

Abstract
J. D. F. Wadsworth, E. A. Asante and J. Collinge (2010) Neuropathology and Applied Neurobiology36, 576–597Contribution of transgenic models to understanding human prion disease
Transgenic mice expressing human prion protein in the absence of endogenous mouse prion protein faithfully replicate human prions. These models reproduce all of the key features of human disease, including long clinically silent incubation periods prior to fatal neurodegeneration with neuropathological phenotypes that mirror human prion strain diversity. Critical contributions to our understanding of human prion disease pathogenesis and aetiology have only been possible through the use of transgenic mice. These models have provided the basis for the conformational selection model of prion transmission barriers and have causally linked bovine spongiform encephalopathy with variant Creutzfeldt-Jakob disease. In the future these models will be essential for evaluating newly identified potentially zoonotic prion strains, for validating effective methods of prion decontamination and for developing effective therapeutic treatments for human prion disease.
Keywords: Creutzfeldt-Jakob disease, fatal familial insomnia, Gerstmann-Sträussler-Scheinker disease, kuru, prion, variant Creutzfeldt-Jakob disease
Introduction
Prions have attracted immense research interest for many years because of their unique composition and properties – being apparently devoid of significant nucleic acid [1–5]. According to the widely accepted ‘protein-only’ hypothesis [6], host-encoded cellular prion protein (PrPC) is converted to an alternative form designated PrPSc[1–5]. It is proposed that PrPSc is the infectious agent acting to replicate itself with high fidelity by recruiting endogenous PrPC and that the difference between these isoforms lies purely in the monomer conformation and its state of aggregation [1–5,7–9]. Notably however, although abnormal isoforms of PrP are undoubtedly the major constituent of mammalian prions, it has not yet been excluded that other molecules may contribute to infectious prion composition or may be required to direct the assembly of PrPSc[10–12]. In this regard the precise identification of minor ‘contaminants’ that co-purify with PrPSc may still be of critical importance to understanding infectious prion composition, the determinants of prion strain or the ability of a prion to infect a host [13].
Central to understanding prion propagation remains the conundrum of prion strains – how a protein-only infectious agent can encode information required to specify distinct disease phenotypes – and also the so-called species barrier effect which limits cross species infection. While originally considered different aspects of the prion problem it is now clear that species barriers and prion strains are intimately related by ‘conformational selection’[5,14]. This hypothesis proposes that although a wide range of mammalian PrPSc conformations may be possible, only a subset will be compatible with each individual PrP primary structure. Ease of transmission of prions between species (or also within species as a result of PrP polymorphisms), therefore relates to overlap of permissible PrPSc conformations between the structures of PrP from the source and recipient as well as heterogeneity in cellular mechanisms affecting prion propagation and clearance kinetics [5,14]. Importantly, conformational selection has now been strongly supported by elegant studies of prions in yeast and other fungi [15–17] and intriguingly evidence for strains in Alzheimer's disease is also now emerging, with self-propagating variations in the structure of amyloid-β fibrils appearing to correlate with differences in cyto-toxicity [18] and patterns of amyloid deposition in transgenic mice [19]. Elucidation of the composition and structure of infectious mammalian prions will therefore not only provide a major advance to understanding the molecular mechanism of prion replication, with direct translational benefits for both diagnosis and rational therapeutics, but will also be of great relevance to a wide range of other neurodegenerative diseases involving accumulation of misfolded host proteins [5,20,21]. Indeed, evidence for commonality of structural features in protein misfolding diseases is provided already by antibodies raised against oligomeric forms of PrP which detect soluble oligomeric forms of a number of other amyloid proteins [22]. More recently it has been proposed that PrPC may play a critical role in the pathogenesis of Alzheimer's disease by mediating amyloid-β oligomer induced synaptic dysfunction [23].
Aside from the intrinsic biological interest of studying prions, human prion disease is a strategic priority for public health protection. The occurrence of variant Creutzfeldt-Jakob disease (vCJD) [24] and the experimental confirmation that it is caused by the same prion strain as that causing bovine spongiform encephalopathy (BSE) in cattle [25–28], has dramatically established the zoonotic potential of animal prion diseases. The extremely prolonged and variable incubation periods seen in human prion disease and the possibility of subclinical carrier states means that it will be some years before the full consequences of human exposure to BSE prions are known [14,29–33]. In the meantime, we are faced with the possibility that significant numbers in the population may be incubating this disease and that they might pass it on to others via blood transfusion, blood products, tissue and organ transplantation and other iatrogenic routes [14,33–40]. Notably, while cattle BSE is now effectively controlled, the emergence of other new or newly recognized potentially zoonotic animal prion strains remains a key issue for public health. A number of novel isolates of bovine prion disease have now been identified which appear to be distinct prion strains [41–44] and the host range of atypical sheep prions [43,45,46] has not been established. Because prion strains can adapt and mutate on passage in new species [5,43,47], and also within species as a result of PrP polymorphisms and other genetic factors [28,48–51], the evaluation of their risks to public health is complex. The demonstration of subclinical carrier states of prion infection in animal models is also relevant to public health, both with respect to prion zoonoses and iatrogenic transmission of human prions [28,33,39,52]. Prions resist many conventional sterilization procedures and effective methods for prion decontamination of surgical instruments and medical equipment although reported have yet to be effectively implemented [53,54].
In order to understand the molecular basis of human prion disease, develop rational therapeutics, improved decontamination methods and diagnostic tools, effective and appropriate experimental models are essential. However, very few alternative experimental approaches are available for studying prion diseases as incubation time, clinical phenotype, neuropathology, immune responses and behaviour can only be studied in an animal. Early studies of human prions used primates [55–57]; however, following the demonstration in 1995 that the species barrier limiting transmission of human prions to wild-type mice can be obviated by expression of human PrP in the absence of endogenous mouse PrP [58,59] such ‘humanised’ transgenic mice have become key experimental models for studying human prion disease [43,60–65]. Two types of genetic modification can be used to generate human PrP-expressing mice, either transgenic expression of human PrP on a mouse PrP knockout background [62] or direct replacement of mouse PrP with human PrP using gene knock-in technology [61].
Determinants of phenotypic variability in human prion disease
Human prion diseases include Creutzfeldt-Jakob disease (CJD), Gerstmann-Sträussler-Scheinker disease (GSS), fatal familial insomnia, kuru and vCJD in humans [1,2,39]. They are associated with a range of clinical presentations and are classified by both clinico-pathological syndrome and aetiology with subclassification according to molecular criteria [39,66] (Table 1). The clinical presentation of human prion disease varies enormously and there is considerable overlap observed between individuals with different disease aetiologies [33,39,66,67] and even in family members with the same pathogenic PRNP mutation [67–74]. Remarkably, kuru demonstrates that incubation periods of infection with human prions can exceed 50 years [32,75]. Progressive dementia, cerebellar ataxia, pyramidal signs, chorea, myoclonus, extrapyramidal features, pseudobulbar signs, seizures and amyotrophic features can be seen in variable combinations. Criteria used for diagnosis of human prion disease have been defined [39,76] and definite diagnosis of sporadic and acquired prion disease relies upon neuropathological examination and the demonstration of pathological PrP deposition in the central nervous system by either immunoblotting or immunohistochemistry [39,76–79] (Figure 1). Polymorphism at residue 129 of human PrP [encoding either methionine (M) or valine (V)] powerfully affects susceptibility to human prion diseases [80–85]. About 38% of northern Europeans are homozygous for the more frequent methionine allele, 51% are heterozygous, and 11% homozygous for valine. Homozygosity at PRNP codon 129 predisposes to the development of sporadic and acquired CJD [80–85] and is most strikingly observed in vCJD where all neuropathologically confirmed cases studied so far have been homozygous for codon 129 methionine of PRNP[38,39,49,50].
Table 1.
Classification of human prion disease
| Aetiology | Phenotype | Frequency | References |
|---|---|---|---|
| Sporadic | |||
| Unknown: random distribution worldwide; incidence of 1–2 per million per annum | Sporadic CJD: subacute myoclonic form and range of atypical forms; multiple distinct prion strains associated with distinct clinicopathological phenotypes which include sporadic fatal insomnia | Approximately 85% | [39,66,85,217] |
| Inherited | |||
| Autosomal dominantly inherited conditions with high penetrance; all forms have germline PRNP coding mutations | Extremely variable: readily mimics familial Alzheimer's disease and other neurodegenerative conditions; over 30 mutations identified; includes GSS, familial CJD and FFI | 10–15% | [39,66,67,72] |
| Acquired | |||
| Iatrogenic infection with human prions via medical or surgical procedures; cadaveric derived pituitary hormones, tissue grafts, and contaminated neurosurgical instruments | Iatrogenic CJD: typical CJD when direct central nervous system human exposure; ataxic onset when peripheral infection | <5% (most from USA, UK, France and Japan) | [39,120,225] |
| Exposure to human prions via endocannibalism | Kuru | Unique to small area of Papua New Guinea; major epidemic in 1950s with gradual decline since cessation of cannibalism | [32,39,75,226,227] |
| Exposure (presumed dietary) to BSE prion strain; probable secondary transmission via blood transfusion and possibly blood products | Variant CJD | Mainly UK with patients in several other countries | [14,24,33,39,40] |
BSE, bovine spongiform encephalopathy; CJD, Creutzfeldt-Jakob disease; FFI, fatal familial insomnia; GSS, Gerstmann-Sträussler-Scheinker disease.
Figure 1.
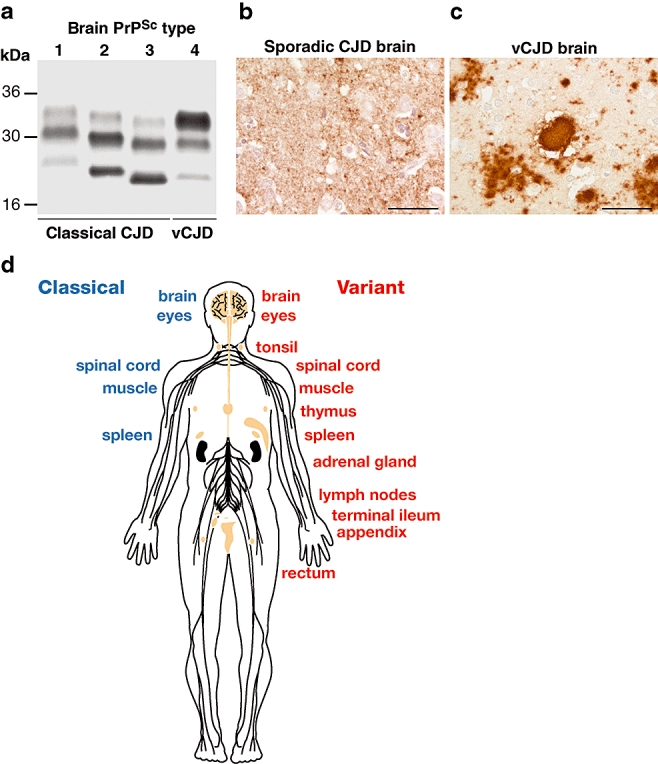
Variant Creutzfeldt-Jakob disease (vCJD) is a distinct human prion strain. (a) Immunoblot of proteinase K digested brain homogenates using antiPrP monoclonal antibody 3F4 showing PrPSc types 1–4 in human brain according to the London classification [90]. Types 1–3 PrPSc are seen in the brain of classical forms of CJD (either sporadic or iatrogenic CJD) and kuru, while type 4 PrPSc is uniquely seen in vCJD brain [25,90,122]. (b,c) Brain sections from sporadic CJD (b) and vCJD (c) showing abnormal PrP accumulation following immunohistochemistry using antiPrP monoclonal antibody ICSM35. Abnormal PrP deposition in sporadic CJD most commonly presents as diffuse, synaptic staining, whereas vCJD is distinguished by the presence of florid PrP plaques consisting of a round amyloid core surrounded by a ring of spongiform vacuoles. Scale bars: 50 µm. (d) Distribution of PrPSc in human tissues. The schematic diagram shows tissues in which PrPSc has been detected using high sensitivity immunoblotting. The vCJD has a peripheral pathogenesis distinct from classical forms of CJD, with a prominent and uniform involvement of lymphoreticular tissues.
Prion strains are classically distinguished by distinct incubation periods and by patterns of neuropathological targeting (so-called lesion profiles) in a panel of defined inbred mouse lines [86]. Common histopathological features involve a classical triad of spongiform vacuolation (affecting any part of the cerebral grey matter), neuronal loss, and astrocytic and microglial proliferation and may be accompanied by amyloid plaques composed of insoluble aggregates of PrP [77,87]. Amyloid plaques are a notable feature of kuru and GSS [77,88,89] but they are less frequently found in the brains of patients with sporadic CJD which typically show a diffuse pattern of PrP deposition [77,90] (Figure 1). The histopathological features of vCJD are relatively consistent when compared to sporadic CJD and distinguish it from other human prion diseases. The most distinctive feature is the presence of large numbers of PrP-positive amyloid plaques that differ in morphology from the plaques seen in kuru and GSS in that the surrounding tissue takes on a microvacuolated appearance, giving the plaques a florid appearance [24,91] (Figures 1 and 2).
Difficulties in assigning human prion strains
The hypothesis that alternative conformations or assembly states of PrPSc provide the molecular substrate for a significant part of the clinicopathological heterogeneity seen in human prion diseases and that this relates to the existence of distinct human prion strains is supported by considerable experimental evidence [1–5,25,92–95] and also by the demonstration of protein conformation-based inheritance mechanisms of yeast prions [15–17]. Despite these advances, the precise molecular basis of mammalian prion strain diversity is unknown. A major confounding issue in this regard has been in resolving whether relatively subtle biochemical differences in PrPSc are of biological importance and accurately reflect the propagation of distinct human prion strains. This is particularly true in sporadic CJD [25,90,93,96–100] where progress has been severely hampered by a lack of transgenic modelling data to firmly distinguish the identity of distinct prion strains and their defining molecular and neuropathological phenotypes. This fundamental problem coupled with the difficulties and variability of the biochemical methods used to distinguish PrPSc types [90,96–98,100–102] has so far precluded an internationally accepted classification system for human prion strains. In this regard, the increasingly recognized co-occurrence of different PrPSc types in the same brain [74,85,90,93,102–108] and the recognition that protease-sensitive pathological isoforms of PrP (distinct from prototypical PrPSc) may have a significant role in both animal and human prion disease [94,99,109–116] has further confounded progress. All of these factors, together with the known ability of genetic background to influence prion strain selection [28,50,51,117–119] and knowledge that route of transmission in acquired human prion disease may dramatically influence clinical and neuropathological presentation [120–123], has re-emphasized the requirement to remove host variability by identifying distinct prion strains in appropriate transgenic models.
