrogen bond in the native state (Q164, K166, N184). The two tryptophan (W180 and W200) fluorophores and the disulfide bond (C188–C201) are also shown.
Ca2+-activated gelsolin adopts an altered structure. The crystal structure of human gelsolin S4–S6 (believed to be a genetic duplication of S1–S3) bound to actin and Ca2+ reveals substantial domain movements (20). S5, which forms only bridging contacts between S4 and S6 in the nonactivated form, establishes significant new contacts with S6. This movement exposes areas of S5 that are buried in the nonactivated form and suggests that domain 2 may be similarly affected on activation and binding. However, dynamic light scattering indicates that the C-terminal half of the molecule (S4–S6) undergoes significantly more bulk structural changes on Ca2+ binding than does the N-terminal half (S1–S3; ref. 30). The threshold concentration of Ca2+ that causes domain movement is not known.
Several studies to elucidate the effects of these mutations have been reported. Maury et al. (31) compared the amyloidogenicity of short gelsolin peptides, concluding that the mutations induced fibril formation under conditions where the wild type (WT) did not. However, a similar study (32) using the entire 71-residue fragment (173–243) found that the amyloidogenicity of the WT and FAF variants are indistinguishable and concluded that it is the ability of the FAF variants to facilitate the 172–173 cleavage that allows amyloid formation in vivo.
Fersht et al. (33) evaluated the thermodynamic stabilities of Ca2+-free WT, D187N, and D187Y domain 2 sequences defined by proteolysis (151–266; see ref. 26). Their results indicate that the D187N and D187Y mutations are destabilizing by 1.22 kcal mol−1 (25°C) and 2.16 kcal mol−1 (15°C), respectively. The authors propose that the instability of the mutants alters the native to denatured state equilibrium and presents more misfolded or unfolded proteins to the protease (33). Puius et al. (34) found that the stability of this WT construct (151–266) in the presence of 2 mM Ca2+ increases almost two-fold over the Ca2+-free form. However, the 151–266 construct studied lacks strand A′ (Fig. 2), which is integral to the structure of domain 2 through interactions with the C′ strand of domain 1 and the B strand that contains the aberrant proteolysis site at its N terminus (29).
The Fersht group (35) also studied the Ca2+-free form of this construct (WT vs. D187N) by NMR and observed few significant structural or dynamic differences. The C-terminal region of D187N was less structured than that of the WT, and the authors propose that the disordered tail region of the mutant exposes the 172–173 cleavage site enabling proteolysis (35). However, this construct lacks the A′ strand, which may complicate these conclusions because the C terminus interacts with both the A′ and B strands (Fig. 2).
This work examines the stability of the FAF mutants in domain 2 constructs of nonactivated gelsolin that include the A′ strand. The first, S2, includes the domain defined by crystallography (134–250), whereas the second, S2loop, adds the loop connecting domains 2 and 3 (134–266; see ref. 29).
Materials and Methods
Protein Expression and Purification.
The proteins were cloned from full-length human plasma gelsolin cDNA and were recombinantly expressed in E. coli BL21(DE3) pLysS cells (Stratagene). Detailed expression and purification procedures are published as supplemental data on the PNAS web site, www.pnas.org.
HPLC Analysis.
Reverse phase HPLC was used to confirm the oxidized state of the purified proteins (see supplemental data for details).
Gel Filtration Assay for S2 and S2loop Function.
The gel filtration assay outlined by Heiss and Cooper was used to test PIP2 binding to the refolded gelsolin constructs to evaluate the functional properties of the proteins (ref. 36; see supplemental data for details).
Ultracentrifugation Analysis.
Sedimentation equilibrium data on all constructs were collected on a temperature-controlled Beckman XL-I analytical ultracentrifuge (Beckman Instruments; see supplemental data for details).
Thermal Denaturation Studies.
CD studies were performed on an Aviv Model 202SF Stopped Flow CD Spectrometer (Aviv Associates, Lakewood, NJ) equipped with a Peltier temperature-controlled cell holder. Far-UV CD spectra were collected on WT and D187N S2 and WT, D187N, and D187Y S2loop gelsolin solutions [25 μM protein, PBS pH 7.4, 4°C (10% glycerol for S2 only)] in a 0.1 cm path length quartz cell. All spectra represent averages of three consecutive steady-state scans.
