

Abstract
The pentalenolactone biosynthetic gene clusters have been cloned and sequenced from two known producers of the sesquiterpenoid antibiotic pentalenolactone, Streptomyces exfoliatus UC5319 and S. arenae TÜ469. The recombinant enzymes PenE and PntE, from S. exfoliatus and S. arenae, respectively, catalyze the flavin-dependent Baeyer-Villiger oxidation of 1-deoxy-11-oxopentalenic acid (7) to pentalenolactone D (8). Recombinant PenD, PntD, and PtlD, the latter from S. avermitilis, each catalyze the Fe2+-α-ketoglutarate-dependent oxidation of pentalenolactone D (8) to pentalenolactone E (15) and pentalenolactone F (16). Incubation of PenD, PntD, or PtlD with the isomeric neopentalenolactone D (9) gave PL308 (12) and a compound tentatively identified as neopentalenolactone E (14). These results are corroborated by analysis of the ΔpenD and ΔpntD mutants of S. exfoliatus and S. arenae, respectively, both of which accumulate pentalenolactone D but are blocked in production of pentalenolactone as well as the precursors pentalenolactones E and F. Finally, complementation of the previously described S. avermitilis ΔptlE ΔptlD deletion mutant with either penE or pntE gave pentalenolactone D (8), while complemention of the ΔptlE ΔptlD double mutant with pntE plus pntD or penE plus pntD gave pentalenolactone F (16).
Terpenoid compounds are ubiquitous in Nature, being widely distributed in terrestial and marine plants, fungi, liverworts, and, as is becoming increasingly common, many bacteria. Of the tens of thousands of known monoterpenes, sequiterpenes, and diterpenes, the several hundred parent cyclic hydrocarbon and alcohol products are formed from the universal acyclic C10, C15, and C20 precursors geranyl, farnesyl, and geranylgeranyl diphosphate. More than a thousand presumptive terpene cyclase genes from plants and microorganisms have been identified, of which about 10% have been experimentally assigned a confirmed biochemical function by identification of their native substrate and characteristic cyclization product. Building on earlier classical isotopic precursor incorporation studies with intact organisms, probing mechanistic investigations using purified, often recombinant, terpene synthases have established many of the key mechanistic details of the intricate cyclization pathways themselves (1–3). Such studies have been powerfully supported by an increasing number of detailed crystallographic investigations (4, 5) and recent computational analyses of cyclization mechanisms (6, 7).
The vast majority of terpenoid metabolites are in fact derived by one or more oxidations and other late stage modifications of the parent hydrocarbons or alcohols initially generated by terpene synthases (3, 8–16). Nature uses an impressive variety of biochemical strategies for the oxidative cleavage of carboncarbon bonds, mediated not just by P450s but also by non-heme-Fe2+-α-ketoglutarate-dependent dioxygenases and flavin-dependent monooxygenases. Little is known about the biosynthetic logic governing this common late-stage oxidative metabolism, including the roles of specific oxygenases and the sequential order of such transformations.
Pentalenolactone (1) is a sesquiterpenoid antibiotic that has been isolated from more than 30 species of Streptomyces (17–20). The antibiotic action of pentalenolactone is due to the presence of an electrophilic epoxylactone moiety that alkylates the active site cysteine of the target glycolytic enzyme glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (21–24). Self-resistance in the pentalenolactone producer Streptomyces arenae TÜ469 is due to an inducible, pentalenolactone-insensitive GAPDH enocoded by the gapR gene (25–28). The biosynthesis of pentalenolactone been studied in considerable detail (29–31) (Scheme 1). Pentalenene synthase, which catalyzes the cyclization of farnesyl diphosphate (2, FPP) to the parent sesquiterpene hydrocarbon pentalenene (3) (32), has been cloned from S. exfoliatus UC5319 and expressed in Escherichia coli (33) and its crystal structure has been determined (34). The detailed mechanism and stereochemistry of the cyclization itself has been elucidated (31, 35). In addition, a variety of oxidized metabolites that represent plausible intermediates and shunt metabolites in the conversion of pentalenene to pentalenolactone have been isolated from several pentalenolactone-producing strains of Streptomyces (36–40).
Scheme 1.

Biosynthesis of Neopentalenolactone D and Proposed Biosynthesis of Pentalenolactone
We have recently reported that the 13.4-kb ptl gene cluster centered at 3.75 MB of the 9.03-Mb linear genome of S. avermitilis harbors 13 unidirectionally transcribed open reading frames (ORFs) that encode several enzymes involved in the early stages of pentalenolactone biosynthesis (41) (Figure 1). We have determined the biochemical function of several of these ORFs, including a pentalenene synthase (PtlA, SAV_2998) (41), a P450-dependent monooxygenase (PtlI, SAV_2999) that catalyzes the oxidative conversion of pentalenene (3) to pentalenal (4) and, most likely, to 1-deoxypentalenic acid (5) (42), an Fe2+-α-ketoglutarate-dependent dioxygenase (PtlH, SAV_2991) that hydroxylates 5 to 1-deoxy-11β-hydroxypentalenic acid (6) (43, 44), and an NAD+-dependent dehydrogenase (PtlF, SAV_2993) that oxidizes 6 to the corresponding ketone, 1-deoxy-11-oxopentalenic acid (7) (45) (Scheme 1, Figure 1). At the 5′-end of the cluster is the resistance gene gap1 (sav2990) which encodes a pentalenolactone-insensitive glyceraldehyde-3-phosphate dehydrogenase (41). Each of these biochemical reactions has been directly demonstrated using the corresponding recombinant proteins and the results have been supported using engineered deletion mutants of S. avermitilis that harbor defined segments of the ptl gene cluster under control of the ermE promoter. In addition, ptlB (sav2997) encodes a presumptive prenyl transferase responsible for the dedicated formation of FPP, while ptlG (sav2992) is predicted to encode a transmembrane efflux protein proposed to be responsible for export of the final antibiotic.
FIGURE 1.

Neopentalenolactone and pentalenolactone biosynthetic gene clusters. (A) S. avermitilis ptl gene cluster for neopentalenolactone biosynthesis. (B) S. arenae pnt gene cluster for pentalenolactone biosynthesis. (C) S. exfoliatus pen gene cluster for pentalenolactone biosynthesis. (D) Proposed S. bingchenggensis pentalenolactone biosynthetic gene cluster.
Conversion of 1-deoxy-11-oxopentalenic acid (7) to pentalenolactone should require four distinct oxidative tranformations, with the intial step expected to be a Baeyer-Villiger-type oxidation of 7 to the corresponding lactone, pentalenolactone D (8) (Scheme 1). In our investigations of the S. avermitilis ptl cluster, however, we unexpectedly found that while recombinant PtlE did indeed catalyze a flavin-dependent Baeyer-Villiger reaction of 7, instead of generating 8 the product of the reaction was the previously unknown isomeric lactone, neopentalenolactone D (9) (46). In support of this finding, the S. avermitilis ΔptlE ΔptlD double deletion mutant accumulated 1-deoxy-11-oxopentalenic acid (7) while the ΔptlD deletion mutant accumulated 9 (Figure 2). In fact, it is now clear that wild-type S. avermitilis does not produce pentalenolactone itself, but instead a group of new metabolites that are neopentalenolactone derivatives. For example, cultures of S. avermitilis SUKA5, from which >1.5 Mb of DNA had been deleted from one end of the 9.05-Mb linear genome (46, 47) but in which the ptl cluster was still intact, produced the novel metabolite neopentalenoketolactone (10), a presumed rearrangement product of the hypothetical labile neopentalenolactone F (11). Similarly, S. avermitilis SUKA16, a derivative of SUKA5 in which a portion of the ptl cluster had been placed under control of the strong constitutive ermE promoter, produced two previously unknown metabolites, characterized as the corresponding methyl esters, PL308 (12) and PL324 (13). Both 12 and 13 are thought to result from hydrolysis of the hypothetical products of oxidation of 9, neopentalenolactones E (14) and F (11). Significantly, neither 10, 12, nor 13 are produced by the S. avermitilis ΔptlD mutant, indicating that each of these metabolites must result from oxidation of neopentalenolactone D (9), presumably mediated by the ptlD gene product.
FIGURE 2.

Production of neopentalenolactone metabolites and intermediates by S. avermitilis mutants.
Although we had discovered a previously unknown branch of the classical pentalenolactone pathway, we were still faced with elucidating the final oxidative steps in the biosynthesis of pentalenolactone itself. We now report the cloning and characterization of the complete pentalenolactone biosynthetic gene clusters from two known producers of pentalenolactone, S. exfoliatus UC5319 (18) and S. arenae TÜ469 (19). We have further established that the orthologous enzymes from the pen and pnt clusters, PenE and PntE respectively, catalyze the flavin-dependent Baeyer-Villiger oxidation of 1-deoxy-11-oxopentalenic acid (7) to pentalenolactone D (8). We also demonstrate that the orthologous proteins PenD and PntD each then mediate the two-stage Fe2+-α-ketoglutarate-dependent oxidation of 8 to pentalenolactones E (15) and F (16), a reaction that we show can also be catalyzed by the closely related S. avermitilis enzyme, PtlD. Simlarly, incubation of PtlD with its natural substrate neopentalenolactone D (9) gives a mixture of neopentalenolactone E (14) and its derived hydrolysis product PL308 (12). These biochemical results are strongly corroborated by analysis of the products of blocked mutants of both S. exfoliatus and S. arenae, as well as by systematic mutant complementation experiments in S. exfoliatus, S. arenae, and S. avermitilis.
EXPERIMENTAL PROCEDURES
Bacterial Strains and Plasmids
Streptomyces and Escherichia coli strains, plasmids and cosmids, are listed in Table S1 (Supplementary Information).
Materials
Reagents and solvents purchased from Sigma-Aldrich or Fisher Scientific were of the highest quality available and were used without further purification. Recombinant 1-deoxy-11β-hydroxypentalenic acid dehydrogenase (PtlF) was expressed and purified as previously described (45). Restriction enzymes and T4 DNA ligase were purchased from New England Biolabs and used according to the manufacturer’s specifications. Isopropylthio-β-D-galactopyranoside (IPTG) was purchased from Invitrogen. Ni-NTA affinity resin was purchased from Qiagen. Amicon Ultra Centrifugal Filter Units (Amicon Ultra-15, 10,000 and 30,000 MWCO) were purchased from Millipore. DNA primers were synthesized by Integrated DNA Technologies. Synthetic genes encoding PenE, PntE, PenD, PntD, and PtlD, optimized for expression in Escherichia coli, were prepared by DNA2.0 and supplied in the vectors pJexpress401 and pJ201.
Methods
General methods were as previously described (41, 46). Growth media and conditions used for E. coli and Streptomyces strains and standard methods for handling E. coli and Streptomyces in vivo and in vitro were those described previously (48, 49), unless otherwise noted. All DNA manipulations were performed following standard procedures (49). DNA sequencing was carried out at the U. C. Davis Sequencing Facility, Davis, CA. All proteins were handled at 4 °C unless otherwise stated. Protein concentrations were determined according to the method of Bradford, using a Hewlett Packard 8452A Diode Array UV/Vis spectrophotometer with bovine serum albumin as the standard (50). Protein purity was estimated using SDS-PAGE gel electrophoresis and visualized using Coomassie Blue stain. GC-MS analyses were carried out using either GC-MS Method 1: Hewlett Packard Series 2 GC-MSD, at 70 eV electron impact (EI), operating in positive ion mode, using a HP5MS capillary column (30 m × 0.25 mm) with a solvent delay of 3 min and a temperature program of 60 °C for 2 min, followed by a termperature gradient of 60–280 °C for 11 min at 20 °C/min, and a hold at 280 °C for 2 min; or GC-MS Method 2: Shimadzu GC-17A, 70 eV EI, operating in positive ion mode, using a neutral bond-5 capillary column (5% phenylmethylsilicon; 30 m × 0.25 mm) with a temperature program of 50–280 °C, 20 °C/min. MALDI-TOF measurements were performed on an Applied Biosystems Voyager DE PRO MALDI-TOF bench top mass spectrometer. LC-ESI-MS analysis of recombinant proteins was carried out on a Thermo LXQ LC-ESI-MS equipped with Surveyor HPLC system and Waters Symmetry C18 column (2.1 mm × 50 mm, 3.5 μm).
Plasmid p56 Harboring the Pentalenene Synthase Gene from S. exfoliatus UC5319
The primer pair MJ-1 (5′-CTGGCCTCCCGTTTCTACCC-3′) and MJ-2 (5′-AGACGCTGCGGCTCTTGGAC-3′) derived from the internal sequence of S. exfoliatus UC5319 pentalenene synthase (GenBank Accession No. U05213) was used to amplify genomic DNA from S. exfoliatus UC5319. The resultant 601-bp PCR product was used as a probe for Southern hybridization with genomic DNA isolated from S. exfoliatus UC5319 that had been digested with different restriction endonucleases, using the Roche DIG High- Prime DNA Labeling and Detection Starter Kit I. A positively-hybrizing band was located at 7 kb in the lane derived from PstI-digested chromosomal DNA. Genomic DNA of S. exfoliatus UC5319 was digested with PstI and the ca. 7-kb DNA fragments were recovered and inserted into the PstI site of pBluescriptII SK(+) to generate a library that was screened by PCR with primer pairs MJ-1 and MJ-2 to identify the positive plasmid p56. Sequencing of the ~7-kb insert of p56 confirmed the presence of the complete pentalenene synthase gene, penA.
Generation and Probing of S. exfoliatus UC5319 and S. arenae TÜ469 Cosmid Libraries
Genomic DNA from S. exfoliatus UC5319 and from S. arenae TÜ469 was isolated by salting out (48) and partially digested with BfuCI to obtain 30–60 kb DNA fragments. The DNA fragments were dephosphorylated with CIAP and cloned into the vector pHZ1357 (51) that had been treated with XbaI, then CIAP, and then BamHI. Each of the libraries was packaged using the Stratagene Gigapack III XL packaging extract and transduced into E. coli DH10B. LB agar plates with 100 μg/ml ampicillin were used to select transductants. The genomic library of S. exfoliatus UC5319 was made up of 2000 colonies and the genomic library of S. arenae TÜ469 consisted of 2880 colonies. The primer pairs MJ-1 and MJ-2 were used to screen the S. exfoliatus UC5319 cosmid library by PCR, giving positive cosmids G21, K5, and O34. The primer pair DQ66F, 5′-CGCCTACACGCAGGACCAGA-3′, and DQ66R, 5′-ACAGGGACGACCCGATGAGC-3′ based on an internal region of the known DNA sequence of S. arenae gapR (GenBank Accession No. U44856) was used to screen the S. arenae TÜ469 cosmid library. Six positive cosmids, 1E2, 7D7, 12B2, 21A5, 21F7, 27H6 were selected by PCR based on the generation of the 461-bp PCR product.