Transgenic modelling has made key contributions to understanding prion biology
Considerable evidence argues that prions are composed largely, if not entirely, of abnormal isoforms of PrP [1–3,5,8]. The essential role of host PrP for prion propagation and pathogenesis is demonstrated by the fact that knockout mice lacking the PrP gene (Prnp°/° mice) are entirely resistant to prion infection [124,125] and that reintroduction of PrP transgenes restores susceptibility to infection in a species-specific manner that allows reverse genetics approaches to studying structure-function relationships in PrP (for reviews see [62,65,126]). PrP in its entirety is unnecessary for prion propagation. Not only can the unstructured N-terminal 90 amino acids be deleted [127,128], but also the first α-helix, the second β-strand and part of helix 2. In transgenic animals, a 106 amino acid fragment of the protein comprising PrPΔ23–88 and Δ141–176 conferred susceptibility to and propagation of prions [129,130]. Notably while expression of PrP N-terminal deletion mutants to residue 106 are tolerated and support prion propagation [127,128], deletion beyond this leads to severe ataxia and neuronal loss in the granular cell layer of the cerebellum [62,131]. Intriguingly, the Doppel protein (Dpl) [132], which has a similar structure to N-terminally truncated PrP, causes a similar cerebellar effect when ectopically expressed in the brain [133]. The severity of neurotoxicity correlates with the level of Dpl expression [134] and can be rescued by PrPC expression [135], indicating that PrPC, Dpl and ΔPrP might compete for a common hypothetical receptor or ligand LPrP that transduces neuroprotective signals when bound to PrPC but not when bound to Dpl or ΔPrP [62,131,136,137]. This model also proposes the existence of a PrPC-like protein termed Π that is capable of compensating for the absence of PrPC in Prnp°/° mice. Recently the protein Sho has been demonstrated to be a glycosylphosphatidylinositol (GPI)-anchored neuronal glycoprotein present in the central nervous system (CNS) from early postnatal life that can counteract the neurotoxic effects of either Dpl or ΔPrP and is therefore a candidate for Π[138].
While PrP expression is absolutely required for prion propagation and neurotoxicity [124] knockout of PrPC in embryonic models [139,140] or in adult brain [141] has no overt phenotypic effect that influences lifespan or fertility. These findings demonstrate that acute loss of PrPC in neurones in adulthood is tolerated, and that the neuropathophysiology of prion diseases is not due to loss of PrPC function [142,143]. Prnp°/° mice are not normal, however (for reviews see [2,62,144,145]). In particular, in addition to a role for PrPC in providing neuroprotective signals, abnormalities in synaptic physiology, circadian rhythms, cognition and olfactory physiology have been reported [146–153]. Notably, a reduction of slow afterhyperpolarizations evoked by trains of action potentials in hippocampal neurones in Prnp°/° mice due to disruption of calcium-activated potassium currents is also affected by the conditional knockout of PrPC suggesting that this phenotype is specifically due to the absence of PrPC, reflecting loss of a differentiated neuronal function, rather than a developmental deficit arising from congenital knockout of PrPC[141]. Important functional correlates of abnormalities of synaptic transmission in Prnp°/° mice include cognitive deficits [152] and impairment of olfactory physiology and behaviour [153] which can be rescued by transgenic neuronal expression of PrPC. Very recently it has been revealed that axonal PrPC expression is required for peripheral myelin maintenance [154] and this finding correlates strongly with earlier demonstrations of extensive demyelination in transgenic mice expressing PrP with deletion mutants in the central domain [155–157]. Importantly, despite current uncertainties regarding the conversion of PrPC to PrPSc and possible mechanisms of neurotoxicity [5], the prevention of this conversion in neurones by conditional knock out of PrPC has been shown to prevent disease progression and reverse early degenerative changes [142]. These data have firmly established PrPC as the prime target for rational therapeutics in prion disease [143,158,159]. Conversely, although PrPSc has long been considered as a target for chemotherapy [158], drugs interacting with PrPSc are likely to be prion strain-specific and may only target a specific subset of PrPSc conformers resulting in propagation of drug resistant prions [143,160].
Human PrP transgenic mouse models
Sporadic CJD prions transmit disease only occasionally to wild-type mice with long and variable incubation periods [26,58,59,122,161]. Early attempts to transmit human prions to transgenic mice met with varied success. Tg(HuPrP)110 and Tg(HuPrP)152 transgenic lines were made by co-expressing wild-type human PrP with valine at codon 129 in mice also expressing endogenous mouse PrPC[161]. However after inoculation with brain homogenates from patients with GSS and sporadic and iatrogenic CJD these transgenic recipients showed no higher frequency of disease than inoculated nontransgenic control mice. This lack of susceptibility of Tg(HuPrP) mice, together with the earlier pioneering work of Scott et al. [162], subsequently led Telling and colleagues to generate mice that expressed a chimeric PrP protein in which a segment of mouse PrPC was replaced with the corresponding human PrP sequence [161]. When similarly inoculated with CJD prions, all the Tg(MHu2MPrP) mice developed neurological disease around 200 days post-inoculation [161]. Concomitant with this chimeric transgene approach, the endogenous mouse PrP allele was also removed by breeding Tg(HuPrP)152 mice to PrP null mice (Prnp°/°) to produce homozygous Tg(HuPrP)152/Prnp°/°[58,59]. These mice were found to be highly susceptible to CJD prions, with all inoculated mice succumbing to disease at short incubation periods [58,59]. Although the chimeric transgene approach has provided extremely important advances to our understanding of human prion strain propagation [59,92,161,163,164], the demonstration that the human to mouse transmission barrier is overcome simply by expressing human PrPC in the absence of endogenous mouse PrPC[58,59] has established this as the most straightforward approach for investigating human prion strain diversity. Importantly, this approach enables the effects of genotype in the inoculum and recipient transgenic mouse to be modelled definitively to provide key information on the role of human PrP primary structure in influencing prion strain propagation [2,5,33,60]. More recently, other human PrPC-expressing mouse models have been generated using knock-in technology which allows transgene integration at the normal genomic location and endogenous levels of expression under the control of normal gene regulatory elements [61,63,165–167]. Both transgenic and knock-in modelling of prion diseases provide complimentary results in most cases; however, over-expression of human PrP transgenes may be desirable. Over-expression results in considerable truncation in incubation periods such that these models are more convenient in practice and in some instances transmission may require over-expression. For instance, transmission of BSE prions to human PrP knock-in mice resulted in no infection [63] whereas the transgenic approach has clearly demonstrated that BSE infection results in complete recapitulation of the vCJD phenotype [28] (Figure 3). Further, it is known that human PrPC functions less efficiently in mice than mouse PrPC, as over-expression is required to rescue a PrP null phenotype [168] and so ‘endogenous’ levels of heterologous PrPC expression may not in fact be the best model of susceptibility.
Figure 3.
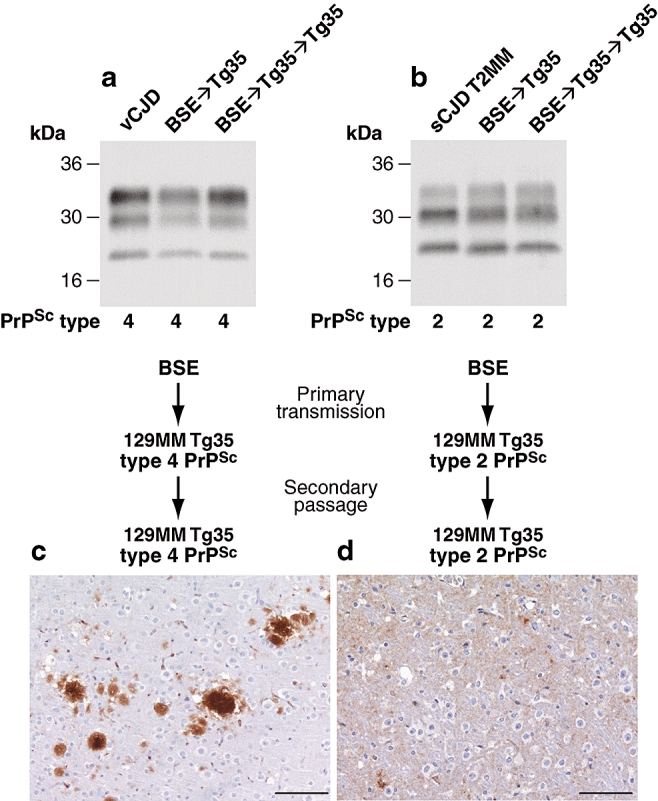
Bovine spongiform encephalopathy (BSE) prions propagate as either variant Creutzfeldt-Jakob disease (vCJD)-like or sporadic CJD (sCJD)-like strains in transgenic mice expressing human prion protein. Primary transmission of BSE prions in transgenic Tg(HuPrP129M+/+Prnp°/°)-35 mice (Tg35) results in the propagation of either type 4 PrPSc and the occurrence of abundant florid PrP plaques that are the neuropathological hallmark of vCJD or type 2 PrPSc and the occurrence of diffuse PrP deposition that is typically seen in sporadic CJD [28]. Molecular and neuropathological characteristics of these distinct prion strains remain stable after secondary passage in the same line of transgenic mice [49]. (a,b) Representative immuno-blots of proteinase-K treated brain homogenates from vCJD and sCJD (PRNP 129 MM genotype with type 2 PrPSc; sCJD T2MM) and transgenic mice analysed with antiPrP monoclonal antibody 3F4. The identity of the brain sample is designated above each lane with the type of PrPSc present in the sample designated below, using the London classification [90]. (c) Representative immunohistochemical analysis of transgenic mouse brain (thalamus) at secondary passage showing abnormal PrP immunoreactivity, including PrP-positive plaques, stained with antiPrP monoclonal antibody ICSM 35. Scale bars: 100 µm.
Transgenic modelling of sporadic and acquired human prion disease
Humanized transgenic mice expressing human PrP 129 valine on a Prnp null background are highly susceptible to sporadic CJD prions regardless of the PrPSc type or codon 129 genotype of the inoculum [25,26,58,59,122,164,169]. These transmissions are typically characterized by 100% attack rates of prion infection producing uniform clinical prion disease after similar short incubation periods of around 200 days [25,26,58,59,122,164,169]. In isolates that have been examined, no fall in mean incubation period is seen after secondary passage in the same mice indicative of the lack of a transmission barrier [58]. The absence of a transmission barrier to sporadic CJD prions is not, however, uniformly observed in transgenic mice expressing human PrP 129 methionine on a Prnp null background. Here mismatch at residue 129 between the inoculum and host can significantly affect transmission. Thus while there appears to be no barrier to transmission of sporadic CJD prions from codon 129 methionine homozygous patients [28,41,164,170], transmission of sporadic CJD prions from codon 129 heterozygous patients and 129 valine homozygous patients is often associated with more prolonged and variable incubation periods and reduced attack rates [28,164,169]. Consistent with both aetiology and the occurrence of the same PrPSc types that are seen in the brain of sporadic CJD patients, iatrogenic CJD prions [25,26,164,169,171] and kuru prions [122] have transmission properties equivalent to those of sporadic CJD prions. Although the precise number of distinct prion strains that are propagated in sporadic CJD remains unknown, Manson and colleagues have recently presented evidence for four distinct prion strains from a limited number of sporadic CJD patients using human PrP knock-in mice [167].
In contrast, to prions propagated in classical CJD and kuru the transmission properties of vCJD prions are strikingly distinct and have established vCJD as a distinct human prion strain (Figures 1 and 2). Our research was the first to demonstrate transmission of BSE prions to transgenic mice expressing human PrP and these data confirmed that vCJD was caused by human exposure to the BSE prion strain [26,28] (Figure 3). The vCJD prions transmit disease to wild-type mice far more efficiently than any other form of human prion disease [26,27,49,122] and in transgenic mice faithful propagation of the vCJD phenotype is dependent upon homozygous expression of human PrP 129 methionine [28,49,63,170,171] (Figure 2). Transgenic mice homozygous for human PrP 129 valine show a pronounced transmission barrier to vCJD prions [26,49,63,166] and propagate a distinct prion strain that has not yet been observed in humans [26,49,171] (Figure 2). Because human PrP with 129 valine appears to be incompatible with the PrPSc conformation propagated in vCJD [49] (Figure 2), and may have a dominant negative influence on the propagation of the vCJD prion strain in codon 129 heterozygous mice [171,172], this could explain why all neuropathologically confirmed cases of vCJD have been in individuals homozygous for 129 methionine. While caution must be exercised in extrapolating from animal models, even where faithful recapitulation of molecular and pathological phenotypes is possible [28,49,122,171], our findings, together with more recent studies from other laboratories [63,170], argue that primary human BSE prion infection, and secondary infection through iatrogenic routes, will not be restricted to a single disease phenotype. Dependent upon the genotype of the prion source and the recipient, the propagation of prion strains seen in sporadic CJD or other novel prion strain types are anticipated [28,33,49,122,171] (Figures 2 and 3). These data reiterate the need to continue to stratify all human prion disease patients at the molecular level to facilitate rapid recognition of novel subtypes and change in the relative frequencies of particular subtypes due to either BSE exposure patterns or iatrogenic sources of human prions.
Figure 2.