Thermal melts were performed in triplicate by monitoring the change in ellipticity at 213 nm as a freshly purified protein sample was heated from 4–64°C and cooled back to 4°C (4°C steps at 20°C/min, 2.5 min equilibration). Steady-state far-UV CD scans were obtained before and after the melt at 4°C to check for reversibility, and at 64°C to confirm the temperature-induced loss of secondary structure.
The data were analyzed assuming two-state behavior for the unfolding transition. The fraction of unfolded protein (Fu) at each temperature was calculated from the raw thermal data by using Eq. 1 and plotted as a function of temperature.
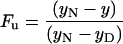 |
1 |
yN and yD are the native and denatured state values of the measured ellipticity at 213 nm and y is the measured signal at that temperature. The data from the triplicate experiments were fit simultaneously to Eq. 2 by using a nonlinear least squares regression analysis (37).
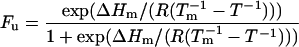 |
2 |
ΔHm is the enthalpy at the folding transition in calories, Tm and T are the midpoint of transition and the temperature in K, respectively, and R is the universal gas constant (1.987 cal deg−1⋅mol−1). ΔHm and Tm are varied and the errors reported are those obtained directly from the fit.
Urea Denaturation Studies.
Preparation of WT and D187N S2 gelsolin for fluorescence-based denaturation.
Eight molar urea was prepared as reported (37). The samples for each data point in the denaturation curve were prepared from stock solutions of protein, 8 M urea and 60% glycerol in PBS pH 7.4 and those for the renaturation by dilution from a stock solution of gelsolin in 5 M urea. The final concentration in each sample was 1 μM protein and 10% glycerol (required to observe a pretransition region in the S2 construct). The samples were prepared in triplicate and incubated for 18 h at the experimental temperature before measurements were made. Fluorescence measurements at 4°C were performed on an Aviv Model ATF105 Automated Titrating Differential/Ratio Spectrofluorometer. Emission scans were recorded from 310–400 nm (excitation at 295 nm).
Preparation of WT, D187N, and D187Y S2loop for CD-based denaturation.
The fluorescence-based urea denaturation curves for the S2loop constructs could not be reliably fit because of steep pretransition baselines. In a manner similar to the S2 fluorescence samples, but without glycerol, 25-μM S2loop samples were prepared for CD. Data were obtained at 4°C and 25°C.
Data analysis for urea denaturation.
The raw data were processed by using excel (Microsoft) and graphed by using kaleidagraph (Synergy Software, Reading, PA). Urea blanks were subtracted from each scan. The fraction of unfolded protein (Fu) at each urea concentration was calculated from the fluorescence intensity at 341 nm (S2) or the ellipticity at 217 nm (S2loop) by using Eq. 1, and fit to Eq. 3 by using a nonlinear least squares analysis (38).
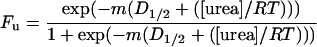 |
3 |
m is the dependence of free energy of unfolding on
urea, D1/2 is the concentration of
urea at the midpoint of the transition, R is the universal
gas constant, and T is the temperature in K. Values of
m and D1/2 were obtained
from the fit. The free energy differences between the native and
unfolded forms of domain 2 in the absence of denaturant,
ΔG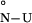 ,†
were extrapolated by using Eqs. 4 and 5.
,†
were extrapolated by using Eqs. 4 and 5.
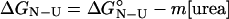 |
4 |
At the midpoint of the unfolding transition ΔGN-U is equal to zero, therefore,
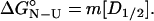 |
5 |
Results
Protein Expression and Purification.
The proteins were expressed and purified as described. A redox buffer was used to promote disulfide bond formation between Cys-188 and -201 (39). Protein purities were determined by SDS/PAGE and electrospray ionization (ESI)-MS (supplementary Table 4). S2 D187Y was too prone to aggregation during purification to be analyzed as described here.
PIP2 binding demonstrated that the structures of the refolded constructs were functionally representative of domain 2 in the full-length protein. Two putative binding sites for PIP2 in domain 2 of gelsolin have been identified (22). Heiss & Cooper's gel filtration chromatography assay was used to discern the difference between the free and PIP2-bound proteins (36). All peaks of all constructs in the presence of PIP2 eluted after 9 min, whereas each protein alone eluted after 14 min (supplementary Fig. 7).
