

Abstract
Aims/hypothesis
Chronically elevated blood glucose (hyperglycaemia) is the primary indicator of type 2 diabetes, which has a prevalence that varies considerably by ethnicity in the USA, with African-Americans disproportionately affected. Genome-wide association studies (GWASs) have significantly enhanced our understanding of the genetic basis of diabetes and related traits, including fasting plasma glucose (FPG). However, the majority of GWASs have been conducted in populations of European ancestry. Thus, it is important to conduct replication analyses in populations with non-European ancestry to identify shared loci associated with FPG across populations.
Methods
We used data collected from non-diabetic unrelated African-American individuals (n = 927) who participated in the Howard University Family Study to attempt to replicate previously published GWASs of FPG. Of the 29 single nucleotide polymorphisms (SNPs) previously reported, we directly tested 20 in this study. In addition to the direct test, we queried a 500 kb window centred on all 29 reported SNPs for local replication of additional markers in linkage disequilibrium (LD).
Results
Using direct SNP and LD-based comparisons, we replicated multiple SNPs previously associated with FPG and strongly associated with type 2 diabetes in populations with European ancestry. The replicated SNPs included those in or near TCF7L2, SLC30A8, G6PC2, MTNR1B, DGKB-TMEM195 and GCKR. We also replicated additional variants in LD with the reported SNPs in ZMAT4 and adjacent to IRS1.
Conclusions/interpretation
We identified multiple GWAS variants for FPG in our cohort of African-Americans. Using an LD-based strategy we also identified SNPs not previously reported, demonstrating the utility of using diverse populations for replication analysis.
Electronic supplementary material
The online version of this article (doi:10.1007/s00125-010-2002-7) contains supplementary material, which is available to authorised users.
Keywords: African-American, Association, GWAS, Replication, Type 2 diabetes
Introduction
Genome-wide association studies (GWASs) have significantly added to our understanding of the genetic basis of type 2 diabetes and related traits, including fasting plasma glucose (FPG), by identifying a number of genes potentially involved in the pathophysiology of this common complex disease [1]. However, the majority of FPG GWASs have been conducted in individuals of European descent, many of which were included in a recent meta-analysis [2]. While this information has laid an important foundation, it is important to investigate whether identified loci transfer across populations with different ancestral backgrounds [3] and whether novel variants could be identified as recently demonstrated in populations of East Asian and Indian backgrounds [4, 5]. Here, we conducted replication of published GWAS results for FPG in African-Americans from the metropolitan area of Washington DC, USA.
Methods
Ethics statement
Ethical approval for the Howard University Family Study (HUFS) was obtained from the Howard University Institutional Review Board and written informed consent was obtained from each participant.
Study design
The individuals studied were unrelated non-diabetic participants over the age of 20 years (n = 927) enrolled in the HUFS. This population-based study of African-Americans in the Washington, DC metropolitan area has been previously described by Adeyemo et al. [6]. For the present study, participants with FPG ≥7 mmol/l or who were receiving treatment for diabetes were excluded. Additional characteristics of the cohort can be found in Electronic supplementary material (ESM) Table 1.
Genotyping
All 927 DNA samples were prepared and genotyped as described by Adeyemo et al. [6]. Briefly, all samples passed a sample success rate of 95%. Single nucleotide polymorphisms (SNPs) were excluded if they had a success rate of less than 95% (41,885 SNPs excluded), a minor allele frequency (MAF) ≤0.01 (19,154 SNPs excluded), or had a p value for the Hardy-Weinberg test of equilibrium <10−3 (6,317 SNPs excluded). The current analysis focuses on the 808,465 autosomal SNPs that passed these filters. In addition, imputation was performed as reported by Shriner et al. [3]. We successfully imputed 1,506,100 SNPs using the Yoruba in Ibadan, Nigeria (YRI) reference panel and an additional 52,291 SNPs using the Centre d’Etude du Polymorphisme (Utah residents with northern and western European ancestry) (CEU) reference panel, for a total of 2,366,856 experimentally determined and imputed SNPs.