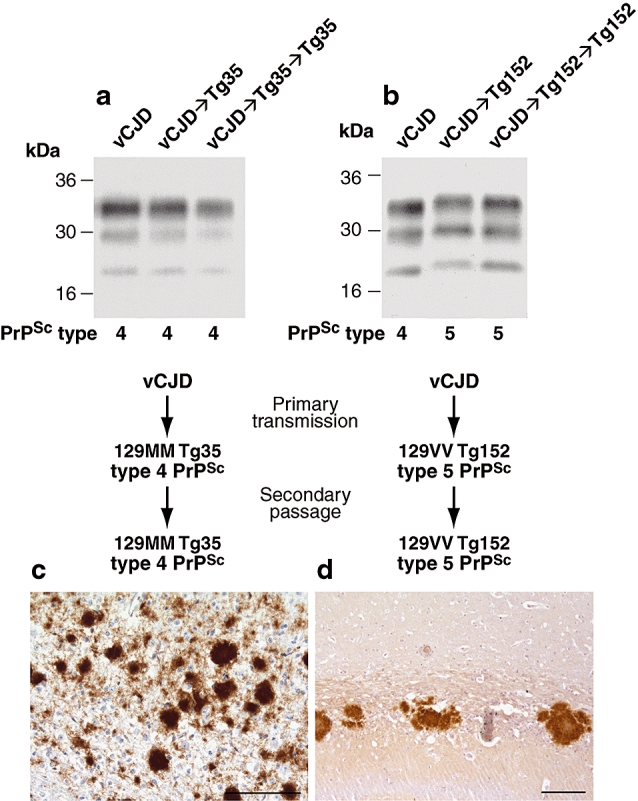
Human prion protein with valine at residue 129 prevents expression of the variant Creutzfeldt-Jakob disease (vCJD) phenotype. Primary and secondary transmission of vCJD prions to transgenic Tg(HuPrP129M+/+Prnp°/°)-35 mice (Tg35) results in faithful propagation of type 4 PrPSc and the occurrence of abundant florid PrP plaques throughout the cortex that are the neuropathological hallmark of vCJD [49]. In contrast, primary transmission of vCJD prions to transgenic Tg(HuPrP129V+/+Prnp°/°)-152 mice (Tg152) produces a novel prion strain that is maintained on secondary passage in the same mice distinguished by the propagation of type 5 PrPSc and a distinct pattern of neuropathology characterized by large nonflorid PrP plaques restricted to the corpus callosum [26,49]. (a,b) Representative immuno-blots of proteinase-K treated brain homogenates from variant CJD and transgenic mice analysed with antiPrP monoclonal antibody 3F4. The identity of the brain sample is designated above each lane with the type of PrPSc present in the sample designated below using the London classification [90]. (c,d) Representative immunohistochemical analysis of transgenic mouse brain at secondary passage showing abnormal PrP plaques stained with antiPrP monoclonal antibodies ICSM 35 (a) or 3F4 (b). Scale bars: 100 µm.
Conformational selection dictates human prion strain propagation
Homozygosity at polymorphic residue 129 of human PrPC remains the key genetic susceptibility factor for sporadic and acquired prion disease [50,80–84] and in vCJD it represents the strongest known common genetic susceptibility polymorphism in any human disease [39,50,173]. The transgenic studies described above have established the molecular basis for this effect by showing that this polymorphism constrains both the propagation of distinct human PrPSc conformers and the occurrence of associated patterns of neuropathology [25,26,28,49,122,171]. Biophysical measurements suggest that this powerful effect of residue 129 on prion strain selection is likely to be mediated via its effect on the conformation of PrPSc or its precursors or on the kinetics of their formation, as it has no measurable effect on the folding, dynamics or stability of PrPC[5,174]. Heterozygosity at codon 129 is thought to confer resistance to prion disease by inhibiting homologous protein-protein interactions essential for efficient prion replication [80,81,171,172] while the presence of methionine or valine at residue 129 controls the propagation of distinct human prion strains via conformational selection [2,5,14,49]. To date, the repertoire of PrPSc isoforms that can be stably propagated by human PrP with 129 methionine or 129 valine remains unknown.
Transgenic modelling of inherited prion disease
How pathogenic mutations in PRNP cause prion disease has yet to be resolved; however, in most cases the mutation is thought to lead to an increased tendency of PrPC to form PrPSc. However, there is now considerable evidence that different mutations may have different structural consequences in the expressed protein, including acting to destabilize the native PrPC fold, to increase aggregation propensity, to alter cellular trafficking, or to stabilize alternative protein (PrPSc) structures [175–180]. While a wealth of data from acquired or sporadic CJD indicates that residue 129 polymorphism critically dictates thermodynamic preferences for PrPSc[2,5,49,52,90], the full spectrum of effects that different pathogenic PRNP mutations have remains unclear. However, molecular strain typing has provided important insights into the phenotypic heterogeneity seen in inherited human prion diseases. In agreement with existing evidence that human prion strain diversity may be generated through variance in PrPSc conformation and glycosylation, cases of inherited prion disease caused by point mutations have glycoform ratios of PrPSc fragments distinct from those seen in both classical CJD [103,177,181–183] and vCJD [177]. Individuals with the same PRNP mutation can also propagate PrPSc with distinct fragment sizes [103,177,184]. However, the detection of PrPSc in the molecular mass range of c. 21–30 kDa is by no means a consistent feature and some PRNP mutations, in particular those in which amyloid plaques are a prominent feature, show smaller protease resistant fragments of ca. 7–15 kDa [72,103,177,181,184,185] while other PRNP mutations show a consistent absence of detectable PrPSc[2,72]. Collectively, these data indicate that pathogenic PRNP mutations have diverse and direct effects on dictating the preferred structure or assembly state of mutant PrPSc isoforms resulting in physicochemical properties that are very different from the PrPSc types propagated in sporadic and acquired forms of human prion disease [74,177,186]. Notably, variable propagation of PrPSc generated from wild-type PrPC may also contribute to phenotypic variability in inherited prion disease [74,187–189]. Co-propagation of distinct PrPSc types combined with differences in their neuropathological targeting, abundance and potential neurotoxicity, provides a general molecular mechanism for generating phenotypic heterogeneity in patients with the same PRNP mutation.
Attempts to transmit inherited prion diseases to nonhuman primates [57] and wild-type mice [190–192] have been inconclusive in answering whether all inherited prion diseases are experimentally transmissible as nonhuman hosts may not be susceptible. While some inherited prion diseases may indeed not be transmissible, and may represent prion proteinopathies [193–195], many pathogenic mutations have yet to be tested in transgenic mice expressing the homotypic human mutant protein. This may be critical as only the human mutant protein may be conformationally susceptible to the mutant prion strain involved [5,186]. Much of the transgenic modelling of inherited prion disease has however focused on superimposing human PrP mutations onto rodent PrPC in order to establish whether infectious prions can be generated de novo. To date, spontaneous neurological dysfunction has been reported in multiple transgenic models expressing mutated rodent PrP. These include mice expressing mouse PrP P101L [196], mouse PrP with octapeptide repeat insertions [194,197–199], truncated mouse PrP [131,200], mouse PrP with D177N and M128V substitutions [201], mouse PrP with L108M, V111M and D177N substitutions [202] and transgenic mice expressing a counterpart of the human A117V mutation or experimental mutations that favour the generation of a transmembrane form of PrP [193,203].
Of the various PRNP mutations studied, the proline to leucine substitution at codon 102 (P102L) of human PrP has been extensively investigated in different laboratories. However, these data have been difficult to interpret. Tg(GSSPrP)174 mice expressing high levels of mouse PrP 101L spontaneously develop neurological dysfunction at 166 days of age [196], however, PrPSc levels are low or undetectable, and brain extracts from affected mice do not transmit CNS degeneration to wild-type mice, but transmission to hamsters and Tg(GSSPrP)196 mice, expressing lower levels of the same mutant transgene product, was reported [204,205]. However, these Tg(GSSPrP)196 mice have subsequently been reported to develop spontaneous disease at advanced age [110,112] and it therefore remains inconclusive whether transmissible prions were generated in these transgenic mice or that the illness observed on secondary passage simply represents acceleration of a spontaneous neurodegenerative disease that is already poised to occur [65,112]. Importantly, in this regard, transgenic mice expressing endogenous levels of mouse PrP 101L (generated by the gene knock-in approach) do not develop spontaneous neurodegeneration [165,192] while mice over-expressing wild-type PrPC have been found to develop spontaneous neurological dysfunction without generating infectious prions [116,206–208].
Collectively, the existing data have yet to conclusively establish whether authentic high titre infectious prions have been generated de novo in mice expressing mouse PrP containing only human pathogenic mutations. In this regard, the critical step of showing transmission to wild-type mice on primary passage remains. An extremely important consideration in such studies is whether superimposition of pathogenic human PrP mutations into rodent PrP will have the same structural consequences. Indeed, there are now examples of inherited prion disease where the amino acid change thought to be pathogenic is found as a normal variant in other mammalian species [209–211] and critically there is now direct experimental evidence indicating that a single analogous amino acid change in human or mouse PrP has extremely different structural consequences for the expressed protein. The introduction of a tryptophan residue at amino acid position 175 in place of the native phenylalanine has been successfully used as an optical probe for studying the folding dynamics of recombinant mouse PrP with no measurable effect on the stability of the protein [212]. However, in complete contrast, introduction of the same mutation into human recombinant PrP renders the protein unable to fold into the native conformation [213]. These findings clearly raise doubt about modelling human pathogenic PrP mutations on nonhomologous PrP sequences from other species. The possibility of propagating novel prion strains that do not recapitulate the molecular and neuropathological phenotype of the original human disease appears probable and for this reason it seems clear that future transgenic models of inherited prion disease should focus on expressing mutated human PrP [186,214].
Of course, studies of the effects of experimental mutations on mouse PrP should also continue. A recent report of de novo generation of prion disease in such models involved the introduction of 2-point mutations into mouse PrP (170N and 174T) that are found as normal variants in the rigid loop of elk PrP [215]. Transgenic mice mPrP(170N,174T), moderately over-expressing these mutations spontaneously develop spongiform encephalopathy and PrP plaque deposition in the brain [215]. Repeated subpassages in Tg20 mice showed transmission of disease, after adaptation, to wild-type mice by the fourth passage, and propagation of protease resistant PrPSc[215]. Recently, Lindquist and colleagues have reported that mice expressing mouse PrP with L108M, V111M and D177N substitutions generated by knock-in technology spontaneously produce transmissible prions [202].
Difficulties associated with modelling human prion disease in human PrP expressing transgenic mice
Whether the full diversity of neuropathological phenotypes seen in human prion disease can be faithfully recapitulated by transgenic modelling remains an open question. In this regard the issue of prion strain selection or mutation will be a major factor. As recently hypothesized [5] prion strains may not exist as previously thought as molecular clones with a single PrPSc type (where strain mutation in a different host would involve generation of a distinct PrPSc type) but may consist of an ensemble of molecular species (containing a dominant PrPSc type that is preferentially propagated by its usual host) from which a less populous subspecies may be selected by an alternative host, resulting in a strain shift or mutation. Different cellular populations and tissues within a single host would provide different environments for strain selection as recently demonstrated in vitro[216]. In addition, the known ability of genetic background to influence prion strain selection [28,50,51,117–119] means that it may be extremely difficult to isolate the full complement of human prion strains in transgenic mice having a single genetic background.
Future perspectives
To date, the conformational repertoire of pathological isoforms of wild-type human PrP and the various forms of mutant human PrP has not been fully defined. Biochemical investigation of PrPSc isoforms in patients allied with detailed clinical and neuropathological analysis will continue to inform on the diversity of phenotypes seen in human prion disease. As it has now become clear that prion strain type, host genetic makeup and other factors (e.g. route of transmission) may significantly influence prion disease phenotype it is expected that the actual number of distinct human prion strains may be far less than the number of identified phenotypes. Continued transgenic modelling will therefore be crucial to establishing how many human prion strains exist, and what the defining molecular features of PrPSc are for each strain. This information allied with comprehensive transgenic modelling of human BSE infection and other relevant, potentially zoonotic, prion strains will inform on how many human prion strains may have an animal origin. Understanding the risks that existing and emerging animal prion diseases pose will have direct translation to protecting public health.
Development of an accurate classification for human prion disease will have major implications for epidemiological research into the causes of sporadic CJD, whose aetiology remains obscure. While spontaneous conversion of PrPC to PrPSc as a rare stochastic event, or somatic mutation of the PrP gene, resulting in expression of a pathogenic PrP mutant are plausible explanations for sporadic CJD [2,74,217,218], other causes for at least some cases, include environmental exposure to human prions [219–221] or exposure to animal prions. In this regard, the number of prion strains causing sheep scrapie has yet to be established [43,45,46] and epidemiological data cannot exclude this as a cause of a proportion of cases. As future research begins to provide a more precise understanding of the origins of human prion disease, this will facilitate re-analysis of epidemiological data, to reveal important risk factors that might have been obscured by analysing sporadic CJD as a single entity.
While much remains to be done in addressing fundamental questions about human prion strains, transmission barriers, subclinical carrier states and the role of PrP polymorphisms and mutations in the aetiology of these diseases and the production of prion strains, a key development in the future will be application of these highly characterized models to evaluate candidate therapeutic drugs and antibodies [158,159,222]. In addition, it is now becoming increasingly clear that genetic loci other than PRNP may play a significant role in prion pathogenesis and strain selection [117,118,223,224]. Long-term genomic studies in both mouse and human have recently identified a number of genes affecting prion disease incubation period or susceptibility [50,51]. The characterization of such genes in new transgenic models is expected to cast significant light on pathogenic mechanisms, including prion co-factors, and may identify new therapeutic strategies.
Acknowledgments
We are grateful to Jacqueline Linehan, Sebastian Brandner and Ray Young for their help in preparing the figures.
