

Abstract
Understanding the mechanism of protein folding requires a detailed knowledge of the structural properties of the barriers separating unfolded from native conformations. The S-peptide from ribonuclease S forms its α-helical structure only upon binding to the folded S-protein. We characterized the transition state for this binding-induced folding reaction at high resolution by determining the effect of site-specific backbone thioxylation and side-chain modifications on the kinetics and thermodynamics of the reaction, which allows us to monitor formation of backbone hydrogen bonds and side-chain interactions in the transition state. The experiments reveal that α-helical structure in the S-peptide is absent in the transition state of binding. Recognition between the unfolded S-peptide and the S-protein is mediated by loosely packed hydrophobic side-chain interactions in two well defined regions on the S-peptide. Close packing and helix formation occurs rapidly after binding. Introducing hydrophobic residues at positions outside the recognition region can drastically slow down association.
Keywords: phi-value analysis, protein-protein interaction, encounter complex formation, thioxo peptide bond
Conformational changes in proteins play an important role in many biological processes. A detailed understanding of the mechanism of conformational transitions requires knowledge of the structural and dynamic properties of the initial and final states as well as the characterization of the transition barrier separating them. Site-directed mutagenesis proved a powerful tool to determine the properties of transition states for reactions involving proteins. Comparing the effect of amino acid replacements on kinetics and thermodynamics of a reaction identifies the interactions that are important for the rate-limiting step of a process. This method has been successfully applied to characterize transition states for protein folding (1, 2), for protein-protein interactions (3–5) and for conformational changes in folded proteins (6). Protein folding transition states were shown to have native-like topology in the whole protein or in major parts of the structure (7–9) and it is commonly assumed that this includes the presence of native-like secondary structure (10, 11). Because site-directed mutagenesis can only modify amino acid side chains, it is still unclear at which stage of a folding reaction backbone hydrogen bonds in secondary structure elements are formed. Several approaches have been applied to test for secondary structure in protein folding transition states. Replacing amino acids by glycyl and prolyl residues leads to changes in backbone conformation, which was used to probe α-helix formation (12), but these replacements introduce additional effects due to altered side-chain interactions. Introducing ester bonds into the polypeptide backbone (13) also has multiple effects because it leads to the loss of a backbone hydrogen bonding donor and strongly increases conformational flexibility of the polypeptide backbone. Deuteration of amide protons destabilizes helical structures, but the effect is very small (14). A near isosteric change in the polypeptide backbone can be achieved by introducing a thioxo-peptide bond, i.e., by replacing an amide oxygen by sulfur (15). Thioxo-peptide bonds slightly restrict conformational space of the polypeptide backbone (16–18) but are tolerated in α-helices and β-sheets (19–21). Thioxylation destabilizes α-helices by about 7 kJ/mol relative to an oxo-amide bond (21), which makes thioxo-peptide bonds a perfect tool to monitor formation of individual backbone hydrogen bonds during protein folding reactions.
We introduced thioxo-peptide bonds and side-chain replacements into the S-peptide from ribonuclease S (RNase S) to monitor formation of α-helical hydrogen bonds and of side-chain interactions in the transition state of a protein folding reaction (Fig. 1). RNase S is obtained by subtilisin cleavage of RNase A and comprises the S-peptide (residues 1–20) and the S-protein (residues 21–124) (22). S-protein is inactive but folded in isolation whereas the conformational equilibrium in the isolated S-peptide favors the unfolded state. The native, α-helical structure from residues 3 to 13 only forms upon binding to the S-protein, which yields a catalytically active complex. Studying the association reaction of ribonuclease S gives information on the folding reaction of a single α-helix induced by binding to a folded protein. Such reactions occur in a large number of intrinsically disordered proteins upon binding to their target proteins (23). It is an unsolved question, whether S-protein recognizes the small fraction of helical conformations of free S-peptide (conformational selection) or, whether S-protein interacts with unfolded S-peptide and folding occurs after binding (12, 24) (Scheme 1).
Fig. 1.

Stereo views of the structure of RNase S M13Nle variant consisting of the S-protein (white) and the S-peptide (green) X-ray structure. A shows side chains that were replaced for φ-value analysis. The positions are color coded according to their response to an increase in hydrophobicity with either a strong increase in kon (red), a slight increase in kon (orange), no effect on kon (yellow), or a strong decrease in kon (blue). B shows a ball-and-stick representation of S-peptide bound to S-protein. Oxygens replaced by sulfur for φ-value analysis and the corresponding H-bond donors are marked as balls and the respective helical H-bonds are shown. Coordinates were taken from the X-ray structure (25). The figure was prepared using Molscript (37).
Scheme 1.

The small size of the S-peptide makes it amenable to chemical synthesis, which expands the chemical repertoire by introducing nonnatural side chain and backbone modifications to yield information on interactions that is not accessible to proteins from natural sources. The combination of backbone thioxylation and amino acid replacements enabled us to obtain a high resolution picture of a protein folding transition state by monitoring the formation of specific intra and intermolecular backbone and side-chain interactions.
Results and Discussion
Kinetic and Thermodynamic Characterization of the S-Peptide/S-Protein Interaction.