Statistical analyses
FPG was log-transformed and values greater than ±3 SDs from the mean value were winsorised (n = 8). All regression models were adjusted for age, sex, BMI and one EIGENSTRAT axis under an additive model. In separate analyses, hypertension was also adjusted for the known association with insulin resistance [7], but the effect was inconsistent, with the magnitude of the p value marginally increasing or decreasing significance for some SNPs (data not shown).
Replication analysis was performed on SNPs identified in GWASs of FPG based on information in the National Human Genome Research Institute’s catalogue of published GWASs (www.genome.gov/gwastudies/). The query returned hits indicating reported SNPs, their respective p values and associated genes (ESM Table 2). If multiple studies reported the same SNP, the SNPs with the lowest p value were included in the present study. The returned results included 16 SNPs associated with FPG in the Meta-Analyses of Glucose and Insulin-related traits Consortium (MAGIC) study [2]. The MAGIC SNPs were supplemented by 13 additional SNPs from previously published GWASs (ESM Table 2) for a total of 29 SNPs that we attempted to replicate in our African-American cohort.
Our replication effort occurred in two stages. In the first stage, we attempted to replicate 20 of the 29 exact published SNPs (i.e. direct replication) available in the HUFS dataset. For this stage, SNPs were considered replicated if the same HUFS SNP had a p value <0.05. For the second stage, we performed a local replication analysis based on a 500 kb linkage disequilibrium (LD) block containing a query SNP determined by the SNPs most distant from the query SNP with r
2 ≥ 0.3. We used the HapMap CEU LD data (http://hapmap.ncbi.nlm.nih.gov/downloads/ld_data/2008-06/00README.TXT) for all SNPs except for rs2166706 where the Gujarati Indians in Houston, Texas, USA (GIH) reference dataset (http://hapmap.ncbi.nlm.nih.gov/downloads/ld_data/2008-09_phaseIII/00README.txt) was used to match the original reported GWAS population [5]. Second, we estimated the covariance matrix for the block of SNPs using the HUFS genotype data. Third, the covariance matrix was spectrally decomposed and the effective degrees of freedom, Neff, were estimated using the relationship 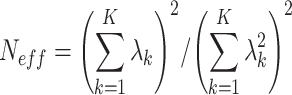 , in which λ
k is the kth eigenvalue of the K × K covariance matrix for the K SNPs [8]. Fourth, the nominal significance threshold α = 0.05 was divided by Neff.
, in which λ
k is the kth eigenvalue of the K × K covariance matrix for the K SNPs [8]. Fourth, the nominal significance threshold α = 0.05 was divided by Neff.
Power calculations were carried out using the Quanto software package (Version 1.2.3, http://hydra.usc.edu/gxe/). Calculations were based on: continuous outcome; an independent individuals design; and a gene-only hypothesis. An additive inheritance model was applied for varying MAFs. MAFs were calculated based on HUFS data; for SNPs with no associated HUFS data, HapMap- or Perlegen-reported MAFs were used. The power for the present study was determined based on reported effect estimates for FPG for each reported MAGIC SNP [2].
Results
Of the 16 SNPs recently reported in the MAGIC meta-analysis of over 122,000 participants [2], 12 were available for testing in the HUFS dataset (ESM Table 2). We directly replicated three SNPs (rs2191349, rs11558471 and rs4506565) located in or near DGKB-TMEM195, SLC30A8 and TCF7L2 genes respectively (Table 1). We also replicated SNPs from other GWASs for FPG: rs2722425 within ZMAT4 (p value = 0.024) as well as rs625643 (p value = 0.048), which is located in a functionally unknown region on chromosome 1 (Table 1). SNPs from the remaining studies that did not directly replicate are not shown. We note that SNPs in C2CD4B, FADS1, GCK and G6PC2 from the MAGIC study and IRS1, PDE4B, and ATP8B4 from other GWASs were not directly compared in the HUFS dataset owing to quality-control filters or lack of genotyping or imputation data.
Table 1.