References
- 1.Prusiner SB. Prions. Proc Natl Acad Sci U S A. 1998;95:13363–83. doi: 10.1073/pnas.95.23.13363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Collinge J. Prion diseases of humans and animals: their causes and molecular basis. Annu Rev Neurosci. 2001;24:519–50. doi: 10.1146/annurev.neuro.24.1.519. [DOI] [PubMed] [Google Scholar]
- 3.Weissmann C. The state of the prion. Nat Rev Microbiol. 2004;2:861–71. doi: 10.1038/nrmicro1025. [DOI] [PubMed] [Google Scholar]
- 4.Caughey B, Baron GS. Prions and their partners in crime. Nature. 2006;443:803–10. doi: 10.1038/nature05294. [DOI] [PubMed] [Google Scholar]
- 5.Collinge J, Clarke A. A general model of prion strains and their pathogenicity. Science. 2007;318:930–6. doi: 10.1126/science.1138718. [DOI] [PubMed] [Google Scholar]
- 6.Griffith JS. Self replication and scrapie. Nature. 1967;215:1043–4. doi: 10.1038/2151043a0. [DOI] [PubMed] [Google Scholar]
- 7.Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science. 1982;216:136–44. doi: 10.1126/science.6801762. [DOI] [PubMed] [Google Scholar]
- 8.Riesner D. Biochemistry and structure of PrP(C) and PrP(Sc) Br Med Bull. 2003;66:21–33. doi: 10.1093/bmb/66.1.21. [DOI] [PubMed] [Google Scholar]
- 9.Silveira JR, Raymond GJ, Hughson AG, Race RE, Sim VL, Hayes SF, Caughey B. The most infectious prion protein particles. Nature. 2005;437:257–61. doi: 10.1038/nature03989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Deleault NR, Harris BT, Rees JR, Supattapone S. Formation of native prions from minimal components in vitro. Proc Natl Acad Sci U S A. 2007;104:9741–6. doi: 10.1073/pnas.0702662104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Geoghegan JC, Valdes PA, Orem NR, Deleault NR, Williamson RA, Harris BT, Supattapone S. Selective incorporation of polyanionic molecules into hamster prions. J Biol Chem. 2007;282:36341–53. doi: 10.1074/jbc.M704447200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang F, Wang X, Yuan CG, Ma J. Generating a prion with bacterially expressed recombinant prion protein. Science. 2010;327:1132–5. doi: 10.1126/science.1183748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Supattapone S. What makes a prion infectious? Science. 2010;327:1091–2. doi: 10.1126/science.1187790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Collinge J. Variant Creutzfeldt-Jakob disease. Lancet. 1999;354:317–23. doi: 10.1016/S0140-6736(99)05128-4. [DOI] [PubMed] [Google Scholar]
- 15.Shorter J, Lindquist S. Prions as adaptive conduits of memory and inheritance. Nat Rev Genet. 2005;6:435–50. doi: 10.1038/nrg1616. [DOI] [PubMed] [Google Scholar]
- 16.Tanaka M, Chien P, Yonekura K, Weissman JS. Mechanism of cross-species prion transmission an infectious conformation compatible with two highly divergent yeast prion proteins. Cell. 2005;121:49–62. doi: 10.1016/j.cell.2005.03.008. [DOI] [PubMed] [Google Scholar]
- 17.Wickner RB, Edskes HK, Shewmaker F, Nakayashiki T. Prions of fungi: inherited structures and biological roles. Nat Rev Microbiol. 2007;5:611–18. doi: 10.1038/nrmicro1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Petkova AT, Leapman RD, Guo Z, Yau WM, Mattson MP, Tycko R. Self-propagating, molecular-level polymorphism in Alzheimer's beta-amyloid fibrils. Science. 2005;307:262–5. doi: 10.1126/science.1105850. [DOI] [PubMed] [Google Scholar]
- 19.Meyer-Luehmann M, Coomaraswamy J, Bolmont T, Kaeser S, Schaefer C, Kilger E, Neuenschwander A, Abramowski D, Frey P, Jaton AL, Vigouret JM, Paganetti P, Walsh DM, Mathews PM, Ghiso J, Staufenbiel M, Walker LC, Jucker M. Exogenous induction of cerebral beta-amyloidogenesis is governed by agent and host. Science. 2006;313:1781–4. doi: 10.1126/science.1131864. [DOI] [PubMed] [Google Scholar]
- 20.Olanow CW, Prusiner SB. Is Parkinson's disease a prion disorder? Proc Natl Acad Sci U S A. 2009;106:12571–2. doi: 10.1073/pnas.0906759106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Miller G. Neurodegeneration. Could they all be prion diseases? Science. 2009;326:1337–9. doi: 10.1126/science.326.5958.1337. [DOI] [PubMed] [Google Scholar]
- 22.Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003;300:486–9. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- 23.Lauren J, Gimbel DA, Nygaard HB, Gilbert JW, Strittmatter SM. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature. 2009;457:1128–32. doi: 10.1038/nature07761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Will RG, Ironside JW, Zeidler M, Cousens SN, Estibeiro K, Alperovitch A, Poser S, Pocchiari M, Hofman A, Smith PG. A new variant of Creutzfeldt-Jakob disease in the UK. Lancet. 1996;347:921–5. doi: 10.1016/s0140-6736(96)91412-9. [DOI] [PubMed] [Google Scholar]
- 25.Collinge J, Sidle KC, Meads J, Ironside J, Hill AF. Molecular analysis of prion strain variation and the aetiology of ‘new variant’ CJD. Nature. 1996;383:685–90. doi: 10.1038/383685a0. [DOI] [PubMed] [Google Scholar]
- 26.Hill AF, Desbruslais M, Joiner S, Sidle KCL, Gowland I, Collinge J. The same prion strain causes vCJD and BSE. Nature. 1997;389:448–50. doi: 10.1038/38925. [DOI] [PubMed] [Google Scholar]
- 27.Bruce ME, Will RG, Ironside JW, McConnell I, Drummond D, Suttie A, McCardle L, Chree A, Hope J, Birkett C, Cousens S, Fraser H, Bostock CJ. Transmissions to mice indicate that ‘new variant’ CJD is caused by the BSE agent. Nature. 1997;389:498–501. doi: 10.1038/39057. [DOI] [PubMed] [Google Scholar]
- 28.Asante E, Linehan J, Desbruslais M, Joiner S, Gowland I, Wood A, Welch J, Hill AF, Lloyd S, Wadsworth JD, Collinge J. BSE prions propagate as either variant CJD-like or sporadic CJD-like prion strains in transgenic mice expressing human prion protein. EMBO J. 2002;21:6358–66. doi: 10.1093/emboj/cdf653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hilton DA, Ghani AC, Conyers L, Edwards P, McCardle L, Ritchie D, Penney M, Hegazy D, Ironside JW. Prevalence of lymphoreticular prion protein accumulation in UK tissue samples. J Pathol. 2004;203:733–9. doi: 10.1002/path.1580. [DOI] [PubMed] [Google Scholar]
- 30.Frosh A, Smith LC, Jackson CJ, Linehan J, Brandner S, Wadsworth JD, Collinge J. Analysis of 2000 consecutive UK tonsillectomy specimens for disease-related prion protein. Lancet. 2004;364:1260–2. doi: 10.1016/S0140-6736(04)17143-2. [DOI] [PubMed] [Google Scholar]
- 31.Hilton DA. Pathogenesis and prevalence of variant Creutzfeldt-Jakob disease. J Pathol. 2005;208:134–41. doi: 10.1002/path.1880. [DOI] [PubMed] [Google Scholar]
- 32.Collinge J, Whitfield J, McKintosh E, Beck J, Mead S, Thomas DJ, Alpers MP. Kuru in the 21st century-an acquired human prion disease with very long incubation periods. Lancet. 2006;367:2068–74. doi: 10.1016/S0140-6736(06)68930-7. [DOI] [PubMed] [Google Scholar]
- 33.Wadsworth JD, Collinge J. Update on human prion disease. Biochim Biophys Acta. 2007;1772:598–609. doi: 10.1016/j.bbadis.2007.02.010. [DOI] [PubMed] [Google Scholar]
- 34.Wadsworth JD, Joiner S, Hill AF, Campbell TA, Desbruslais M, Luthert PJ, Collinge J. Tissue distribution of protease resistant prion protein in variant CJD using a highly sensitive immuno-blotting assay. Lancet. 2001;358:171–80. doi: 10.1016/s0140-6736(01)05403-4. [DOI] [PubMed] [Google Scholar]
- 35.Joiner S, Linehan J, Brandner S, Wadsworth JD, Collinge J. Irregular presence of abnormal prion protein in appendix in variant Creutzfeldt-Jakob disease. J Neurol Neurosurg Psychiatry. 2002;73:597–8. doi: 10.1136/jnnp.73.5.597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Llewelyn CA, Hewitt PE, Knight RS, Amar K, Cousens S, Mackenzie J, Will RG. Possible transmission of variant Creutzfeldt-Jakob disease by blood transfusion. Lancet. 2004;363:417–21. doi: 10.1016/S0140-6736(04)15486-X. [DOI] [PubMed] [Google Scholar]
- 37.Peden AH, Head MW, Ritchie DL, Bell JE, Ironside JW. Preclinical vCJD after blood transfusion in a PRNP codon 129 heterozygous patient. Lancet. 2004;364:527–9. doi: 10.1016/S0140-6736(04)16811-6. [DOI] [PubMed] [Google Scholar]
- 38.Wroe SJ, Pal S, Siddique D, Hyare H, Macfarlane R, Joiner S, Linehan J, Brandner S, Wadsworth JD, Hewitt P, Collinge J. Clinical presentation and pre-mortem diagnosis of variant Creutzfeldt-Jakob disease associated with blood transfusion: a case report. Lancet. 2006;368:2061–7. doi: 10.1016/S0140-6736(06)69835-8. [DOI] [PubMed] [Google Scholar]
- 39.Collinge J. Molecular neurology of prion disease. J Neurol Neurosurg Psychiatry. 2005;76:906–19. doi: 10.1136/jnnp.2004.048660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Peden A, McCardle L, Head MW, Love S, Ward HJ, Cousens SN, Keeling DM, Millar CM, Hill FG, Ironside JW. Variant CJD infection in the spleen of a neurologically asymptomatic UK adult patient with haemophilia. Haemophilia. 2010;16:296–304. doi: 10.1111/j.1365-2516.2009.02181.x. [DOI] [PubMed] [Google Scholar]
- 41.Kong Q, Zheng M, Casalone C, Qing L, Huang S, Chakraborty B, Wang P, Chen F, Cali I, Corona C, Martucci F, Iulini B, Acutis P, Wang L, Liang J, Wang M, Li X, Monaco S, Zanusso G, Zou WQ, Caramelli M, Gambetti P. Evaluation of the human transmission risk of an atypical bovine spongiform encephalopathy prion strain. J Virol. 2008;82:3697–701. doi: 10.1128/JVI.02561-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Beringue V, Andreoletti O, Le Dur A, Essalmani R, Vilotte JL, Lacroux C, Reine F, Herzog L, Biacabe AG, Baron T, Caramelli M, Casalone C, Laude H. A bovine prion acquires an epidemic bovine spongiform encephalopathy strain-like phenotype on interspecies transmission. J Neurosci. 2007;27:6965–71. doi: 10.1523/JNEUROSCI.0693-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Beringue V, Vilotte JL, Laude H. Prion agents diversity and species barrier. Vet Res. 2008;39:47. doi: 10.1051/vetres:2008024. [DOI] [PubMed] [Google Scholar]
- 44.Beringue V, Herzog L, Reine F, Le Dur A, Casalone C, Vilotte JL, Laude H. Transmission of atypical bovine prions to mice transgenic for human prion protein. Emerg Infect Dis. 2008;14:1898–901. doi: 10.3201/eid1412.080941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Baron T, Biacabe AG, Arsac JN, Benestad S, Groschup MH. Atypical transmissible spongiform encephalopathies (TSEs) in ruminants. Vaccine. 2007;25:5625–30. doi: 10.1016/j.vaccine.2006.10.058. [DOI] [PubMed] [Google Scholar]
- 46.Benestad SL, Arsac JN, Goldmann W, Noremark M. Atypical/Nor98 scrapie: properties of the agent, genetics, and epidemiology. Vet Res. 2008;39:19. doi: 10.1051/vetres:2007056. [DOI] [PubMed] [Google Scholar]
- 47.Castilla J, Gonzalez-Romero D, Saa P, Morales R, De Castro J, Soto C. Crossing the species barrier by PrP(Sc) replication in vitro generates unique infectious prions. Cell. 2008;134:757–68. doi: 10.1016/j.cell.2008.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lloyd S, Linehan J, Desbruslais M, Joiner S, Buckell J, Brandner S, Wadsworth JD, Collinge J. Characterization of two distinct prion strains derived from bovine spongiform encephalopathy transmissions to inbred mice. J Gen Virol. 2004;85:2471–8. doi: 10.1099/vir.0.79889-0. [DOI] [PubMed] [Google Scholar]
- 49.Wadsworth JD, Asante EA, Desbruslais M, Linehan J, Joiner S, Gowland I, Welch J, Stone L, Lloyd S, Hill AF, Brandner S, Collinge J. Human prion protein with valine 129 prevents expression of variant CJD phenotype. Science. 2004;306:1793–6. doi: 10.1126/science.1103932. [DOI] [PubMed] [Google Scholar]
- 50.Mead S, Poulter M, Uphill J, Beck J, Whitfield J, Webb TE, Campbell T, Adamson G, Deriziotis P, Tabrizi SJ, Hummerich H, Verzilli C, Alpers MP, Whittaker JC, Collinge J. Genetic risk factors for variant Creutzfeldt-Jakob disease: a genome-wide association study. Lancet Neurol. 2009;8:57–66. doi: 10.1016/S1474-4422(08)70265-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lloyd SE, Maytham EG, Pota H, Grizenkova J, Molou E, Uphill J, Hummerich H, Whitfield J, Alpers MP, Mead S, Collinge J. HECTD2 is associated with susceptibility to mouse and human prion disease. PLoS Genet. 2009;5:e1000383. doi: 10.1371/journal.pgen.1000383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hill AF, Collinge J. Subclinical prion infection. Trends Microbiol. 2003;11:578–84. doi: 10.1016/j.tim.2003.10.007. [DOI] [PubMed] [Google Scholar]
