

Abstract
F1-ATP synthase (F1-ATPase) is equipped with a special mechanism that prevents the wasteful reverse reaction, ATP hydrolysis, when there is insufficient proton motive force to drive ATP synthesis. Chloroplast F1-ATPase is subject to redox regulation, whereby ATP hydrolysis activity is regulated by formation and reduction of the disulfide bond located on the γ subunit. To understand the molecular mechanism of this redox regulation, we constructed a chimeric F1 complex (α3β3γredox) using cyanobacterial F1, which mimics the regulatory properties of the chloroplast F1-ATPase, allowing the study of its regulation at the single molecule level. The redox state of the γ subunit did not affect the ATP binding rate to the catalytic site(s) and the torque for rotation. However, the long pauses caused by ADP inhibition were frequently observed in the oxidized state. In addition, the duration of continuous rotation was relatively shorter in the oxidized α3β3γredox complex. These findings lead us to conclude that redox regulation of CF1-ATPase is achieved by controlling the probability of ADP inhibition via the γ subunit inserted region, a sequence feature observed in both cyanobacterial and chloroplast ATPase γ subunits, which is important for ADP inhibition (Sunamura, E., Konno, H., Imashimizu-Kobayashi, M., Sugano, Y., and Hisabori, T. (2010) Plant Cell Physiol. 51, 855–865).
Keywords: ATP Synthase, ATPases, Chloroplast, Coupling Factors, Molecular Motors, ADP-inhibition, Redox Regulation
Introduction
The ATP synthase complex is ubiquitously found in energy-transducing membranes such as bacterial plasma membranes, mitochondrial inner membranes, and chloroplast thylakoid membranes, where its basic architecture is highly conserved. ATP synthase consists of two motor units: the F1 portion which is powered by ATP, and the Fo portion powered by the proton motive force formed across the energy transducing membranes (1). Fuelling of ATP synthase by ATP and proton motive force results in rotational motion of F1 and Fo, respectively, though they rotate in opposite directions. On the membranes, these two motors are directly connected by protein-protein interaction, and their functions are coupled to each other: the F1 portion catalyzes ATP hydrolysis and ATP synthesis, and the Fo portion catalyzes proton translocation. Upon formation of a proton motive force across the membranes as a result of respiratory or photosynthetic electron transport, the Fo portion is forced to rotate by the proton motive force. Rotation of Fo induces rotation of the central axis subunits, γ and ϵ, of F1, and finally ATP is formed at the catalytic sites on β subunits, presumably due to forced conformational change at the catalytic sites. Vice versa, rotation of F1 forced by ATP hydrolysis induces rotation of Fo and consequently protons are transferred in the opposite direction, where a proton motive force is generated. This enzyme may therefore hydrolyze ATP and transport protons in the opposite direction when there is insufficient proton motive force to drive ATP synthesis.
However this reverse reaction catalyzed by the enzyme is highly restricted in vivo because the reaction results in the wasteful consumption of ATP. To avoid this unfavorable reaction in vivo, several regulatory systems are known to maintain the ATP synthesis reaction. The bacterial and chloroplast ATP synthases are equipped with an intrinsic inhibitory subunit ϵ, which strongly suppresses the ATP hydrolysis reaction (2–4). The C-terminal α-helical part of the ϵ subunit has been shown to be critical for this inhibitory function (5). Another regulatory mechanism is known as ADP-inhibition (6). The ATP hydrolysis reaction of F1-ATPase is strongly inhibited by tight binding of ADP to the catalytic site(s). This inhibition is particularly strong under low concentrations of substrate ATP (7).
In addition to those described above, higher plant chloroplast ATP synthase displays an additional regulatory system, a thiol modulation system (8, 9), whereupon it receives reducing equivalents from the photosynthetic electron transport system via thioredoxin-f, a disulfide-dithiol exchange mediator protein (10). The γ subunit of the chloroplast ATP synthase contains two regulatory cysteines (see Fig. 1A), which are able to form a disulfide bond (8, 11), and the reduction of this disulfide bond by the reduced form thioredoxin (9) activates the enzyme. Inversely, the formation of the disulfide bond on the γ subunit renders the enzyme inactive (11). This regulation is likely to be critical for photosynthetic organisms, because photosynthesis is subject to highly variable environmental conditions, fuelled by an energy source which is both oscillatory (day and night) and fluctuating (irregular weather changes).
FIGURE 1.

Sequence of the γ subunits and the construction of the redox sensitive γ subunit. A, sequences around the redox region of the γ subunit are displayed. The redox region of 9 amino acids unique to the chloroplast γ subunit is shown in green and two cysteines involved in redox reaction in red. So-called insertion region in cyanobacteria (27) is shown in blue. The introduced region from S. oleracea for construction of α3β3γredox is underlined. C. reinhartii, C. reinhardtii; Syn. Sp. PCC 6803, Synechococcus sp. PCC6803; Anab. sp. PCC 7120, Anabaena sp. PCC 7120. B, γ subunit (black) of T. elongates BP-1 ATP synthase was used as the basis for the chimera subunit. The redox region on the γ subunit of CF1 from S. oleracea (underlined) was inserted into the corresponding position. To attach streptavidin-coated-beads via biotin-avidin interaction for single molecule experiments, two additional Cys-112 and Cys-125 (purple) were introduced in the appropriate position (19).
The regulatory cysteines on the γ subunit of chloroplast FoF1, Cys-199 and Cys-205 (see Fig. 1A), are conserved in different photosynthetic organisms, from green algae such as Chlamydomonas reinhardtii (12) to higher plants (8). Although at present nothing is known about the potential γ subunit conformational changes associated with the redox state, the redox switch region seems to be independent from other parts of the γ subunit. Several studies have shown that the insertion of a chloroplast-like regulatory domain including the two cysteines into the ATP synthase γ subunit of non-regulated organisms enables the enzyme to respond to redox change (13–15); removal of the cysteines from this added domain leads to complete lose of redox regulation (16–17). In a previous study, we reported that the capacity for redox regulation could be conferred to non-redox regulated bacterial F1 by substitution of the central region (residues 92–239) of the γ subunit with the counterpart of the γ subunit of chloroplast F1 (14). By using this chimeric protein, we successfully showed redox-regulation of rotation of the γ subunit in the complex, and that an oxidized γ subunit results in an increase in the frequency of long pauses which in turn leads to a decrease in ATPase activity (18).
