

Abstract
We analyzed HIV-1 genome sequences from 68 newly-infected volunteers in the Step HIV-1 vaccine trial. To determine whether the vaccine exerted selective T-cell pressure on breakthrough viruses, we identified potential T-cell epitopes in the founder sequences and compared them to epitopes in the vaccine. We found greater distances for sequences from vaccine recipients than from placebo recipients (p-values ranging from < 0.0001 to 0.09). The most significant signature site distinguishing vaccine from placebo recipients was Gag-84, a site encompassed by several epitopes contained in the vaccine and restricted by HLA alleles common in the cohort. Moreover, the extended divergence was confined to the vaccine components of the virus (Gag, Pol, Nef) and not found in other HIV-1 proteins. These results represent the first evidence of selective pressure from vaccine-induced T-cell responses on HIV-1 infection.
Introduction
The Step trial was a double-blind phase IIB test-of-concept study of the Merck Adenovirus 5 (MRKAd5) HIV-1 subtype B Gag/Pol/Nef vaccine. It was conducted at 34 sites in North America, the Caribbean, South America and Australia, where HIV-1 subtype B is predominant; and enrolled 3,000 individuals. Immunizations were halted after the first interim analysis showed that the vaccine neither prevented HIV-1 infection nor reduced viral load setpoint1,2.
Preliminary analyses showed that the MRKAd5 vaccine was immunogenic: more than 75% of vaccinated participants elicited HIV-specific T cells, yet there was no distinction between volunteers who subsequently became infected and those who remained seronegative1,2.
Although cytotoxic T lymphocyte (CTL) responses were not sufficient to prevent infection, we explored whether vaccine-elicited T cells had an impact on HIV-1 strains that established infection in volunteers. We compared HIV-1 sequences isolated from vaccine and placebo recipients to test for a ‘sieve effect’, i.e., that immunization with MRKAd5 affected founder HIV-1 population(s).
Results
Characterization of founder HIV-1 sequences
Single template-derived and directly sequenced HIV-1 amplicons were obtained from 40 vaccine and 28 placebo recipients, echoing the higher rate of HIV-1 acquisition among vaccinees (Table 1). Near full-length genome (nflg) sequences (~9.1 Kb) were obtained from 66 volunteers, and half-genomes from two individuals. We amplified 429 nflg and 36 additional half-genomes, with up to 14 nflg per specimen. All specimens corresponded to the HIV-1 sample from the time of diagnosis (except for one obtained one month later); including 18 individuals who were seronegative.
Table 1. Study volunteers with available sequence data.
This includes only subjects with infections identified before January 1, 2008. NA= not available.
| Gender |
Circumcision |
Country |
White |
Ad5 Ab titer |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| M | F | Yes | No | NA | US | CAN | Peru | Yes | No | NA | <200 | >200 | |
| All | 67 | 1 | 35 | 23 | 10 | 52 | 3 | 13 | 32 | 27 | 9 | 40 | 28 |
| Vaccine | 40 | 0 | 18 | 17 | 5 | 30 | 1 | 9 | 18 | 18 | 4 | 22 | 18 |
| Placebo | 27 | 1 | 17 | 6 | 5 | 22 | 2 | 4 | 14 | 9 | 5 | 18 | 10 |
For each intra-host dataset, we estimated the number of viral variants that established infection by inspecting nflg alignments, phylogenetic tree topologies and nucleotide sequence diversity; nflg- and gene-specific results were evaluated for congruence (env results: Supplementary Table 1). Distinct clusters were found for each subject, and the divergence between founder variants was never sufficient to suggest multiple source partners. HIV-1 infections were established by a single variant in 75% of individuals, while 2 founder variants were found in 15 individuals and four variants in one individual (Fig. 1). The proportion of multiple founders was similar between vaccine (25%) and placebo (24%) recipients.
Fig. 1. Maximum-Likelihood phylogenetic tree of gag sequences.