- 53.Taylor DM. Resistance of transmissible spongiform encephalopathy agents to decontamination. Contrib Microbiol. 2004;11:136–45. doi: 10.1159/000077054. [DOI] [PubMed] [Google Scholar]
- 54.Jackson GS, McKintosh E, Flechsig E, Prodromidou K, Hirsch P, Linehan J, Brandner S, Clarke A, Weissmann C, Collinge J. An enzyme-detergent method for effective prion decontamination of surgical steel. J Gen Virol. 2005;86:869–78. doi: 10.1099/vir.0.80484-0. [DOI] [PubMed] [Google Scholar]
- 55.Gajdusek DC, Gibbs CJJr, Alpers M. Experimental transmission of a kuru-like syndrome to chimpanzees. Nature. 1966;209:794–6. doi: 10.1038/209794a0. [DOI] [PubMed] [Google Scholar]
- 56.Gibbs CJJr, Gajdusek DC, Asher DM, Alpers MP, Beck E, Daniel PM, Matthews WB. Creutzfeldt-Jakob disease (spongiform encephalopathy): transmission to the chimpanzee. Science. 1968;161:388–9. doi: 10.1126/science.161.3839.388. [DOI] [PubMed] [Google Scholar]
- 57.Brown P, Gibbs CJJr, Rodgers Johnson P, Asher DM, Sulima MP, Bacote A, Goldfarb LG, Gajdusek DC. Human spongiform encephalopathy: the National Institutes of Health series of 300 cases of experimentally transmitted disease. Ann Neurol. 1994;35:513–29. doi: 10.1002/ana.410350504. [DOI] [PubMed] [Google Scholar]
- 58.Collinge J, Palmer MS, Sidle KCL, Hill AF, Gowland I, Meads J, Asante EA, Bradley R, Doey LJ, Lantos PL. Unaltered susceptibility to BSE in transgenic mice expressing human prion protein. Nature. 1995;378:779–83. doi: 10.1038/378779a0. [DOI] [PubMed] [Google Scholar]
- 59.Telling GC, Scott M, Mastrianni J, Gabizon R, Torchia M, Cohen FE, DeArmond SJ, Prusiner SB. Prion propagation in mice expressing human and chimeric PrP transgenes implicates the interaction of cellular PrP with another protein. Cell. 1995;83:79–90. doi: 10.1016/0092-8674(95)90236-8. [DOI] [PubMed] [Google Scholar]
- 60.Asante E, Collinge J. Transgenic studies of the influence of the PrP structure on TSE diseases. Adv Protein Chem. 2001;57:273–311. doi: 10.1016/s0065-3233(01)57025-4. [DOI] [PubMed] [Google Scholar]
- 61.Manson JC, Tuzi NL. Transgenic models of the transmissible spongiform encephalopathies. Expert Rev Mol Med. 2001;2001:1–15. doi: 10.1017/S1462399401002952. [DOI] [PubMed] [Google Scholar]
- 62.Weissmann C, Flechsig E. PrP knock-out and PrP transgenic mice in prion research. Br Med Bull. 2003;66:43–60. doi: 10.1093/bmb/66.1.43. [DOI] [PubMed] [Google Scholar]
- 63.Bishop MT, Hart P, Aitchison L, Baybutt HN, Plinston C, Thomson V, Tuzi NL, Head MW, Ironside JW, Will RG, Manson JC. Predicting susceptibility and incubation time of human-to-human transmission of vCJD. Lancet Neurol. 2006;5:393–8. doi: 10.1016/S1474-4422(06)70413-6. [DOI] [PubMed] [Google Scholar]
- 64.Groschup MH, Buschmann A. Rodent models for prion diseases. Vet Res. 2008;39:32. doi: 10.1051/vetres:2008008. [DOI] [PubMed] [Google Scholar]
- 65.Telling GC. Transgenic mouse models of prion diseases. Methods Mol Biol. 2008;459:249–63. doi: 10.1007/978-1-59745-234-2_17. [DOI] [PubMed] [Google Scholar]
- 66.Wadsworth JD, Hill AF, Beck J, Collinge J. Molecular and clinical classification of human prion disease. Br Med Bull. 2003;66:241–54. doi: 10.1093/bmb/66.1.241. [DOI] [PubMed] [Google Scholar]
- 67.Mead S. Prion disease genetics. Eur J Hum Genet. 2006;14:273–81. doi: 10.1038/sj.ejhg.5201544. [DOI] [PubMed] [Google Scholar]
- 68.Collinge J, Harding AE, Owen F, Poulter M, Lofthouse R, Boughey AM, Shah T, Crow TJ. Diagnosis of Gerstmann-Straussler syndrome in familial dementia with prion protein gene analysis. Lancet. 1989;2:15–17. doi: 10.1016/s0140-6736(89)90256-0. [DOI] [PubMed] [Google Scholar]
- 69.Collinge J, Owen F, Poulter M, Leach M, Crow TJ, Rossor MN, Hardy J, Mullan MJ, Janota I, Lantos PL. Prion dementia without characteristic pathology. Lancet. 1990;336:7–9. doi: 10.1016/0140-6736(90)91518-f. [DOI] [PubMed] [Google Scholar]
- 70.Collinge J, Brown J, Hardy J, Mullan M, Rossor MN, Baker H, Crow TJ, Lofthouse R, Poulter M, Ridley R, Owen F, Bennett C, Dunn G, Harding AE, Quinn N, Doshi B, Roberts GW, Honavar M, Janota I, Lantos PL. Inherited prion disease with 144 base pair gene insertion: II: clinical and pathological features. Brain. 1992;115:687–710. doi: 10.1093/brain/115.3.687. [DOI] [PubMed] [Google Scholar]
- 71.Mallucci G, Campbell TA, Dickinson A, Beck J, Holt M, Plant G, De Pauw KW, Hakin RN, Clarke CE, Howell S, Davies-Jones GAB, Lawden M, Smith CML, Ince P, Ironside JW, Bridges LR, Dean A, Weeks I, Collinge J. Inherited prion disease with an alanine to valine mutation at codon 117 in the prion protein gene. Brain. 1999;122:1823–37. doi: 10.1093/brain/122.10.1823. [DOI] [PubMed] [Google Scholar]
- 72.Kovacs GG, Trabattoni G, Hainfellner JA, Ironside JW, Knight RS, Budka H. Mutations of the prion protein gene phenotypic spectrum. J Neurol. 2002;249:1567–82. doi: 10.1007/s00415-002-0896-9. [DOI] [PubMed] [Google Scholar]
- 73.Mead S, Poulter M, Beck J, Webb T, Campbell T, Linehan J, Desbruslais M, Joiner S, Wadsworth JD, King A, Lantos P, Collinge J. Inherited prion disease with six octapeptide repeat insertional mutation-molecular analysis of phenotypic heterogeneity. Brain. 2006;129:2297–317. doi: 10.1093/brain/awl226. [DOI] [PubMed] [Google Scholar]
- 74.Wadsworth JD, Joiner S, Linehan J, Cooper S, Powell C, Mallinson G, Buckell J, Gowland I, Asante EA, Budka H, Brandner S, Collinge J. Phenotypic heterogeneity in inherited prion disease (P102L) is associated with differential propagation of protease-resistant wild-type and mutant prion protein. Brain. 2006;129:1557–69. doi: 10.1093/brain/awl076. [DOI] [PubMed] [Google Scholar]
- 75.Collinge J, Whitfield J, McKintosh E, Frosh A, Mead S, Hill AF, Brandner S, Thomas D, Alpers MP. A clinical study of kuru patients with long incubation periods at the end of the epidemic in Papua New Guinea. Philos Trans R Soc Lond B Biol Sci. 2008;363:3725–39. doi: 10.1098/rstb.2008.0068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.WHO. WHO Manual for Surveillance of Human Transmissible Spongiform Encephalopathies. Geneva: World Health Organisation Press; 2003. [Google Scholar]
- 77.Budka H, Aguzzi A, Brown P, Brucher JM, Bugiani O, Gullotta F, Haltia M, Hauw JJ, Ironside JW, Jellinger K, Kretzschmar HA, Lantos PL, Masullo C, Schlote W, Tateishi J, Weller RO. Neuropathological diagnostic criteria for Creutzfeldt-Jakob disease (CJD) and other human spongiform encephalopathies (Prion diseases) Brain Pathol. 1995;5:459–66. doi: 10.1111/j.1750-3639.1995.tb00625.x. [DOI] [PubMed] [Google Scholar]
- 78.Ironside JW, Head MW, Bell JE, McCardle L, Will RG. Laboratory diagnosis of variant Creutzfeldt-Jakob disease. Histopathology. 2000;37:1–9. doi: 10.1046/j.1365-2559.2000.00946.x. [DOI] [PubMed] [Google Scholar]
- 79.Wadsworth JD, Powell C, Beck JA, Joiner S, Linehan JM, Brandner S, Mead S, Collinge J. Molecular diagnosis of human prion disease. Methods Mol Biol. 2008;459:197–227. doi: 10.1007/978-1-59745-234-2_14. [DOI] [PubMed] [Google Scholar]
- 80.Collinge J, Palmer MS, Dryden AJ. Genetic predisposition to iatrogenic Creutzfeldt-Jakob disease. Lancet. 1991;337:1441–2. doi: 10.1016/0140-6736(91)93128-v. [DOI] [PubMed] [Google Scholar]
- 81.Palmer MS, Dryden AJ, Hughes JT, Collinge J. Homozygous prion protein genotype predisposes to sporadic Creutzfeldt-Jakob disease. Nature. 1991;352:340–2. doi: 10.1038/352340a0. [DOI] [PubMed] [Google Scholar]
- 82.Windl O, Dempster M, Estibeiro JP, Lathe R, De Silva R, Esmonde T, Will R, Springbett A, Campbell TA, Sidle KCL, Palmer MS, Collinge J. Genetic basis of Creutzfeldt-Jakob disease in the United Kingom: a systematic analysis of predisposing mutations and allelic variation in the PRNP gene. Hum Genet. 1996;98:259–64. doi: 10.1007/s004390050204. [DOI] [PubMed] [Google Scholar]
- 83.Lee HS, Brown P, Cervenáková L, Garruto RM, Alpers MP, Gajdusek DC, Goldfarb LG. Increased susceptibility to Kuru of carriers of the PRNP 129 methionine/methionine genotype. J Infect Dis. 2001;183:192–6. doi: 10.1086/317935. [DOI] [PubMed] [Google Scholar]
- 84.Mead S, Stumpf MP, Whitfield J, Beck J, Poulter M, Campbell T, Uphill J, Goldstein D, Alpers MP, Fisher E, Collinge J. Balancing selection at the prion protein gene consistent with prehistoric kuru-like epidemics. Science. 2003;300:640–3. doi: 10.1126/science.1083320. [DOI] [PubMed] [Google Scholar]
- 85.Collins SJ, Sanchez-Juan P, Masters CL, Klug GM, van Duijn C, Poleggi A, Pocchiari M, Almonti S, Cuadrado-Corrales N, Pedro-Cuesta J, Budka H, Gelpi E, Glatzel M, Tolnay M, Hewer E, Zerr I, Heinemann U, Kretszchmar HA, Jansen GH, Olsen E, Mitrova E, Alperovitch A, Brandel JP, Mackenzie J, Murray K, Will RG. Determinants of diagnostic investigation sensitivities across the clinical spectrum of sporadic Creutzfeldt-Jakob disease. Brain. 2006;129:2278–87. doi: 10.1093/brain/awl159. [DOI] [PubMed] [Google Scholar]
- 86.Bruce ME. TSE strain variation. Br Med Bull. 2003;66:99–108. doi: 10.1093/bmb/66.1.99. [DOI] [PubMed] [Google Scholar]
- 87.Budka H. Neuropathology of prion diseases. Br Med Bull. 2003;66:121–30. doi: 10.1093/bmb/66.1.121. [DOI] [PubMed] [Google Scholar]
- 88.Hainfellner JA, Brantner-Inthaler S, Cervenáková L, Brown P, Kitamoto T, Tateishi J, Diringer H, Liberski PP, Regele H, Feucht R, Mayr N, Wessely P, Summer K, Seitelberger F, Budka H. The original Gerstmann-Straussler-Scheinker family of Austria: divergent clinicopathological phenotypes but constant PrP genotype. Brain Pathol. 1995;5:201–11. doi: 10.1111/j.1750-3639.1995.tb00596.x. [DOI] [PubMed] [Google Scholar]
- 89.Brandner S, Whitfield J, Boone K, Puwa A, O'Malley C, Linehan JM, Joiner S, Scaravilli F, Calder I, Alpers P, Wadsworth JD, Collinge J. Central and peripheral pathology of kuru: pathological analysis of a recent case and comparison with other forms of human prion disease. Philos Trans R Soc Lond B Biol Sci. 2008;363:3755–63. doi: 10.1098/rstb.2008.0091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hill AF, Joiner S, Wadsworth JD, Sidle KC, Bell JE, Budka H, Ironside JW, Collinge J. Molecular classification of sporadic Creutzfeldt-Jakob disease. Brain. 2003;126:1333–46. doi: 10.1093/brain/awg125. [DOI] [PubMed] [Google Scholar]
- 91.Ironside JW, Head MW. Neuropathology and molecular biology of variant Creutzfeldt-Jakob disease. Curr Top Microbiol Immunol. 2004;284:133–59. doi: 10.1007/978-3-662-08441-0_6. [DOI] [PubMed] [Google Scholar]
- 92.Telling GC, Parchi P, DeArmond SJ, Cortelli P, Montagna P, Gabizon R, Mastrianni J, Lugaresi E, Gambetti P, Prusiner SB. Evidence for the conformation of the pathologic isoform of the prion protein enciphering and propagating prion diversity. Science. 1996;274:2079–82. doi: 10.1126/science.274.5295.2079. [DOI] [PubMed] [Google Scholar]
- 93.Parchi P, Giese A, Capellari S, Brown P, Schulz-Schaeffer W, Windl O, Zerr I, Budka H, Kopp N, Piccardo P, Poser S, Rojiani A, Streichenberger N, Julien J, Vital C, Ghetti B, Gambetti P, Kretzschmar H. Classification of sporadic Creutzfeldt-Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann Neurol. 1999;46:224–33. [PubMed] [Google Scholar]
- 94.Safar J, Wille H, Itri V, Groth D, Serban H, Torchia M, Cohen FE, Prusiner SB. Eight prion strains have PrPSc molecules with different conformations. Nat Med. 1998;4:1157–65. doi: 10.1038/2654. [DOI] [PubMed] [Google Scholar]
- 95.Castilla J, Morales R, Saa P, Barria M, Gambetti P, Soto C. Cell-free propagation of prion strains. EMBO J. 2008;27:2557–66. doi: 10.1038/emboj.2008.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Parchi P, Castellani R, Capellari S, Ghetti B, Young K, Chen SG, Farlow M, Dickson DW, Sims AAF, Trojanowski JQ, Petersen RB, Gambetti P. Molecular basis of phenotypic variability in sporadic Creutzfeldt-Jakob disease. Ann Neurol. 1996;39:767–78. doi: 10.1002/ana.410390613. [DOI] [PubMed] [Google Scholar]
- 97.Zanusso G, Farinazzo A, Fiorini M, Gelati M, Castagna A, Righetti PG, Rizzuto N, Monaco S. pH-dependent prion protein conformation in classical Creutzfeldt-Jakob disease. J Biol Chem. 2001;276:40377–80. doi: 10.1074/jbc.C100458200. [DOI] [PubMed] [Google Scholar]
- 98.Cali I, Castellani R, Yuan J, Al Shekhlee A, Cohen ML, Xiao X, Moleres FJ, Parchi P, Zou WQ, Gambetti P. Classification of sporadic Creutzfeldt-Jakob disease revisited. Brain. 2006;129:2266–77. doi: 10.1093/brain/awl224. [DOI] [PubMed] [Google Scholar]
