

Abstract
Knowledge of the genetic relatedness between individuals is important in many research areas in quantitative genetics, conservation genetics, forensics, evolution, and ecology. In the absence of pedigree records, relatedness can be estimated from genetic marker data using a number of estimators. These estimators, however, make the critical assumption of a large random mating population without genetic structures. The assumption is frequently violated in the real world where geographic/social structures or nonrandom mating usually lead to genetic structures. In this study, I investigated two approaches to the estimation of relatedness between a pair of individuals from a subpopulation due to recent common ancestors (i.e., relatedness is defined and measured with the current focal subpopulation as reference). The indirect approach uses the allele frequencies of the entire population with and without accounting for the population structure, and the direct approach uses the allele frequencies of the current focal subpopulation. I found by simulations that currently widely applied relatedness estimators are upwardly biased under the indirect approach, but can be modified to become unbiased and more accurate by using Wright's Fst to account for population structures. However, the modified unbiased estimators under the indirect approach are clearly inferior to the unmodified original estimators under the direct approach, even when small samples are used in estimating both allele frequencies and relatedness.
Keywords: relatedness, inbreeding, genetic markers, population structure, moment estimator, probability of identity-by-descent (IBD), likelihood
KNOWING the degree of relatedness between individuals is essential in many research areas in quantitative genetics, conservation genetics, forensics, evolution, and ecology (Ritland 1996; Lynch and Ritland 1999; Weir et al. 2006). The expected value of relatedness between two individuals (e.g., 0.5 for parent–offspring in a large random mating population) can be easily calculated from their pedigree records. When pedigree is unavailable, incomplete, or unreliable, genetic marker information can be used instead to obtain an estimate of the realized value of relatedness, using a number of estimators developed for this purpose (e.g., Lynch 1988; Queller and Goodnight 1989; Li et al. 1993; Ritland 1996; Lynch and Ritland 1999; Wang 2002; Milligan 2003). When the assumptions are met, these estimators yield unbiased estimates of the expected relatedness calculated from pedigrees. Recently, these estimators were compared for accuracy extensively, using both simulated and empirical data sets (e.g., Lynch and Ritland 1999; Van de Casteele et al. 2001; Wang 2002; Milligan 2003; Csilléry et al. 2006; Anderson and Weir 2007).
The current marker-based methods make the critical assumption of a large random mating population, which implies the absence of close inbreeding (due to mating between closely related individuals, such as siblings) and pervasive inbreeding (due to genetic drift from the finite size or structure of a population). Unfortunately, most real populations are small and genetically structured, and matings are usually confined to individuals in a small area or a social group. Thus both forms of inbreeding may exist, leading to a background level of relatedness that can be quantified by Wright's Fst. For human populations, Fst values ∼0.1 have been reported in some cases (e.g., Hinds et al. 2005; Weir et al. 2005), although typical values for within-continent populations are much smaller. Furthermore, consanguineous marriages are found commonly in some countries or communities (e.g., Bittles et al. 1991; Khoury and Massad 2005). For plant and animal populations, Fst values are often higher (e.g., Marshall and Ritland 2002) and more extreme forms of close inbreeding, such as selfing, could be present.
In a subdivided population, relatedness can be defined and measured with respect to either the entire population or just the focal subpopulation from which individuals are drawn for relatedness estimation (see below). For the same pair of individuals, relatedness is always higher when the reference is the entire population than that when the reference is the focal subpopulation. Which reference is more appropriate depends on the particular applications of relatedness estimates.
In some contexts, a researcher may be interested in the total relatedness due to coalescences in both the recent and the remote past. An example in conservation is to find the set of individuals as breeders that have the minimum average relatedness among them (Ballou and Lacy 1995) and to determine the best mating pairs that are the least related and thus result in the highest average heterozygosity of the next generation (Caballero and Toro 2000). For a given relationship defined within a certain number of generations in the past, pervasive or close inbreeding that occurred in the more remote past will lead to an increased relatedness beyond that expected without inbreeding. Ignoring inbreeding effectively moves the reference point forward in time, resulting in an underestimation of relatedness. In practice, inbreeding is ignored when relatedness is estimated using the current allele frequencies of a subpopulation and assuming the absence of identity-by-descent (IBD) between genes at a locus within individuals. At present, there are no moment estimators of relatedness that can account for inbreeding, but a likelihood method based on the estimation of the nine condensed IBD coefficients (Jacquard 1972) was proposed (Wang 2007) to infer relatedness in populations with inbreeding.
In some other contexts, a researcher may be interested in the relatedness due to coalescences in the immediate past few generations, and that due to coalescences in the remote past is irrelevant. This is true when one wants to sort the pairs of individuals into a few simple relationship categories (such as full-sibs, half-sibs, parent–offspring, and unrelated) from the relatedness estimates. An example is the study of extrapair paternity, where one is interested in knowing whether a social father of an offspring is in fact the biological father (Anderson and Weir 2007). Similarly, in inferring the female mating system, one is interested in knowing whether the offspring from a mother are full-sibs and if not, how many half-sibships (fathers) the offspring fall into. In such cases, background relatedness acts as a noise and needs to be filtered out.
In a structured population, two approaches can be adopted to estimate the relatedness due to recent coalescences (i.e., using the focal subpopulation as reference for IBD). The direct approach is to take the focal subpopulation as reference by using its allele frequencies in relatedness inference. The indirect approach is to take the entire population as reference by using the population allele frequencies in relatedness inference and correct for the population structure statistically using Fst. The latter approach, adopted by Anderson and Weir (2007), is deemed essential when the allele frequencies of the subpopulation from which the focal sample of individuals is taken are unavailable or cannot be accurately estimated due to small sample sizes.
In this investigation, I focus on the estimation of relatedness due to recent coalescences in a structured population, using both approaches. Under a genetic model in the indirect approach, Anderson and Weir (2007) developed and implemented a reduced-likelihood method that accounts for pervasive inbreeding (population structure) but not close inbreeding. They also derived the expectations of several moment estimators in the simple cases of a locus with two alleles or with multiple equifrequency alleles. In this study, I extend Anderson and Weir's work to develop and implement a likelihood method that estimates all of the nine condensed IBD coefficients between two individuals in a genetically structured population. The method can therefore provide unbiased estimates of relatedness in the presence of both pervasive and close inbreeding, and it reduces to that of Anderson and Weir when close inbreeding is absent by assuming a large subpopulation with random mating. I also derive the expectations of several moment estimators in the general case of an arbitrary allele-frequency distribution, which lead to unbiased moment estimators in the presence of pervasive inbreeding. I compared the direct and indirect approaches for different estimators by simulations and found that in realistic situations the direct approach is not only simpler but also more accurate than the indirect approach.
THEORY AND METHODS
In this section, I outline Jacquard's (1972) nine condensed IBD coefficients that fully describe the relationship among the four genes possessed by two diploid individuals at a locus. I then describe briefly Anderson and Weir's model of relatedness in a structured population. On the basis of this model, I show that a full-likelihood method can be developed and implemented to estimate all of the nine condensed IBD coefficients in a population with pervasive and close inbreeding. I also show that, strictly under Anderson and Weir's model, unbiased moment estimators can be derived to account for pervasive inbreeding. Finally, I describe the simulations that are used to compare the performances of different estimators in both the direct and the indirect approaches.
Identity-by-descent between alleles and relatedness between individuals:
A set of two or more alleles at a locus are IBD if they are identical copies of the same ancestral allele. IBD (and IBD-based parameters such as relatedness, inbreeding, and coancestry coefficients) is defined and measured implicitly relative to a particular reference population in which all alleles are designated as nonidentical by descent. Therefore, the IBD status of a set of alleles changes with an alteration of the reference. Alleles non-identical-by-descent may become IBD when the reference moves backward in time or shifts to a larger geographic range and vice versa. The relative nature of IBD (and its derived parameters) is irrelevant for some applications (such as correlation analyses in which IBD is correlated with other quantities) but is important for other applications (such as distinguishing genealogical relationships).
Traditionally in a pedigree analysis, IBD is defined with respect to a specific focal pedigree. Alleles are either IBD from a common ancestor within the pedigree or non-IBD if they trace back to distinct founders of the pedigree, where founders are typically assumed to be random draws from a large gene pool and are thus both noninbred and unrelated. In the absence of a pedigree, IBD can be defined with respect to a reference population x generations in the past, for some specified x. Alleles are either IBD if they are from a common ancestor found within the x generations or non-IBD otherwise. In marker-based inferences of IBD, it is the population whose allele frequencies are used in the inference that acts as the reference (Ritland 1996). By definition, two alleles taken at random from the reference population are non-IBD. When the reference is a large random mating population, we arrive at the familiar probabilities of IBD sharing patterns between family members. For example, parent and offspring share one allele IBD at a probability of 1, and full-sibs share one allele and two alleles IBD at probabilities of 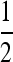 and
and 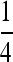 , respectively. In both cases, the coancestry coefficient is
, respectively. In both cases, the coancestry coefficient is 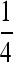 . In practice, the allele frequencies in a population are usually unknown but are estimated from a sample of individuals taken from the population. In such a case, more precisely it is this sample of individuals that actually acts as the reference. When the sample is sufficiently large and taken at random from the population, the population- and sample-based references are similar, as the population is well represented by the sample. Otherwise, they can be very different. Consider a simple example. Suppose a sample of full siblings is taken from a single family in a population and is used to estimate allele frequencies that are then used for IBD (relatedness) inferences. By definition, two alleles taken at random (without replacement) from the sample are non-IBD, and the average relatedness between individuals within the sample should be zero. Indeed, when the sample allele frequencies are used, different estimators yield relatedness estimates between individuals in the sample that are on average zero or very close to zero. Different from the first two definitions of IBD based on pedigrees or generations that both have a clear time cutoff point beyond which all alleles are assumed non-IBD, the population- or sample-based definition does not specify explicitly this time horizon. By this definition, two individuals having many remote common ancestors may be more related than two individuals having few recent common ancestors. This is true with the pedigree- or generation-based definitions only when both remote and recent ancestors fall within the time horizon. Removing the artificial time limit from the IBD definition is an advantage of the population- or sample-based definition as it avoids potential biases brought about by the artificial cutoff point. In the present study, the reference in the direct approach is either the current focal subpopulation or a sample taken at random from it, and the reference in the indirect approach is either the current entire population or a sample taken at random from it.