Sedimentation equilibrium studies were performed to assure the monomeric nature of the constructs at the concentrations used for the biophysical studies. The molecular weights obtained from the single ideal species model for the S2 constructs are 13.56 ± 0.06 and 12.88 ± 0.16 kDa for the WT and the D187N mutant, respectively, and 12.23 ± 0.96, 15.63 ± 1.1, and 10.11 ± 0.53 for WT, D187N, and D187Y S2loop constructs respectively (supplementary Fig. 8 A–E). All values agree with the expected solution molecular weights.
Thermal Denaturation of Gelsolin Domain 2.
The far-UV CD spectra of the S2 constructs (25 μM in PBS, pH 7.4, 10% glycerol) recorded at 4°C are shown in supplementary Fig. 9A. The proteins are folded and composed of both α-helices and β-strands as confirmed by spectra exhibiting broad minima from 206–220 nm. Analogous spectra of the S2loop constructs in the absence of 10% glycerol are shown in supplementary Fig. 9B.
The thermal melts plotted as the fraction of unfolded protein as a function of temperature for the S2 and S2loop constructs are shown in Fig. 3 A and B. The solid lines are the least squares fits to the data determined by using Eq. 2 with the extracted data summarized in Table 1. In the S2 construct, the Tm for the WT protein is 6.0°C lower than that for the D187N mutant, whereas the Tm values for the S2loop WT and D187N are identical. The data obtained for the D187Y mutant were not fit because this transition was not completely reversible; however, estimation suggests a Tm 5°C lower than the WT protein. The D187Y S2loop variant appears to fold much more cooperatively than the WT or D187N proteins. The relative stabilities of the proteins cannot be quantified accurately without determining the temperature dependence of the heat capacity, but their Tm values suggest similar thermal stabilities.
Figure 3.

Thermal Denaturation of WT and D187N S2 as well as WT, D187N, and D187Y S2loop gelsolin monitored by CD at 213 nm. WT data are shown in black circles, D187N data in red triangles, and D187Y data in blue squares. Solid lines represent the best fits by a nonlinear least squares analysis to Eq. 2. (A) Gelsolin S2 WT and D187N (10% glycerol). (B) WT, D187N, and D187Y gelsolin S2loop.
Table 1.
Thermodynamic data from thermal denaturation studies
| S2
|
S2loop
|
||||
|---|---|---|---|---|---|
| WT | D187N | WT | D187N | D187Y | |
| Tm (K) | 307 ± 0.3 | 313 ± 0.1 | 319 ± 0.2 | 319 ± 0.2 | 314* |
| ΔHm (kcal) | 40.333 ± 2.276 | 49.143 ± 1.618 | 52.856 ± 1.638 | 56.806 ± 2.134 | not reversible |
Estimated Tm—transition not reversible.
Urea-Induced Unfolding and Refolding Monitored by Fluorescence and CD Spectroscopies.
Solutions of native S2 WT and D187N (1 μM) exhibit a broad fluorescence emission with a maximum at 341 nm (Fig. 4A). In 5 M urea, the emission intensity is increased by 10–35% and the maximum is shifted to 355 nm. The native S2loop constructs exhibit emission maxima around 330 nm and, like the S2 proteins, have their emission shifted to 355 nm on denaturation in 5 M urea with a 46% (D187Y) to 185% (WT) increase in emission intensity (Fig. 4B). This suggests that the native state S2loop tryptophans are in a more hydrophobic environment than those of the S2 and are strongly quenched by neighboring residues.
Figure 4.

Trypotophan fluorescence emission intensity of native and urea-denatured states at 4°C of WT and D187N S2 (A) and WT, D187N, and D187Y S2loop (B). WT data are shown in black circles, D187N data in red triangles, and D187Y data in blue squares. Native state traces are depicted by filled symbols and denatured state traces by open symbols. Excitation was at 295 nm.