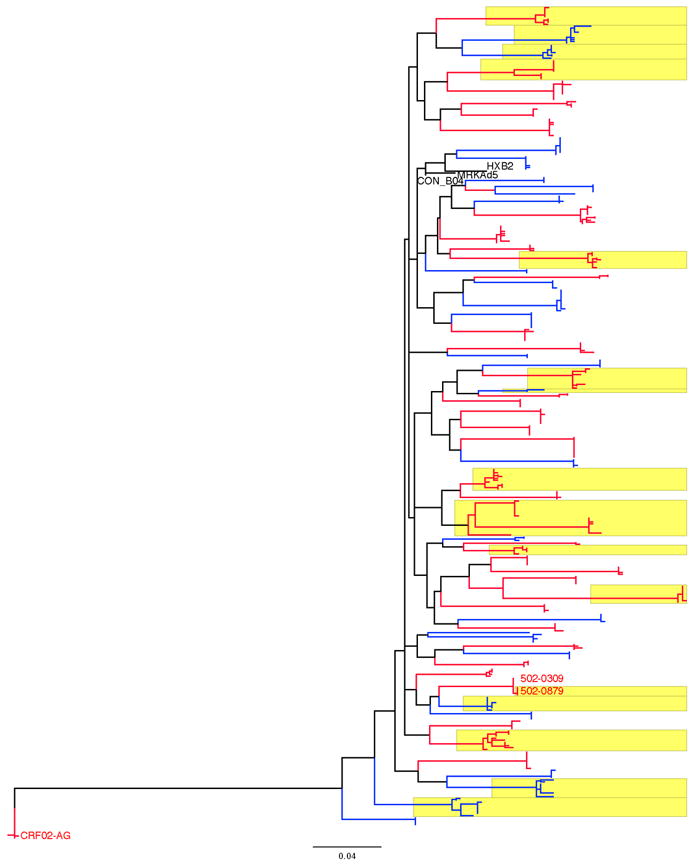
The tree comprises nucleotide sequences from each individual along with the MRKAd5, HXB2 and CON_B04 sequences, and is rooted with sequences from the only subject not infected with a subtype B virus (CRF02-AG). Sequences from placebo recipients are in blue, while sequences from vaccine recipients are in red. Sequences from individuals with two or more founder variants are highlighted in yellow. The sequences from related viruses found in two individuals are labeled with the two subjects’s identification numbers
Phylogenetic analysis of founder viruses
Phylogenetic trees were reconstructed for gag, pol and nef using either all volunteer-derived sequences or consensus sequences corresponding to the founder variant(s), which accurately represented the homogeneous populations found in acute/early HIV-1 infection. We found no evidence of phylogenetic clustering based on vaccine/placebo status (Fig. 1). Phylogenetic analyses were also performed using all volunteer-derived env nucleotide sequences (n = 459) along with 243 circulating sequences isolated since 2000 in Canada, Peru and the US3 (Supplementary Fig. 1) – env was chosen to maximize the phylogenetic signal and the inclusion of contemporary circulating sequences. Sequences from vaccine and placebo recipients were interspersed among contemporary circulating sequences irrespective of vaccine/placebo status, and there was limited geographic clustering. A possible linkage was identified between two vaccine recipients, and confirmed with later specimens.
Whole-gene/protein sieve analysis
Measures of viral sequence diversity and divergence from the MRKAd5 insert sequences showed higher values for gag, pol and nef nucleotide sequences from vaccinees than from placebo recipients, yet these differences were generally not significant, indicating that the volunteer sequences were not globally distinguishable by treatment assignment. Only in gag did the mean population diversity differ significantly: 0.0026 among vaccinees vs 0.0014 among placebo recipients (p = 0.022). Protein divergence from the MRKAd5 vaccine insert sequences also tended to be higher among vaccinees. Next, we compared private and phylogenetically-informative mutations: private mutations are found in only one sequence in an intra-individual set while phylogenetically-informative mutations occur in at least two sequences. There was no distinction between vaccine and placebo groups, except for private mutations in Gag. The proportion of private mutations (number of private mutations divided by the number of sequences for each individual) was higher among vaccinees compared to placebo recipients (mean 0.331 vs. 0.178; p = 0.026), implying that private mutations, i.e., those found at the tips of the trees and that are not yet fixed or not fixed because they are deleterious, were more frequent among vaccinees.
Predicted CTL epitope sieve analysis
Using the peptide HLA-binding predictor NetMHC4 on all consensus founder variant(s), 2,061 potential epitopes were predicted in Gag, Pol and Nef for distance comparisons with MRKAd5. The number of predicted epitopes per individual did not differ between vaccine and placebo recipients (median 29 vs. 31; p = 0.9). Overall, mean epitopic distances to MRKAd5 were significantly larger for vaccine than for placebo recipients (0.061 vs. 0.049; p = 0.007), indicating that, within probable epitopes, founder sequences were more divergent from the vaccine among vaccinees (Fig. 2a). On a protein level, only Gag-specific epitopic distances were significantly larger for vaccine than placebo recipients (mean 0.076 vs 0.034; p < 0.0001) (Fig. 2b-b).
Fig. 2. Epitope-specific protein distances between epitopes from founder sequences and the MRKAd5 HIV-1 vaccine sequence.
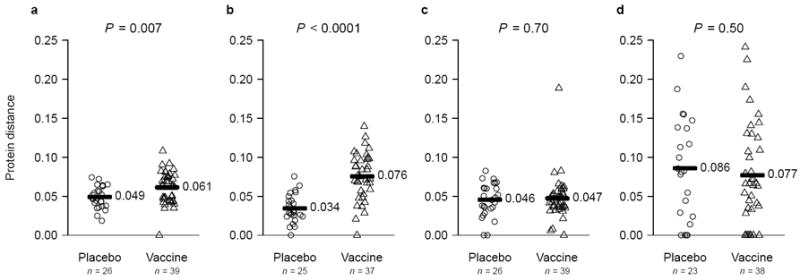
Comparison of epitopic distances across treatment assignment based on NetMHC predictions using Gag, Pol and Nef epitopes combined (a), or separately (b-d).