. In practice, the allele frequencies in a population are usually unknown but are estimated from a sample of individuals taken from the population. In such a case, more precisely it is this sample of individuals that actually acts as the reference. When the sample is sufficiently large and taken at random from the population, the population- and sample-based references are similar, as the population is well represented by the sample. Otherwise, they can be very different. Consider a simple example. Suppose a sample of full siblings is taken from a single family in a population and is used to estimate allele frequencies that are then used for IBD (relatedness) inferences. By definition, two alleles taken at random (without replacement) from the sample are non-IBD, and the average relatedness between individuals within the sample should be zero. Indeed, when the sample allele frequencies are used, different estimators yield relatedness estimates between individuals in the sample that are on average zero or very close to zero. Different from the first two definitions of IBD based on pedigrees or generations that both have a clear time cutoff point beyond which all alleles are assumed non-IBD, the population- or sample-based definition does not specify explicitly this time horizon. By this definition, two individuals having many remote common ancestors may be more related than two individuals having few recent common ancestors. This is true with the pedigree- or generation-based definitions only when both remote and recent ancestors fall within the time horizon. Removing the artificial time limit from the IBD definition is an advantage of the population- or sample-based definition as it avoids potential biases brought about by the artificial cutoff point. In the present study, the reference in the direct approach is either the current focal subpopulation or a sample taken at random from it, and the reference in the indirect approach is either the current entire population or a sample taken at random from it.
A set of two or more alleles at a locus is identical-in-state (IIS) if they all have the same phenotype. For example, they have the same base type for a SNP or the same number of repeat units for a microsatellite. Barring the rare events of mutations in the short timescale in the definition of IBD above, alleles IBD are always IIS. However, alleles IIS are not necessarily IBD, although they are more likely to be IBD than alleles that are not IIS. IBD is invisible, but can be inferred probabilistically from IIS. For example, for a father–mother–offspring trio with genotypes A1A2–A2A2–A1A2, the two A1 alleles are IBD with probability 1, and the A2 allele in the child is IBD with each A2 allele in the mother with a probability of 0.5.
Traditionally, relatedness is defined and estimated for a large random mating population without close and pervasive inbreeding (e.g., Lynch and Ritland 1999). In such a simplified situation, alleles within an individual are always non-IBD while alleles between individuals may or may not be IBD. To accommodate inbreeding, however, the IBD status of alleles both within and between individuals needs to be considered. Among the two genes from individual X and two genes from individual Y, 15 mutually exclusive and exhaustive IBD modes exist (Jacquard 1972). When paternal and maternal genes are not distinguished, the 15 IBD modes reduce to 9 condensed identity modes (Jacquard 1972; Lynch and Walsh 1998) as defined in Figure 1. The relationships among the four alleles are determined by the probabilities of the 9 IBD modes, Δ = {Δ1, Δ2,…, Δ9}, where Δi is the probability of IBD mode Di (i = 1, 2,…, 9). The relationship between X and Y is also fully described by Δ. X and Y are more related when they share more alleles IBD (e.g., D7 vs. D8) and/or share alleles IBD at a higher probability. A widely used parameter that combines Δi values to measure the total degree of relatedness between X and Y is the coancestry coefficient, θXY, which is the probability that two alleles at a locus, one taken at random from X and one from Y, are IBD. By definition,
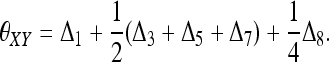 |
(1) |
An equivalent parameter is the relatedness coefficient, 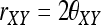 , as adopted in the literature (e.g., Lynch and Ritland 1999). Note, however, rXY as defined above can be >1 when there is inbreeding. The inbreeding coefficient of X (or Y), which is the probability that the two alleles of X (or Y) are IBD, can also be calculated from Δ,
, as adopted in the literature (e.g., Lynch and Ritland 1999). Note, however, rXY as defined above can be >1 when there is inbreeding. The inbreeding coefficient of X (or Y), which is the probability that the two alleles of X (or Y) are IBD, can also be calculated from Δ,
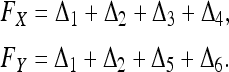 |
(2) |
Figure 1.—

Identity-by-descent modes of the four genes at a locus of two diploid individuals. Each group of four dots represents an IBD mode, with the top pair of dots representing the two genes in individual X and the bottom pair of dots representing the two genes in individual Y. Genes connected by lines are IBD.
When both X and Y are noninbred, an assumption made by most moment and likelihood estimators of relatedness, then only three IBD modes are possible, D7, D8, and D9. In this simple case, 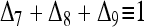 and Δi = 0 for i = 1, 2,…, 6.
and Δi = 0 for i = 1, 2,…, 6.
Corresponding to each of the nine IBD modes Di (i = 1, 2,…, 9) among the four alleles of X and Y, there is an IIS mode Si. Si is defined similarly to Di in Figure 1, except that the alleles connected by a line are IIS rather than IBD. All relatedness estimators infer rXY directly (likelihood methods) or indirectly (moment methods) from Si together with other information such as allele frequencies.
Relatedness in structured populations:
Traditional relatedness models assume that individuals are sampled from a large random mating population. Recently, Anderson and Weir (2007) proposed a model that assumes that individuals come from one of the subpopulations of a population whose allele frequencies are known. Two individuals from a subpopulation are related, relative to two individuals taken at random from the entire population, because of the pervasive inbreeding that leads to the differentiation of the subpopulation from other subpopulations (or from the ancestral population) and because of any close inbreeding or recent coalescences within the subpopulation. Anderson and Weir are interested in estimating the relatedness due to recent coalescences. The relatedness due to pervasive inbreeding or population structure can be filtered out by using the focal subpopulation as reference, realized by estimating relatedness from the allele frequencies of the focal subpopulation (direct approach). Alternatively, this can also be achieved by using the allele frequencies of the entire population and accounting for pervasive inbreeding by Fst (indirect approach). The latter approach, adopted by Anderson and Weir, is deemed desirable when the allele frequencies of the subpopulation are unavailable or cannot be estimated reliably because of small sample sizes.
Let us consider a marker with n codominant alleles, A = {A1, A2,…, An}, whose frequencies in the entire population are p = {p1, p2,…, pn}. The genotypes of two individuals from a subpopulation must fall into one of the nine IIS modes. The probability of each IIS mode, given p, Δ, and the differentiation of the subpopulation from the ancestral population (denoted by θ from here on for simplicity), was derived by Anderson and Weir as listed in Table 1. When θ = 0, allele frequencies in the subpopulation are equal to those in the ancestral population, and the genotype pair probabilities listed in Table 1 reduce to those in a large random mating population as derived before (Milligan 2003).
TABLE 1.
Probability of identity-in-state modes Si given identity-by-descent modes Di
| IBD modes |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| IIS mode | Allelic state | D1 | D2 | D3 | D4 | D5 | D6 | D7 | D8 | D9 |
| S1 | AiAi,AiAi | 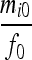 |
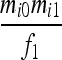 |
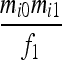 |
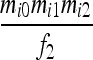 |
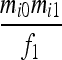 |
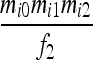 |
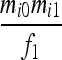 |
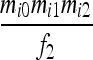 |
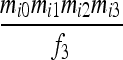 |
| S2 | AiAi,AjAj | 0 | 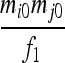 |
0 | 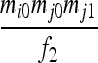 |
0 | 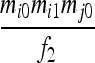 |
0 | 0 | 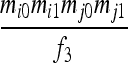 |
| S3 | AiAi,AiAj | 0 | 0 | 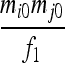 |
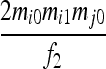 |
0 | 0 | 0 | 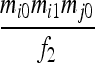 |
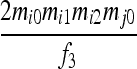 |
| S4 | AiAi,AjAk | 0 | 0 | 0 | 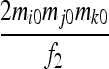 |
0 | 0 | 0 | 0 | 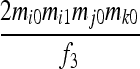 |
| S5 | AiAj,AiAi | 0 | 0 | 0 | 0 | 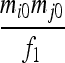 |
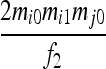 |
0 | 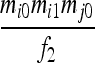 |
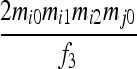 |
| S6 | AjAk,AiAi | 0 | 0 | 0 | 0 | 0 | 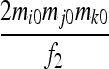 |
0 | 0 | 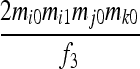 |
| S7 | AiAj,AiAj | 0 | 0 | 0 | 0 | 0 | 0 | 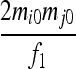 |
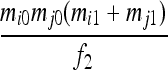 |
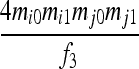 |
| S8 | AiAj,AiAk | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 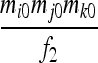 |
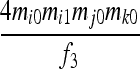 |
| S9 | AiAj,AkAl | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 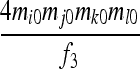 |
Alleles with different subscripts are distinct. Parameters 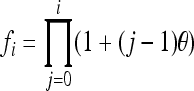 and
and 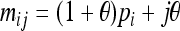 .