The free energy of folding for all constructs was evaluated by urea denaturation. Fig. 5A shows the fraction of unfolded S2 WT and D187N as function of urea concentration in the presence of 10% glycerol at 4°C, as measured by fluorescence. For the S2loop variants, a strictly analogous thermodynamic analysis without added glycerol was performed by CD at 4 and 25°C (Fig. 5B, Table 2). Fluorescence-based denaturation curves (not shown) were also obtained for S2loop and found to be consistent with the CD data, although the pre- and post-transition baselines were very steep. The CD data were used for fitting. The linear extrapolation model (LEM) was used to extract free energy values at 0 M urea (ΔG°) from the denaturation curves (see Table 2 and ref. 38). This model assumes that the unfolding transition exhibits two-state behavior and is reversible.
Figure 5.

Denaturation of gelsolin WT and D187N S2 and WT, D187N, and D187Y S2loop as a function of urea concentration. Data are shown in black circles for the WT, in red triangles for D187N, and in blue squares for D187Y. Solid lines represent the best fits to Eq. 3 via a nonlinear least squares analysis. (A) Denaturation of gelsolin S2 WT and D187N monitored by fluorescence intensity at 341 nm (10% glycerol, 4°C). (B) Urea-induced denaturation of gelsolin S2loop WT, D187N, and D187Y monitored by CD ellipticity at 217 nm (4°C).
Table 2.
Urea denaturation of S2 proteins at 4°C and S2loop proteins at 4°C and 25°C
| S2,
4°C
|
S2loop
|
||||||
|---|---|---|---|---|---|---|---|
| WT | D187N | 4°C
|
25°C
|
||||
| WT | D187N | D187Y | WT | D187N | |||
| [urea]1/2 (M) | 2.25 ± 0.02 | 2.45 ± 0.03 | 2.42 ± 0.01 | 2.33 ± 0.01 | 1.80 ± 0.01 | 2.32 ± 0.01 | 1.79 ± 0.01 |
| m (cal mol−1⋅M−1) | 1054 ± 54* | 1159 ± 61* | 1464 ± 32* | 1453 ± 20* | 2115 ± 42* | 1393 ± 53* | 1630 ± 60* |
| ΔG° (cal mol−1) | 2371 ± 118 | 2839 ± 171 | 3544 ± 78 | 3385 ± 48 | 3807 ± 78 | 3232 ± 124 | 2918 ± 109 |
| Δ(ΔG°) (cal mol−1) | −468 ± 207 | 159 ± 92 | −263 ± 110 | 314 ± 165 | |||
The calculated m value is 1471 cal mol−1⋅M−1 for S2 and 1635 cal mol−1⋅M−1 for S2loop.
The data for the D187Y S2loop protein at 25°C were not analyzed because a pretransition baseline was not observed; however, at 4°C, pretransition baselines were evident. The m values of the WT and D187N proteins in each construct at 4°C are similar, but the D187Y S2loop m value is significantly higher. The differences in free energy [Δ(ΔG°)] between the WT and FAF variant proteins in 0 M urea for each construct were calculated by using data derived from the LEM. The S2loop D187Y variant was also compared in this fashion, although the LEM is most accurate for systems with similar m values (40, 41).
Another strategy for comparison is to use an average m value (1677 cal mol−1⋅M−1) of the S2loop constructs, which gives [Δ(ΔG°)] of −1040 cal mol−1 for the D187Y variant. This technique also is best suited to proteins with similar m values (41). Both of these methods indicate that S2loop D187Y is more stable than WT (Table 2). An additional way to compare the WT S2loop construct with the D187N and D187Y variants is to examine them at a fixed urea concentration where an equilibrium between the folded and unfolded states exists for all three proteins (Table 3). This method suggests that D187Y is less stable (≈1 kcal/mol) than WT to the denaturing conditions provided by 2 M urea at 4°C.
Table 3.
Thermodynamic comparison of S2loop constructs in 2 M Urea at 4°C
| [urea]1/2 M | m cal mol−1⋅M−1 | In 2 M urea
|
|||
|---|---|---|---|---|---|
| Fu | Keq | ΔG cal | |||
| WT | 2.4 | 1464 | 0.24 | 0.32 | 626* |
| D187N | 2.3 | 1453 | 0.31 | 0.45 | 439* |
| D187Y | 1.8 | 2115 | 0.67 | 2.03 | −389* |
These values are opposite in sign from those in Table 2 because of standard differences in calculation methods.