We evaluated the presence of known HLA class-I polymorphisms in predicted epitopes from volunteers’ sequences mismatched compared to MRKAd5, using as reference HLA-associated sites described by Brumme and colleagues5. Among all volunteers’ predicted epitopes, 642 NetMHC epitopes differed from the MRKAd5 and 449 of these (70%) showed mutations typical of known CTL-mediated pressure in subtype B sequences.
Since MRKAd5 expressed gag, pol and nef, a vaccine-mediated impact on founder sequences could be confined to these proteins and not found in other HIV-1 proteins. We therefore conducted the above analysis using the full proteomes of HXB2 and the subtype B 2004 consensus sequence (CON_B04)3 as references. HXB2 is a typical ‘ancestral’ strain and corresponds to one of the vaccine inserts (constituted of CAM-1, HXB2 and JRFL), and CON_B04 is contemporary to the viruses with which volunteers were infected. We expected distances to be higher against MRKAd5 and HXB2, than against CON_B04. Distance comparisons comprised 3,235 NetMHC predicted epitopes with HXB2 and 3,318 with CON_B04. Considering HXB2-derived epitopes for Gag-Pol-Nef, epitopic distances were larger among vaccinees than among placebo recipients (mean 0.069 vs 0.060; p = 0.02) (Fig. 3a). In contrast, there was no distinction if epitopes from Env-Rev-Tat-Vif-Vpr-Vpu were considered (mean 0.126 vs 0.118; p = 0.42) (Fig. 3b). The largest difference was seen in Gag: 0.085 among vaccine vs 0.045 among placebo recipients (p < 0.0001). Also showing that the reference did not bias our results, mean epitopic distances to CON_B04 were larger among vaccinees in the vaccine insert proteins but not in the remainder of the proteome: 0.053 vs 0.044 (p = 0.04) for Gag-Pol-Nef and 0.097 vs 0.098 (p = 0.97) for Env-Rev-Tat-Vif-Vpr-Vpu (Fig. 3c-d). Again, the largest distance between vaccine and placebo against CON_B04 resided in Gag: 0.073 vs. 0.031 (p < 0.0001).
Fig. 3. Protein distances between epitopes from founder sequences and HXB2 or HIV-1 CON_B04.
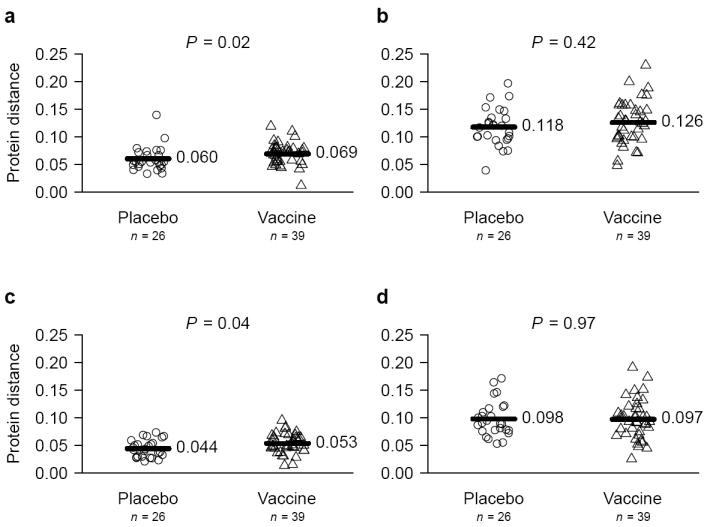
Epitopic distances based on NetMHC predictions are compared across treatment groups, separately for epitopes derived from Gag, Pol and Nef (vaccine insert), and for epitopes derived from the six other HIV-1 proteins.
Comparable results were obtained using Epipred for epitope predictions, the main difference was that the signal was stronger in Nef than in Gag (Online Supplement and Supplementary Fig.2).
K-mers sieve analysis
The above sieve analyses compared only predicted CTL epitopes: any founder sequence peptide with mutations that precluded its identification as an epitope was excluded. Therefore, we calculated the ‘epitope mismatch distance’, which measures the percentage of founder sequences with mismatches when compared to NetMHC-derived epitopes in MRKAd5. We found greater percentages of mismatches among vaccinees than among placebo recipients (p = 0.01) (Supplementary Fig. 3). Mirroring the epitope distance results, only Gag-specific mismatch distances were significantly larger for vaccine than placebo recipients (p < 0.0001) (Supplementary Fig. 3b). With HXB2 as reference, the distances calculated based on Gag-Pol-Nef tended to be higher among vaccinees (p = 0.06), whereas there was no difference for distances from proteins not in MRKAd5 (Supplementary Fig. 3e-f).