SNPs that were reported in the MAGIC study and other GWASs of FPG that were directly analysed for replication in a cohort of African-Americans (the HUFS)
| SNP | Nearest gene | Region | Position | Reported p value | HUFS | ||
|---|---|---|---|---|---|---|---|
| Allele (effect/other) | β | p value | |||||
| HUFS | |||||||
| rs340874 | PROX1 | 1q41 | 212225879 | 7.1 × 10−8 | C/T | −0.0124 | 0.191 |
| rs780094 | GCKR | 2p23.2 | 27594741 | 2.5 × 10−12 | T/C | 0.0034 | 0.66a |
| rs11708067 | ADCY5 | 3q13.33 | 124548468 | 8.7 × 10−9 | G/A | −0.0028 | 0.73a |
| rs11920090 | SLC2A2 | 3q26.2 | 172200215 | 1.9 × 10−6 | A/T | 0.0001 | 1.0 |
| rs2191349 | DGKB-TMEM195 | 7p21.2 | 15030834 | 7.8 × 10−17 | G/T | −0.0136 | 0.023b |
| rs11558471 | SLC30A8 | 8q24.11 | 118254914 | 2.6 × 10−11 | G/A | −0.0255 | 0.0074b |
| rs7034200 | GLIS3 | 9p24.2 | 4279050 | 1.2 × 10−4 | C/A | 0.0056 | 0.35 |
| rs10885122 | ADRA2A | 10q25.2 | 113032083 | 8.4 × 10−11 | G/T | 0.0079 | 0.2 |
| rs4506565 | TCF7L2 | 10q25.2 | 114746031 | 1.2 × 10−8 | T/A | −0.0117 | 0.036a,b |
| rs10830963 | MTNR1B | 11q21 | 92348358 | 1.2 × 10−68 | G/C | −0.0046 | 0.67a |
| rs11605924 | CRY2 | 11p11.2 | 45829667 | 1.5 × 10−9 | C/A | 0.0048 | 0.57 |
| rs7944584 | MADD | 11p11.2 | 47292896 | 1.5 × 10−9 | T/A | 0.0026 | 0.84a |
| From other FPG GWASs | |||||||
| rs625643 | AK024684 | 1p32.3 | 54409755 | 4.0 × 10−3 | T/C | 0.01141 | 0.05a,b |
| rs2722425 | ZMAT4 | 8p11.21 | 40603396 | 9.0 × 10−6 | T/C | 0.01326 | 0.024b |
aGenotyped HUFS SNPs (all other SNPs were imputed)
bSNPs from reported studies that are also statistically significant (p < 0.05) in the HUFS dataset
We also analysed SNPs that were in LD (r 2 ≥ 0.3) with each discovery SNP (ESM Fig. 1). This replication strategy, which queried a 500 kb window centred on the index SNP, yielded a total of 317 SNPs located in or near nine different genes or unknown gene region (G6PC2, GCKR, MTNR1B, DGKB-TMEM195, TCF7L2, SLC30A8, AK024684, ZMAT4 and IRS1). Thirty-eight SNPs distributed across all nine gene regions of the 317 SNPs tested were significantly associated with FPG after Bonferroni correction for multiple comparisons (Table 2).
Table 2.
Significant SNPs and their effects (β) after Bonferroni correction that were in LD (r 2 ≥ 0.3) with reported SNPs from the MAGIC study and other GWASs of FPG
| Region | Nearest gene | Reported | SNP in LD | Study results | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| SNP | p value | Allele (effect/other) | β | r 2 | p value | df a | Corrected p value | |||
| HUFS | ||||||||||
| 2q31.1 | G6PC2 | rs560887 | 4.4 × 10−35 | rs575671 | A/G | −0.0185 | 0.50 | 0.003 | 2.76 | 0.01 |
| 2p23.2 | GCKR | rs780094 | 2.5 × 10−12 | rs780100 | G/T | 0.0142 | 0.46 | 0.020 | 2.55 | 0.05 |
| rs704791 | T/C | 0.0142 | 0.46 | 0.020 | 2.55 | 0.05 | ||||
| rs780102 | T/C | 0.0142 | 0.46 | 0.020 | 2.55 | 0.05 | ||||
| rs704795 | G/A | 0.0154 | 0.46 | 0.011 | 2.55 | 0.03 | ||||
| rs10208529 | T/A | −0.0198 | 0.54 | 0.021 | 2.55 | 0.05 | ||||
| 7p21.2 | DGKB-TMEM195 | rs2191349 | 7.8 × 10−17 | rs2215383 | T/C | −0.0175 | 1.0 | 0.003 | 2.38 | 0.007 |
