

Abstract
Mouse B2 RNA represses RNA polymerase II (Pol II) transcription during the cellular heat shock response. B2 RNA binds Pol II, enters complexes at promoters, and keeps the polymerase from engaging the DNA. Here we show that phosphorylation of Ser5 residues in the Pol II carboxy terminal domain (CTD) decreases after heat shock at the promoter of the repressed actin gene in mouse cells, despite the continued presence of Cdk7 and cyclin H. Biochemical assays revealed that B2 RNA, when present with Pol II in promoter-bound complexes, specifically represses CTD phosphorylation by TFIIH.
Key words: RNA polymerase II, carboxy terminal domain, phosphorylation, TFIIH, transcription, B2 RNA, repression, heat shock, non-coding RNA
Transcription of mRNA genes by Pol II is a tightly regulated and highly coordinated event that requires numerous factors working in conjunction to control gene-specific transcription with spatial and temporal specificity. Pol II transcription is aided by a set of general factors that function at core promoters, including TFIIA, TFIIB, TFIID, TFIIE, TFIIF, TFIIH and mediator.1 These factors, along with promoter-specific transcriptional regulator proteins and chromatin-modifying factors, coordinate the assembly of pre-initiation complexes at the promoters of genes. More recently, a model has emerged in which Pol II transcription at many genes is also tightly regulated at steps that occur subsequent to the recruitment of Pol II and general factors to promoters, such as promoter proximal pausing.2 Central to regulating post-initiation events is phosphorylation of Ser2, Ser5 and Ser7 residues in the heptapeptide repeats of the CTD of the largest subunit of Pol II (Rpb1).3 Of the general transcription factors, TFIIH, a 10 subunit complex that contains both kinase and helicase activities, can phosphorylate Ser5 and Ser7 at promoters.1,4–7 The kinase activity is contained within the CAK submodule, which consists of the three subunits Cdk7, cyclin H and Mat1. The helicase subunits, ERCC2 and ERCC3, are thought to be critical for promoter melting, transcriptional initiation and promoter escape.1
Transcription is not only orchestrated by a multitude of protein factors and post-translational modifications, but also by non-coding RNAs (ncRNAs), which are an emerging class of regulators of Pol II transcription.8 We found that B2 RNA, a ncRNA in mouse cells, functions as a transcriptional regulator of Pol II.9 Transcribed by RNA polymerase III from short interspersed elements (SINEs), B2 RNA is upregulated upon heat shock and other cellular stresses.10 Also upregulated is the other predominant mouse SINE RNA, B1 RNA. B2 RNA binds directly to Pol II and potently represses transcription; by contrast, B1 RNA can bind tightly to Pol II but is not capable of transcriptional repression in vitro.11,12
We found that in heat-shocked mouse cells, B2 RNA mediates the transcriptional repression of genes such as actin.9 Biochemical experiments to investigate the mechanism of repression showed that B2 RNA, via its interaction with Pol II, assembles into complexes at the promoters of genes and renders the complexes transcriptionally inactive.11 Within these inhibited complexes, B2 RNA disrupts the contacts between Pol II and the promoter DNA.13 Consistent with this in vitro model for repression, cell-based experiments have co-localized Pol II and B2 RNA at the promoter of the repressed actin gene, specifically after heat shock in mouse cells.12
We wondered what effect heat shock and B2 RNA would have on phosphorylation of the CTD of Pol II. We prepared nuclei from NIH 3T3 cells and performed western blots with four different antibodies that recognize different epitopes of the Rpb1 subunit of Pol II (Fig. 1). An antibody that recognizes the N-terminus of Rpb1 showed upper and lower bands corresponding to a hyper-phosphorylated CTD and hypophosphorylated CTD, respectively. Upon heat shock, there was a decrease in the hyperphosphorylated form of Rpb1. A similar decrease was seen using the 8WG16 antibody, which recognizes the CTD of Rpb1. An antibody that specifically recognizes phosphorylated Ser5 (Ser5-P) showed that this form of Rpb1 was substantially reduced after heat shock. The same was true using an antibody that specifically recognizes phosphorylated Ser2 (Ser2-P). Because B2 RNA represses transcription at an early point in the reaction and Ser2-P is thought to occur after Ser5-P,3 we focused on the decrease in Ser5-P that occurs upon heat shock. The decrease in Ser5 phosphorylation could be due to a decrease in levels of TFIIH, or the CAK submodule. We found, however, that nuclear levels of the ERCC3, Cdk7 and cyclin H subunits of TFIIH did not decrease upon heat shock; also, Cdk7 reproducibly exhibited a slight increase upon heat shock (Fig. 1).