Discussion
Interactions between domains of multidomain proteins have been shown to play important roles in modulating stability and structure. Studying individual domains is often the only practical method for characterizing the thermodynamic stability of large proteins, because multidomain proteins commonly exhibit complex transitions (42).
The definition of a domain—the sequence extracted from a protein that represents both its functional and structural features in the context of the full-length protein—is critical. In the case of nonactivated gelsolin, domain 2 is positioned at one end of the molecule as seen in Fig. 1. Solvent accessible surface area calculations show that 83% of the domain is exposed in the Ca2+-free full-length protein (insight ii, Micron Separations). Two areas of domain 2 make interdomain contacts: strand A′ at the edge of the sheet contacts the C′ strand from domain 1, and two strands in the β-sheet (on the face opposite the loop) interact with a helix from domain 6. The FAF-associated mutations are located on strand C, which is not involved in interdomain interactions. The conclusions drawn here assume that the mutations will affect the energetics of domain 2 in a similar fashion whether studied as an independent domain or in the context of the six-domain protein.
It is difficult to predict a priori the sequence that defines an autonomous stable folding unit. Here the crystal structure was used to choose the domain 2 constructs studied within: S2, based on the common folding topology (residues 134–250), and S2loop (134–266), which adds a C-terminal extension of 16 residues. Although this loop does not adopt a regular secondary structure, it is significant because it packs against domain 2, covering the 172–173 proteolysis site and burying part of D187, W180, and the residues involved in the hydrogen bonding network. The side chain of L251 (in the loop) appears to interact with the residues that form the hydrophobic core of domain 2 (Fig. 6). The S2 construct studied herein and the 151–266 construct studied previously (33) may not faithfully represent domain 2 in the context of the full-length protein as they lack the loop and strand A′, respectively. Additionally, the 151–266 construct was not demonstrated to be functional (33, 35).
Figure 6.

Ribbon diagram representation of the hydrophobic core of horse plasma gelsolin domain 2. The other five domains and the remainder of domain 2 are omitted for clarity. The polypeptide backbone of the S2 domain (134–250) is represented in blue. The side chains of the residues that form the hydrophobic core of domain 2 are shown in blue. In orange is the polypeptide backbone of the loop connecting S2 to S3 (251–266), including the side chain of residue L251.
The S2loop construct appears to best represent domain 2 in the context of the entire protein. Thermal denaturation studies show that the WT and D187N Tm values increase in the S2loop construct relative to their S2 counterparts (Table 1); however, these values cannot be directly compared because the S2 proteins were studied in the presence of 10% glycerol. The D187Y S2 construct was not stable enough to study, whereas the S2loop construct is amenable to biophysical examination. The higher level of protein expression, lower sensitivity to aggregation, and increased stability without glycerol suggest that the S2loop construct is more relevant to domain 2 as it exists in the entire Ca2+-free protein.
These results must be interpreted considering the evidence for the possible structural (20) and stability (34) differences between the active and inactive forms of gelsolin. If gelsolin is in an inactive form when it encounters the 172–173 protease, these results suggest that a factor other than thermodynamic instability allows proteolysis.
Effects of the Mutations on the S2 and the S2loop Constructs.
D187N.
In the S2 constructs, the D187N mutation stabilizes the protein by 0.47 kcal mol−1 (in 10% glycerol), whereas in S2loop the same mutation is destabilizing by 0.16 kcal mol−1 (Table 2). A comparison of the S2loop D187N and WT at 4°C in 2.0 M urea agrees with the LEM values, revealing a destabilization of 0.19 kcal mol−1 for the mutant (Table 3). It is clear that the stability difference between the WT and D187N variant is less than the 1.22 kcal mol−1 derived from studying the 151–266 construct (33).
These findings suggest that the D187N protein is very similar to the WT. Native state fluorescence, m values, minor differences in stability, and similar general behavior in our hands indicate that WT and D187N are almost identical. It is unlikely that the small differences in stability found here would shift the equilibrium from the native state to the denatured state sufficiently to completely explain D187N proteolysis and amyloidogenicity. Other mechanisms, such as the effects of Ca2+ activation, must be considered.