S-peptide corresponding to residues 1–20 in ribonuclease A was chemically synthesized by solid-phase peptide synthesis. To avoid oxidation Met13 was replaced by norleucine (Nle) which has only minor effects on stability of the S-protein/S-peptide complex (25) and results in the canonical S-peptide sequence
which represents the reference wild-type S-peptide. Isothermal titration calorimetry (ITC) yields a free energy of binding (ΔG0) of -37.4 ± 0.1 kJ/mol at 25 °C (Table 1 and Fig. S1), which is in good agreement with previous results on a truncated S-peptide (amino acids 1–15) that also contained Nle at position 13 (ΔG0 = -36.7 ± 0.4 kJ/mol) (25).
Table 1.
Effect of single thioxo-bonds on stability and association kinetics of the S-peptide/S-protein complex at pH 6.0, 50 mM NaOAc, 100 mM NaCl, 25 °C
| Position of thioxo-bond | Keq (M-1) | ΔΔG0* (kJ/mol) | kon (·105 M-1 s-1) | ΔΔG0‡* (kJ/mol) | ϕ-value† |
| WT | (3.6 ± 0.1)·106 | - | (4.4 ± 0.2)·105 | - | |
| Lys1 | (1.3 ± 0.1)·106 | 2.5 | n.d. | ||
| Glu2 | (1.1 ± 0.1)·106 | 2.9 | n.d. | ||
| Thr3 | (6.1 ± 0.1)·104 | 10.1 | (3.0 ± 0.1)·105 | 0.90 | 0.09 |
| Ala4‡ | (1.4 ± 0.1)·106 | 2.3 | (4.4 ± 0.1)·105 | 0 | - |
| Lys7 | (1.3 ± 0.1)·104 | 13.9 | (4.1 ± 0.3)·105 | 0.17 | 0.01 |
| Phe8§ | (6.8 ± 0.9)·103 | 15.5 | (2.2 ± 0.1)·105 | 1.7 | 0.10 |
| Glu9 | (4.0 ± 0.5)·104 | 11.1 | (5.1 ± 0.4)·105 | −0.41 | −0.04 |
| Arg10 | (6.5 ± 0.1)·104 | 9.9 | (4.1 ± 0.3)·105 | 0.14 | 0.01 |
| His12§,¶ | (1.3 ± 0.3)·103 | 19.6 | n.d. | - | n.d. |
| Ser15 | (2.9 ± 0.1)·105 | 6.2 | (3.6 ± 0.4)·105 | 0.46 | 0.07 |
*ΔΔG0 represents the difference in free energy between a variant and the WT protein, which has a ΔG0 of -37.4 kJ/mol. ΔG0 was calculated from the equilibrium constant using ΔG0 = -RT ln Keq.
†Errors in ϕ-values depend on the errors in Keq and kon as well as on ΔΔG0 and are ≤ 0.05 for the variants used for ϕ-value analysis.
‡ΔΔG0 too small to determine an accurate ϕ-value (8).
¶Thioxo His12 is strongly destabilized, which does not allow measurement of the association kinetics by stopped-flow fluorescence.
Kinetics of S-peptide/S-protein association were measured under pseudo-first-order conditions with > 5-fold excess of S-peptide over S-protein. Fig. 2A compares folding kinetics of S-peptide onto S-protein measured by the change in circular dichroism (CD) at 225 nm, which monitors α-helix formation in the S-peptide, and by the change in fluorescence intensity above 305 nm, which is sensitive for changes in the environment of tyrosine residues in the S-protein. Both probes detect the same process with a time constant (τ = 1/kobs) of 29 ± 1 ms, which shows that helix formation in the S-peptide and changes in tertiary structure in the S-protein have the same rate-limiting step. Due to its higher sensitivity florescence-detection was used in all further studies. Fig. 2B shows that complex formation becomes faster with increasing S-peptide concentration under pseudo-first-order conditions which reveals a rate-limiting bimolecular association step with a second-order rate constant (kon) of (4.4 ± 0.1)·105 M-1 s-1 at 25 °C (Fig. 2B). This value is similar to kon-values found for other site-specific protein-protein interactions but significantly lower than the maximum diffusion-controlled limit for bimolecular reactions (kmax = 5 × 109 M-1 s-1), which is expected due to steric and orientational constraints (26) and because only a fraction of the protein surface is reactive. At high S-peptide concentrations the reaction becomes concentration-independent in some variants. These results show that the concentration-dependent reaction represents formation of an encounter complex between S-peptide and S-protein that is followed by a concentration-independent structural rearrangement in the bound state (12, 24), which may become rate-limiting at high S-peptide concentrations. All measurements were performed under conditions where association is rate-limiting. The rate constant for dissociation, koff, is obtained from the intercept of the pseudo-first-order plot with the y-axis and can be used to calculate Keq exclusively from kinetic measurements according to Keq = kon/koff. Fig. 2B shows, however, that koff is very small and thus the extrapolation to [S-peptide] = 0 is error-prone. Therefore Keq was determined independently by ITC measurements (Fig. S1).
Fig. 2.

Kinetics of the association reaction between unfolded S-peptide and folded S-protein. (A) Comparison of the reaction monitored by Tyr fluorescence > 305 nm (black) and by CD at 222 nm (red) reveals identical pseudo-first-order kinetics with a rate constant, kobs, of 35 ± 1 s-1 (τ = 29 ms). Concentration of S-protein was 10 μM, concentration S-peptide was 100 μM. The fluorescence-detected kinetics show an additional slow, concentration-independent reaction with a time constant of 5 s at 25 °C, which may arise from refolding of a small fraction of unfolded S-protein, which is at the edge of its stability at 25 °C. (B) Pseudo-first-order plot of RNase S association in the presence of increasing S-peptide concentrations. S-protein concentration was 5 μM. Association was monitored by the change in Tyr fluorescence. The slope of the plot yields a bimolecular association rate constant of kon = (4.4 ± 0.2)·105 M-1 s-1 using kobs = kon·[S-peptide]. Conditions were pH 6.0, 50 mM NaOAc, 100 mM NaCl at 25 °C.