To gain a better understanding of the redox regulation of γ subunit rotation, we introduced the redox-regulation segment of the higher plant ATP synthase into the γ subunit of cyanobacterial F1 (19) (see Fig. 1B). The enhanced stability demonstrated by this new chimeric complex provides the chance to thoroughly characterize the change in rotational motion conferred by a change in the redox state of the γ subunit.
EXPERIMENTAL PROCEDURES
Materials
Maleimide-PEG11-biotin was purchased from Thermoscientific Co. Ltd (Yokohama, Japan). Carboxylated polystyrene beads were from Polysciences. Other chemicals were of the highest grade commercially available.
Preparation of the Chimeric Complex
The redox sensitive chimera complex (termed as α3β3γredox)2 was constructed using an expression plasmid for α3β3γ complex of Thermosynechoccus elongatus BP-1 (19). The redox region of the γ subunit of CF1 from Spinacia oleracea (CF1 γ187–210, IHTLLPLSPKGEICDINGKCVDA) was inserted into the corresponding position of the γ subunit of T. elongatus BP-1 (T. elongatus BP-1 γ188–201, VQTLLPLDPQGLET) using the mega-primer method (20) with the following mutation primer: 5′-GGCGGAAAATTTCATCATCGGCTGCGTCGACACATTTACCATTGATGTCGCAAATTTCTCCTTTGGGTGAGAGGGGGAGCAGGG-3′. The α3β3γredox was then expressed and purified by Ni-NTA chromatography as described (19). During the purification steps, 2 mm DTT was added to prevent unexpected disulfide bond formation between the four cysteines introduced. Prior to the specific oxidation process as described, DTT was removed by gel filtration using SuperdexTM-200 column (GE Healthcare) equilibrated with 50 mm MOPS-KOH (pH 7.5), and 100 mm KCl. 4 μm α3β3γredox was incubated with 40 μm thioredoxin-f, 200 μm GSSG, 2 mm GSH, 50 mm MOPS-KOH (pH 7.5), and 100 mm KCl at 25 °C for 10 min to form a disulfide bond between the two cysteines located within the redox region of the chimera complex. This procedure allowed specific biotinylation of Cys-112 and Cys-125 on the γ subunit for observation of α3β3γredox rotation. Following removal of the oxidizing reagents, 9 μm α3β3γredox was biotinylated with 5 mm maleimide-PEG11-Biotin, 50 mm MOPS-KOH, pH 7.5, and 100 mm KCl at 25 °C for 20 min. Unreacted maleimide-PEG11-biotin was removed from the solution by gel-filtration, and the resulting chimera complex was stored at −80 °C in 10% glycerol.
Confirmation of the Specific Biotinylation of Cys-112 and Cys-125 of the γ Subunit by Mass Spectrometry and SDS-PAGE
30 μl of oxidized and reduced F1 (0.5 mg/ml) complexes were precipitated with trichloroacetic acid (final 5%), washed with ice-cold acetone, and finally dissolved in 1 mm maleimide-PEG11-biotin, 100 mm Tris-HCl, pH 7.5, and 1% (w/v) SDS. The protein samples were then analyzed by 12% non-reducing SDS-PAGE. Gels from the non-reducing SDS-PAGE analysis were stained with Coomassie Brilliant Blue R-250 and the γ subunit bands manually excised. The proteins in the gel were digested by “in-gel” cleavage (21) at 37 °C with 10 μg/ml sequencing grade modified trypsin (Promega, Tokyo) in 50 mm NH4HCO3, pH 8.0. Gels were reduced with 10 mm DTT at 56 °C for 1 h and alkylated with 1% (w/v) ICH2CONH2 in 50 mm NH4HCO3, pH 8.0 at room temperature for 45 min. Peptides were extracted from the gel with 5% (w/v) trifluoroacetic acid and 50% (v/v) acetonitrile solution. All digests were analyzed in a MALDI-TOF mass spectrometer (AXIMA-CFR Plus, Shimazu, Kyoto) using α-cyano-hydroxy-trans-cinnamic acid as the matrix. The instrument was calibrated with angiotensin II (m/z values, 1046.542) and adrenocorticotropic hormone fragment 18–39 (m/z value, 2465.199). The MS data obtained were analyzed with the data set of expected digested products of the γ subunit with allowance of missed cleavages of 3 and with carbamidomethyl as a variable modification.
ATP Hydrolysis Activity Assay
ATP hydrolysis activity was measured by an ATP regenerating method as described (22). The reaction was initiated by adding 20 nm (final) of enzyme solution into the 1.2 ml of the assay solution. The assay solution contained 50 mm MOPS-KOH (pH 7.5), 100 mm KCl, 2 mm MgCl2, 200 μm, or 2 mm MgATP, 0.2 mm NADH, 2 mm phosphoenolpyruvate, 50 μg/ml pyruvate kinase, and 50 μg/ml lactate dehydrogenase. The rates of ATP hydrolysis were measured at the steady state phase, 200 s after addition of the enzyme.
Rotation Assay
The rotation assay was performed as described (22) with some modifications. Previously oxidized or reduced α3β3γredox complex was fixed on the glass surface and then the streptavidin-coated duplex beads (φ = 291 nm) attached to the immobilized α3β3γredox complex. Rotation of the complex was initiated by addition of 60 μl of 50 mm MOPS-KOH (pH 7.5), 100 mm KCl, 2 mm MgCl2, 200 μm MgATP, 2 mm phosphoenolpyruvate, and 100 μg/ml pyruvate kinase. To prevent re-oxidation of the complex during the measurement of the reduced complex, 0.5 μm reduced thioredoxin-f and 5 μm of DTT was added into the assay buffer. Rotation of the duplex beads attached to the immobilized α3β3γredox complex was observed by dark field microscopy at a frame rate of 200 frames/s, and the recorded data were analyzed by custom-made software (created by Yusung Kim).