Figure 1.
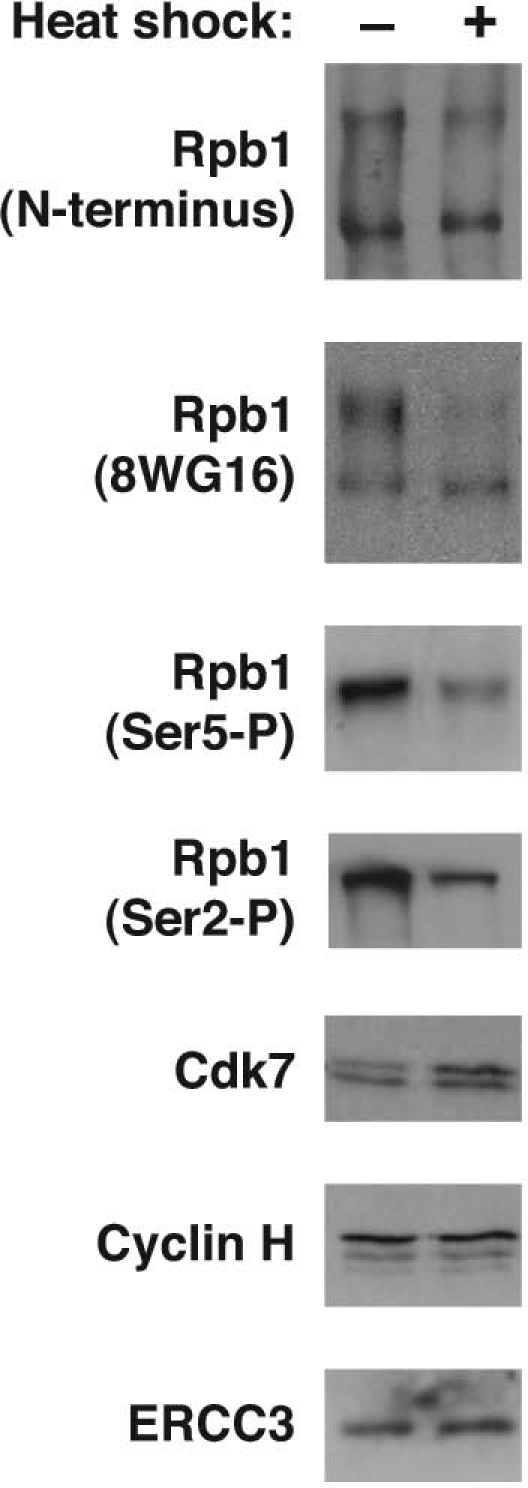
The level of Ser5-P and Ser2-P in nuclei decreases upon heat shock. Western blots were performed to detect total Rpb1, Ser5-P, Ser2-P and three subunits of TFIIH in nuclei isolated from NIH 3T3 cells. Representative data are shown.