To obtain structural information on the transition state of the S-peptide/S-protein interaction we related the effect of chemical modifications in the S-peptide on kon to the respective effects on Keq according to the Leffler equation (1, 27, 28)
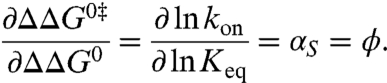 |
[1] |
In the extreme cases, the interaction that was perturbed by the modification is either completely present (ϕ = 1) or absent (ϕ = 0) in the transition state.
Thioxo Scan of RNase S Recombination.
Backbone hydrogen bond formation in the transition state of S-peptide/S-protein association was monitored by introducing single thioxo-peptide bonds at different positions between residues 1 and 15 of the S-peptide (Fig. 1 and Table 1). This region becomes structured upon complex formation and includes the α-helix from residues 3 to 13 (Fig. 1). All investigated S-peptide variants are unfolded at 25 °C (Fig. S2) and yield catalytically active RNase S upon recombination with S-protein indicating native structure of the complex. Thioxo groups in the region from Thr3 to Glu9 are hydrogen bonding acceptors for amide protons of residues 7–13, respectively, and are thus expected to destabilize the native, helical conformation of S-peptide by at least 7 kJ/mol, as observed for helical peptides (21). To probe hydrogen bond formation in the helical region of the S-peptide we introduced thioxo-peptide bonds at different positions throughout the helix (Fig. 1B and Table 1). All thioxo bonds in the helical region lead to reduced stability of RNase S compared to the wild type (Table 1). A thioxo-peptide bond at position 4 destabilizes the native complex by only 2.3 kJ/mol, whereas thioxo-groups at positions 7 to 9 lead to a stronger destabilization than expected from model peptides. Carbonyl oxygens of these residues form close intramolecular contacts within the S-peptide and/or with the S-protein (Fig. 1B and Table S1). A larger thioxo group at positions 7 to 9 is thus expected to interfere with close-packing.
To test for backbone interactions outside the helical region of the S-peptide we introduced thioxo-peptide bonds at several positions N- and C-terminal of the helix (Fig. 1 and Table 1). Amino acids Arg10 to Nle13 are located at the C-terminal end of the helix and thus their carbonyl oxygens do not participate in intrahelical hydrogen bonds. Thioxo-peptide bonds at positions 10 and 12 nevertheless lead to a large destabilization of RNase S which is probably due to destabilization of intermolecular hydrogen bonds to amide protons of the S-protein and to steric effects induced by the larger sulfur atom in this tightly packed region (Fig. 1 and Table S1). Further, the ϕ-,ψ-angles of the His12-Nle13 peptide bond are energetically unfavorable for thioxo groups compared to carbonyl groups (Table S1) (18), which may explain the drastic destabilization of the thioxo His12 RNase S. Thioxo-peptide bonds at positions Lys1 and Glu2 are located N-terminal of the helix and lead to only a minor destabilization of 2.5 kJ/mol and 2.9 kJ/mol, respectively (Table 1).
To characterize backbone interactions in the transition state of RNase S recombination we measured the effect of thioxylation on kon and compared the results to the respective effects on Keq according to Eq. 1. For a reliable ϕ-value analysis ΔΔG0 has to be sufficiently large (> 5–7 kJ/mol) (8, 11). Table 1 shows that all thioxo-peptide bonds in the helical region except thioxo Ala4 fulfill this criterion. To test for pure kinetic effects of thioxylation we also determined kon for this variant. The association kinetics revealed that thioxylation does not significantly affect kon at any investigated position resulting in uniform ϕ-values between 0 and 0.1 (Fig. 3 and Table 1). The absolute changes in ΔG0‡ are ≤ 1.7 kJ/mol at all positions (Table 1), which is much smaller than the observed effect of a thioxo-peptide bond on helix stability (ΔG0 = 7 kJ/mol). This result shows that native intrahelical backbone hydrogen bonds are not yet formed in the transition state of S-peptide/S-protein association. In addition, the low ϕ-value for thioxo Arg10 indicates the absence of this intermolecular hydrogen bond and of close-packing interactions around this thioxo group in the transition state. This low ϕ-value further rules out nonnative helical structure formation C-terminal of the native helix, which is supported by the low ϕ-value of 0.1 observed for the thioxo Ser15 variant. These results reveal the absence of helical structure in the transition state of binding and indicate the formation of only loosely packed intermolecular interactions because O → S substitutions at carbonyl groups in the region from Lys7 to His12, which are located in the tightly packed core of the complex, have no effect on association kinetics, although they drastically destabilize the native complex.
Fig. 3.

Effect of single thioxo-peptide bonds at the indicated positions on the association kinetics between S-peptide and S-protein under pseudo-first-order conditions. For comparison the result for wild-type S-peptide is shown as dashed line. The resulting kon-values are given in Table 1. Experimental conditions and data analysis as described in Fig. 2.