- 99.Gambetti P, Dong Z, Yuan J, Xiao X, Zheng M, Alshekhlee A, Castellani R, Cohen M, Barria MA, Gonzalez-Romero D, Belay ED, Schonberger LB, Marder K, Harris C, Burke JR, Montine T, Wisniewski T, Dickson DW, Soto C, Hulette CM, Mastrianni JA, Kong Q, Zou WQ. A novel human disease with abnormal prion protein sensitive to protease. Ann Neurol. 2008;63:697–708. doi: 10.1002/ana.21420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Parchi P, Notari S, Weber P, Schimmel H, Budka H, Ferrer I, Haik S, Hauw JJ, Head MW, Ironside JW, Limido L, Rodriguez A, Strobel T, Tagliavini F, Inter-Laboratory KHA. Assessment of PrP(Sc) Typing in Creutzfeldt-Jakob disease: a western blot study within the NeuroPrion consortium. Brain Pathol. 2008;19:384–91. doi: 10.1111/j.1750-3639.2008.00187.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wadsworth JD, Hill AF, Joiner S, Jackson GS, Clarke A, Collinge J. Strain-specific prion-protein conformation determined by metal ions. Nat Cell Biol. 1999;1:55–9. doi: 10.1038/9030. [DOI] [PubMed] [Google Scholar]
- 102.Uro-Coste E, Cassard H, Simon S, Lugan S, Bilheude JM, Perret-Liaudet A, Ironside JW, Haik S, Basset-Leobon C, Lacroux C, Peoch K, Streichenberger N, Langeveld J, Head MW, Grassi J, Hauw JJ, Schelcher F, Delisle MB, Andreoletti O. Beyond PrP type 1/type 2 dichotomy in Creutzfeldt-Jakob disease. PLoS Pathog. 2008;4:e1000029. [PubMed] [Google Scholar]
- 103.Piccardo P, Dlouhy SR, Lievens PMJ, Young K, Bird TD, Nochlin D, Dickson DW, Vinters HV, Zimmerman TR, Mackenzie IRA, Kish SJ, Ang LC, De Carli C, Pocchiari M, Brown P, Gibbs CJ, Gajdusek DC, Bugiani O, Ironside J, Tagliavini F, Ghetti B. Phenotypic variability of Gerstmann-Straussler-Scheinker disease is associated with prion protein heterogeneity. J Neuropathol Exp Neurol. 1998;57:979–88. doi: 10.1097/00005072-199810000-00010. [DOI] [PubMed] [Google Scholar]
- 104.Puoti G, Giaccone G, Rossi G, Canciani B, Bugiani O, Tagliavini F. Sporadic Creutzfeldt-Jakob disease: co-occurrence of different types of PrPSc in the same brain. Neurology. 1999;53:2173–6. doi: 10.1212/wnl.53.9.2173. [DOI] [PubMed] [Google Scholar]
- 105.Head MW, Bunn TJ, Bishop MT, McLoughlin V, Lowrie S, McKimmie CS, Williams MC, McCardle L, Mackenzie J, Knight R, Will RG, Ironside JW. Prion protein heterogeneity in sporadic but not variant Creutzfeldt-Jakob disease: U.K. cases 1991–2002. Ann Neurol. 2004;55:851–9. doi: 10.1002/ana.20127. [DOI] [PubMed] [Google Scholar]
- 106.Polymenidou M, Stoeck K, Glatzel M, Vey M, Bellon A, Aguzzi A. Coexistence of multiple PrP(Sc) types in individuals with Creutzfeldt-Jakob disease. Lancet Neurol. 2005;4:805–14. doi: 10.1016/S1474-4422(05)70225-8. [DOI] [PubMed] [Google Scholar]
- 107.Schoch G, Seeger H, Bogousslavsky J, Tolnay M, Janzer RC, Aguzzi A, Glatzel M. Analysis of prion strains by PrP(Sc) profiling in sporadic Creutzfeldt-Jakob disease. PLoS Med. 2005;3:e14. doi: 10.1371/journal.pmed.0030014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Yull HM, Ritchie DL, Langeveld JP, van Zijderveld FG, Bruce ME, Ironside JW, Head MW. Detection of type 1 prion protein in variant Creutzfeldt-Jakob disease. Am J Pathol. 2006;168:151–7. doi: 10.2353/ajpath.2006.050766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Tzaban S, Friedlander G, Schonberger O, Horonchik L, Yedidia Y, Shaked G, Gabizon R, Taraboulos A. Protease-sensitive scrapie prion protein in aggregates of heterogeneous sizes. Biochemistry. 2002;41:12868–75. doi: 10.1021/bi025958g. [DOI] [PubMed] [Google Scholar]
- 110.Tremblay P, Ball HL, Kaneko K, Groth D, Hegde RS, Cohen FE, DeArmond SJ, Prusiner SB, Safar JG. Mutant PrP(Sc) conformers induced by a synthetic peptide and several prion strains. J Virol. 2004;78:2088–99. doi: 10.1128/JVI.78.4.2088-2099.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Safar JG, Geschwind MD, Deering C, Didorenko S, Sattavat M, Sanchez H, Serban A, Vey M, Baron H, Giles K, Miller BL, DeArmond SJ, Prusiner SB. Diagnosis of human prion disease. Proc Natl Acad Sci U S A. 2005;102:3501–6. doi: 10.1073/pnas.0409651102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Nazor KE, Kuhn F, Seward T, Green M, Zwald D, Purro M, Schmid J, Biffiger K, Power AM, Oesch B, Raeber AJ, Telling GC. Immunodetection of disease-associated mutant PrP, which accelerates disease in GSS transgenic mice. EMBO J. 2005;24:2472–80. doi: 10.1038/sj.emboj.7600717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Thackray AM, Hopkins L, Bujdoso R. Proteinase K-sensitive disease-associated ovine prion protein revealed by conformation-dependent immunoassay. Biochem J. 2007;401:475–83. doi: 10.1042/BJ20061264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Barron RM, Campbell SL, King D, Bellon A, Chapman KE, Williamson RA, Manson JC. High titres of TSE infectivity associated with extremely low levels of PrPSc in vivo. J Biol Chem. 2007;282:35878–86. doi: 10.1074/jbc.M704329200. [DOI] [PubMed] [Google Scholar]
- 115.Cronier S, Gros N, Tattum MH, Jackson GS, Clarke AR, Collinge J, Wadsworth JD. Detection and characterization of proteinase K-sensitive disease-related prion protein with thermolysin. Biochem J. 2008;416:297–305. doi: 10.1042/BJ20081235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Colby DW, Wain R, Baskakov IV, Legname G, Palmer CG, Nguyen HO, Lemus A, Cohen FE, DeArmond SJ, Prusiner SB. Protease-sensitive synthetic prions. PLoS Pathog. 2010;6:e1000736. doi: 10.1371/journal.ppat.1000736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Stephenson DA, Chiotti K, Ebeling C, Groth D, DeArmond SJ, Prusiner SB, Carlson GA. Quantitative trait loci affecting prion incubation time in mice. Genomics. 2000;69:47–53. doi: 10.1006/geno.2000.6320. [DOI] [PubMed] [Google Scholar]
- 118.Lloyd S, Onwuazor ON, Beck J, Mallinson G, Farrall M, Targonski P, Collinge J, Fisher E. Identification of multiple quantitative trait loci linked to prion disease incubation period in mice. Proc Natl Acad Sci U S A. 2001;98:6279–83. doi: 10.1073/pnas.101130398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Lloyd S, Collinge J. Genetic susceptibility to prion diseases in humans and mice. Current Genomics. 2005;6:1–11. [Google Scholar]
- 120.Brown P, Preece M, Brandel JP, Sato T, McShane L, Zerr I, Fletcher A, Will RG, Pocchiari M, Cashman NR, D'Aignaux JH, Cervenáková L, Fradkin J, Schonberger LB, Collins SJ. Iatrogenic Creutzfeldt-Jakob disease at the millennium. Neurology. 2000;55:1075–81. doi: 10.1212/wnl.55.8.1075. [DOI] [PubMed] [Google Scholar]
- 121.Will RG. Acquired prion disease: iatrogenic CJD, variant CJD, kuru. Br Med Bull. 2003;66:255–65. doi: 10.1093/bmb/66.1.255. [DOI] [PubMed] [Google Scholar]
- 122.Wadsworth JD, Joiner S, Linehan JM, Desbruslais M, Fox K, Cooper S, Cronier S, Asante EA, Mead S, Brandner S, Hill AF, Collinge J. Kuru prions and sporadic Creutzfeldt-Jakob disease prions have equivalent transmission properties in transgenic and wild-type mice. Proc Natl Acad Sci U S A. 2008;105:3885–90. doi: 10.1073/pnas.0800190105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Wadsworth JD, Joiner S, Linehan JM, Asante EA, Brandner S, Collinge J. Review. The origin of the prion agent of kuru: molecular and biological strain typing. Philos Trans R Soc Lond B Biol Sci. 2008;363:3747–53. doi: 10.1098/rstb.2008.0069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Bueler H, Aguzzi A, Sailer A, Greiner RA, Autenried P, Aguet M, Weissmann C. Mice devoid of PrP are resistant to scrapie. Cell. 1993;73:1339–47. doi: 10.1016/0092-8674(93)90360-3. [DOI] [PubMed] [Google Scholar]
- 125.Sailer A, Bueler H, Fischer M, Aguzzi A, Weissmann C. No propagation of prions in mice devoid of PrP. Cell. 1994;77:967–8. doi: 10.1016/0092-8674(94)90436-7. [DOI] [PubMed] [Google Scholar]
- 126.Scott MRD, Telling GC, Prusiner SB. Transgenetics and gene targeting in studies of prion diseases. Curr Top Microbiol Immunol. 1996;207:95–123. doi: 10.1007/978-3-642-60983-1_8. [DOI] [PubMed] [Google Scholar]
- 127.Fischer M, Rulicke T, Raeber A, Sailer A, Moser M, Oesch B, Brandner S, Aguzzi A, Weissmann C. Prion protein (PrP) with amino-proximal deletions restoring susceptibility of PrP knockout mice to scrapie. EMBO J. 1996;15:1255–64. [PMC free article] [PubMed] [Google Scholar]
- 128.Flechsig E, Shmerling D, Hegyi I, Raeber AJ, Fischer M, Cozzio A, von Mering C, Aguzzi A, Weissmann C. Prion protein devoid of the octapeptide repeat region restores susceptibility to scrapie in PrP knockout mice. Neuron. 2000;27:399–408. doi: 10.1016/s0896-6273(00)00046-5. [DOI] [PubMed] [Google Scholar]
- 129.Muramoto T, Scott M, Cohen FE, Prusiner SB. Recombinant scrapie-like prion protein of 106 amino acids is soluble. Proc Natl Acad Sci U S A. 1996;93:15457–62. doi: 10.1073/pnas.93.26.15457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Supattapone S, Bosque P, Muramoto T, Wille H, Aagaard C, Peretz D, Nguyen HOB, Heinrich C, Torchia M, Safar J, Cohen FE, DeArmond SJ, Prusiner SB, Scott M. Prion protein of 106 residues creates an artificial transmission barrier for prion replication in transgenic mice. Cell. 1999;96:869–78. doi: 10.1016/s0092-8674(00)80596-6. [DOI] [PubMed] [Google Scholar]
- 131.Shmerling D, Hegyi I, Fischer M, Blättler T, Brandner S, Götz J, Rülicke T, Flechsig E, Cozzio A, von Mering C, Hangartner C, Aguzzi A, Weissmann C. Expression of amino-terminally truncated PrP in the mouse leading to ataxia and specific cerebellar lesions. Cell. 1998;93:203–14. doi: 10.1016/s0092-8674(00)81572-x. [DOI] [PubMed] [Google Scholar]
- 132.Moore RC, Lee IY, Silverman GL, Harrison PM, Strome R, Heinrich C, Karunaratne A, Pasternak SH, Chishti MA, Liang Y, Mastrangelo P, Wang K, Smit AFA, Katamine S, Carlson GA, Cohen FE, Prusiner SB, Melton DW, Tremblay P, Hood LE, Westaway D. Ataxia in prion protein (PrP)-deficient mice is associated with upregulation of the novel PrP-like protein Doppel. J Mol Biol. 1999;292:797–817. doi: 10.1006/jmbi.1999.3108. [DOI] [PubMed] [Google Scholar]
- 133.Silverman GL, Qin K, Moore RC, Yang Y, Mastrangelo P, Tremblay P, Prusiner SB, Cohen FE, Westaway D. Doppel is an N-glycosylated, glycosylphosphatidylinositol-anchored protein. J Biol Chem. 2000;275:26834–41. doi: 10.1074/jbc.M003888200. [DOI] [PubMed] [Google Scholar]
- 134.Rossi D, Cozzio A, Flechsig E, Klein MA, Rülicke T, Aguzzi A, Weissmann C. Onset of ataxia and Purkinje cell loss in PrP null mice inversely correlated with Dpl level in brain. EMBO J. 2001;20:694–702. doi: 10.1093/emboj/20.4.694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Nishida N, Tremblay P, Sugimoto T, Shigematsu K, Shirabe S, Petromilli C, Erpel SP, Nakaoke R, Atarashi R, Houtani T, Torchia M, Sakaguchi S, DeArmond SJ, Prusiner SB, Katamine S. A mouse prion protein transgene rescues mice deficient for the prion protein gene from Purkinje cell degeneration and demyelination. Lab Invest. 1999;79:689–97. [PubMed] [Google Scholar]
- 136.Weissmann C, Aguzzi A. Perspectives: neurobiology. PrP's double causes trouble. Science. 1999;286:914–15. doi: 10.1126/science.286.5441.914. [DOI] [PubMed] [Google Scholar]
- 137.Flechsig E, Weissmann C. The role of PrP in health and disease. Curr Mol Med. 2004;4:337–53. doi: 10.2174/1566524043360645. [DOI] [PubMed] [Google Scholar]
- 138.Watts JC, Drisaldi B, Ng V, Yang J, Strome B, Horne P, Sy MS, Yoong L, Young R, Mastrangelo P, Bergeron C, Fraser PE, Carlson GA, Mount HT, Schmitt-Ulms G, Westaway D. The CNS glycoprotein Shadoo has PrP(C)-like protective properties and displays reduced levels in prion infections. EMBO J. 2007;26:4038–50. doi: 10.1038/sj.emboj.7601830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Bueler H, Fischer M, Lang Y, Bluethmann H, Lipp H-P, DeArmond SJ, Prusiner SB, Aguet M, Weissmann C. Normal development and behaviour of mice lacking the neuronal cell-surface PrP protein. Nature. 1992;356:577–82. doi: 10.1038/356577a0. [DOI] [PubMed] [Google Scholar]
- 140.Manson JC, Clarke A, Hooper ML, Aitchison L, McConnell I, Hope J. 129/Ola mice carrying a null mutation in PrP that abolishes mRNA production are developmentally normal. Mol Neurobiol. 1994;8:121–7. doi: 10.1007/BF02780662. [DOI] [PubMed] [Google Scholar]