.
Likelihood estimator:
The probability of observing a particular IIS mode, Si, for two individuals at a single locus, given their IBD coefficients Δ and the allele frequencies, is equal to the likelihood of Δ,
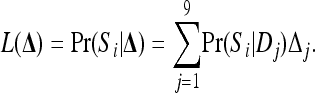 |
(3) |
In (3), 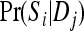 is listed in Table 1, Δ = {Δ1, Δ2,…, Δ9} are the parameters being estimated, and θ is treated as a known constant. In practice, θ can be estimated from allele-frequency data using methods, such as that of Weir and Cockerham (1984), that assume unrelated and noninbred individuals. More desirably, a likelihood method that jointly estimates θ and pairwise relatedness could be more accurate for both. Such a method is not available yet and is out of the scope of this study. For multiple loci in linkage equilibrium, the likelihood is simply the product of the single-locus likelihoods.
is listed in Table 1, Δ = {Δ1, Δ2,…, Δ9} are the parameters being estimated, and θ is treated as a known constant. In practice, θ can be estimated from allele-frequency data using methods, such as that of Weir and Cockerham (1984), that assume unrelated and noninbred individuals. More desirably, a likelihood method that jointly estimates θ and pairwise relatedness could be more accurate for both. Such a method is not available yet and is out of the scope of this study. For multiple loci in linkage equilibrium, the likelihood is simply the product of the single-locus likelihoods.
Given a set of markers with known allele frequencies in the entire population and the value of parameter θ, the maximum-likelihood estimates of Δ for two individuals conditional on their observed IIS modes can be obtained by maximizing function (3) over the legitimate parameter space (i.e., 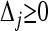 for j = 1, 2,…, 9, subject to constraint
for j = 1, 2,…, 9, subject to constraint 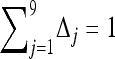 ). Relatedness between the two individuals is then calculated by (1) using the estimated Δ-values. It is impossible to solve (3) analytically. I use Powell's quadratically convergent method (Press et al. 1996) with slight modifications to solve this nine-dimentional constrained maximization problem. This method is chosen because it is derivative free and simple to implement. Yet, tests using numerous simulated and empirical data sets indicate that the method converges reliably, with different initial values and different initial searching directions of Δ leading to the same maximum-likelihood estimates. Hereafter, this estimator is referred as the full-likelihood estimator as it estimates the full set of nine IBD coefficients between two individuals. It is denoted as
). Relatedness between the two individuals is then calculated by (1) using the estimated Δ-values. It is impossible to solve (3) analytically. I use Powell's quadratically convergent method (Press et al. 1996) with slight modifications to solve this nine-dimentional constrained maximization problem. This method is chosen because it is derivative free and simple to implement. Yet, tests using numerous simulated and empirical data sets indicate that the method converges reliably, with different initial values and different initial searching directions of Δ leading to the same maximum-likelihood estimates. Hereafter, this estimator is referred as the full-likelihood estimator as it estimates the full set of nine IBD coefficients between two individuals. It is denoted as 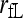 and
and 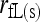 when population structure is ignored (assuming θ = 0) and taken into account (assuming θ > 0), respectively.
when population structure is ignored (assuming θ = 0) and taken into account (assuming θ > 0), respectively.
When the subpopulation from which individuals are sampled is large and at random mating, then Δj = 0 for j = 1, 2,…, 6. In such a case, as was considered by Anderson and Weir, only three IBD coefficients, Δ7, Δ8, and Δ9, need to be obtained from (3) while the rest are constrained to be zero. Hereafter, this is referred as the reduced-likelihood estimator, denoted as 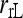 and
and 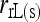 when population structure is ignored (assuming θ = 0) and taken into account (assuming θ > 0), respectively.
when population structure is ignored (assuming θ = 0) and taken into account (assuming θ > 0), respectively.
Moment estimators:
Current moment estimators are biased when applied to individuals sampled from a subpopulation and when the allele frequencies of the entire population are used in the estimation, as shown by Anderson and Weir (2007). They derived analytical functions of the biases of several moment estimators in the special cases of a biallelic locus and a locus with equi-frequency alleles. Below I study the biases of these moment estimators in the general case of a locus with an arbitrary number and frequency distribution of alleles. This allows me to derive unbiased moment estimators of relatedness in a structured population.
Estimator by Queller and Goodnight (1989):
One of the earliest and most commonly used moment estimator is that published by Queller and Goodnight (1989). There are a number of variants to this estimator, and I choose to use the symmetric one obtained by averaging the estimates using each of the two individuals as reference. For individuals X and Y with genotypes {a, b} and {c, d}, respectively, at a locus, the estimator is
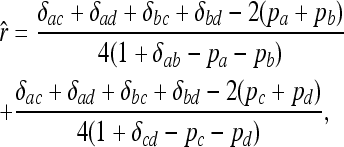 |
(4) |
where alleles A1, A2,…, An are denoted by a, b, c, d to avoid subscripts, and 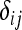 is the Kronecker delta variable (
is the Kronecker delta variable (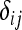 =1 if i = j and
=1 if i = j and 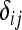 = 0 otherwise).
= 0 otherwise).
In the appendix, I show that the expectation of (4) is
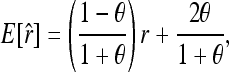 |
(5) |
where E is the expectation operator and r is the true relatedness between X and Y. As can be seen, (4) is unbiased only when θ = 0, which is true if the individuals come from a large random mating population (or subpopulation) and the allele frequencies of the population (subpopulation) are used in the estimation. Otherwise, (4) is upwardly biased. The more differentiated the subpopulations are, the larger the biases.
Replacing the left side of (5) by the original estimator (4) and solving for r, I obtain an unbiased estimator in the presence of population structure,
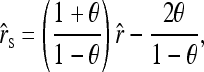 |
(6) |
where 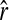 is given by (4). The unbiased estimator can be regarded as the original estimator corrected for the population structure or the misspecification of allele frequencies, using the differentiation parameter θ. It is unbiased regardless of the value of θ. When θ = 0, it reduces to the original estimator.
is given by (4). The unbiased estimator can be regarded as the original estimator corrected for the population structure or the misspecification of allele frequencies, using the differentiation parameter θ. It is unbiased regardless of the value of θ. When θ = 0, it reduces to the original estimator.
For multiple loci, the Queller and Goodnight estimators, (4) and (6), are calculated by averaging the single-locus estimates, following the literature (e.g., Milligan 2003; Anderson and Weir 2007). For simplicity, hereafter estimators (4) and (6) are denoted as 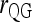 and
and 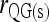 , respectively, where subscript “s” indicates structured populations.
, respectively, where subscript “s” indicates structured populations.
Estimator by Lynch (1988) and Li et al. (1993):
Another widely applied moment estimator is based on a similarity index SXY, defined as the arithmetic average fraction of alleles at a locus in a reference individual (either X or Y) for which there is another allele in the other individual (either Y or X) that is identical in state (Lynch 1988; Li et al. 1993). Thus, 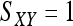 for genotype pairs { AiAi, AiAi } or {AiAj, AiAj },
for genotype pairs { AiAi, AiAi } or {AiAj, AiAj }, 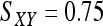 for genotype pairs { AiAi, AiAj },
for genotype pairs { AiAi, AiAj }, 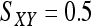 for genotype pairs { AiAj, AiAk }, and
for genotype pairs { AiAj, AiAk }, and 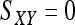 for genotype pairs { AiAj, AkAl }. For a single locus, the estimator is
for genotype pairs { AiAj, AkAl }. For a single locus, the estimator is
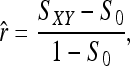 |
(7) |
where 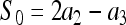 (with
(with 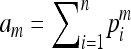 for m = 2, 3) is the expected similarity index for unrelated individuals in a large random mating population. In a structured population, the average similarity index for individuals X and Y with relatedness r can be derived (see appendix) as
for m = 2, 3) is the expected similarity index for unrelated individuals in a large random mating population. In a structured population, the average similarity index for individuals X and Y with relatedness r can be derived (see appendix) as
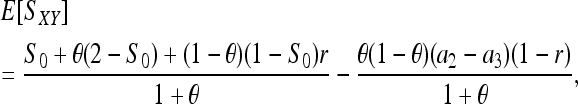 |
(8) |
where S0 is as defined in (7). When θ = 0, (8) reduces to 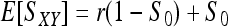 as derived before (e.g., Li et al. 1993). Equation 8 indicates that two individuals taken from a subpopulation are similar in genotypes because of θ (common ancestry) as well as r (relatedness) and S0 (chances). Inserting (8) into (7) yields the expected value of (7):
as derived before (e.g., Li et al. 1993). Equation 8 indicates that two individuals taken from a subpopulation are similar in genotypes because of θ (common ancestry) as well as r (relatedness) and S0 (chances). Inserting (8) into (7) yields the expected value of (7):
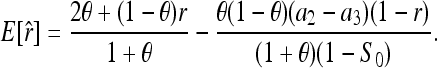 |
(9) |
Equation 9 shows that (7) is unbiased only when θ = 0. Otherwise, it is upwardly biased, giving relatedness estimates larger than the true value. Unlike the Queller and Goodnight estimator, however, the extent of the bias depends not only on parameters of θ and r, but also on the marker allele-frequency distributions that determine a2 and a3. For the special case of equi-frequency alleles, (9) reduces to
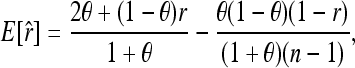 |
which is identical to Equation A16 of Anderson and Weir (2007), noting that 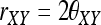 .
