

Abstract
Large-conductance, Ca2+- and voltage-sensitive K+ (BK) channels regulate neuronal functions such as spike frequency adaptation and transmitter release. BK channels are composed of four Slo1 subunits, which contain the voltage-sensing and pore-gate domains in the membrane and Ca2+ binding sites in the cytoplasmic domain, and accessory β subunits. Four types of BK channel β subunits (β1–β4) show differential tissue distribution and unique functional modulation, resulting in diverse phenotypes of BK channels. Previous studies show that both the β1 and β2 subunits increase Ca2+ sensitivity, but different mechanisms may underline these modulations. However, the structural domains in Slo1 that are critical for Ca2+-dependent activation and targeted by these β subunits are not known. Here, we report that the N termini of both the transmembrane (including S0) and cytoplasmic domains of Slo1 are critical for β2 modulation based on the study of differential effects of the β2 subunit on two orthologs, mouse Slo1 and Drosophila Slo1. The N terminus of the cytoplasmic domain of Slo1, including the AC region (βA–αC) of the RCK1 (regulator of K+ conductance) domain and the peptide linking it to S6, both of which have been shown previously to mediate the coupling between Ca2+ binding and channel opening, is specifically required for the β2 but not for the β1 modulation. These results suggest that the β2 subunit modulates the coupling between Ca2+ binding and channel opening, and, although sharing structural homology, the BK channel β subunits interact with structural domains in the Slo1 subunit differently to enhance channel activity.
Introduction
Large-conductance, Ca2+- and voltage-sensitive K+ (BK) channels activate in response to depolarizing membrane potentials and increases in intracellular Ca2+ (Marty, 1981). The opening of BK channels effectively repolarizes the membrane and shuts Ca2+ entry through voltage-dependent Ca2+ channels, thereby reducing intracellular Ca2+. As a consequence, BK channels are negative feedback regulators of membrane excitability and Ca2+ entry in neurons (Lancaster and Nicoll, 1987; Storm, 1987) and smooth muscle cells (Brayden and Nelson, 1992).
Functional BK channels are tetramers of the pore-forming Slo1 subunit that is composed of seven membrane-spanning segments (S0–S6), in which S1–S4 form the voltage-sensing domain (VSD) and S5 and S6 form the pore-gate domain, and a large cytoplasmic domain (Fig. 1) (Meera et al., 1997). This cytoplasmic domain is composed of two RCK (regulator of K+ conductance) domains and contains two putative Ca2+ binding sites, D367 in RCK1 and the Ca2+ bowl in RCK2 (Fig. 1) (Schreiber and Salkoff, 1997; Xia et al., 2002; Wu et al., 2010; Yuan et al., 2010).
Figure 1.

Schematic of BK channel structure. Top, S0–S6 are transmembrane segments, and RCK1 and RCK2 are located in cytoplasm. PGD, Pore-gate domain. Star (★) represents the location of Ca2+ binding sites. The sequence and secondary structure of the N terminus including S0 and the C-Linker and AC region are shown below. Asterisk (*) identifies conserved residues.
BK channels also associate with β subunits that are composed of two transmembrane (TM) segments with cytoplasmic termini and a large extracellular loop. There are four types of β subunits, β1–β4, that have tissue-specific distributions and impart unique effect on voltage- and Ca2+-dependent activation of BK channels (Orio et al., 2002). The β1 and β2 subunits have the highest sequence homology and both increase apparent Ca2+ sensitivity (Brenner et al., 2000), but different mechanisms may be responsible for their functions; the β1 subunit was shown to primarily modify the voltage-dependent activation, whereas the β2 subunit shows less effects on the voltage sensor, although both affect the Ca2+ binding affinity and the allosteric coupling between sensors, and sensors and gate (Cox and Aldrich, 2000; Orio and Latorre, 2005; Yang et al., 2008).
Previous studies have identified the N terminus, S0 segment, and voltage-sensing residues in Slo1 that play important roles for the function of the β1 subunit (Wallner et al., 1996; Morrow et al., 2006; Yang et al., 2008). However, the structural components in Slo1 that are important for the function of the β2 subunit are not known. In this study, we take advantage of differential effects of the β2 subunit on two BK channel orthologs to make chimeras between them and test which structural component confers the properties of its parent channel in response to the modulation by the β2 subunit. These experiments identified two regions of the Slo1 subunit that are critical for the β2-dependent increase of Ca2+ sensitivity. The first region contains the extracellular N terminus and S0 segment. The second region, which is specifically important for the β2-dependent increase of Ca2+ sensitivity, contains the linker connecting S6 and the cytoplasmic domain, the C-Linker, and the N terminus of RCK1, the AC region (βA–αC) (Fig. 1). This segment has been shown previously to mediate the allosteric coupling between Ca2+ binding and the opening of the activation gate (Niu et al., 2004; Krishnamoorthy et al., 2005; Yang et al., 2010). Our results in this study support the role of this segment in Ca2+-dependent activation of BK channels and suggest that the β2 subunit enhances the allosteric coupling between Ca2+ binding and the opening of the activation gate.
Materials and Methods
Mutagenesis and expression.
All chimeras and point mutations were generated from the mbr5 splice variant of mouse Slo1 (mSlo1) (Butler et al., 1993) and the A1C2E1G3I0 splice variant of Drosophila Slo1 (dSlo1) (Adelman et al., 1992) in the pBSC–MXT vector. The protein sequences of mSlo1 and dSlo1 used in the design of each chimera are shown in Table 1. Human β1 and β2 (KCNMB1 and KCNMB2; GenBank accession numbers U25138 and AF209747, respectively) cDNAs were subcloned into pcDNA3.1 (+). The β2 N terminus-deleted (β2ND) subunit was created by removing amino acids from positions 2 through 20. All chimeras and mutations were made using overlap-extension PCR (Shi et al., 2002) with Pfu polymerase (Stratagene). The PCR-amplified regions for all constructs were verified by sequencing. cRNA was transcribed in vitro using T3 polymerase (Ambion) for all Slo1 constructs and T7 polymerase (Ambion) for β1 and β2ND subunits. A total of 0.05–20 ng of Slo1 cRNA or a mixture of 5–15 ng of Slo1 and 25–50 ng of β subunit cRNAs were injected into each Xenopus laevis oocyte. Oocytes were incubated in ND96 solution (in mm: 96 NaCl, 2 KCl, 1.8 CaCl2, 1 MgCl2, and 5 HEPES, pH 7.6) at 18°C for 3–6 d before recording.
Table 1.
Residue information of mSlo1–dSlo1 chimeras
| Chimera name | Residues from |
|
|---|---|---|
| dSlo1 | mSlo1 | |
| C1 | 1-596 | 580-1169 |
| C2 | 1-336 | 323-1169 |
| C3 | 1-248 | 234-1169 |
| C4 | 1-215 | 201-1169 |
| C5 | 1-147 | 134-1169 |
| m[dS0] | 1-70 | 44-1169 |
| m[dLinker + dAC] | 340-432 | 1-325 and 419-1169 |
| m[dS0 + dLinker + dAC] | 1-70 and 340-432 | 44-325 and 419-1169 |
| d[mS0] | 71-1164 | 1-43 |
| d[mLinker + mAC] | 1-339 and 433-1164 | 326-418 |
| d[mS0 + mLinker + mRCK1c] | 71-346 and 559-1164 | 1-43 and 320-543 |
Electrophysiology.