Probing Side-Chain Interactions in the Transition State for Association.
To identify interactions between the S-peptide and the S-protein that are important during formation of the encounter complex we measured ϕ-values for side-chain replacements at various positions in the S-peptide. Side chains of the amino acids in the region from Phe8 to Asp14 of the S-peptide form interactions with the S-protein (see Table S2). Close packing at the interface involves side chains of Phe8, His12, and Nle13, which contribute 64% to the S-peptide surface area that becomes buried in the complex (29) (Table S2) and were shown to be important for complex stability, which makes them well suited for ϕ-value analysis. Because Phe8 is the only aromatic residue in the S-peptide it is essential for concentration determination and could only be replaced by other aromatic amino acids. We introduced Tyr and Trp as well as the nonnatural amino acid 1-naphthylalanine (Nal) at position 8 to probe the effect of changes in hydrophobicity on the association kinetics. Replacing Phe8 by Tyr, slightly decreases hydrophobicity and leads to a major destabilization of the complex by 12.5 kJ/mol, whereas kon only slightly decreased (Fig. 4A). Trp and Nal at position 8 increase the hydrophobic surface area and destabilize the complex by 7.7 and 3.5 kJ/mol, respectively. Trp slightly accelerates association whereas Nal leads to a fourfold increase in kon (Fig. 4A). An increase in kon despite destabilization of the complex results in negative ϕ-values for these two variants (Fig. 4A and Table 2) which shows that increasing hydrophobic surface area at position 8 is favorable for formation of the encounter complex but unfavorable for the stability of the close-packed native complex.
Fig. 4.

Effect of various side-chain replacements on the association kinetics between S-peptide and S-protein. For comparison the result for the wild-type S-peptide is shown as dashed line. The resulting kon-values are given in Table 2. Experimental conditions and data analysis as described in Fig. 2.
Table 2.
Effect of side-chain replacements on stability and association kinetics of the S-peptide/S-protein complex at pH 6.0, 50 mM NaOAc, 100 mM NaCl, 25 °C
| S-peptide | Keq (M-1) | ΔΔG0 (kJ/mol) | kon (M-1 s-1) | ΔΔG0‡ (kJ/mol) | ϕ-value |
| WT | (3.6 ± 0.1)·106 | (4.4 ± 0.2)·105 | |||
| WT pH 8.5 | (1.2 ± 0.1)·107 | −3.0 | (9.5 ± 0.6)·105 | ||
| WT 1% DMSO | (2.2 ± 0.1)·106 | 1.2 | (3.4 ± 0.1)·105 | ||
| WT, low I* | (3.0 ± 0.1)·106 | 0.4 | (3.7 ± 0.3)·105 | - | |
| A5F | (2.6 ± 0.1)·106 | 0.8 | (4.2 ± 0.2)·105 | 0.12 | n.w.d. |
| A6F | (3.6 ± 0.2)·105 | 5.7 | (3.9 ± 0.2)·105 | 0.25 | 0.05 |
| K7F | (1.9 ± 0.1)·106 | 1.7 | (5.7 ± 0.4)·105 | −0.66 | < 0 |
| F8Y† | (2.3 ± 0.3)·104 | 12.5 | (3.4 ± 0.1)·105 | 0.61 | 0.05 |
| F8W | (1.6 ± 0.2)·105 | 7.7 | (5.0 ± 0.2)·105 | −1.79 | < 0 |
| F8Nal | (1.1 ± 0.1)·106 | 3.5 | (1.5 ± 0.1)·106 | −2.42 | < 0 |
| E9Q | (5.1 ± 0.2)·106 | −0.9 | (5.7 ± 0.4)·105 | −0.67 | n.w.d. |
| E9F | (4.0 ± 0.2)·106 | −0.3 | (1.6 ± 0.1)·106 | −3.16 | < 0 |
| R10F‡ | (7.7 ± 0.3)·105 | 2.6 | (5.8 ± 0.3)·105 | −0.70 | < 0 |
| Q11F‡ | (1.3 ± 0.1)·106 | 1.3 | (3.4 ± 0.1)·105 | 0 | 0 |
| H12A | (3.1 ± 0.1)·104 | 11.8 | (2.7 ± 0.5)·106 | −4.52 | < 0 |
| H12F | (1.4 ± 0.2)·105 | 8.0 | (3.5 ± 0.9)·106 | −5.15 | < 0 |
| Nle13A | (2.2 ± 0.8)·104 | 12.6 | (2.3 ± 0.1)·105 | 1.59 | 0.14 |
| Nle13F | (2.4 ± 0.1)·105 | 6.7 | (5.7 ± 0.1)·105 | −0.65 | < 0 |
| D14F | (1.1 ± 0.1)·105 | 8.6 | 38 ± 9 | 28.9 | 2.7 |
The protonated state of His12 forms a helix-stabilizing stacking interaction with Phe8 (30). Replacing His12 by Ala or Phe destabilizes the complex by 11.8 and 8.0 kJ/mol, respectively, but accelerates association by a factor of 4.5 and 8, respectively, which also results in negative ϕ-values (Fig. 4B and Table 2) and indicates that a His at position 12 is important for complex stability but unfavorable for the association reaction. Because the experiments were carried out at pH 6, where His is mainly protonated, we tested the role of the positive charge by studying the interaction with a deprotonated His at pH 8.5. At pH 8.5 the complex is slightly stabilized and kon is increased, but to a much lesser extent compared to replacing His by Ala or Phe (Fig. 4B), which shows that removal of the positive charge contributes to the increase in kon but it is not the major source. Rather increased hydrophobicity at position 12 seems to be the main determinant for the acceleration of association.