.
An unbiased estimator can be derived from (9), which is
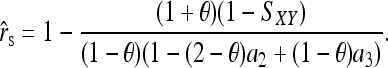 |
(10) |
For simplicity, hereafter estimators (7) and (10) are denoted as 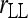 and
and 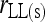 , respectively.
, respectively.
Estimator by Ritland (1996):
An estimator derived in Ritland (1996) and Li and Horvitz (1953) is
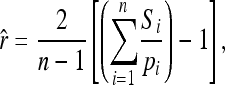 |
(11) |
where Si, the similarity for allele i between individuals X and Y, takes a value of 0 (if X and Y do not share any i allele), 0.25 (if both X and Y have a single i allele), 0.5 (if one individual has two and the other individual has one i allele), and 1 (if both X and Y have two i alleles).
As shown in the appendix, the expected value of (11) is
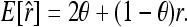 |
(12) |
Like other moment estimators shown above, therefore, estimator (11) is unbiased only when θ = 0. Otherwise, it is upwardly biased. An unbiased estimator accounting for the population structure can be obtained from (12) as
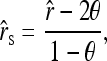 |
(13) |
where 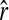 is the original estimator given by (11). For simplicity hereafter, estimators (11) and (13) are denoted as
is the original estimator given by (11). For simplicity hereafter, estimators (11) and (13) are denoted as 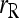 and
and 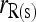 , respectively.
, respectively.
Estimator by Lynch and Ritland (1999):
The estimator of relatedness between individuals X and Y with genotypes {a, b} and {c, d}, respectively, is
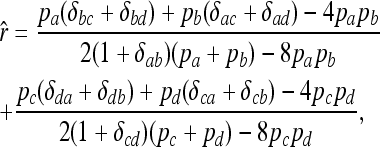 |
(14) |
where the delta variables are defined as in (4). For multiple loci, the overall estimate is obtained by weighting single-locus estimates, using the weights of Lynch and Ritland (their Equation 7a).
In a structured population, it is shown in the appendix that (14) has the same expectation as rQG. Therefore, the bias is given by (5) and the unbiased estimator is given by (6), where 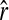 is calculated by (14) instead of (4). From here on, the multilocus symmetrical estimator of Lynch and Ritland (1999) is denoted as
is calculated by (14) instead of (4). From here on, the multilocus symmetrical estimator of Lynch and Ritland (1999) is denoted as 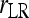 and
and 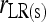 for unstructured and structured populations, respectively.
for unstructured and structured populations, respectively.
Estimator by Wang (2002):
Wang (2002) proposed an estimator that uses the similarity index of Lynch (1988) and Li et al. (1993) to estimate both Δ7 and Δ8 and thus r. He classified genotype pairs into four exclusive similarity categories, with categories 1, 2, 3, and 4 containing genotype pairs that have similarity index values [as defined in (7) above] of 1, 0.75, 0.5, and 0, respectively. The genotype data of individuals X and Y are summarized into a set of four indicator variables, 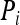 , for i = 1, 2, 3, and 4. If the genotype pair of X and Y falls into category i, then
, for i = 1, 2, 3, and 4. If the genotype pair of X and Y falls into category i, then 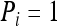 and
and 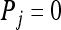 for j = 1, 2, 3, 4 and j ≠ i. The relatedness estimator turns out to be the same as that of Lynch (1988) and Li et al. (1993) for the case of biallelic loci (Wang 2002). For loci with three or more alleles, the estimator is
for j = 1, 2, 3, 4 and j ≠ i. The relatedness estimator turns out to be the same as that of Lynch (1988) and Li et al. (1993) for the case of biallelic loci (Wang 2002). For loci with three or more alleles, the estimator is
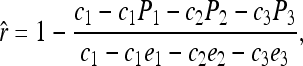 |
(15) |
where constants 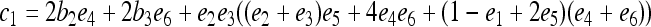 ,
, 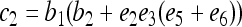 ,
, 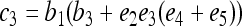 ,
, 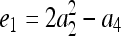 ,
, 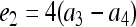 , and
, and 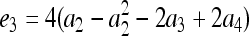 , in which
, in which 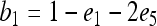 ,
, 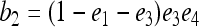 ,
, 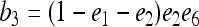 ,
, 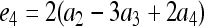 ,
, 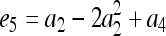 , and
, and 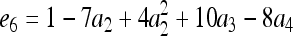 .
.
In a structured population, the expectation of (15) is a complicated function of the moments of allele-frequency distribution (am, m = 2 ∼ 4), r and Δ8, as shown in the appendix. This relatedness estimator is unbiased when θ = 0, but is upwardly biased when θ > 0, like the other moment estimators. Furthermore, the extent of bias depends not only on θ, but also on Δ8 (appendix). However, the coefficient of Δ8 in the expectation is always very small (<0.05). Therefore an almost unbiased estimator when θ > 0 can be derived by ignoring the term of Δ8,
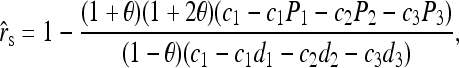 |
(16) |
where three constants additional to those in (15) are 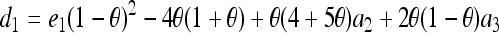 ,
, 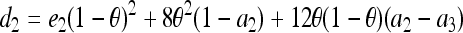 , and
, and 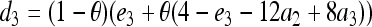 . Hereafter, (15) and (16) are denoted as
. Hereafter, (15) and (16) are denoted as 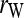 and
and  for unstructured and structured populations, respectively.
for unstructured and structured populations, respectively.
For easy reference, the abbreviations, original sources, equations (in this article), some properties, and plotting symbols of the estimators compared in the present study are listed in Table 2.
TABLE 2.
A list of the estimators compared in this study
| Estimator abbreviation | Source | Equation | Allowing for population structure | Allowing for close inbreeding | Plotting symbol |
|---|---|---|---|---|---|
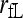 |
This study | (3) | No | Yes | ♦ |
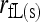 |
This study | (3) | Yes | Yes | ♦ |
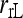 |
Anderson and Weir (2007) | (3) | No | No | ★ |
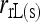 |
Anderson and Weir (2007) | (3) | Yes | No | ★ |
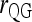 |
Queller and Goodnight (1989) | (4) | No | No | ▴ |
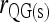 |
This study | (6) | Yes | No | ▴ |
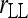 |
Lynch (1988); Li et al. (1993) | (7) | No | No | ⋆ |
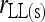 |
This study | (10) | Yes | No | ⋆ |
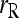 |
Li and Horvitz (1953); Ritland (1996) | (11) | No | Yes | □ |
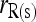 |
This study | (13) | Yes | Yes | □ |
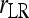 |
Lynch and Ritland (1999) | (14) | No | No | ⋄ |
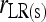 |
This study | (6) | Yes | No | ⋄ |
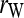 |
Wang (2002) | (15) | No | No | ▵ |
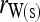 |
This study | (16) | Yes | No | ▵ |
The original source and equation (in this article) are listed in columns 2 and 3, respectively.
Simulations:
Simulations were conducted to check and compare the accuracies of different estimators in various circumstances. In the first set of simulations, I assumed both the allele frequencies of the entire population and the parameter θ were known and were used in estimating the relatedness between individuals taken from within a subpopulation. The simulation began with the generation of allele frequencies of the entire population, assuming an equal allele frequency (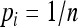 for i = 1, 2,…, n), a uniform Dirichlet frequency distribution, or a triangular frequency distribution [
for i = 1, 2,…, n), a uniform Dirichlet frequency distribution, or a triangular frequency distribution [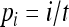 for i = 1, 2,…, n, where
for i = 1, 2,…, n, where 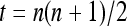 ]. The allele frequencies of a subpopulation were generated from the Dirichlet distribution using the parameter θ and the allele frequencies of the entire population, as described in Weir (2003). The genotypes of two individuals with a given relationship, parent–offspring (PO), full-sib (FS), half-sib (HS), and unrelated (UR), are then generated at each of a number of marker loci using the allele frequencies of the subpopulation. The individual genotypes, together with the allele frequencies of the entire population and the known parameter θ, were then used as data in estimating relatedness by various estimators. The factors considered in this set of simulations are the level of differentiation (θ) and the number of markers, each having either 10 or 2 alleles to mimic microsatellites and SNPs, respectively.