Since Ser5-P occurs at active promoters, it seemed possible that the decrease in Ser5-P upon heat shock would occur at promoters of genes repressed upon heat shock. To test this we performed ChIP assays to detect total Pol II and Ser5-P before and after heat shock at the actin promoter, which we have previously shown to be occupied by B2 RNA after heat shock. As shown in Figure 2A, the occupancy of Ser5-P Pol II decreased substantially more than that of total Pol II after heat shock. In fact, the decrease of Ser5-P at the promoter was nearly equivalent to the decrease in Pol II occupancy in a downstream region of the actin gene, which reflects the level of transcriptional repression observed at this gene in response to heat shock.
Figure 2.

After heat shock, levels of Ser5-P substantially decrease at the actin promoter, but levels of Cdk7 and cyclin H remain relatively stable. (A) Phosphorylation of Ser5 is significantly decreased at the actin promoter after heat shock. ChIP assays were performed with antibodies against the N-terminus of Rpb1 and Ser5-P at the actin promoter, as well as the N-terminus of Rpb1 in a downstream region of the actin gene. (B) Levels of general transcription factors, including the Cdk7 and cyclin H subunits of TFIIH, do not substantially decrease at the actin promoter after heat shock. ChIP assays were performed with antibodies against the indicated factors.
The decrease in Ser5-P at the actin promoter could be due to a decrease in the occupancy of TFIIH, specifically the Cdk7 and cyclin H subunits. To test this we performed ChIP assays with antibodies against subunits of the general transcription machinery. As shown in Figure 2B, Cdk7 and cyclin H occupancy only decreased 2–3-fold upon heat shock, which was similar to the decrease in TBP, TFIIAγ, and the ERCC3 subunit of TFIIH. Hence, the 18-fold decrease in Ser5-P at the actin promoter is not due to a decrease in Cdk7 and cyclin H. Our data is most consistent with a model in which the kinase activity of TFIIH is inhibited at the actin promoter after heat shock.
Given that repression of actin transcription upon heat shock is due to promoter-bound B2 RNA,9,12 it seemed possible that B2 RNA also represses the kinase activity of Cdk7/cyclin H. To test this we performed in vitro kinase assays by adding TFIIE/TFIIH to purified Pol II in the presence and absence of B2 RNA or B1 RNA as a control. As shown in Figure 3A, B2 RNA did not repress the phosphorylation of Rpb1 by TFIIH. Rather, there was an increase in Rpb1 phosphorylation in the presence of B2 RNA; however, this was not specific to B2 RNA because B1 RNA caused a similar increase. Neither B2 RNA nor B1 RNA affected phosphorylation of the CTD by ERK2, a mitogen-activated protein kinase that also phosphorylates Ser5 on the CTD.14
Figure 3.

B2 RNA inhibits TFIIH phosphorylation of the CTD only when the ncRNA is incorporated with Pol II into complexes on promoter DNA. (A) B2 RNA and B1 RNA stimulate phosphorylation of the CTD by TFIIH in the absence of promoter DNA. Phosphorylation of the CTD by TFIIE/TFIIH or ERK2 as a specificity control, is shown in the absence or presence B2 RNA or B1 RNA. (B) B2 RNA, but not B1 RNA, specifically inhibits phosphorylation of the CTD by TFIIH in the presence of the general transcription machinery and promoter DNA. Pre-initiation complexes were formed in the absence or presence of B2 RNA or B1 RNA. TFIIE and TFIIH were then added and phosphorylation of the CTD was monitored. ERK2 was used in place of TFIIE/TFIIH in separate assays. (C) Pol II and TFIIH levels on promoter DNA do not significantly change in the presence of B2 RNA. Complexes were assembled on immobilized promoter DNA in the presence and absence of B2 RNA. After washing, Rpb1 and ERCC3 subunits bound to DNA were probed by western blotting.