Macroscopic currents were recorded from inside-out patches formed with borosilicate pipettes of ∼0.9–1.5 MΩ resistance. The data were acquired using an Axopatch 200-B patch-clamp amplifier (Molecular Devices) and Pulse acquisition software (HEKA). Recordings were digitized at 20 μs intervals and low-pass filtered at 10 kHz with the four-pole Bessel filter built in the amplifier. Capacitive transients and leak currents were subtracted using a P/5 protocol. Experiments were conducted at room temperature (20–22°C). The pipette solution contained the following (in mm): 140 KMeSO3, 20 HEPES, 2 KCl, and 2 MgCl2, pH 7.2. The internal solution contained the following (in mm): 140 KMeSO3, 20 HEPES, 2 KCl, and 1 N-(2-hydroxyethyl) ethylenediamine-N,N,N-triacetic acid (HEDTA), pH 7.2. CaCl2 was added to the internal solution to give the appropriate free [Ca2+]i, which was measured with a calcium-sensitive electrode (Orion Research). 18-Crown-6-tetracarboylic acid (50 μm; Sigma-Aldrich) was added to internal solutions to chelate Ba2+. For nominal 0 μm [Ca2+]i, the same internal solution was used except that HEDTA was substituted by 5 mm EGTA and no CaCl2 was added. The free [Ca2+] in nominal 0 μm [Ca2+]i solution is 0.5 nm. Experiments were conducted over a period of time in which multiple different [Ca2+]i were made and their concentrations varied. For the sake of simplicity, the 2 μm Ca2+ is referred to as ∼2 μm when the concentration varied from 1.8 to 2.3 μm, and the saturating [Ca2+]i is referred to as ∼100 μm when the concentration varied from 87.1 and 118.4 μm.
Data analysis.
The relative conductance was determined by measuring tail current amplitudes at indicated voltages for all channels with and without β subunits. The conductance–voltage (G–V) relationships for all channels with and without β subunits were fitted with the Boltzmann equation: , where G/Gmax is the ratio of conductance to maximum conductance in the same [Ca2+]i, z is the number of equivalent charges, V½ is the voltage at which the channel is 50% activated, e is the elementary charge, k is Boltzmann's constant, and T is the absolute temperature. Curve fittings were done with Igor Pro software (WaveMetrics) using the Levenberg–Marquardt algorithm to perform nonlinear least-squares fits. Statistics were performed using Origin 6.1 (OriginLab Corp.), and independent/unpaired t test was performed. A p value of <0.05 was considered significant.
Monod–Wyman–Changeux model.
Monod–Wyman–Changeux (MWC) model fits were performed using the following equation:
 |
where Popen is the open probability of the channel, L0 is the steady-state equilibrium constant from open to closed channels ([C0]/[O0]) in the absence of Ca2+ binding at 0 mV, z, e, k, and T are same as in the Boltzmann equation (see above), and KC and KO are the dissociation constants of Ca2+ in the closed and open states, respectively. MWC model code was written and executed in MATLAB version 7.4 (MathWorks).
Because BK channels are activated by both voltage and Ca2+, the ideal condition for measuring Ca2+ sensitivity of channel activation would be in the absence of voltage sensor movements. Such measurements have been done at very negative voltages (less than −130 mV) at which the voltage sensor of BK channels is kept at the resting state (Horrigan and Aldrich, 2002). Ca2+-dependent activation of BK channels under such a condition could be fitted by the MWC model, and the results show that the parameters KC and KO have very similar values as those obtained by the MWC model fitting to the G–V relations (Cox et al., 1997; Horrigan and Aldrich, 2002; Sweet and Cox, 2008; Yang et al., 2010), which is the method we used in this study. The lack of a large influence of voltage on Ca2+ affinity measurement is not surprising because it has been shown that voltage and Ca2+ activate the channel through distinct mechanisms that have little interaction (Cui and Aldrich, 2000; Horrigan and Aldrich, 2002; Sweet and Cox, 2008). In the MWC model used in this study, each Slo1 subunit is assumed to contain a single Ca2+ binding site, although previous studies have proposed two putative Ca2+ binding sites in each Slo1 subunit (Schreiber and Salkoff, 1997; Xia et al., 2002). A model composed of two Ca2+ binding sites would not provide any additional information because we are interested in the effect on the overall Ca2+ sensitivity of the BK channel. Despite this simplification, the MWC model used in this study provides an appropriate account of the Ca2+ sensitivity changes in BK channels.
Results
The β2ND subunit modulates Ca2+ sensitivity of mSlo1 and dSlo1 differently
Besides increasing Ca2+ sensitivity of mSlo1 channels, the β2 subunit also inactivates the channel by a “ball and chain” mechanism such that the N terminus of the β2 subunit blocks the open channel (Ding et al., 1998; Hicks and Marrion, 1998; Wallner et al., 1999; Xia et al., 1999, 2003) (Fig. 2A). To study increases in Ca2+ sensitivity conferred by the β2 subunit in the absence of inactivation, we deleted the amino acids 2–20 of the human β2 subunit, which resulted in the β2ND subunit that no longer causes inactivation (Fig. 2B) (Wallner et al., 1999; Xia et al., 1999, 2003; Brenner et al., 2000). We coexpressed the β2ND subunit with mSlo1 and dSlo1 and observed the changes to their properties the (Fig. 2). The association of β2ND increases the rate of activation of mSlo1 (Brenner et al., 2000; Orio and Latorre, 2005; Orio et al., 2006; Lee and Cui, 2009) and decreases the rate of deactivation of both mSlo1 and dSlo1 (Fig. 2B). Because the association of β2ND with mSlo1 shifts the G–V relation to more negative voltages, the deactivation time course was measured at a more negative voltage with β2ND than without β2ND. However, mSlo1 with β2ND still deactivates more slowly than mSlo1 alone (τDact, 5.7 ± 0.3 ms at −120 mV and 1.8 ± 0.1 ms at −50 mV, respectively when prepulsed to +100 mV, p < 0.001 with n = 14 and 55, respectively). Measured at the same voltage, dSlo1 with β2ND also deactivates significantly more slowly than dSlo1 alone (τDact, 1.5 ± 0.2 and 0.8 ± 0.1 ms at −50 mV, respectively, when prepulsed to +200 mV, p < 0.001 with n = 7 and 23, respectively). These changes in the macroscopic currents indicate a physical association between the β2ND subunit and Slo1 orthologs. Coexpression of the wild-type (WT) β2 subunit with both mSlo1 and dSlo1 confer inactivation in the channels (Fig. 2A), further indicating that the β2 subunits associate with both Slo1 orthologs. However, the β2ND subunit has different effects on the Ca2+-dependent activation of the two Slo1 orthologs. The association of the β2ND subunit shifts the G–V relation of mSlo1 channels to more negative potentials by ∼100 mV at the saturating [Ca2+]i of ∼100 μm but has little effect on the G–V of dSlo1 channels (V½ of dSlo1, −19.3 ± 2.5 mV and V½ dSlo1 + β2ND, −23.8 ± 2.7 mV, p > 0.05; V½ is the voltage at which the G–V relation is half-maximum) (Fig. 2C). At nominal 0 [Ca2+]i, the β2ND association does not alter the G–V relation of the mSlo1 channels (V½ of mSlo1, 178.3 ± 1.1 mV and V½ mSlo1+β2ND, 181.9 ± 1.8 mV, p > 0.05) (Fig. 2C) (Orio and Latorre, 2005), indicating that β2ND modulates Ca2+-dependent activation of mSlo1. In contrast, the association of β2ND with dSlo1 has no significant effect at any [Ca2+]i tested in our study. For instance, at ∼2 μm Ca2+i, V½ of dSlo1 and dSlo1+β2ND is 243.4 ± 6.5 and 241.5 ± 5.2 mV, respectively (p > 0.05) (Fig. 2C). The plot of V½ versus [Ca2+]i shows that the association of the β2ND subunit results in a gradual increase in the negative shift of the mSlo1 G–V relation with increasing [Ca2+]i but no shift of the dSlo1 G–V relation (Fig. 2D). In [Ca2+]i ≤ 1 μm, few dSlo1 channels activate at voltages within the experimental limit (up to +300 mV) so that the G–V relations cannot be determined in these [Ca2+]i. The results in Figure 2 clearly show that the β2ND subunit modulates Ca2+-dependent activation of mSlo1 but not dSlo1.
Figure 2.
