

Abstract
Aflatoxins are polyaromatic mycotoxins that contaminate a range of food crops as a result of fungal growth and contribute to serious health problems in the developing world because of their toxicity and mutagenicity. Although relatively resistant to biotic degradation, aflatoxins can be metabolized by certain species of Actinomycetales. However, the enzymatic basis for their breakdown has not been reported until now. We have identified nine Mycobacterium smegmatis enzymes that utilize the deazaflavin cofactor F420H2 to catalyse the reduction of the α,β-unsaturated ester moiety of aflatoxins, activating the molecules for spontaneous hydrolysis and detoxification. These enzymes belong to two previously uncharacterized F420H2 dependent reductase (FDR-A and -B) families that are distantly related to the flavin mononucleotide (FMN) dependent pyridoxamine 5′-phosphate oxidases (PNPOxs). We have solved crystal structures of an enzyme from each FDR family and show that they, like the PNPOxs, adopt a split barrel protein fold, although the FDRs also possess an extended and highly charged F420H2 binding groove. A general role for these enzymes in xenobiotic metabolism is discussed, including the observation that the nitro-reductase Rv3547 from Mycobacterium tuberculosis that is responsible for the activation of bicyclic nitroimidazole prodrugs belongs to the FDR-A family.
Introduction
Aflatoxins are secondary metabolites of various Aspergillus species, particularly A. flavus and A. parasiticus, and are generally separated into two groups: the difurocoumarocyclopentenones (including AFB1 and AFB2) and the difurocoumarolactones (including AFG1 and AFG2) (Fig. 1). Humans can be exposed to aflatoxins via consumption of Aspergillus-contaminated maize or nuts, or the milk of animals fed contaminated crops. Recurrent consumption of aflatoxin-contaminated food has been shown to contribute to an increased risk of nutritional deficiencies, immune suppression and hepatocellular carcinoma in several developing nations (Wagacha and Muthomi, 2008). The levels of aflatoxins in internationally traded commodities are tightly regulated by the FAO/WHO and other regulatory bodies (van Egmond et al., 2007).
Fig. 1.
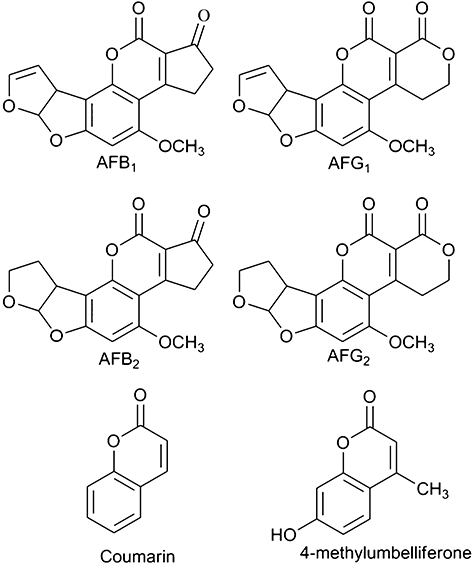
Chemical structures of compounds used in this study.
Although higher organisms and many microorganisms have little ability to degrade aflatoxins, they are known to be metabolized by certain fungi and bacteria (Ciegler et al., 1966). The bacteria known to degrade aflatoxins are all Actinomycetales of the suborder Corynebacterineae, such as Nocardia corynebacterioides (Ciegler et al., 1966; Hormisch et al., 2004), Rhodococcus erythropolis and Mycobacterium fluorantheniorans sp. Nov DSM44556T (Teniola et al., 2005). There is evidence that aflatoxin degradation by these species is enzyme catalysed (Smiley and Draughon, 2000; Alberts et al., 2006), although the reaction products and the enzymes involved have not previously been described. Because the degradation products of aflatoxins are non-toxic (Ciegler et al., 1966; Teniola et al., 2005), there is considerable interest in identifying and characterizing the enzymes involved.
Herein, we describe the isolation and characterization of nine F420H2-dependent reductases (FDRs) from Mycobacterium smegmatis that utilize an F420H2 cofactor to catalyse the reduction of aflatoxins, leading to their spontaneous breakdown. Structural and phylogenetic analysis suggests that these nine enzymes fall into two hitherto unclassified enzyme families that are distantly related to the well characterized FMN-dependent pyridoxamine 5′-phosphate oxidase (PNPOx) family (di Salvo et al., 2003). The mechanism of aflatoxin breakdown appears to involve an initial, enzyme-catalysed reduction of the double bond of the α,β-unsaturated ester moiety, followed by spontaneous hydrolysis. The possible cellular functions of these enzymes in xenobiotic metabolism are discussed, as are their potential uses, both as an enzymatic means by which aflatoxins may be detoxified and as potential drug targets, owing to their exclusive presence in Archea, Actinomycetales and some species of γ-Proteobacteria.
Results
Identification of aflatoxin degrading bacterial strains
Seven bacterial strains (Mycobacterium smegmatis mc2155, Mycobacterium sp. ESD, Arthrobacter KW-ES, Arthrobacter RW thio, Arthrobacter RLH#41, Agrobacterium radiobacter P230 and Pseudomonas monteilii C11) known to degrade various xenobiotics (Sutherland et al., 2002; Horne et al., 2002a,b) were tested for their ability to degrade aflatoxin AFG1 in liquid culture. Live cultures and cell lysates of the two Mycobacterium strains were found to degrade AFG1, while no aflatoxin degradation was observed in live cultures or cell lysates of the other strains tested. The activity of the Mycobacterium lysates was lost upon heating, consistent with an enzyme-catalysed reaction, as shown by thin-layer chromatography (TLC; Fig. S1). Although other Actinomycetales have previously been shown to degrade aflatoxins (Ciegler et al., 1966; Hormisch et al., 2004; Teniola et al., 2005; Alberts et al., 2006), the failure of the Arthrobacter cultures tested to degrade AFG1 demonstrates that aflatoxins are not degraded by all Actinomycetales. Because the genome of M. smegmatis mc2155 has been sequenced and annotated, and methodologies for its manipulation in the laboratory are well established, it was used in all subsequent experiments to isolate and identify the gene/enzyme systems responsible for aflatoxin degradation.
Aflatoxin degradation is cofactor F420-dependent
Transposon mutagenesis was used as a first approach to identify the genes/enzymes involved in the degradation of aflatoxin by M. smegmatis. After mutagenesis with the EZ::TN <R6Kγori/KAN-2> transposon, 3160 viable isolates were assayed by TLC for their ability to degrade AFG1 over 4 days (Fig. S1). Only five of the 3160 isolates did not degrade AFG1. Sequencing of the region surrounding the transposon insertion site in these colonies showed that four of the insertions disrupted the fbiC gene, which catalyses the final step in the biosynthesis of the deazaflavin, Fo, from intermediates of the riboflavin synthesis and tyrosine metabolic pathways (Fig. S2) (Choi et al., 2002; Graham et al., 2003). Fo is the precursor for the production of the cofactor F420 (Graupner and White, 2001). The fifth transposon insertion disrupted the fgd gene, which encodes the F420-dependent glucose 6-phosphate dehydrogenase (FGD) (Purwantini and Daniels, 1998; Bashiri et al., 2008), which is known to recycle F420 from its oxidized to its reduced F420H2 form in Mycobacteria (Fig. S2). These data are consistent with an essential role for F420 in the degradation of aflatoxins in M. smegmatis. Significantly, all the bacterial genera previously shown to degrade aflatoxins (see above) are also known to synthesize F420 (Isabelle et al., 2002).
To test whether FGD catalysed aflatoxin degradation, it was heterologously expressed in and purified from Escherichia coli. As expected, FGD could catalyse the reduction of F420 to F420H2 in the presence of glucose 6-phosphate (Purwantini and Daniels, 1998; Bashiri et al., 2008). However, neither FGD in the presence of F420/F420H2, nor F420H2 in isolation, were able to catalyse aflatoxin breakdown at detectable levels. Thus, although F420H2 is clearly essential to the reaction, it is most likely involved as a cofactor for another enzyme not identified in this screen.
Identification and kinetic analysis of two families of F420H2-dependent aflatoxin degrading enzymes
The enzymes responsible for aflatoxin degradation in M. smegmatis were purified from soluble cell extracts. Two protocols were used, one involved ammonium sulphate precipitation, hydrophobic interaction chromatography, anion exchange chromatography and SDS-PAGE (Fig. 2). The second involved ammonium sulphate precipitation, hydrophobic interaction, gel filtration and anion exchange chromatography. Proteins were then identified from active fractions in the final purification steps via tandem mass spectrometry of peptides obtained by tryptic digests and comparison of these fragments to predicted proteins from the M. smegmatis genome (Table S1). Only four proteins were identified in both purifications; MSMEG_3380, 3004, 2027 and 5717. The observation that multiple enzymes with the same activity were identified provides an explanation as to why they were not identified by transposon mutagenesis, since they would all need to be simultaneously inactivated to prevent AFG1 degradation.
Fig. 2.