To further explore the effect of B2 RNA and B1 RNA on CTD phosphorylation we assembled minimal pre-initiation complexes from TBP, TFIIB, TFIIF, Pol II and promoter DNA in the absence or presence of the ncRNAs. TFIIE and TFIIH were then added along with [γ-32P]ATP. Surprisingly, in this context, B2 RNA substantially reduced CTD phosphorylation by TFIIH, whereas B1 RNA had no effect (Fig. 3B). Phosphorylation of the CTD in pre-initiation complexes by ERK2 was not inhibited by B2 RNA; therefore, inhibition is specific for the TFIIH kinase. Moreover, levels of promoter-associated Pol II and TFIIH were approximately the same in immobilized pre-initiation complexes compared to immobilized inhibited complexes containing B2 RNA (Fig. 3C). These results show that B2 RNA represses CTD phosphorylation by TFIIH, but only when Pol II is in transcriptionally repressed complexes on promoter DNA.
To repress transcription, B2 RNA must build into complexes at promoters along with Pol II: if B2 RNA is added to pre-formed pre-initiation complexes, it is unable to repress transcription.11 To determine whether the mechanism by which B2 RNA represses CTD phosphorylation is related to that of transcriptional repression we performed order-of-addition experiments (Fig. 4A). Complexes on promoter DNA were formed in the absence or presence of B2 RNA, or B1 RNA as a control. TFIIE and TFIIH were added later, followed by ATP. When B2 RNA was present during complex formation, it blocked phosphorylation of the CTD (compare lane 2 to lane 1 in Fig. 4A); however, when B2 RNA was added after pre-initiation complex formation, it was unable to repress phosphorylation of the CTD, even though TFIIE and TFIIH were added to reactions after B2 RNA (Fig. 4A and lane 4). Moreover, when B2 RNA was incorporated into complexes and then the complexes were treated with an RNase, CTD phosphorylation was no longer repressed. B1 RNA did not repress CTD phosphorylation by TFIIH under any of the tested conditions. These data show that B2 RNA must enter complexes with Pol II at promoters in order to repress phosphorylation of the CTD by TFIIH and that this repression can be reversed by RNase treatment, which are properties shared with the mechanism by which B2 RNA represses transcription.
Figure 4.

To repress phosphorylation of the CTD by TFIIH, B2 RNA must build into complexes on promoter DNA along with Pol II. (A) The time course indicates the points at which general transcription factors, Pol II and B2 RNA (or B1 RNA) were added to reactions. B2 RNA was added either before (point 1) or after (point 2) pre-initiation complex formation. Where indicated, complexes were treated with RNase 1 (10 U, NEB) prior to adding TFIIE and TFIIH. (B) Model depicting that the presence of B2 RNA at a promoter prevents phosphorylation of Ser5 residues on the CTD by TFIIH.
Our findings reveal an additional means by which B2 RNA controls steps in the transcription reaction: it blocks phosphorylation of the CTD by TFIIH. Previously, we had found that B2 RNA incorporates into complexes at the promoters of genes and represses transcription by keeping the polymerase from engaging promoter DNA.13 Moreover, in heat-shocked cells we observed co-localization of B2 RNA and Pol II at the actin promoter.12 Here we found that upon heat shock, Ser5 phosphorylation sharply decreases at the actin promoter, while the levels of TFIIH and total Pol II decrease to a much lower extent. In vitro experiments showed that B2 RNA, when assembled into complexes at promoters, inhibits Ser5 phosphorylation by TFIIH. We propose a model in which the presence of B2 RNA at the promoters of repressed genes in response to heat shock prevents Ser5 phosphorylation, thereby providing an additional level at which this ncRNA represses transcription (Fig. 4B).
The repression of Ser5 phosphorylation by B2 RNA mirrors features of the previously documented mechanism of transcriptional repression by B2 RNA.13 Namely, inhibition of Ser5 phosphorylation by TFIIH only occurs when B2 RNA is added to reactions prior to the formation of closed complexes, can be reversed by RNase degradation of B2 RNA, and does not occur with B1 RNA. These observations support a model in which repression of Ser5 phosphorylation is due to the B2 RNA/Pol II interaction and not due to B2 RNA targeting TFIIH in addition to Pol II. If the latter were true, B2 RNA would likely inhibit TFIIH kinase activity in the absence of forming complexes on promoter DNA and also when added to pre-initiation complexes, neither of which was observed. In fact, we observed that B2 RNA and B1 RNA both stimulated phosphorylation of the CTD by TFIIE/TFIIH in the absence of other general factors and promoter DNA. Given that U1 RNA has previously been shown to stimulate Cdk7/cyclin H kinase activity,15 it appears that the TFIIH kinase can be stimulated by multiple different structured RNAs.