Other Proteins
Recombinant thioredoxin-f of S. oleracea was expressed in E. coli and purified as described (23). Chloroplast F1-ATPase (α3β3γδϵ) was purified from fresh market spinach leaves (24) and the δ and ϵ subunits were removed by the following procedures. DEAE-Sephacel (12 ml, Amersham Biosciences) was previously equilibrated with 25 mm Tris-HCl (pH 8.0), 2 mm ATP, and 5 mm DTT. The crude extract dissolved in the same buffer was loaded on the column and the δ and ϵ subunits were eluted from the column with 25 mm Tris-HCl (pH 8.0), 2 mm ATP, 5 mm DTT, 30% glycerol, 20% ethanol, and 100 mm NaCl. The column was then washed with 25 mm Tris-HCl (pH 8.0) and 2 mm ATP. CF1 (α3β3γ) was eluted with 25 mm Tris-HCl (pH 8.0), 2 mm ATP, and 400 mm NaCl. The purified CF1 was stored as ammonium sulfate precipitates at 4 °C. Before use, the preparation was desalted by gel-filtration (NAPTM-5 column, GE Healthcare) using 50 mm Tricine-KOH (pH 8.0) and 100 mm KCl.
RESULTS
Redox Regulation of Cyanobacterial F1-ATPase
To investigate redox regulation of chloroplast-type F1-ATPase, we first designed a new chimera F1 complex, α3β3γredox complex, consisting of cyanobacterial F1 subunits obtained from T. elongatus BP-1, and the redox regulatory region of the spinach CF1 γ subunit as the counterpart (see Fig. 1B). The biochemical properties of cyanobacterial F1-ATPase are very close to that of higher plant chloroplasts, though the enzyme complex of the former lacks the specific redox regulatory region (see Fig. 1A) (19). By introduction of the 24 amino acid sequence of the spinach CF1- γ subunit, including the nine missing amino acid residues EICDINGKC, into the cyanobacterial counterpart, we successfully conferred redox regulatory capacity to the complex. After specific biotinylation of cysteines required for the probe fixation as described below, the activities of the oxidized enzyme and the reduced enzyme were measured. The ratio between the enzyme activity of the reduced form (ActR) and that of the oxidized one (ActO) (ActR/ActO = 2.8) was similar to that of spinach CF1 (ActR/ActO = 2.7). In addition, our new chimeric enzyme was rapidly reduced by recombinant thioredoxin-f obtained from spinach chloroplasts (Fig. 2A), implying that the chimeric enzyme complex successfully mimics the redox regulatory system of CF1. Compared with the previously synthesized chimeric F1, α3β3γTCT (14), the present chimeric complex, α3β3γredox, showed higher stability for purification and assay experiments.
FIGURE 2.
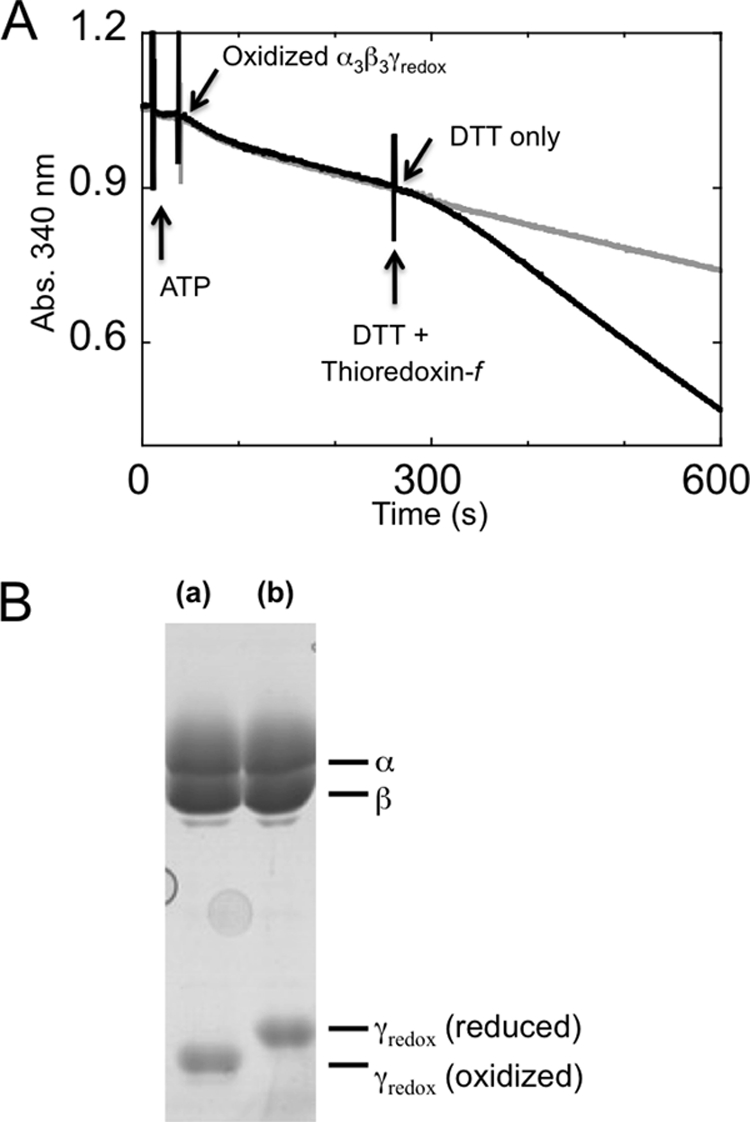
Redox sensitivity of biotinylated α3β3γredox. After biotinylation of the α3β3γredox complex, the redox sensitivity was confirmed in both regulation of the activity (A) and disulfide bond formation ability by oxidation (B). A, activation by reduction of the γ subunit of α3β3γredox. ATPase activity was measured using an ATP-regenerating system as a decrease in NADH absorption at 340 nm. A final concentration of 200 μm ATP was used, and the reaction was initiated by adding oxidized α3β3γredox. When the reaction reached the steady state (210 s from the start of reaction), a final concentration of 10 μm DTT with (black trace) or without (gray trace) 5 μm of reduced spinach thioredoxin-f was added. B, the disulfide bond formation ability of the biotinylated α3β3γredox complex was confirmed by further labeling with 4-acetamido-4′-maleimidylstilbene-2,2′-disulfonic acid following oxidation (a) or reduction (b) of the redox sensitive Cys-200 and Cys-206.