| rs10244051 | T/G | −0.0163 | 1.0 | 0.004 | 2.38 | 0.01 | ||||
| rs10258074 | A/T | −0.0163 | 0.97 | 0.004 | 2.38 | 0.01 | ||||
| rs2191348 | G/T | −0.0164 | 1.0 | 0.004 | 2.38 | 0.009 | ||||
| rs6947830 | G/A | −0.0131 | 1.0 | 0.022 | 2.38 | 0.05b | ||||
| rs4719433 | T/C | −0.0131 | 1.0 | 0.022 | 2.38 | 0.05b | ||||
| rs1974620 | C/T | −0.0147 | 0.97 | 0.011 | 2.38 | 0.03b | ||||
| rs1558317 | T/A | −0.0147 | 0.96 | 0.011 | 2.38 | 0.03 | ||||
| rs1558318 | A/T | −0.0147 | 0.97 | 0.011 | 2.38 | 0.03 | ||||
| 8q24.11 | SLC30A8 | rs11558471 | 2.6 × 10−11 | rs11774700 | C/T | −0.0249 | 0.78 | 0.004 | 1.74 | 0.006 |
| 10q25.2 | TCF7L2 | rs4506565 | 1.2 × 10−8 | rs7901695 | C/T | −0.0127 | 0.88 | 0.022 | 1.72 | 0.04 |
| 11q21 | MTNR1B | rs10830963 | 1.2 × 10−68 | rs1447350 | C/G | −0.0141 | 0.43 | 0.014 | 2.46 | 0.03 |
| rs1447351 | A/G | −0.0131 | 0.43 | 0.022 | 2.46 | 0.05 | ||||
| rs4611171 | T/G | −0.0131 | 0.42 | 0.022 | 2.46 | 0.05b | ||||
| rs1597023 | G/A | −0.0131 | 0.42 | 0.022 | 2.46 | 0.05 | ||||
| rs4406791 | A/C | −0.0131 | 0.42 | 0.022 | 2.46 | 0.05 | ||||
| rs1447352 | G/A | −0.0131 | 0.43 | 0.022 | 2.46 | 0.05 | ||||
| rs9666752 | G/A | −0.0148 | 0.36 | 0.009 | 2.46 | 0.02 | ||||
| From other FPG GWASs | ||||||||||
| 1p32.1 | AK024684 | rs625643 | 4 × 10−3 | rs671431 | G/C | 0.0143 | 0.62 | 0.022 | 1.66 | 0.04 |
| 2q36.1 | IRS1 | rs2943641 | 5 × 10−2 | rs6713510 | A/T | 0.0169 | 0.43 | 0.005 | 5.13 | 0.03 |
| rs4072096 | G/A | 0.0171 | 0.43 | 0.005 | 5.13 | 0.03 | ||||
| rs4645008 | A/C | 0.0181 | 0.43 | 0.003 | 5.13 | 0.02 | ||||
| rs2943644 | T/C | 0.0181 | 0.41 | 0.003 | 5.13 | 0.02 | ||||
| rs2943633 | C/T | 0.0167 | 0.45 | 0.005 | 5.13 | 0.03 | ||||
| rs2972150 | C/A | 0.0161 | 0.45 | 0.007 | 5.13 | 0.04 | ||||
| rs2943636 | C/T | 0.0165 | 0.45 | 0.006 | 5.13 | 0.03 | ||||
| rs2972149 | A/G | 0.0201 | 0.50 | 0.0007 | 5.13 | 0.004b | ||||
| rs1515114 | A/T | 0.0196 | 0.60 | 0.0009 | 5.13 | 0.005b | ||||
| rs2722429 | G/A | 0.0186 | 0.60 | 0.001 | 5.13 | 0.006 | ||||
| 8p11.21 | ZMAT4 | rs2722425 | 9 × 10−6 | rs1823643 | T/C | 0.0192 | 0.89 | 0.002 | 1.53 | 0.007 |
| C/A | 0.0145 | 0.65 | 0.021 | 1.53 | 0.03b | |||||
| C/T | 0.0146 | 0.68 | 0.018 | 1.53 | 0.03 | |||||
a df calculated from covariance matrix of SNPs in LD (r 2 ≥ 0.3) with query SNP in 500 kb window; df were used for Bonferroni corrections
bGenotyped HUFS SNPs (all other SNPs were imputed)
Based on reported effect sizes of the 14 MAGIC loci (excluding TCF7L2 and SLC30A8), the power was calculated for each SNP using the African-American MAFs where available (ESM Table 3). The estimated power for this study ranged from a low of 0.25 to a high of 0.99. The SNPs in the four genes most strongly powered (i.e. > 90% power) in this study were either directly replicated (DGKB-TMEM195) or locally replicated (G6PC2, MTNR1B and GCKR) with markers in moderate LD (r 2 ≥ 0.4). The effect sizes for SNPs rs4506565 and rs11558471 (previously reported loci TCF7L2 and SLC30A8, respectively) were not reported in the MAGIC study.