A correlation between hydrophobicity and kon is also observed at position 13. Phe and Ala at position 13 both destabilize the S-peptide/S-protein complex relative to Nle, but have different effects on kon. The more hydrophobic Phe slightly accelerates association, whereas the less hydrophobic Ala leads to a decrease in kon with a ϕ-value of 0.14 (Fig. 4C and Table 2).
Position-Dependence of Hydrophobic Interactions in the Transition State.
To test, whether increased hydrophobicity at positions outside the major interaction sites on the S-protein also accelerates association, we increased hydrophobicity in different regions of the S-peptide. Ala5 and Asp14 also form packing interactions with the S-protein (Fig. 1A and Table S2). An Ala5Phe substitution affects neither complex stability nor association kinetics, indicating that this region is not important for recognition during complex formation (Fig. 4D). Asp14 is located C-terminal of the tightly packed interface of the S-peptide helix with the S-protein and forms interactions with Val118, Phe120, and Tyr25 of the S-protein (Table S2). Increased hydrophobicity in the Asp14Phe variant yields catalytically active RNase S, which is destabilized by 8.6 kJ/mol. The association kinetics of this variant are decelerated more than 10,000-fold (ΔΔG0‡ = 28.9 kJ/mol) leading to a nonclassical ϕ-value of 2.7 (Fig. S4) which shows that the destabilizing effect of the Asp → Phe replacement on the transition state of association is much larger than the effect on the native complex. However, multiphasic association kinetics are observed for this variant indicating a change in the reaction mechanism which does not allow a quantitative comparison with the wild-type protein. These results show that a hydrophobic residue at position 14 does not allow efficient formation of site-specific interactions between S-protein and S-peptide, which is surprising because the complementary region on the S-protein is mainly hydrophobic. The effect of the D14F replacement is not due to the removal of favorable electrostatic interactions, because the association kinetics of the wild-type protein become slightly slower with decreasing ionic strength (Table 2). It is further very unlikely that the strong decrease in kon is due to native and/or nonnative interactions in the unfolded state of the D14F variant. These interactions would stabilize the unfolded state of the S-peptide by nearly 30 kJ/mol, which is about 80% of the free energy obtained upon complex formation (Table 2). In addition, the D14F replacement has no effect on the CD spectrum at 25 °C and on the thermal unfolding curve of the free S-peptide (Fig. S2).
To test for the effect of increased hydrophobicity at more solvent-exposed positions in the S-peptide on kon we replaced side chains of Ala6, Lys7, Glu9, Arg10, and Gln11 by Phe. The side chain of Glu9 is facing the S-protein in native RNase S but it is not in direct contact with any other residues in the S-peptide or S-protein (Fig. 1). Replacing Glu9 by Phe has virtually no effect on complex stability, but increases kon fourfold (Fig. 4D). This effect is not due to the removal of the negative charge, because the E9Q replacement increases both stability and kon slightly (Table 2). A small destabilization of the complex and a slight increase in kon is observed when Lys7, which does not interact with the S-protein, and Arg10, which is partially solvent-exposed (see Table S2), are replaced by Phe (Fig. 4D, Fig. S5, and Table 2). These results show that also increased hydrophobicity in the vicinity of the binding sites in the close-packed core can accelerate association slightly which suggests some structural plasticity in the location of the hydrophobic interactions between S-protein and S-peptide in the transition state (Fig. 1). However, increased hydrophobicity near the S-peptide binding site does not generally accelerate association. The Q11F replacement destabilizes the complex slightly but has no effect on kon (Fig. S5 and Table 2), although Gln11 is interacting with the S-protein. Similarly, replacing Ala6 by Phe, which increases the hydrophobicity of the S-peptide on the solvent-exposed side of the helix, destabilizes the complex by about 6 kJ/mol but has no effect on kon (Fig. S5 and Table 2).
Structure of the Transition State for Association.
Our results enable us to map both side-chain and backbone interactions in the rate-limiting step for S-protein/S-peptide recognition and association. S-protein was shown to be folded and have native secondary structure even in the absence of S-peptide (31). S-peptide is unfolded in isolation (Fig. S2). Thioxylation at all investigated positions in the S-peptide gives ϕ-values around zero, which indicates that thioxylation has no effect on the energetics on the rate-limiting step for binding/folding and demonstrates the absence of interactions involving the carbonyl oxygens in the transition state. Isolated model α-helical peptides are destabilized by 7 kJ/mol upon introduction of a thioxo-peptide bond (21). This comparison suggests that neither native nor nonnative helical structure in the S-peptide is important for the binding process. The absence of helical structure in the transition state is supported by the increase in kon in the R10F and H12A mutants, despite the removal of the helix-stabilizing salt bridge between Glu2 and Arg10 and a helix-stabilizing Phe8/His12+ interaction, respectively (30, 32). The S-protein/S-peptide recognition during formation of the encounter complex is rather mediated by loosely packed hydrophobic side-chain interactions. Increased side-chain hydrophobicity facilitates association mainly in two regions of the S-peptide located at positions 8/9 and 12/13 which directly face S-protein in the native-complex (Fig. 1A). The effect on association is most pronounced when polar or charged residues are replaced by hydrophobic residues (Fig. 4 and Table 2). The nature of the hydrophobic side chain seems to be of minor importance for recognition, because there is only a small difference in kon between various nonpolar side chains in the recognition regions (Fig. 4 and Table 2). Phe, one of the most hydrophobic natural amino acids (33) leads to only slightly faster association than the significantly less hydrophobic Ala. The largest difference between hydrophobic amino acids is observed for the Phe to Nal replacement at position 8. The hydrophobic interactions seem to be required to correctly position S-peptide and S-protein relative to each other, which allows subsequent rapid formation of the α-helix and of the close-packed core.
