

Abstract
Neurogenesis is a long and winding journey. A neural progenitor cell migrates long distances, differentiates by forming a single axon and multiple dendrites, undergoes maturation, and ultimately survives. The initial formation of neurites during neuronal differentiation, commonly referred to as “neurite outgrowth,” can be induced by a large repertoire of signals that stimulate an array of receptors and downstream signaling pathways. The Gi/o family of heterotrimeric G-proteins are abundantly expressed in the brain and enriched at neuronal growth cones. Recent evidence has uncovered several Gi/o-coupled receptors that induce neurite outgrowth and has begun to elucidate the underlying molecular mechanisms. Emerging data suggests that signals from several Gi/o-coupled receptors converge at the transcription factor STAT3 to regulate neurite outgrowth and at Rac1 and Cdc42 to regulate cytoskeletal reorganization. Physiologically, signaling through Gi/o-coupled cannabinoid receptors is critical for proper central nervous system development. As the mechanisms by which Gi/o-coupled receptors regulate neurite outgrowth are clarified, it is becoming evident that modulating signals from Gi/o and their receptors has great potential for the treatment of neurodegenerative diseases.
Keywords: G-protein, Neurite Outgrowth, Cell Signaling, Cannabinoid Receptor, Neurodegeneration, Review
2. INTRODUCTION
Neuronal differentiation is a complex process that integrates many signals to drive electrophysiological, morphological, and transcriptional changes (1,2,3,4). This process is characterized by the initial formation of immature neurites, commonly referred to as “neurite outgrowth.” The neurites then further develop into a single axon and multiple dendrites, followed by maturation of the neuron and the formation of dendritic spines (1). Many signals at the cell surface are integrated to shape axonal and dendritic outgrowths as well as their directionality and maturation (5,6,7,8). Not surprisingly, neurite outgrowth is precisely regulated due to its importance in the proper development of the organism. During neurite outgrowth, signals at the membrane are transduced to a large repertoire of enzymes to ultimately trigger changes in gene transcription in the nucleus. In addition, the signals produce vast changes in the actin and microtubule cytoskeletal networks in order to generate and stabilize the growing neurites (1,8).
A diverse array of ligands including neurotrophins, cytokines, hormones, and neurotransmitters can stimulate neurite outgrowth upon binding to their cognate receptors (5,7,9,10,11,12,13,14). Heterotrimeric guanine nucleotide-binding proteins (G-proteins) are one of the most widely used signal transduction systems in mammals, and signaling through the Gi/o family of G-proteins regulates neurite outgrowth. In cortical neurons, dopamine-mediated stimulation of the D2 dopamine receptor can induce axonal neurite elongation (10). Similarly, activation of the serotonin (5-HT) 1B receptor by serotonin can enhance neurite outgrowth in thalamic neurons (11). While the activation of the adenosine 1A receptor in striatal neuronal precursor cells can induce neurite outgrowth (15), the activation of this receptor in hippocampal neurons appears to have an inhibitory role (16). G-proteins and their coupled receptors relay signals from the plasma membrane to downstream effectors in order to shape cell signaling pathways (17,18,19). By doing so, they regulate a vast array of cellular processes including metabolic enzyme activity, ion channel function, motility, transcription, and differentiation (17,18,19,20). This review will focus on signals from Gi/o-coupled receptors that regulate neurite outgrowth during neuronal differentiation and their physiological and pathological implications.
3. Gi/o SIGNALING
The molecular signals of many hormones, neurotransmitters, and chemokines are converted into intracellular responses by G-protein coupled receptors (17,18,19). These receptors generally consist of a seven transmembrane protein that is associated with a heterotrimeric G-protein which, as its name implies, is composed of three subunits: an alpha subunit (Gα) containing a Ras-like domain that can strongly bind the guanine nucleotides GTP and GDP, and beta and gamma subunits (Gβγ) that function as a dimer and cannot be dissociated under non-denaturing conditions (21,22). G-proteins not only process and sort signals but also define the sensitivity to the signal. As a result, G-protein signaling modulates many important physiological functions such as the pacemaker activity in the heart, development, and learning and memory (17,20,23). Heterotrimeric G-proteins act as molecular switches (Figure 1). In the inactive conformation, Gα is bound to GDP at the seven transmembrane receptor and associates with the Gβγ dimer, which prevents the association of the α subunit with downstream effectors. Upon ligand binding to the seven transmembrane receptor, GDP is exchanged for GTP on the α subunit. This switches Gα to the active conformation and results in the release of Gα and Gβγ from the receptor. The conformational change in Gα also leads to its dissociation from Gβγ and both molecules signal to downstream partners. The switch is turned off by the intrinsic GTP hydrolysis (GTPase) activity of Gα, which leads to its reassociation with Gβγ and the receptor. Two families of proteins, the regulators of G-protein signaling (RGS) and the activators of G-protein signaling (AGS), provide another layer of regulation of heterotrimeric G-protein signaling. RGS proteins greatly enhance the intrinsic GTPase activity of Gα and act as GAPs (GTPase activating proteins) specific for Gα (24). Thus, RGS proteins are crucial in inactivating G-protein signaling and are important regulators of the many processes controlled by G-proteins. In contrast, AGS proteins can activate G-proteins by two mechanisms, and their mode of action has not been fully clarified (25). While some AGS proteins such as AGS1 promote the exchange of GTP for GDP (26), others activate G-proteins independently of nucleotide exchange. AGS3, for instance, binds to GDP-bound Gα and appears to prevent Gα from reassociating with Gβγ (27,28).
Figure 1.
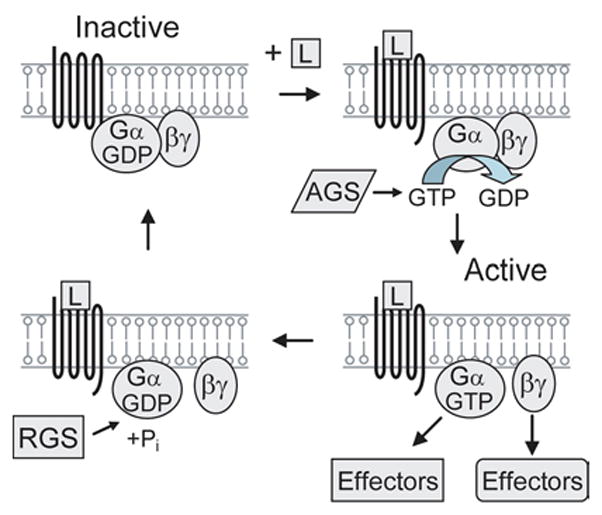
Heterotrimeric G-protein mechanism. The activation of the G-protein coupled receptor by a ligand (L) causes the exchange of GDP for GTP on the α subunit. This switches Gα to the active conformation and results in the release of Gα and Gβγ from the receptor to signal to downstream effectors. The switch is turned off by the intrinsic GTP hydrolysis (GTPase) activity of Gα, which leads to its reassociation with Gβγ and the receptor. The regulators of G-protein signaling (RGS) play key roles in inactivating G-protein signaling. The activators of G-protein signaling (AGS) activate G-proteins by several mechanisms.
Heterotrimeric G-proteins transduce many diverse signals into a wide assortment of cellular responses by coupling to their downstream effectors (23). Over 20 different Gα proteins have been identified in the mammalian heterotrimeric G-protein superfamily, and the α subunits can be divided into four main families: Gs, Gq, Gi/o, and G12/13 (20,29,30). Several Gα proteins (Gs, Gi, and Gq) are expressed ubiquitously, while others such as Go are restricted to specific tissues. Eight members of the Gαi/o family have been identified: Gαi1, Gαi2, Gαi3, Gαo, Gαz, Gαgust, Gαt-c, and Gαt-r. All of the members of the Gαi/o family except Gαz are pertussis toxin sensitive. Several bacterial toxins including pertussis toxin disrupt the G-protein cycle shown in Figure 1 and are useful in dissecting the mechanism of G-proteins. These toxins ADP ribosylate the α subunit of specific G-proteins (31). For Gαi/o, treatment with pertussis toxin leads to uncoupling of the receptor and the G-protein and inactivation of signaling. Under normal conditions, the binding of an extracellular ligand to its receptor results in activation of signaling pathways downstream of the G-proteins in order to elicit the desired cellular response (21). It is often difficult to functionally separate the downstream pathways activated by Gαi and Gαo. As a result, they are commonly named together as Gαi/o.
In contrast to the well characterized pathways transduced by other G-proteins such as Gs, the mechanisms by which Gi/o signals to its downstream effectors and induces cellular responses are not as well understood. The ubiquitously expressed Gαi family members have been shown to inhibit several adenylyl cyclases (32). Gαo is expressed abundantly in the brain and central nervous system, and it constitutes 0.5% of membrane protein in neurons (33,34,35). Gαo is also expressed in lower levels in the pituitary, heart, and pancreas (33). In cellular and biochemical experiments, the Gβγ subunit of Go has been shown to regulate numerous cellular processes including the inhibition of neuronal voltage-dependent Ca2+ channels (N-, P/Q-, and R-type) and the activation of inward rectifying K+ channels (36,37,38). Go has also been shown to activate phospholipase C (PLC)-β (39), adenylyl cyclases 2 and 4 (40), and phosphoinositol-3-kinase (PI3K)-β and -γ (41,42); it has inhibitory effects on adenylyl cyclase 1 (40) and several voltage-gated Ca2+ channels (43,44).
In contrast, the function of the α subunit of Go has remained elusive despite the fact that it is the most abundant G-protein in the brain. In Chinese Hamster Ovary (CHO) cells, Gαo was shown to signal to p42/44 mitogen activated protein kinase (MAPK) through protein kinase C (PKC), although the functional significance of this was not known (45). It was also determined that Gαo was highly expressed in the neuronal growth cone (46), the specialized structure at the tip of the growing neurite that is generated during neuronal differentiation (47,48). Strittmatter et al. (49) extended this finding by demonstrating that an activated mutant of Gαo can induce neurite outgrowth in PC12 cells. The generation of Gαo-deficient mice provided further insight into its function (50,51). Gαo knockout mice were smaller than their wild-type littermates and displayed a marked decrease in lifespan. They also exhibited hypersensitivity to pain, severe motor impairment, and occasional tremors and seizures. Still, the animals were hyperactive and displayed abnormal motor behavior as they ran in circles for extended periods of time. Studies of Gαo knockout mice also indicated that Gαo is important for muscarinic inhibition of L-type Ca2+ channels in the heart (50) and the regulation of Ca2+ and K+ channels in hippocampal neurons (52). Finally, Gαo is required for cell survival in the accessory olfactory system, as the loss of Gαo caused neuronal apoptosis and a decrease in Go receptor containing neurons in the basal vomeronasal organ (53).
Only recently have studies begun to elucidate the mechanistic details of Gαo signaling, and it has proven difficult to identify direct effectors of Gαo. Several studies have demonstrated that Gαo signaling activates a Src-STAT3 pathway that triggers cell transformation in NIH-3T3 fibroblasts (54,55). In addition, Gαi2 is important for inducing cell transformation in a Src- and STAT3-dependent manner in NIH-3T3 cells when these cells are transfected v-fms, an oncogenic form of colony stimulating factor (CSF)-1 receptor (56). In both of these cases, direct effectors of Gα remained unknown. To identify proteins that directly interact with Gαo, our laboratory utilized a yeast two hybrid system (57). We found that Gαo interacted with a Gz-GTPase activating protein (Gz-GAP), RGS protein 17 (RGS-17), a GTPase protein for the small G-protein Rap1 (Rap1GAPII), and the G-protein regulator of neurite outgrowth (GRIN). Gz-GAP (also called RGSZ1) plays a role in attenuating mu opioid receptor inhibition of cAMP (58). RGS-17 has been shown to regulate Gi/o, Gz, and Gq signaling (59,60). Rap1GapII also interacts with Gαi2, and this interaction leads to a decrease in Rap1 activity (61). In thyroid cells, withdrawal of thyroid-stimulating hormone causes Rap1GAPII proteasomal degradation that is glycogen synthase kinase (GSK)-3β dependent (62). The interaction between Gαo and Rap1GAPII was confirmed in vitro and in cultured cells, and Rap1GAPII appears to preferentially bind to the inactivated form of Gαo (63). In the case of Gαo, the interaction with Rap1GAPII leads to the activation of Rap1. Expression of Gαo induces the degradation of Rap1GAPII and treatment with proteasomal inhibitors blocks this effect, suggesting the degradation is ubiquitin-proteasome dependent. GRIN was concurrently identified in a mouse embryo expression library screen for proteins that bound to GTPγS-bound Gαz (64). Two GRIN family members GRIN1 and GRIN2 bind specifically to Gαo, and GRIN1 also binds to Gαi and Gαz. Similar to Gαo, GRIN1 and 2 are also enriched in neuronal growth cones. The roles of Rap1GAPII and GRIN in Go signaling during neurite outgrowth are discussed below. The ligands that stimulate Gi/o-coupled receptors and the effectors that are activated are summarized in Figure 2.