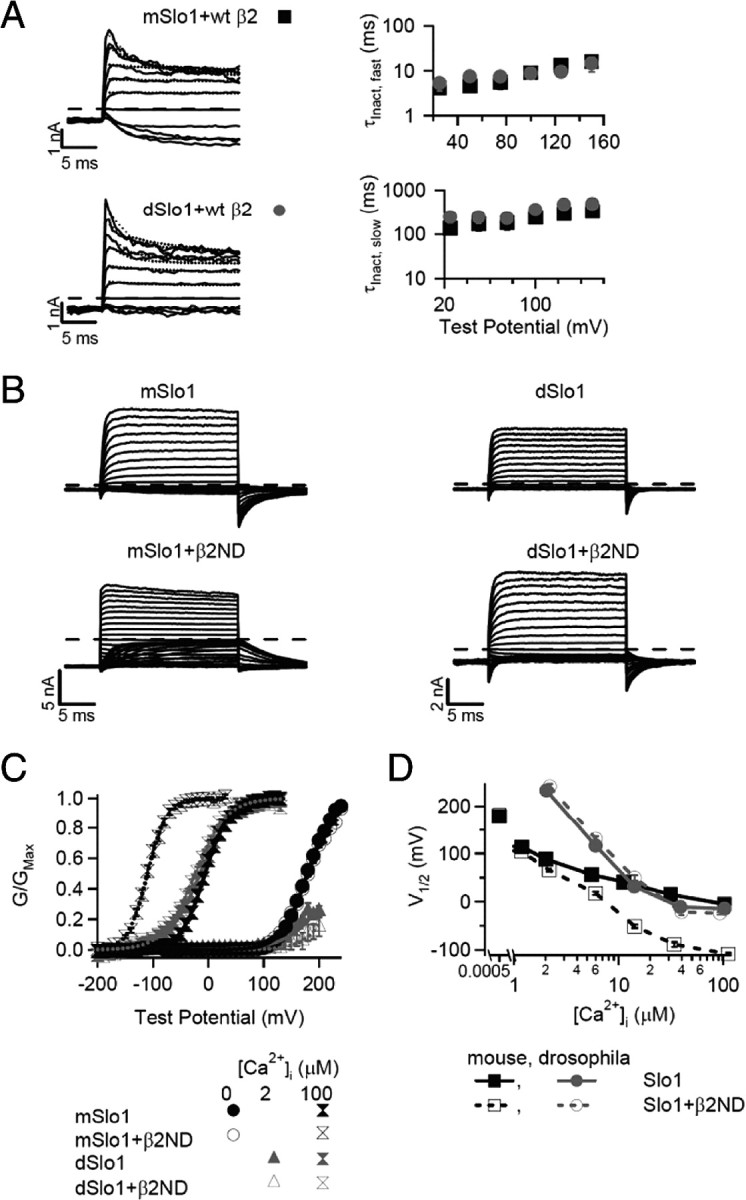
Differential modulation by β2ND on Ca2+ sensitivity of mSlo1 and dSlo1. A, Macroscopic currents of mSlo1 and dSlo1 with WT hβ2 at ∼100 μm [Ca2+]i (left). The voltage pulses are from −100 to +150 mV for 1 s with 25 mV increments (shown only the first 20 ms), and the prepulse potential is −140 mV for 195 ms (shown only the last 5 ms). The dotted line represents the biexponential fit of the inactivation profile, and the long dashed line represents the zero current line for each current trace. On the right, voltage dependence of the inactivation time constant is shown. The fast and slow components of τInact were obtained by fitting current traces with a biexponential function from the peak amplitude to steady state (n = 10 for mSlo1 + WT β2, except at +150 mV, n = 9; n = 5 for dSlo1 + WT β2, except at +150 mV, n = 4). B, Macroscopic currents of mSlo1 and dSlo1 with and without β2ND at ∼100 μm [Ca2+]i. Voltage pulses are from −200 to 100 mV with 10 mV increments, and the repolarizing potential is −50 mV, except for mSlo1 + β2ND, which is −80 mV. The dashed line represents the zero current line for each current trace. C, Mean G–V relationship of mSlo1/dSlo1 with and without β2ND in 0, ∼2, and ∼100 μm [Ca2+]i, fitted with Boltzmann equation (smooth lines). The error bars in this and other figures show the SEM. D, V½ versus [Ca2+]i plot of mSlo1/dSlo1 with and without β2ND. The number of patches for each dataset are for the following (μm): [Ca2+]i, 0, 1, 2, 5, 10, 30, 100; mSlo1, 118, 33, 29, 29, 42, 25, 56; mSlo1 + β2ND, 44, 8, 8, 11, 9, 12, 28; dSlo1, 0, 0, 11, 19, 15, 9, 27; dSlo1 + β2ND, 0, 0, 3, 4, 7, 8, 14.
To further illustrate the differences in Ca2+ sensitivity imparted by β2ND on mSlo1 and dSlo1 channels, a voltage-dependent MWC model (Cox et al., 1997) was used to fit the G–V relations at different [Ca2+]i (see Fig. 6; Table 2). In mSlo1 channels, the association of β2ND subunit increases the dissociation constant for Ca2+ binding in the closed state (KC) by >300% and decreases the dissociation constant in the open state (KO) by 14% (Table 2), same as reported previously (Orio and Latorre, 2005). Because of the disproportional change in the two dissociation constants, the ratio c = KO/KC is smaller in mSlo1 + β2ND than in mSlo1 channels, which indicates that Ca2+ favors the open state in the closed–open equilibrium more with the association of β2ND (Table 2) (Cox et al., 1997). The association of β2ND does not alter voltage dependence of mSlo1 channels, which is reflected in similar values of the parameters z and L0, where z is the number of gating charges in the voltage sensor of the channels, and L0 is the equilibrium constant of channel from the open to the closed state at 0 mV and in the absence of Ca2+ (Table 2). In dSlo1 channels, the association of β2ND does not change KC, KO, or voltage dependence (Table 2). Collectively, these results show that the β2ND subunit specifically enhances Ca2+ sensitivity in mSlo1 channels but results in no such changes in dSlo1 channels, although β2ND is associated with dSlo1 subunit.
Figure 6.
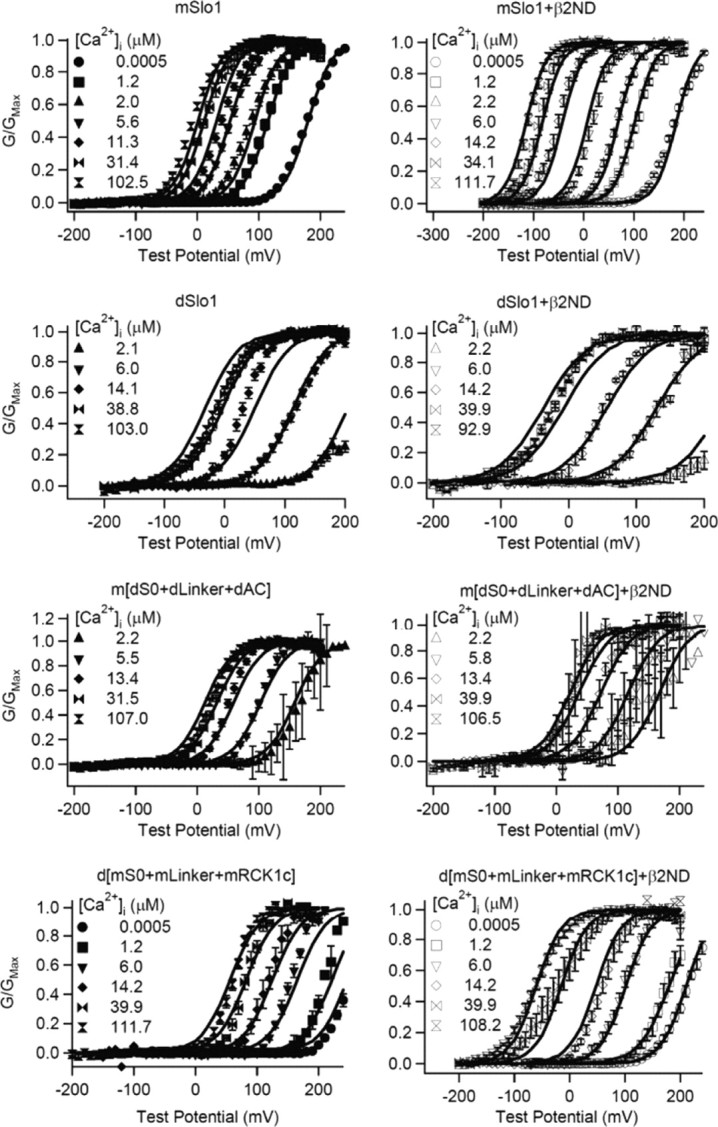
MWC model fittings of WT and chimera channels. G–V relationships (symbols) for mSlo1, dSlo1, m[dS0 + dLinker + dAC], and d[mS0 + mLinker + mRCK1c] with and without β2ND fitted with the MWC model (lines). The [Ca2+]i for each symbol is shown.