D187Y.
The m value derived from urea denaturation of D187Y is significantly higher than that of the WT or D187N (Table 2). The m value correlates with the change in accessible surface area (ASA) on unfolding (ΔA) (41). The higher m value for the D187Y mutant likely reflects an increase in ΔA over the WT protein; this could result from either an increase in the ASA of the unfolded form or a decrease in the ASA of the native form (40, 41). Several mutants of staphylococcal nuclease with large m values were shown to be unfolded to a greater extent in the denatured state, a factor that may contribute in this case (40, 43). The 44% increase in ASA is hard to explain by native state changes; however, the dramatic differences in the native state fluorescence of the D187Y variant relative to WT and D187N suggest that native state differences may exist. The presence of a folding intermediate usually lowers the m value and thus is not applicable here (44, 45). Another possible explanation for the large m value is aggregation during denaturation; however, excellent fits to a monomer in 1.75 M urea for all three S2loop proteins were observed by equilibrium ultracentrifugation studies (data not shown).
The LEM estimates D187Y to be more stable than WT in 0 M urea and thus appears to break down in this case. All other data indicate that D187Y is less stable than the WT. Each method of analysis used argues that any destabilization is considerably less than the 2.16 kcal mol−1 derived from biophysical studies on the construct missing the A′ strand at 15°C (151–266; see ref. 33). The minor free energy change found for D187Y may not be sufficient to explain protease sensitivity during export; thus, it is important to keep open the possibility that other factors, such as Ca2+-induced structural or stability changes, may contribute.
All of the data describing the D187Y mutant argue that the D187Y native state differs from that of the WT in a manner currently unknown. D187Y expression levels are lower, it is more sensitive to concentration-dependent aggregation, and its thermal denaturation is irreversible. Native state fluorescence of the D187Y protein is red shifted and significantly less quenched than the WT or D187N. The D187Y m value differs considerably from that of the WT, indicating differences between the native or denatured states of the proteins.
The data suggest that the D187N protein is similar to the WT in the native state, whereas the D187Y mutant may be significantly different. Despite this, both mutations enable aberrant proteolysis and subsequent amyloid deposition leading to FAF. That the two mutant proteins may have significantly different native state structures, but are both proteolyzed and deposited as fibrils, indicates that these two mutants may follow significantly different pathways to amyloidosis. Thus, the mechanism that allows proteolysis and deposition for one mutant may not operate in the case of the other.
Conclusions
Based on the structure of gelsolin and the biophysical data presented within, the S2loop construct (134–266) appears to be appropriate to represent domain 2 in the context of the full-length protein. Data indicate that the D187N construct is similar in structure and stability to the WT domain whereas D187Y may adopt a significantly different native state structure. It is unlikely that the destabilization of the D187N or D187Y variants in the absence of added calcium is sufficient to explain the protease sensitivity and amyloidosis associated with FAF.
Supplementary Material
Acknowledgments
We thank Hans Purkey, Mike Petrassi, and Songpon Deechongkit for assistance with analytical ultracentrifugation and Dr. Nick Pace for helpful discussions. We also thank the National Institutes of Health (DH 46335), the Skaggs Institute for Chemical Biology, the Lita Annanberg-Hazen Foundation for financial support, and the reviewers for insightful comments.
Abbreviations
- FAF
Familial Amyloidosis of Finnish Type
- WT
wild type
- PIP2
phosphatidylinositol 4,5-bisphosphate
- LEM
linear extrapolation model
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
See commentary on page 2117.
ΔG° refers here to data collected at 4°C.