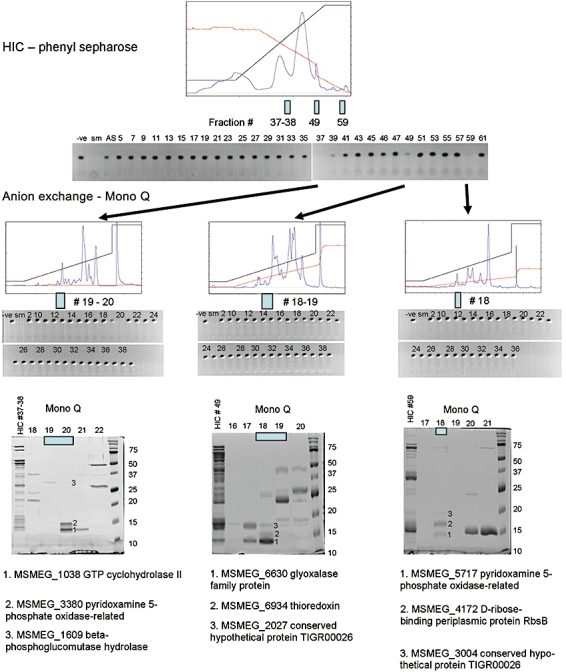
Purification protocol. Protein fractions that showed F420H2 dependent AFG1 degradation as measured by TLC were separated from M. smegmatis extracts. The ammonium sulphate precipitated proteins were first purified by hydrophobic interaction chromatography (HIC). Active fractions were further purified by anion exchange chromatography before separation by SDS-PAGE. Bands were cut from SDS-PAGE gels, digested with trypsin and analysed by LC/MS/MS. Peptides were identified from the annotated M. smegmatis genome sequence and corresponding results are shown for some of the excised bands. In the TLCs ‘-ve’ denotes no enzyme negative control and ‘sm’ denotes M. smegmatis cell extract positive control.
The four proteins identified above were all predicted to be related to the PNPOx family by the protein fold recognition server PHYRE (Kelley and Sternberg, 2009), although their relationship is distant (less than 15% amino acid identity). The four proteins have predicted molecular weights, based on their sequences, of approximately 16kDa (Table S1), while proteins in the active M. smegmatis fractions were calculated to be approximately 30 kDa by size exclusion chromatography (Fig. S3), suggesting that these four proteins most likely oligomerize as dimers in solution. These results are consistent with the observation that the PNPOx enzymes form homodimers (di Salvo et al., 1998). Phylogenetic analysis of 146 PNPOx-like sequences, including 28 present in M. smegmatis (Fig. 3 and Fig. S4), shows that the four proteins belong to two distinct families (< 12% amino acid identity), hereafter denoted FDR-A and -B (F420H2 dependent reductase -A and -B), which are both clearly differentiated from the functionally defined PNPOx family. Although the PNPOx family is widely distributed among both prokaryotes and eukaryotes, the FDR-A and FDR-B families appear to be limited to the Actinomycetales, Archaea and some Proteobacteria, all of which possess F420. Furthermore, the two families are unrelated to other previously characterized F420H2 dependent reductases, including sulphite reductase (Johnson and Mukhopadhyay, 2005), dinitrophenol reductase (Ebert et al., 2001) and the methylenetetrahydromethanopterin reductase complex involved in methanogenesis in Archaea (Deppenmeier, 2002; Graham and White, 2002). Interestingly, the only other functionally characterized enzyme that is related to either of these families is the bicyclic nitroimidazole prodrug activating reductase from M. tuberculosis, Rv3547 (Manjunatha et al., 2006), which has 49% sequence identity to MSMEG_5998 of the FDR-A family (Fig. 3).
Fig. 3.
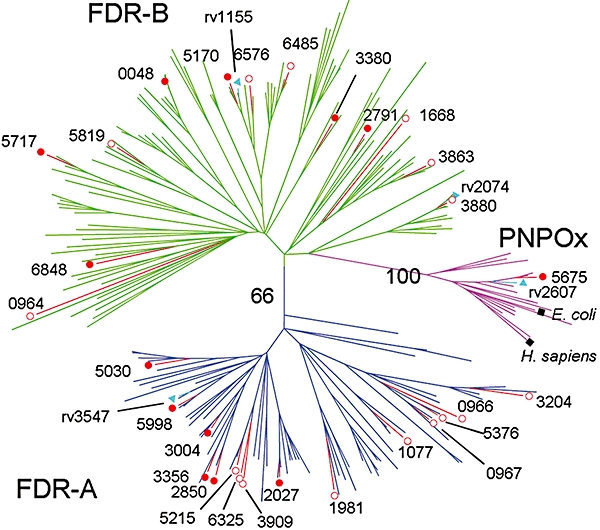
Phylogenetic relationship between the PNPOx, FDR-A and FDR-B families. A condensed tree was constructed from 146 protein sequences of FDRs/PNPOxs from seven species of Actinomycetales (M. smegmatis, M. tuberculosis H37Rv, M. vanbaalenii, Rhodococcus sp. RH1, Arthrobacter sp. FB24, S. coelicolor, Frankia alni and Nocardioides sp. JS614), plus the known PNPOx enzymes from H. sapiens, E. coli, Saccharomyces cerevisiae, Caenorhabditis elegans and Mus musculus. Solid red circles denote the FDRs described in this paper, and open red circles denote other potential FDRs from M. smegmatis (TIGR locus number shown). For clarity, only enzymes from other species that have been previously characterized are labelled: blue triangles denote M. tuberculosis enzymes with known structures or previously described functions and black squares denote previously described PNPOxs.
The next phase of our work focused on the four M. smegmatis enzymes identified by chromatography plus five other members of the FDR-A and FDR-B families in the M. smegmatis genome. These five enzymes were identified as close paralogues both by blast analysis and by their retention of a highly conserved loop motif and a putative phosphate binding motif (Fig. S4). The nine genes were cloned, expressed, purified and assayed against the four aflatoxins in the presence of F420H2 (Table 1). All nine enzymes catalysed measurable degradation of all four aflatoxins tested (AFG1, AFG2, AFB1 and AFB2). AFG1 was found to be the best substrate for all nine enzymes: for example MSMEG_3380 has nearly 50-fold more activity with AFG1 than AFB2. However, the specific activities of the various enzymes differed by more than four orders of magnitude for AFG1. The four enzymes originally identified in the chromatographic experiments were not the most active, perhaps because of purification, expression or stability issues. The three most active enzymes were from the FDR-A family (MSMEG_5998, 2850 and 3356). Kinetic parameters were obtained for MSMEG_5998 with AFB1 (KM = 47 ± 6 µM, Kcat = 63 ± 4 min−1); these values could not be obtained for other enzymes because enzyme activity with saturating substrate concentrations could not be measured, owing to the low solubility of the aflatoxins. The fact that all nine enzymes tested had aflatoxin degrading activity suggests that some, if not all, of the other 19 representatives of the two FDR families which we have found by blast analysis of the M. smegmatis genome might also catalyse aflatoxin breakdown.
Table 1.
Specific activities of MSMEG proteins for aflatoxins
| Specific activity (nmol min−1 µmol−1 enzyme)a | |||||
|---|---|---|---|---|---|
| Protein family | TIGR Locusb | AFG1 | AFG2 | AFB1 | AFB2 |
| FDR-A | 5998 | 83 000 ± 2000 | 6100 ± 200 | 9100 ± 700 | 7700 ± 700 |
| 2850 | 12 000 ± 2000 | 8000 ± 500 | 1300 ± 200 | 410 ± 60 | |
| 3356 | 8080 ± 70 | 3100 ± 500 | 1200 ± 200 | 1300 ± 200 | |
| 3004 | 1600 ± 100 | 1260 ± 50 | 310 ± 60 | 620 ± 20 | |
| 2027 | 660 ± 40 | 310 ± 50 | 220 ± 50 | 160 ± 20 | |
| FDR-B | 3380 | 1600 ± 100 | 320 ± 40 | 130 ± 10 | 33 ± 4 |
| 6848 | 900 ± 200 | 430 ± 50 | 100 ± 10 | 90 ± 3 | |
| 5170 | 240 ± 40 | 140 ± 6 | 330 ± 50 | 90 ± 2 | |
| 5717 | 3 ± 0.7 | 3 ± 0.5 | 3 ± 0.1 | 2 ± 0.2 | |
Units are expressed per µmole of the monomer for both FDR-A and -B.
The TIGR locus number is preceded by ‘MSMEG_’.
Although genome sequences of other Actinomycete strains that metabolize aflatoxins, such as R. erythropolis, N. corynebacterioides and M. fluoranthenivorans (Ciegler et al., 1966; Hormisch et al., 2004; Teniola et al., 2005), have not been published, our preliminary screening of several related Actinomycete genome sequences (Table S2) shows that each has at least 10 putative FDR-As and FDR-Bs. It thus seems likely that the aflatoxin degrading abilities of the FDRs shown in M. smegmatis, and quite likely many other Actinomycetales, may originate from FDRs of the A and B families. It is notable that there is only one annotated FDR-B and no annotated FDR-A sequences present in Arthrobacter genomes so far published (Table S2), which may explain the lack of aflatoxin degradation observed in the three Arthrobacter strains analysed in this study.