Table 2.
Parameters for MWC model fits
| L0 | z | KC (μm) | KO (μm) | 1/c | |
|---|---|---|---|---|---|
| mSlo1 | 9.20E+03 | 1.28 | 7.36 | 0.72 | 10.25 |
| mSlo1 + β2ND | 9.41E+03 | 1.25 | 32.51 | 0.62 | 52.35 |
| dSlo1 | 5.50E+05 | 0.88 | 22.00 | 0.51 | 43.48 |
| dSlo1 + β2ND | 5.51E+05 | 0.78 | 23.95 | 0.53 | 45.28 |
| m[dS0 + dLinker + dAC] | 9.98E+05 | 1.12 | 12.36 | 0.40 | 30.60 |
| m[dS0 + dLinker + dAC] + β2ND | 9.80E+05 | 1.02 | 9.45 | 0.36 | 26.18 |
| d[mS0 + mLinker + mRCK1c] | 3.50E+04 | 1.07 | 30.76 | 3.30 | 9.32 |
| d[mS0 + mLinker + mRCK1c] + β2ND | 7.90E+03 | 1.08 | 71.00 | 2.45 | 28.98 |
The N termini of the transmembrane and cytoplasmic domains of mSlo1 and dSlo1 are necessary and sufficient to account for the different effects of the β2ND subunit
Taking advantage of the differences in the response of the Slo1 orthologs to β2ND modulation, a chimera strategy to exchange the secondary structural regions between mSlo1 and dSlo1 subunits was devised to find the structural components responsible for the β2-dependent enhancement of Ca2+ sensitivity. To examine whether the association of β2ND alters Ca2+ sensitivity of a channel, two criteria were used. First, Ca2+ sensitivity of a channel was estimated by the shift of its G–V relation in ∼100 μm [Ca2+]i in response to the association of β2ND, ΔVhalf = V½(100Ca) with β2ND − V½(100Ca) without β2ND. Second, Ca2+ sensitivity of a channel was computed as the G–V shift ΔV½(0/2-100Ca), in response to an increase of [Ca2+]i from 0 or ∼2 to ∼100 μm, the change in Ca2+ sensitivity resulted from the association of β2ND was ΔΔVhalf = ΔV½(0/2-100Ca) with β2ND − ΔV½(0/2-100Ca) without β2ND, which was then normalized to ΔΔVhalf of the WT mSlo1 obtained under the same condition (Fig. 3). Using these two criteria, mSlo1 exhibited greater modulation by the association of β2ND than dSlo1 (Fig. 3).
Figure 3.

Effects of chimeras of mSlo1/dSlo1 on the β2ND modulation of Ca2+ dependence. The black bars show the change in Ca2+ sensitivity between with and without β2ND, normalized to mSlo1 (see Results for definitions). The gray bars show the Ca2+ response at ∼100 μm [Ca2+]i between with and without β2ND. The symbol * means that ∼2 μm [Ca2+]i was used for comparison, and ** means that the G–V could not be determined at 0 or ∼2 μm [Ca2+]i. Bottom shows the schematic of different chimeras of mSlo1 and dSlo1 used in the top. The number of patches for each dataset are as follows for either ∼0/2 and 100 μm [Ca2+]i: C1, 3, 5; C1 + β2ND, 9, 11; C2, 4, 3; C2 + β2ND, 5, 4; C3, 7, 4; C3 + β2ND, 5, 5; C4, 5, 5; C4 + β2ND, 8, 3; C5, 7, 5; C5 + β2ND, 3, 4; m[dS0]: 9, 6; m[dS0] + β2ND, 4, 7; m[dLinker + dAC], 7, 20; m[dLinker + dAC] + β2ND, 0, 20; m[dS0 + dLinker + dAC], 5, 14; m[dS0 + dLinker + dAC] + β2ND: 2, 9; d[mS0], 3, 20; d]mS0] + β2ND, 0,12; d[mLinker + mAC], 13, 7; d[mLinker + mAC] + β2ND, 23, 13; d[mS0 + mLinker + mRCK1c], 11, 16; d[mS0 + mLinker + mRCK1c] + β2ND, 15, 14.
The chimeras developed for this study are shown in the bottom of Figure 3, where the initial designs (chimeras C1 through m[dS0]) were composed of dSlo1 channels with increasing mSlo1 regions substituting the dSlo1 counterparts. In chimeras C1–C5, the addition of mSlo1 regions did not confer β2ND modulation; rather, the Ca2+ sensitivity of these channels and their responses to the association of β2ND were similar to that of dSlo1 (Fig. 3). Even the chimera containing only the N terminus of dSlo1, including the transmembrane segment S0 (m[dS0]), shows an ∼40% reduced response to the β2ND modulation, indicating that the N terminus of mSlo1 is important for the β2ND modulation of Ca2+ sensitivity (Fig. 3). The result of m[dS0] in Figure 3 is computed from the current measurements and G–V relations shown in Figure 4, A and B. Figure 4C shows additional studies of the Ca2+ sensitivity of this chimera where the V½ of G–V relations of the channel with (black unfilled circles) and without (black filled circles) β2ND in various [Ca2+]i from nominal 0 to saturating ∼100 μm are plotted versus [Ca2+]i. For comparison, similar data from dSlo1 (blue dotted and solid lines) and mSlo1 (orange dotted and solid lines) are also shown (Fig. 4C). The β2ND subunit enhances Ca2+ sensitivity of mSlo1 channels and gradually shifts its G–V relation as [Ca2+]i increases (Fig. 2) so that the V½–[Ca2+]i relations of mSlo1 channels with and without β2ND bifurcate (Fig. 4C, orange). On the contrary, the β2ND subunit does not alter Ca2+ sensitivity of dSlo1 channels so that the V½–[Ca2+]i relations of dSlo1 channels with and without β2ND superimpose (Fig. 4C, blue). Although the V½–[Ca2+]i relations of m[dS0] with and without β2ND bifurcate (Fig. 4C, black), the difference between the two curves is smaller than that of mSlo1 at all non-zero [Ca2+]i, clearly showing that the N terminus of mSlo1 is important for the β2ND-dependent enhancement of Ca2+ sensitivity.
Figure 4.

Effects of the N terminus and the C-Linker/AC region of dSlo1 in mSlo1 background on the β2ND modulation of Ca2+ sensitivity. A, Macroscopic currents of m[dS0], m[dLinker + dAC], and m[dS0 + dLinker + dAC] with and without β2ND at ∼100 μm [Ca2+]i. Voltage pulses are from −200 to 200 mV with 10 mV increments, and the repolarizing potential is −50 mV, except for m[dS0] and m[dLinker + dAC] with β2ND, which is −80 mV. The dashed line represents the zero current line for each current trace. B, G–V relations of chimeras compared with that of mSlo1 (orange) at low (0, ∼2, or ∼10 μm) and ∼100 μm [Ca2+]i. Because of the apparent inactivation of the channels, the G–V relations were measured from both the peak currents and tail currents, and both methods resulted in the same V½. C, V½ versus [Ca2+]i plots of chimeras compared with that of mSlo1 and dSlo1 with and without β2ND. The number of patches for each dataset are as follows (in μm): [Ca2+]i, 0, 1, 2, 5, 10, 30, 100; m[dS0], 9, 6, 4, 4, 4, 4, 6; m[dS0] + β2ND, 4, 3, 3, 4, 3, 5, 7; m[dLinker + dAC], 0, 0, 7, 14, 15, 6, 20; m[dLinker + dAC] + β2ND, 0, 0, 0, 0, 4, 5, 20; m[dS0 + dLinker + dAC], 0, 0, 5, 4, 4, 4, 14; m[dS0 + dLinker + dAC] + β2ND, 0, 0, 2, 3, 4, 2, 9.