Figure 2.
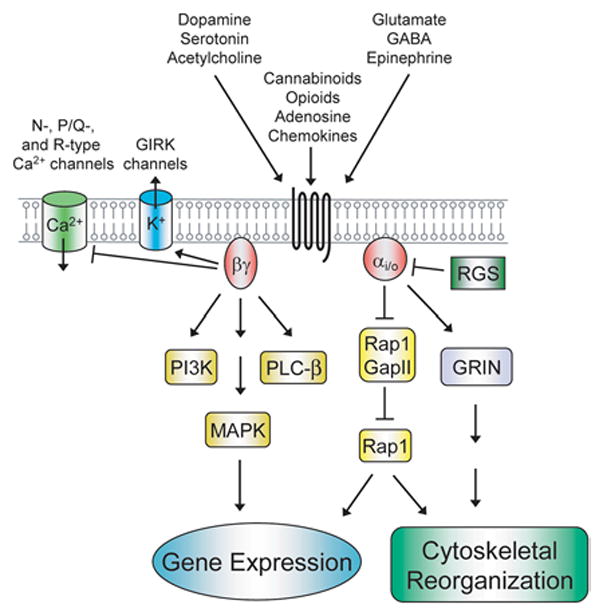
Effector pathways activated by Gi/o signaling. Signals from a wide array of hormones, neurotransmitters, and chemokines are transduced into intracellular responses by Gi/o-coupled receptors. Depicted are pathways that are stimulated by Gα and Gβγ and lead to changes in gene expression and cytoskeletal reorganization. See text for further details. GIRK, G-protein-coupled inward rectifying potassium channels.
4. INDUCTION OF NEURITE OUTGROWTH BY Gi/o
The identification of direct interactors with the α subunit of Go raised several questions. What ligands might stimulate these interactions? What downstream signaling pathways are subsequently activated? What is the cellular functional output of the signaling cascades? It was likely that Gαo signaling stimulated neurite outgrowth. Gαo and one of its interactors GRIN are both enriched in the growth cone of neurites (46,64). The collapse of the growth cone, which occurs when the extending neurite contacts variety of molecules (65,66), can be inhibited by pertussis toxin (67). Additionally, the expression of an activated mutant of Gαo as well as the expression of GRIN induced neurite outgrowth (49,64). Rap1 had also been implicated in neurite outgrowth. Stable expression of the Src family member tyrosine kinase GTK stimulated neurite outgrowth in PC12 cells (68). This appears to be induced by signaling to focal adhesion kinase (FAK) through CrkII, and this in turn leads to phosphorylation of the adaptor protein Shb. Expression of Rap1GAP in this context inhibits GTK-induced neurite outgrowth, linking Rap1 to this pathway. Finally, NGF and EGF can induce neurite outgrowth in a Rap1 dependent manner in PC12 cells that overexpress the adaptor Shb (69).
Neurite outgrowth had been shown to be induced by several ligands that activate Gi/o-coupled receptors. Dopamine stimulation of the D2 dopamine receptor induces neurite outgrowth in cortical neurons (10) and serotonin activation of the serotonin-1B receptor enhances neurite outgrowth in thalamic neurons (11). In these cases, however, the effectors and signaling pathways downstream of Gαo had not been elucidated. Recently, our laboratory demonstrated that the cannabinoid receptor 1 (CB1R) stimulates Gαo and activates downstream signaling converging on STAT3 that ultimately leads to neurite outgrowth in Neuro2A cells (14,63). In order to induce neurite outgrowth, signals from Gαo are transduced to the nucleus to regulate gene expression and to the actin and microtubule cytoskeletal networks. Our current knowledge of Gαo signaling to nucleus during neurite outgrowth is shown in Figure 3 and is discussed below. The following two sections will focus primarily on signal flow through the Gi/o-coupled CB1R, which is one of the best described systems for Gi/o-mediated neurite outgrowth.
Figure 3.
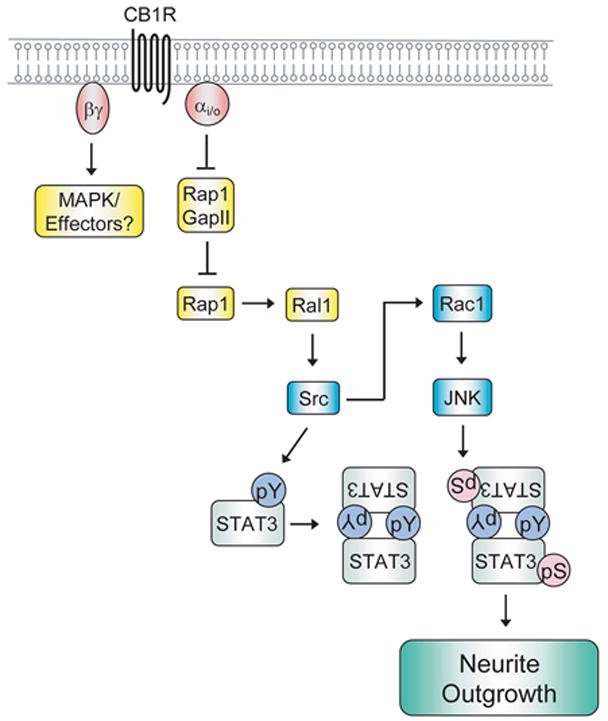
Gi/o signaling to the nucleus during the induction of neurite outgrowth. Signal flow emanating from stimulation of the Gi/o-coupled cannabinoid receptor 1 (CB1R) to the activation of the transcription factor STAT3 is depicted in the schematic. It is likely that Gβγ also signals to downstream effectors to change patterns of gene expression, possibly through p42/44 mitogen activated protein kinase (MAPK). See text for further details. pY, phospho-tyrosine; pS, phospho-serine.
4.1. Signaling to the nucleus
Heterotrimeric G-proteins transduce signals from the plasma membrane to the nucleus to change patterns of gene expression. During the past few years, several studies have begun to clarify the mechanisms by which activation of the Gi/o-coupled CB1R induces neurite outgrowth. Stimulation of CB1R with HU-210, a potent agonist for this receptor, induces neurite outgrowth (63). Either siRNA targeting Rap1 or dominant negative Rap1 is able to block this effect. This is consistent with the finding that activation of CB1R stimulates the interaction between Gαo and Rap1GAPII, which results in the ubiquitin-proteasome mediated degradation of Rap1GAPII. Treatment with pertussis toxin or lactacystin also blocks Rap1GAPII degradation and inhibits CB1R-mediated neurite outgrowth. These results suggest that CB1R stimulates neurite outgrowth via the activation of Rap1 primarily through the attenuation of its inhibition by Rap1GAPII. However, the mechanistic details of Rap1GAPII degradation remain to be determined. Future studies will address whether Gαo bridges an interaction between a currently unknown ubiquitin E3 ligase and Rap1GAPII. It is also possible that the binding to Gαo causes a conformational change in Rap1GAPII which enables it to be recognized and degraded by the ubiquitin proteasome system.
Downstream of the activation of Rap1, Gi/o-coupled receptors activate Src and STAT3 (14). Both Src and STAT3 are phosphorylated in response to CB1R activation by HU-210, and this activation is pertussis toxin sensitive. In addition, dominant negative Src and STAT3 both strongly inhibit CB1R-mediated neurite outgrowth. Src and STAT3 appear to be activated by Rap1 as dominant negative Rap1 inhibits the phosphorylation of both proteins. Dominant negative Ral1 also inhibits Src and STAT3 activation and neurite outgrowth, suggesting a role for Ral in this pathway. Ral1 acts downstream of Rap1, as dominant negative Ral1 inhibits activated Rap-mediated neurite outgrowth. Src is downstream of Ral1 since dominant negative Ral1 does not inhibit STAT3 activation mediated by v-Src. CB1R activation also stimulates the small GTPase Rac1 (discussed below in “4.2. Signaling to the cytoskeleton”) and c-Jun N-terminal kinase (JNK). Both of these proteins are activated downstream of Src, and JNK enhances STAT3 activation. Lastly, inhibition of JNK blocked the activation of STAT3 by Src and CB1R as well as CB1R-mediated neurite outgrowth.
In addition to CB1R signaling, activation of the Gi/o-coupled Serotonin (5-HT) 1 receptor induces neurite outgrowth in Neuro2A cells transfected with the 5-HT1A receptor and enhances cell survival in SK-N-SH cells endogenously expressing the receptor (70). Similar to CB1R, neurite outgrowth induced by the 5-HT1 receptor was pertussis toxin sensitive, required Rap1, and correlated with the phosphorylation of Src and STAT3. Dominant negative STAT3 strongly inhibited neurite outgrowth, whereas inhibition of p42/44 MAPK and PI3K led to a partial inhibition. Thus, it appears that several ligands that bind to distinct Gi/o-coupled receptors may induce neurite outgrowth by similar mechanisms.
While the signaling networks emanating from the CB1R and 5-HT1 receptors provide insight into Gi/o signaling during neurite outgrowth, it also opens several new avenues to explore. The genes that are transcriptional targets of STAT3 during neurite outgrowth remain to be identified. It is likely that CB1R stimulation leads to the activation of other transcription factors in addition to STAT3 and that different cell types may use different effectors to induce neurite outgrowth. STAT3 appears to play an important role in neurite outgrowth in response to Gi/o signaling in Neuro2A and SK-N-SH cells, but its contribution to neurite outgrowth in PC12 cells is less clear. IL-6 stimulation induces neurite outgrowth in PC12 cells in a STAT3 dependent manner, and co-treatment of IL-6 and the neurotrophin NGF (nerve growth factor) leads to synergistic neurite outgrowth (12,71). However, a separate study found p42/44 MAPK activation downstream of the IL-6 receptor is essential for neurite outgrowth, while STAT3 appeared to have a negative regulatory role on neurite outgrowth (72). Recently, NGF stimulation has been shown to lead to the activation of STAT3 DNA binding and transcriptional activities (73). Inhibition of STAT3 expression led to a decrease in NGF induced gene transcription in PC12 cells and a decrease in neurite outgrowth induced by brain-derived neurotrophic factor (BDNF) in primary hippocampal neuron cultures. Similar to STAT3, the role of p42/44 MAPK in neurite outgrowth may vary with cell type. While stimulation of CB1R activates p42/44 MAPK in Neuro2A cells, its role in neurite outgrowth is unclear as p42/44 MAPK does not appear to be required for neurite outgrowth (14). However, p42/44 MAPK does appear to have an important role in response to 5-HT1 receptor signaling during neurite outgrowth (70). While p42/44 MAPK can also be activated by signaling through other Gi/o-coupled receptors such as the M1 muscarinic acetylcholine receptor and the platelet-activating factor receptor (45), the functional relevance is not known. Thus, it appears likely that different signaling pathways may mediate similar effects in different cell types. Future studies will be necessary to elucidate the underlying molecular mechanisms and to delineate how multiple signals are integrated into transcriptional responses during neurite outgrowth. In addition to the α subunit, Gβγ also signals to downstream effectors upon receptor stimulation. The βγ subunit can activate several effector pathways, including MAPK, PI3K, and PLC (74). Currently, the contribution of βγ and its effector pathways that may be activated during neurite outgrowth remain to be determined.