The PNPOx and FDR families are functionally distinct
Because the FDR families have significant sequence similarity to the PNPOxs and the respective cofactors F420H2 and FMN/FMNH2 are structurally related (Fig. S2), we investigated whether there was any cofactor, or catalytic, promiscuity between these enzyme families. Phylogenetic analysis (Fig. 3) identified MSMEG_5675 as the probable M. smegmatis PNPOx. In contrast to the expressed FDR-A and -B enzymes, preparations of purified MSMEG_5675 were an intense yellow colour with an absorption maximum at 450 nm, indicative of protein bound to FMN. MSMEG_5675 was confirmed to be a PNPOx by its capacity to catalyse the conversion of pyridoxamine 5′-phosphate (PMP) to pyridoxal 5′-phosphate (PLP) with a specific activity of 0.18 s−1, consistent with the catalytic activity of other PNPOxs (di Salvo et al., 1998; di Salvo et al., 2002). No activity was observed after incubating the FDRs overnight with 100 µM FMN and PMP, suggesting that these enzymes exclusively utilize F420H2 as a cofactor. Moreover, once FMN was stripped from MSMEG_5675, the apo-enzyme was found to be unable to catalyse aflatoxin degradation in the presence of F420H2 (data not shown). Thus, our data suggest that the PNPOx and FDR families are functionally distinct and they do not appear to have overlapping activities or cofactor preferences.
Structures of the FDR-A and FDR-B enzymes
To gain further insight into the function of these newly identified enzymes, crystal structures of members of the FDR-A (MSMEG_3356) and FDR-B families (MSMEG_3380) were solved to 2.0 and 1.2 Å resolution respectively. MSMEG_3356 and MSMEG_3380 both adopt the PNPOx-like split barrel fold, comprising a central split barrel surrounded by four helices (Fig. 4). MSMEG_3380 exists as a homodimer, while MSMEG_3356 is monomeric.
Fig. 4.
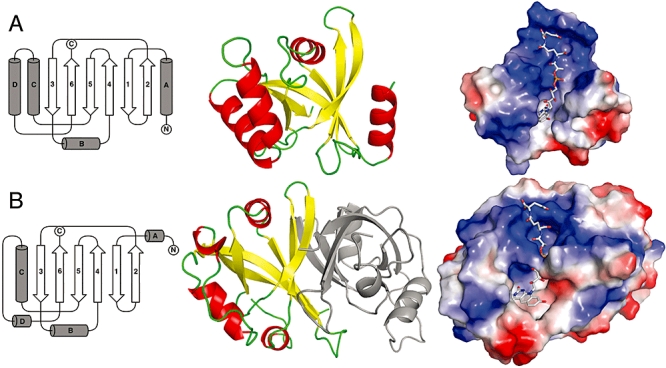
The structures of FDR-A and FDR-B enzymes.
A. MSMEG_3356; from left to right, a topology diagram, showing the arrangement of the split barrel of anti-parallel β-sheets, surrounded by three large α-helices at the side and a short α-helix above, a cartoon diagram, and an electrostatic surface potential representation with F420H2 docked.
B. MSMEG_3380; from left to right, a topology diagram of the structure, showing the arrangement of the split barrel of anti-parallel β-sheets, surrounded by one large α-helix at the side and three short α-helices above and below, a cartoon diagram of the 3380 dimer, and an electrostatic surface potential representation with F420H2 docked.
The structures of MSMEG_3380 and MSMEG_3356 differ significantly, as would be expected given their low sequence similarity; only 88 amino acids could be structurally aligned with an r.m.s.d of 2.2 Å. A DALI (Holm et al., 2008) search indicated that the closest known structural homologue to the FDR-A MSMEG_3356 is a putative PNPOx from Agrobacterium tumefaciens (3DNH), with just 15% amino acid identity. Thus, it appears that MSMEG_3356 is the first characterized structure of a member of the FDR-A family. The closest relatives of MSMEG_3380 with known structures are the functionally uncharacterized proteins from M. tuberculosis Rv1155 and Rv2074 (Biswal et al., 2005; 2006; Canaan et al., 2005), with amino acid identities of 18% and 24% respectively. These proteins have been putatively classified as PNPOxs, owing to their similarity to the PNPOx family, but no functional analysis has been performed. As shown in our phylogenetic analysis (Fig. 3), they have greater similarity to the FDR-B family than the PNPOxs and we propose that these proteins and MSMEG_3380 comprise the first structures of the FDR-B family.
Despite their low similarity, the structures of both the FDRs described here are characterized by large hydrophobic pockets formed by several aromatic residues and a positively charged groove that is principally formed by several lysine and arginine side-chains (Fig. 4). In terms of shape and electrostatic character, these pockets/channels are highly complementary to the F420H2 cofactor, which possesses an aromatic deazaflavin group and a long, negatively charged, side-chain consisting of a phospho-lactate and a variable number of glutamates. In the absence of a structure of the holoenzyme, we have docked the cofactor, as shown in Fig. 4. Consistent with the level of electrostatic and hydrophobic interactions implied by the docking results, titration curves of activity versus cofactor concentration (Fig. 5) show that both MSMEG_3356 and MSMEG_3380 bind F420H2 tightly, with apparent KM values in the nanomolar range (260 nM and 130 nM, respectively).
Fig. 5.
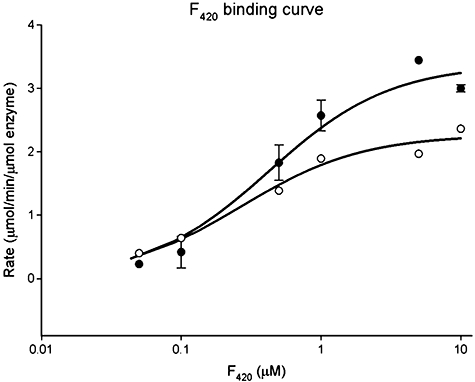
F420H2 cofactor binding to enzymes 3356 and 3380. The apparent KM's for F420H2 were calculated by measuring the velocity of the reaction with varying concentrations of F420H2 from 0.05 to 10 µM while maintaining all other components at 100 µM aflatoxin, 1 µM enzyme. For clarity the velocity is expressed as percent of maximum velocity. Empty circles, 3356; filled circles, 3380.
Although the residues forming the putative F420H2 binding channels and substrate binding pockets occur in homologous regions of the two families, the identities of the residues involved are not conserved (Fig. 6 and Fig. S4). In MSMEG_3356, the deazaflavin/substrate pocket is formed by W7, N25, F26, M31, P48, M49, M50, F64, A68, W76, F121 and Y124, and the positively charged groove is formed by the side-chains of N8, H36, R39, K40, T41, K43, T47 and K67. In MSMEG_3380 the deazaflavin/substrate pocket is formed by M34, W35, Y95, L98 and L106 of monomer A and Y74 and Y76 of monomer B. The positively charged groove is formed by the side-chains of R24, Q30, N32, H47, R51, Q52, K53 and R55 from monomer A and R80 from monomer B. The hydrophobic aflatoxin/deazaflavin binding cavity of MSMEG_3380 is significantly larger than that of MSMEG_3356 (649 Å3 versus 411 Å3), as measured by the 3V server (Voss and Gerstein, 2010), suggesting that these enzymes are likely to have different substrate ranges.
Fig. 6.
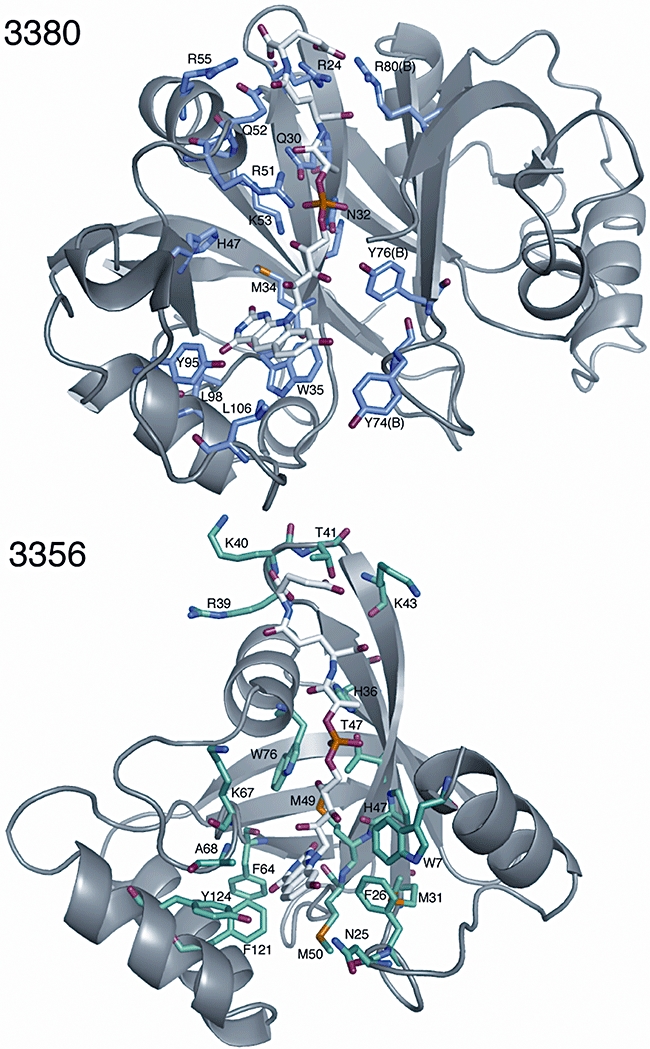
The F420H2 binding grooves of an FDR-A, MSMEG_3356, and an FDR-B, MSMEG_3380. The deazaflavin pockets are shown to be rich in aromatic and hydrophobic residues, while the side-chain binding grooves are rich in basic residues.