To examine whether the N terminus of mSlo1 alone is responsible for the β2ND-dependent enhancement of Ca2+ sensitivity, we made the converse chimera of m[dS0], d[mS0], in which the N terminus to the end of S0 of mSlo1 is transplanted into dSlo1. Because few d[mS0] channels open in [Ca2+]i <∼2 μm (Fig. 5A,B), the change of Ca2+ sensitivity attributable to the association of β2ND is only estimated by ΔVhalf in Figure 3, which suggests that the N terminus of mSlo1 is not sufficient to render dSlo1 the same response to β2ND as mSlo1. Additional studies of Ca2+ sensitivity of this chimera in various [Ca2+]i show that the V½–[Ca2+]i relations of d[mS0] with and without β2ND only bifurcate at high [Ca2+]i (more than ∼30 μm), and the difference between the two curves is smaller than that of mSlo1, demonstrating that the N terminus of mSlo1 is not the only structural component important for the β2ND-dependent enhancement of Ca2+ sensitivity (Fig. 5C).
Figure 5.

Effects of the N terminus and C-Linker/AC region of mSlo1 in dSlo1 background on the β2ND modulation of Ca2+ sensitivity. A, Macroscopic currents of d[mS0], d[mLinker + mAC], and d[mS0 + mLinker + mRCK1c] with and without β2ND at ∼100 μm [Ca2+]i. Voltage pulses are from −200 to 200 mV, except for d[mS0 + mLinker + mRCK1c] + β2ND, which are from −200 to 100 mV, with 10 mV increments. The repolarizing potential is −50 mV. The dashed line represents the zero current line for each current trace. B, G–V relations of chimeras with and without β2ND at low (0 or ∼2 μm) and ∼100 μm [Ca2+]i compared with that of dSlo1 (blue). C, V½ versus [Ca2+]i plots of chimeras compared with that of mSlo1 and dSlo1 with and without β2ND. The number of patches for each dataset are as follows (in μm): [Ca2+]i, 0, 1, 2, 5, 10, 30, 100; d[mS0], 0, 0, 3, 10, 6, 4, 20; d[mS0] + β2ND, 0, 0, 0, 3, 2, 3, 16; d[mLinker + mAC], 13, 5, 4, 4, 4, 4, 7; d[mLinker + mAC] + β2ND, 23, 5, 3, 3, 5, 5, 13; d[mS0 + mLinker + mRCK1c], 11, 2, 4, 7, 6, 16; d[mS0 + mLinker + mRCK1c] + β2ND, 15, 5, 5, 5, 6, 14.
Previous studies have shown that the AC region (the βA–αC of RCK1 domain) (Krishnamoorthy et al., 2005; Yang et al., 2010) and the C-Linker (Niu et al., 2004; Krishnamoorthy et al., 2005) are important in coupling Ca2+ binding to opening of the activation gate. To examine whether this segment is also important for the β2ND-dependent enhancement of Ca2+ sensitivity, we studied the chimera channels in which this segment from dSlo1 is transplanted into mSlo1, m[dLinker + dAC]. Its response to β2ND is estimated using ΔVhalf (Fig. 3), which shows a dramatic reduction from that of mSlo1 and is similar to that of dSlo1, suggesting that the N terminus of the cytoplasmic domain is important for the β2ND-dependent enhancement of Ca2+ sensitivity. Surprisingly, although the inactivation “ball” is deleted in β2ND, its association with m[dLinker + dAC] causes inactivation of the channel (Fig. 4A). The mechanism of this inactivation is not clear at this time and needs additional study. Nevertheless, additional studies of the Ca2+ sensitivity of this chimera in several [Ca2+]i shows that the V½–[Ca2+]i relations with and without β2ND superimpose (Fig. 4C), confirming that β2ND can no longer enhance Ca2+ sensitivity of this channel. The converse chimera in the dSlo1 background, d[mLinker + mAC] showed no increase in Ca2+ sensitivity with the association of β2ND either (Fig. 5). Together, these results suggest that the N terminus of the cytoplasmic domain of mSlo1 is also necessary but not sufficient to account for the difference in the β2ND-dependent increase of Ca2+ sensitivity between mSlo1 and dSlo1.
Either terminus of the transmembrane or cytoplasmic domain of mSlo1 is important for the β2ND-dependent increase of Ca2+ sensitivity, and, as expected, the chimera containing both termini from dSlo1 in the mSlo1 background, m[dS0 + dLinker + dAC], showed no increase in Ca2+ sensitivity in response to β2ND, similar to dSlo1 (Figs. 3, 4). We studied the converse chimera in dSlo1 background, d[mS0 + mLinker + mAC], to examine whether N termini of both the transmembrane and cytoplasmic domains of mSlo1 are sufficient to render the β2ND-dependent increase of Ca2+ sensitivity in dSlo1. However, this chimera failed to express any functional channel with β2ND subunit for reasons that are currently not understood. Therefore, we modified this chimera by extending the AC region to include βG of the RCK1 domain that results in d[mS0 + mLinker + mRCK1c]. The coexpression of this chimera with β2ND showed increased Ca2+ sensitivity that is more like mSlo1 than dSlo1 (Figs. 3, 5), suggesting that the N termini of the transmembrane and cytoplasmic domains of mSlo1 are critical for the differential β2ND-dependent enhancement of Ca2+ sensitivity in dSlo1 and mSlo1.
The swapping of channel properties is further illustrated by fitting the G–V relations of chimeras m[dS0 + dLinker + dAC] and d[mS0 + mLinker + mRCK1c] to the MWC model (Fig. 6). The association of β2ND with m[dS0 + dLinker + dAC] produces a minor reduction in both KC and KO which does not change the c factor (Fig. 6; Table 2), and consequently there was a lack of G–V shift to negative potentials in the presence of Ca2+ (Fig. 4). However, the association of β2ND with d[mS0 + mLinker + mRCK1c] increases KC and decreases KO. These changes are in the same direction as those when β2ND is associated with mSlo1, which results in reduction in c factor and enhanced G–V shifts in response to [Ca2+]i increase (Fig. 6; Table 2).
Different molecular mechanisms for the β1- and β2-dependent enhancement of Ca2+ sensitivity
Similar to the β2 subunit, the β1 subunit also enhances the apparent Ca2+ sensitivity of BK channels to cause a G–V shift to more negative potentials in the presence of Ca2+, and the two β subunits share a >60% sequence homology (Brenner et al., 2000; Orio and Latorre, 2005). Furthermore, previous studies have shown that the β1 subunit also specifically modulates Ca2+ sensitivity of the mSlo1 channel but not that of dSlo1, and the N terminus of mSlo1 including S0 is critical for such modulation (Wallner et al., 1996; Morrow et al., 2006). To examine whether the β1-dependent enhancement of Ca2+ sensitivity is also sensitive to the difference between mSlo1 and dSlo1 in the N terminus of the cytoplasmic domain, we studied the coexpression of the β1 subunit with the chimera m[dLinker + dAC] (Fig. 7). In the saturating [Ca2+]i of ∼100 μm, the association of either the β1 or β2ND subunit shifts the G–V relation of mSlo1 to more negative potentials (Fig. 7A). The mutation m[dLinker + dAC] nearly abolishes the response of G–V relation to the association of β2ND but not β1 (Fig. 7A). Similar results are observed in various [Ca2+]i (Fig. 7B), indicating that the differences in the structure of N-terminal part of the cytoplasmic domain between mSlo1 and dSlo1 are critical for β2 modulation but inconsequential for β1 function. Conversely, for chimera m[dS0]/h[dS0], there was a lack of G–V shift in response to the association of both β2ND and β1 (Wallner et al., 1996), indicating that the N terminus of the membrane-spanning domain is critical for the function of both the β1 and β2 subunits.
Figure 7.

The role of the C-Linker and AC region of mSlo1 in the β1 and β2ND modulation of Ca2+ sensitivity. A, G–V relations of mSlo1 and m[dLinker + dAC] with and without β1 or β2ND in ∼100 μm [Ca2+]i. B, V½ versus [Ca2+]i plots of mSlo1 and m[dLinker + dAC] with β1 (left) or β2ND (right). The number of patches are as follows (in μm): [Ca2+]i, 0, 1, 2, 5, 10, 30, 100: mSlo1 + β1, 38, 13, 15, 11, 12, 8, 21; m[dLinker + dAC] + β1, 0, 0, 0, 10, 6, 9, 26.