4.2. Signaling to the cytoskeleton
During neurite outgrowth, the actin and microtubule cytoskeletal networks work in a coordinated fashion to generate and stabilize the growing neurites (1,8). The actin cytoskeleton reorganizes to allow formation of the growth cone and the microtubules re-align into bundles to stabilize the growing neurite. The Rho family of small G-proteins plays a key role in the cytoskeletal reorganization that occurs during the initiation, guidance, and elongation of the neurite (1). Twenty two genes encoding mammalian Rho GTPase family members have been identified (1,75). The three best described Rho GTPases RhoA, Rac1, and Cdc42 regulate the cytoskeleton and a myriad of cellular functions including cell polarity, gene transcription, cell cycle, enzyme activity, apoptosis and vesicle transport (75,76). On the biochemical level, Rho regulates the actin-myosin contractile apparatus and stress fiber formation, Rac regulates the formation of web-like lamellopodia protrusions and membrane ruffling, and Cdc42 regulates the protrusions of finger-like filopodia. In order to generate the growth cone and initiate neurite formation, signals are transduced at the membrane through the Rho family of GTPases to the actin cytoskeleton. In general, neurite outgrowth is stimulated by Rac and Cdc42 activation, while Rho activation appears to promote neurite retraction (1). The mechanisms by which Gi/o may signal to the actin cytoskeleton are discussed below and summarized in Figure 4.
Figure 4.
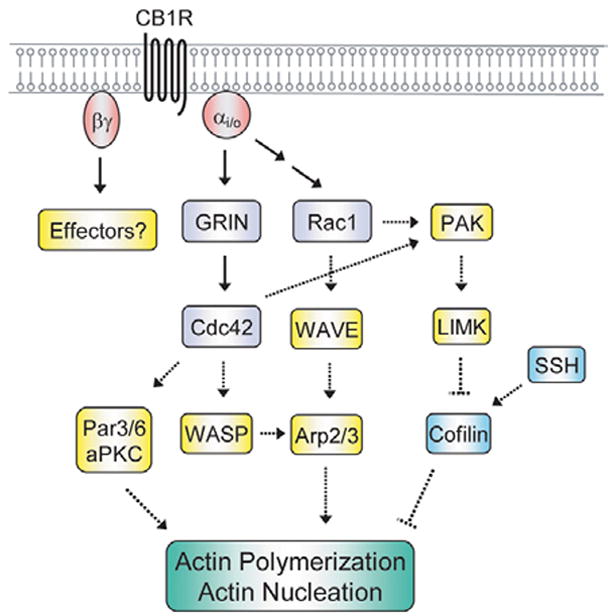
Gi/o signaling to the actin cytoskeleton during the induction of neurite outgrowth. Signaling from the Gi/o-coupled CB1R to its effectors GRIN, Cdc42 and Rac1 and their potential downstream targets is shown in the schematic. The intermediate molecules between CB1R and Rac1 have been omitted for clarity. It is likely that Gβγ also signals to yet to be identified downstream effectors to reorganize the actin cytoskeleton. Known interactions are depicted by solid arrows, putative interactions by dashed arrows. See text for further details. PAK, p21-activated kinase; SSH, Slingshot; WASP, Wiskott-Aldrich-syndrome protein.
The molecular mechanism of Gi/o stimulated cytoskeletal signaling is just beginning to be elucidated. The Gαo interactor GRIN is enriched in the growth cone, and co-expression of GRIN with an activated mutant of Gαo enhances neurite outgrowth in Neuro2A cells (64). Co-expression of these two proteins also leads to activation of Cdc42, and neurite outgrowth under these conditions is blocked by dominant negative Cdc42 and dominant negative Rac1 (77). It is possible Cdc42 may mediate its effects by activating the Par6-Par3-atypical PKC (aPKC) complex (78). In hippocampal neurons that are plated in culture, Cdc42 and the Par6-Par3-aPKC complex are enriched in the developing axon. Alterations in this signaling axis lead to defects in neuronal differentiation, causing cells to produce either no or multiple axons (79). Other signaling molecules may also be downstream of Cdc42, and the signals that lead to Cdc42 activation and the downstream molecules that lead to cytoskeletal reorganization remain to be elucidated.
Our laboratory has linked Rac1 to neurite outgrowth induced by CB1R activation (14). Stimulation of CB1R leads to the activation of Rac1 that is Src dependent and expression of dominant negative Rac1 inhibits neurite outgrowth in Neuro2A cells. Rac1 is important for JNK activation, which in turn enhances Stat3 transcriptional activation and neurite outgrowth. In addition to these effects, Rac1 is likely to play an important role in cytoskeletal signaling during CB1R-mediated neurite outgrowth. Both Rac1 and Cdc42 can activate the p21-activated kinase (PAK) family of serine/threonine kinases (1,75). PAK can signal to the actin cytoskeleton by phosphorylating and activating the LIM kinases (LIMKs), which in turn phosphorylate and deactivate cofilin and its closely related protein, actin depolymerizing factor (ADF). Cofilin and ADF act by stimulating the depolymerization and severing of actin filaments (80,81) and their phosphorylation at Serine-3 by LIMKs results in actin polymerization. The Slingshot (SSH) phosphatases alleviate the inhibition of cofilin and ADF through dephosphorylation (82). Cofilin and ADF as well as their regulators LIMKs and SSHs are enriched in neuronal growth cones (1,83,84,85), and thus have been implicated in regulating actin cytoskeletal reorganization during neurite outgrowth. This regulation of actin dynamics is tightly regulated as the inhibition of cofilin/ADF, LIMK, or SSH inhibits neurite outgrowth (86). Rac1 and Cdc42 also can mediate their effects through the Wiskott-Aldrich-syndrome family of proteins (WASP). Rac1 can indirectly trigger the WASP family member WAVE to activate Arp2/3, which stimulates actin nucleation and the formation of new actin filaments (87,88). Similarly, Cdc42 can directly activate WASP to interact with and stimulate Arp2/3 (89). Future studies will examine whether Gi/o-coupled receptor stimulation activates Cdc42 and Rac1 to regulate PAK signaling to cofilin/ADF and WASP signaling to Arp2/3.
Might Gi/o also regulate RhoA activity during neurite outgrowth? The Gi/o effector Rap1 has been shown to activate the RhoGAP RA-RhoGAP (90). RA-RhoGAP is localized in the neuronal growth cone and Rap1 enhances the GAP activity of RA-RhoGAP toward RhoA. Rap1 activation of RA-RhoGAP also promotes neurite outgrowth in NG108 cells. Overexpression of another RhoGAP, p190 RhoGAP can induce neurite outgrowth in Neuro2A cells (91). In the developing and mature nervous system, p190 RhoGAP is phosphorylated by Src. This modification is likely to activate p190 RhoGAP, as phosphorylation of p190 RhoGAP is important for its binding to RhoA (92). It remains to be determined whether signals from Gi/o modulate RhoA during neurite outgrowth and whether Rap1 or other signaling components regulate this activity. The precise mechanisms of actin cytoskeletal regulation by Gi/o as well as deciphering Gi/o signaling to the microtubule network will be addressed by future studies.
5. PHYSIOLOGICAL AND PATHOLOGICAL IMPLICATIONS
5.1. Cannabinoid signaling during development
Cannabinoid signaling has important functions during central nervous system (CNS) development and in the postnatal brain. Endogenous cannabinoids (endocannabinoids) regulate synapse activity postnatally and regulate progenitor cell proliferation and the migration, differentiation, and survival of neurons during development (93,94). Recent studies of endocannabinoid ligands, their receptors, and downstream signaling have begun to clarify the mechanisms of cannabinoid signaling in vivo and provide insight into how exogenous cannabinoids affect these functions and may be used as therapeutics for several chronic diseases. The mechanisms by which endocannabinoids signal through the CB1R to shape CNS development are discussed below.
The endocannabinoids anandamide (AEA) and 2-arachidonoylglycerol (2-AG) are endogenous ligands for the Gi/o-coupled CB1R (reviewed in (94)). In the developing mouse CNS, CB1R expression appears to begin around embryonic day 11 (E11) in early progenitors and increases throughout the whole brain until birth. At E13.5, CB1R is expressed on pyramidal cells in the hippocampus and developing cerebral cortex primarily on distal parts of the axons (95). CB1R is also highly expressed in migrating GABAergic interneurons in the hippocampus and cortex. By E18, migrating GABAergic interneurons express CB1R on axons and at axonal growth cones (95). In this context, signaling through CB1R helps to shape axon guidance in the developing cortex. A recent study has determined that CB1R-deficient mice display defects in post-synaptic targeting in cortical interneurons (95). Endocannabinoids may regulate axon guidance by causing the internalization and retrograde transport of CB1R, which is concomitant with the activation of p42/44 MAPK. RhoA is also activated and its activation induces growth cone collapse. Inhibition of the Rho effector ROCK during this process can convert repulsion and collapse of the growth cone into chemoattraction. Thus, there may be fine-tuning of endocannabinoid signaling to precisely regulate axonal target selection. In addition to regulating synaptic targeting, endocannabinoid signaling plays an important role in neuronal migration and morphogenesis. During morphogenesis, GABAergic cortical interneurons migrate long distances before reaching specific cortical layers, and it has been shown that BDNF helps determine the specification of cortical interneurons (96,97). The endocannabinoid AEA cooperates with BDNF to induce interneuron migration by transactivating the TrkB receptor through Src kinase activation (98). In fact, CB1R stimulation leads to TrkB receptor activation in a Src-dependent manner in the absence of BDNF, and CB1R and TrkB form complexes during AEA stimulation. In contrast, AEA inhibits BDNF-induced differentiation. Thus, signaling through CB1R helps to govern BDNF-induced migration and differentiation during corticogenesis.
Signaling through CB1R also regulates neural progenitor cell proliferation and differentiation. Proliferation of progenitor cells is promoted by CB1R activation, and this effect is attenuated in CB1R knockout mice (99,100). Endocannabinoids also regulate differentiation of progenitors into glia as loss of CB1R negatively impacts gliogenesis in vivo (100). In contrast, CB1R signaling may inhibit progenitor differentiation into neurons (101). Treatment with AEA inhibits cortical neuron progenitor differentiation and NGF-induced differentiation in PC12 cells. CB1R stimulation may inhibit neural progenitor cell differentiation by dampening signaling through the Rap1-B-Raf-p42/44 MAPK pathway as AEA blocked sustained p44/22 MAPK activation. Thus, in contrast to morphogenesis, CB1R stimulation can block Trk receptor activation during differentiation. The importance of proper regulation of endocannabinoid signaling during development is underscored by the prenatal exposure to Δ9-tetrahydrocannabinol (THC), the major psychoactive component of marijuana (Cannabis sativa). Several studies have indicated that maternal consumption of marijuana influences behavior in offspring, causing hyperactivity, inattention and cognitive defects by adolescence (102,103,104). THC also increases the synthesis and release of endocannabinoids in a pertussis sensitive manner, thus linking Gi/o-coupled receptors to its effects (105). In the developing hippocampus, blocking CB1R stimulation can lead to epileptic discharges, while over-activation can reduce network activity (106). Disturbing this balance has the potential for profound consequences during development. Indeed, sustained prenatal exposure to THC in vivo increases the density of CB1R-positive GABAergic interneurons in the hippocampus (98). These studies demonstrate that THC can alter the patterning of CB1R expressing cells and provide the framework to delineate the mechanism by which prenatal exposure to THC causes long-lasting effects.