At first glance the formation of hydrophobic interactions with i, i + 4 spacing at positions 8/9 and 12/13 in the absence of helical structure in the region between the interaction sites seems paradoxical. This conundrum can be resolved if we assume that initial recognition and binding occur at either of the two regions—but not both simultaneously—prior to formation of the encounter complex. Interaction at the second site together with α-helix formation would then occur rapidly after this rate-limiting step. An alternative explanation would involve a distorted helical structure that is bound in the transition state and that has the same free energy for an oxo and a thioxo-peptide bond. However, this explanation seems unlikely because ϕ, ψ angles in the range of -60 ± 15°, -43 ± 15° give rise to undistorted helical H-bonds. Thioxo groups would even destabilize a loosely packed helix, as observed for isolated model peptides (21), which is not in agreement with the results from the thioxo S-peptide variants (Table 1). Conversely, if ϕ, ψ angles are outside of this fairly narrow helix basin, the segment would no longer be consistent with either an α-helical structure or the 8/9 and 12/13 spacing of the hydrophobic interactions.
Increasing hydrophobicity on the solvent-exposed side of the helix has no effect on kon at positions Ala6 and Gln11 (Fig. 1B), which shows that a general increase in hydrophobicity is not sufficient to accelerate association. The deceleration of association upon increasing hydrophobicity at position 14 suggests that the negative charge at this position of the wild-type S-peptide prevents strong interactions of this position with the S-protein during formation of the encounter complex and thus directs binding to the loosely packed hydrophobic interactions involving the regions around Phe8 and His12/Nle13. Strong interactions between extended regions of the S-protein and S-peptide already during formation of the encounter complex may prevent subsequent rapid helix formation and rapid formation of the tightly packed native interface in the encounter complex. The transition state structure is independent of temperature, because nearly identical changes in ΔG° and kon are obtained at 10 °C (Table S3).
Stability and dynamics of RNase S have previously been investigated by hydrogen exchange which suggested that helix unzipping without unbinding of the S-peptide occurs as a native-state fluctuation (24). This result is in agreement with our findings and indicates that the helical structure of the S-peptide forms only in the final folding reaction from the encounter complex to native RNase S. Partly contradictory results on the structure of the transition state for association were obtained from a fluorescein-labeled S-peptide, which binds 40-times faster than the wild-type S-peptide in a process that is accelerated by electrostatic interactions (12, 34, 35). In contrast, electrostatic contributions slightly slow down association of the unlabeled peptide as indicated by the slightly decreased kon at low ionic strength (Table 2). This discrepancy can be attributed to the differences in net charge between wild-type S-peptide and the fluorescein-labeled variant. Fluorescein labeling of Lys7 requires acetylation of the N terminus and replacement of Lys1. In addition, a truncated variant with an amidated C terminus at position S15 was used for labeling. These modifications change the net charge of the S-peptide from +1 to -1, which accelerates association most likely by a favorable interaction with a positively charged RNA binding region on the S-protein. At high salt concentrations these interactions are shielded and the fluoresceine-labeled variant and the wild-type S-peptide have a similar kon-values (34).
Role of Hydrophobic Interactions in Protein Folding Transition States.
Nonclassical ϕ-values < 0 upon increasing side-chain hydrophobicity are a surprising feature of the transition state for RNase S recombination. In ϕ-value analysis of protein folding transition states a larger side chain is typically replaced by a smaller side chain, commonly by alanine. This replacement often leads to fractional ϕ-values around 0.2–0.3 (8, 11). In the S-peptide/S-protein system this type of replacement would also lead to classical ϕ-values at most positions, due to decreased hydrophobicity (see Table 2). A study on the effect of hydrophobicity on the folding kinetics of the small monomeric protein NTL9 also revealed that increased hydrophobicity can accelerate folding despite a destabilization of the native state (36). Together with our results these observations suggest that loosely packed hydrophobic interactions are sufficient to position the different parts of the polypeptide chain correctly relative to each other in the transition state for protein folding and for protein-protein interactions. Increasing hydrophobicity at the interaction sites and even at neighboring sites can stabilize the transition state and thus lead to faster folding or association. This native-like orientation of the different parts of a protein by hydrophobic side-chain interactions seems to be more important for the rate-limiting step of folding than the presence of native secondary structure, which can occur fast after the rate-limiting step.