BK channels contain two high-affinity Ca2+ binding sites located at D367 in the RCK1 domain and the Ca2+ bowl in the RCK2 domain (Fig. 1). Previous studies show that the two binding sites have slightly different affinities for Ca2+ and they contribute independently with only a small cooperativity to Ca2+-dependent activation (Xia et al., 2002; Qian et al., 2006; Sweet and Cox, 2008). A recent study shows that mutations in the AC region specifically affect Ca2+-dependent activation derived from the D367 site but not that from the Ca2+ bowl (Yang et al., 2010). These results indicate that the two Ca2+ binding sites may couple to the activation gate through distinct structural pathways. To examine which of these pathways is affected by the β subunits, we studied the effects of β2ND and β1 on each of the pathways by mutating the Ca2+ binding site in one pathway. Mutating either Ca2+ binding site of mSlo1 by D367A or 5D5N (five consecutive Asp residues in the Ca2+ bowl are mutated to Asn) reduced overall G–V shift in response to a [Ca2+]i increase from 0 to saturating ∼100 μm in both the absence and presence of β2ND subunit (Fig. 8A). Nevertheless, the G–V shift is larger in the presence of β2ND (Fig. 8A, gray bars). When both Ca2+ binding sites are mutated, channel activation is no longer sensitive to Ca2+ in either the presence or absence of β2ND (Fig. 8). These results indicate that the β2 subunit modulates mSlo1 activation as long as at least one Ca2+ binding site is intact. Interestingly, regardless of whether one or both Ca2+ binding sites are intact, the association of β2ND results in similar increase of G–V shift (Fig. 8B). To the contrary, although the β1 subunit also modulates mSlo1 activation as long as at least one Ca2+ binding site is intact (Fig. 8A, white bars), the mutation of either Ca2+ binding site reduces the ability of β1 to increase G–V shift (Fig. 8B). This result suggests that, although β1 subunit is primarily known to affect the voltage-dependent activation (Cox and Aldrich, 2000; Bao and Cox, 2005; Orio and Latorre, 2005; Yang et al., 2008), it may also be sensitive to the integrity of the Ca2+ binding site, which is supported by Bao and Cox (2005). Together, these experiments indicate that the β subunits modulate both pathways of Ca2+-dependent activation, but the β1 and β2 subunits may target different points in the Ca2+-dependent activation. Based on the recently published structures of the BK channel gating ring, the Linker and AC region reside closest to the pore (Wu et al., 2010; Yuan et al., 2010), which is the sole structural gateway to couple both Ca2+ binding sites to the activation gate (Fig. 9). The β2 subunit may directly modify the allosteric mechanism in this region to affect the coupling between Ca2+ binding and channel opening. The β1 subunit, conversely, may alter the coupling between Ca2+ binding and channel opening via different mechanisms, possibly involving the VSD. The difference of the two β subunits in response to Ca2+ binding site mutations may be the manifestations of such mechanistic differences. However, the exact nature of these allosteric mechanisms are not fully understood.
Figure 8.

Dependence of the β1 and β2 modulation of Ca2+ sensitivity on Ca2+ binding sites in mSlo1. A, Bar graph of the G–V shift in response to a [Ca2+]i change from 0–100 μm WT, single Ca2+ binding site mutations (D367A and 5D5N), double Ca2+ binding site mutation (D367A/5D5N), and double Ca2+ binding site plus the Mg2+ binding site mutation (D367A/5D5N/E399N) without (black) and with β1 (white) or β2ND (gray). B, Differences in ΔV½ from A of β1 (white) and β2ND (gray) with WT, single, double, and triple binding site mutations. The number of patches for each dataset are as follows for ∼0 and ∼100 μm [Ca2+]I, respectively: mD367A, 10, 9; mD367A + β1, 9, 9; mD367A + β2ND, 5, 7; 5D5N, 6, 11; 5D5N + β1, 8, 9; 5D5N + β2ND, 4, 8; mD367A/5D5N, 14, 6; mD367A/5D5N + β1, 6, 5; mD367A/5D5N + β2ND, 5, 5; mD367A/5D5N/E399N, 15, 15; mD367A/5D5N/E399N + β1, 5, 9; mD367A/5D5N/E399N + β2ND, 8, 12.
Figure 9.

A structural model for the β2 subunit modulation. Two opposing mSlo1 and β2 (orange) subunits of BK channels are shown in which the diagrams for the VSD and S0 segment are constructed around the pore of the MthK channel (Protein Data Bank identification number 1LNQ) (Jiang et al., 2002). The gating ring of the BK channel (Protein Data Bank identification number 3MT5) (Yuan et al., 2010) is aligned to the MthK channel using Chimera version 1.4.1 (University of California, San Francisco). VMD (Visual Molecular Dynamics) version 1.8.7 (University of Illinois at Urbana-Champaign) was used to show the aligned structure in diagram and surface representation. The helices in the VSD are shown as cylinders and are positioned according to the KV1.2 channel. The TM segments of the β2 subunit are positioned according to Zakharov et al. (2009). Green regions in the Slo1 subunit (S0, C-Linker and AC) are involved in the β2 modulation of Ca2+ sensitivity.
Discussion
The β subunits of BK channels modulate specific properties of the pore-forming Slo1 subunit. In the case of the β2 subunit, the BK channels exhibit increased Ca2+ sensitivity by shifting the V½ to more negative potentials with increasing [Ca2+]i. In this study, we identified the regions in Slo1 targeted by the β2 subunit modulation by taking advantage of different responses of two BK channel homologs, mSlo1 and dSlo1, to the β2 subunit modulation. We found two regions in mSlo1 that are both necessary and sufficient to account for the enhanced Ca2+ sensitivity with the β2 subunit compared with dSlo1: the N terminus of the channel to S0 and the N terminus of the cytoplasmic domain, C-Linker/AC region. Both of these structural regions from mSlo1 are required for conferring Ca2+ sensitivity in dSlo1, but the replacement of either one in mSlo1 by the counterpart in dSlo1 reduced or abolished the β2-dependent Ca2+ sensitivity increase.
The N terminus of mSlo1 including S0 is important for both the β1 and β2 subunits to enhance Ca2+ sensitivity of channel activation (Figs. 3, 5) (Wallner et al., 1996; Morrow et al., 2006), which is consistent with the structural and functional role of S0. Using disulfide cross-linking, Liu et al. (2008a, 2010) showed that S0 is an integral part of the voltage-sensing domain of BK channels; its N-terminal extracellular end is close to that of S3 and S4. Consistently, mutations in S0 alter voltage dependence of channel activation, suggesting that it may interact with other membrane-spanning segments of the VSD (Koval et al., 2007). Similar disulfide cross-linking studies revealed that, when the β1 (Liu et al., 2008b, 2010), β2 (Zakharov et al., 2009), and β4 (Wu et al., 2009) subunits associate with Slo1, TM2 is located close to S0 while TM1 is located close to S2. These results suggest that S0 may directly interact with the TM2 of β subunits, whereas the VSD may serve as a scaffold for the conformation of β subunits. Both the β1 and β2 subunits are unable to enhance Ca2+ sensitivity of dSlo1 (Wallner et al., 1996), and a switch of the N terminus and S0 of mSlo1 to that of dSlo1 reduces the function of both β subunits. These results may arise because, although the β subunits can associate with dSlo1 (Fig. 2), the S0 of dSlo1 may alter either the direct TM2–S0 interaction or the structure of the VSD that leads to a conformational change in β subunits.