5.2. Neurodegeneration
Beyond its regulation of neurite outgrowth and neurogenesis during development, signaling through Gi/o and its coupled receptors may possess neuroprotective and anti-inflammatory properties. Cannabinoid receptor stimulation can protect hippocampal neurons from excitotoxicity (107,108,109). Furthermore, CB1R stimulation in vivo decreases hippocampal neuron loss after cerebral ischemia and acute brain trauma (110). Cannabinoids may also help protect neurons from alteration in glucose levels (111). Hyperglycemia plays a critical role in the development and progression of diabetic neuropathy, which is a complication that arises from diabetes mellitus and causes cognitive deficits. In a mouse model of diabetes, cannabinoid treatment ameliorates the cognitive impairment in these mice without altering the underlying hyperglycemia. However, these effects appear to be independent of CB1R, as the CB1R antagonist SR141716A did not affect the cognitive improvements (112). Cannabinoids are also important in modulating inflammation (113) and are being pursued as potential therapies for obesity (114,115). In addition, signaling through several Gi/o-coupled receptors has been shown to modulate a spectrum of neurodegenerative disease and may represent an attractive target for therapeutic intervention (116,117). The roles of Gi/o-coupled CB1R and adenosine receptors in neurological disorders are discussed below and summarized in Table 1.
Table 1.
Potential therapeutics targeting Gi/o-coupled receptor signaling for the treatment of neurological disorders
| Compound(s) | Gi/o-coupled Receptor | Indication | Mechanism of Action | Clinical Status | Reference |
|---|---|---|---|---|---|
| 2-AG1; WIN 55212-2 | CB1R | Cerebral ischemia/acute brain trauma | Receptor agonists | Animal Model | 110,111 |
| HU-210 | ND6 | Diabetic neuropathy | CB1R independent | Animal Model | 112 |
| Δ9-THC2 | CB1R | Parkinson’s Disease | Receptor agonist | Animal Model | 119 |
| Cannabidiol | ND | Parkinson’s Disease | CB1R independent | Animal Model | 119 |
| HU-210; WIN 55212-2; JWH-133 | CB1R | Alzheimer’s Disease | Receptor agonists | Animal Model | 123 |
| VDM-11 | CB1R | Alzheimer’s Disease | endocannabinoid cellular reuptake inhibitor | Animal Model | 124 |
| Δ9-THC; WIN 55212-2 | CB2R | Amyotrophic Lateral Sclerosis | Receptor agonists | Animal Model | 121,122 |
| Δ9-THC | CB1R | Multiple Sclerosis | Receptor agonist | Animal Model | 125 |
| OMDM1; OMDM2 | CB1R | Multiple Sclerosis | endocannabinoid cellular reuptake inhibitors | Animal Model | 126,127 |
| CP55,940 | CB1R | Huntington’s Disease | Receptor agonist | Animal Model | 128 |
| AM404 | CB1R | Huntington’s Disease | endocannabinoid cellular reuptake inhibitor | Animal Model | 128,129 |
| FR194921 | A1AR | Anxiety Dementia | Receptor antagonist | Animal Model | 137 |
| T-62 | A1AR | Migraine headaches, chronic pain | Allosteric enhancer | Phase II Clinical Trials in U.S.A. | 140 |
| ADAC3; NNC 21-0041,9; NNC 90-1515,4 | A1AR | Cerebral ischemia | Receptor Agonists | Animal Model | 142–144 |
| IB-MECA4 | A3AR | Cerebral ischemia | Receptor agonist | Animal Model | 145 |
| CHA5; adenosine | A1AR | Seizure | Receptor agonists | Animal Model | 147,150 |
2-AG, 2-arachidonoylglycerol;
Δ9-THC, delta9-tetrahydrocannabinol;
ADAC, adenosine amine congener;
IB-MECA, N6-(3-iodobenzyl)adenosine-5′-N-methylcarboxamide;
CHA, N6-cyclohexyladenosine;
ND, not determined
Even though the endocannabinoid system is just beginning to be unraveled, it is being pursued as a therapy for many CNS disorders. In addition to cannabinoid signaling, several drugs targeting cannabinoid metabolism have shown efficacy at alleviating the symptoms of several animal models of neurodegenerative diseases (reviewed in (117,118). In Parkinson’s disease, CB1R agonists can prevent dopaminergic neuron loss (119). However, agonist-mediated stimulation of non-cannabinoid receptors can also promote neuronal apoptosis, demonstrating the need for strong receptor selectivity in potential therapeutic applications (120). Similarly, CB1R stimulation is detrimental to motor neuron survival in Amyotrophic Lateral Sclerosis while activation of the related family member type 2 cannabinoid receptor (CB2R) is neuroprotective (121,122). In Alzheimer’s disease, stimulation of CB1R is protective against neurodegeneration (123). A similar prevention of degeneration is also observed by inhibiting the catabolism of AEA, thus increasing the pool of available endocannabinoids (124). Activation of CB1R is neuroprotective in animal models of multiple sclerosis and animals with CB2R genetically ablated in T cells display a severe disease phenotype (125). Neuroprotective effects in multiple sclerosis disease models are also observed by inhibiting the catabolism of AEA (126,127). Lastly, CB1R agonists as well as inhibitors of cannabinoid catabolism exhibit neuroprotective properties in models of Huntington’s disease (118,128,129). As our understanding of the endocannabinoid system and the molecular details of the downstream signaling from the cannabinoid receptors continues to expand, new agents that selectively target the signaling through specific receptors as well as the synthesis and degradation of endocannabinoids will be developed for the treatment of neurodegenerative diseases.
Similar to CB1R, the Gi/o-coupled A1 adenosine receptor regulates neurite outgrowth and also represents a potential target for the treatment of CNS disorders. Adenosine receptors are one of the main targets in the brain for caffeine, and there are several agonists and antagonists currently being evaluated for clinical use (116). Four subtypes of adenosine receptors have been identified, A1, A2A, A2B, and A3. In addition to A1, A3 also couples to Gi/o. Stimulation of A1AR and A3AR have long been known to inhibit adenylyl cyclase and subsequent cAMP production (130), and more recently they have been shown to activate other signaling molecules including phospholipases C and D, RhoA, p42/44 MAPK, and PI3K (131,132,133,134). On the cellular level, both A1AR and A3AR inhibit adenylyl cyclase and activate phospholipase C and p42/44 MAPK pathways (130,133). For A1AR, this results in the activation of pertussis toxin-sensitive K+ channels and inhibition of Q-, P- and N-type Ca2+ channels (135), which as discussed above are known effects of Gi/o signaling. Signaling through A3AR has protective functions against ischemia in cardiac tissue (136) and both A1- and A3AR regulate cell growth, survival, and differentiation (reviewed in (116,133).
The discovery that caffeine exerts its stimulatory effects upon the CNS via antagonism of adenosine receptors has spurred the pursuit of modulators of these receptors to treat neurological disorders (116). Many agonists and antagonists of adenosine receptors have been generated as well as allosteric enhancers of receptor agonists. The latter molecules bind to the receptor in an area that is distinct from the agonist binding site and enhance the response of the receptor in the presence of the agonist. Antagonism of A1AR has shown promise in the treatment of anxiety and dementia as treatment with an A1AR antagonist reduced memory deficits in an animal model of dementia and had anxiolytic properties (137). Signaling through A1AR has also been shown to modulate pain by inhibiting cAMP production. A1AR-deficient mice display hypersensitivity to pain and increased anxiety, suggesting that A1AR may be important in regulating chronic pain (138,139). Stimulation of A1AR may be useful in the treatment of migraine headaches (140), and the A1AR allosteric enhancer T-62 is in Phase II clinical trials for the treatment of chronic pain. The selective modulation of A1AR may also have neuroprotective effects as adenosine can counteract the toxic effects of glutamate-mediated excitotoxicity (141). Indeed, several A1AR agonists display neuroprotective properties in animal models of cerebral ischemia (142,143,144). In addition, stimulation of A3AR is also neuroprotective in a gerbil model of ischemia (145). Finally, as adenosine can inhibit excitatory neural activity, it is thought that signaling through adenosine receptors may inhibit seizures in the healthy brain (146). A1AR agonists can inhibit seizures in rats, and this effect is markedly decreased by an A1AR antagonist (147). Thus, the improper regulation of adenosine signaling can potentially aggravate existing seizures, and adenosine receptor agonists represent an attractive drug for therapeutic intervention (146,148). While efforts at the potential treatment of seizures have been hampered by side effects on the cardiovascular system including heart rate and blood pressure, several studies using local delivery methods have shown promise for the future (149,150).
6. PERSPECTIVE
Since the discovery over a decade ago that Gαo induces neurite outgrowth, there has been rapid progress in elucidating the Gi/o-coupled receptors and the ligands that stimulate signaling through the system. The identification of Rap1GAPII as a direct interactor of Gαo led to the delineation of a central signaling network from CB1R to STAT3 that controls cannabinoid-induced neurite outgrowth. Several Gi/o-coupled receptors have now been identified that upon stimulation induce neurite outgrowth with STAT3 emerging as a key signaling focal point. While it is clear that Rac1 and Cdc42 are central players in cytoskeletal reorganization during neurite outgrowth, future studies will elucidate the mechanistic details leading to actin and microtubule reorganization. These findings provide the foundation for tackling the next set of challenges. These include defining the target genes activated in response to Gi/o signaling, delineating their transcriptional regulation, and determining how signals that stimulate other receptor-based signaling systems are integrated into this network to induce neurite outgrowth. Indeed, steps have already been taken for the latter, as signals through both the Gi/o-coupled CB1R and the TrkB receptor are integrated for proper corticogenesis during development (98). Clarifying these signaling networks will also provide a framework for delineating Gi/o signaling during axonal growth cone pathfinding and collapse as well as axonal regeneration. While several studies have implicated Gi/o-coupled receptors in these processes (46,64,67,151,152,153), the molecular mechanisms remain mostly undiscovered. Deciphering the signaling networks that regulate Gi/o-mediated neurite outgrowth from the level of receptor activation to gene expression will yield great insight into the regulation of neurite outgrowth, how this system is modulated physiologically during development and in the adult, and how it can be exploited for therapeutic intervention.
Acknowledgments
This work was supported by National Institutes of Health Grants DK-65495 to J.C.H. and GM-54508 and CA-81050 to R.I. K.D.B is supported by an individual American Cancer Society Spirit of Birmingham and Johnson Memorial Postdoctoral Fellowship Award.