Sequencing and Sequence Assembly
For the S. exfoliatus UC5319 cluster, plasmid p56 harboring the pentalenene synthase gene and flanked by a partial ptl-like gene cluster of ~6.9 kb was sequenced by primer walking. Cosmids G21, K5, and O34 from the genomic library of S. exfoliatus UC5319 were further screened by PCR using the primer pair MJ-3 (5′-TGGCGACGATCCAGGCGGTC-3′) and MJ-4 (5′-CCATCACCACGCCGGATTCG-3′) derived from plasmid p56 and corresponding to the partial penG gene. Only Cosmid G21 gave an amplicon of the predicted size (573 bp). Cosmid G21 was therefore selected for sequencing by primer walking in order to obtain complete DNA sequence information for the S. exfoliatus UC5319 pentalenolactone biosynthetic gene cluster. Each of the six positive cosmids of S. arenae TÜ469 were digested with different restriction endonuclease enzymes to build up restriction enzyme patterns. Several BamHI DNA fragments were then inserted into the BamHI site of vector pSET152 to generate subclones that were sequenced at each end. Four BamHI DNA fragments of about 18 kb were believed to harbor the complete ptl-like gene cluster according to the restriction enzyme pattern and primary DNA sequence information. Four subclones, pDQ19 (or pDQ9 from 21F7), pDQ20, pDQ22 (or pDQ7 from 21F7), and pDQ23 containing the four BamHI DNA fragments from cosmid 1E2 and a subclone pDQ31 containing a KpnI DNA fragment about 7 kb from cosmid 7D7 as a supplement were sequenced. The DNA fragments recovered from these plasmids were partially digested with BfuCI to obtain 1–2 kb DNA fragments that were each inserted into the BamHI site of pBluescriptII SK(+) to generate a small library that was submitted for sequencing. Multiple alignments of sequences were constructed by using the BioEdit sequence alignment editor. The gaps were finished by primer walking. The sequence data were analyzed with the online FramePlot program (http://www.nih.go.jp/~jun/cgi-bin/frameplot.pl). DNA and deduced protein sequence homology searches were performed by using the NCBI BLAST server. The ptl-like gene cluster of S. exfoliatus UC5319 was named the pen gene cluster and the ptl-like gene cluster of S. arenae TÜ469 was named pnt gene cluster (Figure 1). Both sequences have been deposited in GenBank with Accession Numbers HQ292066 and HQ292065, respectively.
Expression and Purification of Recombinant PenE and PntE Proteins
The synthetic penE and pntE genes optimized for expression in E. coli carried a 5′-NdeI site including the ATG start codon and a XhoI site immediately downstream of the stop codon. The plasmid pJexpress401:penE_opt_alt2 was digested with NdeI and XhoI and sub-cloned into the corresponding sites of pET-28a(+) vector to give the expression plasmid pET28a-penE which was then transformed into E. coli BL21(DE3). For expression of synthetic pntE, a pair of primers DQ75F3 (5′-CGGCAGCCATATGGTTGATCTGGAG-3′ [NdeI underlined]) and DQ75R3 (5′-GGCTCCTCGAGGCGCAGCTCCAGGC-3′ [XhoI underlined]) was used to amplify the synthetic gene pntE in pJexpress401:pntE_opt_alt2. The PCR product was digested with NdeI and XhoI and inserted into the corresponding site of pET-26b to generate expression plasmid pDQ58 (pET26b-pntE) that was sequenced to confirm that no mutations had been introduced during PCR amplification. For overexpression of recombinant PenE with an N-terminal His6-tag and PntE with a C-terminal His6-tag, E. coli BL21(DE3) harboring pET28a-penE or pDQ58 (pET26b-pntE) was grown in Terrific Broth (TB) media supplemented with 50 μg/mL of kanamycin at 37 °C until the OD600 reached 0.6–0.8. IPTG was added to a final concentration of 0.4 mM and the culture was further incubated at 18 °C overnight. The cells were then harvested by centrifugation at 5,000g for 15 min and resuspended in Lysis Buffer A (50 mM Tris-HCl, 10% glycerol, 300 mM NaCl, 0.1 mM DTT, 2.7 mM β-mercaptoethanol, 10 mM imidazole, pH 8.0) containing 10 mg/l pepstatin, 10 mg/l phenylmethylsulfonyl fluoride (PMSF), and 0.2 mg/ml benzamidine. After cell-disruption by sonication, the cell debris was removed by centrifugation at 20,000g for 30 min and the supernatant was loaded into a Ni-NTA column pre-equilibrated with lysis buffer. After collecting the flow-through and washing with 20 mM imidazole in Lysis Buffer A at a flow rate of 2 mL/min, the PenE protein was eluted with Elution Buffer A (50 mM Tris-HCl, 10% glycerol, 300 mM NaCl, 0.1 mM DTT, 2.7 mM β-mercaptoethanol, 200 mM imidazole, pH 8.0) at a flow rate of 1 mL/min. The fractions were analyzed by SDS-PAGE. The pooled PenE protein was concentrated and buffer-exchanged into storage buffer (50 mM NaH2PO4, 0.1 mM DTT, pH 7.0) using an Amicon Ultra Centrifugal Filter (Ultracel- 30K) while the recombinant PntE was concentrated and buffer-exchanged using a PD-10 gel filtration column into the storage buffer containing 10% glycerol (50 mM NaH2PO4, 0.1 mM DTT, pH 7.0 containing 10% glycerol). The yield of purified recombinant PenE was 35 mg/L of culture and of PntE 27 mg/L culture. UV, Pen E, λmax 375, 450 nm; PntE, λmax 380, 450 nm; MALDI-TOF, His6-PenE 67480±30 Da (predicted P-Met, 67518); PntE-His6-tag 66915±30 Da (predicted 67018). ESI-MS, PntE-His6- tag, m/z 67008.
Expression and Purification of Recombinant PenD, PntD, and PtlD Proteins
The synthetic penD and pntD genes in the pJexpress401 vector and synthetic ptlD in pJ201 were prepared by DNA2.0 with codons optimized for expression in E. coli. Each of the plasmids were digested with NdeI and XhoI and the penD, pntD, and ptlD products ligated into doubly-digested pET-28a(+) to give the expression vectors pET28a-penD, pET28a-pntD, and pET28a-ptlD, respectively, which were then individually transformed into E. coli BL21(DE3). Cultures of each of the recombinant E. coli transformants were grown in TB media containing 50 μg/mL of kanamycin at 37 °C to OD600 of 0.6–0.8. After addition of 0.4 mM IPTG, the culture was further incubated at 18 °C overnight. The harvested cells were resuspended in Lysis Buffer B (50 mM NaH2PO4, 300 mM NaCl, 10 mM imidazole, pH 8.0) and the suspension was then sonicated. After removal of cell debris, the cell-free extract was applied to a Ni- NTA column and washed with 20 mM imidazole in Lysis Buffer B. The proteins were finally eluted with Elution Buffer B (50 mM NaH2PO4, 300 mM NaCl, 250 mM imidazole, pH 8.0). The concentrated protein was exchanged into the storage buffer (20 mM Tris-HCl, 20% glycerol, pH 7.5) using an Amicon Ultracentrifugal Filter (Ultracel-10K). The purified PenD, PntD, PtlD concentrations were 42, 36, and 48 mg/L of culture, respectively. ESI-MS for His6-tag-PenD, m/z 35472 (predicted 35532); ESIMS for His6-tag-PntD, m/z 35626 (predicted 35624); ESI-MS for His6-tag-PtlD, m/z 36268) (predicted P-Met, 36269).
Incubations of PenE and PntE with 1-Deoxy-11-oxopentalenic Acid (7)
1-Deoxy-11β-hydroxypentalenic acid (6) (0.1 mM), prepared as described below, was incubated at room temperature with recombinant PtlF (14.4 μM) and β-NAD+ (1.6 mM) in 1 mL of Tris buffer (100 mM Tris-HCl, 1.5 mM DTT, 2% DMSO, pH 8.0) to generate 1-deoxy-11-oxopentalenic acid (7). After 20 min, purified PenE (24.5 μM) or PntE (5.4 μM) protein was added along with β-NADH (1 mM) and FAD (50 μM). After an additional 2 h, the reaction mixture was quenched by addition of 10% HCl to adjust the pH to 2.0, followed by extraction with 3× 1 mL of dichloromethane. The combined organic extracts were dried over with anhydrous Na2SO4 and concentrated on a rotovap. The residue was resuspended in methanol and treated with trimethylsilydiazomethane (TMS-CHN2) to the yield pentalenolactone D methyl ester (8-Me), which was identified by direct GC-MS comparison with an authentic sample of 8-Me (GC-MS Method 1).
Incubations of PenD, PntD, and PtlD with pentalenolactone D (8), neopentalenolactone D (9), or pentalenolactone E (15)
Enzymatic reactions were performed in 10 mM imidazole buffer (5 ml, pH 6.5) containing 1 mM α-ketoglutarate, 1 mM sodium ascorbate, 0.1 mM Fe(NH4)2(SO4)2, 0.5 mM DTT, 1 mg/ml bovine catalase, and 0.1 mM pentalenolactone D (8), neopentalenolactone D (9), or pentalenolactone E (15). The reaction was initiated by addition of purified PntD, PenD, or PtlD protein (8.4 μM) and the mixture was incubated at 30 °C for 1.5 h. After quenching by addition of 10% HCl to adjust the pH to 2.0, the reaction mixture was extracted with dichloromethane. The combined organic extracts were dried over anhydrous Na2SO4 and concentrated in vacuo. The residue was resuspended in methanol and treated with TMS-CHN2 followed by GC-MS analysis of the derived methyl esters (GC-MS Method 1).
Construction of pntD Mutant S. arenae ZD18
The plasmid pJTU1278 is a derivative of pHZ1358 with a cassette carrying multiple cloning sites and a marker for lacZ selection (52). The XbaI and SpeI sites were removed by digestion of pJTU1278 with XbaI and SpeI and religation to generate the vector pDQ44. The method of PCR-targeted Streptomyces gene replacement was used to generate mutants of S. arenae TÜ469 (53). A 6412-bp KpnI DNA fragment from pDQ34 harboring pntD was inserted into the KpnI site of pDQ44 to generate pDQ45. The plasmid pDQ45 was transformed into E. coli BW25113/pIJ790. A pair of primers, DQ79F (5′-ATGGAAGTGACACCGATACCCGGCGCCCCTCTCGGCGCCATTCCGGGGATCCGTCGACC -3′ and DQ79R (5′-TCATGCGGCGGCCTCCGCGCGCCAGGGGCCGGCCGGGTCTGTAGG-CTGGAGCTGCTTC- 3′), was used for PCR amplification, using as template the 1382-bp EcoRI + HindIII DNA fragment from pIJ773 carrying oriT and aac(3)IV to give a 1447-bp product that was transformed into E. coli BW25113/pIJ790 containing pDQ45 to generate plasmid pDQ46 in which the 822-bp DNA fragment of pntD from nt 40 to nt 861 nt had been replaced by oriT and aac(3)IV (~1369 bp) (Figure 4). The plasmid pDQ46 recovered from E. coli BW25113 was transformed into E. coli ET12567/pUZ8002 and the reisolated unmethylated pDQ46 was digested with XbaI and religated to generate pDQ47 from which the oriT and aac(3)IV DNA fragment had been deleted leaving a 81-bp scar.
FIGURE 4.
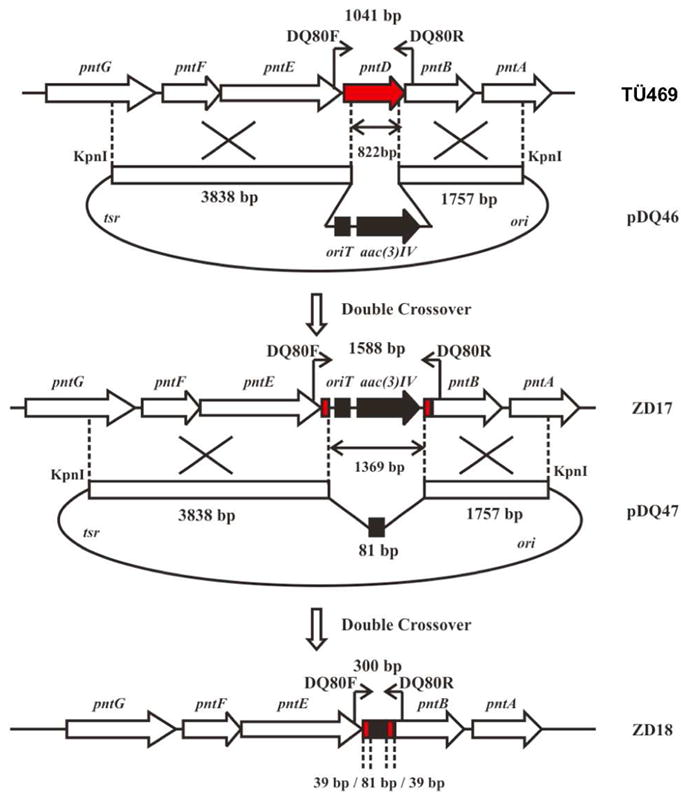
Construction of ΔpntD deletion mutant S. arenae ZD18. See Figure S5.