]. The allele frequencies of a subpopulation were generated from the Dirichlet distribution using the parameter θ and the allele frequencies of the entire population, as described in Weir (2003). The genotypes of two individuals with a given relationship, parent–offspring (PO), full-sib (FS), half-sib (HS), and unrelated (UR), are then generated at each of a number of marker loci using the allele frequencies of the subpopulation. The individual genotypes, together with the allele frequencies of the entire population and the known parameter θ, were then used as data in estimating relatedness by various estimators. The factors considered in this set of simulations are the level of differentiation (θ) and the number of markers, each having either 10 or 2 alleles to mimic microsatellites and SNPs, respectively.
The second set of simulations was conducted similarly, except for the relationships and the factors considered. Instead of the four relationships (UR, PO, FS, and HS) without close inbreeding, I considered full-sibs from parents who are themselves full-sibs (FSFS). The IBD coefficients for this relationship are 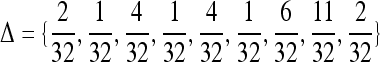 , and the true relatedness between and inbreeding coefficient of individuals are
, and the true relatedness between and inbreeding coefficient of individuals are 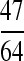 and
and 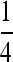 , respectively. The factor considered in this set of simulations is the number of markers, each having 10 alleles with frequencies in a subpopulation drawn from the Dirichlet distribution given θ, which is fixed at 0.1. Relatedness was estimated by the moment estimators and the likelihood estimator with and without close inbreeding taken into account, using the allele frequencies of the entire population and the known parameter θ to account for pervasive inbreeding.
, respectively. The factor considered in this set of simulations is the number of markers, each having 10 alleles with frequencies in a subpopulation drawn from the Dirichlet distribution given θ, which is fixed at 0.1. Relatedness was estimated by the moment estimators and the likelihood estimator with and without close inbreeding taken into account, using the allele frequencies of the entire population and the known parameter θ to account for pervasive inbreeding.
The third set of simulations dealt with the more realistic situation where allele frequencies are unknown. What one has is a sample of individuals (i.e., multilocus genotypes) from each of one or more subpopulations. In such a situation, two approaches can be adopted in estimating the relatedness between two (focal) individuals taken from within a (focal) subpopulation. The direct approach is to estimate the allele frequencies of the focal subpopulation, using the sample from it. The estimated allele frequencies are then used in estimating the relatedness between the focal individuals. The indirect approach is to use the samples from each subpopulation to estimate the allele frequencies of the entire population and parameter θ, which are then used in estimating relatedness as shown in simulation set 1. In both approaches, allele frequencies can be estimated by simple allele counting, assuming all sampled individuals are unrelated. Parameter θ in the indirect approach can be estimated by several methods, such as that of Weir and Cockerham (1984). In this set of simulations, however, I use the simulated parameter value of θ in relatedness estimation to obtain the maximally achievable accuracy for this approach. Simulations were conducted similarly to those in the first set, except for allele-frequency estimation (as briefed above) and the factors considered. I considered UR and FS relationships and different sample sizes and different numbers of subpopulations that were sampled. A fixed value of θ = 0.1 was used in simulations and was used in relatedness estimations in the indirect approach.
In each set of the simulations described above, k = 100,000 replicates were conducted for each set of parameters. The performance of an estimator was measured by the mean of the k replicate estimates, which informs the bias when it was compared with the true simulated value of relatedness. The overall performance was measured by mean squared error (MSE), calculated as 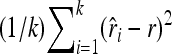 , where
, where 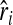 is the estimate in replicate i (1 ∼ k), and r is the simulated value of relatedness. Likelihood estimates are constrained to the legitimate range of [0, 1], while moment estimates are not. For a fair comparison in MSE between moment and likelihood estimators, moment estimates are truncated to force into the range of [0, 1] before calculating MSEs.
is the estimate in replicate i (1 ∼ k), and r is the simulated value of relatedness. Likelihood estimates are constrained to the legitimate range of [0, 1], while moment estimates are not. For a fair comparison in MSE between moment and likelihood estimators, moment estimates are truncated to force into the range of [0, 1] before calculating MSEs.
RESULTS
Consequences of ignoring population structure:
The bias and MSE of different estimators with and without population structure taken into account are compared in Figure 2. It is clear from Figure 2 that all estimators are upwardly biased when they assume a large random mating population (θ = 0) but are applied to a structured population (θ > 0). The extent of bias increases roughly linearly with an increasing value of θ. The highest bias occurs to estimator 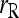 , while the other estimators (including
, while the other estimators (including 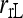 ) have a similar degree of bias. When population structure is taken into account, all moment estimators become unbiased irrespective of the value of θ. The likelihood estimator,
) have a similar degree of bias. When population structure is taken into account, all moment estimators become unbiased irrespective of the value of θ. The likelihood estimator, 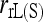 , has a small upward bias because it is constrained to be nonnegative.
, has a small upward bias because it is constrained to be nonnegative.
Figure 2.—

Bias and MSE of relatedness estimates for unrelated (UR), parent–offspring (PO), full-sib (FS), and half-sib (HS) individuals drawn from a subpopulation, as a function of the differentiation (θ) of the subpopulation from the ancestral population. Relatedness was estimated assuming either a large random mating population (θ = 0, rows 1 and 3) or a structured population with known θ-value (rows 2 and 4). In both cases, the allele frequencies in the entire population are assumed known and used in the estimation. Ten markers, each having 10 alleles with frequencies in a uniform Dirichlet distribution, are used for estimating relatedness. The symbols in the plot are listed in Table 2, which are as follows: ▵, 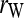 and
and 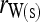 ; ⋆,
; ⋆, 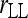 and
and 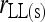 ; ⋄,
; ⋄, 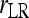 and
and 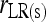 ; □,
; □, 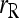 and
and 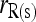 ; ▴,
; ▴, 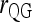 and
and 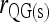 ; and ★,
; and ★, 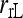 and
and 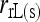 .
.
The MSE of each estimator increases with an increasing value of θ, when a large random mating population is assumed (θ = 0). This is true for all four relationships considered. 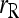 has the highest MSE while the likelihood estimator (
has the highest MSE while the likelihood estimator (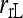 ) has usually the lowest or close to the lowest MSE. When population structure is taken into account, the MSE is reduced for all estimators, except for
) has usually the lowest or close to the lowest MSE. When population structure is taken into account, the MSE is reduced for all estimators, except for 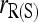 in the cases of PO and FS pairs. The reduction in MSE is expected as the bias component in MSE is removed when population structure is accounted for. The reason that
in the cases of PO and FS pairs. The reduction in MSE is expected as the bias component in MSE is removed when population structure is accounted for. The reason that 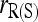 has a larger MSE than
has a larger MSE than 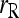 is that the former has an increased variance over the latter. On the basis of MSE,
is that the former has an increased variance over the latter. On the basis of MSE, 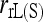 ,
, 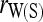 ,
, 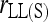 , and
, and 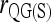 have the best performance across the four relationships and the range of θ values considered.
have the best performance across the four relationships and the range of θ values considered.
Among the six estimators compared, 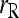 and
and 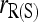 are the least accurate as measured by MSE when θ is substantial. This is because this estimator is sensitive to rare alleles that could lead to extreme estimates. Under the uniform Dirichlet distribution, some alleles at a 10-allele locus may have low frequencies. Both
are the least accurate as measured by MSE when θ is substantial. This is because this estimator is sensitive to rare alleles that could lead to extreme estimates. Under the uniform Dirichlet distribution, some alleles at a 10-allele locus may have low frequencies. Both 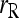 and
and 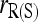 become increasingly sensitive to rare alleles with an increasing θ. This is because, for a given allele with a low frequency in the entire population, the probability that it has a substantial frequency in the focal subpopulation and thus appears in the genotypes of the focal individuals increases with θ. The advantage of this estimator over others in the case of unrelated or loosely related individuals in a large random mating population (θ = 0), as demonstrated before (e.g., Wang 2002), is lost when it is applied to a structured population (θ > 0).
become increasingly sensitive to rare alleles with an increasing θ. This is because, for a given allele with a low frequency in the entire population, the probability that it has a substantial frequency in the focal subpopulation and thus appears in the genotypes of the focal individuals increases with θ. The advantage of this estimator over others in the case of unrelated or loosely related individuals in a large random mating population (θ = 0), as demonstrated before (e.g., Wang 2002), is lost when it is applied to a structured population (θ > 0).
Similar results to those shown in Figure 2 are obtained for an equal or a triangular allele-frequency distribution. The main difference is that 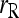 and
and 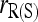 do not perform as poorly in comparison with the other estimators when θ is substantial.
do not perform as poorly in comparison with the other estimators when θ is substantial.
Effect of type and number of markers:
Figure 3 compares the MSEs among estimators when a variable number of microsatellites and SNPs are used in relatedness estimation. Confirming the results in Figure 2, accounting for population structure leads to a decrease in MSE for both microsatellites and SNPs and for all estimators except for that of Ritland (1996). When population structure is ignored, MSE tends to attenuate with an increasing number of markers. This is because MSE is increasingly dominated by the bias rather than sampling errors of the estimators with an increasing number of markers. Because of the misspecification of the relatedness model, more markers lead to just a smaller sampling variance but have no effect on the bias. Therefore, using unbiased estimators is especially important now that more and more markers are routinely genotyped at ease and used in relatedness analyses.