- 141.Mallucci G, Ratté S, Asante E, Linehan J, Gowland I, Jefferys JGR, Collinge J. Post-natal knockout of prion protein alters hippocampal CA1 properties, but does not result in neurodegeneration. EMBO J. 2002;21:202–10. doi: 10.1093/emboj/21.3.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Mallucci G, Dickinson A, Linehan J, Klohn P, Brandner S, Collinge J. Depleting neuronal PrP in prion infection prevents disease and reverses spongiosis. Science. 2003;302:871–4. doi: 10.1126/science.1090187. [DOI] [PubMed] [Google Scholar]
- 143.Mallucci G, Collinge J. Rational targeting for prion therapeutics. Nat Rev Neurosci. 2005;6:23–34. doi: 10.1038/nrn1584. [DOI] [PubMed] [Google Scholar]
- 144.Westergard L, Christensen HM, Harris DA. The cellular prion protein (PrP(C) ): Its physiological function and role in disease. Biochim Biophys Acta. 2007;1772:629–44. doi: 10.1016/j.bbadis.2007.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Aguzzi A, Calella AM. Prions: protein aggregation and infectious diseases. Physiol Rev. 2009;89:1105–52. doi: 10.1152/physrev.00006.2009. [DOI] [PubMed] [Google Scholar]
- 146.Collinge J, Whittington MA, Sidle KCL, Smith CJ, Palmer MS, Clarke A, Jefferys JGR. Prion protein is necessary for normal synaptic function. Nature. 1994;370:295–7. doi: 10.1038/370295a0. [DOI] [PubMed] [Google Scholar]
- 147.Manson JC, Hope J, Clarke A, Johnston A, Black C, MacLeod N. PrP gene dosage and long term potentiation. Neurodegeneration. 1995;4:113–14. doi: 10.1006/neur.1995.0014. [DOI] [PubMed] [Google Scholar]
- 148.Tobler I, Gaus SE, Deboer T, Achermann P, Fischer M, Rulicke T, Moser M, Oesch B, McBride PA, Manson JC. Altered circadian activity rhythms and sleep in mice devoid of prion protein. Nature. 1996;380:639–42. doi: 10.1038/380639a0. [DOI] [PubMed] [Google Scholar]
- 149.Colling SB, Collinge J, Jefferys JGR. Hippocampal slices from prion protein null mice: disrupted Ca2+-activated K+ currents. Neurosci Lett. 1996;209:49–52. doi: 10.1016/0304-3940(96)12596-9. [DOI] [PubMed] [Google Scholar]
- 150.Carleton A, Tremblay P, Vincent JD, Lledo PM. Dose-dependent, prion protein (PrP)-mediated facilitation of excitatory synaptic transmission in the mouse hippocampus. Pflugers Arch. 2001;442:223–9. doi: 10.1007/s004240100523. [DOI] [PubMed] [Google Scholar]
- 151.Herms JW, Tings T, Dunker S, Kretzschmar HA. Prion protein affects Ca2+-activated K+ currents in cerebellar purkinje cells. Neurobiol Dis. 2001;8:324–30. doi: 10.1006/nbdi.2000.0369. [DOI] [PubMed] [Google Scholar]
- 152.Criado JR, Sanchez-Alavez M, Conti B, Giacchino JL, Wills DN, Henriksen SJ, Race R, Manson JC, Chesebro B, Oldstone MB. Mice devoid of prion protein have cognitive deficits that are rescued by reconstitution of PrP in neurons. Neurobiol Dis. 2005;19:255–65. doi: 10.1016/j.nbd.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 153.Le Pichon CE, Valley MT, Polymenidou M, Chesler AT, Sagdullaev BT, Aguzzi A, Firestein S. Olfactory behavior and physiology are disrupted in prion protein knockout mice. Nat Neurosci. 2009;12:60–9. doi: 10.1038/nn.2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Bremer J, Baumann F, Tiberi C, Wessig C, Fischer H, Schwarz P, Steele AD, Toyka KV, Nave KA, Weis J, Aguzzi A. Axonal prion protein is required for peripheral myelin maintenance. Nat Neurosci. 2010;13:310–18. doi: 10.1038/nn.2483. [DOI] [PubMed] [Google Scholar]
- 155.Radovanovic I, Braun N, Giger OT, Mertz K, Miele G, Prinz M, Navarro B, Aguzzi A. Truncated prion protein and Doppel are myelinotoxic in the absence of oligodendrocytic PrPC. J Neurosci. 2005;25:4879–88. doi: 10.1523/JNEUROSCI.0328-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Baumann F, Tolnay M, Brabeck C, Pahnke J, Kloz U, Niemann HH, Heikenwalder M, Rulicke T, Burkle A, Aguzzi A. Lethal recessive myelin toxicity of prion protein lacking its central domain. EMBO J. 2007;26:538–47. doi: 10.1038/sj.emboj.7601510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Li A, Christensen HM, Stewart LR, Roth KA, Chiesa R, Harris DA. Neonatal lethality in transgenic mice expressing prion protein with a deletion of residues 105-125. EMBO J. 2007;26:548–58. doi: 10.1038/sj.emboj.7601507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Trevitt CR, Collinge J. A systematic review of prion therapeutics in experimental models. Brain. 2006;129:2241–65. doi: 10.1093/brain/awl150. [DOI] [PubMed] [Google Scholar]
- 159.Nicoll AJ, Collinge J. Preventing prion pathogenicity by targeting the cellular prion protein. Infect Disord Drug Targets. 2009;9:48–57. doi: 10.2174/1871526510909010048. [DOI] [PubMed] [Google Scholar]
- 160.Ghaemmaghami S, Ahn M, Lessard P, Giles K, Legname G, DeArmond SJ, Prusiner SB. Continuous quinacrine treatment results in the formation of drug-resistant prions. PLoS Pathog. 2009;5:e1000673. doi: 10.1371/journal.ppat.1000673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Telling GC, Scott M, Hsiao KK, Foster D, Yang S-L, Torchia M, Sidle KCL, Collinge J, DeArmond SJ, Prusiner SB. Transmission of Creutzfeldt-Jakob disease from humans to transgenic mice expressing chimeric human-mouse prion protein. Proc Natl Acad Sci U S A. 1994;91:9936–40. doi: 10.1073/pnas.91.21.9936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Scott M, Groth D, Foster D, Torchia M, Yang SL, DeArmond SJ, Prusiner SB. Propagation of prions with artificial properties in transgenic mice expressing chimeric PrP genes. Cell. 1993;73:979–88. doi: 10.1016/0092-8674(93)90275-u. [DOI] [PubMed] [Google Scholar]
- 163.Mastrianni JA, Capellari S, Telling GC, Han D, Bosque P, Prusiner SB, DeArmond SJ. Inherited prion disease caused by the V210I mutation – Transmission to transgenic mice. Neurology. 2001;57:2198–205. doi: 10.1212/wnl.57.12.2198. [DOI] [PubMed] [Google Scholar]
- 164.Korth C, Kaneko K, Groth D, Heye N, Telling G, Mastrianni J, Parchi P, Gambetti P, Will R, Ironside J, Heinrich C, Tremblay P, DeArmond SJ, Prusiner SB. Abbreviated incubation times for human prions in mice expressing a chimeric mouse-human prion protein transgene. Proc Natl Acad Sci U S A. 2003;100:4784–9. doi: 10.1073/pnas.2627989100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Manson JC, Jamieson E, Baybutt H, Tuzi NL, Barron R, McConnell I, Somerville R, Ironside J, Will R, Sy MS, Melton DW, Hope J, Bostock C. A single amino acid alteration (101L) introduced into murine PrP dramatically alters incubation time of transmissible spongiform encephalopathy. EMBO J. 1999;18:6855–64. doi: 10.1093/emboj/18.23.6855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Asano M, Mohri S, Ironside JW, Ito M, Tamaoki N, Kitamoto T. vCJD prion acquires altered virulence through trans-species infection. Biochem Biophys Res Commun. 2006;342:293–9. doi: 10.1016/j.bbrc.2006.01.149. [DOI] [PubMed] [Google Scholar]
- 167.Bishop MT, Will RG, Manson JC. Defining sporadic Creutzfeldt-Jakob disease strains and their transmission properties. Proc Natl Acad Sci U S A. 2010;107:12005–10. doi: 10.1073/pnas.1004688107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Whittington MA, Sidle KCL, Gowland I, Meads J, Hill AF, Palmer MS, Jefferys JGR, Collinge J. Rescue of neurophysiological phenotype seen in PrP null mice by transgene encoding human prion protein. Nat Genet. 1995;9:197–201. doi: 10.1038/ng0295-197. [DOI] [PubMed] [Google Scholar]
- 169.Kobayashi A, Asano M, Mohri S, Kitamoto T. Cross-sequence transmission of sporadic Creutzfeldt-Jakob disease creates a new prion strain. J Biol Chem. 2007;282:30022–8. doi: 10.1074/jbc.M704597200. [DOI] [PubMed] [Google Scholar]
- 170.Beringue V, Le Dur A, Tixador P, Reine F, Lepourry L, Perret-Liaudet A, Haik S, Vilotte JL, Fontes M, Laude H. Prominent and persistent extraneural infection in human PrP transgenic mice infected with variant CJD. PLoS ONE. 2008;3:e1419. doi: 10.1371/journal.pone.0001419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Asante E, Linehan J, Gowland I, Joiner S, Fox K, Cooper S, Osiguwa O, Gorry M, Welch J, Houghton R, Desbruslais M, Brandner S, Wadsworth JD, Collinge J. Dissociation of pathological and molecular phenotype of variant Creutzfeldt-Jakob disease in transgenic human prion protein 129 heterozygous mice. Proc Natl Acad Sci U S A. 2006;103:10759–64. doi: 10.1073/pnas.0604292103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Hizume M, Kobayashi A, Teruya K, Ohashi H, Ironside JW, Mohri S, Kitamoto T. Human prion protein (PrP) 219K is converted to PrPSc but shows heterozygous inhibition in variant Creutzfeldt-Jakob disease infection. J Biol Chem. 2009;284:3603–9. doi: 10.1074/jbc.M809254200. [DOI] [PubMed] [Google Scholar]
- 173.Mead S, Whitfield J, Poulter M, Shah P, Uphill J, Beck J, Campbell T, Al Dujaily H, Hummerich H, Alpers MP, Collinge J. Genetic susceptibility, evolution and the kuru epidemic. Philos Trans R Soc Lond B Biol Sci. 2008;363:3741–6. doi: 10.1098/rstb.2008.0087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Hosszu LL, Jackson GS, Trevitt C, Jones S, Batchelor M, Bhelt D, Prodromidou K, Clarke A, Waltho JP, Collinge J. The residue 129 polymorphism in human prion protein does not confer susceptibility to CJD by altering the structure or global stability of PrPC. J Biol Chem. 2004;279:28515–21. doi: 10.1074/jbc.M313762200. [DOI] [PubMed] [Google Scholar]
- 175.Riek R, Wider G, Billeter M, Hornemann S, Glockshuber R, Wuthrich K. Prion protein NMR structure and familial human spongiform encephalopathies. Proc Natl Acad Sci U S A. 1998;95:11667–72. doi: 10.1073/pnas.95.20.11667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Liemann S, Glockshuber R. Influence of amino acid substitutions related to inherited human prion diseases on the thermodynamic stability of the cellular prion protein. Biochemistry. 1999;38:3258–67. doi: 10.1021/bi982714g. [DOI] [PubMed] [Google Scholar]
- 177.Hill AF, Joiner S, Beck J, Campbell TA, Dickinson A, Poulter M, Wadsworth JD, Collinge J. Distinct glycoform ratios of protease resistant prion protein associated with PRNP point mutations. Brain. 2006;129:676–85. doi: 10.1093/brain/awl013. [DOI] [PubMed] [Google Scholar]
- 178.Ashok A, Hegde RS. Selective processing and metabolism of disease-causing mutant prion proteins. PLoS Pathog. 2009;5:e1000479. doi: 10.1371/journal.ppat.1000479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Hornemann S, Von Schroetter C, Damberger FF, Wuthrich K. Prion protein-detergent micelle interactions studied by NMR in solution. J Biol Chem. 2009;284:22713–21. doi: 10.1074/jbc.M109.000430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.van der Kamp MW, Daggett V. The consequences of pathogenic mutations to the human prion protein. Protein Eng Des Sel. 2009;22:461–8. doi: 10.1093/protein/gzp039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Parchi P, Chen SG, Brown P, Zou W, Capellari S, Budka H, Hainfellner J, Reyes PF, Golden GT, Hauw JJ, Gajdusek DC, Gambetti P. Different patterns of truncated prion protein fragments correlate with distinct phenotypes in P102L Gerstmann-Sträussler-Scheinker disease. Proc Natl Acad Sci U S A. 1998;95:8322–7. doi: 10.1073/pnas.95.14.8322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Furukawa H, Doh-ura K, Kikuchi H, Tateishi J, Iwaki T. A comparative study of abnormal prion protein isoforms between Gerstmann-Sträussler-Scheinker syndrome and Creutzfeldt-Jakob disease. J Neurol Sci. 1998;158:71–5. doi: 10.1016/s0022-510x(98)00096-3. [DOI] [PubMed] [Google Scholar]
- 183.Cardone F, Liu QG, Petraroli R, Ladogana A, D'Alessandro M, Arpino C, Di Bari M, Macchi G, Pocchiari M. Prion protein glycotype analysis in familial and sporadic Creutzfeldt-Jakob disease patients. Brain Res Bull. 1999;49:429–33. doi: 10.1016/s0361-9230(99)00077-5. [DOI] [PubMed] [Google Scholar]
- 184.Piccardo P, Liepnieks JJ, William A, Dlouhy SR, Farlow MR, Young K, Nochlin D, Bird TD, Nixon RR, Ball MJ, DeCarli C, Bugiani O, Tagliavini F, Benson MD, Ghetti B. Prion proteins with different conformations accumulate in Geustmann-Straussler-Scheinker disease caused by A117V and F198S mutations. Am J Pathol. 2001;158:2201–7. doi: 10.1016/S0002-9440(10)64692-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Tagliavini F, Lievens PMJ, Tranchant C, Warter JM, Mohr M, Giaccone G, Perini F, Rossi G, Salmona M, Piccardo P, Ghetti B, Beavis RC, Bugiani O, Frangione B, Prelli F. A 7-kDa prion protein (PrP) fragment, an integral component of the PrP region required for infectivity, is the major amyloid protein in Gerstmann-Straussler-Scheinker disease A117V. J Biol Chem. 2001;276:6009–15. doi: 10.1074/jbc.M007062200. [DOI] [PubMed] [Google Scholar]