Plasmid pDQ46 was conjugated into S. arenae TÜ469 using apramycin to select exconjugants. The single crossover strains were inoculated onto SFM agar plates (20 g soy flour, 20 g mannitol, 20 g agar in 1 L water (48)) without any added antibiotic in order to obtain double crossover strains under relaxed conditions with apramycin resistance and without thiostrepton resistance. The resultant mutants were confirmed by PCR using the primer pair DQ80F (5′-TGCGAAAGGAGGCAACGC-3′) and DQ80R (5′-GGTGATCCAGCCGAAGTGGTAG-3′). The PCR product of the wild type S. arenae strain is 1041 bp and the PCR product of the pntD mutant, S arenae ZD17, was 1588 bp in which an internal 822 bp of TÜ469 pntD from 40 nt to 861 nt had been replaced by the 1369-bp fragment harboring oriT and aac(3)IV (Figure S5).
Plasmid pDQ47 was conjugated into S. arenae ZD17 and thiostrepton was used to select exconjugants. The single crossover strains were inoculated on SFM plates without antibiotic in order to obtain double crossover strains that had lost both apramycin and thiostrepton resistance. PCR amplification with the primer pair DQ80F/R gave a 300-bp product from the double crossover strain S. arenae ZD18 in which the 1369-bp oriT and aac(3)IV DNA fragment in ZD17 has been deleted leaving a 81-bp scar corresponding to an in-frame deletion mutant of pntD (Figure S5).
Construction of penD Mutant S. exfoliatus ZD20
A ca. 7 kb BamHI + HindIII DNA fragment harboring penD from plasmid p56 was inserted into doubly digested pDQ44 to generate pDQ49. pDQ49 was transformed into E. coli BW25113/pIJ790. The 1382-bp EcoRI + HindIII DNA fragment from pIJ773 carrying oriT and aac(3)IV was used as tempate for PCR amplification with the primer pair MJ-26 (5′-ATGGACGTAACGCCGATACCCGGCGCAGCTCTCGGGGCAATTCCGGGGATCCGTCGAC C-3′) and MJ-27 (5′-TCATGCGGCGACCTCCGCGTGCCAGGGGTCGGTGGGGTCTGTAGGCTGGAGCTGCTTC -3′) to give a 1447-bp DNA fragment that was transformed into E. coli BW25113/pIJ790 containing pDQ49 to generate plasmid pDQ50 in which an internal 819 bp of penD from nt 40 to nt 858 had been replaced by the ca. 1369-bp oriT and aac(3)IV. The plasmid pDQ50 isolated from E. coli BW25113 was transformed into E. coli ET12567/pUZ8002 and the resultant unmethylated pDQ50 was digested with XbaI and religated to generate pDQ51 in which the 1369 bp oriT and aac(3)IV DNA fragment had been deleted and replaced with a 81-bp scar. Plasmid pDQ51 was conjugated into S. exfoliatus UC5319 and thiostrepton was used to select exconjugants (Figure 3). The single crossover strains were inoculated on SFM plates without any antibiotic under relaxed conditions in order to get double crossover strains that had lost thiostrepton resistance. The primer pair DQ81F (5′-GGCAGGCGAGAAGGAATGG-3′) and DQ81R (5′-TGGTGACG TGAGCGGTGGTC-3′) was used to select the penD mutant, S. exfoliatus ZD20, which gave a PCR product of 503 bp compared to 1241 bp with wild type strain S. exfoliatus UC5319. The in-frame deletion mutant S. exfoliatus ZD20 has an 81-bp scar that has replaced an internal 819 bp of penD from nt 40 to nt 858 (Figure S5).
FIGURE 3.

Construction of ΔpenD deletion mutant S. exfoliatus ZD20. (See also Figure S5).
Analysis of Pentalenolactone Production in Wild-Type S. arenae TÜ469 and Deletion Mutant ZD18(ΔpntD)
The liquid synthetic medium (28) used for fermentation of S arenae TÜ469 and ZD18 contained 4% mannitol, 0.25% asparagines, 0.2% (NH4)2SO4, 0.1% NaCl, 0.3% K2HPO4•3H2O, 0.1% MgSO4•7H2O, 0.04% CaCl2•2H2O, 0.002% FeSO4•7H2O, 0.001% ZnSO4•7H2O, pH 6.2. The strains of S. arenae TÜ469 and ZD18 were fermented at 30 °C for 6 days. The fermentation broth was adjusted to pH 2.5 with HCl and extracted with chloroform. The organic layers were dried over anhydrous Na2SO4, concentrated, methylated with TMS-CHN2 and analyzed by GC-MS.
Analysis of Pentalenolactone Production in Wild-Type S. exfoliatus UC5319 and Deletion Mutant ZD20(ΔpenD)
The liquid medium used in fermentation of S. exfoliatus UC5319 and ZD20 contained 0.2% NaCl, 0.5% CaCO3, 1% corn gluten meal, 0.1125% Bactodextrose, 0.2% blackstrap molasses, and 2% corn starch, pH 7.2 (37). After incubation at 30 °C for 6 days, the culture was acidified to pH 2.4 with H2SO4 and extracted with chloroform. The organic layer was dried over anhydrous Na2SO4, concentrated, methylated with TMS-CHN2 and analyzed by GC-MS.
Complementation of S. avermitilis ΔptlE ΔptlD Double Deletion Mutant with penE, pntE, penD, and pntD
The construction of the S. avermitilis SUKA16 pKU462::ermEp-ptl-cluster ΔptlE ΔptlD double deletion mutant has been previously described (46). The recombinant plasmid pDQ34 (pnt-cluster) was digested with Acc65I and the largest 6,441-bp fragment carrying the pnt cluster was introduced into the unique Acc65I site of pKU464aac(3)IV to generate pKU464aac(3)IV::pnt-cluster. A second recombinant plasmid p56 (pen-cluster) was digested with PstI and the largest 6906-bp fragment carrying the pen cluster was introduced into the unique PstI site of pKU464aac(3)IV to generate pKU464aac(3)IV::pen-cluster. To express pntE-pntD or penE-penD under the control of an alternative constitutive promoter, ermEp was introduced upstream of pntE or penE using the λ recombination system (46). The ermEp promoter with aac(3)I was amplified by PCR using the primer pair, forward: 5′-TCGCGCTCCTGCCGGTACTTCTCCCTCACCGCTTCCAGGTCCATATGTAGATCCTACCA ACCGGC-3′ (bold characters indicate the N-terminal region of pntE and underlined characters correspond to the start condon of pntE) and reverse: 5′-GCAGGTGCTGGCGACGATCCAGGCGGTCACGGCGGGGCAGGGAAGCTAGCGATCTCG GCTTGAAC-3′ (bold characters indicate a part of the region for pntG) for DNA from the pnt cluster and the primer pair, forward: 5′-CCTACGCTGCGCTTGTCGCGTTCCTGCCGGTATTTCTCCCTCATATGTAGATCCTACCA ACCGGC-3′ (bold characters indicate N-terminal region of penE and underlined characters correspond to the start codon of penE) and reverse: 5′-GAAGTTAGCAGCCCTTGCGCCCTGAGTGCTTGCGGCAGCGTGAAGCTAGCGATCTCGG CTTGAAC-3′ (bold characters indicate the region for lacZα of the vector at upstream of cloning site) for the pen cluster, respectively. The initial denaturation step (95 °C, 3 min) was followed by 25 cycles of amplification (95 °C, 30 s; 65 °C, 30 s; 72 °C, 30 s) and final incubation at 72 °C (10 min) using Phusion DNA polymerase (New England Biolobs, USA). Each amplicon was co-transformed into L-arabinose-induced E. coli BW25113 with linearized (SstI-digestion) pKU464aac(3)IV::pnt-cluster or pKU464aac(3)IV::pen-cluster by electroporation as described previously (46). The desired recombinant plasmids were obtained by the selection for resistance with 25 μg/mL of apramycin [aac(3)IV] and 25 μg/mL of fortimicin [aac(3)I] and the isolated plasmids were confirmed by restriction digestion to obtain pKU464aac(3)IV::ermEp::pnt-cluster and pKU464aac(3)IV::ermEp::pen-cluster, respectively. To construct expression cassettes for pntE, pntE-pntD, penE and penE-penD, the downstream region of each plasmid was removed by the λ-recombination system and Cre/loxP site-specific recombination. An antibiotic gene aad(3″), was amplified for each targeted deletion derivative using the designated primer pairs. For pntE expression: forward primer 5′-GACCGTCCCCCACCGTCTCTGCGAAAGGAGGCAACGCCATGCAGTGAGTTCGAGCGAC TCGAGT-3′ (bold characters indicate downstream of pntE and underlined characters correspond to the start codon of pntD) and reverse primer-pnt 5′-TCGAAGCGGCCCGCGCGCTCGGCGAGGTCGACCGTGGGCTGCTGGTACCGAGCGAAC GCGTT-3′ (bold characters indicate the region for pntA) for the construction of the ermEp-pntE cassette. For pntE-pntD expression: forward primer 5′-TGGCGCGCGGAGGCCGCCGCATGAGCGCCGCTCCCGCCTGCCAGTGAGTTCGAGCGAC TCGAGT-3′ (bold characters indicate the region between pntD and pntB, and underlined characters correspond to the stop codon of pntD) and the above reverse primer-pen for the construction of ermEppntE- pntD cassette. For penE expression: forward primer 5′-GCTGACCACCCCACTCTCAGCAAAAGGCGGCAACGCCCATGCAGTGAGTTCGAGCGAC TCGAGT-3′ (bold characters indicate the region between penD and penE, underlined characters correspond to the stop codon of penE and italic characters are the start codon of penD) and reverse primer-pen 5′-GGTCATGAGTTTCTCAACTTCCCGTCTCTACAGATCTGTTGCTGGTACCGAGCGAACGC GTT-3′ (bold characters indicate the downstream of penA) for the construction of the ermEp-penE cassette. For penE-penD expression: 5′-CCGACCCCTGGCACGCGGAGGTCGCCGCATGAGCACCGCTCCAGTGAGTTCGAGCGAC TCGAGT-3′ (bold characters indicate the region between penB and penD and underlined characters correspond to the stop codon of penD) and reverse primer-pen for the construct of ermEp-penE-penD cassette. Each pair of primers was used for the amplification of the loxP-aad(3″)-loxP segment using pKU473 as template DNA. The initial denaturation step (95 °C, 3 min) was followed by 25 cycles of amplification (95 °C, 30 s; 65 °C, 30 s; 72 °C, 60 s) and then a final incubation at 72 °C (10 min). Each amplified segment was introduced by electroporation into L-arabinose-induced E. coli BW25113 carrying the pKU464aac(3)IV::ermEp::pnt-cluster or pKU464aac(3)IV::ermEp::pen-cluster. Transformants were selected by resistance to 25 μg/mL of apramycin, 25 μg/mL of fortimicin, 50 μg/mL of streptomycin and 100 μg/mL of spectinomycin. To remove the resistance gene aad(3″), pTH19cs1::cre, which is a derivative of pSC101 in which cre is controlled by the lacZ promoter and whose replication is temperature-sensitive (54), was introduced into the transformants by the selection of chloramphenicol resistance (30 μg/mL). The chloramphenicol-resistant transformants were spread on LB agar containing 0.1 mM IPTG and 25 μg/mL of apramycin and the plates were incubated overnight at 37 °C to express cre and to cure pTH19cs1::cre. The plates were replicated onto LB agar containing 25 μg/ml of apramycin, 50 μg/mL of streptomycin and 100 μg/mL of spectinomycin, and LB agar- 20 containing 25 μg/mL of apramycin and 30 μg/mL of chloramphenicol, respectively. After plates were incubated overnight at 30 °C, chloramphenicol, streptomycin and spectinomycin-sensitive clones were selected. The desired recombinant plasmids were confirmed by restriction digestion to obtain pKU464aac(3)IV::ermEp::pntE, pKU464aac(3)IV::ermEp::pntE-pntD, pKU464aac(3)IV::ermEp::penE, and pKU464aac(3)IV::ermEp::penE-penD. To introduce each plasmid into the S. avermitilis deletion mutants, each recombinant plasmid was first introduced into E. coli GM2929 hsdS::Tn10 to obtain unmethylated DNA preparations. Protoplasts of S. avermitilis SUKA16 ΔptlE or ΔptlE ΔptlD were transformed by each unmethylated recombinant plasmid preparation using polyethylene glycol as described previously (55). Each transformant was selected by overlaying apramycin. Spores of each transformant were prepared from growth on YMS medium (56) and each spore suspension in 20% glycerol (v/v) was stored at −30 °C.
Analysis of Products from Complementation of the S. avermitilis ΔptlE ΔptlD Double Deletion Mutants
The cultivation of transformants was performed as previously described (41, 46) and the products were isolated as previously described (41, 46, 57). For the analysis of the ratio of pentalenolactone D (8) and neopentalenolactone D (9) of the S. avermtilis SUKA16 ΔptlE ΔptlD double mutant carrying pntE or a ptlE/pntE hybrid gene, cultures of each transformant were centrifuged at 3,000 rpm for 5 min to obtain the fermentation broth which was adjusted to pH 2.0 with 2 N HCl and extracted with chloroform. The organic layers were dried over anhydrous Na2SO4, concentrated and methylated with TMS-CHN2. The extracts were analyzed by GC-MS (Method 2). For HPLC analysis, the culture extracts were subjected to ODS-HPLC (Pegasil 5 μm, 4.6φ × 250 mm) developed with 50% CH3CN in water at a flow rate of 0.8 mL/min (detection at 210 nm).
Isolation of 1-Deoxy-11β-hydroxypentalenic acid (6), pentalenolactone D (8) and pentalenolactone E (15)
The metabolites 1-deoxy-11β-hydroxypentalenic acid (6) and pentalenolactone D (8) were each isolated as the derived methyl esters, as previously described, from large-scale cultures of the appropriate S. avermitilis SUKA16 mutant (46). After treatment of the acidic organic extracts of each culture with TMS-CHN2, the resulting mixtures of methyl esters were purified chromatographically (preparative silica gel and ODS column chromatography) as previously described (46) to yield 6-Me (from the S. avermitilis SUKA16 pKU462::ermEp-ptl-cluster ΔptlF deletion mutant) and 8-Me (from the penE-complemented S. avermitilis SUKA16 pKU462::ermEp-ptl-cluster ΔptlE-ΔptlD/penE+ mutant). Pentalenolactone E methyl ester (15-Me) was isolated from S. exfoliatus UC5319 as previously described (58).