Sieve analysis of distances and stratification of the cohort
A sieve effect might be strongest in vaccinees who adhered to the vaccine regimen and had measurable T-cell responses prior to infection. Hence, sieve analyses were repeated for adherent vaccinees with a week 8 IFN-γ ELISpot response, but distances were not greater in the restricted population. There was also no evidence that the interval from last vaccination to sequence sampling (range 22-549 days), or the number of immunizations prior to infection (range 1-3), biased the assessment of the sieve effect (Supplementary Fig. 4).
Amino acid signature site sieve analysis
At each amino acid (AA) site, we compared the residues in founder HIV-1 sequences to the residue in MRKAd5 and measured the rate of AA mismatches to identify signature sites distinguishing vaccine and placebo recipients. Ten signature sites were identified with a q-value < 0.20: four in Gag, two in Pol, and four in Nef. No signature sites were identified in the control protein Env (Fig. 4).
Fig. 4. Amino Acid Signature Sites.
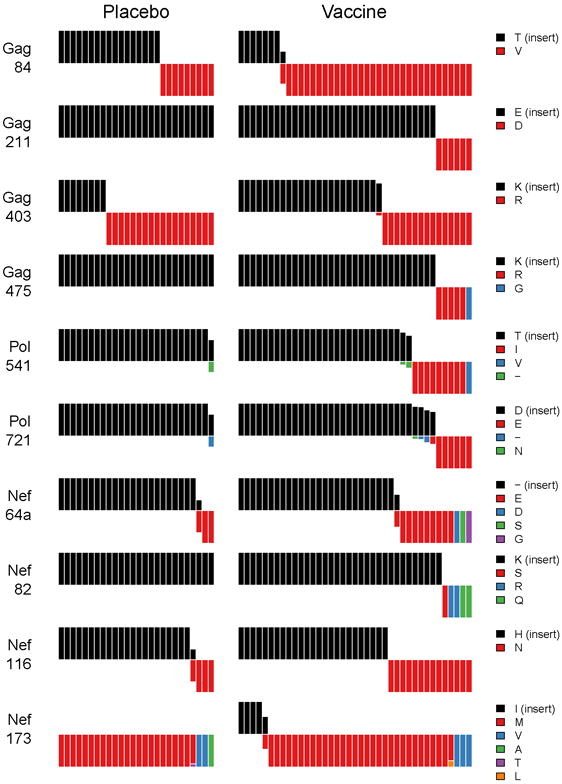
Ten AA signature sites distinguishing vaccine and placebo recipients are represented with bar graphs. Each bar represents the AA found in one individual: the upper/black bars represent the individuals with an AA matching the MRKAd5 HIV-1 insert at that position and the lower/red bars correspond to individuals with mismatched AA.
Signature site Gag84 showed the strongest evidence (q = 0.012): it is encompassed by several known CTL epitopes, including the well-characterized A*02-epitope SLYNTVATL (77-85). Thirty-six of 64 subjects had an HLA class I allele restricting epitopes that spanned Gag846. The signature at Gag84 was more pronounced among individuals with an HLA allele matching an “A-list” epitope7 (79%:17% mismatch vaccine:placebo compared to 80%:46% mismatch in the 28 subjects without an A-list restricting allele), supporting that vaccine-induced T-cell pressure led to the high rate of mismatch among vaccine recipient founder sequences. Additional signature sites included Gag211 located in the A*2501 epitope ETINEEAAEW (203-212) (q = 0.09). The consensus residue E211, found in 93% of circulating sequences, was found in all sequences from placebo recipients and in 33 of 39 vaccinees. However, five of the latter 6 vaccinees had an allele restricting a potential epitope covering position 211. Gag84 was the only significant signature site after stringent Holm-Bonferroni correction (p = 0.01), certainly due to the high frequency of HLA-A*02 among volunteers and the multiplicity of epitopes encompassing that site. The corollary is that we had limited power to detect AA mutations associated with rare HLA alleles in our cohort.
As CD8+ T-cell epitopes are generally 9-mers, a similar antigen scanning analysis was performed to identify 9-mers with significantly different distance distributions in vaccinees compared to placebo recipients. Fourteen 9-mers had greater distances among vaccinees: ten in Gag (two encompassed signature sites described above) and four in the epitope-rich region Nef121-132 (Supplementary Table 2). Interestingly, some of the identified k-mers mapped to protein regions with no predicted epitopes.
CTL responses in vaccinees prior to infection
To relate the above date to detectable CTL responses, IFN-γ ELISpot assays were conducted following vaccination but prior to HIV-1 infection using PBMC from 27 of the 39 vaccinees with sequence data, and peptides corresponding to the immunogen (Fig. 5). Of the 27, only 21 had at least one response to Gag, Pol or Nef. CTL responses elicited by the vaccine generally corresponded to predicted epitopes, but few responses were detected, thus precluding our ability to detect a sieve effect using only the epitope recognition data. (Predictions based on Epipred are shown in Supplementary Fig. 5).