References
- 1.Govek EE, Newey SE, Van Aelst L. The role of the Rho GTPases in neuronal development. Genes Dev. 2005;19(1):1–49. doi: 10.1101/gad.1256405. [DOI] [PubMed] [Google Scholar]
- 2.Ohnuma S, Harris WA. Neurogenesis and the cell cycle. Neuron. 2003;40(2):199–208. doi: 10.1016/s0896-6273(03)00632-9. [DOI] [PubMed] [Google Scholar]
- 3.Diez del Corral R, Storey KG. Markers in vertebrate neurogenesis. Nat Rev Neurosci. 2001;2(11):835–9. doi: 10.1038/35097587. [DOI] [PubMed] [Google Scholar]
- 4.da Silva JS, Dotti CG. Breaking the neuronal sphere: regulation of the actin cytoskeleton in neuritogenesis. Nat Rev Neurosci. 2002;3(9):694–704. doi: 10.1038/nrn918. [DOI] [PubMed] [Google Scholar]
- 5.Huang EJ, Reichardt LF. Neurotrophins: roles in neuronal development and function. Annu Rev Neurosci. 2001;24:677–736. doi: 10.1146/annurev.neuro.24.1.677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yoshikawa S, Thomas JB. Secreted cell signaling molecules in axon guidance. Curr Opin Neurobiol. 2004;14(1):45–50. doi: 10.1016/j.conb.2004.01.005. [DOI] [PubMed] [Google Scholar]
- 7.Huber AB, Kolodkin AL, Ginty DD, Cloutier JF. Signaling at the growth cone: ligand-receptor complexes and the control of axon growth and guidance. Annu Rev Neurosci. 2003;26:509–63. doi: 10.1146/annurev.neuro.26.010302.081139. [DOI] [PubMed] [Google Scholar]
- 8.Rodriguez OC, Schaefer AW, Mandato CA, Forscher P, Bement WM, Waterman-Storer CM. Conserved microtubule-actin interactions in cell movement and morphogenesis. Nat Cell Biol. 2003;5(7):599–609. doi: 10.1038/ncb0703-599. [DOI] [PubMed] [Google Scholar]
- 9.He JC, Neves SR, Jordan JD, Iyengar R. Role of the Go/i signaling network in the regulation of neurite outgrowth. Can J Physiol Pharmacol. 2006;84(7):687–94. doi: 10.1139/y06-025. [DOI] [PubMed] [Google Scholar]
- 10.Reinoso BS, Undie AS, Levitt P. Dopamine receptors mediate differential morphological effects on cerebral cortical neurons in vitro. J Neurosci Res. 1996;43(4):439–53. doi: 10.1002/(SICI)1097-4547(19960215)43:4<439::AID-JNR5>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 11.Lotto B, Upton L, Price DJ, Gaspar P. Serotonin receptor activation enhances neurite outgrowth of thalamic neurones in rodents. Neurosci Lett. 1999;269(2):87–90. doi: 10.1016/s0304-3940(99)00422-x. [DOI] [PubMed] [Google Scholar]
- 12.Wu YY, Bradshaw RA. Activation of the Stat3 signaling pathway is required for differentiation by interleukin-6 in PC12-E2 cells. J Biol Chem. 2000;275(3):2147–56. doi: 10.1074/jbc.275.3.2147. [DOI] [PubMed] [Google Scholar]
- 13.Xiang Y, Li Y, Zhang Z, Cui K, Wang S, Yuan XB, Wu CP, Poo MM, Duan S. Nerve growth cone guidance mediated by G protein-coupled receptors. Nat Neurosci. 2002;5(9):843–8. doi: 10.1038/nn899. [DOI] [PubMed] [Google Scholar]
- 14.He JC, Gomes I, Nguyen T, Jayaram G, Ram PT, Devi LA, Iyengar R. The Galpha o/i coupled cannabinoid receptor mediated neurite outgrowth involves Rap regulation of Src and Stat 3. J Biol Chem. 2005;280(39):33426–33434. doi: 10.1074/jbc.M502812200. [DOI] [PubMed] [Google Scholar]
- 15.Canals M, Angulo E, Casado V, Canela EI, Mallol J, Vinals F, Staines W, Tinner B, Hillion J, Agnati L, Fuxe K, Ferre S, Lluis C, Franco R. Molecular mechanisms involved in the adenosine A and A receptor-induced neuronal differentiation in neuroblastoma cells and striatal primary cultures. J Neurochem. 2005;92(2):337–48. doi: 10.1111/j.1471-4159.2004.02856.x. [DOI] [PubMed] [Google Scholar]
- 16.Thevananther S, Rivera A, Rivkees SA. A1 adenosine receptor activation inhibits neurite process formation by Rho kinase-mediated pathways. Neuroreport. 2001;12(14):3057–63. doi: 10.1097/00001756-200110080-00015. [DOI] [PubMed] [Google Scholar]
- 17.Neves SR, Ram PT, Iyengar R. G protein pathways. Science. 2002;296(5573):1636–9. doi: 10.1126/science.1071550. [DOI] [PubMed] [Google Scholar]
- 18.Gudermann T, Schoneberg T, Schultz G. Functional and structural complexity of signal transduction via G-protein-coupled receptors. Annu Rev Neurosci. 1997;20:399–427. doi: 10.1146/annurev.neuro.20.1.399. [DOI] [PubMed] [Google Scholar]
- 19.Gilman AG. G proteins transducers of receptor-generated signals. Annu Rev Biochem. 1987;56:615–49. doi: 10.1146/annurev.bi.56.070187.003151. [DOI] [PubMed] [Google Scholar]
- 20.Offermanns S. G-proteins as transducers in transmembrane signalling. Prog Biophys Mol Biol. 2003;83(2):101–30. doi: 10.1016/s0079-6107(03)00052-x. [DOI] [PubMed] [Google Scholar]
- 21.Neer EJ. Heterotrimeric G proteins: organizers of transmembrane signals. Cell. 1995;80(2):249–57. doi: 10.1016/0092-8674(95)90407-7. [DOI] [PubMed] [Google Scholar]
- 22.Hamm HE. The many faces of G protein signaling. J Biol Chem. 1998;273(2):669–72. doi: 10.1074/jbc.273.2.669. [DOI] [PubMed] [Google Scholar]
- 23.Birnbaumer L, Abramowitz J, Brown AM. Receptor-effector coupling by G proteins. Biochim Biophys Acta. 1990;1031(2):163–224. doi: 10.1016/0304-4157(90)90007-y. [DOI] [PubMed] [Google Scholar]
- 24.Ishii M, Kurachi Y. Physiological actions of regulators of G-protein signaling (RGS) proteins. Life Sci. 2003;74(2–3):163–71. doi: 10.1016/j.lfs.2003.09.004. [DOI] [PubMed] [Google Scholar]
- 25.Lanier SM. AGS proteins, GPR motifs and the signals processed by heterotrimeric G proteins. Biol Cell. 2004;96(5):369–72. doi: 10.1016/j.biolcel.2004.03.012. [DOI] [PubMed] [Google Scholar]
- 26.Cismowski MJ, Ma C, Ribas C, Xie X, Spruyt M, Lizano JS, Lanier SM, Duzic E. Activation of heterotrimeric G-protein signaling by a ras-related protein. Implications for signal integration. J Biol Chem. 2000;275(31):23421–4. doi: 10.1074/jbc.C000322200. [DOI] [PubMed] [Google Scholar]
- 27.Peterson YK, Bernard ML, Ma H, Hazard S, 3rd, Graber SG, Lanier SM. Stabilization of the GDP-bound conformation of Gialpha by a peptide derived from the G-protein regulatory motif of AGS 3. J Biol Chem. 2000;275(43):33193–6. doi: 10.1074/jbc.C000509200. [DOI] [PubMed] [Google Scholar]
- 28.Ghosh M, Peterson YK, Lanier SM, Smrcka AV. Receptor- and nucleotide exchange-independent mechanisms for promoting G protein subunit dissociation. J Biol Chem. 2003;278(37):34747–50. doi: 10.1074/jbc.C300271200. [DOI] [PubMed] [Google Scholar]
- 29.Simon MI, Strathmann MP, Gautam N. Diversity of G proteins in signal transduction. Science. 1991;252(5007):802–8. doi: 10.1126/science.1902986. [DOI] [PubMed] [Google Scholar]
- 30.Hurowitz EH, Melnyk JM, Chen YJ, Kouros-Mehr H, Simon MI, Shizuya H. Genomic characterization of the human heterotrimeric G protein alpha, beta, and gamma subunit genes. DNA Res. 2000;7(2):111–20. doi: 10.1093/dnares/7.2.111. [DOI] [PubMed] [Google Scholar]
- 31.Nurnberg B. In: Bacterial Toxins. Aktories K, editor. Chapman & Hall; London: 1997. pp. 33–45. [Google Scholar]
- 32.Sunahara RK, Dessauer CW, Gilman AG. Complexity and diversity of mammalian adenylyl cyclases. Annu Rev Pharmacol Toxicol. 1996;36:461–80. doi: 10.1146/annurev.pa.36.040196.002333. [DOI] [PubMed] [Google Scholar]
- 33.Strathmann M, Wilkie TM, Simon MI. Alternative splicing produces transcripts encoding two forms of the alpha subunit of GTP-binding protein Go. Proc Natl Acad Sci USA. 1990;87(17):6477–81. doi: 10.1073/pnas.87.17.6477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Strathmann M, Simon MI. G protein diversity: a distinct class of alpha subunits is present in vertebrates and invertebrates. Proc Natl Acad Sci USA. 1990;87(23):9113–7. doi: 10.1073/pnas.87.23.9113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tsukamoto T, Toyama R, Itoh H, Kozasa T, Matsuoka M, Kaziro Y. Structure of the human gene and two rat cDNAs encoding the alpha chain of GTP-binding regulatory protein Go: two different mRNAs are generated by alternative splicing. Proc Natl Acad Sci USA. 1991;88(8):2974–8. doi: 10.1073/pnas.88.8.2974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Logothetis DE, Kurachi Y, Galper J, Neer EJ, Clapham DE. The beta gamma subunits of GTP-binding proteins activate the muscarinic K+ channel in heart. Nature. 1987;325(6102):321–6. doi: 10.1038/325321a0. [DOI] [PubMed] [Google Scholar]
- 37.Catterall WA. Structure and regulation of voltage-gated Ca2+ channels. Annu Rev Cell Dev Biol. 2000;16:521–55. doi: 10.1146/annurev.cellbio.16.1.521. [DOI] [PubMed] [Google Scholar]
- 38.Mark MD, Herlitze S. G-protein mediated gating of inward-rectifier K+ channels. Eur J Biochem. 2000;267(19):5830–6. doi: 10.1046/j.1432-1327.2000.01670.x. [DOI] [PubMed] [Google Scholar]
- 39.Camps M, Carozzi A, Schnabel P, Scheer A, Parker PJ, Gierschik P. Isozyme-selective stimulation of phospholipase C-beta 2 by G protein beta gamma-subunits. Nature. 1992;360(6405):684–6. doi: 10.1038/360684a0. [DOI] [PubMed] [Google Scholar]
- 40.Taussig R, Gilman AG. Mammalian membrane-bound adenylyl cyclases. J Biol Chem. 1995;270(1):1–4. doi: 10.1074/jbc.270.1.1. [DOI] [PubMed] [Google Scholar]
- 41.Tang X, Downes CP. Purification and characterization of Gbetagamma-responsive phosphoinositide 3-kinases from pig platelet cytosol. J Biol Chem. 1997;272(22):14193–9. doi: 10.1074/jbc.272.22.14193. [DOI] [PubMed] [Google Scholar]
- 42.Vanhaesebroeck B, Leevers SJ, Panayotou G, Waterfield MD. Phosphoinositide 3-kinases: a conserved family of signal transducers. Trends Biochem Sci. 1997;22(7):267–72. doi: 10.1016/s0968-0004(97)01061-x. [DOI] [PubMed] [Google Scholar]
- 43.Herlitze S, Garcia DE, Mackie K, Hille B, Scheuer T, Catterall WA. Modulation of Ca2+ channels by G-protein beta gamma subunits. Nature. 1996;380(6571):258–62. doi: 10.1038/380258a0. [DOI] [PubMed] [Google Scholar]
- 44.Qin N, Olcese R, Zhou J, Cabello OA, Birnbaumer L, Stefani E. Identification of a second region of the beta-subunit involved in regulation of calcium channel inactivation. Am J Physiol. 1996;271(5 Pt 1):C1539–45. doi: 10.1152/ajpcell.1996.271.5.C1539. [DOI] [PubMed] [Google Scholar]