To obtain the corresponding free acids, individual solutions of 6-Me (1.2 mg), 8-Me (5 mg), 9-Me (5 mg) and 15-Me (1.5 mg) in 2 mL of 5% aqueous K2CO3 and 4 mL of methanol were heated at reflux for 24 h. After evaporation of the methanol via rotovap, the aqueous solution was acidified to pH 2.0 with 10% HCl and then extracted with chloroform. The combined organic phase was dried over anhydrous Na2SO4 and concentrated to give, respectively, 1.0 mg of 1-deoxy-11β-hydroxypentalenic acid (6), 4.0 mg of pentalenolactone D (8), 4.2 mg of neopentalenolactone D (9), and 1.2 mg of pentalenolactone E (15).
Construction of ptlE/pntE hybrid genes
Chimeric ptlE and pntE hybrids were prepared by the TAP method (59). The hybrid genes containing ptlE and pntE were constructed by PCR using overlapped primer pairs as follows: a part of the ptlE gene was amplified by PCR using template DNA from pKU462::ptl-cluster and the primer pairs, ptl-pnt_fwd0 forward: 5′-GGCCGGCCATATGGTGGATATCGAGGCAGTGAGAGCGAAGTACCGGGAGGAACGCGA C-3′ (bold characters correspond to the N-terminal region of ptlE, underlined characters indicate NdeI site and italic characters correspond to the start codon of ptlE) and either ptl-pnt_rev300 reverse: 5′-GTAGCTCTCCACGTCGCACCGGACCCCGGGAAAACGGTTCCAGTACCAGGTCCC-3′ (bold characters indicate pntE at 295 to 318 nt and underlined characters are ptlE at 277 to 306 nt), or ptl-pnt_rev600 reverse: 5′-GGTGCTGTCGCCGCCGGTGTAGCCGAAATCCCAGCGGCTGGTGTGGAACG-3′ (bold characters indicate pntE at 637 to 660 nt and underlined characters are ptlE at 623 to 648 nt), or ptl-pnt_rev1200 reverse: 5′-CGCGGTGAGCCGTTCGACGCCCTGTCCCTGGGTGTCCACCAGGGTGA-3′ (bold characters indicate pntE at 1,198 to 1,221 nt and underlined characters are ptlE at 1,184 to 1,206 nt). The corresponding 3′-portions of the pntE gene were amplified by PCR with template DNA from pKU464aac(3)IV::ermEp::pntE using the primer pairs, either ptl-pen_fwd300 forward: 5′-TGGTACTGGAACCGTTTTCCCGGGGTCCGGTGCGACGTGGAGAGCTACGTCTACATGCC- 3′ (bold characters indicate ptlE at 283 to 306 nt and underlined characters are pntE at 295 to 329 nt), or ptl-pnt_fwd600 forward: 5′-TTCCACACCAGCCGCTGGGATTTCGGCTACACCGGCGGCGACAGCACCGG-3′ (bold characters indicate ptlE at 625 to 648 nt and underlined characters are pntE at 637 to 662 nt), or ptl-pnt_fwd1200 forward: 5′-GTCACCCTGGTGGACACCCAGGGACAGGGCGTCGAACGGCTCACCGC-3′ (bold characters indicate ptlE at 1,183 to 1,206 nt and underlined characters are pntE at 1,198 to 1,220 nt), and ptl-pnt_rev0 reverse: 5′-GCTCTAGAGGTGGGGGACGGTCAGCGCAGTTCGAGGCCCGCCAGG-3′ (bold characters indicate C-terminal region of pntE, underlined characters are XbaI site and italic characters correspond to the stop codon of pntE). The initial denaturation step (95 °C, 3 min) was followed by 5 cycles of amplification (95 °C, 30 s; 50 °C, 30 s; 72 °C, 60 s) followed by 15 cycles using an annealing temperature of 65 °C, and then a final incubation at 72 °C (10 min). Each amplicon was treated with T4 DNA polymerase in the presence of dNTPs to remove 3′-nucleotide overhangs at both ends. The second PCR amplification used template DNA from 50-fold dilution of both amplification mixtures, (amplicons derived from ptl-pnt_fwd0/ptl-pnt_rev300 primer pair and ptl-pnt_fwd300/ptl-pnt_rev0 primer pair, ptl-pnt_fwd0/ptl-pnt_rev600 primer pair and ptl-pnt_fwd600/ptl-pnt_rev0 primer pair or ptl-pnt_fwd0/ptl-pnt_rev1200 primer pair and ptl-pnt_fwd1200/ptl-pnt_rev0 primer pair), and Phusion DNA polymerase using 10-fold concentrated primer pair, ptl-pnt_fwd0 and ptl-pnt_rev0. The initial denaturation step (95 °C, 3 min) was followed by 20 cycles of amplification (95 °C, 30 s; 65 °C, 30 s; 72 °C, 100 s) and then a final incubation at 72 °C (10 min). Each amplified fragment was digested with NdeI and XbaI and the resulting fragment was ligated with the large NdeI/XbaI fragment of pKU460aac(3)IV::ermEp. After confirmation of the sequence of each hybrid gene, the desired recombinant plasmids (pKU460aac(3)IV::ermEp::ptlE/pntE300, pKU460aac(3)IV::ermEp::ptlE/pntE600, and pKU460aac(3)IV::ermEp::ptlE/pntE1200, respectively) were propagated in E. coli GM2929 hsdS::Tn10 to prepare unmethylated DNA. The S. avermitilis SUKA16 ΔptlE ΔptlD double-deletion mutant was transformed by PEG-mediated protoplast transformation using the unmethylated DNA preparation (55).
RESULTS
Pentalenolactone biosynthetic gene clusters of S. exfoliatus UC5319 and S. arenae TÜ469
To isolate the pentalenolactone biosynthetic gene clusters of the known pentalenolactone producers S. exfoliatus UC5319 and S. arenae TÜ469, we used probes based on the previously cloned S. exfoliatus pentalenene synthase gene and the S. arenae gapR resistance gene to screen DNA libraries obtained from the corresponding parent organisms. Thus PCR screening of an S. arenae TÜ469 cosmid library with a primer pair based on the sequence of the S. arenae gapR gene led to isolation of 6 candidate cosmids that together were found to harbor the 13-kb pnt cluster encoding 11 ORFs (Figure 1B). Similarly, screening of an S. exfoliatus UC5319 plasmid library with a 601-bp PCR probe derived from the internal sequence of the pentalenene synthase gene allowed isolation of plasmid p56 harboring a 7-kb insert that included the complete pentalenene synthase gene, penA. PCR screening of an S. exfoliatus cosmid library then served to identify a single Cosmid G21 harboring the entire ~13-kb pen biosynthetic cluster consisting of 11 ORFs that were identical in organization to those in the pnt biosynthetic cluster (Figure 1C).
Analysis of the sequence of the S. exfoliatus pen biosynthetic gene cluster (Figure 1C) revealed that the furthest upstream gene, designated penR, encodes a 153-aa MarR-family transcriptional regulator with strong similarity to the deduced SAV_2989 protein that is found just upstream of the previously characterized gap1 (sav2990) pentalenolactone resistance gene of S. avermitilis (Table 1). All but one of the remaining 10 unidirectionally transcribed ORFs in the pen cluster correspond closely in gene organization and deduced amino acid sequence to the corresponding ORFs of the previously characterized S. avermitilis ptl cluster. Thus the S. exfoliatus gapN gene has a high level of similarity to S. avermitilis gap1 that has been shown to encode a pentalenolactone-insensitive GAPDH. The downstream 8 ORFs from penH to penI have the same gene order as ptlH to ptlI and exhibit 65–78% identity and 76–87% similarity at the deduced amino acid level to the corresponding ptl gene products. Only the penM gene, which encodes a predicted 398-aa cytochrome P450 monooxygenase, has no homolog in the ptl cluster. In addition to PenA, the previously characterized S. exfoliatus pentalenene synthase, PenI is assigned as a cytochrome P450 monooxygenase, PenH as a Fe2+-α-ketoglutaratedependent hydroxylase, and PenF as an NAD+-dependent dehydrogenase, based on their close matches to the corresponding PtlI, PtlH, and PtlF proteins, each of established biochemical function (42–45). Together PenI, PenH, and PenF are expected to catalyze the oxidative conversion of pentalenene (3) to 1-deoxy-11-oxopentalenic acid (7), as previously established for the othologous S. avermitilis proteins (Scheme 1). The penB gene is predicted to encode a polyprenyl diphosphate synthase, based on its similarity to ptlB and other prenyltransferase-encoding genes, while penG encodes an apparent transmembrane efflux protein, by analogy to the predicted function of ptlG. Interestingly, the S. exfoliatus pen cluster carries no genes homologous to the S. avermitilis genes ptlC, ptlR, ptlJ and ptlL which have as yet no assigned function. Most importantly, the S. exfoliatus penE gene is predicted to encode a 584-aa protein that is a homolog of the known Baeyer-Villiger monooxygenase from S. avermitilis, PtlE (70% identity and 81% similarity) while penD encodes a 298-aa protein that is a presumptive Fe2+-α-ketoglutarate-dependent dioxygenase with 65% identity and 82% similarity to the S. avermitilis enzyme PtlD.
Table 1.
Comparison of predicted proteins of the ptl, pen, and pnt biosynthetic gene clusters.
| Ptl (aa)a | Pen (aa, % identity/similarity to Ptl) | Pnt (aa, % identity/similarity to Ptl)) | Pen:Pnt (% identity/similarity | Known or predicted function |
|---|---|---|---|---|
| Sav_2989 (161) | PenR (153, 75/88) | PntR (151, 73/85) | 82/89 | predicted MarR family transcriptional regulator |
| Gap1 (334) | GapN (336, 63/76) | GapR (334, 88/92) | 60/75 | glyceraldehyde-3-phosphate dehydrogenase |
| PenM (398) | PntM (398) | 81/87 | predicted cytochrome P450 monooxygenase | |
| PtlH (285) | PenH (283, 73/83) | PntH (283, 73/84) | 89/94 | 1-deoxypentalenic acid 11β-hydroxylase |
| PtlG (484) | PenG (484, 65/76) | PntG (484, 65/77) | 84/90 | predicted transmembrane efflux protein |
| PtlF (270) | PenF (275, 69/80) | PntF (282, 70/80) | 91/94 | 1-deoxy-11β-hydroxy-pentalenic acid dehydrogenase |
| PtlE (594) | PenE (584, 70/81) | PntE (589, 69/82) | 93/96 | Baeyer-Villiger monooxygenase |
| PtlD (306) | PenD (298, 65/82) | PntD (299, 64/81) | 86/93 | predicted dioxygenase |
| PtlC (58) | hypothetical protein | |||
| PtlB (337) | PenB (337, 78/84) | PntB (337, 76/83) | 93/94 | predicted polyprenyl diphosphate synthase |
| PtlA (336) | PenA (337, 76/86) | PntA (337, 76/87) | 91/95 | pentalenene synthase |
| PtlI (449) | PenI (463, 74/87) | PntI (457, 74/86) | 89/95 | cytochrome P450 monooxygenase |
| PtlR (153) | putative AraC-family transcriptional regulator | |||
| PtlJ (144) | putative lyase | |||
| PtlL (256) | hypothetical protein |
For annotations of the ptl cluster, see cited papers as well as ref. (60,61) and the S. avermitilis Genome Project website http://avermitilis.ls.kitasato-u.ac.jp/metabolite/.
The 11 ORFs of the 13-kb S. arenae pnt pentalenolactone biosynthetic gene cluster are identical in organization to those of the closely related pen cluster (Figure 1). Thus the divergently transcribed pntR gene encodes a 153-aa MarR-family transcriptional regulator with 80% identity and 87% similarity to PenR and 73% identity and 85% similarity to S. avermitilis SAV_2989 (Table 1). Besides the previously described gapR resistance gene encoding a pentalenolactone-insensitive GAPDH, the remaining 9 ORFs from PntM to PntI correspond closely in amino acid sequence to the orthologous ORFs of the pen cluster, with levels of amino acid sequence identity from 81–93% and similarity from 87–96%. Interestingly, the levels of identity and similarity between the orthologous Pnt and Pen proteins are higher by about 15–24% and 8–15%, respectively, than the corresponding relationships between the individual Pnt or Pen proteins and the homologous proteins of the ptl cluster. No matches were found in the pnt cluster to any of the predicted proteins encoded by the ptlC, ptlR, ptlJ and ptlL genes of S. avermitilis.
Biochemical characterization of the PenE and PntE Baeyer-Villiger monooxygenases
With the full sequences in hand for the individual pentalenolactone biosynthetic gene clusters from the two pentalenolactone producers, S. exfoliatus UC5319 and S. arenae TÜ469, we turned our attention to the enzymatic Baeyer-Villiger oxidation of the common intermediate 1-deoxy-11-oxopentalenic acid (7), the key branch-point that distinguishes the pentalenolactone and neopentalenolactone biosynthetic pathways. A synthetic penE gene with codons optimized for E. coli was inserted into pET-28a and expressed in E. coli BL21(DE3) to give recombinant PenE carrying an N-terminal His6-tag. Purification by metal ion affinity chromatography using a Ni2+-NTA resin, gave recombinant PenE protein, >95% pure by SDS-PAGE (Figure S1). PenE carried a bound FAD cofactor, as deduced from the characteristic UV maxima at 375 and 450 nm. MALDI-TOF MS analysis of His6-tag-PenE gave the expected MD value for the parent flavin-free protein lacking the N-terminal Met. In similar manner, a synthetic pntE gene was inserted into pET-26b and the resultant recombinant PntE with an appended C-terminal His6- tag was purified by immobilized metal ion affinity chromatography. The purified recombinant PntE-His6- tag protein, >95% pure by SDS-PAGE (Figure S1), carried a bound FAD (λmax 380 and 450 nm) and exhibited the expected MD values for the cofactor-free protein by both MALDI-TOF and ESI-MS. To identify the heretofore cryptic enzymatic activity of PenE and PntE, each recombinant enzyme was incubated with 1-deoxy-11-oxopentalenic acid (7) (Scheme 2). The requisite substrate 7 was generated in situ from 1-deoxy-11β-hydroxypentalenic acid (6) using recombinant PtlF dehydrogenase, due to the tendency of 7 to undergo facile epimerization at C-9 during attempted purification. The reduced flavin cofactor was generated continuously with excess NADPH and catalytic FAD. Treatment of the organic extracts with TMS-CHN2 followed by capillary GC-MS analysis of the derived methyl esters showed in each case exclusive formation of the Baeyer-Villiger oxidation product, pentalenolactone D methyl ester (8-Me) (Scheme 2), which was identical by direct comparison an authentic sample of 8-Me in both retention time and mass spectrum (Figure S2). Control incubations with boiled PenE or PntE gave only unoxidized 7-Me and 9-epi-7-Me. Neopentalenolactone D methyl ester (9-Me) could not be detected in any of the incubation mixtures.