Fig. 5. Summary of sequence analyses and IFN-g ELISpot data from Gag (a,b), Nef (c,d) and Pol (e,f).
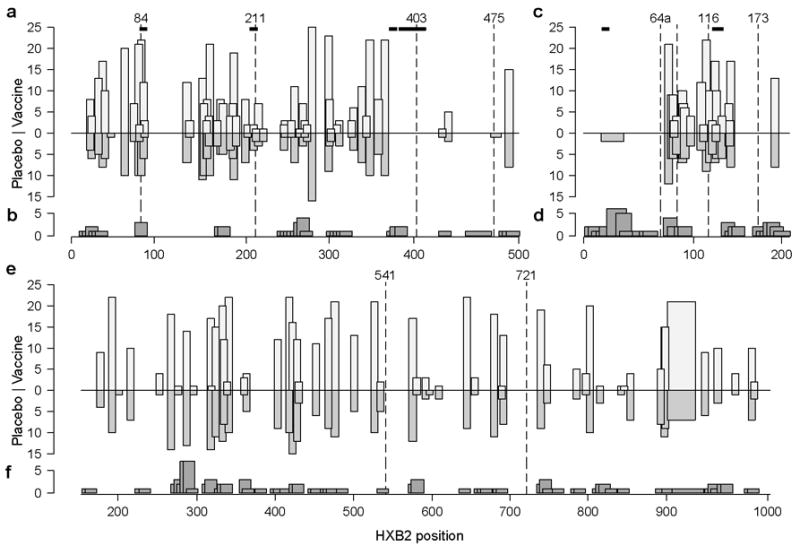
For each protein, the upper panel (a,c,e) represents the location of predicted epitopes based on NetMHC in vaccine and placebo groups (light and medium gray columns, respectively), amino acid signature sites (dashed lines), and signature k-mers (horizontal bars). The lower panel (b,d,f) corresponds to IFN-γ ELISpot responses detected in 21 vaccine recipients prior to infection. 15mer peptides overlapping by 11 amino acids were used to assess responses. The region covered by each responsive peptide is shown as the box width, whereas the number of subjects reacting with that peptide is given by the box height.
Discussion
Ideally, our analyses would have focused on CTL responses induced by the vaccine and detected in vaccine recipients pre-infection. However, the paucity of CTL responses identified, and only in a subset of vaccine recipients (21 individuals), precluded statistically powered analyses. To evaluate more subjects than the immune reactivity data authorized, our sequence analyses included both the vaccine and placebo groups. Because the clinical trial was randomized and double-blinded, and the analysis restricted to infections diagnosed during the blinded period, the observed sequence differences between vaccine vs. placebo are causally attributable to vaccine assignment. Our results showed that the MRKAd5 vaccine had an impact on breakthrough viral populations: viruses infecting vaccinees were more likely to encode epitopes that differed from those present in the vaccine. Moreover, the difference between vaccine and placebo recipients was confined to the segments of the virus included in the vaccine, i.e., Gag, Pol and Nef, while other proteins did not differ by treatment assignment.
As the vaccine lacked clinical efficacy, it is remarkable that it left a genetic imprint on the founder viral strains. A sieve effect for CTL-based vaccines can be understood either as 1) the exclusion of specific HIV-1 variants (the ones that are the most similar to the vaccine inserts) from establishing a sustained infection due to killing by vaccine-elicited CTL, or as 2) the ‘diversion’ of the founder variants, driven to accumulate more CTL-mediated mutations and/or to accumulate them faster, a process that requires escape followed by selective outgrowth of the escape mutant during acute infection. It is difficult to distinguish these hypotheses with the current data, and both processes could act concomitantly. We found no direct evidence of a selective exclusion at transmission since: i) vaccinees were more likely to become infected than placebo recipients, ii) single or multiple founder variants were equally likely to be found in both groups, and iii) phylogenetic analyses showed no clustering of founder viruses according to vaccine/placebo status. The fact that the sieve effect was seen only in predicted epitopes and only in proteins that were components of the vaccine suggests the possibility of T-cell mediated selection occurring post-infection. Additional factors (albeit weaker) concur with the hypothesis of T-cell mediated selection post-infection: iv) phylogenetic analyses of gag showed that genetic distances calculated between volunteers’ sequences (or between volunteers’ sequences and the vaccine sequence) tended to be higher among vaccinees, while genetic distances calculated from the Most Recent Common Ancestor (MRCA) were not, and v) the proportion of private mutations was higher among gag nucleotide sequences from vaccine recipients. These two related observations suggest that the variation corresponds to recent events during infection that have not been fixed (as opposed to more basal effects that could have reflected founder events with more divergent strains among vaccinees). Such evidence support that CTL responses primed by the vaccine could have elicited anamnestic CTL responses, i.e., the responses could have occurred sooner or more frequently during breakthrough infection than in placebo recipients, leading to accelerated CTL escape. The possibility of early CTL escape following HIV-1 vaccination has been reported in an HLA-B27+ vaccine recipient8. The mutation R264G was found in the KK10 epitope and caused viral escape in the third year of infection, a much shorter time frame than typical for a KK10 escape9-11, but much later than the time span evaluated here. Reece and colleagues recently compared the fate of macaques vaccinated either with a single Gag epitope or with a full Gag insert12. They reported fast escape mutations and no control of viremia for macaques vaccinated with the single epitope construct as opposed to no escape mutations in that same epitope and control of viremia for macaques immunized with full-length Gag. This suggests that a vaccine that does not elicit a broad enough response could provoke rapid escape mutations, a pattern that is reminiscent of the Step immunizations that elicited only narrow CTL responses. Counterbalancing findings in favor of post-infection selection, and arguing in favor of restriction of specific variants from establishing infection (despite overall higher infection rate in vaccinees) is our failure to significantly detect simultaneously both the unmutated and escape forms of epitopes in vaccinees’s sequences.