Figure 3.—

MSE of relatedness estimates for unrelated (UR) and full-sib (FS) individuals drawn from a subpopulation, as a function of the number of markers used in the estimation. The subpopulations are differentiated with a parameter value of θ = 0.1. Relatedness was estimated assuming either a large random mating population (θ = 0, columns 1 and 3) or a structured population with known θ-value (columns 2 and 4). In both cases, the allele frequencies in the entire population are assumed known and used in the estimation. For microsatellites and SNPs, each marker has 10 and 2 alleles, respectively, whose frequencies in the entire population are drawn from a uniform Dirichlet distribution. The plotting symbols for the different estimators are listed in Table 2. For SNPs, the estimator of Wang (2002) and that of Lynch (1988) and Li et al. (1993) are identical and thus only the latter is shown.
Figure 3 also shows that a microsatellite gives much more information than a SNP. However, given a sufficient number of SNPs, they can still yield accurate relatedness estimates. For SNPs that have two alleles per locus, several moment estimators have peculiar and undesirable properties as discussed in the literature (e.g., Lynch and Ritland 1999; Wang 2002). This is especially obvious for 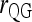 and
and 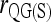 , which become undefined if the reference individual is a heterozygote. Although
, which become undefined if the reference individual is a heterozygote. Although 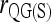 performs well for microsatellites, it no longer falls into the top performance group of estimators for SNPs.
performs well for microsatellites, it no longer falls into the top performance group of estimators for SNPs.
When population structure is accounted for, the best estimators are 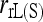 ,
, 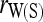 , and
, and 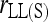 regardless of the actual relatedness (UR or FS) and the marker types and numbers. The likelihood estimator,
regardless of the actual relatedness (UR or FS) and the marker types and numbers. The likelihood estimator, 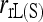 , is not obviously more accurate than the best moment estimators, even when hundreds of markers are used.
, is not obviously more accurate than the best moment estimators, even when hundreds of markers are used.
Close inbreeding:
Figure 4 compares the bias and accuracy of moment and likelihood estimators for closely inbred individuals (FSFS) taken from a subpopulation, as obtained from the second set of simulations. As can be seen, 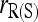 is the only one of the moment estimators that provides unbiased estimates of relatedness for closely inbred individuals (Wang 2007). The remaining four moment estimators are downward biased, and the bias is constant irrespective of the number of loci. When close inbreeding is unaccounted for, the likelihood estimator,
is the only one of the moment estimators that provides unbiased estimates of relatedness for closely inbred individuals (Wang 2007). The remaining four moment estimators are downward biased, and the bias is constant irrespective of the number of loci. When close inbreeding is unaccounted for, the likelihood estimator, 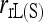 , is even more biased than moment estimators. However, when close inbreeding is taken into account, the likelihood estimator,
, is even more biased than moment estimators. However, when close inbreeding is taken into account, the likelihood estimator, 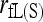 , quickly becomes unbiased with an increasing number of markers. Although unbiased,
, quickly becomes unbiased with an increasing number of markers. Although unbiased, 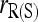 is the least accurate estimator, except when the number of markers is extremely large. This is because, as discussed before,
is the least accurate estimator, except when the number of markers is extremely large. This is because, as discussed before, 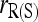 is very sensitive to rare alleles and could yield extreme estimates. Its high MSE is dominated by its high sampling variance. The likelihood estimator is the most inaccurate when close inbreeding is ignored [
is very sensitive to rare alleles and could yield extreme estimates. Its high MSE is dominated by its high sampling variance. The likelihood estimator is the most inaccurate when close inbreeding is ignored [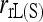 ], but becomes the most accurate when close inbreeding is accounted for [
], but becomes the most accurate when close inbreeding is accounted for [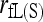 ] and the markers are numerous. With just a few markers (say, ≤20),
] and the markers are numerous. With just a few markers (say, ≤20), 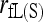 is outperformed by the best moment estimators [
is outperformed by the best moment estimators [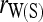 ,
, 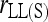 , and
, and 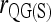 ]. This is because the full-likelihood model is overparameterized and thus is data hungry. Only with sufficient marker information does it give satisfactory estimation of the full set of nine IBD coefficients and thus accurate estimation of inbreeding and relatedness coefficients. This implies that, in the absence of sufficient marker information in practice, inbreeding is better ignored in estimating relatedness. The estimates thus obtained may be slightly biased, but are more accurate than the unbiased estimates obtained by using the full-likelihood method that takes inbreeding into account.
]. This is because the full-likelihood model is overparameterized and thus is data hungry. Only with sufficient marker information does it give satisfactory estimation of the full set of nine IBD coefficients and thus accurate estimation of inbreeding and relatedness coefficients. This implies that, in the absence of sufficient marker information in practice, inbreeding is better ignored in estimating relatedness. The estimates thus obtained may be slightly biased, but are more accurate than the unbiased estimates obtained by using the full-likelihood method that takes inbreeding into account.
Figure 4.—

Bias and MSE of relatedness estimates for full-sibs whose parents are also full-sibs (FSFS) drawn from a subpopulation, as a function of the number of markers. Each marker has 10 alleles with frequencies in a uniform Dirichlet distribution in the entire population, and the frequencies together with the known parameter value of θ = 0.1 are used in estimating relatedness. The symbols in the plot are listed in Table 2.
Comparison of two approaches:
Figure 5 compares the bias and accuracy of different estimators when allele frequencies are unknown but estimated from samples, as obtained from the third set of simulations. As can be seen, three moment estimators, 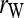 ,
, 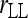 , and
, and 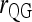 , quickly become unbiased with an increasing sample size for both FS and UR relationships when the direct approach was adopted. In contrast, all estimators are biased for both relationships when the indirect approach was adopted. In general, the direct approach is less biased than the indirect approach for each estimator, except when Ritland's (1996) estimator is used for FS. For the overall accuracy measured by MSE, the direct approach is always better than the indirect approach for all estimators and relationships considered. For UR, MSE increases with an increasing sample size for all estimators in both approaches. This is counterintuitive and occurs because MSE is calculated from relatedness estimates that are truncated to the range of [0, 1]. The proportion of negative estimates increases with a decreasing sample size. The truncation in both likelihood (automatic) and moment (artificial) estimators leads to a decrease in sampling variance and thus a reduction in MSE. Because truncation is more frequent with a smaller sample size, MSE increases with an increasing sample size.
, quickly become unbiased with an increasing sample size for both FS and UR relationships when the direct approach was adopted. In contrast, all estimators are biased for both relationships when the indirect approach was adopted. In general, the direct approach is less biased than the indirect approach for each estimator, except when Ritland's (1996) estimator is used for FS. For the overall accuracy measured by MSE, the direct approach is always better than the indirect approach for all estimators and relationships considered. For UR, MSE increases with an increasing sample size for all estimators in both approaches. This is counterintuitive and occurs because MSE is calculated from relatedness estimates that are truncated to the range of [0, 1]. The proportion of negative estimates increases with a decreasing sample size. The truncation in both likelihood (automatic) and moment (artificial) estimators leads to a decrease in sampling variance and thus a reduction in MSE. Because truncation is more frequent with a smaller sample size, MSE increases with an increasing sample size.
Figure 5.—

Bias and MSE of relatedness estimates for unrelated (UR) and full-sib (FS) individuals drawn from a subpopulation, as a function of sample size (number of individuals) used in estimating allele frequencies. The subpopulations are differentiated with a parameter value of θ = 0.1. Relatedness was estimated using the estimated allele frequencies of either the focal subpopulation (the direct approach, first row) or the entire population together with the parameter value of θ = 0.1 (the indirect approach, second row). In the latter case, allele frequencies were estimated from samples from 10 subpopulations. Ten markers, each having 10 alleles with frequencies in a uniform Dirichlet distribution, are used for estimating relatedness. The plotting symbols for the different estimators are listed in Table 2.
Overall, the direct approach is clearly less biased and more accurate than the indirect approach. This is true even when the simulated parameter value of θ is used in the indirect approach. In reality, however, θ is unknown and has to be estimated from samples. Using estimated rather than true values of θ is expected to make the indirect approach even worse. The main cause of the inaccuracy of the indirect approach is that allele frequencies of the entire population are difficult to estimate. The more differentiated a population is, the more subpopulations need to be sampled to estimate its allele frequencies accurately. Figure 6 illustrates the effect of the number of subpopulations that are sampled to estimate population allele frequencies in the indirect approach. A sample of 80 individuals was taken from each of a number of subpopulations in a population with a differentiation parameter value of θ = 0.1 and was used to estimate the population allele frequencies. The estimated population allele frequencies together with the parameter value of θ = 0.1 are then used to estimate relatedness by different estimators, using 10 markers each having 10 alleles with a uniform Dirichlet frequency distribution. Figure 6 shows that all estimators, except for 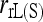 in the case of UR, become less biased with an increasing number of sampled subpopulations. All estimators become more accurate for FS but less accurate for UR with an increasing number of sampled subpopulations. The counterintuitive result with UR is again caused by truncation. MSE always decreases with an increasing number of sampled subpopulations for all five moment estimators, when it is calculated from the original relatedness estimates without truncation (data not shown).
in the case of UR, become less biased with an increasing number of sampled subpopulations. All estimators become more accurate for FS but less accurate for UR with an increasing number of sampled subpopulations. The counterintuitive result with UR is again caused by truncation. MSE always decreases with an increasing number of sampled subpopulations for all five moment estimators, when it is calculated from the original relatedness estimates without truncation (data not shown).