Specific Biotinylation of the α3β3γredox Complex for Single Molecule Observations
The α3β3γredox complex has 4 cysteines on its γ subunit: two are originally located in the redox region and two were introduced to enable optical probe fixation. For single molecule experiments, two cysteines (Cys-112 and Cys-125) located on the γ subunit had to be biotinylated to attach them to the avidin-coated-plastic beads for observation of rotation with high angular resolution; the two cysteines (Cys-200 and Cys-206) located within the γ subunit redox region are expected to have remained unmodified for redox regulation. To carry out this specific modification, we first introduced a disulfide bond between Cys-200 and Cys-206 to protect these residues from biotin modification. This disulfide bond was successfully formed between these residues alone by using the oxidized form of thioredoxin-f and a glutathione pool as an electron acceptor (Fig. 3, lane d). The specific labeling of the desired cysteines was confirmed by MALDI TOF-Mass spectrometry following fragmentation of the γ subunit by trypsin (Fig. 4, A–C). Under the conditions in which two cysteines were biotinylated, two isotropic signals containing Cys-112 and Cys-125 were detected as biotinylated fragments (Fig. 4, A and B). The biotinylated fragment containing Cys-206 was only observed when four cysteines were biotinylated without oxidation (Fig. 4C). The γ subunit located within the complex remained redox sensitive after biotinylation of Cys-112 and Cys-125, and full reduction and oxidation of the complex was confirmed by SDS-PAGE following the thiol modification of Cys-200 and Cys-206 using 4-acetamido-4′-maleimidylstilbene-2,2′-disulfonic acid (Fig. 2B).
FIGURE 3.
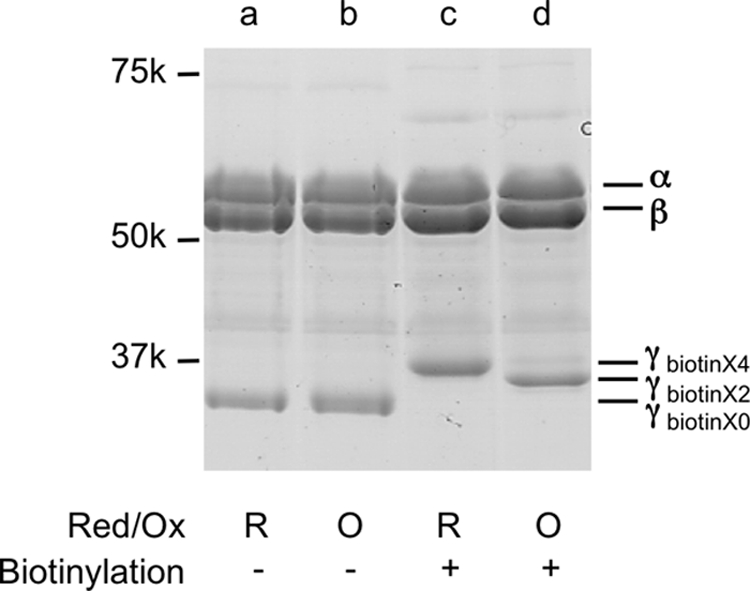
Biotinylation of α3β3γredox. To prevent biotinylation of the cysteines involved in redox regulation (Cys-200, Cys-206), 4 μm F1 was oxidized with 40 μm thioredoxin-f, 200 μm GSSG (oxidized form of glutathione), 2 mm GSH (reduced form of glutathione), 50 mm MOPS-KOH, pH 7.5, and 100 mm KCl at 25 °C for 10 min. Reduced (c) and oxidized (d) F1 was then biotinylated using maleimide-PEG11-biotin (M.W.: 922.1, net mass addition: 921.5). Reduced (a) and oxidized (b) form F1s without biotinylation are shown as controls.
FIGURE 4.

Peptide analysis of the biotinylated α3β3γredox. A–C, mass spectrometry of fragmented γ subunit. The γ subunit separated by SDS-PAGE as shown in Fig. 3, was digested by trypsin in the gel and the resulting peptide fragments were obtained. The molecular weights of the fragments were measured by MALDI-TOF-MS. The fragments containing biotinylated (Bt) cysteines (A; Cys-112, B; Cys-125, and C; Cys-206) of the γ subunit were shown. Molecular weights of each fragment were 2091.7, 1837.0, and 2175.0, respectively. The non-cysteine-containing fragment (M.W. 2176.4) of γ is shown in C.
Effect of LDAO: Relief from ADP Inhibition
By reduction, both the α3β3γredox complex and the purified CF1 showed ∼2.7-fold activation, as mentioned. We then studied the extent of reduction induced ATPase activation under various assay conditions. As expected, this was affected by addition of lauryl dimethylamine-N-oxide (LDAO) (Fig. 5). LDAO is known to be effective in recovering F1-ATPase from ADP inhibition (25); the α3β3γredox complex ATPase activity also increased remarkably when LDAO was added to both the reduced and oxidized forms of the complexes. However, the activity ratio between the reduced and oxidized form of the enzyme decreased remarkably depending on the concentration of LDAO, indicating that ADP-inhibition of the oxidized form is more pronounced than the reduced form (Fig. 5).
FIGURE 5.

Relationship between redox regulation of the complex and LDAO activation. The α3β3γredox complex was biotinylated in advance and ATP hydrolysis rates were measured using an ATP regenerating system (see “Experimental Procedures”), and the results obtained from three independent experiments were averaged. The ATP hydrolysis activity of the reduced α3β3γredox complex (open circle) and that of oxidized α3β3γredox (closed circle) are plotted against various concentrations of LDAO. LDAO was added 200 s after reaction initiation and the effect of LDAO on the activity was assessed for 50–100 s after addition of LDAO. The ratio of activities obtained from the data for the reduced α3β3γredox complex over those for the oxidized α3β3γredox complex is shown (triangle).