Scheme 2.

PenE- and PntE-Catalyzed Baeyer-Villiger Oxidation of 7 to Pentalenolactone D (8)
Biochemical characterization of the Fe2+-α-ketoglutarate-dependent dioxygenases PenD, PntD, and PtlD
Synthetic genes for penD, pntD, and ptlD, each optimized for expression in E. coli, were individually cloned into pET-28a and the derived expression vectors were used to transform E. coli BL21(DE3). The resulting recombinant PenD, PntD, and PtlD proteins, each carrying an N-terminal His6-tag, were each purified to >95% purity by Ni-NTA chromatography (Figure S1). Each of the three proteins exhibited the predicted molecular mass MD upon ESI-MS analysis.
Individual incubations of recombinant PenD or PntD with pentalenolactone D (8) in the presence of Fe2+ and α-ketoglutarate, followed by treatment of the organic products with TMS-CHN2, gave predominantly the double oxidation product pentalenolactone F methyl ester (16-Me) accompanied by small quantities of pentalenolactone E methyl ester (15-Me) (Scheme 3A and Figure S3). Each of the products was identical to authentic reference standards in both retention time and mass spectrum by direct GC-MS comparison. Under the incubation conditions, the substrate was consumed within 90 min. Control incubations using boiled PenD or PntD gave only recovered (8-Me). Incubation of pentalenolactone E (15) with either PenD or PntD followed by treatment with TMS-CHN2 gave pentalenolactone F methyl ester (16-Me), as established by GC-MS analysis and comparison with authentic 16-Me (ret. time 12.84 min, M+ m/z 292) (Scheme 3B, Figure S3).
Scheme 3.

Oxidation of pentalenolactone D (8) and neopentalenolactone D (9). A) Conversion of pentalenolactone D (8) to pentalenolactone E (15) and pentalenolactone F (16) catalyzed by PenD, PntD, or PtlD. B) Epoxidation of 15 to 16 catalyzed by PenD or PntD. C) Oxidation of neopentalenolactone D (9) catalyzed by PtlD, PenD, or PntD.
The natural substrate for the corresponding S. avermitilis dioxygenase, PtlD, is expected to be the isomeric lactone neopentalenolactone D (9), as suggested by the accumulation of 9 in S. avermitilis mutants lacking the ptlD gene, as well as the observed formation of 9 by PtlE-catalyzed Baeyer-Villiger oxidation of the common cyclopentanone intermediate 7 (46) (Scheme 1). Very interestingly, we found that recombinant S. avermitilis PtlD could also catalyze the two-step oxidation of its unnatural substrate pentalenolactone D (8) to pentalenolactone F (16), as established by GC-MS analysis of the derived methyl esters (Scheme 3A and Figure S4). Together, these results establish that PtlD, PenD and PntD not only share a high level of mutual sequence similarity but utilize a common mechanism of action as well, in spite of the isomeric structures of their native substrates.
To examine directly the proposed native biochemical function of PtlD, we incubated recombinant PtlD with its natural substrate 9 in the presence of Fe2+ and α-ketoglutarate (Scheme 3C). After 90 min incubation, extraction with ethyl acetate, and methylation of the crude organic extract, the major product was PL308 (12), which was identical in retention time and mass spectrum (ret. time 11.92 min, m/z 308) by direct capillary GC-MS comparison with authentic 12 (46) (Figure S4). Although neither the more highly oxidized PL324 (13, 10-hydroxy-PL308) nor the closely related ketolactone 10-Me could be detected by GC-MS analysis of the resulting methylated organic extracts, an unstable coproduct, m/z 276 (ret. time 12.10 min) could be isolated which has tentatively been assigned the structure of neopentalenolactone E methyl ester (14-Me). The same mixture of PL308 (12) and 14-Me was also generated upon incubation of neopentalenolactone D (9) under identical conditions with either recombinant PenD or PntD. To confirm the structures of the two neopentalenolactone D-derived oxidation products, we carried out a preparative scale incubation of 9 with recombinant PtlD and examined the individual products by 1H NMR after column chromatographic separation of the derived the methyl esters. The major component was confirmed to be PL308 (12), as established by direct comparison with the spectrum of authentic 12 (46). The second, component, 14-Me (m/z 276), exhibited several key 1H NMR signals strongly supporting the proposed neopentalenolactone E methyl ester structure. Besides the methyl ester singlet at δ 3.74, a characteristic pair of upfield-shifted olefinic singlets at δ 4.88 (H-10syn) and 4.73 (H-10anti) indicated the presence of the enol lactone (62), while the conjugated olefinic proton signal at δ 6.81 (H-7) is typical of all pentalenolactone metabolites (37). Importantly a proton signal at δ 4.39 corresponding to the saturated H-9 carbinyl proton of neopentalenolactone D methyl ester (9-Me) was entirely absent from the NMR spectrum of 14-Me. Due to the intrinsic chemical instability of the isolated sub-milligram quantities of 14-Me the remaining signals were insufficiently intense to allow complete assignment of the proposed structure or were obscured by residual solvent or other impurities. In a GC-MS time-course study, after 10-min incubation with PtlD, 23% of the neopentalenolactone D substrate had been consumed, with 15% formation of the seco-acid 17 corresponding to PL308 and 8% neopentalenolactone E. After 45 min, the products consisted of an equal mixture of PL308 and neopentalenolactone E, with only 2% of residual 9. Finally, after 90 min, the fraction of PL308 increased to 73% while the proportion of neopentalenolactone E fell to 26%, consistent with the proposed PtlD-catalyzed oxidation of 9 to the unstable enollactone 14 followed by its non-enzymatic hydrolysis to 17.
Characterization of the penD and pntD deletion mutants
To investigate the in vivo role of the penD and pntD genes we generated the corresponding in-frame S. exfoliatus/ΔpenD ZD20 and S. arenae/ΔpntD ZD18 deletion mutants, using PCR-targeted Streptomyces gene replacement. To this end, an internal 819-bp fragment of penD carried in a 7-kb segment of the pen cluster was first replaced by DNA from oriT and aac(3)IV, most of which was then deleted by digestion with XbaI and religation to leave an in-frame 81-bp DNA scar within the orginal penD gene (Figure 3). The derived plasmid, pDQ51, was then conjugated into S. exfoliatus UC5319 and single crossover exconjugants were selected with thiostrepton. Double crossover mutant strains were then obtained by growth in the absence of antibiotic and the desired ΔpenD deletion mutant, S. exfoliatus ZD20, was identified by PCR screening using primer pairs based on the remaining upstream and downstream portions of penD (Figure S5).
In like manner, a 822-bp fragment of pntD harbored within a 6.4-kb segment of the pnt cluster was replaced by a 81-bp scar (Figure 4). The desired in-frame ΔpntD deletion mutant, S. arenae ZD18, was then obtained by two successive rounds of homologous recombination, using PCR to screen the exconjugants and to confirm the targeted deletion (Figure S5).
Wild-type S. exfoliatus UC5319 and S. arenae TÜ469 and the corresponding ΔpenD and ΔpntD deletion mutants, S. exfoliatus ZD20 and S. arenae ZD18, were each grown in liquid production cultures at 30 °C for 6 days. After acidification and chloroform extraction, analysis of the derived mixture of methyl esters by capillary GC-MS confirmed that each of the wild-type parent strains produced pentalenolactone (1) as well as varying proportions of pentalenolactones D (8), E (15), and F (16). By contrast, neither S. exfoliatus ZD20 (ΔpenD) nor S. arenae ZD18 (ΔpntD) produced any detectable pentalenolactone (1) or either of the late-stage intermediates 15 and 16. Instead, both deletion mutants accumulated the Baeyer-Villiger oxidation product pentalenolactone D (8), the demonstrated in vitro substrate of both PenD and PntD (Figure 5, Figure S6). As expected, neither of the two wild-type strains nor the ΔpenD and ΔpntD deletion mutants produced any detectable neopentalenolactone D (9).
FIGURE 5.

Production of pentalenolactone D (8) by ΔpenD deletion mutant S. exfoliatus ZD20 and ΔpntD deletion mutant S. arenae ZD18.
Complementation of the S. avermitilis ΔptlE ΔptlD deletion mutant with penE, pntE, penE-penD and pntE-pntD
The previously described S. avermitilis SUKA16 ΔptlE ΔptlD double deletion mutant accumulates 1-deoxy-11-oxopentalenic acid (7) (Figure 2) but not neopentalenolactone D (9) (46). In the wild-type strain, 7 serves as the natural substrate for the PtlE-catalyzed Baeyer-Villiger oxidation to 9 (46). To test the in vivo function of the corresponding Baeyer-Villiger oxygenase genes from the pentalenolactone biosynthetic clusters, penE and pntE under control of the constitutive ermEp promoter were each introduced by protoplast transformation into the S. avermitilis ΔptlE ΔptlD double deletion mutant. Complementation with either penE or pntE resulted in formation of pentalenolactone D (8), as established by GC-MS analysis of the methylated organic extracts of the corresponding transformants (Figure 6, Figures S7 and S8). Both GC-MS and LC-MS analysis of a concentrated sample from cultures of the S. avermitilis double deletion mutant complemented with pntE (S. avermitilis ermEp-ptl cluster ΔptlEΔptlD ermEp-pntE) also revealed the presence of a very low level of the isomeric Baeyer-Villiger product neopentalenolactone D (9) as no more than 2–3% of the mixture (Figure S8). Trace amounts of 9 were also observed in the culture of the S. avermitilis double deletion mutant complemented with penE.
FIGURE 6.

Complementation of S. avermitilis ΔptlEΔptlD deletion mutants.
We also complemented the S. avermitilis double deletion mutants with both penE and penD or pntE and pntD, respectively, again under control of the ermE promoter. GC-MS analysis of the derived methyl esters and comparison with authentic standards established that the resulting transformants produced the expected product pentalenolactone F (16) resulting from two-step PenD- or PntD-catalyzed desaturation and epoxidation of 8, accompanied by the isomeric epoxylactone, 9,10-epi-pentalenolactone F (18) (Figure 6, Figure S7, S7-2, and S9). The observed ratio of 16 to 18 was ~3:2. The complementation by penE-penD was less efficient and only very small amounts of 16 and 18 were detected as the derived methyl esters. Notably, the intermediate pentalenolactone E (15) that was observed in the enzymatic reaction of PtlD, PenD and PntD with 8 could not be detected in the extracts of the mutants complemented with either penE-penD or pntE-pntD.
In vivo analysis of Baeyer-Villiger products generated by hybrid PtlE/PntE proteins
Although the amino acid sequences of PtlE and of PntE were extremely similar, each enzyme catalyzed a distinct regio-specific Baeyer-Villiger reaction. To explore the protein structural basis for this difference in function, we constructed a progressive series of three different ptlE/pntE hybrid genes, in which the crossover points were engineered by the TAP method between the NADPH-binding and FAD-binding motifs (91 – 102 aa; ptl-pntE300), inside the FAD-binding motif (206 – 216 aa; ptlE/pntE600) and between the FAD-binding motif and C-terminus region (395 – 407 aa; ptlE/pntE1200), respectively (Fig. S10) (59). Each hybrid gene was expressed under the control of the ermE promoter in S. avermitilis ΔptlE ΔptlD double mutants. Transformants carrying ptlE/pntE300 or ptlE/pntE600 produced both pentalenolactone D (8) and neopentalenolactone D (9). The production of 9 in the S. avermitilis ΔptlE ΔptlD double mutant carrying ptlE/pntE300 increased slightly in comparison to that of the double mutant carrying pntE alone, with a ratio of 8 to 9 of ca. 20:1. The double mutant carrying ptlE/pntE600 produced increased levels of 9, resulting in a ratio of 8 to 9 of ca. 5:1. By contrast, 8 was not observed at all in the extracts of the double mutant carrying ptlE/pntE1200, while the overall productivity of this latter transformant was reduced (Fig. S11).
DISCUSSION
Pentalenolactone biosynthetic gene clusters
The 11 ORFS of the pen and pnt pentalenolactone biosynthetic gene clusters of S. exfoliatus and S. arenae are not only identical in organization but exhibit a very high degree of mutual sequence identity (Figure 1 and Table 1). Both the pentalenolactone and the neopentalenolactone biosynthetic pathways involve the multistep conversion of FPP (2) to a common intermediate, 1-deoxy-11-oxopentalenic acid (7), mediated in turn by the orthologous PenB/PntB/PtlB, PenA/PntA/PtlA, PenI/PntI/PtlI, PenH/PntH/PtlH, and PenF/PntF/PtlF gene products (Scheme 1). At this point the two pathways branch. PenE and PntE each catalyze the Baeyer-Villiger oxidation of 7 to pentalenolactone D (8) (Schemes 1 and 2) while PtlE catalyzes the complementary oxidation of 7 to the isomeric lactone, neopentalenolactone D (9) (Scheme 1).