Figure 6.—

Bias and MSE of relatedness estimates for unrelated (UR) and full-sib (FS) individuals drawn from a subpopulation, as a function of the number of subpopulations sampled in estimating population allele frequencies. The subpopulations are differentiated with a parameter value of θ = 0.1. Relatedness was estimated using the estimated allele frequencies of the entire population together with the parameter value of θ = 0.1. Ten markers, each having 10 alleles with frequencies in a uniform Dirichlet distribution, are used for estimating relatedness. The symbols in the plot are listed in Table 2.
DISCUSSION
The coefficients of inbreeding of and relatedness between individuals have an implicit reference population in which they are defined as zero (Rousset 2002). In other words, genes at a locus, whether they are found within or between diploid individuals, are expected to be nonidentical by descent in the reference population. Without this initial condition as reference, it is impossible to measure IBD between genes and thus relatedness between individuals. Therefore, the magnitudes of both inbreeding and relatedness coefficients change with a shift in the reference. For a single isolated population, the relatedness between two individuals increases when the reference moves backward in generations (time). For a subdivided (or structured) population, the relatedness between two individuals in a subpopulation increases as the reference moves backward in time and/or moves from the focal subpopulation to a larger number of subpopulations. In practice, it is the population whose allele frequencies are used in estimating relatedness that acts as the reference. When the same sample is used in estimating both allele frequencies and relatedness, therefore, the average relatedness across all possible pairs of individuals in the sample is expected to be close to zero.
In the real world, the assumption of a large random mating population made by most estimators is rarely satisfied. In a structured population, the relatedness between two individuals within a subpopulation can be measured with respect to the focal subpopulation or the entire population. The former gauges the relatedness due to recent coalescences that occurred within the subpopulation, while the latter gauges the total relatedness due to both recent and ancient coalescences that occurred within the entire population. In this investigation, I focused on the former, since most applications of relatedness are found in fine-scale genetic studies of populations in the immediate or short timescale.
Following the model of Anderson and Weir (2007), I studied the estimation of relatedness of two individuals within a subpopulation and with respect to it. The estimation was made using the allele frequencies of the entire population and the differentiation among subpopulations, θ. Confirming the model of Anderson and Weir (2007), I found that all current estimators assuming a large random mating population are upwardly biased when applied to a structured population. The extent of the bias depends on θ and the estimator (Figure 1). I derived the expectations of different moment relatedness estimators for a marker with an arbitrary allele-frequency distribution, which led to modified moment estimators that are unbiased for a structured population. Simulations confirm that these modified moment estimators are not only unbiased regardless of θ, but also more accurate than the original estimators for all pairwise relationships considered (Figures 1 and 2). The likelihood estimator is upwardly biased for unrelated or slightly related individuals even when population structure is taken into account. This is because the estimator is constrained to the range of [0, 1], and the true relatedness is at or close to the lower bound of the range. Simulations indicate that its accuracy is close to but never substantially higher than that of the best moment estimator for all pairwise relationships. This is true even when hundreds of SSRs or SNPs are used, confirming previous studies (e.g., Lynch and Ritland 1999; Wang 2002). It could become the most accurate estimator when many markers of very different information content are used.
An advantage of the likelihood method is its flexibility. As shown in this study, the likelihood method can be made to estimate the full set of nine IBD coefficients (and thus the inbreeding coefficients of and relatedness between individuals) by accounting for both population structure (pervasive inbreeding) and close inbreeding. In contrast, among the available moment estimators only that of Ritland (1996) can accommodate both types of inbreeding. Unfortunately, this estimator is very sensitive to rare alleles. Although it is among the top estimators for unrelated or slightly related individuals when θ is small, it performs poorly for close relationships such as sibship and parentage or for highly differentiated populations. My simulations in Figure 4 show that with close inbreeding, all estimators that ignore inbreeding become downwardly biased, while estimators that allow for inbreeding are either always unbiased [moment estimator, 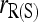 ] or quickly becoming unbiased with an increasing amount of marker information [likelihood estimator,
] or quickly becoming unbiased with an increasing amount of marker information [likelihood estimator, 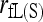 ]. In terms of accuracy,
]. In terms of accuracy, 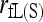 becomes the best only when a sufficient number of markers are used in the analysis. Otherwise, it is less accurate than estimators that ignore close inbreeding. This is because the full-likelihood model has to estimate six more parameters than the reduced (noninbreeding) model. The results imply that, in practical situations where <15 SSRs are available, it is better to ignore close inbreeding even if it is present.
becomes the best only when a sufficient number of markers are used in the analysis. Otherwise, it is less accurate than estimators that ignore close inbreeding. This is because the full-likelihood model has to estimate six more parameters than the reduced (noninbreeding) model. The results imply that, in practical situations where <15 SSRs are available, it is better to ignore close inbreeding even if it is present.
An alternative and direct approach to estimating the relatedness between individuals from a subpopulation with respect to it is to use the allele frequencies of the focal subpopulation. In the common case of unknown allele frequencies, they can be estimated from the same sample that is analyzed for relatedness. Indeed, simulations show clearly that this direct approach is more accurate than the indirect one that uses the entire population as the reference and accounts for population structure by using θ. This is true even when the size of the sample from the focal subpopulation is small (∼40 individuals) and the true value of θ is used in the indirect approach (Figure 5). Estimating the allele frequencies of the entire population is much more difficult than estimating that of a single subpopulation, because sampling errors occur both between and within subpopulations in the former but just within a subpopulation in the latter. Therefore, a large number of individuals from each of a large number of subpopulations must be sampled to obtain accurate allele-frequency estimates of the population (Figures 5 and 6). The sampling of subpopulations is expected to have an especially large impact when the population is highly differentiated. In the real world, a population may have a cryptic structure, may have a continuous geographic distribution, or may have subpopulations with different sizes and extents of differentiation. These complexities make the estimation of allele frequencies of the entire population difficult in practice. It is even more difficult to accurately estimate Fst, because it depends also on factors other than population structure. Highly polymorphic microsatellites usually lead to a much lower estimate of Fst than the biallelic marker SNPs, for example (Hedrick 1999).
APPENDIX
Here I consider the biases of several moment estimators when they are applied to estimating the relatedness of two individuals taken from a large random mating subpopulation of the population whose allele frequencies are used in the estimation. The biases are caused by the population structure, or the background relatedness θ, which is ignored by these estimators. Since the subpopulation is assumed large and with random mating, only three IBD coefficients, 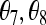 , and
, and 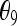 , are relevant.
, are relevant.
Estimator by Queller and Goodnight (1989):
The expected value of estimator (4) can be derived by considering all possible genotype combinations at a locus of two individuals from a subpopulation and averaging their relatedness as calculated by (4). The genotype pairs are in nine IIS modes, as listed in the nine rows of Table 1. For each IIS mode, only three IBD modes D7, D8, and D9, listed in the last three columns of Table 1, need to be considered. Denoting the estimated relatedness between X and Y with genotypes {AiAj} and {AkAl} as 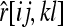 , its expected value is
, its expected value is
 |
(A1) |
where  in which the Kronecker delta variable
in which the Kronecker delta variable  = 1 if a = b and
= 1 if a = b and  = 0 otherwise. Term i on the right side of (A1) corresponds to IIS mode i (i = 1, 2,…, 9). Inserting
= 0 otherwise. Term i on the right side of (A1) corresponds to IIS mode i (i = 1, 2,…, 9). Inserting 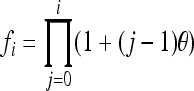 ,
, 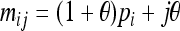 , and (4) into (A1) and after some tedious algebra, I obtain (5), which is identical to that derived by Anderson and Weir (2007) for the special case of equi-frequency alleles.
, and (4) into (A1) and after some tedious algebra, I obtain (5), which is identical to that derived by Anderson and Weir (2007) for the special case of equi-frequency alleles.
Estimator by Lynch (1988) and Li et al. (1993):
Using Table 1, the expected similarity index between individuals X and Y taken from a subpopulation is derived as
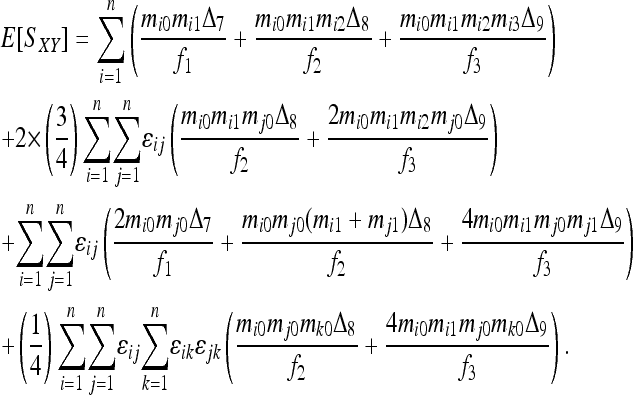 |
(A2) |
Terms 1, 2, 3, and 4 on the right side of (A2) correspond to IIS modes S1, S3 + S5, S7, and S8, respectively. It leads to (8) when 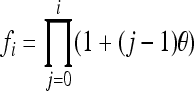 and
and 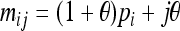 are inserted and is simplified after some tedious algebra.
are inserted and is simplified after some tedious algebra.