Methods
S-peptide variants were synthesized by Fmoc solid-phase peptide synthesis and purified by reversed-phase HPLC-chromatography. Thioxo-peptide bonds were introduced as described (15). S-protein was obtained by cleaving RNase A with subtilisin and subsequent purification with cation-exchange and size-exclusion chromatography. Keq for RNaseS complex formation was determined by isothermal titration calorimetry (ITC). Keq-values for very instable complexes were determined by activity assays. Kinetics of complex formation were induced by stopped-flow mixing of S-protein and S-peptide under pseudo-first-order conditions with > 5-fold excess of S-peptide and monitored by the change in Tyr fluorescence or by the change in CD at 222 nm, which gave identical kinetics. Standard conditions were 50 mM NaOAc pH 6.0, 100 mM NaCl. Fig. 1 was prepared using the program Molscript (37). More detailed descriptions of the experimental procedures are given in the SI Text.
Supplementary Material
Acknowledgments.
We thank Buzz Baldwin and George Rose for discussion, Jörg Fanghaenel for assistance in ITC measurements, and Therese Schulthess for S-protein preparations. This work was supported by the Bundesministerium für Bildung und Forschung (ProNet-T3) and the Munich Center for Integrated Protein Science.
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1012668108/-/DCSupplemental.
References
- 1.Matouschek A, Kellis JJ, Serrano L, Fersht AR. Mapping the transition state and pathway of protein folding by protein engineering. Nature. 1989;340:122–126. doi: 10.1038/340122a0. [DOI] [PubMed] [Google Scholar]
- 2.Matthews CR. Effect of point mutations on the folding of globular proteins. Method Enzymol. 1987;154:498–511. doi: 10.1016/0076-6879(87)54092-7. [DOI] [PubMed] [Google Scholar]
- 3.Kiel C, Selzer T, Shaul Y, Schreiber G, Herrmann C. Electrostatically optimized Ras-binding Ral guanine dissociation stimulator mutants increase the rate of association by stabilizing the encounter complex. Proc Natl Acad Sci USA. 2004;101:9223–9228. doi: 10.1073/pnas.0401160101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mateu MG, Manuel M, Del Pino S, Fersht AR. Mechanism of folding and assembly of a small tetrameric protein domain from tumor suppressor p53. Nat Struct Biol. 1999;6:191–198. doi: 10.1038/5880. [DOI] [PubMed] [Google Scholar]
- 5.Taylor MG, Kirsch JF, Rajpal A. Kinetic epitope mapping of the chicken lysozyme. HyHEL-10 fab complex: delineation of docking trajectories. Protein Sci. 1998;7:1857–1867. doi: 10.1002/pro.5560070902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Grosman C, Zhou M, Auerbach A. Mapping the conformational wave of acetylcholine receptor channel gating. Nature. 2000;403:773–776. doi: 10.1038/35001586. [DOI] [PubMed] [Google Scholar]
- 7.Itzhaki LS, Otzen DE, Fersht AR. The structure of the transition state for folding of chymotrypsin inhibitor 2 analyzed by protein engineering methods: evidence for a nucleation-condesation mechanism for protein folding. J Mol Biol. 1995;254:260–288. doi: 10.1006/jmbi.1995.0616. [DOI] [PubMed] [Google Scholar]
- 8.Sánchez IE, Kiefhaber T. Origin of unusual phi-values in protein folding: evidence against specific nucleation sites. J Mol Biol. 2003;334:1077–1085. doi: 10.1016/j.jmb.2003.10.016. [DOI] [PubMed] [Google Scholar]
- 9.Nickson AA, Stoll KE, Clarke J. Folding of a LysM domain: entropy-enthalpy compensation in the transition state of an ideal two-state folder. J Mol Biol. 2008;380:557–569. doi: 10.1016/j.jmb.2008.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Otzen DE, Itzhaki LS, elMasry NF, Jackson SE, Fersht AR. Structure of the transition state for the folding/unfolding of the barley chymotrypsin inhibitor 2 and its implications for mechanisms of protein folding. Proc Natl Acad Sci USA. 1994;91:10422–10425. doi: 10.1073/pnas.91.22.10422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Naganathan AN, Munoz V. Insights into protein folding mechanisms from large scale analysis of mutational effects. Proc Natl Acad Sci USA. 2010;107:8611–8616. doi: 10.1073/pnas.1000988107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goldberg JM, Baldwin RL. A specific transition state for S-peptide combining with folded S-protein and then refolding. Proc Natl Acad Sci USA. 1999;96:2019–2024. doi: 10.1073/pnas.96.5.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Deechongkit S, et al. Context-dependent contributions of backbone hydrogen bonding to beta-sheet formation. Nature. 2004;430:101–105. doi: 10.1038/nature02611. [DOI] [PubMed] [Google Scholar]
- 14.Krantz BA, et al. Understanding protein hydrogen bond formation with kinetic H/D amide proton effects. Nat Struc Biol. 2002;9:458–463. doi: 10.1038/nsb794. [DOI] [PubMed] [Google Scholar]