Our studies provide additional insight into how B2 RNA represses CTD phosphorylation by TFIIH. First, since our in vitro kinase assays with complexes on promoter DNA were performed under conditions in which transcription could not initiate, phosphorylation of the CTD by TFIIH can occur in pre-initiation complexes and inhibition of this phosphorylation by B2 RNA occurs prior to transcription initiation. Second, since ERK2 was able to phosphorylate the CTD in promoter-bound complexes, B2 RNA does not mask the CTD in this context. Instead, the unique conformation of inhibited complexes induced by B2 RNA could prevent Cdk7/cyclin H from interacting with the CTD. B2 RNA blocks the formation of closed complexes by disrupting polymerase-promoter contacts and, hence, also inhibits the melting of promoter DNA to form open complexes.13 Given our data, it is conceivable that TFIIH phosphorylation of Ser5 is coupled to Pol II engaging the promoter DNA or to promoter melting, which is intriguing given the role of the TFIIH helicase in forming open complexes. We also believe that the lack of polymerase-promoter contacts in inhibited complexes could explain the observation by ChIP that total Pol II at the actin promoter after heat shock decreased approximately 2.5-fold more than the general factors: B2 RNA, disrupting contacts between Pol II and the promoter, might decrease the efficiency of formaldehyde cross-linking and hence the apparent occupancy of Pol II at the actin promoter. It is also possible that the sequence of the promoter might influence the manner in which B2 RNA inhibits Pol II-promoter contacts.
It was previously observed that after heat shock TFIIH-specific phosphorylation of Pol II decreased in HeLa cells, and that nuclear extracts from these cells had lower TFIIH-associated kinase activity, likely due to increased association of TFIIH with chromatin.16 We also observed, by western blot, a general decrease in the level of Ser5-P in nuclei after heat shock, while the levels of TFIIH subunits and Pol II remained relatively constant. The global decrease in Ser5 phosphorylation after heat shock is in contrast to the well documented transcriptional activation of classical heat shock genes (e.g., hsp70), which requires Ser5 phosphorylation by Cdk7.17 Widespread but gene-specific repression by B2 RNA could cause an overall decrease in Ser5-P in response to heat shock, while still allowing phosphorylation of Ser5 at promoters of genes that are active.
Materials and Methods
The following antibodies were used in western blots and ChIP assays where indicated: Rpb1 N-terminus (sc899, Santa Cruz Biotechnology), Rpb1 Ser5-P (A300-655A, Bethyl Laboratories), TBP (sc273, Santa Cruz Biotechnology), TFIIA-γ (sc25365x, Santa Cruz Biotechnology), ERCC3 (sc293x, Santa Cruz Biotechnology), Cdk7 in ChIP assays (A300–405A, Bethyl Laboratories), Cdk7 in western blots (sc856x, Santa Cruz Biotechnology), and cyclin H (sc609, Santa Cruz Biotechnology).
Samples for western blots were prepared by heat shocking NIH 3T3 cells at 50–60% confluence for 15 min at 45°C and recovering for 30 min at 37°C. Cells were harvested in ice cold PBS with protease and phosphatase inhibitors (Complete cocktail tablets, Roche; 10 mM NaF). Nuclei were isolated as described previously in reference 12, with the addition of 10 mM NaF. Nuclei from approximately 6 × 106 cells were resuspended in 400 µl RIPA buffer [150 mM NaCl, 1% NP40, 0.5% sodium deoxycholate, 0.1% SDS, 50 mM Tris (pH 7.9), 10 mM NaF, PMSF and 1× protease inhibitors (Complete cocktail tablets, Roche)] and nutated for 30 min at 4°C. Samples were briefly sonicated using a cup sonicator filled with ice water (one 50 s burst at a setting of 4.5). After centrifugation, proteins were resolved using 5 or 8% SDS-PAGE and detected by western blotting.