A BLASTP search of the NCBI non-redundant (nr) protein database with the S. exfoliatus PenA sequence revealed that the predicted SBI_09679 protein (UniProt ID D7CBC0; GenBank ADI12797.1) from the recently reported genome sequence of S. bingchenggensis BCW-1 (63) corresponds to a pentalenene synthase with a high degree of certainty. In addition to the convincing 91 % identity and 96% similarity between the predicted SBI_09679 gene product and the amino acid sequence of PenA, SBI_09679 is also found within a cluster of 9 unidirectionally transcribed ORFs (GenBank ADI2790.1- ADI2798.1) that is identical in organization to both the pen and pnt biosynthetic gene clusters, with typically >90% pairwise predicted amino acid sequence identity and similarity to the orthologous pen or pnt proteins (Figure 1D). Interestingly, the individual ORFs of the predicted S. bingchenggensis pentalenolactone biosynthetic cluster more closely resemble those of the validated pentalenolactone producers, S. exfoliatus and S. arenae, than they do the corresponding ORFs of the S. avermitilis neopentalenolactone ptl cluster. The only significant differences in the organization of the presumptive S. bingchenggensis biosynthetic cluster from that of the corresponding pen, pnt, and ptl biosynthetic gene clusters are the absence of an upstream MarR-like penR/pntR/sav2989 homolog and the fact that the apparent pentalenolactone resistance gene SBI_00942 (UniProt D7C675, GenBank ADI04063.1), with 64–68% amino acid sequence identity and 77–80% similarity to Gap1, GapR, and GapN, is separated by some 2.0 Mb from the pentalenolactone biosynthetic cluster in the 11.9-Mb S. bingchenggensis linear genome. Although S. bingchenggensis is known to be a prolific producer of a wide variety of natural products, including several anthelminthic milbemycins, the polyether ionophore nanchangmycin, and the cyclic bingchamide pentapeptides (63), there are no reports of the isolation of pentalenolactone from this organism.
The BLASTP search also turned up nearly a dozen additional bacterial terpene synthases that have been erroneously annotated as “pentalenene synthase” in spite of the fact that they have much lower overall sequence similarity (<35%) to any of the validated pentalenene synthases. For example, the recently reported genome sequence of S. clavuligerus ATCC 27064 alone contains 7 such misidentified “pentalenene synthase” genes (64, 65). Additional incorrectly attributed “pentalenene synthases” can be found in the reported genome sequences of Streptomyces hygroscopicus ATCC 53653, Streptomyces bingchenggensis BCW-1, Streptomyces flavogriseus ATCC 33331, and Sorangium cellulosum ‘So ce 56’ as well as the fungus Aspergillus flavus NRRL3357. Besides the obviously low level of overall pairwise similarity to either of the previously reported pentalenene synthase sequences for PenA or PtlA, two apparent Mg2+-binding domains in all of these predicted proteins also deviate significantly from the highly conserved DDFLD and NDIASLEKE motifs found only in the three bona fide pentalenene synthases, PenA, PtlA, and PntA, as well as SBI_09679. Of equal importance, none of the numerous missannotated “pentalenene synthases” are located within biosynthetic gene clusters containing orthologs of any of the 10 remaining functional ORFs that are found in the pen, pnt, and ptl biosynthetic gene clusters or the presumptive pentalenolactone biosynthetic cluster of S. bingchenggensis. These erroneously inferred electronic annotations might originally have been due to the fact that, until relatively recently, the S. exfoliatus pentalenene synthase was the only bacterial terpene synthase sequence of experimentally assigned biochemical function. Unfortunately, such annotation errors, once they are in the public databases, are readily propagated and will be difficult to correct in uncurated databases in the absence of agreed standards for annotation of terpene synthases of unknown function (66, 67).
The role of the both PenE and PntE in mediating the Baeyer-Villiger oxidation of 1-deoxy-11- oxopentalenic acid (7) to pentalenolactone D (8) is conclusively supported by three independent but complementary lines of evidence: 1) the demonstrated flavin-dependent enzymatic conversion of 7 to 8 catalyzed by both recombinant PenE and PntE (Scheme 2); 2) the accumulation of 8 by the in-frame deletion mutants of S. exfoliatus and S. arenae lacking penD or pntD, respectively, and the concurrent abolition of formation of pentalenolactone (1) and both of the late-stage intermediates 15 and 16 (Figure 6); and 3) the production of the heterologous natural product pentalenolactone D (8) by S. avermitilis deletion mutants lacking both ptlE and ptlD that have been complemented by either penE or pntE (Figure 6). PenE and PntE should therefore be considered as paralogs of S. avermitilis PtlE, which catalyzes the analogous Baeyer-Villiger oxidation of the common substrate 7 to a distinct product, the isomeric lactone, neopentalenolactone D (9) (46). Each of the three flavin-dependent monooxygenases, PenE, PntE, and PtlE, catalyze the committed step at the branch point of the pentalenolactone and neopentalenolactone pathways. Notably all three Baeyer-Villigerases are highly regiospecific with respect to product formation, generating no more than minor amounts (2–3% or less) of the unnatural isomeric lactone. Detailed sequence comparisons do not reveal any obvious residues that can account for the differences in product specificity. Interestingly, the 70% levels of pairwise identity and 80% similarity between either PenE or PntE and the paralogous oxygenase PtlE are not meaningfully distinguishable from the comparable levels of identity (69–73%) and similarity (80–84%) between the orthologous sets of proteins PenH or PntH compared to PtlH that each catalyze identical biochemical reactions on identical substrates or from the comparable levels of identity between the PenF or PntF proteins and PtlF. Nonetheless, by constructing a series of hybrid ptlE-pntE genes and in vivo complementation of the S. avermitilis ΔptlE ΔptlD double mutant with these ptlE/pntE hybrids, we have provided important clues regarding the protein structural basis for the regiospecificity of the individual Baeyer-Villiger reactions. The two transformants carrying either ptlE/pntE300 or ptlE/pntE600 each produced both neopentalenolactone (8) and its isomer neopentalenolactone D (9). The ratio of 8 to 9 was only slightly perturbed in the former transformant compared to wild-type while the productivity of 9 was dramatically increased in the ptlE/pntE600 transformant. By contrast, the transformant carrying ptlE/pntE1200 produced only 9. These results indicate that the N-terminal region of these Baeyer- Villigerases may influence the regiospecificity of the Baeyer-Villiger oxidation reaction, especially the region around the FAD-binding motif.
Baeyer-Villiger monooxygenases
The majority of known or predicted biochemical Baeyer-Villiger monooxygenases (BVMOs) appear to be opportunistic enzymes that catalyze catabolic degradation of exogenous ketone substrates such as cyclohexanone, cyclopentanone, cyclododecanone, or 4-hydroxyacetophenone to the corresponding lactones or esters with very high chemo, regio- and stereospecificity (68–70). The PtlE, PenE, and PntE proteins, which are Class B BVMOs characterized by a tightly bound FAD cofactor and a dependence on external NADPH (71), are among the first such enzymes that have been demonstrated to play a role in a specific biosynthetic pathway and for which the natural substrate has been defined (46, 72). All three proteins harbor several conserved BVMO fingerprint motifs, including FXGXXXHXXXW(D/P) (68). The enzymatic formation of 8 or 9 is presumably controlled by differences in the conformation of the peroxide bond in the common Criegeetype ketone–4a-hydroperoxyflavin adduct 19, leading to migration of either the methylene or methane carbon of 7 (70, 73–75) (Scheme 4). Although we detected minor amounts (2–3%) of neopentalenolactone D (9) along with the major product 8 in cultures extracts of S. avermitilis double deletion mutants that had been complemented with pntE, we could not detect this minor isomer in the GC-MS analysis of the in vitro incubations with recombinant PenE or PntE, possibly due to the substantially smaller quantities of lactone that were analyzed in these experiments.
Scheme 4.

Baeyer-Villiger Oxidation of 7 to Pentalenolactone D (8) and Neopentalenolactone D (9) by Fragmentation of Discrete Conformers of the Common Criegee Adduct 19
Desaturation–epoxidation catalyzed by the Fe2+-α-ketoglutarate-dependent dioxygenases PenD, PntD, and PtlD
The role of PenD and PntD in the conversion of pentalenolactone D (8) to the epoxylactone 16 is also supported by three interlocking lines of experimental evidence: 1) direct demonstration that both PenD and PntD catalyze the 2-step, Fe2+-α-ketoglutarate-dependent, oxidation of 8 to 16 through the intermediacy of the exomethylenelactone, pentalenolactone E (15) (Scheme 3A); 2) the accumulation of 8 and the absence of pentalenolactone (1) as well as both 15 and 16 in cultures of the in-frame deletion mutants S. exfoliatus ZD20 and S. arenae ZD18 that lack penD or pntD, respectively (Figure 5); and 3) the production of pentalenolactone F (16) by S. avermitilis deletion mutants lacking both ptlE and ptlD that have been complemented by penE plus penD or by pntE plus pntD. (Figure 6).
Accompanying 16 in both of the latter S. avermitilis mutant complementation experiments is the stereoisomeric epoxide metabolite, 9,10-epi-pentalenolactone F (18). Although 18 has previously been isolated from cultures of wild-type S. exfoliatus UC5319 as a co-metabolite of 16 (37, 38, 40, 76), this epoxide epimer is not produced by the enzymatic incubations of pentalenolactone D (8) with either PenD, PntD, or PtlD. The in vivo formation of the epimeric epoxylactone 18 as a shunt metabolite is therefore most likely due to oxidation of the intermediate α-methylenelactone pentalenolactone E (15) by an oxygenase that is distinct from both PenD or PntD and that is encoded by a gene that is external to either the pen or pnt biosynthetic gene clusters (Scheme 5). In a similar manner, we have recently reported that the hydroxylation of the normal biosynthetic pathway intermediate 1-deoxypentalenic acid (5) to the commonly occurring shunt metabolite pentalenic acid (20) is catalyzed by CYP105D7, a typical heme-dependent monooxygenase that is encoded by the sav7469 gene, located in the 9.05-Mb S. avermitilis linear chromosome more than 5 Mb from the ptl biosynthetic gene cluster (77).
Scheme 5.

Formation of Shunt Metabolites 9,10-epi-Pentalenolactone F (18) and Pentalenic Acid (20)
The PenD-, PntD-, and PtlD-catalyzed dehydrogenation of the α-methyl substituent of pentalenolactone D (8) to generate the intermediate α-methylenelactone 15 is reminiscent of the desaturase activity of the multifunctional Fe2+-α-ketoglutarate-dependent clavaminate synthase (CAS), which catalyzes the oxidative conversion dihydroclavaminate to clavaminate (78). Although PenD, PntD, and PtlD all belong to the CAS superfamily, these enzymes have no significant linear sequence similarity to CAS itself. Moreover, although the PenD and PntD desaturatase–epoxidases on the one hand and the PenH and PntH hydroxylases on the other are all Fe2+-α-ketoglutarate-dependent dioxygenases that act on structurally related substrates, pentalenolactone D (8) and 1-deoxypentalenic acid (5), respectively, the two groups of enzymes have no primary sequence similarity and catalyze two completely different biochemical reactions – 2-step desaturation/epoxidation versus simple hydroxylation.
While most known biochemical epoxidations are catalyzed by flavin-dependent or cytochrome P450- dependent monooxygenases (73, 79), direct epoxidation catalyzed by a Fe2+-α-ketoglutarate-dependent dioxygenase is exceedingly rare (80), the only prior example being the recently discovered epoxidation of the unsaturated tripeptide dapdiamide D catalyzed by the DdaC protein of Pantoea agglomerans (GenBank Accession No. ADN39482) (81). In fact the DdaC protein has no sequence similarity to either PenD or PntD. Although henbane hyoscamine-6β-hydroxylase is another Fe2+-α-ketoglutaratedependent dioxygenase that catalyzes a consecutive two-step epoxidation of the saturated substrate hyoscyamine to the corresponding epoxide scopolamine, the reaction differs significantly from the PenD- and PntD-catalyzed transformation since it involves the intermediacy of 6β-hydroxyhyoscyamine which is oxidized directly to scopolamine without the involvement of 6,7-dehydrohyoscyamine (82, 83). An analogous hydroxylation–epoxidation mechanism for the PenD-, PntD-, or PtlD-catalyzed conversion of pentalenolactone D (8) to the epoxylactone 16 is firmly excluded by the demonstrated formation of the unsaturated α-methylenelactone 15, as well as the absence of any detectable 9- hydroxypentalenolactone D in the incubation mixtures.
The natural substrate for S. avermitilis PtlD is neopentalenolactone D (9), as established by both in vivo accumulation of 9 in cultures of S. avermitilis ΔptlD deletion mutants and the results of in vitro incubation of 9 with recombinant PtlD. Intriguingly, PtlD has a broad enough substrate specificity to permit oxidative desaturation of the isomeric substrate pentalenolactone D (8) to pentalenolactone E (15) and thence further oxidation to the epoxylactone 16. Although we have not carried out detailed kinetic comparisons, PenD, PntD, and PtlD each catalyzed the consecutive two step dehydrogenation– epoxidation of 8 with approximately equal efficiency.
In contrast to the observed 2-step desaturation-epoxidation of pentalenolactone D (8), in vitro incubation of recombinant S. avermitilis PtlD or the surrogate enzymes PenD and PntD with neopentalenolactone D (9) resulted only in a single-step 2-electron dehydrogenation to give a product tentatively identified as neopentalenolactone E (14), which underwent facile hydrolysis to the seco-acid 17, which upon methylation yielded PL308 (12). Interestingly, we were unable to detect either the expected epoxidation product, neopentalenolactone F (11) or either of the corresponding hydrolysis or rearrangement products, PL324 (13, 10-hydroxy-PL308) or neopentalenoketolactone (10) that have previously been isolated from engineered mutants of S. avermitilis harboring an induced ptl biosynthetic cluster (46). Whether this difference in product profile is due to the intrinsic instability of the neopentalenolactone E intermediate under the incubation conditions or the absence of an essential cofactor or protein that is present in S. avermitilis is not yet known. It is curious, however, that the PenD-, PntD-, or PtlD-catalyzed epoxidation of the electron-poor conjugated double bond of the α-methylene lactone 15 appears to be proceed more favorably than oxidation of the electron-rich double bond of the isomeric enollactone neopentalenolactone E (14).
It is also noteworthy that while the PenE, PntE, and PtlE proteins each catalyze the flavin-dependent oxidation of the identical ketone substrate 7, the individual Baeyer-Villiger oxidations yield isomeric lactone products 8 or 9. By comparison, although PenD and PntD on the one hand and PtlD on the other normally act on a specific lactone substrate, 8 or 9, each of these three dioxygenases is able to catalyze the oxidation of either 8 or 9, giving in each case the same mixture of corresponding products.