- 45.van Biesen T, Hawes BE, Raymond JR, Luttrell LM, Koch WJ, Lefkowitz RJ. G(o)-protein alpha-subunits activate mitogen-activated protein kinase via a novel protein kinase C-dependent mechanism. J Biol Chem. 1996;271(3):1266–9. doi: 10.1074/jbc.271.3.1266. [DOI] [PubMed] [Google Scholar]
- 46.Strittmatter SM, Valenzuela D, Kennedy TE, Neer EJ, Fishman MC. G0 is a major growth cone protein subject to regulation by GAP-43. Nature. 1990;344(6269):836–41. doi: 10.1038/344836a0. [DOI] [PubMed] [Google Scholar]
- 47.Dickson BJ. Molecular mechanisms of axon guidance. Science. 2002;298(5600):1959–64. doi: 10.1126/science.1072165. [DOI] [PubMed] [Google Scholar]
- 48.Chotard C, Salecker I. Neurons and glia: team players in axon guidance. Trends Neurosci. 2004;27(11):655–61. doi: 10.1016/j.tins.2004.09.001. [DOI] [PubMed] [Google Scholar]
- 49.Strittmatter SM, Fishman MC, Zhu XP. Activated mutants of the alpha subunit of G(o) promote an increased number of neurites per cell. J Neurosci. 1994;14(4):2327–38. doi: 10.1523/JNEUROSCI.14-04-02327.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Valenzuela D, Han X, Mende U, Fankhauser C, Mashimo H, Huang P, Pfeffer J, Neer EJ, Fishman MC. G alpha(o) is necessary for muscarinic regulation of Ca2+ channels in mouse heart. Proc Natl Acad Sci USA. 1997;94(5):1727–32. doi: 10.1073/pnas.94.5.1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jiang M, Gold MS, Boulay G, Spicher K, Peyton M, Brabet P, Srinivasan Y, Rudolph U, Ellison G, Birnbaumer L. Multiple neurological abnormalities in mice deficient in the G protein Go. Proc Natl Acad Sci USA. 1998;95(6):3269–74. doi: 10.1073/pnas.95.6.3269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Greif GJ, Sodickson DL, Bean BP, Neer EJ, Mende U. Altered regulation of potassium and calcium channels by GABA(B) and adenosine receptors in hippocampal neurons from mice lacking Galpha(o) J Neurophysiol. 2000;83(2):1010–8. doi: 10.1152/jn.2000.83.2.1010. [DOI] [PubMed] [Google Scholar]
- 53.Tanaka M, Treloar H, Kalb RG, Greer CA, Strittmatter SM. G(o) protein-dependent survival of primary accessory olfactory neurons. Proc Natl Acad Sci U S A. 1999;96(24):14106–11. doi: 10.1073/pnas.96.24.14106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kroll SD, Chen J, De Vivo M, Carty DJ, Buku A, Premont RT, Iyengar R. The Q205LGo-alpha subunit expressed in NIH-3T3 cells induces transformation. J Biol Chem. 1992;267(32):23183–8. [PubMed] [Google Scholar]
- 55.Ram PT, Horvath CM, Iyengar R. Stat3-mediated transformation of NIH-3T3 cells by the constitutively active Q205L Galphao protein. Science. 2000;287(5450):142–4. doi: 10.1126/science.287.5450.142. [DOI] [PubMed] [Google Scholar]
- 56.Corre I, Baumann H, Hermouet S. Regulation by Gi2 proteins of v-fms-induced proliferation and transformation via Src-kinase and STAT 3. Oncogene. 1999;18(46):6335–42. doi: 10.1038/sj.onc.1203010. [DOI] [PubMed] [Google Scholar]
- 57.Jordan JD, Carey KD, Stork PJ, Iyengar R. Modulation of rap activity by direct interaction of Galpha(o) with Rap1 GTPase-activating protein. J Biol Chem. 1999;274(31):21507–10. doi: 10.1074/jbc.274.31.21507. [DOI] [PubMed] [Google Scholar]
- 58.Ajit SK, Ramineni S, Edris W, Hunt RA, Hum WT, Hepler JR, Young KH. RGSZ1 interacts with protein kinase C interacting protein PKCI-1 and modulates mu opioid receptor signaling. Cell Signal. 2007;19(4):723–30. doi: 10.1016/j.cellsig.2006.09.008. [DOI] [PubMed] [Google Scholar]
- 59.Mao H, Zhao Q, Daigle M, Ghahremani MH, Chidiac P, Albert PR. RGS17/RGSZ2, a novel regulator of Gi/o, Gz, and Gq signaling. J Biol Chem. 2004;279(25):26314–22. doi: 10.1074/jbc.M401800200. [DOI] [PubMed] [Google Scholar]
- 60.Nunn C, Mao H, Chidiac P, Albert PR. RGS17/RGSZ2 and the RZ/A family of regulators of G-protein signaling. Semin Cell Dev Biol. 2006;17(3):390–9. doi: 10.1016/j.semcdb.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 61.Mochizuki N, Ohba Y, Kiyokawa E, Kurata T, Murakami T, Ozaki T, Kitabatake A, Nagashima K, Matsuda M. Activation of the ERK/MAPK pathway by an isoform of rap1GAP associated with G alpha(i) Nature. 1999;400(6747):891–4. doi: 10.1038/23738. [DOI] [PubMed] [Google Scholar]
- 62.Tsygankova OM, Feshchenko E, Klein PS, Meinkoth JL. Thyroid-stimulating hormone/cAMP and glycogen synthase kinase 3beta elicit opposing effects on Rap1GAP stability. J Biol Chem. 2004;279(7):5501–7. doi: 10.1074/jbc.M305824200. [DOI] [PubMed] [Google Scholar]
- 63.Jordan JD, He JC, Eungdamrong NJ, Gomes I, Ali W, Nguyen T, Bivona TG, Philips MR, Devi LA, Iyengar R. Cannabinoid receptor induced neurite outgrowth is mediated by Rap1 activation through Galphao/I-triggered proteasomal degradation of Rap1GAPII. J Biol Chem. 2005;280(12):11413–11421. doi: 10.1074/jbc.M411521200. [DOI] [PubMed] [Google Scholar]
- 64.Chen LT, Gilman AG, Kozasa T. A candidate target for G protein action in brain. J Biol Chem. 1999;274(38):26931–8. doi: 10.1074/jbc.274.38.26931. [DOI] [PubMed] [Google Scholar]
- 65.Negishi M, Oinuma I, Katoh H. Plexins: axon guidance and signal transduction. Cell Mol Life Sci. 2005;62(12):1363–71. doi: 10.1007/s00018-005-5018-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Giniger E. How do Rho family GTPases direct axon growth and guidance? A proposal relating signaling pathways to growth cone mechanics. Differentiation. 2002;70(8):385–96. doi: 10.1046/j.1432-0436.2002.700801.x. [DOI] [PubMed] [Google Scholar]
- 67.Igarashi M, Strittmatter SM, Vartanian T, Fishman MC. Mediation by G proteins of signals that cause collapse of growth cones. Science. 1993;259(5091):77–9. doi: 10.1126/science.8418498. [DOI] [PubMed] [Google Scholar]
- 68.Anneren C, Reedquist KA, Bos JL, Welsh M. GTK, a Src-related tyrosine kinase, induces nerve growth factor-independent neurite outgrowth in PC12 cells through activation of the Rap1 pathway, Relationship to Shb tyrosine phosphorylation and elevated levels of focal adhesion kinase. J Biol Chem. 2000;275(37):29153–61. doi: 10.1074/jbc.M003926200. [DOI] [PubMed] [Google Scholar]
- 69.Lu L, Anneren C, Reedquist KA, Bos JL, Welsh M. NGF-Dependent neurite outgrowth in PC12 cells overexpressing the Src homology 2-domain protein shb requires activation of the Rap1 pathway. Exp Cell Res. 2000;259(2):370–7. doi: 10.1006/excr.2000.4984. [DOI] [PubMed] [Google Scholar]
- 70.Fricker AD, Rios C, Devi LA, Gomes I. Serotonin receptor activation leads to neurite outgrowth and neuronal survival. Brain Res Mol Brain Res. 2005;138(2):228–35. doi: 10.1016/j.molbrainres.2005.04.016. [DOI] [PubMed] [Google Scholar]
- 71.Wu YY, Bradshaw RA. Synergistic induction of neurite outgrowth by nerve growth factor or epidermal growth factor and interleukin-6 in PC12 cells. J Biol Chem. 1996;271(22):13033–9. doi: 10.1074/jbc.271.22.13033. [DOI] [PubMed] [Google Scholar]
- 72.Ihara S, Nakajima K, Fukada T, Hibi M, Nagata S, Hirano T, Fukui Y. Dual control of neurite outgrowth by STAT3 and MAP kinase in PC12 cells stimulated with interleukin-6. Embo J. 1997;16(17):5345–52. doi: 10.1093/emboj/16.17.5345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ng YP, Cheung ZH, Ip NY. STAT3 as a downstream mediator of Trk signaling and functions. J Biol Chem. 2006;281(23):15636–44. doi: 10.1074/jbc.M601863200. [DOI] [PubMed] [Google Scholar]
- 74.Marinissen MJ, Gutkind JS. G-protein-coupled receptors and signaling networks: emerging paradigms. Trends Pharmacol Sci. 2001;22(7):368–76. doi: 10.1016/s0165-6147(00)01678-3. [DOI] [PubMed] [Google Scholar]
- 75.Jaffe AB, Hall A. Rho GTPases: biochemistry and biology. Annu Rev Cell Dev Biol. 2005;21:247–69. doi: 10.1146/annurev.cellbio.21.020604.150721. [DOI] [PubMed] [Google Scholar]
- 76.Hall A. Rho GTPases and the control of cell behaviour. Biochem Soc Trans. 2005;33(Pt 5):891–5. doi: 10.1042/BST20050891. [DOI] [PubMed] [Google Scholar]
- 77.Nakata H, Kozasa T. Functional characterization of Galphao signaling through G protein-regulated inducer of neurite outgrowth 1. Mol Pharmacol. 2005;67(3):695–702. doi: 10.1124/mol.104.003913. [DOI] [PubMed] [Google Scholar]
- 78.Gibson MC, Perrimon N. Apicobasal polarization: epithelial form and function. Curr Opin Cell Biol. 2003;15(6):747–52. doi: 10.1016/j.ceb.2003.10.008. [DOI] [PubMed] [Google Scholar]
- 79.Schwamborn JC, Puschel AW. The sequential activity of the GTPases Rap1B and Cdc42 determines neuronal polarity. Nat Neurosci. 2004;7(9):923–9. doi: 10.1038/nn1295. [DOI] [PubMed] [Google Scholar]
- 80.Bamburg JR. Proteins of the ADF/cofilin family: essential regulators of actin dynamics. Annu Rev Cell Dev Biol. 1999;15:185–230. doi: 10.1146/annurev.cellbio.15.1.185. [DOI] [PubMed] [Google Scholar]
- 81.Bamburg JR, Wiggan OP. ADF/cofilin and actin dynamics in disease. Trends Cell Biol. 2002;12(12):598–605. doi: 10.1016/s0962-8924(02)02404-2. [DOI] [PubMed] [Google Scholar]
- 82.Niwa R, Nagata-Ohashi K, Takeichi M, Mizuno K, Uemura T. Control of actin reorganization by Slingshot, a family of phosphatases that dephosphorylate ADF/cofilin. Cell. 2002;108(2):233–46. doi: 10.1016/s0092-8674(01)00638-9. [DOI] [PubMed] [Google Scholar]