Our finding of a sieve effect on breakthrough viruses provides impetus for designing novel vaccine inserts: we postulate that vaccine pressure should be elicited in a controlled manner for a vaccine to be beneficial, i.e., the vaccine should trigger mutations in regions of the virus known to be associated with viral control and should avoid prematurely creating a cycle of escape variants that could act as immunodominant decoys13. Therefore, we propose that ‘cornering’ the virus to potentially debilitated forms should be a goal for novel designs of CTL-based vaccines.
The results presented here are the first to demonstrate an impact of vaccination on viruses establishing HIV-1 infection, and Gag was the principally correlated vaccine target. The differences in genetic distances across HIV-1 genes reflected their intrinsic variability, and differences based on reference strains were also expected, with shorter distances to the contemporary CON_B04 than to the ancestral HXB2 sequence. The fact that Pol is subjected to the strongest purifying selection renders the detection of differences more difficult and may explain the lack of significance in Pol. Imprinting on Gag was most robustly supported in both our phylogenetic and sieve analyses; yet, based on Epipred-derived epitopes, the signal appeared to be mostly driven by differences in Nef. Given that the epitope prediction tools we used were trained on partially different datasets and assumptions, they rely on potentially different features of epitope-MHC binding; therefore we may expect non-redundant information and possibly discrepancies between prediction methods. It is recognized that the predictors are not extremely accurate because i) our catalogue of known epitopes is limited, and ii) multiple operating pathways intersect between the degradation of a protein and the presentation of an epitope14. Numerous studies have shown varying performances of predictive algorithms depending on datasets and alleles, yet, there has been no critical assessment of the different methods, and, as such, no consensus on an ideal epitope prediction method15. Importantly, we note that the sieve analyses based on K-mers, which are free of epitope predictions, corroborated the analyses based on predicted epitopes.
It is significant that hallmarks of CTL pressure were identified over broader epitopic regions than estimated by the CTL responses measured prior to infection, suggesting that vaccine-elicited T cells may be sufficient to impact genetic features of breakthrough viruses without detection of responses using conventional IFN-γ-ELISpot assays (N.F. & M.J.M, in prep.). It would appear, however, that the vaccine-elicited responses lacked sufficient immunogenicity, were not targeting protective epitopes, or were weak and thus easily evaded. Although the selective forces we detected were not sufficient to prevent infection or reduce viral loads, they do provide a new benchmark to evaluate the impact of forthcoming vaccines. The selective impact on founding viruses may have resulted from CTL below current levels of detection, or potentially from other selective forces such as targeting by CD4+ T cells or the innate immune system. It remains to be established what the impact of anamnestic responses is on the kinetics of emergence of escape mutants, and whether vaccine-mediated selection had a prolonged effect on viral sequence evolution or an impact on viral replication capacity. Assessing whether vaccine-mediated polymorphisms in founding viruses affected viral fitness is also crucial to explore, as a fitness-impaired virus could translate into a reduction in viral loads and attenuation of disease progression.
Methods
HIV-1 near full-length genome sequencing
PCR products corresponded to single amplifiable viral genomes derived from plasma collected at HIV-1 diagnosis. GenBank accession numbers: JF320002-JF320643.
Our strategy was to obtain 5 to 10 nflg sequences per specimen, depending on intra-host sequence variation assessments: if 5 or more phylogenetically-informative sites were found in the first 5 nflg, then 5 additional nflg were sequenced. Phylogenetically-informative sites correspond to mutations that are found in at least two sequences (http://indra.mullins.microbiol.washington.edu/cgi-bin/InSites/index.cgi).