- 186.Asante EA, Gowland I, Grimshaw A, Linehan JM, Smidak M, Houghton R, Osiguwa O, Tomlinson A, Joiner S, Brandner S, Wadsworth JD, Collinge J. Absence of spontaneous disease and comparative prion susceptibility of transgenic mice expressing mutant human prion proteins. J Gen Virol. 2009;90:546–58. doi: 10.1099/vir.0.007930-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Gabizon R, Telling G, Meiner Z, Halimi M, Kahana I, Prusiner SB. Insoluble wild-type and protease-resistant mutant prion protein in brains of patients with inherited prion disease. Nat Med. 1996;2:59–64. doi: 10.1038/nm0196-59. [DOI] [PubMed] [Google Scholar]
- 188.Silvestrini MC, Cardone F, Maras B, Pucci P, Barra D, Brunori M, Pocchiari M. Identification of the prion protein allotypes which accumulate in the brain of sporadic and familial Creutzfeldt-Jakob disease patients. Nat Med. 1997;3:521–5. doi: 10.1038/nm0597-521. [DOI] [PubMed] [Google Scholar]
- 189.Chen SG, Parchi P, Brown P, Capellari S, Zou WQ, Cochran EJ, Vnencak-Jones CL, Julien J, Vital C, Mikol J, Lugaresi E, Autilio-Gambetti L, Gambetti P. Allelic origin of the abnormal prion protein isoform in familial prion diseases. Nat Med. 1997;3:1009–15. doi: 10.1038/nm0997-1009. [DOI] [PubMed] [Google Scholar]
- 190.Tateishi J, Kitamoto T. Inherited prion diseases and transmission to rodents. Brain Pathol. 1995;5:53–9. doi: 10.1111/j.1750-3639.1995.tb00577.x. [DOI] [PubMed] [Google Scholar]
- 191.Tateishi J, Kitamoto T, Hoque MZ, Furukawa H. Experimental transmission of Creutzfeldt-Jakob disease and related diseases to rodents. Neurology. 1996;46:532–7. doi: 10.1212/wnl.46.2.532. [DOI] [PubMed] [Google Scholar]
- 192.Barron RM, Thomson V, Jamieson E, Melton DW, Ironside J, Will R, Manson JC. Changing a single amino acid in the N-terminus of murine PrP alters TSE incubation time across three species barriers. EMBO J. 2001;20:5070–8. doi: 10.1093/emboj/20.18.5070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Hegde RS, Tremblay P, Groth D, DeArmond S, Prusiner SB, Lingappa VR. Transmissible and genetic prion diseases share a common pathway of neurodegeneration. Nature. 1999;402:822–6. doi: 10.1038/45574. [DOI] [PubMed] [Google Scholar]
- 194.Chiesa R, Piccardo P, Quaglio E, Drisaldi B, Si-Hoe SL, Takao M, Ghetti B, Harris DA. Molecular distinction between pathogenic and infectious properties of the prion protein. J Virol. 2003;77:7611–22. doi: 10.1128/JVI.77.13.7611-7622.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Chakrabarti O, Hegde RS. Functional depletion of mahogunin by cytosolically exposed prion protein contributes to neurodegeneration. Cell. 2009;137:1136–147. doi: 10.1016/j.cell.2009.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Hsiao KK, Scott M, Foster D, Groth DF, DeArmond SJ, Prusiner SB. Spontaneous neurodegeneration in transgenic mice with mutant prion protein. Science. 1990;250:1587–90. doi: 10.1126/science.1980379. [DOI] [PubMed] [Google Scholar]
- 197.Chiesa R, Piccardo P, Ghetti B, Harris DA. Neurological illness in transgenic mice expressing a prion protein with an insertional mutation. Neuron. 1998;21:1339–51. doi: 10.1016/s0896-6273(00)80653-4. [DOI] [PubMed] [Google Scholar]
- 198.Chiesa R, Pestronk A, Schmidt RE, Tourtellotte WG, Ghetti B, Piccardo P, Harris DA. Primary myopathy and accumulation of PrPSc-like molecules in peripheral tissues of transgenic mice expressing a prion protein insertional mutation. Neurobiol Dis. 2001;8:279–88. doi: 10.1006/nbdi.2001.0400. [DOI] [PubMed] [Google Scholar]
- 199.Harris DA, Chiesa R, Drisaldi B, Quaglio E, Migheli A, Piccardo P, Ghetti B. A murine model of a familial prion disease. Clin Lab Med. 2003;23:175–86. doi: 10.1016/s0272-2712(02)00069-0. [DOI] [PubMed] [Google Scholar]
- 200.Muramoto T, DeArmond S, Scott M, Telling GC, Cohen FE, Prusiner SB. Heritable disorder reseming neuronal storage disease in mice expressing prion protein with deletion of an alpha-helix. Nat Med. 1997;3:750–5. doi: 10.1038/nm0797-750. [DOI] [PubMed] [Google Scholar]
- 201.Dossena S, Imeri L, Mangieri M, Garofoli A, Ferrari L, Senatore A, Restelli E, Balducci C, Fiordaliso F, Salio M, Bianchi S, Fioriti L, Morbin M, Pincherle A, Marcon G, Villani F, Carli M, Tagliavini F, Forloni G, Chiesa R. Mutant prion protein expression causes motor and memory deficits and abnormal sleep patterns in a transgenic mouse model. Neuron. 2008;60:598–609. doi: 10.1016/j.neuron.2008.09.008. [DOI] [PubMed] [Google Scholar]
- 202.Jackson WS, Borkowski AW, Faas H, Steele AD, King OD, Watson N, Jasanoff A, Lindquist S. Spontaneous generation of prion infectivity in fatal familial insomnia knockin mice. Neuron. 2009;63:438–50. doi: 10.1016/j.neuron.2009.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Hegde RS, Mastrianni JA, Scott MR, DeFea KA, Tremblay P, Torchia M, DeArmond SJ, Prusiner SB, Lingappa VR. A transmembrane from of the prion protein in neurodegenerative disease. Science. 1998;279:827–34. doi: 10.1126/science.279.5352.827. [DOI] [PubMed] [Google Scholar]
- 204.Hsiao KK, Groth D, Scott M, Yang S-L, Serban H, Rapp D, Foster D, Torchia M, DeArmond SJ, Prusiner SB. Serial transmission in rodents of neurodegeneration from transgenic mice expressing mutant prion protein. Proc Natl Acad Sci U S A. 1994;91:9126–130. doi: 10.1073/pnas.91.19.9126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Telling GC, Haga T, Torchia M, Tremblay P, DeArmond SJ, Prusiner SB. Interactions between wild-type and mutant prion proteins modulate neurodegeneration transgenic mice. Genes Dev. 1996;10:1736–50. doi: 10.1101/gad.10.14.1736. [DOI] [PubMed] [Google Scholar]
- 206.Westaway D, DeArmond SJ, Cayetano-Canlas J, Groth D, Foster D, Yang S-L, Torchia M, Carlson GA, Degeneration PSB. of skeletal muscle, peripheral nerves and the central nervous system in transgenic mice overexpressing wild-type prion proteins. Cell. 1994;76:117–29. doi: 10.1016/0092-8674(94)90177-5. [DOI] [PubMed] [Google Scholar]
- 207.Chiesa R, Piccardo P, Biasini E, Ghetti B, Harris DA. Aggregated, wild-type prion protein causes neurological dysfunction and synaptic abnormalities. J Neurosci. 2008;28:13258–67. doi: 10.1523/JNEUROSCI.3109-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Colby DW, Giles K, Legname G, Wille H, Baskakov IV, DeArmond SJ, Prusiner SB. Design and construction of diverse mammalian prion strains. Proc Natl Acad Sci U S A. 2009;106:20417–22. doi: 10.1073/pnas.0910350106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Butefisch CM, Gambetti P, Cervenakova L, Park KY, Hallett M, Goldfarb LG. Inherited prion encephalopathy associated with the novel PRNP H187R mutation: a clinical study. Neurology. 2000;55:517–22. doi: 10.1212/wnl.55.4.517. [DOI] [PubMed] [Google Scholar]
- 210.Lysek DA, Nivon LG, Wuthrich K. Amino acid sequence of the Felis catus prion protein. Gene. 2004;341:249–53. doi: 10.1016/j.gene.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 211.Colucci M, Moleres FJ, Xie ZL, Ray-Chaudhury A, Gutti S, Butefisch CM, Cervenakova L, Wang W, Goldfarb LG, Kong Q, Ghetti B, Chen SG, Gambetti P. Gerstmann-Straussler-Scheinker: a new phenotype with ‘curly’ PrP deposits. J Neuropathol Exp Neurol. 2006;65:642–51. doi: 10.1097/01.jnen.0000228198.81797.4d. [DOI] [PubMed] [Google Scholar]
- 212.Wildegger G, Liemann S, Glockshuber R. Extremely rapid folding of the C-terminal domain of the prion protein without kinetic intermediates. Nat Struct Biol. 1999;6:550–3. doi: 10.1038/9323. [DOI] [PubMed] [Google Scholar]
- 213.Hart T, Hosszu LL, Trevitt CR, Jackson GS, Waltho JP, Collinge J, Clarke AR. Folding kinetics of the human prion protein probed by temperature jump. Proc Natl Acad Sci U S A. 2009;106:5651–6. doi: 10.1073/pnas.0811457106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Asante E, Li YG, Gowland I, Jefferys JG, Collinge J. Pathogenic human prion protein rescues PrP null phenotype in transgenic mice. Neuroscience Lett. 2004;360:33–6. doi: 10.1016/j.neulet.2004.01.049. [DOI] [PubMed] [Google Scholar]
- 215.Sigurdson CJ, Nilsson KP, Hornemann S, Heikenwalder M, Manco G, Schwarz P, Ott D, Rulicke T, Liberski PP, Julius C, Falsig J, Stitz L, Wuthrich K, Aguzzi A. De novo generation of a transmissible spongiform encephalopathy by mouse transgenesis. Proc Natl Acad Sci U S A. 2009;106:304–9. doi: 10.1073/pnas.0810680105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Li J, Browning S, Mahal SP, Oelschlegel AM, Weissmann C. Darwinian evolution of prions in cell culture. Science. 2010;327:869–72. doi: 10.1126/science.1183218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Brown P, Cathala F, Raubertas RF, Gajdusek DC, Castaigne P. The epidemiology of Creutzfeldt-Jakob disease: conclusion of a 15-year investigation in France and review of the world literature. Neurology. 1987;37:895–904. doi: 10.1212/wnl.37.6.895. [DOI] [PubMed] [Google Scholar]
- 218.Mead S, Webb TE, Campbell TA, Beck J, Linehan J, Rutherfoord S, Joiner S, Wadsworth JD, Heckmann J, Wroe S, Doey L, King A, Collinge J. Inherited prion disease with 5-OPRI: phenotype modification by repeat length and codon 129. Neurology. 2007;69:730–8. doi: 10.1212/01.wnl.0000267642.41594.9d. [DOI] [PubMed] [Google Scholar]
- 219.Collins S, Law MG, Fletcher A, Boyd A, Kaldor J, Masters CL. Surgical treatment and risk of sporadic Creutzfeldt-Jakob disease: a case-control study. Lancet. 1999;353:693–7. doi: 10.1016/s0140-6736(98)08138-0. [DOI] [PubMed] [Google Scholar]
- 220.Mahillo-Fernandez I, Pedro-Cuesta J, Bleda MJ, Cruz M, Molbak K, Laursen H, Falkenhorst G, Martinez-Martin P, Siden A. Surgery and risk of sporadic Creutzfeldt-Jakob disease in Denmark and Sweden: registry-based case-control studies. Neuroepidemiology. 2008;31:229–40. doi: 10.1159/000163097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Pedro-Cuesta J, Mahillo-Fernandez I, Rabano A, Calero M, Cruz M, Siden A, Laursen H, Falkenhorst G, Molbak K. Nosocomial transmission of sporadic Creutzfeldt-Jakob disease: results from a risk-based assessment of surgical interventions. J Neurol Neurosurg Psychiatry. 2010 doi: 10.1136/jnnp.2009.188425. E-pub Jun 14; doi:10.1136/jnnp.2009.188425 (Epub ahead of print) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Collinge J, Gorham M, Hudson F, Kennedy A, Keogh G, Pal S, Rossor M, Rudge P, Siddique D, Spyer M, Thomas D, Walker S, Webb T, Wroe S, Darbyshire J. Safety and efficacy of quinacrine in human prion disease (PRION-1 study): a patient-preference trial. Lancet Neurology. 2009;2009:334–44. doi: 10.1016/S1474-4422(09)70049-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Lloyd S, Uphill JB, Targonski PV, Fisher E, Collinge J. Identification of genetic loci affecting mouse-adapted bovine spongiform encephalopathy incubation time in mice. Neurogenetics. 2002;4:77–81. doi: 10.1007/s10048-002-0133-9. [DOI] [PubMed] [Google Scholar]
- 224.Moreno CR, Lantier F, Lantier I, Sarradin P, Elsen JM. Detection of new quantitative trait loci for susceptibility to transmissible spongiform encephalopathies in mice. Genetics. 2003;165:2085–91. doi: 10.1093/genetics/165.4.2085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Brown P, Preece MA, Will RG. ‘Friendly fire’ in medicine: hormones, homografts, and Creutzfeldt-Jakob disease. Lancet. 1992;340:24–7. doi: 10.1016/0140-6736(92)92431-e. [DOI] [PubMed] [Google Scholar]
- 226.Alpers MP. Epidemiology and clinical aspects of kuru. In: Prusiner SB, McKinley MP, editors. Prions: Novel Infectious Pathogens Causing Scrapie and Creutzfeldt-Jakob Disease. San Diego: Academic Press; 1987. pp. 451–65. [Google Scholar]
- 227.Alpers MP. The epidemiology of kuru: monitoring the epidemic from its peak to its end. Philos Trans R Soc Lond B Biol Sci. 2008;363:3707–13. doi: 10.1098/rstb.2008.0071. [DOI] [PMC free article] [PubMed] [Google Scholar]