- 83.Endo M, Ohashi K, Sasaki Y, Goshima Y, Niwa R, Uemura T, Mizuno K. Control of growth cone motility and morphology by LIM kinase and Slingshot via phosphorylation and dephosphorylation of cofilin. J Neurosci. 2003;23(7):2527–37. doi: 10.1523/JNEUROSCI.23-07-02527.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gehler S, Shaw AE, Sarmiere PD, Bamburg JR, Letourneau PC. Brain-derived neurotrophic factor regulation of retinal growth cone filopodial dynamics is mediated through actin depolymerizing factor/cofilin. J Neurosci. 2004;24(47):10741–9. doi: 10.1523/JNEUROSCI.2836-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Rosso S, Bollati F, Bisbal M, Peretti D, Sumi T, Nakamura T, Quiroga S, Ferreira A, Caceres A. LIMK1 regulates Golgi dynamics, traffic of Golgi-derived vesicles, and process extension in primary cultured neurons. Mol Biol Cell. 2004;15(7):3433–49. doi: 10.1091/mbc.E03-05-0328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Endo M, Ohashi K, Mizuno K. LIM kinase and slingshot are critical for neurite extension. J Biol Chem. 2007;282(18):13692–702. doi: 10.1074/jbc.M610873200. [DOI] [PubMed] [Google Scholar]
- 87.Smith LG, Li R. Actin polymerization: riding the wave. Curr Biol. 2004;14(3):R109–11. [PubMed] [Google Scholar]
- 88.Ho HY, Rohatgi R, Lebensohn AM, Le M, Li J, Gygi SP, Kirschner MW. Toca-1 mediates Cdc42-dependent actin nucleation by activating the N-WASP-WIP complex. Cell. 2004;118(2):203–16. doi: 10.1016/j.cell.2004.06.027. [DOI] [PubMed] [Google Scholar]
- 89.Leung DW, Rosen MK. The nucleotide switch in Cdc42 modulates coupling between the GTPase-binding and allosteric equilibria of Wiskott-Aldrich syndrome protein. Proc Natl Acad Sci U S A. 2005;102(16):5685–90. doi: 10.1073/pnas.0406472102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Yamada T, Sakisaka T, Hisata S, Baba T, Takai Y. RA-RhoGAP, Rap-activated Rho GTPase-activating protein implicated in neurite outgrowth through Rho. J Biol Chem. 2005;280(38):33026–34. doi: 10.1074/jbc.M504587200. [DOI] [PubMed] [Google Scholar]
- 91.Brouns MR, Matheson SF, Settleman J. p190 RhoGAP is the principal Src substrate in brain and regulates axon outgrowth, guidance and fasciculation. Nat Cell Biol. 2001;3(4):361–7. doi: 10.1038/35070042. [DOI] [PubMed] [Google Scholar]
- 92.Dumenil G, Sansonetti P, Tran Van Nhieu G. Src tyrosine kinase activity down-regulates Rho-dependent responses during Shigella entry into epithelial cells and stress fibre formation. J Cell Sci. 2000;113(Pt 1):71–80. doi: 10.1242/jcs.113.1.71. [DOI] [PubMed] [Google Scholar]
- 93.Mackie K, Stella N. Cannabinoid receptors and endocannabinoids: evidence for new players. Aaps J. 2006;8(2):E298–306. doi: 10.1007/BF02854900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Harkany T, Guzman M, Galve-Roperh I, Berghuis P, Devi LA, Mackie K. The emerging functions of endocannabinoid signaling during CNS development. Trends Pharmacol Sci. 2007;28(2):83–92. doi: 10.1016/j.tips.2006.12.004. [DOI] [PubMed] [Google Scholar]
- 95.Berghuis P, Rajnicek AM, Morozov YM, Ross RA, Mulder J, Urban GM, Monory K, Marsicano G, Matteoli M, Canty A, Irving AJ, Katona I, Yanagawa Y, Rakic P, Lutz B, Mackie K, Harkany T. Hardwiring the brain: endocannabinoids shape neuronal connectivity. Science. 2007;316(5828):1212–6. doi: 10.1126/science.1137406. [DOI] [PubMed] [Google Scholar]
- 96.Marty S, Berninger B, Carroll P, Thoenen H. GABAergic stimulation regulates the phenotype of hippocampal interneurons through the regulation of brain-derived neurotrophic factor. Neuron. 1996;16(3):565–70. doi: 10.1016/s0896-6273(00)80075-6. [DOI] [PubMed] [Google Scholar]
- 97.Berghuis P, Dobszay MB, Sousa KM, Schulte G, Mager PP, Hartig W, Gorcs TJ, Zilberter Y, Ernfors P, Harkany T. Brain-derived neurotrophic factor controls functional differentiation and microcircuit formation of selectively isolated fast-spiking GABAergic interneurons. Eur J Neurosci. 2004;20(5):1290–306. doi: 10.1111/j.1460-9568.2004.03561.x. [DOI] [PubMed] [Google Scholar]
- 98.Berghuis P, Dobszay MB, Wang X, Spano S, Ledda F, Sousa KM, Schulte G, Ernfors P, Mackie K, Paratcha G, Hurd YL, Harkany T. Endocannabinoids regulate interneuron migration and morphogenesis by transactivating the TrkB receptor. Proc Natl Acad Sci U S A. 2005;102(52):19115–20. doi: 10.1073/pnas.0509494102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Aguado T, Monory K, Palazuelos J, Stella N, Cravatt B, Lutz B, Marsicano G, Kokaia Z, Guzman M, Galve-Roperh I. The endocannabinoid system drives neural progenitor proliferation. Faseb J. 2005;19(12):1704–6. doi: 10.1096/fj.05-3995fje. [DOI] [PubMed] [Google Scholar]
- 100.Aguado T, Palazuelos J, Monory K, Stella N, Cravatt B, Lutz B, Marsicano G, Kokaia Z, Guzman M, Galve-Roperh I. The endocannabinoid system promotes astroglial differentiation by acting on neural progenitor cells. J Neurosci. 2006;26(5):1551–61. doi: 10.1523/JNEUROSCI.3101-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Rueda D, Navarro B, Martinez-Serrano A, Guzman M, Galve-Roperh I. The endocannabinoid anandamide inhibits neuronal progenitor cell differentiation through attenuation of the Rap1/B-Raf/ERK pathway. J Biol Chem. 2002;277(48):46645–50. doi: 10.1074/jbc.M206590200. [DOI] [PubMed] [Google Scholar]
- 102.Richardson GA, Day NL, Goldschmidt L. Prenatal alcohol, marijuana, and tobacco use: infant mental and motor development. Neurotoxicol Teratol. 1995;17(4):479–87. doi: 10.1016/0892-0362(95)00006-d. [DOI] [PubMed] [Google Scholar]
- 103.Fried PA, Watkinson B, Gray R. Differential effects on cognitive functioning in 13- to 16-year-olds prenatally exposed to cigarettes and marihuana. Neurotoxicol Teratol. 2003;25(4):427–36. doi: 10.1016/s0892-0362(03)00029-1. [DOI] [PubMed] [Google Scholar]
- 104.Huizink AC, Mulder EJ. Maternal smoking, drinking or cannabis use during pregnancy and neurobehavioral and cognitive functioning in human offspring. Neurosci Biobehav Rev. 2006;30(1):24–41. doi: 10.1016/j.neubiorev.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 105.Hunter SA, Burstein SH. Receptor mediation in cannabinoid stimulated arachidonic acid mobilization and anandamide synthesis. Life Sci. 1997;60(18):1563–73. doi: 10.1016/s0024-3205(97)00122-7. [DOI] [PubMed] [Google Scholar]
- 106.Bernard C, Milh M, Morozov YM, Ben-Ari Y, Freund TF, Gozlan H. Altering cannabinoid signaling during development disrupts neuronal activity. Proc Natl Acad Sci U S A. 2005;102(26):9388–93. doi: 10.1073/pnas.0409641102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Skaper SD, Buriani A, Dal Toso R, Petrelli L, Romanello S, Facci L, Leon A. The ALIAmide palmitoylethanolamide and cannabinoids, but not anandamide, are protective in a delayed postglutamate paradigm of excitotoxic death in cerebellar granule neurons. Proc Natl Acad Sci USA. 1996;93(9):3984–9. doi: 10.1073/pnas.93.9.3984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Shen M, Thayer SA. Cannabinoid receptor agonists protect cultured rat hippocampal neurons from excitotoxicity. Mol Pharmacol. 1998;54(3):459–62. doi: 10.1124/mol.54.3.459. [DOI] [PubMed] [Google Scholar]
- 109.Hampson AJ, Grimaldi M. Cannabinoid receptor activation and elevated cyclic AMP reduce glutamate neurotoxicity. Eur J Neurosci. 2001;13(8):1529–36. doi: 10.1046/j.0953-816x.2001.01536.x. [DOI] [PubMed] [Google Scholar]
- 110.Panikashvili D, Simeonidou C, Ben-Shabat S, Hanus L, Breuer A, Mechoulam R, Shohami E. An endogenous cannabinoid (2-AG) is neuroprotective after brain injury. Nature. 2001;413(6855):527–31. doi: 10.1038/35097089. [DOI] [PubMed] [Google Scholar]
- 111.Nagayama T, Sinor AD, Simon RP, Chen J, Graham SH, Jin K, Greenberg DA. Cannabinoids and neuroprotection in global and focal cerebral ischemia and in neuronal cultures. J Neurosci. 1999;19(8):2987–95. doi: 10.1523/JNEUROSCI.19-08-02987.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Dagon Y, Avraham Y, Link G, Zolotarev O, Mechoulam R, Berry EM. The synthetic cannabinoid HU-210 attenuates neural damage in diabetic mice and hyperglycemic pheochromocytoma PC12 cells. Neurobiol Dis. 2007 doi: 10.1016/j.nbd.2007.04.017. [DOI] [PubMed] [Google Scholar]
- 113.Cheng Y, Hitchcock SA. Targeting cannabinoid agonists for inflammatory and neuropathic pain. Expert Opin Investig Drugs. 2007;16(7):951–65. doi: 10.1517/13543784.16.7.951. [DOI] [PubMed] [Google Scholar]
- 114.Patel PN, Pathak R. Rimonabant: a novel selective cannabinoid-1 receptor antagonist for treatment of obesity. Am J Health Syst Pharm. 2007;64(5):481–9. doi: 10.2146/060258. [DOI] [PubMed] [Google Scholar]
- 115.Armstrong HE, Galka A, Lin LS, Lanza TJ, Jr, Jewell JP, Shah SK, Guthikonda R, Truong Q, Chang LL, Quaker G, Colandrea VJ, Tong X, Wang J, Xu S, Fong TM, Shen CP, Lao J, Chen J, Shearman LP, Stribling DS, Rosko K, Strack A, Ha S, Van der Ploeg L, Goulet MT, Hagmann WK. Substituted acyclic sulfonamides as human cannabinoid-1 receptor inverse agonists. Bioorg Med Chem Lett. 2007;17(8):2184–7. doi: 10.1016/j.bmcl.2007.01.087. [DOI] [PubMed] [Google Scholar]
- 116.Jacobson KA, Gao ZG. Adenosine receptors as therapeutic targets. Nat Rev Drug Discov. 2006;5(3):247–64. doi: 10.1038/nrd1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Centonze D, Finazzi-Agro A, Bernardi G, Maccarrone M. The endocannabinoid system in targeting inflammatory neurodegenerative diseases. Trends Pharmacol Sci. 2007;28(4):180–7. doi: 10.1016/j.tips.2007.02.004. [DOI] [PubMed] [Google Scholar]
- 118.Maccarrone M, Battista N, Centonze D. The endocannabinoid pathway in Huntington’s disease: a comparison with other neurodegenerative diseases. Prog Neurobiol. 2007;81(5–6):349–79. doi: 10.1016/j.pneurobio.2006.11.006. [DOI] [PubMed] [Google Scholar]
- 119.Lastres-Becker I, Molina-Holgado F, Ramos JA, Mechoulam R, Fernandez-Ruiz J. Cannabinoids provide neuroprotection against 6-hydroxydopamine toxicity in vivo and in vitro: relevance to Parkinson’s disease. Neurobiol Dis. 2005;19(1–2):96–107. doi: 10.1016/j.nbd.2004.11.009. [DOI] [PubMed] [Google Scholar]