ChIP assays were performed as follows. NIH 3T3 cells were heat shocked for 15 min at 45°C and recovered for 30 min at 37°C. Prior to harvesting, cells were treated with 0.25 or 1% formaldehyde for 10 min at room temperature. Glycine (0.125 M) was added for 5 min. Cells were harvested and nuclei were isolated by resuspending 4–6 × 106 cells in 400 µl of buffer A [4 mM MgCl2, 10 mM Tris-HCl (pH 7.4), 10 mM NaCl and 0.5% (v/v) NP40, 0.4 mM PMSF, 1× protease inhibitors (Complete cocktail tablets, Roche)] and nutating for 5 min at 4°C. Nuclei were harvested by centrifugation and washed once in an equal volume of buffer A. Nuclei were then resuspended in 800 µl of buffer B (50 mM Tris (pH 7.9), 10 mM EDTA, 0.4 mM PMSF, 1% SDS and protease inhibitors) and nutated for 10 min at 4°C. A 1.2 ml of buffer C (15 mM Tris (pH 7.9), 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.4 mM PMSF and protease inhibitors) was added and samples were sonicated using a cup sonicator filled with ice water (5–7 50 sec bursts at a setting of 4.5, with 1.75 min between bursts). Samples were centrifuged and the supernatants stored at −80°C. For each immunoprecipitation, 300 µl of sonicated chromatin was pre-cleared with 18 µl of protein A/G beads (Santa Cruz Biotechnology) that were equilibrated in buffer D (15 mM Tris (pH 7.9), 150 mM NaCl, 1 mM EDTA, 0.5% NP-40, 0.4 mM PMSF). Antibody was added and samples were nutated at 4°C overnight. 25 µl of pre-blocked protein A/G beads (pre-blocking occurred by nutating the beads overnight at 4°C in buffer D containing 0.4 mg/ml yeast RNA and 0.5 mg/ml BSA) were added and samples were nutated an additional 1–2 h at 4°C. Beads were washed sequentially with 400 µl of low salt buffer [20 mM Tris (pH 7.9), 150 mM NaCl, 2 mM EDTA, 1% Triton X-100, 0.1% SDS], 400 µl of high salt buffer [20 mM Tris (pH 7.9), 500 mM NaCl, 2 mM EDTA, 1% Triton X-100, 0.1% SDS], 400 µl of LiCl buffer [10 mM Tris (pH 7.9), 1 mM EDTA, 1% deoxycholate, 1% NP-40, 250 mM LiCl] and twice with 400 µl of TE [10 mM Tris (pH 7.9), 1 mM EDTA]. Two hundred and fifty µl of elution buffer containing 1% SDS, 50 mM Tris (pH 7.9) and 10 mM EDTA was added to the beads and the mixture was nutated for 1 h at 37°C. NaCl was added to 200 mM and crosslinks were reversed by incubation at 65°C for 12 h. Beads were precipitated and supernatants were transferred to new tubes. Samples were diluted with water to 0.5% SDS and then treated with Proteinase K (60 µg) for 1 h at 55°C. After phenol extraction and ethanol precipitation samples were resuspended in 100 µl Tris (pH 7.9) and subjected to qPCR using sybr green detection. The primers used were: actin promoter, 5′-AAG GAG CTG CAA AGA AGC TGT and 5′-GTT CCG AAA GTT GCC TTT TAT G; actin downstream, 5′-CTC TGC TCT CCC TGG ATT TG and 5′-CAG ATG TTC ACC TGC CTT CA.