Redox Regulation of Rotation in the Presence of 200 μm ATP
To characterize the substep responsible for CF1 redox regulation during rotation, the rotational behavior of the α3β3γredox complex was analyzed by single molecule observation. For this purpose, polystyrene duplex beads with a diameter of ∼291 nm were attached to the γ subunit of the α3β3γredox complex, and rotation of the γ subunit in the complex observed for 10–30 min by dark field microscopy. Fig. 6, A (oxidized enzyme) and B (reduced enzyme) show the typical time course of rotation of the γ subunit in the presence of 200 μm ATP at room temperature. Torques calculated based on the fluctuation theorem as described (26) were 47.3 ± 6.2 pNnm (n = 3) and 48.6 ± 4.1 pNnm (n = 5), for the oxidized and reduced α3β3γredox complex, respectively. The maximal rotational speeds, determined from 100 continuous rotations, were 18.5 ± 2.8 rps (n = 19) for oxidized complex and 19.5 ± 4.2 rps (n = 17) for reduced one, respectively (Fig. 6, A and B), due to large bead friction, though the averaged rotation speeds during 600 s observation showed substantial difference; 2.7 ± 2.0 rps (n = 16) for the oxidized complex and 7.0 ± 2.6 rps (n = 10) for the reduced one, respectively. The pause duration was then analyzed using the pause length histogram, which was obtained from pauses longer than 1 s (Fig. 6, C and D). These relatively long pauses are unlikely to originate from the ATP binding state, because ATP binding takes in the order of submilliseconds in the presence of 200 μm ATP according to the binding rate constant for ATP, kon of 1.8 × 107 ∼1.9 × 107 m−1 s−1 (Fig. 7). For curve fitting to the histograms of pause durations, double exponential equations gave adequate results, implying that the observed rotation behavior contained at least two independent pauses. Consequently, the results gave two independent lifetimes (τ), the time constants of the marked events. The short lifetimes (τsp) for the oxidized and reduced states of the chimeric complex were 6.6 and 8.6 s, respectively. These short pauses were, however, not sufficiently reliable since the bin width (= 10 s) of these histograms was longer than the results obtained. Several studies have been conducted on ADP inhibition including analysis of the relevance between ADP inhibition and rotation of TF1 ATPase (7), and an exploration of the correlation between the inserted region of the γ subunit and ADP inhibition of cyanobacterial F1 (27). Though the conclusions drawn from these reports were very similar, the origin of the short pause remains to be determined. In contrast, the long pauses with longer lifetimes (τlp) of oxidized and reduced state of α3β3γredox complex were significantly different, 124 and 78 s, respectively (Fig. 6, C and D). These long pauses can be interpreted as the ADP-inhibited state, which occurs at the 80° position (7) when the ATP binding position was settled as 0°. The long pause position during rotation was then determined (Fig. 8) by using a medium exchange method from 0.2 μm to 200 μm ATP. Consequently, the pause position was determined as 79.3 ± 12.4° (n = 22) ahead from the ATP binding position, which is close to the position for ADP inhibition. These pause duration analyses suggest that the oxidized α3β3γredox complex is more likely to be dropped into an ADP inhibited state. Data relating to the duration of rotation which appeared between the two pauses, were also collected and analyzed (Fig. 6, E and F). The histograms relating to the continuous rotating duration were fitted to a single exponential equation. The lifetime (τrot) for oxidized and reduced chimera were 29 and 36 s, respectively. Reduction of α3β3γredox is likely to result in a relatively longer continuous rotation.
FIGURE 6.

Single molecule analysis of rotation of the γ subunit under redox conditions. A and B, the typical time courses of the revolutions of the oxidized (A) and the reduced (B) α3β3γredox complex. Rotation of the γ subunit was recorded as the movement of the 291-nm diameter beads attached to γ and the data obtained were analyzed using custom-made software. Rotation observation was performed at 25 °C in the presence of 200 μm ATP. The histogram of pauses (>1 s) of the oxidized α3β3γredox complex (C) and the reduced α3β3γredox complex (D) were constructed from data for 27 and 20 molecules, respectively. In the inset, the vertical axis is drawn with a finer scale, and the bars for the data less than 20 s are omitted. The histograms are fitted with a double exponential equation, Nsp × exp(−t/τsp)+Nlp × exp(−t/τlp). The histograms of rotation duration between a pause and a next pause of the oxidized (E) and the reduced (F) α3β3γredox complexes were constructed from data for 28 and 20 molecules, respectively. The histograms were fitted with a single exponential equation, Nrot × exp (−t/τrot).
FIGURE 7.
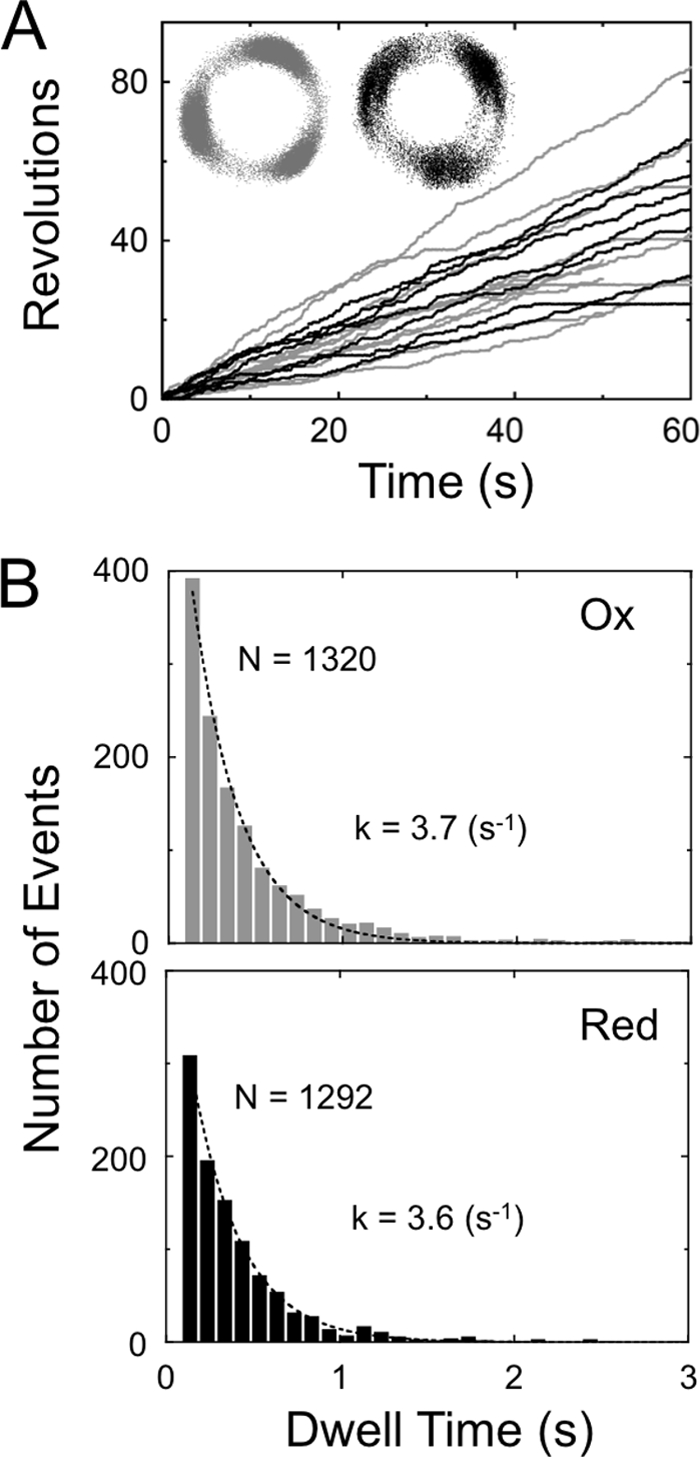
Rotation of α3β3γredox at 200 nm ATP. A, typical time courses of rotation of the oxidized (gray trace) and reduced (black trace) α3β3γredox. Average rotating speeds were 0.87 ± 0.44 rps (n = 15) for oxidized and 0.81 ± 0.32 rps (n = 7) for reduced α3β3γredox. Insets represent the traces of centroids of the beads. B, the dwell time periods during rotation observed in the trace shown in A were collected and constructed into histograms. The data for each histogram are from 13 molecules for the oxidized complex (gray) and 6 molecules for the reduced one (black). Each histogram was fitted with a single exponential equation, Const. × exp(−kt). The binding rate constants obtained ATP were 1.9 × 107 m−1 s−1 for the oxidized complex and 1.8 × 107 m−1 s−1 for the reduced complex.