Contrary to the N terminus of the channel, the C-Linker and AC region of mSlo1 are specifically important for the β2, but not β1, subunit to modulate Ca2+ sensitivity (Fig. 7). This result is consistent with previous findings that the cytoplasmic termini of the β1 and β2 subunits are important in determining their specific effects on channel gating (Orio et al., 2006; Wang and Brenner, 2006). Taking these results together, it is likely that the C-Linker and AC region of Slo1 may interact with the cytosolic domain of the β2 subunit either directly or through an allosteric mechanism. Previous studies show that changes in the length of the C-Linker alter Ca2+ sensitivity of the channel (Niu et al., 2004), whereas mutations in the AC region revealed an allosteric network important for Ca2+ dependence of channel gating (Krishnamoorthy et al., 2005; Yang et al., 2010). Our results further support the important role of the C-Linker and AC region in mediating the coupling between Ca2+ binding and channel opening and show that the β2 subunit may modulate Ca2+ sensitivity by altering such coupling. Conversely, the switch of the C-Linker and AC region from mSlo1 to that of dSlo1 has no effect on the β1 modulation of Ca2+ sensitivity (Fig. 7). This result could be attributable to the fact that β1 does not interact with the C-Linker or AC region so that it is not sensitive to the switch of these regions between mSlo1 and dSlo1. Alternatively, β1 may interact with these regions but the interaction differs from how β2ND interacts with these regions such that β1 is insensitive to the differences between mSlo1 and dSlo1 in these regions. Because of the inherited limitations of chimera studies, our results cannot distinguish these two possibilities. Nevertheless, it is clear that the two β subunits interact with the structural domains in Slo1 differently, which is consistent with previous studies showing that the β1 subunit alters the voltage sensor movements (Cox and Aldrich, 2000), and mutations in the VSD of Slo1 alter the modulation of the β1 but not the β2 subunit (Yang et al., 2008). These results provide a molecular mechanism for the previous results obtained by fitting of functional data to an allosteric model for channel activation, which show that the β2 subunit alters Ca2+ binding affinities and allosteric coupling factors, whereas the β1 subunit has less effects on parameters for Ca2+-dependent activation (Bao and Cox, 2005; Orio and Latorre, 2005).
These experimental results suggest a model in which the TM1 and TM2 segments of the β2 subunit associate with the VSD of mSlo1, whereas the cytoplasmic termini of the β2 subunit may interact with the C-Linker and AC region in mSlo1 to alter the coupling between Ca2+ binding and channel opening, thereby enhancing Ca2+ sensitivity of channel gating (Fig. 9). The interaction between the β2 subunit with S0 and other parts of the VSD may help the β2 subunit adopt a correct conformation and a proper orientation toward the mSlo1 that are critical for its cytoplasmic termini to interact with the C-Linker and AC region. Therefore, mutations in either S0 and the N terminus of the channel or the C-Linker and AC region may interrupt the interactions between the cytoplasmic termini of β2 with the C-Linker and AC region to reduce or abolish the β2 modulation of Ca2+ sensitivity.
Besides S0, other structures in Slo1 may also affect the conformation of the β2 subunit and its orientation toward Slo1. For instance, for chimera m[dS0], in which the N terminus including S0 of mSlo1 is replaced by the counterpart of dSlo1, the β2 subunit still increases Ca2+ sensitivity, although the effect is mostly reduced (Fig. 3), indicating that the β2 subunit remains in a conformation and orientation toward Slo1 that still allows some interaction with the C-Linker and AC region. Conversely, chimera C5, in which an additional structure of mSlo1 is replaced by the counterpart of dSlo1, further reduces the β2 modulation of Ca2+ sensitivity (Fig. 3). This result indicates that the S0–S1 linker and S1 of mSlo1 may also participate in the β2-dependent enhancement of Ca2+ sensitivity (Fig. 3), possibly being involved in maintaining the conformation of the β2 subunit. To test this theory, N terminus to S1 segment of mSlo1 were transplanted in dSlo1 to test whether the C-Linker and AC region of mSlo1 is still required for the β2 modulation of Ca2+ sensitivity. However, because of a large shift in its G–V relation to positive voltages, it is difficult to determine the modulation of Ca2+ sensitivity by the β2 subunit (data not shown).
The β2 subunit enhances Ca2+ sensitivity of mSlo1 channels but has little effect on channel activation in the absence of Ca2+ (Figs. 2, 6; Table 2). Interestingly, for d[mS0 + mLinker + mRCK1c] channels, the association of the β2ND subunit results in a large shift of the G–V relation in the absence of intracellular Ca2+ (Fig. 5), indicating a change in the equilibrium between the open and closed conformation (Fig. 6; Table 2). This result indicates that the β2 subunit not only alters the Ca2+-dependent mechanism to enhance Ca2+ sensitivity but also affects the structure and mechanism important for channel opening in the absence of Ca2+. These results are consistent with the previously reported results of an allosteric model fitting of BK channel gating, which showed that the enhancement of Ca2+ sensitivity by the β2 subunit requires changes to the allosteric coupling factors and intrinsic channel opening (Orio and Latorre, 2005). Such an effect of the β2 subunit may arise from its interactions with the membrane-spanning segments and VSD of Slo1 or with the C-Linker and RCK domains. It could also arise from the possibility that the association of the β2 subunit alters the interactions between the membrane-spanning and cytoplasmic domains of Slo1 that are critical for BK channel activation. These mechanisms may also contribute to the modulation of the β2 subunit on Ca2+ sensitivity.
Footnotes
This work was supported by Epilepsy Foundation (U.S.L.) and National Institutes of Health Grants R01-HL70393 (J.C.) and R01-NS060706 (J.C.). J.C. is the Spencer T. Olin Professor of Biomedical Engineering. The mSlo1 clone was kindly provided to us by Larry Salkoff (Washington University, St. Louis, MO). The β1 and β2 clones were kindly provided by Robert Brenner (University of Texas Health Science Center, San Antonio, TX).
References
- Adelman JP, Shen KZ, Kavanaugh MP, Warren RA, Wu YN, Lagrutta A, Bond CT, North RA. Calcium-activated potassium channels expressed from cloned complementary DNAs. Neuron. 1992;9:209–216. doi: 10.1016/0896-6273(92)90160-f. [DOI] [PubMed] [Google Scholar]
- Bao L, Cox DH. Gating and ionic currents reveal how the BKCa channel's Ca2+ sensitivity is enhanced by its beta1 subunit. J Gen Physiol. 2005;126:393–412. doi: 10.1085/jgp.200509346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brayden JE, Nelson MT. Regulation of arterial tone by activation of calcium-dependent potassium channels. Science. 1992;256:532–535. doi: 10.1126/science.1373909. [DOI] [PubMed] [Google Scholar]
- Brenner R, Jegla TJ, Wickenden A, Liu Y, Aldrich RW. Cloning and functional characterization of novel large conductance calcium-activated potassium channel beta subunits, hKCNMB3 and hKCNMB4. J Biol Chem. 2000;275:6453–6461. doi: 10.1074/jbc.275.9.6453. [DOI] [PubMed] [Google Scholar]
- Butler A, Tsunoda S, McCobb DP, Wei A, Salkoff L. mSlo, a complex mouse gene encoding “maxi” calcium-activated potassium channels. Science. 1993;261:221–224. doi: 10.1126/science.7687074. [DOI] [PubMed] [Google Scholar]
- Cox DH, Aldrich RW. Role of the beta1 subunit in large-conductance Ca2+-activated K+ channel gating energetics. Mechanisms of enhanced Ca2+ sensitivity. J Gen Physiol. 2000;116:411–432. doi: 10.1085/jgp.116.3.411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox DH, Cui J, Aldrich RW. Allosteric gating of a large conductance Ca-activated K+ channel. J Gen Physiol. 1997;110:257–281. doi: 10.1085/jgp.110.3.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui J, Aldrich RW. Allosteric linkage between voltage and Ca2+-dependent activation of BK-type mslo1 K+ channels. Biochemistry (Mosc) 2000;39:15612–15619. doi: 10.1021/bi001509+. [DOI] [PubMed] [Google Scholar]