Phylogenetic Analyses
Maximum-likelihood phylogenetic trees were reconstructed by estimating the GTR-I-G nucleotide substitution model and included sequence(s) for all volunteers as well as MRKAd5, HXB2 and CON_B04 sequences. Pairwise diversity and tree-based divergence measures were calculated from the most recent common ancestor, MRKAd5, HXB2 and CON_B04 sequences.
CTL Epitope Prediction
Known HIV-1 epitopes were included and potential CTL epitopes were predicted using NetMHC4 (http://www.cbs.dtu.dk/services/NetMHC/) and Epipred16 (http://atom.research.microsoft.com/bio/epipred.aspx/). NetMHC predicts binding of peptides to 4-digit HLA alleles; we accepted known epitopes reported at the Los Alamos National Laboratory HIV database (HIVDB) and variant epitopes that had identical HXB2 coordinates and were strong or weak binders. Epipred identifies known and potential HIV-1 CTL epitope motifs using 2-digit HLA information. HLA-specific epitopes were predicted in all HIV-1 proteins derived from the volunteers’ sequences and in the corresponding consensus founder sequences, based on each individual’s HLA genotype. Sequences from 3 volunteers were excluded (non-B subtype virus; non-male volunteer; no HLA-genotype). Epitopes were also identified in MRKAd5, HXB2 and CON_B04 sequences.
Sieve analyses
We performed both ‘global’ sieve analyses, which are based on summary measures of distances between volunteer sequences and a reference sequence, and ‘local’ sieve analyses, which identify signature positions/peptides between the vaccine and placebo group.
Whole-Protein Tree-based Sieve Analysis
Tree-based distances were calculated using an HIV-specific substitution model of protein evolution (HIV-10) 17. For each individual, the average of the distances between the MRKAd5 HIV-1 sequence and each volunteer sequence was computed, and comparisons between the vaccine and placebo groups were done using a Wilcoxon/Mann-Whitney test.
Global sieve effects restricted to CTL epitopes from founder variants
Two T cell epitope-based distance measures were used: the ‘CTL epitope’ and ‘K-mer’ distances. For the subset of predicted epitopes that were shared in the volunteers’ and reference sequence (with at most 2 AA differences), we computed pairwise distances using the HIV-10 model. For each subject, the CTL epitope distance was the average of the different epitope-specific pairwise distances.
K-mer distances are based on epitopes in the reference sequence and allow inclusion of mutated forms of the epitopes in the volunteers’ sequences that would not be recognized as epitopes. Using NetMHC predictions, we defined the distance as the estimated percent of mismatched epitopes, i.e. the number of k-mers in the reference sequence that had mismatches against the corresponding peptide in the founder sequence(s).
Global sieve effects restricted to CTL epitopes in all individual sequences
Parallel to above but based on all volunteer-derived sequences.
Signature positions in founder variants
For each site in Gag, Pol, and Nef, we compared the rate of AA mismatch to the MRKAd5 insert between the vaccine group and placebo group. The same analysis was done for Env with HXB2 as reference. We used the t-statistic numerator-type statistic of Gilbert, Wu, and Jobes18, and their permutation procedure, to compute an unadjusted p-value for each position. To account for multiple tests, we applied the Holm-Bonferroni multiplicity adjustment procedure with Tarone’s19 modification to improve power, and also estimated q-values.
Signature positions in all individual sequences
For each position we estimated the probability of a mismatch between an individual sequence and the reference sequence (either MRKAd5 or HXB2) for the vaccine and placebo groups. If a position is a signature position, the parameter estimated by this difference will be significantly different from zero. Unadjusted p-values were computed using the nonparametric bootstrap (robust with different numbers of sequences per subject). The analyses were performed on all positions for which at least four subjects had a non-consensus AA in at least one of their sequences. Adjusted p-values and q-values were computed as for Analysis 4.
Signature K-mer peptides based on all individual sequences
For each K-mer peptide in the MRKAd5 and HXB2 reference sequences we compared the distribution of similarity scores for all of the individual sequences between the vaccine and placebo groups. For each sequence we computed a similarity score to the reference K-mer by selecting the closest similarity score across all K-mers in that sequence. The estimated means were computed using generalized estimating equations to fit restricted-moment models (marginal mean models), using a Gaussian model with exchangeable correlation structure.
Further details can be found in the Online Supplement.
Supplementary Material
Acknowledgments
We thank the study participants for their time and dedication; the HVTN Laboratory Program, SCHARP, and Core staff who contributed to the study implementation and analysis; the Merck functional teams: the Clinical Research Specialist Organization, Worldwide Clinical Data Management Operation, Clinical Research Operations, and the Clinical Assay and Sample Receiving Operations. This work was supported by US Public Health Service grant AI41505.