The catalytic mechanism of aflatoxin reduction
Liquid chromatography-mass spectrometry (LCMS) analysis of the aflatoxin degradation reaction showed that all of the FDRs tested appeared to catalyse aflatoxin breakdown via the same mechanism for all four substrates, with the ion species of the product differing from the corresponding reactant by 2.02 m/z in each instance (Fig. 7). This is consistent with the reduction of aflatoxin via transfer of two electrons from F420H2. The reaction product is unstable over time and several low abundance metabolites (possibly generated by spontaneous hydrolysis) appear after an hour. Previously characterized reactions involving AFB1 and AFG1 involved oxidation of the unsaturated carbons of the furan ring by mammalian liver CYP450's (Mishra and Das, 2003), or their spontaneous reduction to form AFB2 and AFG2 (Fig. 1). As the FDRs described here also catalyse reduction of AFB2 and AFG2, it is unlikely that it is the furan moiety that is reduced by these enzymes.
Fig. 7.
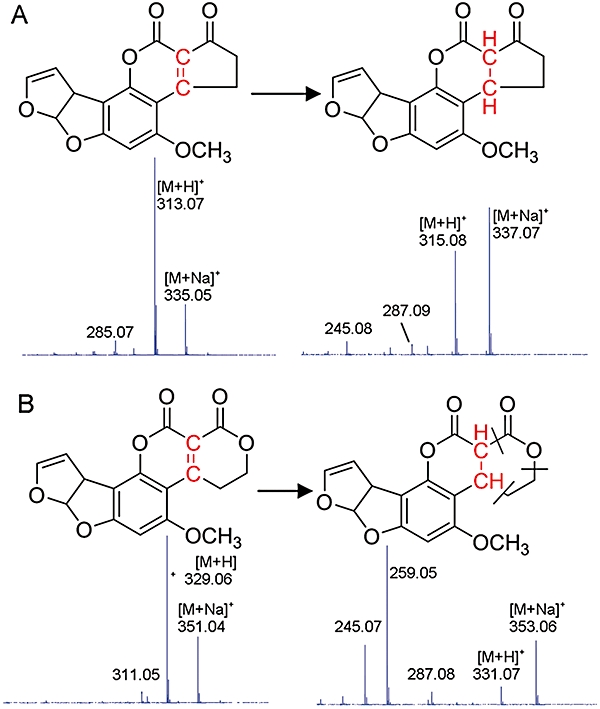
Proposed reaction mechanism of AFB1 and AFG1 reduction by reduced F420.
A. The proposed reduction mechanism of AFB1, with the mass spectrum of the substrate and product below the chemical structures.
B. The mass spectrum and chemical structure of AFG1 and the reduced product.
A second candidate moiety for the FDR catalysed reduction is the α,β-unsaturated ester between the lactone rings in AFG1/G2 and the lactone and cyclopentenone rings in AFB1/B2. Indeed, the LCMS-induced fragmentation of AFG1 (Fig. 7) produces ion species (287.08 and 259.05 m/z) that would result from a retro Diels-Alder fragmentation of the lactone ring (loss of CO2 and C2H4 respectively), consistent with destabilization of the ring resulting from reduction of the α,β-unsaturated ester. To test this hypothesis, MSMEG_3380 was incubated with coumarin and 4-methylumbelliferone. Coumarin and 4-methylumbelliferone both contain α,β-unsaturated ester moieties, and lack furan rings. However, the α,β-unsaturated ester moiety in 4-methylumbelliferone is stabilized by a methyl group (Fig. 1). Degradation of coumarin was observed at rates similar to that for aflatoxin (0.5 µmol min−1 µmol−1), while no degradation of 4-methylumbelliferone was observed. Further experiments will be required to establish details of the catalytic mechanism, but these results suggest that it is the α,β-unsaturated ester moiety that is reduced by the FDRs.
Discussion
The possible physiological role of FDRs
Herein we describe two families of enzymes that utilize F420H2 to degrade aflatoxin. F420/F420H2 is a deazaflavin hydride carrier found in the Archea, Actinomycetales and some species of γ-Proteobacteria. Although non-essential in Actinomycetales in a laboratory setting (Nakano et al., 2004), an important role for the cofactor has been documented in a wide variety of metabolic processes. These include methanogenesis (Graham and White, 2002), sulphite reduction (Johnson and Mukhopadhyay, 2005), the degradation of nitroaromatic compounds (Ebert et al., 2001; Manjunatha et al., 2006; Guerra-Lopez et al., 2007; Singh et al., 2008), antibiotic resistance (Hasan et al., 2010), malachite green decolourization (Guerra-Lopez et al., 2007) and the biosynthesis of chlortetracyclines in Streptomyces (Nakano et al., 2004). It may be that the highly negative redox potential of F420H2 (Jacobson and Walsh, 1984) is enabling for some of these energetically demanding reactions, including the degradation of aflatoxins by the FDR-A and -B families described here.
It seems unlikely that M. smegmatis would need between nine and 28 FDR enzymes specifically to degrade aflatoxin. Indeed, it is questionable as to whether aflatoxins are the primary physiological substrates for any of them; M. smegmatis has been found in soil where it may encounter aflatoxins but it is also known to be a human saprophyte where it would be very unlikely to encounter these carcinogens. The KM values of these enzymes for aflatoxin were also higher than what might be expected for a physiological substrate; they were above the solubility limit for aflatoxin (approximately 100 µM) for all the enzymes except for the most active aflatoxin degrading FDR that we characterized, MSMEG_5998, which had a KM of 47 ± 6 µM for AFB1. By comparison the KM value for E. coli PNPOx for pyridoxine 5′-phosphate is 2 µM (di Salvo et al., 2003). Furthermore, there are significant differences between the active site structure of the FDR-A and -B enzymes, which suggest that they may have a broad substrate range. Our analysis of the substrate binding pockets of MSMEG_3356 and MSMEG_3380 reveal that while the pockets are formed by homologous regions the residues are not conserved. Also, the MSMEG_3380 cavity is significantly larger than that of MSMEG_3356 (649 Å3 versus 411 Å3). The aflatoxin degrading activity we have characterized may thus reflect inherently broad substrate specificities of the FDR-A and -B families for polyaromatic compounds containing α,β-unsaturated lactones.
Our phylogenetic and genomic analyses (Fig. 3 and Table S2) show that the FDR-A and -B families are widespread among the Actinomycetales. Many taxa in this order inhabit environments where aflatoxins are unlikely to be found. However, the FDRs may be involved in other metabolic processes. We can suggest three possibilities. First, we note that other Actinomycetales, including some previously found to degrade aflatoxins (Hormisch et al., 2004; Teniola et al., 2005), have been isolated from environments contaminated with structurally similar polyaromatic hydrocarbons (Willumsen et al., 2001; Miller et al., 2004). The FDR enzymes may play a role in the degradation of these compounds and hence their utilization as potential carbon and energy sources. Second, some Actinomycetales are plant pathogens/symbionts (El-Tarabily and Sivasithamparam, 2006; Schrey and Tarkka, 2008) and many plant antimycobacterials are coumarinyl compounds that are structurally similar to aflatoxins and could be substrates for FDR-A and -B enzymes (Stavri and Gibbons, 2005). Finally, many antibiotic intermediates are potential substrates for these enzymes; for instance the aclacinomycin biosynthetic gene cluster in Streptomyces galilaeus contains an uncharacterized gene, AclJ, which our phylogenetic analysis suggests is an FDR-A enzyme (Chung et al., 2002). Some FDR-A and -B enzymes may therefore be involved in antibiotic biosynthesis.
Potential industrial and medical applications
The FDR-catalysed aflatoxin breakdown described in this work constitutes significant detoxification of these compounds, as previous studies have shown that removal of the lactone ring of AFB1 reduces its mutagenicity by 450-fold and its acute toxicity by 18-fold (Lee et al., 1981). Furthermore, pre-incubation with various Actinomycetales has been demonstrated to reduce the mutagenicity and toxicity of aflatoxin to animals (Ciegler et al., 1966; Alberts et al., 2006; Tejada-Castaneda et al., 2008). Accordingly, there is a potential use for these enzymes in decontaminating aflatoxin-contaminated food crops, either as a transgenic input trait for the crop or in a post-harvest application. There are precedents for the effective decontamination of two other mycotoxins (zearalenone and deoxynivalenol) in maize and rice by transgenic expression of the corresponding bacterial detoxifying enzymes (Igawa et al., 2007; Ohsato et al., 2007). Coexpression of the genes required for the biosynthesis of reduced F420 would also be required for the FDRs to be effective as an input trait and there are now many examples of plants being engineered with multiple genes (Taverniers et al., 2008). The identification of non-pathogenic strains of Actinomycetales that have the capacity to degrade aflatoxin may provide a less technologically intensive biocontrol approach. Indeed, inoculation of Aspergillus growth media with Streptomyces species has been shown to protect peanuts from aflatoxin contamination in the laboratory (Zucchi et al., 2008).
The exclusivity of these FDRs to certain Actinobacteria is an important ‘point of difference’ that can be targeted in the development of drugs, particularly considering the presence of several human pathogens within this phylum, including M. tuberculosis. For instance, the candidate anti-tuberculosis drug PA-824 is now known to be a prodrug that is metabolized to its active form through the activity of an FDR-A (Rv3547) (Manjunatha et al., 2006). Our identification and structural characterization of the first members of these FDR families thus raises the possibility of rational design of improved prodrugs with even greater activity and specificity. However, before detailed analysis of the catalytic mechanism is possible, structures of the holoenzymes (F420H2 : Enz) and, ideally, a ternary complex will be required, since substantial conformational changes are often seen in cofactor and substrate binding in related enzymes, such as PNPOx (Safo et al., 2005).