- Ding JP, Li ZW, Lingle CJ. Inactivating BK channels in rat chromaffin cells may arise from heteromultimeric assembly of distinct inactivation-competent and noninactivating subunits. Biophys J. 1998;74:268–289. doi: 10.1016/S0006-3495(98)77785-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hicks GA, Marrion NV. Ca2+-dependent inactivation of large conductance Ca2+-activated K+ (BK) channels in rat hippocampal neurones produced by pore block from an associated particle. J Physiol. 1998;508:721–734. doi: 10.1111/j.1469-7793.1998.721bp.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horrigan FT, Aldrich RW. Coupling between voltage sensor activation, Ca2+ binding and channel opening in large conductance (BK) potassium channels. J Gen Physiol. 2002;120:267–305. doi: 10.1085/jgp.20028605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Lee A, Chen J, Cadene M, Chait BT, MacKinnon R. Crystal structure and mechanism of a calcium-gated potassium channel. Nature. 2002;417:515–522. doi: 10.1038/417515a. [DOI] [PubMed] [Google Scholar]
- Koval OM, Fan Y, Rothberg BS. A role for the S0 transmembrane segment in voltage-dependent gating of BK channels. J Gen Physiol. 2007;129:209–220. doi: 10.1085/jgp.200609662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnamoorthy G, Shi J, Sept D, Cui J. The NH2 terminus of RCK1 domain regulates Ca2+-dependent BK(Ca) channel gating. J Gen Physiol. 2005;126:227–241. doi: 10.1085/jgp.200509321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancaster B, Nicoll RA. Properties of two calcium-activated hyperpolarizations in rat hippocampal neurones. J Physiol. 1987;389:187–203. doi: 10.1113/jphysiol.1987.sp016653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee US, Cui J. β subunit-specific modulations of BK channel function by a mutation associated with epilepsy and dyskinesia. J Physiol. 2009;587:1481–1498. doi: 10.1113/jphysiol.2009.169243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G, Zakharov SI, Yang L, Deng SX, Landry DW, Karlin A, Marx SO. Position and role of the BK channel alpha subunit S0 helix inferred from disulfide crosslinking. J Gen Physiol. 2008a;131:537–548. doi: 10.1085/jgp.200809968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G, Zakharov SI, Yang L, Wu RS, Deng SX, Landry DW, Karlin A, Marx SO. Locations of the beta1 transmembrane helices in the BK potassium channel. Proc Natl Acad Sci U S A. 2008b;105:10727–10732. doi: 10.1073/pnas.0805212105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G, Niu X, Wu RS, Chudasama N, Yao Y, Jin X, Weinberg R, Zakharov SI, Motoike H, Marx SO, Karlin A. Location of modulatory beta subunits in BK potassium channels. J Gen Physiol. 2010;135:449–459. doi: 10.1085/jgp.201010417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marty A. Ca-dependent K channels with large unitary conductance in chromaffin cell membranes. Nature. 1981;291:497–500. doi: 10.1038/291497a0. [DOI] [PubMed] [Google Scholar]
- Meera P, Wallner M, Song M, Toro L. Large conductance voltage- and calcium-dependent K+ channel, a distinct member of voltage-dependent ion channels with seven N-terminal transmembrane segments (S0–S6), an extracellular N terminus, and an intracellular (S9–S10) C terminus. Proc Natl Acad Sci U S A. 1997;94:14066–14071. doi: 10.1073/pnas.94.25.14066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrow JP, Zakharov SI, Liu G, Yang L, Sok AJ, Marx SO. Defining the BK channel domains required for beta1-subunit modulation. Proc Natl Acad Sci U S A. 2006;103:5096–5101. doi: 10.1073/pnas.0600907103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu X, Qian X, Magleby KL. Linker-gating ring complex as passive spring and Ca2+-dependent machine for a voltage- and Ca2+-activated potassium channel. Neuron. 2004;42:745–756. doi: 10.1016/j.neuron.2004.05.001. [DOI] [PubMed] [Google Scholar]
- Orio P, Latorre R. Differential effects of beta 1 and beta 2 subunits on BK channel activity. J Gen Physiol. 2005;125:395–411. doi: 10.1085/jgp.200409236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orio P, Rojas P, Ferreira G, Latorre R. New disguises for an old channel: MaxiK channel beta-subunits. News Physiol Sci. 2002;17:156–161. doi: 10.1152/nips.01387.2002. [DOI] [PubMed] [Google Scholar]
- Orio P, Torres Y, Rojas P, Carvacho I, Garcia ML, Toro L, Valverde MA, Latorre R. Structural determinants for functional coupling between the beta and alpha subunits in the Ca2+-activated K+ (BK) channel. J Gen Physiol. 2006;127:191–204. doi: 10.1085/jgp.200509370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian X, Niu X, Magleby KL. Intra- and intersubunit cooperativity in activation of BK channels by Ca2+ J Gen Physiol. 2006;128:389–404. doi: 10.1085/jgp.200609486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber M, Salkoff L. A novel calcium-sensing domain in the BK channel. Biophys J. 1997;73:1355–1363. doi: 10.1016/S0006-3495(97)78168-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Krishnamoorthy G, Yang Y, Hu L, Chaturvedi N, Harilal D, Qin J, Cui J. Mechanism of magnesium activation of calcium-activated potassium channels. Nature. 2002;418:876–880. doi: 10.1038/nature00941. [DOI] [PubMed] [Google Scholar]
- Storm JF. Action potential repolarization and a fast after-hyperpolarization in rat hippocampal pyramidal cells. J Physiol. 1987;385:733–759. doi: 10.1113/jphysiol.1987.sp016517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweet TB, Cox DH. Measurements of the BKCa channel's high-affinity Ca2+ binding constants: effects of membrane voltage. J Gen Physiol. 2008;132:491–505. doi: 10.1085/jgp.200810094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallner M, Meera P, Toro L. Determinant for beta-subunit regulation in high-conductance voltage-activated and Ca2+-sensitive K+ channels: an additional transmembrane region at the N terminus. Proc Natl Acad Sci U S A. 1996;93:14922–14927. doi: 10.1073/pnas.93.25.14922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallner M, Meera P, Toro L. Molecular basis of fast inactivation in voltage and Ca2+-activated K+ channels: a transmembrane beta-subunit homolog. Proc Natl Acad Sci U S A. 1999;96:4137–4142. doi: 10.1073/pnas.96.7.4137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Brenner R. An S6 mutation in BK channels reveals beta1 subunit effects on intrinsic and voltage-dependent gating. J Gen Physiol. 2006;128:731–744. doi: 10.1085/jgp.200609596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu RS, Chudasama N, Zakharov SI, Doshi D, Motoike H, Liu G, Yao Y, Niu X, Deng SX, Landry DW, Karlin A, Marx SO. Location of the β4 transmembrane helices in the BK potassium channel. J Neurosci. 2009;29:8321–8328. doi: 10.1523/JNEUROSCI.6191-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Yang Y, Ye S, Jiang Y. Structure of the gating ring from the human large-conductance Ca2+-gated K+ channel. Nature. 2010;466:393–397. doi: 10.1038/nature09252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia XM, Ding JP, Lingle CJ. Molecular basis for the inactivation of Ca2+- and voltage-dependent BK channels in adrenal chromaffin cells and rat insulinoma tumor cells. J Neurosci. 1999;19:5255–5264. doi: 10.1523/JNEUROSCI.19-13-05255.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia XM, Zeng X, Lingle CJ. Multiple regulatory sites in large-conductance calcium-activated potassium channels. Nature. 2002;418:880–884. doi: 10.1038/nature00956. [DOI] [PubMed] [Google Scholar]
- Xia XM, Ding JP, Lingle CJ. Inactivation of BK channels by the NH2 terminus of the beta2 auxiliary subunit: an essential role of a terminal peptide segment of three hydrophobic residues. J Gen Physiol. 2003;121:125–148. doi: 10.1085/jgp.20028667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Zhang G, Shi J, Lee US, Delaloye K, Cui J. Subunit-specific effect of the voltage sensor domain on Ca2+ sensitivity of BK channels. Biophys J. 2008;94:4678–4687. doi: 10.1529/biophysj.107.121590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Krishnamoorthy G, Saxena A, Zhang G, Shi J, Yang H, Delaloye K, Sept D, Cui J. An epilepsy/dyskinesia-associated mutation enhances BK channel activation by potentiating Ca2+ sensing. Neuron. 2010;66:871–883. doi: 10.1016/j.neuron.2010.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan P, Leonetti MD, Pico AR, Hsiung Y, MacKinnon R. Structure of the human BK channel Ca2+-activation apparatus at 3.0 A resolution. Science. 2010;329:182–186. doi: 10.1126/science.1190414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zakharov SI, Wu RS, Liu G, Motoike H, Karlin A, Marx SO. Locations of the beta2 transmembrane helices in the BK potassium channel. Presented at the 53rd Annual Biophysical Society Meeting; February–March; Boston. 2009. [Google Scholar]