FIGURE 8.

Stop angular position of long pauses. A, after observation of rotation at 0.2 μm ATP (blue), the ATP concentration was changed to 200 μm ATP (gray and black). The long pauses (>40 s) are labeled on the traces as p1 to p4. B and C, the histograms of angular distribution of the beads are shown. Blue bars represent the angular distribution of 120° step rotation of the beads at 200 nm ATP. One of the three peak positions at 200 nm ATP is set as 0°. The black bars represent the angular distribution of long pauses observed in the data (p1 to p4) after buffer exchange.
Taken together, we conclude that the oxidized α3β3γredox complex remains in the ADP-inhibited state for longer, and takes longer to recover from the ADP-inhibited state than the reduced α3β3γredox complex. The difference in the lifetimes between these two states eventually leads to a difference in the total active enzyme population size.
DISCUSSION
In a previous study we prepared a chmieric α3β3γTCT complex originating from the thermophilic bacteria Bacillus PS3 and spinach chloroplast, and reported that oxidized α3β3γTCT caused frequent long pauses (> 1 min) (14, 18). However, we could not assign the origin of these long pauses to one of the sub-steps, 80° and further 40° (28), because the significantly modified chimeric mutants had insufficient angular resolution to allow for identification of sub-steps. In this study, a new mutant complex (α3β3γredox) consisting of two distinct benefits was prepared from the thermophilic cyanobacterial F1: the capacity to remain stable for extended observation periods, and the ability to achieve high angular resolution by connection of the γ subunit to an optical probe via two biotin bonds. Because cyanobacterial and chloroplast F1 share common ancestry, only 24 residues (Fig. 1, insertion of 9 chloroplast specific residues and 15 substitutions near the insertion; within them 8 residues are identical to cyanobacterial γ) were required to confer the redox regulatory capability to the complex. In addition, cysteines in the inserted redox region were reduced by their natural electron donor, thioredoxin-f (Fig. 2). A similar mutant (possessing the same insertion and 4 substitutions of the amino acids near the insertion) has already been prepared by Werner-Grüne et al. using direct transformation of Synechocystis 6803 cells (13). The mutant strain showed redox sensitivity to ATPase activity, as well as physiological differences in the dark and light (low O2 consumption in the dark and high O2 production in the light), though this redox switch conferred no growth advantage to the bacteria. Thus the redox regulatory property conferred to the cyanobacterial α3β3γredox must be close to that observed for CF1. We therefore sought to determine whether the behavior of α3β3γredox at the single molecule level reflects the change in activity seen by redox regulation.
By using the newly designed chimeric complex (α3β3γredox) in single molecule studies we successfully confirmed that oxidation of the redox region caused frequent long pauses (Fig. 6, C and D), which was consistent with a previous study (18). In addition, we observed that duration of rotation under oxidized conditions was relatively shorter than that shown under reduced conditions (Fig. 6, E and F). The redox-dependent behaviors of the chimeric complex are attributable to insertion of the redox region, since the wild-type complex lacking the redox region did not show any differences in activity and rotation behavior even when the complex had been previously reduced or oxidized (Fig. 9).
FIGURE 9.