Comparison between the FDRs and PNPOxs
Structurally and phylogenetically, the FDRs and PNPOxs appear to have a common evolutionary origin, yet these enzyme families have evolved to utilize different cofactors. Notwithstanding their structural similarities there are at least two important chemical differences between the two cofactors. One is the lower redox midpoint potential of F420 in solvent at pH 7.5 (−350 mV for F420 versus −230 mV for FMN) (Draper and Ingraham, 1968; Jacobson and Walsh, 1984). The other is that F420 is not stable as a semiquinone and may only catalyse a two electron reduction, while FMN may catalyse one or two electron reduction and oxidation reactions (Goldberg et al., 1981). Indeed, owing to this, the chemistry that is generally catalysed by the two enzyme families is opposite (oxidation by the PNPOxs versus reduction by the FDRs). We suggest it is this difference in catalytic activity that has resulted in the evolutionary expansion of the FDRs in M. smegmatis (28 genes) compared with the single PNPOx gene; the reducing power of F420H2 may have allowed these enzymes to catalyse the reduction of a wide variety of compounds, particularly xenobiotics, and provided a selective pressure for duplication and specialization.
There are also significant differences in the mechanisms of cofactor recycling between the FDRs and the PNPOxs. PNPOxs bind FMN with high affinity, KD 30 nM (Wada and Snell, 1961), and require molecular oxygen as the electron acceptor (Pogell, 1958; Notheis et al., 1995). In contrast, there is a requirement for F420 to be enzymatically reduced by FGD, at least in Mycobacteria and Nocardia (Purwantini et al., 1997). It is currently impossible to determine whether a common evolutionary progenitor of the FDRs and PNPOxs would have used either FMN, F420H2, or perhaps a different molecule again (such as a common phosphorylated intermediate from the biosynthetic pathways of FMN and F420; Fig. S2) as a cofactor, but these families of enzymes have the potential to advance our understanding of how enzymes evolve to utilize the catalytic power that different cofactors provide.
Experimental procedures
Bacterial strains, chemicals
Aflatoxins B1, B2, G1 and G2 were obtained from Sigma-Aldrich and Fermentek (Israel). All bacterial strains were previously isolated in our laboratory (Sutherland et al., 2002; Horne et al., 2002a,b), except for M. smegmatis mc2155, which was obtained from Dr H. Billman-Jacob (University of Melbourne, Australia). F420 was purified from M. smegmatis soluble fractions according to previously published methods (Isabelle et al., 2002).
Identification of aflatoxin degrading bacteria
Bacterial strains were first grown on Luria–Bertani (LB) agar before inoculation into Peptone Yeast extract Broth (PYB; 9 g/l peptone, 4.5 g l−1 yeast extract, 23 mM Na2HPO4, 88 mM KH2PO4, 9 mM NaCl, pH 6.0) supplemented with 4 µg ml−1 AFG1 or 6 µg ml−1 AFB1, and incubation at 28°C for 48 h on an orbital shaker (200 rpm). Five microlitres of each reaction mixture was spotted and dried onto silica gel 60 F254 TLC plates (Merck, Germany). Chloroform/acetone/acetic acid (40:10:1 by volume) was used as the developing solvent and aflatoxin fluorescence was detected by viewing under ultraviolet light (365 nm). Images of TLC plates were recorded by an AlphaImager 2200 Imaging System (Alpha Innotech, USA) fitted with an ethidium bromide bandpass filter.
Transposon mutagenesis of M. smegmatis
Random insertion mutants of M. smegmatis mc2155 were generated with the EZ::TN <R6Kγori/KAN-2> insertion kit (Epicentre, USA). The EZ::TN <R6Kγori/KAN-2> tnp transposase complex (1 µl) was used to electroporate 100 µl of electrocompetent M. smegmatis mc2155 cells, which was then plated onto LB agar plates containing 20 µg ml−1 kanamycin. Approximately 2000 mutants were obtained per transformation. Transposon insertion mutants were individually inoculated into 2 ml square wells of 96-deep-well growth blocks (Axygen, USA) containing 200 µl of PYB supplemented with 20 µg ml−1 kanamycin and 4 µg ml−1 aflatoxin G1. The growth blocks were sealed with silicone mats (Axygen) and incubated for 3 days (37°C at 200 rpm) before 5 µl of each culture was examined for aflatoxin degradation by TLC. Mutants that exhibited detectable growth but had reduced ability to degrade AFG1 compared with wild-type cells were selected for further analysis. The genomic regions of selected mutants containing the EZ::TN <R6Kγori/KAN-2> transposon were isolated by plasmid rescue. Genomic DNA was isolated using the Bactozol DNA isolation kit (Molecular Research Center, USA), digested with EcoRI, self-ligated and used to electroporate E. coli TransforMax EC100D pir-116 (Epicentre, USA). Transformants containing the transposon-interrupted M. smegmatis DNA were selected by plating onto LB agar containing 40 µg ml−1 kanamycin. The resulting plasmids were isolated and the genomic regions flanking the transposon were sequenced using primers supplied with the EZ::TN <R6Kγori/KAN-2> insertion kit.
Identification of AFG1-degrading proteins in M. smegmatis
Protein fractions with AFG1 activity were purified from M. smegmatis by ammonium sulphate precipitation, hydrophobic interaction chromatography, anion exchange chromatography and gel filtration chromatography, as detailed below. Proteins from active fractions were visualized by SDS-PAGE and Coomasie staining. Mass spectrometry of tryptic digests with an Agilent XCT ion trap mass spectrometer (Campbell et al., 2008) was used to identify the proteins from protein bands excised from SDS-PAGE and active fractions from gel filtration chromatography. Agilent's Spectrum Mill software was used to match the data with annotated protein sequences in the M. smegmatis mc2155 CMR database (Peterson et al., 2001).
Chromatography of M. smegmatis proteins
Mycobacterium smegmatis was grown for 4 days before centrifugation at 10 000 g and lysis via sonication with glass beads in 20 mM Tris-HCl, pH 7.5, with 1 mg ml−1 lysozyme, 5 mM DTT and 1 mM PMSF. Saturated (NH4)2SO4 solution (0°C) was added to the soluble fraction to a concentration of 40% before centrifugation at 20 000 g. A second (NH4)2SO4 cut (70%) of the soluble fraction was made and the resulting pellet was resuspended in 1 M (NH4)2SO4, 20 mM Tris-HCl, pH 7.5, and clarified via filtration with a 0.22 µm filter. A Biologic HR FPLC system (Bio-Rad, USA) was used during the subsequent chromatographic separation steps. Hydrophobic interaction chromatography (HIC) was performed using a 200 ml phenyl sepharose high performance column (GE Healthcare, USA) equilibrated with 1 M (NH4)2SO4, 20 mM Tris-HCl, pH 7.5. After the resolubilized (NH4)2SO4 pellet was loaded onto the column, proteins were eluted over a linear gradient [1 M–0 M (NH4)2SO4] over 100 ml. Active fractions were pooled and dialysed against 20 mM Tris-HCl, pH 7.5 prior to anion exchange chromatography with a MonoQ HR 5/5 column (GE Healthcare), equilibrated with 20 mM Tris-HCl, pH 7.5. The dialysed fractions were loaded onto the column and eluted over a linear gradient (0–0.5 M NaCl) over 20 column volumes. For size exclusion chromatography, as used in the second purification, HIC active fractions were pooled and loaded onto a Superdex 200 Hi Load 26/60 gel filtration column (GE Healthcare), equilibrated with 150 mM NaCl, 20 mM Tris-HCl, pH 7.5. Gel filtration chromatography standards (GE Healthcare) were run in parallel to allow estimation of the size of the proteins in solution.
Phylogeny
Homologues of the four aflatoxin degrading enzymes identified by reverse genetics were identified by searching the TIGR CMR peptide and NCBI protein databases, using the blast and CDD (Marchler-Bauer et al., 2007). MEGA4 (Tamura et al., 2007) was used for the phylogenetic analysis of a limited data set of 146 amino acid sequences from seven different Actinomycetales (M. smegmatis, M. tuberculosis H37Rv, M. vanbaalenii, Rhodococcus sp. RH1, Arthrobacter sp. FB24, S. coelicolor, Frankia alni and Nocardioides sp. JS614), plus the known PNPOx enzymes from H. sapiens, E. coli, Saccharomyces cerevisiae, Caenorhabditis elegans and Mus musculus. Protein sequences were aligned by the Gonnett algorithm and the tree constructed from the N-terminal sequences using the neighbour-joining method with pairwise deletion, poisson correction and 1000 bootstrap replicates. The 28 M. smegmatis PNPOx-like sequences are referred to by their TIGR locus numbers (MSMEG_).