The recombinant human TBP, TFIIB, TFIIE, TFIIF and native human Pol II and TFIIH used in kinase assays were prepared as described previously in references 18 and 19. The DNA template consisted of either a plasmid containing the Adenovirus major late core promoter (−53 to +10) fused to a 380 bp G-less cassette, or linear AdMLP immobilized on streptavidin beads via a biotin. Kinase reactions were performed in a buffered solution consisting of 10 mM Tris-HCl, 10 mM Hepes, 10% glycerol, 1 mM DTT, 4 mM MgCl2, 50 mM KCl, 50 µg/ml BSA at pH 7.9. When pre-initiation complexes were formed, the DNA template (1 nM) was pre-incubated with TBP (3.5 nM) at 30°C for 5 min. TFIIB (10 nM), TFIIF (2 nM), Pol II (1–3 nM) and ncRNA (5 nM, when included) were incubated together in a separate tube at 30°C for 5 min. The contents of these two tubes were mixed and incubated at 30°C for 15 min to allow complexes to form. For all kinase reactions, either ERK2 (1 µM final concentration) or a mixture of TFIIE and TFIIH (9 nM TFIIE-34, 5 nM TFIIE-56, 0.25 µl TFIIH) were incubated with other factors and/or ncRNA at 30°C for at least 3 min prior to adding [γ-32P]ATP (30 µM, 12 µCi) and continuing the incubation for 20 min. Samples were resolved by 5% SDS-PAGE and labeled Rpb1 was visualized using phosphorimagery.
Acknowledgements
This work was supported by a Public Health Service grant (R01 GM068414) from the National Institute of General Medical Sciences. We thank Kevin Sours and Natalie Ahn for activated ERK2.
Abbreviations
- Pol II
RNA polymerase II
- CTD
carboxy terminal domain
- Cdk7
cyclin dependent kinase 7
- Ser5-P
phosphorylated serine 5 in the Pol II CTD
- Ser2-P
phosphorylated serine 2 in the Pol II CTD
- TBP
TATA binding protein
- ChIP
chromatin immunoprecipitation
- ncRNA
non-coding RNA
- SINE
short interspersed element
Footnotes
Previously published online: www.landesbioscience.com/journals/transcription/article/14306
References
- 1.Thomas MC, Chiang CM. The general transcription machinery and general cofactors. Crit Rev Biochem Mol Biol. 2006;41:105–178. doi: 10.1080/10409230600648736. [DOI] [PubMed] [Google Scholar]
- 2.Fuda NJ, Ardehali MB, Lis JT. Defining mechanisms that regulate RNA polymerase II transcription in vivo. Nature. 2009;461:186–192. doi: 10.1038/nature08449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Buratowski S. Progression through the RNA polymerase II CTD cycle. Mol Cell. 2009;36:541–546. doi: 10.1016/j.molcel.2009.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lu H, Zawel L, Fisher L, Egly JM, Reinberg D. Human general transcription factor IIH phosphorylates the C-terminal domain of RNA polymerase II. Nature. 1992;358:641–645. doi: 10.1038/358641a0. [DOI] [PubMed] [Google Scholar]
- 5.Shiekhattar R, Mermelstein F, Fisher RP, Drapkin R, Dynlacht B, Wessling HC, et al. Cdk-activating kinase complex is a component of human transcription factor TFIIH. Nature. 1995;374:283–287. doi: 10.1038/374283a0. [DOI] [PubMed] [Google Scholar]