Discussion
Chronically elevated FPG is a primary indicator of diabetes, making it an important barometer of the progression of impaired glucose metabolism. In this paper, we attempted to replicate, in nearly 1,000 African-Americans, significant GWAS loci for FPG in populations of predominantly European ancestry. In light of well-reported increased genetic diversity in populations with African ancestry [9, 10], our replication strategy not only focused on the reported SNPs but also included querying variants in LD with the reported SNPs.
We focused our replication analysis on the MAGIC study of over 122,000 participants to identify FPG-associated SNPs shared across ethnically diverse populations. In addition, we included SNPs from prior GWASs of FPG to add breadth to our replication pool, keeping in mind that potential differences in susceptibility loci between populations may exist [11]. Of the 12 SNPs reported by the MAGIC study that were directly testable in our African-American cohort, we replicated three SNPs within DGKB-TMEM195, TCF7L2 and SLC30A8. We also replicated SNPs in ZMAT4, which encodes a zinc finger, matrin type 4 protein identified in previous GWASs but not replicated in the MAGIC meta-analysis. Using the local (LD-based) replication strategy, we replicated additional SNPs in or near previously reported genes, including the insulin receptor substrate 1 gene.
Interestingly, comparison of the LD structure in HUFS to HapMap reference samples CEU and YRI supports the utility of African-American population samples in refining association loci. For example, the covariance matrix generated for the local replication of rs625643 spans 40 kb and includes 16 SNPs. In HapMap CEU, nearly the entire region is in moderate LD, whereas in HapMap YRI two distinct LD blocks are observed (ESM Fig. 2) and lower (on average) r 2 values are observed between rs625643 and downstream SNPs (0.78 for CEU and 0.5 for YRI). As expected, African-American samples (i.e. HapMap African Ancestry in Southwest, USA [ASW] and this study HUFS) show an LD structure intermediate to CEU and YRI (ESM Fig. 3 and ESM Fig. 2). Furthermore, given the association signals in HUFS, a case can be made for a narrowing of the region of interest from 40 kb to 3 kb between the locally replicated SNP rs671431 and the original discovery SNP (ESM Fig. 3).
We acknowledge the fluid interpretation of r 2 values within the context of establishing variants in LD with each other as well as the concern of being overly conservative in our correction for multiple comparisons. At an r 2 ≥ 0.3, we attempted to first capture a significant portion of SNPs in LD within the reference sample while maintaining confidence in the ability of related SNPs to serve as proxies [12]. In addition, a blanket search window of 500 kb would allow for capture of some unique characteristics of LD and long-range LD associated with admixed populations such as African-Americans [13]. To address the burden of potentially overcorrecting for multiple comparisons, our Bonferroni correction strategy was based on the estimation of effective degrees of freedom [8], which provides an analysis of covariance among HUFS SNPs in the extracted LD block that was based on CEU HapMap samples. We feel this approach better describes the relationship of SNPs in LD within the queried window instead of assuming the very conservative approach of independent effects for all tested SNPs.
We also acknowledge the limitation of our study of about 1,000 participants to detect some of the very small effect sizes reported by the MAGIC study, which included more than 122,000 participants. However, this study had over 80% power to replicate similar effect sizes for 10 of the 14 SNPs reported by the MAGIC study (ESM Table 3); this is evident in this study’s ability to replicate several of the published GWAS variants for FPG. We caution that lack of replication in this study may be due to limited sample size, differences in effect sizes calculated for SNPs and difference in allele frequency between populations of European and African ancestries.