References
- 1.Pepys M B. In: Immunological Diseases. Samter M, editor. Vol. 1. Brown, Boston: Little; 1988. pp. 631–674. [Google Scholar]
- 2.Lansbury P T. Biochemistry. 1992;31:6865–6870. doi: 10.1021/bi00145a001. [DOI] [PubMed] [Google Scholar]
- 3.Kelly J W. Curr Opin Struct Biol. 1996;6:11–17. doi: 10.1016/s0959-440x(96)80089-3. [DOI] [PubMed] [Google Scholar]
- 4.Jacobson D R, Buxbaum J N. Adv Hum Genet. 1991;20:69–123. doi: 10.1007/978-1-4684-5958-6_2. [DOI] [PubMed] [Google Scholar]
- 5.Sipe J D. Crit Rev Clin Lab Sci. 1994;31:325–354. doi: 10.3109/10408369409084679. [DOI] [PubMed] [Google Scholar]
- 6.Chiti F, Webster P, Taddei N, Clark A, Stefani M, Ramponi G, Dobson C M. Proc Natl Acad Sci USA. 1999;96:3590–3594. doi: 10.1073/pnas.96.7.3590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Selkoe D J. Science. 1997;275:630–631. doi: 10.1126/science.275.5300.630. [DOI] [PubMed] [Google Scholar]
- 8.Colon W, Kelly J W. Biochemistry. 1992;31:8654–8660. doi: 10.1021/bi00151a036. [DOI] [PubMed] [Google Scholar]
- 9.Hurle M R, Helms L R, Li L, Chan W, Wetzel R. Proc Natl Acad Sci USA. 1994;91:5446–5450. doi: 10.1073/pnas.91.12.5446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Booth D R, Sunde M, Bellotti V, Robinson C V, Hutchinson W L, Fraser P E, Hawkins P N, Dobson C M, Radford S E, Blake C C F, et al. Nature (London) 1997;385:787–793. doi: 10.1038/385787a0. [DOI] [PubMed] [Google Scholar]
- 11.Wetzel R. Cell. 1996;86:699–702. doi: 10.1016/s0092-8674(00)80143-9. [DOI] [PubMed] [Google Scholar]
- 12.Harper J D, Wong S S, Lieber C M, Lansbury P T., Jr Chem Biol. 1997;4:119–125. doi: 10.1016/s1074-5521(97)90255-6. [DOI] [PubMed] [Google Scholar]
- 13.Harper J D, Wong S S, Lieber C M, Lansbury P T., Jr Biochemistry. 1999;38:8972–8980. doi: 10.1021/bi9904149. [DOI] [PubMed] [Google Scholar]
- 14.Lambert M P, Barlow A K, Chromy B A, Edwards C, Freed R, Liosatos M, Morgon T E, Rozovsky I, Trommer B, Viola K L, et al. Proc Nat Acad Sci USA. 1998;95:6448–6453. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Snyder S W, Ladror U S, Wade W S, Wang G T, Barrett L W, Matayoshi E D, Huffaker H J, Krafft G A, Holzman T F. Biophys J. 1994;67:1216–1228. doi: 10.1016/S0006-3495(94)80591-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maury C P J, Kere J, Tolvanen R, De la Chapelle A. FEBS Lett. 1990;276:75–77. doi: 10.1016/0014-5793(90)80510-p. [DOI] [PubMed] [Google Scholar]
- 17.Maury C P J, Alli K, Baumann M. FEBS Lett. 1990;260:85–87. doi: 10.1016/0014-5793(90)80072-q. [DOI] [PubMed] [Google Scholar]
- 18.Kiuru S. Amyloid. 1998;5:55–66. doi: 10.3109/13506129809007291. [DOI] [PubMed] [Google Scholar]
- 19.Yin H L. BioEssays. 1987;7:176–179. doi: 10.1002/bies.950070409. [DOI] [PubMed] [Google Scholar]
- 20.Robinson R C, Mejillano M, Le V P, Burtnick L D, Yin H L, Choe S. Science. 1999;286:1939–1942. doi: 10.1126/science.286.5446.1939. [DOI] [PubMed] [Google Scholar]
- 21.Xian W. Ph.D. thesis. Lincoln, NE: Univ. of Nebraska; 1996. [Google Scholar]
- 22.Yu F X, Sun H Q, Janmey P A, Yin H L. J Biol Chem. 1992;267:14616–14621. [PubMed] [Google Scholar]
- 23.Kwiatkowski D J, Stossel T P, Orkin S H, Mole J E, Colten H R, Yin H L. Nature (London) 1986;323:455–458. doi: 10.1038/323455a0. [DOI] [PubMed] [Google Scholar]