Footnotes
Author contributions Designed the sequence analysis: MR, ACdC, PBG, JIM
Conducted the analyses: MR, ACdC, PBG, CAM, LH, BSM, WD, FL, JIM
Analyzed the data: MR, ACdC, NF, PBG, JIM
Wrote the manuscript: MR, ACdC, NF, PBG, JIM
Designed lab experiments: JIM, FEM, ST, ESB, NF
Performed lab experiments: MB, AB, AOS, JC, TJ, MN, KW, HZ, DNR, SS, JNS, NF
Conducted the Step trial, provided material, oversaw laboratories: JH, LC, SB, DRC, MNR, AD, MJM, SGS, SD, JS, JIM
References
- 1.Buchbinder SP, et al. Efficacy assessment of a cell-mediated immunity HIV-1 vaccine (the Step Study): a double-blind, randomised, placebo-controlled, test-of-concept trial. Lancet. 2008;372:1881–1893. doi: 10.1016/S0140-6736(08)61591-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McElrath MJ, et al. HIV-1 vaccine-induced immunity in the test-of-concept Step Study: a case-cohort analysis. Lancet. 2008;372:1894–1905. doi: 10.1016/S0140-6736(08)61592-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Leitner T, et al., editors. HIV Sequence Compendium 2005, 648+viii. Theoretical Biology and Biophysics Group, Los Alamos National Laboratory, NM; Los Alamos, NM: 2006. [Google Scholar]
- 4.Buus S, et al. Sensitive quantitative predictions of peptide-MHC binding by a ‘Query by Committee’ artificial neural network approach. Tissue Antigens. 2003;62:378–384. doi: 10.1034/j.1399-0039.2003.00112.x. [DOI] [PubMed] [Google Scholar]
- 5.Brumme ZL, et al. HLA-associated immune escape pathways in HIV-1 subtype B Gag, Pol and Nef proteins. PLoS One. 2009;4:e6687. doi: 10.1371/journal.pone.0006687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Frahm N, et al. Extensive HLA class I allele promiscuity among viral CTL epitopes. Eur J Immunol. 2007;37:2419–2433. doi: 10.1002/eji.200737365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Korber BTM, et al., editors. HIV Molecular Immunology. Los Alamos National Laboratory, Theoretical Biology and Biophysics; Los Alamos, New Mexico: 2006/2007. [Google Scholar]
- 8.Betts MR, et al. Characterization of functional and phenotypic changes in anti-Gag vaccine-induced T cell responses and their role in protection after HIV-1 infection. Proc Natl Acad Sci U S A. 2005;102:4512–4517. doi: 10.1073/pnas.0408773102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kelleher AD, et al. Clustered mutations in HIV-1 gag are consistently required for escape from HLA-B27-restricted cytotoxic T lymphocyte responses. J Exp Med. 2001;193:375–386. doi: 10.1084/jem.193.3.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Feeney ME, et al. Immune escape precedes breakthrough human immunodeficiency virus type 1 viremia and broadening of the cytotoxic T-lymphocyte response in an HLA-B27-positive long-term-nonprogressing child. J Virol. 2004;78:8927–8930. doi: 10.1128/JVI.78.16.8927-8930.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goulder PJ, et al. Late escape from an immunodominant cytotoxic T-lymphocyte response associated with progression to AIDS. Nat Med. 1997;3:212–217. doi: 10.1038/nm0297-212. [DOI] [PubMed] [Google Scholar]
- 12.Reece JC, et al. Timing of immune escape linked to success or failure of vaccination. PLoS One. 2010;5 doi: 10.1371/journal.pone.0012774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rolland M, Nickle DC, Mullins JI. HIV-1 Group M Conserved Elements Vaccine. PLoS pathogens. 2007;3:e157. doi: 10.1371/journal.ppat.0030157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yewdell JW. Confronting complexity: real-world immunodominance in antiviral CD8+ T cell responses. Immunity. 2006;25:533–543. doi: 10.1016/j.immuni.2006.09.005. [DOI] [PubMed] [Google Scholar]
- 15.Lafuente EM, Reche PA. Prediction of MHC-peptide binding: a systematic and comprehensive overview. Curr Pharm Des. 2009;15:3209–3220. doi: 10.2174/138161209789105162. [DOI] [PubMed] [Google Scholar]
- 16.Heckerman D, Kadie C, Listgarten J. Leveraging information across HLA alleles/supertypes improves epitope prediction. J Comput Biol. 2007;14:736–746. doi: 10.1089/cmb.2007.R013. [DOI] [PubMed] [Google Scholar]
- 17.Nickle DC, et al. HIV-specific probabilistic models of protein evolution. PLoS One. 2007;2:e503. doi: 10.1371/journal.pone.0000503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gilbert PB, Wu C, Jobes DV. Genome scanning tests for comparing amino acid sequences between groups. Biometrics. 2008;64:198–207. doi: 10.1111/j.1541-0420.2007.00845.x. [DOI] [PubMed] [Google Scholar]
- 19.Tarone RE. A modified Bonferroni method for discrete data. Biometrics. 1990;46:515–522. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.