The need for understanding differential susceptibility to diseases at the population level makes the identification of risk factors for diabetes and its indicators, including FPG, particularly important in the African-American community and other ethnic groups, given their disproportionate rate of morbidity and mortality from diabetes and associated complications. Unfortunately and for multiple reasons, the majority of GWASs aimed at identifying genetic variants associated with FPG and diabetes have so far focused predominantly on individuals of European ancestry. While the results from these studies provided tremendous insight into the genetic architecture of the disease, recent studies of non-European populations have shown utility in expanding the breadth of populations studied. Specifically, studies in East Asians allowed for a ‘wider net’ to be cast in the identification of type 2 diabetes susceptibility variants [4, 11]. The present study’s focus on individuals self-identified as African-Americans not only widens the net but also underscores the need for directed investigation of under-represented populations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Characteristics of HUFS participants (PDF 48.4 kb)
GWAS catalog search resultsa (XLS 45.5 kb)
Distribution of estimated power for HUFS to replicate reported SNPs in the MAGIC study (PDF 128 kb)
Corrected p–values and linkage disequilibrium in the HUFS sample for locally replicated loci. The green dots indicate replicated SNPs that are significant and the blue diamond indicates the original discovery SNP. If the original discovery SNP was not available in HUFS for genotyping (rs560887 and rs2943641), a blue bar on the x–axis is shown for relative position. The position of genes associated with each query SNP is denoted by a black horizontal bar. Gene position is relative to the listed SNPs for each plot. (PDF 1,586 kb)
Linkage disequilibrium plots from Haploview for a 40–kb region, which was determined by the covariance matrix of the local replication analysis of rs625643 located in an unknown gene region (AK024684). Triangle plots were generated based on information from three different HapMap samples of European, African, and African American ancestries (i.e., CEU, YRI, and ASW, respectively). Pairwise SNP r2 values (x 100) are indicated and LD between markers range from complete or strong (black) to weak or no (white) LD. (PDF 63 kb)
Corrected p–values and linkage disequilibrium in the HUFS sample for rs625643 located in a noncoding region (AK024684). The blue diamond indicates the original discovery SNP rs625643 discovered in individuals of European ancestry. The green dot indicates SNP rs671431 replicated in HUFS (African Americans). The shaded area indicates a region of interest (approximately 3 kb) informed by rs625643 and rs671431 taking into account low association values of flanking SNPs and different LD structure in African Americans compared to CEU (PDF 117 kb)
Acknowledgements
The HUFS was supported by National Institutes of Health grants S06GM008016-320107 to C. Rotimi and S06GM008016-380111 to A. Adeyemo. We thank the participants of the study, for which enrolment was carried out at the Howard University General Clinical Research Center, supported by National Institutes of Health grant 2M01RR010284. The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official view of the National Institutes of Health. This research was supported in part by the Intramural Research Program of the Center for Research on Genomics and Global Health, which is supported by the National Human Genome Research Institute, the National Institute of Diabetes and Digestive and Kidney Diseases, the Center for Information Technology and the Office of the Director at the National Institutes of Health (Z01HG200362). Genotyping support was provided by the Coriell Institute for Medical Research.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Abbreviations
- CEU
Centre d’Etude du Polymorphisme (Utah residents with northern and western European ancestry)
- FPG
Fasting plasma glucose
- GWAS
Genome-wide association study
- HUFS
Howard University Family Study
- LD
Linkage disequilibrium
- MAF
Minor allele frequency
- MAGIC
Meta-analyses of Glucose and Insulin-related traits Consortium
- SNP
Single nucleotide polymorphism
- YRI
Yoruba in Ibadan, Nigeria
References
- 1.O’Rahilly S. Human genetics illuminates the paths to metabolic disease. Nature. 2009;462:307–314. doi: 10.1038/nature08532. [DOI] [PubMed] [Google Scholar]