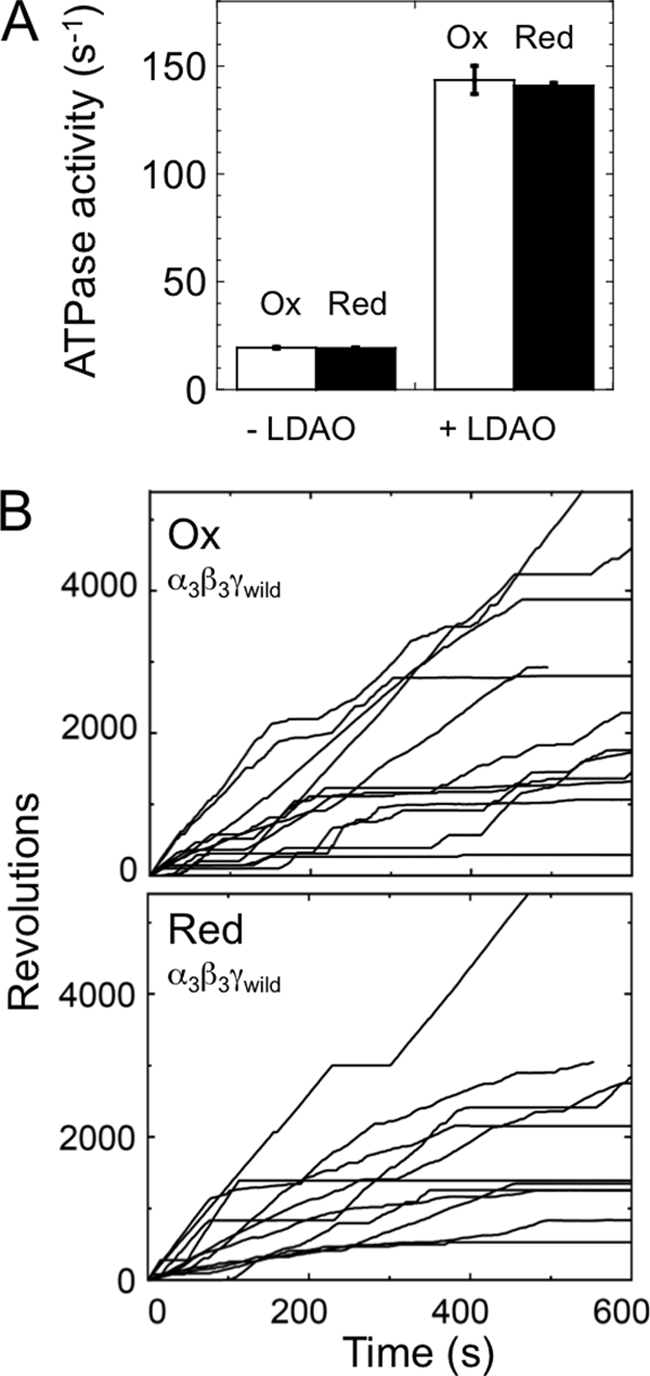
ATPase activity and rotation of α3β3γwild complex under redox conditions. A, ATPase activities of both oxidized (open square) and reduced (closed square) α3β3γwild complexes were measured (n = 3) in the presence of 200 μm ATP with or without 0.3% LDAO. B, the typical rotational behaviors of oxidized and reduced α3β3γwild complex. Rotation of the complex was observed using 291 nm diameter beads at 200 μm ATP. Maximum speeds of 10 continuous rotations for the oxidized and reduced complexes were 16.1 ± 5.7 rps (n = 13) and 15.1 ± 5.4 rps (n = 11), respectively. Averaged speeds of 10 min rotation for the oxidized and reduced complexes were 3.5 ± 2.2 rps (n = 10) and 3.6 ± 3.2 rps (n = 10), respectively.
As shown in Fig. 8, the stop position of the long pause was determined as 80° ahead of the ATP binding position. This position is known to be where the sub-step of ATP hydrolysis (29) and phosphate release (30) occur; innate ADP inhibition also takes place at this position (7). Based on the results showing that relief from ADP inhibition by LDAO (Fig. 5) reduces redox sensitivity, we concluded that ADP inhibition was involved in the prolonged pauses during rotation, especially those observed under oxidized conditions.
The substrate binding rate at the catalytic sites was calculated by measuring the duration of ATP waiting dwell at 200 nm ATP, where F1 spends most of its time waiting for ATP (see Fig. 7). When the enzyme activity is regulated by the redox regulatory system, the on-rate of ATP to the α3β3γredox complex was unchanged, but the ATP binding affinity (∼1/Km) was slightly lower in the reduced state (Fig. 10). The difference in ATP affinity between the reduced and oxidized α3β3γredox complex is unlikely to be a significant element in redox regulation, since the difference in the activity ratio between both states was remarkable and even larger at higher concentrations of ATP (Fig. 10) where the difference of Km is negligible.
FIGURE 10.

ATP hydrolysis kinetics of α3β3γredox. ATPase activities at various MgATP concentrations (open circle for reduced, closed circle for oxidized α3β3γredox), and the activity ratio of the reduced α3β3γredox complex over the oxidized one were plotted (triangle). Each measurement was conducted under 2 mm of excess Mg2+. Activities were fitted with Michaelis-Menten equation. Vmax values of the reduced and oxidized α3β3γredox complexes were 60 s−1 and 23 s−1, respectively and Kms of the reduced and oxidized complexes 35 μm and 22 μm, respectively.
In this study we showed that redox regulation by the redox switch regulates the frequency of ADP inhibition. Considering the position of the redox switch in relation to the inserted region of the γ subunit (Fig. 1A), we propose that the redox switch simply takes advantage of the function of the inserted region (27) by changing conformation via a disulfide bond, though as yet there is no crystal structure for the γ subunit of chloroplast F1.
As shown in this and our previous study (27), the evolutionary tuning of the γ subunit central region is related to ADP inhibition. However the active site on the αβ surface and the expected position of the regulatory region of the γ subunit are located some distance apart, and no direct contact between these two regions has been reported to date. In our previous study using α3β3γTCT, deletion of three negatively charged residues at the regulatory region caused the inverse regulation, higher activity at the oxidized state (31, 32), implying that the activity of the enzyme is very sensitive to small conformational changes in this region. One possible mechanism for the regulatory region is that the redox change of this region slides or results in a change in the angle between the N-terminal α-helix and the C-terminal α-helix of the γ subunit. Since these terminal helices of the γ subunit consist of a coiled-coil structure and have direct contact with the inside of the α3β3 ring (33), distortion between two helices could affect its reaction steps, in this case, the tightness of the ADP-inhibited state, or in other words, the probability of dropping in and reverting out of the ADP inhibition state. Moreover, the redox switch located next to the inserted region could regulate the level of distortion via disulfide bond.
In photosynthetic organisms, the ability to reduce the rate of wasteful hydrolysis activity in the dark confers a significant advantage. However, despite multiple dedicated studies, the physiological significance of this regulatory mechanism has to date not been clearly proven. Several experiments in which the redox regulatory property is abolished by removal of the relevant cysteines have been performed in vivo in Arabidopsis thaliana (16) and Chlamydomonas reinhardtii (17). In each of these studies, ATP synthase was converted to a redox insensitive enzyme resulting in increased ATPase activity (16), but growth was not significantly affected. Furthermore, introduction of the redox region into F1 in Synechocystis had no effect on growth, though the mutant showed reduced oxygen consumption levels in the dark (13). Although to date there has been no strong evidence for viability of these mutants, this regulatory mechanism must play a physiologically important role in evolution, in particular providing a survival advantage under strongly oscillating (day and night) or fluctuating (e.g. irregular weather changes) light conditions. This is likely to be the case because the redox regulation may help minimize energy waste, with redox regulation becoming beneficial to chloroplast type ATP synthesis under insufficient energy conditions such as low light.