Estimator by Ritland (1996):
This estimator was not considered by Anderson and Weir (2007). Similar to the estimator by Queller and Goodnight (1989), the expected value of this estimator is given by (A1), where 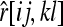 denotes the relatedness estimated by (11) [rather than by (4)] between X and Y with genotypes {AiAj} and {AkAl}. With the same simplifying procedure, the expectation reduces to (12).
denotes the relatedness estimated by (11) [rather than by (4)] between X and Y with genotypes {AiAj} and {AkAl}. With the same simplifying procedure, the expectation reduces to (12).
Estimator by Lynch and Ritland (1999):
The expected value of the Lynch and Ritland estimator is given by (A1), where 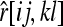 denotes the relatedness estimated by (14) between X and Y with genotypes {AiAj} and {AkAl}. After some tedious algebra, the expectation reduces to (5), the same as the Queller and Goodnight estimator.
denotes the relatedness estimated by (14) between X and Y with genotypes {AiAj} and {AkAl}. After some tedious algebra, the expectation reduces to (5), the same as the Queller and Goodnight estimator.
Estimator by Wang (2002):
The similarity index between the genotypes of individuals X and Y falls into four exclusive categories, with categories 1, 2, 3, and 4 containing genotype pairs that have similarity index values of 1, 0.75, 0.5, and 0, respectively. The expected frequency of category i (i = 1, 2, 3), 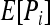 , is derived from Table 1:
, is derived from Table 1:
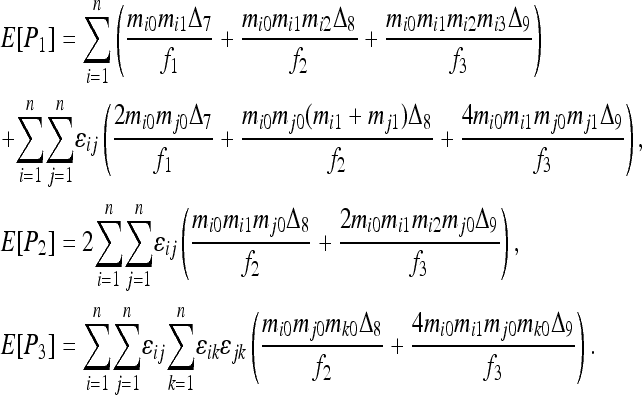 |
Replacing Pi in (15) by its expected value 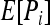 derived above for i = 1, 2, and 3, I obtained (after some algebra) the expected relatedness
derived above for i = 1, 2, and 3, I obtained (after some algebra) the expected relatedness
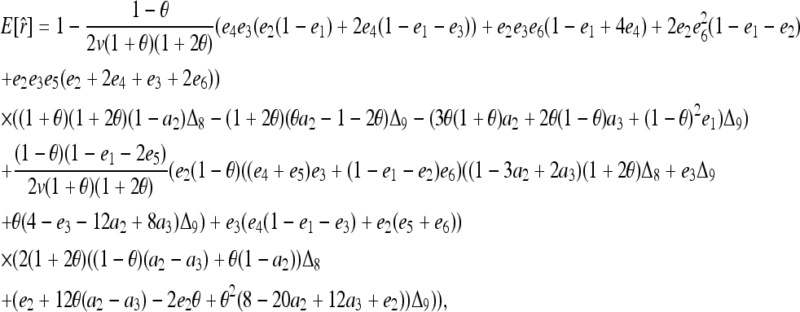 |
(A3) |
where 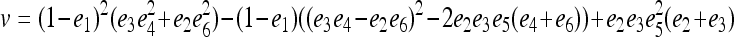 . Inserting
. Inserting 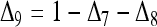 into (A3), it can be rearranged as functions of r =
into (A3), it can be rearranged as functions of r = 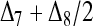 and
and 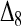 . This means that, when θ > 0, the expectation of the Wang estimator varies with
. This means that, when θ > 0, the expectation of the Wang estimator varies with 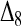 as well as the true relatedness r. However, the coefficient of
as well as the true relatedness r. However, the coefficient of 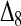 is always very small and can be ignored to obtain an almost unbiased estimator, (16).
is always very small and can be ignored to obtain an almost unbiased estimator, (16).
References
- Anderson, A. D., and B. S. Weir, 2007. A maximum likelihood method for estimation of pairwise relatedness in structured populations. Genetics 176 421–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballou, J. D., and R. C. Lacy, 1995. Identifying genetically important individuals for management of genetic variation in pedigreed populations, pp. 76–111 in Population Management for Survival and Recovery. Analytical Methods and Strategies in Small Population Conservation, edited by J. D. Ballou, M. Gilpin and T. J. Foose. Columbia University Press, New York.
- Bittles, A. H., W. M. Mason, J. Greene and N. A. Rao, 1991. Reproductive behavior and health in consanguineous marriages. Science 252 789–794. [DOI] [PubMed] [Google Scholar]
- Caballero, A., and M. A. Toro, 2000. Interrelations between effective population size and other pedigree tools for the management of conserved populations. Genet. Res. 75 331–343. [DOI] [PubMed] [Google Scholar]
- Csillery, K., T. Johnson, D. Beraldi, T. Clutton-Brock, D. Coltman et al., 2006. Performance of marker-based relatedness estimators in natural populations of outbred vertebrates. Genetics 173 2091–2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedrick, P. W., 1999. Perspective: highly variable loci and their interpretation in evolution and conservation. Evolution 53 313–318. [DOI] [PubMed] [Google Scholar]
- Hinds, D., L. Stuve, G. Nilsen, E. Halperin, E. Eskin et al., 2005. Whole-genome patterns of common DNA variation in three human populations. Science 307 1072–1079. [DOI] [PubMed] [Google Scholar]
- Jacquard, A., 1972. Genetic information given by a relative. Biometrics 28 1101–1114. [PubMed] [Google Scholar]
- Khoury, S. A., and D. Massad, 2005. Consanguineous marriage in Jordan. Am. J. Med. Genet. 43 769–775. [DOI] [PubMed] [Google Scholar]
- Li, C. C., and D. G. Horvitz, 1953. Some methods of estimating the inbreeding coefficient. Am. J. Hum. Genet. 5 107–117. [PMC free article] [PubMed] [Google Scholar]
- Li, C. C., D. E. Weeks and A. Chakravarti, 1993. Similarity of DNA fingerprints due to chance and relatedness. Hum. Hered. 43 45–52. [DOI] [PubMed] [Google Scholar]
- Lynch, M., 1988. Estimation of relatedness by DNA fingerprinting. Mol. Biol. Evol. 5 584–599. [DOI] [PubMed] [Google Scholar]
- Lynch, M., and K. Ritland, 1999. Estimation of pairwise relatedness with molecular markers. Genetics 152 1753–1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch, M., and B. Walsh, 1998. Genetics and Analysis of Quantitative Traits. Sinauer Associates, Sunderland, MA.
- Marshall, H. D., and K. Ritland, 2002. Genetic diversity and differentiation of Kermode bear populations. Mol. Ecol. 11 685–697. [DOI] [PubMed] [Google Scholar]
- Milligan, B. G., 2003. Maximum-likelihood estimation of relatedness. Genetics 163 1153–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Press, W. H., S. A. Teukolsky, W. T. Vetterling and B. P. Flannery, 1996. Numerical Recipes in Fortran 77, Ed. 2. Cambridge University Press, Cambridge, UK.
- Queller, D. C., and K. F. Goodnight, 1989. Estimating relatedness using genetic markers. Evolution 43 258–275. [DOI] [PubMed] [Google Scholar]
- Ritland, K., 1996. Estimators for pairwise relatedness and inbreeding coefficients. Genet. Res. 67 175–186. [Google Scholar]
- Rousset, F., 2002. Inbreeding and relatedness coefficients: What do they measure? Heredity 88 371–380. [DOI] [PubMed] [Google Scholar]
- Van de Casteele, T., P. Galbusera and E. Matthysen, 2001. A comparison of microsatellite-based pairwise relatedness estimators. Mol. Ecol. 10 1539–1549. [DOI] [PubMed] [Google Scholar]
- Wang, J., 2002. An estimator for pairwise relatedness using molecular markers. Genetics 160 1203–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, J., 2007. Triadic IBD coefficients and applications to estimating pairwise relatedness. Genet. Res. 89 135–153. [DOI] [PubMed] [Google Scholar]
- Weir, B. S., 2003. Forensics, pp. 830–852 in Handbook of Statistical Genetics, edited by D. Balding, M. Bishop and C. Cannings. John Wiley & Sons, Chichester, UK.
- Weir, B. S., and C. C. Cockerham, 1984. Estimating F-statistics for the analysis of population structure. Evolution 38 1358–1370. [DOI] [PubMed] [Google Scholar]
- Weir, B. S., L. R. Cardon, A. D. Anderson, D. M. Nielsen and W. G. Hill, 2005. Measures of human population structure show heterogeneity among genomic regions. Genome Res. 15 1468–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weir, B. S., A. D. Anderson and A. B. Hepler, 2006. Genetic relatedness analysis: modern data and new challenges. Nat. Rev. Genet. 7 771–780. [DOI] [PubMed] [Google Scholar]