- 120.Kim SR, Lee DY, Chung ES, Oh UT, Kim SU, Jin BK. Transient receptor potential vanilloid subtype 1 mediates cell death of mesencephalic dopaminergic neurons in vivo and in vitro. J Neurosci. 2005;25(3):662–71. doi: 10.1523/JNEUROSCI.4166-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Raman C, McAllister SD, Rizvi G, Patel SG, Moore DH, Abood ME. Amyotrophic lateral sclerosis: delayed disease progression in mice by treatment with a cannabinoid. Amyotroph Lateral Scler Other Motor Neuron Disord. 2004;5(1):33–9. doi: 10.1080/14660820310016813. [DOI] [PubMed] [Google Scholar]
- 122.Bilsland LG, Dick JR, Pryce G, Petrosino S, Di Marzo V, Baker D, Greensmith L. Increasing cannabinoid levels by pharmacological and genetic manipulation delay disease progression in SOD1 mice. Faseb J. 2006;20(7):1003–5. doi: 10.1096/fj.05-4743fje. [DOI] [PubMed] [Google Scholar]
- 123.Ramirez BG, Blazquez C, Gomez del Pulgar T, Guzman M, de Ceballos ML. Prevention of Alzheimer’s disease pathology by cannabinoids: neuroprotection mediated by blockade of microglial activation. J Neurosci. 2005;25(8):1904–13. doi: 10.1523/JNEUROSCI.4540-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.van der Stelt M, Mazzola C, Esposito G, Matias I, Petrosino S, De Filippis D, Micale V, Steardo L, Drago F, Iuvone T, Di Marzo V. Endocannabinoids and beta-amyloid-induced neurotoxicity in vivo: effect of pharmacological elevation of endocannabinoid levels. Cell Mol Life Sci. 2006;63(12):1410–24. doi: 10.1007/s00018-006-6037-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Maresz K, Pryce G, Ponomarev ED, Marsicano G, Croxford JL, Shriver LP, Ledent C, Cheng X, Carrier EJ, Mann MK, Giovannoni G, Pertwee RG, Yamamura T, Buckley NE, Hillard CJ, Lutz B, Baker D, Dittel BN. Direct suppression of CNS autoimmune inflammation via the cannabinoid receptor CB1 on neurons and CB2 on autoreactive T cells. Nat Med. 2007;13(4):492–7. doi: 10.1038/nm1561. [DOI] [PubMed] [Google Scholar]
- 126.Mestre L, Correa F, Arevalo-Martin A, Molina-Holgado E, Valenti M, Ortar G, Di Marzo V, Guaza C. Pharmacological modulation of the endocannabinoid system in a viral model of multiple sclerosis. J Neurochem. 2005;92(6):1327–39. doi: 10.1111/j.1471-4159.2004.02979.x. [DOI] [PubMed] [Google Scholar]
- 127.Ortega-Gutierrez S, Molina-Holgado E, Arevalo-Martin A, Correa F, Viso A, Lopez-Rodriguez ML, Di Marzo V, Guaza C. Activation of the endocannabinoid system as therapeutic approach in a murine model of multiple sclerosis. Faseb J. 2005;19(10):1338–40. doi: 10.1096/fj.04-2464fje. [DOI] [PubMed] [Google Scholar]
- 128.Lastres-Becker I, de Miguel R, De Petrocellis L, Makriyannis A, Di Marzo V, Fernandez-Ruiz J. Compounds acting at the endocannabinoid and/or endovanilloid systems reduce hyperkinesia in a rat model of Huntington’s disease. J Neurochem. 2003;84(5):1097–109. doi: 10.1046/j.1471-4159.2003.01595.x. [DOI] [PubMed] [Google Scholar]
- 129.Lastres-Becker I, Hansen HH, Berrendero F, De Miguel R, Perez-Rosado A, Manzanares J, Ramos JA, Fernandez-Ruiz J. Alleviation of motor hyperactivity and neurochemical deficits by endocannabinoid uptake inhibition in a rat model of Huntington’s disease. Synapse. 2002;44(1):23–35. doi: 10.1002/syn.10054. [DOI] [PubMed] [Google Scholar]
- 130.van Calker D, Muller M, Hamprecht B. Adenosine regulates via two different types of receptors, the accumulation of cyclic AMP in cultured brain cells. J Neurochem. 1979;33(5):999–1005. doi: 10.1111/j.1471-4159.1979.tb05236.x. [DOI] [PubMed] [Google Scholar]
- 131.Rogel A, Bromberg Y, Sperling O, Zoref-Shani E. Phospholipase C is involved in the adenosine-activated signal transduction pathway conferring protection against iodoacetic acid-induced injury in primary rat neuronal cultures. Neurosci Lett. 2005;373(3):218–21. doi: 10.1016/j.neulet.2004.10.012. [DOI] [PubMed] [Google Scholar]
- 132.Mozzicato S, Joshi BV, Jacobson KA, Liang BT. Role of direct RhoA-phospholipase D1 interaction in mediating adenosine-induced protection from cardiac ischemia. Faseb J. 2004;18(2):406–8. doi: 10.1096/fj.03-0592fje. [DOI] [PubMed] [Google Scholar]
- 133.Schulte G, Fredholm BB. Signalling from adenosine receptors to mitogen-activated protein kinases. Cell Signal. 2003;15(9):813–27. doi: 10.1016/s0898-6568(03)00058-5. [DOI] [PubMed] [Google Scholar]
- 134.Hammarberg C, Fredholm BB, Schulte G. Adenosine A3 receptor-mediated regulation of p38 and extracellular-regulated kinase ERK1/2 via phosphatidylinositol-3′-kinase. Biochem Pharmacol. 2004;67(1):129–34. doi: 10.1016/j.bcp.2003.08.031. [DOI] [PubMed] [Google Scholar]
- 135.Belardinelli L, Shryock JC, Song Y, Wang D, Srinivas M. Ionic basis of the electrophysiological actions of adenosine on cardiomyocytes. Faseb J. 1995;9(5):359–65. doi: 10.1096/fasebj.9.5.7896004. [DOI] [PubMed] [Google Scholar]
- 136.Tracey WR, Magee W, Masamune H, Oleynek JJ, Hill RJ. Selective activation of adenosine A3 receptors with N6-(3-chlorobenzyl)-5′-N-methylcarboxamidoadenosine (CB-MECA) provides cardioprotection via KATP channel activation. Cardiovasc Res. 1998;40(1):138–45. doi: 10.1016/s0008-6363(98)00112-6. [DOI] [PubMed] [Google Scholar]
- 137.Maemoto T, Tada M, Mihara T, Ueyama N, Matsuoka H, Harada K, Yamaji T, Shirakawa K, Kuroda S, Akahane A, Iwashita A, Matsuoka N, Mutoh S. Pharmacological characterization of FR194921, a new potent, selective, and orally active antagonist for central adenosine A1 receptors. J Pharmacol Sci. 2004;96(1):42–52. doi: 10.1254/jphs.fp0040359. [DOI] [PubMed] [Google Scholar]
- 138.Johansson B, Halldner L, Dunwiddie TV, Masino SA, Poelchen W, Gimenez-Llort L, Escorihuela RM, Fernandez-Teruel A, Wiesenfeld-Hallin Z, Xu XJ, Hardemark A, Betsholtz C, Herlenius E, Fredholm BB. Hyperalgesia, anxiety, and decreased hypoxic neuroprotection in mice lacking the adenosine A1 receptor. Proc Natl Acad Sci U S A. 2001;98(16):9407–12. doi: 10.1073/pnas.161292398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Wu WP, Hao JX, Halldner L, Lovdahl C, DeLander GE, Wiesenfeld-Hallin Z, Fredholm BB, Xu XJ. Increased nociceptive response in mice lacking the adenosine A1 receptor. Pain. 2005;113(3):395–404. doi: 10.1016/j.pain.2004.11.020. [DOI] [PubMed] [Google Scholar]
- 140.Li X, Conklin D, Pan HL, Eisenach JC. Allosteric adenosine receptor modulation reduces hypersensitivity following peripheral inflammation by a central mechanism. J Pharmacol Exp Ther. 2003;305(3):950–5. doi: 10.1124/jpet.102.047951. [DOI] [PubMed] [Google Scholar]
- 141.Stone TW. Purines and neuroprotection. Adv Exp Med Biol. 2002;513:249–80. doi: 10.1007/978-1-4615-0123-7_9. [DOI] [PubMed] [Google Scholar]
- 142.Von Lubitz DK, Beenhakker M, Lin RC, Carter MF, Paul IA, Bischofberger N, Jacobson KA. Reduction of postischemic brain damage and memory deficits following treatment with the selective adenosine A1 receptor agonist. Eur J Pharmacol. 1996;302(1–3):43–8. doi: 10.1016/0014-2999(96)00101-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Von Lubitz DK, Lin RC, Paul IA, Beenhakker M, Boyd M, Bischofberger N, Jacobson KA. Postischemic administration of adenosine amine congener (ADAC): analysis of recovery in gerbils. Eur J Pharmacol. 1996;316(2–3):171–9. doi: 10.1016/s0014-2999(96)00667-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Knutsen LJ, Lau J, Petersen H, Thomsen C, Weis JU, Shalmi M, Judge ME, Hansen AJ, Sheardown MJ. N-substituted adenosines as novel neuroprotective A(1) agonists with diminished hypotensive effects. J Med Chem. 1999;42(18):3463–77. doi: 10.1021/jm960682u. [DOI] [PubMed] [Google Scholar]
- 145.Von Lubitz DK, Lin RC, Popik P, Carter MF, Jacobson KA. Adenosine A3 receptor stimulation and cerebral ischemia. Eur J Pharmacol. 1994;263(1–2):59–67. doi: 10.1016/0014-2999(94)90523-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Pagonopoulou O, Efthimiadou A, Asimakopoulos B, Nikolettos NK. Modulatory role of adenosine and its receptors in epilepsy: possible therapeutic approaches. Neurosci Res. 2006;56(1):14–20. doi: 10.1016/j.neures.2006.05.010. [DOI] [PubMed] [Google Scholar]
- 147.Zeraati M, Mirnajafi-Zadeh J, Fathollahi Y, Namvar S, Rezvani ME. Adenosine A1 and A2A receptors of hippocampal CA1 region have opposite effects on piriform cortex kindled seizures in rats. Seizure. 2006;15(1):41–8. doi: 10.1016/j.seizure.2005.10.006. [DOI] [PubMed] [Google Scholar]
- 148.Boison D. Adenosine and epilepsy: from therapeutic rationale to new therapeutic strategies. Neuroscientist. 2005;11(1):25–36. doi: 10.1177/1073858404269112. [DOI] [PubMed] [Google Scholar]
- 149.Huber A, Guttinger M, Mohler H, Boison D. Seizure suppression by adenosine A(2A) receptor activation in a rat model of audiogenic brainstem epilepsy. Neurosci Lett. 2002;329(3):289–92. doi: 10.1016/s0304-3940(02)00684-5. [DOI] [PubMed] [Google Scholar]
- 150.Guttinger M, Padrun V, Pralong WF, Boison D. Seizure suppression and lack of adenosine A1 receptor desensitization after focal long-term delivery of adenosine by encapsulated myoblasts. Exp Neurol. 2005;193(1):53–64. doi: 10.1016/j.expneurol.2004.12.012. [DOI] [PubMed] [Google Scholar]
- 151.Sasaki Y, Hoshi M, Akazawa C, Nakamura Y, Tsuzuki H, Inoue K, Kohsaka S. Selective expression of Gi/o-coupled ATP receptor P2Y12 in microglia in rat brain. Glia. 2003;44(3):242–50. doi: 10.1002/glia.10293. [DOI] [PubMed] [Google Scholar]
- 152.Costigan M, Samad TA, Allchorne A, Lanoue C, Tate S, Woolf CJ. High basal expression and injury-induced down regulation of two regulator of G-protein signaling transcripts, RGS3 and RGS4 in primary sensory neurons. Mol Cell Neurosci. 2003;24(1):106–16. doi: 10.1016/s1044-7431(03)00135-0. [DOI] [PubMed] [Google Scholar]
- 153.Frohnert PW, Stonecypher MS, Carroll SL. Lysophosphatidic acid promotes the proliferation of adult Schwann cells isolated from axotomized sciatic nerve. J Neuropathol Exp Neurol. 2003;62(5):520–9. doi: 10.1093/jnen/62.5.520. [DOI] [PubMed] [Google Scholar]