This work was supported by a Grant-in-Aid for Scientific Research (No. 18074002, to T. H.) from the Ministry of Education, Culture, Sports, Science and Technology, Japan and in part by the Management Expenses Grants for National Universities Corporations from the Ministry of Education, Culture, Sports, Science and Technology, Japan.
- α3β3γredox
- redox sensitive chimera complex derived from the α3β3γ complex of T. elongatus BP-1
- LDAO
- lauryl dimethylamine-N-oxide.
REFERENCES
- 1. Mitchell P. (1961) Nature 191, 144–148 [DOI] [PubMed] [Google Scholar]
- 2. Nelson N., Nelson H., Racker E. (1972) J. Biol. Chem. 247, 7657–7662 [PubMed] [Google Scholar]
- 3. Smith J. B., Sternweis P. C. (1977) Biochemistry 16, 306–311 [DOI] [PubMed] [Google Scholar]
- 4. Weber J., Dunn S. D., Senior A. E. (1999) J. Biol. Chem. 274, 19124–19128 [DOI] [PubMed] [Google Scholar]
- 5. Nowak K. F., McCarty R. E. (2004) Biochemistry 43, 3273–3279 [DOI] [PubMed] [Google Scholar]
- 6. Hirono-Hara Y., Ishizuka K., Kinosita K., Jr., Yoshida M., Noji H. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 4288–4293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hirono-Hara Y., Noji H., Nishiura M., Muneyuki E., Hara K. Y., Yasuda R., Kinosita K., Jr., Yoshida M. (2001) Proc. Natl. Acad. Sci. U.S.A. 98, 13649–13654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Miki J., Maeda M., Mukohata Y., Futai M. (1988) FEBS Lett. 232, 221–226 [DOI] [PubMed] [Google Scholar]
- 9. Schwarz O., Schürmann P., Strotmann H. (1997) J. Biol. Chem. 272, 16924–16927 [DOI] [PubMed] [Google Scholar]
- 10. Wolosiuk R. A., Buchanan B. B. (1977) Nature 266, 565–567 [Google Scholar]
- 11. Nalin C. M., McCarty R. E. (1984) J. Biol. Chem. 259, 7275–7280 [PubMed] [Google Scholar]
- 12. Yu L. M., Selman B. R. (1988) J. Biol. Chem. 263, 19342–19345 [PubMed] [Google Scholar]
- 13. Werner-Grüne S., Gunkel D., Schumann J., Strotmann H. (1994) Mol. Gen. Genet. 244, 144–150 [DOI] [PubMed] [Google Scholar]
- 14. Bald D., Noji H., Stumpp M. T., Yoshida M., Hisabori T. (2000) J. Biol. Chem. 275, 12757–12762 [DOI] [PubMed] [Google Scholar]
- 15. Krenn B. E., Strotmann H., Van Walraven H. S., Scholts M. J., Kraayenhof R. (1997) Biochem. J. 323, 841–845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wu G., Ort D. R. (2008) Photosynth Res. 97, 185–193 [DOI] [PubMed] [Google Scholar]
- 17. Ross S. A., Zhang M. X., Selman B. R. (1995) J. Biol. Chem. 270, 9813–9818 [DOI] [PubMed] [Google Scholar]
- 18. Bald D., Noji H., Yoshida M., Hirono-Hara Y., Hisabori T. (2001) J. Biol. Chem. 276, 39505–39507 [DOI] [PubMed] [Google Scholar]
- 19. Konno H., Murakami-Fuse T., Fujii F., Koyama F., Ueoka-Nakanishi H., Pack C. G., Kinjo M., Hisabori T. (2006) EMBO J. 25, 4596–4604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Landt O., Grunert H. P., Hahn U. (1990) Gene 96, 125–128 [DOI] [PubMed] [Google Scholar]
- 21. Wilm M., Shevchenko A., Houthaeve T., Breit S., Schweigerer L., Fotsis T., Mann M. (1996) Nature 379, 466–469 [DOI] [PubMed] [Google Scholar]
- 22. Meiss E., Konno H., Groth G., Hisabori T. (2008) J. Biol. Chem. 283, 24594–24599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Motohashi K., Hisabori T. (2006) J. Biol. Chem. 281, 35039–35047 [DOI] [PubMed] [Google Scholar]
- 24. Hisabori T., Kondoh A., Yoshida M. (1999) FEBS Lett. 463, 35–38 [DOI] [PubMed] [Google Scholar]
- 25. Dunn S. D., Tozer R. G., Zadorozny V. D. (1990) Biochemistry 29, 4335–4340 [DOI] [PubMed] [Google Scholar]
- 26. Hayashi K., Ueno H., Iino R., Noji H. (2010) Phys. Rev. Lett. 104, 218103. [DOI] [PubMed] [Google Scholar]
- 27. Sunamura E., Konno H., Imashimizu-Kobayashi M., Sugano Y., Hisabori T. (2010) Plant Cell Physiol. 51, 855–865 [DOI] [PubMed] [Google Scholar]
- 28. Yasuda R., Noji H., Yoshida M., Kinosita K., Jr., Itoh H. (2001) Nature 410, 898–904 [DOI] [PubMed] [Google Scholar]
- 29. Shimabukuro K., Yasuda R., Muneyuki E., Hara K. Y., Kinosita K., Jr., Yoshida M. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 14731–14736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Adachi K., Oiwa K., Nishizaka T., Furuike S., Noji H., Itoh H., Yoshida M., Kinosita K., Jr. (2007) Cell 130, 309–321 [DOI] [PubMed] [Google Scholar]
- 31. Konno H., Yodogawa M., Stumpp M. T., Kroth P., Strotmann H., Motohashi K., Amano T., Hisabori T. (2000) Biochem. J. 352 Pt 3, 783–788 [PMC free article] [PubMed] [Google Scholar]
- 32. Ueoka-Nakanishi H., Nakanishi Y., Konno H., Motohashi K., Bald D., Hisabori T. (2004) J. Biol. Chem. 279, 16272–16277 [DOI] [PubMed] [Google Scholar]
- 33. Abrahams J. P., Leslie A. G., Lutter R., Walker J. E. (1994) Nature 370, 621–628 [DOI] [PubMed] [Google Scholar]