Cloning, expression and purification of recombinant proteins
Genes were amplified from M. smegmatis mc2155 genomic DNA using Platinum high fidelity Taq polymerase (Invitrogen, USA) using the primer pairs in Table S3. The amplicons were recombined into the Gateway expression vector, pDEST17 (Invitrogen, USA) and the sequences confirmed by capillary electrophoresis (Micromon, Australia). Proteins were expressed in E. coli BL21-AI (Invitrogen) and purified by nickel agarose affinity chromatography using a 1 ml Ni-NTA superflow column (Qiagen, Germany). Proteins were stored at 4°C in 50 mM Tris-HCl pH 7.5. The FDR MSMEG_5998, which was purified as exclusion bodies, was refolded following the methods of Whitbread et al. (2005). Protein concentrations were determined by measuring absorbance at 280 nm using a NanoDrop Spectrophotometer ND1000 and calculated based on the extinction co-efficient for each protein determined using Vector NTI (Invitrogen). FMN was stripped from MSMEG_5675 by dialysis, as described previously (di Salvo et al., 1998), and this was confirmed by loss of the FMN absorption maxima at 450 nm. For protein crystallography, MSMEG_3380 and MSMEG_3356 were cloned into pDEST17, with a TEV-cleavage site designed into the forward primer (Table S3). Expression and purification were performed as described previously (Jackson et al., 2008). The fgd gene was cloned into the NdeI and BamHI sites of pET14b (Novagen, Germany) and expressed in Tuner cells (Novagen) by induction with 0.4 mM Isopropyl-β-D-thiogalactopyranoside (IPTG).
Enzyme assays
The FGD activity was estimated using the previously described spectrophotometric assay for F420 to F420H2 reduction (Purwantini and Daniels, 1996). One unit of FGD activity was defined as the amount of enzyme required to reduce one µmole of F420 per min. Assays of aflatoxin degradation activity were conducted in either 10 µl or 20 µl reaction volumes, in the dark, at room temperature, using reaction mix (30 µM aflatoxin, 10 µM F420, 0.2 U µl−1 FGD, 2.5 mM glucose 6-phosphate, 20 mM Tris-HCl, pH 7.5) and enzyme. Reactions were incubated for 0.5 min to 24 h depending on the enzyme and aflatoxin used. The reaction was stopped by the addition of formic acid to a final concentration of 2% and incubated on ice for 15 min. Protein was pelleted by centrifugation for 5 min at 14 000 g. Samples were injected onto an Agilent Zorbax XDB C18 column (3.5 µm, 2.1 × 30 mm). Aflatoxin concentrations were quantified using an Agilent 1200 series binary HPLC running isocratically with 30% methanol and 0.5% acetic acid and detected at 365 nm and quantified using Chemstation software (Agilent). Results are given in specific activity units because of the low aqueous solubility of aflatoxin [∼100 µM for AFB1 (Grant and Phillips, 1998)], which prevented the estimation of kinetic parameters.
LC/MS assays
The reaction products of aflatoxin reduction were determined using an Agilent 1100 Series Binary LC with diode array detector and in-line Time of Flight Mass Spectrometer (MSD TOF). Samples were separated on an Agilent Zorbax XDB C18 column (3.5 µm, 2.1 × 30 mm) over a gradient of 5% acetonitrile (v/v) and 0.1% formic acid (v/v) from 0.5 min to 20% acetonitrile at 2 min, and then increased to 50% acetonitrile at 10 min, at a flow rate of 0.3 ml min−1. Reaction products were analysed using Analyst QS software.
Structural analysis
The crystallization and data collection of 3380 have been previously reported (Jackson et al., 2008). The crystallization of Seleno-Methionine 3356 was performed in 2 µl hanging drops with 20 mg ml−1 enzyme and a reservoir solution of 36% PEG-MME 5000, 0.1 M sodium acetate, pH 5.5. Seleno-Methionine labelled crystals were soaked in cryoprotectant [35% PEG 10K, 10 mM Tris-HCl, pH 8.5 for MSMEG_3380 and reservoir solution with 10% glycerol and 10% ethylene glycol (v/v) for MSMEG_3356] before vitrification in liquid nitrogen. Both data sets were collected at the Australian Synchrotron, 3380 on the PX1 beamline and 3356 on the PX2 microfocus beamline. The use of the microfocus beamline for 3356 was necessitated by severe non-merohedral twinning of the crystals; crystals were scanned for regions displaying a minimum of twinning. Intensity data were collected at 100 K using a wavelength corresponding to the SeMet edge at 0.9793 Å and a 1.0° oscillation angle per image. Diffraction data were integrated and scaled using the programs MOSFLM (Leslie, 2006) and SCALA (Evans, 2006). Details of the data collection are given in Table 2. Based on systematic absences, the 3380 and 3356 crystals were assigned to the P212121 and P21 space groups respectively. The SHELX suite of programs (Schneider and Sheldrick, 2002) was used for experimental phasing. Four and eight selenium sites were found in the asymmetric units of 3380 and 3356, respectively, using SHELXD, and were used to calculate phases. After refinement and density modification, a significant amount of both structures, corresponding to a dimer in the asymmetric unit of MSMEG_3380 and a tetramer in the asymmetric unit of MSMEG_3358, could be auto-built using ARP/wARP (Perrakis et al., 2001), yielding a preliminary Rfree values of 0.304 and 0.421 for 3380 and 3356 respectively. Further maximum-likelihood refinement for 3380, or twin-refinement with a twin fraction of 7% for 3356, using REFMAC5 (Murshudov et al., 1997), interspersed with manual building in COOT (Emsley and Cowtan, 2004), addition of water molecules and final anisotropic (for 3380) refinement bought the final Rwork/Rfree values to 16.8/19.1 and 19.0/25.5 for 3380 and 3356 respectively. The F420 cofactor of the FDRs was docked into the structures using the docking program CDOCKER (Wu et al., 2003), as implemented in the Accelrys Discovery Studio. Hydrogens were added to cofactor and enzyme and the CHARMm forcefield was used. Slight manual adjustment of the top hit was made using COOT. Analysis of cavity volumes was performed by submitting the coordinates to the 3V server (http://3vee.molmovdb.org) (Voss and Gerstein, 2010), and all images were created using Pymol (http://www.pymol.org).
Table 2.
Data collection and refinement statistics for the crystallography
| Crystal | MSMEG_3356 | MSMEG_3380 |
|---|---|---|
| PDB ID | 3H96 | 3F7E |
| Space group | P1211 | P212121 |
| Unit cell (Å) | a = 59.65, b = 71.15, c = 63.84 | a = 56.86, b = 65.35, c = 69.74 |
| Unit cell (°) | α = 90, β = 90.39, γ = 90 | α = 90, β = 90, γ = 90 |
| Data collection | ||
| Resolution (Å)a | 45.7–2.0 (2.11–2.00) | 22–1.23 (1.23–1.30) |
| Unique reflections | 34129 | 73957 |
| Redundancy | 7.2 (6.4) | 6.4 (6.2) |
| <I/σ(I)> | 5.7 (3.3) | 6.8 (1.5) |
| Completeness (%) | 94.5 (91.5) | 97.6 (95.4) |
| Rsym (%)b | 10.5 (17.4) | 7.2 (49.4) |
| Refinement | ||
| No. reflections work/free | 31 389/2743 | 70 232/3725 |
| Resolution range | 45.7–2.0 (2.05–2.00) | 20–1.23 (1.26–1.23) |
| Rwork/Rfree (%)c | 19.0/25.5 (20.9/27.7) | 16.8/19.1 (20.0/23.1) |
| R.m.s deviations | ||
| Lengths (Å) | 0.025 | 0.012 |
| Angles (°) | 1.84 | 1.59 |
Values in parenthesis are for the highest resolution shell.
Rsymm = |Ij – Ij|/Ij, where Ij is the averaged intensity for symmetry related reflections.
Rwork = |F(obs) – F(calc)|/F(obs); 5% of the data that were excluded from the refinement were used to calculate Rfree.
Acknowledgments
We thank Dr Helen Billman-Jacob for kindly providing the Mycobacterium smegmatis mc2155 strain. We gratefully acknowledge Professor Chris Easton and Dr Mike Lacey for their assistance in interpreting the data. We thank the CSIRO C3 crystallization facility for assistance in obtaining protein crystals and the Australian Synchrotron for beamtime. This work forms part of the Grain Protection Genes initiative, which has been jointly funded by CSIRO and the Australian Grains R&D Corporation.