- 24.Wen D, Corina K, Chow E P, Miller S, Janmey P A, Pepinsky R B. Biochemistry. 1996;35:9700–9709. doi: 10.1021/bi960920n. [DOI] [PubMed] [Google Scholar]
- 25.Kangas H, Paunio T, Kalkkinen N, Jalanko A, Peltonen L. Hum Mol Genet. 1996;5:1237–1243. doi: 10.1093/hmg/5.9.1237. [DOI] [PubMed] [Google Scholar]
- 26.Pope B J, Gooch J T, Weeds A G. Biochemistry. 1997;36:15848–15855. doi: 10.1021/bi972192p. [DOI] [PubMed] [Google Scholar]
- 27.Way M, Weeds A. J Mol Biol. 1988;203:1127–1133. doi: 10.1016/0022-2836(88)90132-5. [DOI] [PubMed] [Google Scholar]
- 28.Way M, Pope B, Gooch J, Hawkins M, Weeds A G. EMBO J. 1990;9:4103–4109. doi: 10.1002/j.1460-2075.1990.tb07632.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Burtnick L D, Koepf E K, Grimes J, Jones E Y, Stuart D I, Mclaughlin P J, Robinson R C. Cell (Cambridge, Mass) 1997;90:661–670. doi: 10.1016/s0092-8674(00)80527-9. [DOI] [PubMed] [Google Scholar]
- 30.Hellweg T, Hinssen H, Eimer W. Biophys J. 1993;65:799–805. doi: 10.1016/S0006-3495(93)81121-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maury C P J, Nurmiaho-Lassila E-L, Rossi H. Lab Invest. 1994;70:558–564. [PubMed] [Google Scholar]
- 32.Ratnaswamy G, Koepf E, Bekele H, Yin H, Kelly J W. Chem Biol. 1999;6:293–304. doi: 10.1016/S1074-5521(99)80075-1. [DOI] [PubMed] [Google Scholar]
- 33.Isaacson R L, Weeds A G, Fersht A R. Proc Natl Acad Sci USA. 1999;96:11247–11252. doi: 10.1073/pnas.96.20.11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Puius Y A, Fedorov E V, Eichinger L, Schleicher M, Almo S C. Biochemistry. 2000;39:5322–5331. doi: 10.1021/bi992364d. [DOI] [PubMed] [Google Scholar]
- 35.Kazmirski S L, Howard M J, Isaacson R L, Fersht A R. Proc Natl Acad Sci USA. 2000;97:10706–10711. doi: 10.1073/pnas.180310097. . (First Published September 19, 2000; 10.1073/pnas.180310097) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Heiss S G, Cooper J A. Biochemistry. 1991;30:8753–8758. doi: 10.1021/bi00100a006. [DOI] [PubMed] [Google Scholar]
- 37.Pace C N, Scholtz J M. In: Protein Structure. 2nd Ed. Creighton T E, editor. New York: Oxford Univ. Press; 1997. pp. 299–321. [Google Scholar]
- 38.Bolen D W, Santoro M M. Biochemistry. 1988;27:8069–8074. doi: 10.1021/bi00421a015. [DOI] [PubMed] [Google Scholar]
- 39.Wetlaufer D B, Saxena V P. Biochemistry. 1970;9:5015–5023. doi: 10.1021/bi00827a028. [DOI] [PubMed] [Google Scholar]
- 40.Shortle D. Adv Protein Chem. 1995;46:217–247. doi: 10.1016/s0065-3233(08)60336-8. [DOI] [PubMed] [Google Scholar]
- 41.Myers J K, Pace C N, Scholtz J M. Protein Sci. 1995;4:2138–2148. doi: 10.1002/pro.5560041020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jaenicke R. Prog Biophys Mol Biol. 1999;71:155–241. doi: 10.1016/s0079-6107(98)00032-7. [DOI] [PubMed] [Google Scholar]
- 43.Wrabl J, Shortle D. Nat Struct Biol. 1999;6:876–883. doi: 10.1038/12338. [DOI] [PubMed] [Google Scholar]
- 44.Pace C N. Methods Enzymol. 1986;131:266–280. doi: 10.1016/0076-6879(86)31045-0. [DOI] [PubMed] [Google Scholar]
- 45.Spudich G, Marqusee S. Biochemistry. 2000;39:11677–11683. doi: 10.1021/bi000466u. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.