CONCLUSION
For the biosynthesis of pentalenolactone, Nature has orchestrated the oxidative conversion of the parent tricyclic sesquiterpene hydrocarbon pentalenene to the final metabolite pentalenolactone by calling on a full range of the oxidative instruments in its biochemical repertoire. A heme-dependent, P450 monooxygenase (PenI/PntI) first catalyzes the three-step oxidative conversion of an allylic methyl group to a conjugated carboxylic acid, after which an Fe2+-α-ketoglutarate-dependent dioxygenase (PenH/PntH) hydroxylates an unactivated saturated methylene group. Following oxidation to the derived ketone by a NADP+-dependent dehydrogenase (PenF/PntF), an NADPH–FAD-dependent oxygenase (PenE/PntE) mediates the Baeyer-Villiger reaction to yield the corresponding α-methyl-substituted δ-lactone. This intermediate then serves as the substrate for a two-step dehydrogenation–epoxidation that is catalyzed by a second Fe2+-α-ketoglutarate-dependent dioxygenase (PenD/PntD). The final movement in this virtuoso biosynthetic performance is an unprecedented oxidative rearrangement to give pentalenolactone (1), a reaction that we have recently shown to be catalyzed by the heme-dependent P450 monooxygenases PenM and PntM.
Supplementary Material
Acknowledgments
We thank Tun-Li Shen of the Department of Chemistry, Brown University for assistance with MS analysis. Prof. Zixin Deng and Prof. Linquan Bai and their group at Shanghai Jiaotong University kindly provided valuable advice on methods of genomic library construction and Streptomyces gene knockout and provided strains and plasmids. We also thank NBRP of the National Institute of Genetics, Japan for distribution of plasmid vector pTHcs1.
Abbreviations
- BVMO
Baeyer-Villiger monooxygenase
- CIAP
calf intestinal alkaline phosphatase
- DTT
1,4-dithiothreitol
- FAD
flavin adenine dinucleotide
- FPP
farnesyl diphosphate
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- HSV
Herpes simplex virus
- IPTG
isopropyl-β-D-thiogalactopyranoside
- LB
Luria-Bertani medium
- NAD+
β-nicotinamide adenine dinucleotide
- NADPH
β-Nicotinamide adenine dinucleotide 2′-phosphate reduced tetrasodium salt hydrate
- ODS
Octadecyl silane
- ORF
open reading frame
- SFM
soy flour, mannitol
- PMSF
phenylmethylsulfonyl fluoride
- TAP
Transcriptionally Active Polymerase Chain Reaction
- TB
Terrific Broth
Footnotes
This work was supported by National Institutes of Health Grant GM30301 (D.E.C.) and by a Grant-in- Aid for Scientific Research on Innovative Areas from MEXT Japan, from JSPS 20310122 and from the Institute for Fermentation, Osaka, Japan (H.I.).
SUPPORTING INFORMATION AVAILABLE
Table of strains and plasmids, GC-MS and LC-MS data, and construction of deletion mutants and complemented bacterial mutants. This material is available free of charge at http://pubs.acs.org.
Contributor Information
Haruo Ikeda, Email: ikeda@ls.kitasato-u.ac.jp.
David E. Cane, Email: David_Cane@brown.edu.
References
- 1.Cane DE. The enzymatic formation of sesquiterpenes. Chem Rev. 1990;9:1089–1103. [Google Scholar]
- 2.Cane DE. Sesquiterpene Biosynthesis: Cyclization Mechanisms. In: Cane DE, editor. Comprehensive Natural Products Chemistry. Isoprenoids Including Carotenoids and Steroids. Elsevier; Oxford: 1999. pp. 155–200. [Google Scholar]
- 3.Wise ML, Croteau R. Monoterpene Biosynthesis. In: Cane DE, editor. Comprehensive Natural Products Chemistry. Isoprenoids Including Carotenoids and Steroids. Elsevier; Oxford: 1999. pp. 97–153. [Google Scholar]
- 4.Christianson DW. Structural biology and chemistry of the terpenoid cyclases. Chem Rev. 2006;106:3412–3442. doi: 10.1021/cr050286w. [DOI] [PubMed] [Google Scholar]
- 5.Christianson DW. Unearthing the roots of the terpenome. Curr Opin Chem Biol. 2008;12:141–150. doi: 10.1016/j.cbpa.2007.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gutta P, Tantillo DJ. Theoretical studies on farnesyl cation cyclization: pathways to pentalenene. J Am Chem Soc. 2006;128:6172–6179. doi: 10.1021/ja058031n. [DOI] [PubMed] [Google Scholar]
- 7.Hong YJ, Tantillo DJ. Consequences of conformational preorganization in sesquiterpene biosynthesis: theoretical studies on the formation of the bisabolene, curcumene, acoradiene, zizaene, cedrene, duprezianene, and sesquithuriferol sesquiterpenes. J Am Chem Soc. 2009;131:7999–8015. doi: 10.1021/ja9005332. [DOI] [PubMed] [Google Scholar]
- 8.Tudzynski B. Gibberellin biosynthesis in fungi: genes, enzymes, evolution, and impact on biotechnology. Appl Microbiol Biotechnol. 2005;66:597–611. doi: 10.1007/s00253-004-1805-1. [DOI] [PubMed] [Google Scholar]
- 9.Tudzynski B, Mihlan M, Rojas MC, Linnemannstons P, Gaskin P, Hedden P. Characterization of the final two genes of the gibberellin biosynthesis gene cluster of Gibberella fujikuroi: des and P450–3 encode GA4 desaturase and the 13-hydroxylase, respectively. J Biol Chem. 2003;278:28635–28643. doi: 10.1074/jbc.M301927200. [DOI] [PubMed] [Google Scholar]
- 10.Tudzynski B, Rojas MC, Gaskin P, Hedden P. The gibberellin 20-oxidase of Gibberella fujikuroi is a multifunctional monooxygenase. J Biol Chem. 2002;277:21246–21253. doi: 10.1074/jbc.M201651200. [DOI] [PubMed] [Google Scholar]
- 11.Ringer KL, Davis EM, Croteau R. Monoterpene metabolism. Cloning, expression, and characterization of (−)-isopiperitenol/(−)-carveol dehydrogenase of peppermint and spearmint. Plant Physiol. 2005;137:863–872. doi: 10.1104/pp.104.053298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Takahashi S, Zhao Y, O’Maille PE, Greenhagen BT, Noel JP, Coates RM, Chappell J. Kinetic and molecular analysis of 5-epiaristolochene 1,3-dihydroxylase, a cytochrome P450 enzyme catalyzing successive hydroxylations of sesquiterpenes. J Biol Chem. 2005;280:3686–3696. doi: 10.1074/jbc.M411870200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ro DK, Paradise EM, Ouellet M, Fisher KJ, Newman KL, Ndungu JM, Ho KA, Eachus RA, Ham TS, Kirby J, Chang MC, Withers ST, Shiba Y, Sarpong R, Keasling JD. Production of the antimalarial drug precursor artemisinic acid in engineered yeast. Nature. 2006;440:940–943. doi: 10.1038/nature04640. [DOI] [PubMed] [Google Scholar]
- 14.Teoh KH, Polichuk DR, Reed DW, Nowak G, Covello PS. Artemisia annua L. (Asteraceae) trichome-specific cDNAs reveal CYP71AV1, a cytochrome P450 with a key role in the biosynthesis of the antimalarial sesquiterpene lactone artemisinin. FEBS Lett. 2006;580:1411–1416. doi: 10.1016/j.febslet.2006.01.065. [DOI] [PubMed] [Google Scholar]
- 15.Zhao B, Lei L, Vassylyev DG, Lin X, Cane DE, Kelly SL, Yuan H, Lamb DC, Waterman MR. Crystal structure of albaflavenone monooxygenase containing a moonlighting terpene synthase active site. J Biol Chem. 2009;284:36711–36719. doi: 10.1074/jbc.M109.064683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhao B, Lin X, Lei L, Lamb DC, Kelly SL, Waterman MR, Cane DE. Biosynthesis of the sesquiterpene antibiotic albaflavenone in Streptomyces coelicolor A3(2) J Biol Chem. 2008;283:8183–8189. doi: 10.1074/jbc.M710421200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Koe BK, Sobin BA, Celmer WD. PA 132, a new antibiotic. I. Isolation and chemical properties. Antibiot Annu. 1957:672–675. [PubMed] [Google Scholar]
- 18.Martin DG, Slomp G, Mizsak S, Duchamp DJ, Chidester CG. The structure and absolute configuration of pentalenolactone (PA 132) Tetrahedron Lett. 1970:4901–4904. doi: 10.1016/s0040-4039(00)99739-9. [DOI] [PubMed] [Google Scholar]
- 19.Keller-Schierlein W, Lemke J, Nyfeler R, Zähner H. Stoffwechselprodukte von Mikroorganismen. 105 Arenaemycin E, D, und C. Arch Mikrobiol. 1972;84:301–316. [PubMed] [Google Scholar]
- 20.Takeuchi S, Ogawa Y, Yonehara H. The Structure of pentalenolactone (PA-132) Tetrahedron Lett. 1969:2737–2740. doi: 10.1016/s0040-4039(01)88256-3. [DOI] [PubMed] [Google Scholar]
- 21.Duszenko M, Balla H, Mecke D. Specific inactivation of glucose metabolism from eucaryotic cells by pentalenolactone. Biochim Biophys Acta. 1982;714:344–350. doi: 10.1016/0304-4165(82)90343-9. [DOI] [PubMed] [Google Scholar]
- 22.Hartmann S, Neeff J, Heer U, Mecke D. Arenaemycin (pentalenolactone): A specific inhibitor of glycolysis. FEBS Lett. 1978;93:339–342. doi: 10.1016/0014-5793(78)81135-1. [DOI] [PubMed] [Google Scholar]
- 23.Cane DE, Sohng JK. Inhibition of glyceraldehyde-3-phosphate dehydrogenase by pentalenolactone: kinetic and mechanistic Studies. Arch Biochem Biophys. 1989;270:50–61. doi: 10.1016/0003-9861(89)90006-4. [DOI] [PubMed] [Google Scholar]
- 24.Cane DE, Sohng JK. Inhibition of glyceraldehyde-3-phosphate dehydrogenase by pentalenolactone. 2 Identification of the site of alkylation by tetrahydropentalenolactone. Biochemistry. 1994;33:6524–6530. doi: 10.1021/bi00187a020. [DOI] [PubMed] [Google Scholar]
- 25.Fröhlich K-U, Wiedmann M, Lottspeich F, Mecke D. Substitution of a pentalenolactone-sensitive glyceraldehyde-3-phosphate dehydrogenase by a genetically distinct resistant isoform accompanies pentalenolactone production in Streptomyces arenae. J Bacteriol. 1989;171:6696–6702. doi: 10.1128/jb.171.12.6696-6702.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Frohlich KU, Kannwischer R, Rudiger M, Mecke D. Pentalenolactone-insensitive glyceraldehyde-3-phosphate dehydrogenase from Streptomyces arenae is closely related to GAPDH from thermostable eubacteria and plant chloroplasts. Arch Microbiol. 1996;165:179–186. doi: 10.1007/BF01692859. [DOI] [PubMed] [Google Scholar]
- 27.Maurer K-H, Mecke D. Regulation of enzymes involved in the biosynthesis of the sesquiterpene antibiotic pentalenolactone in Streptomyces arenae. J Antibiot. 1986;39:266–271. doi: 10.7164/antibiotics.39.266. [DOI] [PubMed] [Google Scholar]
- 28.Maurer KH, Pfeiffer F, Zehender H, Mecke D. Characterization of two glyceraldehyde-3-phosphate dehydrogenase isoenzymes from the pentalenolactone producer Streptomyces arenae. J Bacteriol. 1983;153:930–936. doi: 10.1128/jb.153.2.930-936.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cane DE, Rossi T, Pachlatko JP. The biosynthesis of pentalenolactone. Tetrahedron Lett. 1979:3639–3642. [Google Scholar]
- 30.Cane DE, Rossi T, Tillman AM, Pachlatko JP. Stereochemical studies of isoprenoid biosynthesis. Biosynthesis of pentalenolactone from [UL-13C6]-glucose and [6–2H2]-glucose. J Am Chem Soc. 1981;103:1838–1843. [Google Scholar]
- 31.Cane DE, Oliver JS, Harrison PHM, Abell C, Hubbard BR, Kane CT, Lattman R. The biosynthesis of pentalenene and pentalenolactone. J Am Chem Soc. 1990;112:4513–4524. [Google Scholar]
- 32.Cane DE, Tillman AM. Pentalenene biosynthesis and the enzymatic cyclization of farnesyl pyrophosphate. J Am Chem Soc. 1983;105:122–124. [Google Scholar]
- 33.Cane DE, Sohng J-K, Lamberson CR, Rudnicki SM, Wu Z, Lloyd MD, Oliver JS, Hubbard BR. Pentalenene synthase. Purification, molecular cloning, sequencing and high-level expression in Escherichia coli of a terpenoid cyclase from Streptomyces UC5319. Biochemistry. 1994;33:5846–5857. doi: 10.1021/bi00185a024. [DOI] [PubMed] [Google Scholar]
- 34.Lesburg CA, Zhai G, Cane DE, Christianson DW. Crystal structure of pentalenene synthase: mechanistic insights on terpenoid cyclization reactions in biology. Science. 1997;277:1820–1824. doi: 10.1126/science.277.5333.1820. [DOI] [PubMed] [Google Scholar]
- 35.Cane DE, Weiner SW. Cyclization of farnesyl diphosphate to pentalenene. Orthogonal stereochemistry in an enzyme-catalyzed SE′ reaction. Can J Chem. 1994;72:118–127. [Google Scholar]
- 36.Takahashi S, Takeuchi M, Arai M, Seto H, Otake N. Studies on the biosynthesis of pentalenolactone. V. Isolation of deoxypentalenylglucuron. J Antibiot. 1983;36:226–228. doi: 10.7164/antibiotics.36.226. [DOI] [PubMed] [Google Scholar]
- 37.Cane DE, Sohng JK, Williard PG. Isolation and structure determination of pentalenolactones A, B, D, and F. J Org Chem. 1992;57:844–852. [Google Scholar]
- 38.Seto H, Noguchi H, Sankawa U, Iitaka Y. Studies on the biosynthesis of pentalenolactone. VI. The X-ray crystal structure investigation of pentalenolactone G and structural revision of pentalenolactone F. J Antibiot. 1984;37:816–817. doi: 10.7164/antibiotics.37.816. [DOI] [PubMed] [Google Scholar]