- 2.Dupuis J, Langenberg C, Prokopenko I, et al. New genetic loci implicated in fasting glucose homeostasis and their impact on type 2 diabetes risk. Nat Genet. 2010;42:105–116. doi: 10.1038/ng.520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shriner D, Adeyemo A, Gerry NP, et al. Transferability and fine-mapping of genome-wide associated loci for adult height across human populations. PLoS One. 2009;4:e8398. doi: 10.1371/journal.pone.0008398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McCarthy MI. Casting a wider net for diabetes susceptibility genes. Nat Genet. 2008;40:1039–1040. doi: 10.1038/ng0908-1039. [DOI] [PubMed] [Google Scholar]
- 5.Chambers JC, Zhang W, Zabaneh D, et al. Common genetic variation near melatonin receptor MTNR1B contributes to raised plasma glucose and increased risk of type 2 diabetes among Indian Asians and European Caucasians. Diabetes. 2009;58:2703–2708. doi: 10.2337/db08-1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Adeyemo A, Gerry N, Chen G, et al. A genome-wide association study of hypertension and blood pressure in African Americans. PLoS Genet. 2009;5:e1000564. doi: 10.1371/journal.pgen.1000564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reaven GM. Insulin resistance, hyperinsulinemia, hypertriglyceridemia, and hypertension. Parallels between human disease and rodent models. Diabetes Care. 1991;14:195–202. doi: 10.2337/diacare.14.3.195. [DOI] [PubMed] [Google Scholar]
- 8.Bretherton CS, Widmann M, Dymnikov VP, Wallace JM, Blade I. The effective number of spatial degrees of freedom of a time-varying field. J Climate. 1999;12:1990–2009. doi: 10.1175/1520-0442(1999)012<1990:TENOSD>2.0.CO;2. [DOI] [Google Scholar]
- 9.Schuster SC, Miller W, Ratan A, et al. Complete Khoisan and Bantu genomes from southern Africa. Nature. 2010;463:943–947. doi: 10.1038/nature08795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tishkoff SA, Reed FA, Friedlaender FR, et al. The genetic structure and history of Africans and African Americans. Science. 2009;324:1035–1044. doi: 10.1126/science.1172257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Unoki H, Takahashi A, Kawaguchi T, et al. SNPs in KCNQ1 are associated with susceptibility to type 2 diabetes in East Asian and European populations. Nat Genet. 2008;40:1098–1102. doi: 10.1038/ng.208. [DOI] [PubMed] [Google Scholar]
- 12.Ardlie KG, Kruglyak L, Seielstad M. Patterns of linkage disequilibrium in the human genome. Nat Rev Genet. 2002;3:299–309. doi: 10.1038/nrg777. [DOI] [PubMed] [Google Scholar]
- 13.Evans DM, Cardon LR. A comparison of linkage disequilibrium patterns and estimated population recombination rates across multiple populations. Am J Hum Genet. 2005;76:681–687. doi: 10.1086/429274. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Characteristics of HUFS participants (PDF 48.4 kb)
GWAS catalog search resultsa (XLS 45.5 kb)
Distribution of estimated power for HUFS to replicate reported SNPs in the MAGIC study (PDF 128 kb)
Corrected p–values and linkage disequilibrium in the HUFS sample for locally replicated loci. The green dots indicate replicated SNPs that are significant and the blue diamond indicates the original discovery SNP. If the original discovery SNP was not available in HUFS for genotyping (rs560887 and rs2943641), a blue bar on the x–axis is shown for relative position. The position of genes associated with each query SNP is denoted by a black horizontal bar. Gene position is relative to the listed SNPs for each plot. (PDF 1,586 kb)
Linkage disequilibrium plots from Haploview for a 40–kb region, which was determined by the covariance matrix of the local replication analysis of rs625643 located in an unknown gene region (AK024684). Triangle plots were generated based on information from three different HapMap samples of European, African, and African American ancestries (i.e., CEU, YRI, and ASW, respectively). Pairwise SNP r2 values (x 100) are indicated and LD between markers range from complete or strong (black) to weak or no (white) LD. (PDF 63 kb)
Corrected p–values and linkage disequilibrium in the HUFS sample for rs625643 located in a noncoding region (AK024684). The blue diamond indicates the original discovery SNP rs625643 discovered in individuals of European ancestry. The green dot indicates SNP rs671431 replicated in HUFS (African Americans). The shaded area indicates a region of interest (approximately 3 kb) informed by rs625643 and rs671431 taking into account low association values of flanking SNPs and different LD structure in African Americans compared to CEU (PDF 117 kb)