Supporting information
Additional supporting information may be found in the online version of this article.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Alberts JF, Engelbrecht Y, Steyn PS, Holzapfel WH, van Zyl WH. Biological degradation of aflatoxin B1 by Rhodococcus erythropolis cultures. Int J Food Microbiol. 2006;109:121–126. doi: 10.1016/j.ijfoodmicro.2006.01.019. [DOI] [PubMed] [Google Scholar]
- Bashiri G, Squire CJ, Moreland NJ, Baker EN. Crystal structures of F420-dependent glucose-6-phosphate dehydrogenase FGD1 involved in the activation of the anti-tuberculosis drug candidate PA-824 reveal the basis of coenzyme and substrate binding. J Biol Chem. 2008;283:17531–17541. doi: 10.1074/jbc.M801854200. [DOI] [PubMed] [Google Scholar]
- Biswal BK, Cherney MM, Wang M, Garen C, James MN. Structures of Mycobacterium tuberculosis pyridoxine 5′-phosphate oxidase and its complexes with flavin mononucleotide and pyridoxal 5′-phosphate. Acta Crystallogr D Biol Crystallogr. 2005;61:1492–1499. doi: 10.1107/S0907444905026673. [DOI] [PubMed] [Google Scholar]
- Biswal BK, Au K, Cherney MM, Garen C, James MN. The molecular structure of Rv2074, a probable pyridoxine 5′-phosphate oxidase from Mycobacterium tuberculosis, at 1.6 angstroms resolution. Acta Crystallograph Sect F Struct Biol Cryst Commun. 2006;62:735–742. doi: 10.1107/S1744309106025012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell PM, Cao AT, Hines ER, East PD, Gordon KH. Proteomic analysis of the peritrophic matrix from the gut of the caterpillar, Helicoverpa armigera. Insect Biochem Mol Biol. 2008;38:950–958. doi: 10.1016/j.ibmb.2008.07.009. [DOI] [PubMed] [Google Scholar]
- Canaan S, Sulzenbacher G, Roig-Zamboni V, Scappuccini-Calvo L, Frassinetti F, Maurin D, et al. Crystal structure of the conserved hypothetical protein Rv1155 from Mycobacterium tuberculosis. FEBS Lett. 2005;579:215–221. doi: 10.1016/j.febslet.2004.11.069. [DOI] [PubMed] [Google Scholar]
- Choi KP, Kendrick N, Daniels L. Demonstration that fbiC is required by Mycobacterium bovis BCG for coenzyme F(420) and FO biosynthesis. J Bacteriol. 2002;184:2420–2428. doi: 10.1128/JB.184.9.2420-2428.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung JY, Fujii I, Harada S, Sankawa U, Ebizuka Y. Expression, purification, and characterization of AknX anthrone oxygenase, which is involved in aklavinone biosynthesis in Streptomyces galilaeus. J Bacteriol. 2002;184:6115–6122. doi: 10.1128/JB.184.22.6115-6122.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciegler A, Lillehoj EB, Peterson RE, Hall HH. Microbial detoxification of aflatoxin. Appl Microbiol. 1966;14:934–939. doi: 10.1128/am.14.6.934-939.1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deppenmeier U. The unique biochemistry of methanogenesis. Prog Nucleic Acid Res Mol Biol. 2002;71:223–283. doi: 10.1016/s0079-6603(02)71045-3. [DOI] [PubMed] [Google Scholar]
- Draper RD, Ingraham LL. A potentiometric study of the flavin semiquinone equilibrium. Arch Biochem Biophys. 1968;125:802–808. doi: 10.1016/0003-9861(68)90517-1. [DOI] [PubMed] [Google Scholar]
- Ebert S, Fischer P, Knackmuss HJ. Converging catabolism of 2,4,6-trinitrophenol (picric acid) and 2,4-dinitrophenol by Nocardioides simplex FJ2-1A. Biodegradation. 2001;12:367–376. doi: 10.1023/a:1014447700775. [DOI] [PubMed] [Google Scholar]
- van Egmond HP, Schothorst RC, Jonker MA. Regulations relating to mycotoxins in food: perspectives in a global and European context. Anal Bioanal Chem. 2007;389:147–157. doi: 10.1007/s00216-007-1317-9. [DOI] [PubMed] [Google Scholar]
- El-Tarabily KA, Sivasithamparam K. Non-streptomycete actinomycetes as biocontrol agents of soil-borne fungal plant pathogens and as plant growth promoters. Soil Biol Biochem. 2006;38:1505–1520. [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Evans P. Scaling and assessment of data quality. Acta Crystallogr D Biol Crystallogr. 2006;62:72–82. doi: 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- Goldberg M, Pecht I, Kramer HE, Traber R, Hemmerich P. Structure and properties of 5-deazaflavin radicals as compared to natural flavosemiquinones. Biochim Biophys Acta. 1981;673:570–593. doi: 10.1016/0304-4165(81)90487-6. [DOI] [PubMed] [Google Scholar]
- Graham DE, White RH. Elucidation of methanogenic coenzyme biosyntheses: from spectroscopy to genomics. Nat Prod Rep. 2002;19:133–147. doi: 10.1039/b103714p. [DOI] [PubMed] [Google Scholar]
- Graham DE, Xu H, White RH. Identification of the 7,8-didemethyl-8-hydroxy-5-deazariboflavin synthase required for coenzyme F(420) biosynthesis. Arch Microbiol. 2003;180:455–464. doi: 10.1007/s00203-003-0614-8. [DOI] [PubMed] [Google Scholar]
- Grant PG, Phillips TD. Isothermal adsorption of aflatoxin B-1 on HSCAS clay. J Agric Food Chem. 1998;46:599–605. doi: 10.1021/jf970604v. [DOI] [PubMed] [Google Scholar]
- Graupner M, White RH. Biosynthesis of the phosphodiester bond in coenzyme F(420) in the methanoarchaea. Biochemistry. 2001;40:10859–10872. doi: 10.1021/bi0107703. [DOI] [PubMed] [Google Scholar]
- Guerra-Lopez D, Daniels L, Rawat M. Mycobacterium smegmatis mc(2) 155 fbiC and MSMEG_2392 are involved in triphenylmethane dye decolorization and coenzyme F-420 biosynthesis. Microbiology-Sgm. 2007;153:2724–2732. doi: 10.1099/mic.0.2006/009241-0. [DOI] [PubMed] [Google Scholar]
- Hasan MR, Rahman M, Jaques S, Purwantini E, Daniels L. Glucose-6-phosphate accumulation in mycobacteria: implications for a novel F420-dependent anti-oxidant defense system. J Biol Chem. 2010;285:19135–19144. doi: 10.1074/jbc.M109.074310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm L, Kaariainen S, Rosenstrom P, Schenkel A. Searching protein structure databases with DaliLite v.3. Bioinformatics. 2008;24:2780–2781. doi: 10.1093/bioinformatics/btn507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hormisch D, Brost I, Kohring GW, Giffhorn F, Kroppenstedt RM, Stackebrandt E, et al. Mycobacterium fluoranthenivorans sp. nov., a fluoranthene and aflatoxin B1 degrading bacterium from contaminated soil of a former coal gas plant. Syst Appl Microbiol. 2004;27:653–660. doi: 10.1078/0723202042369866. [DOI] [PubMed] [Google Scholar]
- Horne I, Sutherland TD, Harcourt RL, Russell RJ, Oakeshott JG. Identification of an opd (organophosphate degradation) gene in an Agrobacterium isolate. Appl Environ Microbiol. 2002a;68:3371–3376. doi: 10.1128/AEM.68.7.3371-3376.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horne I, Sutherland TD, Oakeshott JG, Russell RJ. Cloning and expression of the phosphotriesterase gene hocA from Pseudomonas monteilii C11. Microbiology. 2002b;148:2687–2695. doi: 10.1099/00221287-148-9-2687. [DOI] [PubMed] [Google Scholar]
- Igawa T, Takahashi-Ando N, Ochiai N, Ohsato S, Shimizu T, Kudo T, et al. Reduced contamination by the Fusarium mycotoxin zearalenone in maize kernels through genetic modification with a detoxification gene. Appl Environ Microbiol. 2007;73:1622–1629. doi: 10.1128/AEM.01077-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isabelle D, Simpson DR, Daniels L. Large-scale production of coenzyme F420-5,6 by using Mycobacterium smegmatis. Appl Environ Microbiol. 2002;68:5750–5755. doi: 10.1128/AEM.68.11.5750-5755.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson CJ, Taylor MC, Tattersall DB, French NG, Carr PD, Ollis DL, et al. Cloning, expression, purification, crystallization and preliminary X-ray studies of a pyridoxine 5′-phosphate oxidase from Mycobacterium smegmatis. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2008;64:435–437. doi: 10.1107/S1744309108011512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson F, Walsh C. Properties of 7,8-didemethyl-8-hydroxy-5-deazaflavins relevant to redox coenzyme function in methanogen metabolism. Biochemistry. 1984;23:979–988. [Google Scholar]
- Johnson EF, Mukhopadhyay B. A new type of sulfite reductase, a novel coenzyme F420-dependent enzyme, from the methanarchaeon Methanocaldococcus jannaschii. J Biol Chem. 2005;280:38776–38786. doi: 10.1074/jbc.M503492200. [DOI] [PubMed] [Google Scholar]
- Kelley LA, Sternberg MJ. Protein structure prediction on the Web: a case study using the Phyre server. Nat Protoc. 2009;4:363–371. doi: 10.1038/nprot.2009.2. [DOI] [PubMed] [Google Scholar]
- Lee LS, Dunn JJ, DeLucca AJ, Ciegler A. Role of lactone ring of aflatoxin B1 in toxicity and mutagenicity. Experientia. 1981;37:16–17. doi: 10.1007/BF01965543. [DOI] [PubMed] [Google Scholar]