- 15.Wildemann D, et al. A nearly isosteric photosensitive amide-backbone substitution allows enzyme activity switching in ribonuclease S. J Am Chem Soc. 2007;129:4910–4918. doi: 10.1021/ja069048o. [DOI] [PubMed] [Google Scholar]
- 16.Cour TFM. Stereochemistry of peptides containing a thioacyl group. Int J Pept Prot Res. 1987;30:564–571. doi: 10.1111/j.1399-3011.1987.tb03366.x. [DOI] [PubMed] [Google Scholar]
- 17.Artis DR, Lipton MA. Conformations of thioamide-containing dipeptides: a computational study. J Am Chem Soc. 1998;120:12200–12206. [Google Scholar]
- 18.Tran TT, Treutlein H, Burgess AW. Conformational analysis of thiopeptides: (ϕ,ψ) maps of thio-substituted dipeptides. J Comput Chem. 2001;22:1026–1037. [Google Scholar]
- 19.Miwa JH, Pallivathucal L, Gowda S, Lee KE. Conformational stability of helical peptides containing a thioamide linkage. Org Lett. 2002;4:4655–4657. doi: 10.1021/ol027056d. [DOI] [PubMed] [Google Scholar]
- 20.Miwa JH, Patel AK, Vivatrat N, Popek SM, Meyer AM. Compatibility of the thioamide functional group with β-sheet secondary structure: incorporation of a thioamide linkage into a β-hairpin peptide. Org Lett. 2001;3:3373–3375. doi: 10.1021/ol0166092. [DOI] [PubMed] [Google Scholar]
- 21.Reiner A, Wildemann D, Fischer G, Kiefhaber T. Effect of thioxopeptide bonds on alpha-helix structure and stability. J Am Chem Soc. 2008;130:8079–8084. doi: 10.1021/ja8015044. [DOI] [PubMed] [Google Scholar]
- 22.Richards FM, Vithayathil PJ. The preparation of subtilisin-modified ribonuclease and the separation of the peptide and protein components. J Biol Chem. 1959;234:1459–1465. [PubMed] [Google Scholar]
- 23.Dyson JH, Wright PE. Intrinsically unstructured proteins and their functions. Nat Rev Mol Cell Bio. 2005;6:197–208. doi: 10.1038/nrm1589. [DOI] [PubMed] [Google Scholar]
- 24.Schreier AA, Baldwin RL. Mechanism of dissociation of S-peptide from ribonuclease S. Biochemistry. 1977;16:4203–4209. doi: 10.1021/bi00638a012. [DOI] [PubMed] [Google Scholar]
- 25.Thomson J, Ratnaparkhi GS, Varadarajan R, Sturtevant JM, Richards FM. Thermodynamic and structural consequences of changing a sulfur atom to a methylene group in the M13Nle mutation in ribonuclease-S. Biochemistry. 1994;33:8587–8593. doi: 10.1021/bi00194a025. [DOI] [PubMed] [Google Scholar]
- 26.Schreiber G, Haran G, Zhou H-X. Fundamental aspects of protein-protein association kinetics. Chem Rev. 2009;109:839–860. doi: 10.1021/cr800373w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Leffler JE. Parameters for the description of transition states. Science. 1953;117:340–341. doi: 10.1126/science.117.3039.340. [DOI] [PubMed] [Google Scholar]
- 28.Sánchez IE, Kiefhaber T. Hammond behavior versus ground state effects in protein folding: evidence for narrow free energy barriers and residual structure in unfolded states. J Mol Biol. 2003;327:867–884. doi: 10.1016/s0022-2836(03)00171-2. [DOI] [PubMed] [Google Scholar]
- 29.Kim EE, Varadarajan R, Wyckoff HW, Richards FM. Refinement of the crystal structure of ribonuclease S. Comparison with and between the various ribonuclease A structures. Biochemistry. 1992;31:12304–12314. doi: 10.1021/bi00164a004. [DOI] [PubMed] [Google Scholar]
- 30.Shoemaker KR, et al. Side-chain interactions in the C-peptide helix: Phe 8 … His 12+ Biopolymers. 1990;29:1–11. doi: 10.1002/bip.360290104. [DOI] [PubMed] [Google Scholar]
- 31.Loftus D, Gbenle GO, Kim PS, Baldwin RL. Effects of denaturants on amide proton exchange rates: a test for structure in protein fragments and folding intermediates. Biochemistry. 1986;25:1428–1436. doi: 10.1021/bi00354a036. [DOI] [PubMed] [Google Scholar]
- 32.Fairman R, Shoemaker KR, York EJ, Stewart JM, Baldwin RL. The Glu 2- … Arg 10+ side-chain interaction in the C-peptide helix of ribonuclease A. Biophys Chem. 1990;37:107–119. doi: 10.1016/0301-4622(90)88012-h. [DOI] [PubMed] [Google Scholar]
- 33.Rose GD, Geselowitz AR, Lesser GJ, Lee RH, Zehfus MH. Hydrophobicity of amino acid residues in globular proteins. Science. 1985;229:834–838. doi: 10.1126/science.4023714. [DOI] [PubMed] [Google Scholar]
- 34.Goldberg JM, Baldwin RL. Kinetic mechanism of a partial folding reaction. 1. properties of the reaction and effect of denaturants. Biochemistry. 1998;37:2546–2555. doi: 10.1021/bi972402y. [DOI] [PubMed] [Google Scholar]
- 35.Goldberg JM, Baldwin RL. Kinetic mechanism of a partial folding reactions: 2. nature of the transition state. Biochemistry. 1998;37:2556–2563. doi: 10.1021/bi972403q. [DOI] [PubMed] [Google Scholar]
- 36.Anil B, Sato S, Cho JH, Raleigh DP. Fine structure analysis of a protein folding transition state; distinguishing between hydrophobic stabilization and specific packing. J Mol Biol. 2005;354:693–705. doi: 10.1016/j.jmb.2005.08.054. [DOI] [PubMed] [Google Scholar]
- 37.Kraulis P. MolScript: a program to produce both detailed and schematic plots of protein structures. J Appl Crystallogr. 1991;24:946–950. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.