- 6.Glover-Cutter K, Larochelle S, Erickson B, Zhang C, Shokat K, Fisher RP, et al. TFIIH-associated Cdk7 kinase functions in phosphorylation of C-terminal domain Ser7 residues, promoter-proximal pausing and termination by RNA polymerase II. Mol Cell Biol. 2009;29:5455–5464. doi: 10.1128/MCB.00637-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Akhtar MS, Heidemann M, Tietjen JR, Zhang DW, Chapman RD, Eick D, et al. TFIIH kinase places bivalent marks on the carboxy-terminal domain of RNA polymerase II. Mol Cell. 2009;34:387–393. doi: 10.1016/j.molcel.2009.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goodrich JA, Kugel JF. From bacteria to humans, chromatin to elongation and activation to repression: The expanding roles of noncoding RNAs in regulating transcription. Crit Rev Biochem Mol Biol. 2009;44:3–15. doi: 10.1080/10409230802593995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Allen TA, Von Kaenel S, Goodrich JA, Kugel JF. The SINE-encoded mouse B2 RNA represses mRNA transcription in response to heat shock. Nat Struct Mol Biol. 2004;11:816–821. doi: 10.1038/nsmb813. [DOI] [PubMed] [Google Scholar]
- 10.Liu WM, Chu WM, Choudary PV, Schmid CW. Cell stress and translational inhibitors transiently increase the abundance of mammalian SINE transcripts. Nucl Acids Res. 1995;23:1758–1765. doi: 10.1093/nar/23.10.1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Espinoza CA, Allen TA, Hieb AR, Kugel JF, Goodrich JA. B2 RNA binds directly to RNA polymerase II to repress transcript synthesis. Nat Struct Mol Biol. 2004;11:822–829. doi: 10.1038/nsmb812. [DOI] [PubMed] [Google Scholar]
- 12.Mariner PD, Walters RD, Espinoza CA, Drullinger LF, Wagner SD, Kugel JF, et al. Human Alu RNA is a modular transacting repressor of mRNA transcription during heat shock. Mol Cell. 2008;29:499–509. doi: 10.1016/j.molcel.2007.12.013. [DOI] [PubMed] [Google Scholar]
- 13.Yakovchuk P, Goodrich JA, Kugel JF. B2 RNA and Alu RNA repress transcription by disrupting contacts between RNA polymerase II and promoter DNA within assembled complexes. Proc Natl Acad Sci USA. 2009;106:5569–5574. doi: 10.1073/pnas.0810738106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Trigon S, Serizawa H, Conaway JW, Conaway RC, Jackson SP, Morange M. Characterization of the residues phosphorylated in vitro by different C-terminal domain kinases. J Biol Chem. 1998;273:6769–6775. doi: 10.1074/jbc.273.12.6769. [DOI] [PubMed] [Google Scholar]
- 15.O'Gorman W, Thomas B, Kwek KY, Furger A, Akoulitchev A. Analysis of U1 small nuclear RNA interaction with cyclin H. J Biol Chem. 2005;280:36920–36925. doi: 10.1074/jbc.M505791200. [DOI] [PubMed] [Google Scholar]
- 16.Dubois MF, Vincent M, Vigneron M, Adamczewski J, Egly JM, Bensaude O. Heat-shock inactivation of the TFIIH-associated kinase and change in the phosphorylation sites on the C-terminal domain of RNA polymerase II. Nucleic Acids Res. 1997;25:694–700. doi: 10.1093/nar/25.4.694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schwartz BE, Larochelle S, Suter B, Lis JT. Cdk7 is required for full activation of Drosophila heat shock genes and RNA polymerase II phosphorylation in vivo. Mol Cell Biol. 2003;23:6876–6886. doi: 10.1128/MCB.23.19.6876-6886.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kugel JF, Goodrich JA. Promoter escape limits the rate of transcription from the adenovirus major late promoter on negatively supercoiled templates. Proc Natl Acad Sci USA. 1998;95:9232–9237. doi: 10.1073/pnas.95.16.9232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Weaver JR, Kugel JF, Goodrich JA. The sequence at specific positions in the early transcribed region sets the rate of transcript synthesis by RNA polymerase II in vitro. J Biol Chem. 2005;280:39860–39869. doi: 10.1074/jbc.M509376200. [DOI] [PubMed] [Google Scholar]