- 39.Seto H, Sasaki T, Yonehara H, Takahashi S, Takeuchi M, Kuwano H, Arai M. Studies of the biosynthesis of pentalenolactone. VII. Isolation of pentalenolactones P and O. J Antibiot. 1984;37:1076–1078. doi: 10.7164/antibiotics.37.1076. [DOI] [PubMed] [Google Scholar]
- 40.Williard PG, Sohng JK, Cane DE. The x-ray crystal structure of pentalenolactone F methyl ester (epi-pentalenolactone F) J Antibiot. 1988;41:130–133. doi: 10.7164/antibiotics.41.130. [DOI] [PubMed] [Google Scholar]
- 41.Tetzlaff CN, You Z, Cane DE, Takamatsu S, Omura S, Ikeda H. A gene cluster for biosynthesis of the sesquiterpenoid antibiotic pentalenolactone in Streptomyces avermitilis. Biochemistry. 2006;45:6179–6186. doi: 10.1021/bi060419n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Quaderer R, Omura S, Ikeda H, Cane DE. Pentalenolactone biosynthesis. Molecular cloning and assignment of biochemical function to PtlI, a cytochrome P450 of Streptomyces avermitilis. J Am Chem Soc. 2006;128:13036–13037. doi: 10.1021/ja0639214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.You Z, Omura S, Ikeda H, Cane DE. Pentalenolactone biosynthesis. Molecular cloning and assignment of biochemical function to PtlH, a non-heme iron dioxygenase of Streptomyces avermitilis. J Am Chem Soc. 2006;128:6566–6567. doi: 10.1021/ja061469i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.You Z, Omura S, Ikeda H, Cane DE, Jogl G. Crystal structure of the non-heme iron dioxygenase PtlH in pentalenolactone biosynthesis. J Biol Chem. 2007;282:36552–36560. doi: 10.1074/jbc.M706358200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.You Z, Omura S, Ikeda H, Cane DE. Pentalenolactone biosynthesis: Molecular cloning and assignment of biochemical function to PtlF, a short-chain dehydrogenase from Streptomyces avermitilis, and identification of a new biosynthetic intermediate. Arch Biochem Biophys. 2007;459:233–240. doi: 10.1016/j.abb.2006.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jiang J, Tetzlaff CN, Takamatsu S, Iwatsuki M, Komatsu M, Ikeda H, Cane DE. Genome mining in Streptomyces avermitilis: A biochemical Baeyer-Villiger reaction and discovery of a new branch of the pentalenolactone family tree. Biochemistry. 2009;48:6431–6440. doi: 10.1021/bi900766w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Komatsu M, Uchiyama T, Omura S, Cane DE, Ikeda H. Genome-minimized Streptomyces host for the heterologous expression of secondary metabolism. Proc Natl Acad Sci U S A. 2010;107:2646–2651. doi: 10.1073/pnas.0914833107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kieser T, Bibb MJ, Buttner MJ, Chater KF, Hopwood DA. Practical Streptomyces Genetics. John Innes Foundation; Norwich, UK: 2000. [Google Scholar]
- 49.Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning, A Laboratory Manual. 2. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 1989. [Google Scholar]
- 50.Bradford M. A rapid and sensitive method for the quantitation of microgram quantitites of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 51.Sun Y, Zhou X, Liu J, Bao K, Zhang G, Tu G, Kieser T, Deng Z. ‘Streptomyces nanchangensis’, a producer of the insecticidal polyether antibiotic nanchangmycin and the antiparasitic macrolide meilingmycin, contains multiple polyketide gene clusters. Microbiology. 2002;148:361–371. doi: 10.1099/00221287-148-2-361. [DOI] [PubMed] [Google Scholar]
- 52.He Y, Wang Z, Bai L, Liang J, Zhou X, Deng Z. Two pHZ1358-derivative vectors for efficient gene knockout in Streptomyces. J Microbiol Biotechnol. 2010;20:678–682. doi: 10.4014/jmb.0910.10031. [DOI] [PubMed] [Google Scholar]
- 53.Gust B, Challis GL, Fowler K, Kieser T, Chater KF. PCR-targeted Streptomyces gene replacement identifies a protein domain needed for biosynthesis of the sesquiterpene soil odor geosmin. Proc Natl Acad Sci U S A. 2003;100:1541–1546. doi: 10.1073/pnas.0337542100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hashimoto-Gotoh T, Yamaguchi M, Yasojima K, Tsujimura A, Wakabayashi Y, Watanabe Y. A set of temperature sensitive-replication/-segregation and temperature resistant plasmid vectors with different copy numbers and in an isogenic background (chloramphenicol, kanamycin, lacZ, repA, par, polA) Gene. 2000;241:185–191. doi: 10.1016/s0378-1119(99)00434-5. [DOI] [PubMed] [Google Scholar]
- 55.Ikeda H, Takada Y, Pang CH, Tanaka H, Omura S. Transposon mutagenesis by Tn4560 and applications with avermectin-producing Streptomyces avermitilis. J Bacteriol. 1993;175:2077–2082. doi: 10.1128/jb.175.7.2077-2082.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ikeda H, Kotaki H, Omura S. Genetic studies of avermectin biosynthesis in Streptomyces avermitilis. J Bacteriol. 1987;169:5615–5621. doi: 10.1128/jb.169.12.5615-5621.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Takamatsu S, Lin X, Nara A, Komatsu M, Cane DE, Ikeda H. Activation of a silent sesquiterpenoid biosynthetic pathway in Streptomyces avermitilis controlling epi-isozizaene and albaflavenone biosynthesis and isolation of a new oxidized epi-isozizaene metabolite. Microb Biotech. 2010;3 doi: 10.1111/j.1751-7915.2010.00209.x. doi: 0.1111/j.1751–7915.2010.00209.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cane DE, Rossi T. The isolation and structural elucidation of pentalenolactone E. Tetrahedron Lett. 1979:2973–2974. [Google Scholar]
- 59.Liang X, Teng A, Braun DM, Felgner J, Wang Y, Baker SI, Chen S, Zelphati O, Felgner PL. Transcriptionally active polymerase chain reaction (TAP): high throughput gene expression using genome sequence data. J Biol Chem. 2002;277:3593–3598. doi: 10.1074/jbc.M110652200. [DOI] [PubMed] [Google Scholar]
- 60.Ikeda H, Ishikawa J, Hanamoto A, Shinose M, Kikuchi H, Shiba T, Sakaki Y, Hattori M, Omura S. Complete genome sequence and comparative analysis of the industrial microorganism Streptomyces avermitilis. Nat Biotechnol. 2003;21:526–531. doi: 10.1038/nbt820. [DOI] [PubMed] [Google Scholar]
- 61.Omura S, Ikeda H, Ishikawa J, Hanamoto A, Takahashi C, Shinose M, Takahashi Y, Horikawa H, Nakazawa H, Osonoe T, Kikuchi H, Shiba T, Sakaki Y, Hattori M. Genome sequence of an industrial microorganism Streptomyces avermitilis: deducing the ability of producing secondary metabolites. Proc Natl Acad Sci U S A. 2001;98:12215–12220. doi: 10.1073/pnas.211433198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Harkat H, Dembelé AY, Weibel J-M, Blanc A, Pale P. Cyclization of alkynoic acids with gold catalysts: a surprising dichotomy between AuI and AuIII. Tetrahedron. 2009;65:1871–1879. [Google Scholar]
- 63.Wang XJ, Yan YJ, Zhang B, An J, Wang JJ, Tian J, Jiang L, Chen YH, Huang SX, Yin M, Zhang J, Gao AL, Liu CX, Zhu ZX, Xiang WS. Genome sequence of the milbemycin-producing bacterium Streptomyces bingchenggensis. J Bacteriol. 2010;192:4526–4527. doi: 10.1128/JB.00596-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fischbach M, Ward D, Young S, Jaffe D, Gnerre S, Berlin A, Heiman D, Hepburn T, Sykes S, Alvarado L, Kodira CD, Straight P, Clardy J, Hung D, Kolter R, Mekalanos J, Walker S, Walsh CT, Lander E, Galagan J, Nusbaum C, Birren B. NCBI GenBank Nucleotide Accession Nr ABJH00000000. 2008. Annotation of Streptomyces clavuligerus ATCC 27064; whole genome shotgun sequencing project. [Google Scholar]
- 65.Medema MH, Trefzer A, Kovalchuk A, van den Berg M, Muller U, Heijne W, Wu L, Alam MT, Ronning CM, Nierman WC, Bovenberg RAL, Breitling R, Takano E. The sequence of a 1.8-Mb bacterial linear plasmid reveals a rich evolutionary reservoir of secondary metabolic pathways. Genome Biol Evol. 2010;2:212–224. doi: 10.1093/gbe/evq013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bohlmann J, Gershenzon J. Old substrates for new enzymes of terpenoid biosynthesis. Proc Natl Acad Sci U S A. 2009;106:10402–10403. doi: 10.1073/pnas.0905226106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Degenhardt J, Kollner TG, Gershenzon J. Monoterpene and sesquiterpene synthases and the origin of terpene skeletal diversity in plants. Phytochemistry. 2009;70:1621–1637. doi: 10.1016/j.phytochem.2009.07.030. [DOI] [PubMed] [Google Scholar]
- 68.Fraaije MW, Kamerbeek NM, van Berkel WJ, Janssen DB. Identification of a Baeyer-Villiger monooxygenase sequence motif. FEBS Lett. 2002;518:43–47. doi: 10.1016/s0014-5793(02)02623-6. [DOI] [PubMed] [Google Scholar]
- 69.Fraaije MW, Wu J, Heuts DP, van Hellemond EW, Spelberg JH, Janssen DB. Discovery of a thermostable Baeyer-Villiger monooxygenase by genome mining. Appl Microbiol Biotechnol. 2005;66:393–400. doi: 10.1007/s00253-004-1749-5. [DOI] [PubMed] [Google Scholar]
- 70.de Gonzalo G, Mihovilovic MD, Fraaije MW. Recent developments in the application of Baeyer-Villiger monooxygenases as biocatalysts. Chembiochem. 2010;11:2208–2231. doi: 10.1002/cbic.201000395. [DOI] [PubMed] [Google Scholar]
- 71.van Berkel WJ, Kamerbeek NM, Fraaije MW. Flavoprotein monooxygenases, a diverse class of oxidative biocatalysts. J Biotechnol. 2006;124:670–689. doi: 10.1016/j.jbiotec.2006.03.044. [DOI] [PubMed] [Google Scholar]
- 72.Beam MP, Bosserman MA, Noinaj N, Wehenkel M, Rohr J. Crystal structure of Baeyer-Villiger monooxygenase MtmOIV, the key enzyme of the mithramycin biosynthetic pathway. Biochemistry. 2009;48:4476–4487. doi: 10.1021/bi8023509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Palfey BA, McDonald CA. Control of catalysis in flavin-dependent monooxygenases. Arch Biochem Biophys. 2010;493:26–36. doi: 10.1016/j.abb.2009.11.028. [DOI] [PubMed] [Google Scholar]
- 74.Ryerson CC, Ballou DP, Walsh C. Mechanistic studies on cyclohexanone oxygenase. Biochemistry. 1982;21:2644–2655. doi: 10.1021/bi00540a011. [DOI] [PubMed] [Google Scholar]
- 75.Mihovilovic MD, Kapitan P. Regiodivergent Baeyer-Villiger oxidation of fused ketone substrates by recombinant whole-cells expressing two monooxygenases from Brevibacterium. Tetrahedron Lett. 2004;45:2751–2754. [Google Scholar]
- 76.Tillman AM, Cane DE. Pentalenolactone F, a new metabolite isolated from Streptomyces Isolation and structure elucidation. J Antibiot. 1983;36:170–172. doi: 10.7164/antibiotics.36.170. [DOI] [PubMed] [Google Scholar]
- 77.Takamatsu S, Xu L-H, Fushinobu S, Shoun H, Komatsu M, Cane DE, Ikeda H. Pentalenic acid is a shunt metabolite in the biosynthesis of the pentalenolactone family of metabolites: Hydroxylation of 1-deoxypentalenic acid mediated by CYP105D7 (SAV_7469) of Streptomyces avermitilis. J Antibiot. 2010 doi: 10.1038/ja.2010.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Janc JW, Egan LA, Townsend CA. Purification and characterization of clavaminate synthase from Streptomyces antibioticus A multifunctional enzyme of clavam biosynthesis. J Biol Chem. 1995;270:5399–5404. doi: 10.1074/jbc.270.10.5399. [DOI] [PubMed] [Google Scholar]
- 79.Isin EM, Guengerich FP. Complex reactions catalyzed by cytochrome P450 enzymes. Biochim Biophys Acta. 2007;1770:314–329. doi: 10.1016/j.bbagen.2006.07.003. [DOI] [PubMed] [Google Scholar]
- 80.Hausinger RP. Fe(II)/alpha-ketoglutarate-dependent hydroxylases and related enzymes. Crit Rev Biochem Mol Biol. 2004;39:21–68. doi: 10.1080/10409230490440541. [DOI] [PubMed] [Google Scholar]
- 81.Hollenhorst MA, Bumpus SB, Matthews ML, Bollinger JM, Kelleher NL, Walsh CT. The nonribosomal peptide synthetase enzyme DdaD tethers N-β-Fumaramoyl-l-2,3-diaminopropionate for Fe(II)-α-±-ketoglutarate-dependent epoxidation by DdaC during dapdiamide antibiotic biosynthesis. J Am Chem Soc. 2010;132:15773–15781. doi: 10.1021/ja1072367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hashimoto T, Matsuda J, Yamada Y. Two-step epoxidation of hyoscyamine to scopolamine is catalyzed by bifunctional hyoscyamine 6 beta-hydroxylase. FEBS letters. 1993;329:35–39. doi: 10.1016/0014-5793(93)80187-y. [DOI] [PubMed] [Google Scholar]
- 83.Matsuda J, Okabe S, Hashimoto T, Yamada Y. Molecular cloning of hyoscyamine 6β-hydroxylase, a 2-oxoglutarate-dependent dioxygenase, from cultured roots of Hyoscyamus-Niger. J Biol Chem. 1991;266:9460–9464. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.