- Leslie AG. The integration of macromolecular diffraction data. Acta Crystallogr D Biol Crystallogr. 2006;62:48–57. doi: 10.1107/S0907444905039107. [DOI] [PubMed] [Google Scholar]
- Manjunatha UH, Boshoff H, Dowd CS, Zhang L, Albert TJ, Norton JE, et al. Identification of a nitroimidazo-oxazine-specific protein involved in PA-824 resistance in Mycobacterium tuberculosis. Proc Natl Acad Sci USA. 2006;103:431–436. doi: 10.1073/pnas.0508392103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchler-Bauer A, Anderson JB, Derbyshire MK, DeWeese-Scott C, Gonzales NR, Gwadz M, et al. CDD: a conserved domain database for interactive domain family analysis. Nucleic Acids Res. 2007;35:D237–D240. doi: 10.1093/nar/gkl951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller CD, Hall K, Liang YN, Nieman K, Sorensen D, Issa B, et al. Isolation and characterization of polycyclic aromatic hydrocarbon-degrading Mycobacterium isolates from soil. Microb Ecol. 2004;48:230–238. doi: 10.1007/s00248-003-1044-5. [DOI] [PubMed] [Google Scholar]
- Mishra HN, Das C. A review on biological control and metabolism of aflatoxin. Crit Rev Food Sci Nutr. 2003;43:245–264. doi: 10.1080/10408690390826518. [DOI] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- Nakano T, Miyake K, Endo H, Dairi T, Mizukami T, Katsumata R. Identification and cloning of the gene involved in the final step of chlortetracycline biosynthesis in Streptomyces aureofaciens. Biosci Biotechnol Biochem. 2004;68:1345–1352. doi: 10.1271/bbb.68.1345. [DOI] [PubMed] [Google Scholar]
- Notheis C, Drewke C, Leistner E. Purification and characterization of the pyridoxol-5′-phosphate-oxygen oxidoreductase (deaminating) from Escherichia coli. Biochim Biophy Acta. 1995;1247:265–271. doi: 10.1016/0167-4838(94)00235-9. [DOI] [PubMed] [Google Scholar]
- Ohsato S, Ochiai-Fukuda T, Nishiuchi T, Takahashi-Ando N, Koizumi S, Hamamoto H, et al. Transgenic rice plants expressing trichothecene 3-O-acetyltransferase show resistance to the Fusarium phytotoxin deoxynivalenol. Plant Cell Rep. 2007;26:531–538. doi: 10.1007/s00299-006-0251-1. [DOI] [PubMed] [Google Scholar]
- Perrakis A, Harkiolaki M, Wilson KS, Lamzin VS. ARP/wARP and molecular replacement. Acta Crystallogr D Biol Crystallogr. 2001;57:1445–1450. doi: 10.1107/s0907444901014007. [DOI] [PubMed] [Google Scholar]
- Peterson JD, Umayam LA, Dickinson T, Hickey EK, White O. The comprehensive microbial resource. Nucleic Acids Res. 2001;29:123–125. doi: 10.1093/nar/29.1.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pogell BM. Enzymatic oxidation of pyridoxamine phosphate to pyridoxal phosphate in rabbit liver. J Biol Chem. 1958;232:761–776. [PubMed] [Google Scholar]
- Purwantini E, Daniels L. Purification of a novel coenzyme F420-dependent glucose-6-phosphate dehydrogenase from Mycobacterium smegmatis. J Bacteriol. 1996;178:2861–2866. doi: 10.1128/jb.178.10.2861-2866.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purwantini E, Daniels L. Molecular analysis of the gene encoding F420-dependent glucose-6-phosphate dehydrogenase from Mycobacterium smegmatis. J Bacteriol. 1998;180:2212–2219. doi: 10.1128/jb.180.8.2212-2219.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purwantini E, Gillis TP, Daniels L. Presence of F420-dependent glucose-6-phosphate dehydrogenase in Mycobacterium and Nocardia species, but absence from Streptomyces and Corynebacterium species and methanogenic Archaea. FEMS Microbiol Lett. 1997;146:129–134. doi: 10.1111/j.1574-6968.1997.tb10182.x. [DOI] [PubMed] [Google Scholar]
- Safo MK, Musayev FN, Schirch V. Structure of Escherichia coli pyridoxine 5′-phosphate oxidase in a tetragonal crystal form: insights into the mechanistic pathway of the enzyme. Acta Crystallogr D Biol Crystallogr. 2005;61:599–604. doi: 10.1107/S0907444905005512. [DOI] [PubMed] [Google Scholar]
- di Salvo M, Yang E, Zhao G, Winkler ME, Schirch V. Expression, purification, and characterization of recombinant Escherichia coli pyridoxine 5′-phosphate oxidase. Protein Expr Purif. 1998;13:349–356. doi: 10.1006/prep.1998.0904. [DOI] [PubMed] [Google Scholar]
- di Salvo ML, Ko TP, Musayev FN, Raboni S, Schirch V, Safo MK. Active site structure and stereospecificity of Escherichia coli pyridoxine-5′-phosphate oxidase. J Mol Biol. 2002;315:385–397. doi: 10.1006/jmbi.2001.5254. [DOI] [PubMed] [Google Scholar]
- di Salvo ML, Safo MK, Musayev FN, Bossa F, Schirch V. Structure and mechanism of Escherichia coli pyridoxine 5′-phosphate oxidase. Biochim Biophys Acta. 2003;1647:76–82. doi: 10.1016/s1570-9639(03)00060-8. [DOI] [PubMed] [Google Scholar]
- Schneider TR, Sheldrick GM. Substructure solution with SHELXD. Acta Crystallogr D. 2002;58:1772–1779. doi: 10.1107/s0907444902011678. [DOI] [PubMed] [Google Scholar]
- Schrey SD, Tarkka MT. Friends and foes: streptomycetes as modulators of plant disease and symbiosis. Antonie Van Leeuwenhoek. 2008;94:11–19. doi: 10.1007/s10482-008-9241-3. [DOI] [PubMed] [Google Scholar]
- Singh R, Manjunatha U, Boshoff HI, Ha YH, Niyomrattanakit P, Ledwidge R, et al. PA-824 kills nonreplicating Mycobacterium tuberculosis by intracellular NO release. Science. 2008;322:1392–1395. doi: 10.1126/science.1164571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smiley RD, Draughon FA. Preliminary evidence that degradation of aflatoxin B1 by Flavobacterium aurantiacum is enzymatic. J Food Prot. 2000;63:415–418. doi: 10.4315/0362-028x-63.3.415. [DOI] [PubMed] [Google Scholar]
- Stavri M, Gibbons S. The antimycobacterial constituents of dill (Anethum graveolens) Phytother Res. 2005;19:938–941. doi: 10.1002/ptr.1758. [DOI] [PubMed] [Google Scholar]
- Sutherland TD, Horne I, Harcourt RL, Russell RJ, Oakeshott JG. Isolation and characterization of a Mycobacterium strain that metabolizes the insecticide endosulfan. J Appl Microbiol. 2002;93:380–389. doi: 10.1046/j.1365-2672.2002.01728.x. [DOI] [PubMed] [Google Scholar]
- Tamura K, Dudley J, Nei M, Kumar S. MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol Biol Evol. 2007;24:1596–1599. doi: 10.1093/molbev/msm092. [DOI] [PubMed] [Google Scholar]
- Taverniers I, Papazova N, Bertheau Y, De Loose M, Holst-Jensen A. Gene stacking in transgenic plants: towards compliance between definitions, terminology, and detection within the EU regulatory framework. Environ Biosafety Res. 2008;7:197–218. doi: 10.1051/ebr:2008018. [DOI] [PubMed] [Google Scholar]
- Tejada-Castaneda ZI, Avila-Gonzalez E, Casaubon-Huguenin MT, Cervantes-Olivares RA, Vasquez-Pelaez C, Hernandez-Baumgarten EM, Moreno-Martinez E. Biodetoxification of aflatoxin-contaminated chick feed. Poult Sci. 2008;87:1569–1576. doi: 10.3382/ps.2007-00304. [DOI] [PubMed] [Google Scholar]
- Teniola OD, Addo PA, Brost IM, Farber P, Jany KD, Alberts JF, et al. Degradation of aflatoxin B(1) by cell-free extracts of Rhodococcus erythropolis and Mycobacterium fluoranthenivorans sp. nov. DSM44556(T) Int J Food Microbiol. 2005;105:111–117. doi: 10.1016/j.ijfoodmicro.2005.05.004. [DOI] [PubMed] [Google Scholar]
- Voss NR, Gerstein M. 3V: cavity, channel and cleft volume calculator and extractor. Nucleic Acids Res. 2010;38(Suppl):W555–W562. doi: 10.1093/nar/gkq395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wada H, Snell EE. The enzymatic oxidation of pyridoxine and pyridoxamine phosphates. J Biol Chem. 1961;236:2089–2095. [PubMed] [Google Scholar]
- Wagacha JM, Muthomi JW. Mycotoxin problem in Africa: current status, implications to food safety and health and possible management strategies. Int J Food Microbiol. 2008;124:1–12. doi: 10.1016/j.ijfoodmicro.2008.01.008. [DOI] [PubMed] [Google Scholar]
- Whitbread AK, Masoumi A, Tetlow N, Schmuck E, Coggan M, Board PG. Characterization of the omega class of glutathione transferases. Methods Enzymol. 2005;401:78–99. doi: 10.1016/S0076-6879(05)01005-0. [DOI] [PubMed] [Google Scholar]
- Willumsen PA, Nielsen JK, Karlson U. Degradation of phenanthrene-analogue azaarenes by Mycobacterium gilvum strain LB307T under aerobic conditions. Appl Microbiol Biotechnol. 2001;56:539–544. doi: 10.1007/s002530100640. [DOI] [PubMed] [Google Scholar]
- Wu G, Robertson DH, Brooks CL, 3rd, Vieth M. Detailed analysis of grid-based molecular docking: a case study of CDOCKER-A CHARMm-based MD docking algorithm. J Comput Chem. 2003;24:1549–1562. doi: 10.1002/jcc.10306. [DOI] [PubMed] [Google Scholar]
- Zucchi TD, de Moraes LA, de Melo IS. Streptomyces sp. ASBV-1 reduces aflatoxin accumulation by Aspergillus parasiticus in peanut grains. J Appl Microbiol. 2008;105:2153–2160. doi: 10.1111/j.1365-2672.2008.03940.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.