

Abstract
Interactions between proteins underlie numerous biological functions. Theoretical work suggests that protein interactions initiate with formation of transient intermediates that subsequently relax to specific, stable complexes. However, the nature and roles of these transient intermediates have remained elusive. Here, we characterized the global structure, dynamics, and stability of a transient, on-pathway intermediate during complex assembly between the Signal Recognition Particle (SRP) and its receptor. We show that this intermediate has overlapping but distinct interaction interfaces from that of the final complex, and it is stabilized by long-range electrostatic interactions. A wide distribution of conformations is explored by the intermediate; this distribution becomes more restricted in the final complex and is further regulated by the cargo of SRP. These results suggest a funnel-shaped energy landscape for protein interactions, and they provide a framework for understanding the role of transient intermediates in protein assembly and biological regulation.
Keywords: EPR spectroscopy, fluorescence spectroscopy, molecular recognition, protein targeting, GTPases
Interactions between proteins are central to biology and underlie numerous molecular recognition, regulation, and signaling events (1). A challenge in our understanding of protein interactions is to reconcile their fast association kinetics required for biological function (106–108 M-1 s-1) with the fact that formation of stable protein assemblies often involves extensive short-range, stereospecific interactions that are difficult to accomplish during a single diffusional encounter (2–4). This problem becomes more pronounced in protein interactions that require extensive conformational changes in the interaction partners. Much theoretical work has suggested that assembly of a protein complex initiates with the formation of a transient intermediate held together by solvent cage and long-range electrostatic attractions, followed by relative rotatory diffusions of the binding partners to search for the optimal interaction interface with shape and electrostatic complementarity (4–9). An extreme example of this concept is the “fly-casting mechanism,” in which unstructured protein molecules bind targets weakly at a relatively large distance followed by folding at the target site (10–12). In general, formation of transient intermediates reduces the dimension of translational and rotational search and could significantly accelerate protein association.
Despite significant progress in theoretical work, direct experimental demonstration of this model has been limited, and the structural and dynamic nature of transient intermediates during protein interactions has remained elusive. Experimental studies of transient intermediates are still at the infant stage, because these intermediates have short lifetimes and are rarely populated at equilibrium. Pioneering NMR studies have revealed the structures of rare conformational states in equilibrium with the predominant structure in the apo-protein or the final complex, and provided direct experimental support for the ability of proteins to explore different conformations (13–17). Nevertheless, many of these studies have focused on protein interactions that are inherently weak and nonspecific; whether the same principle applies to the assembly of a stable and stereospecific protein complex remains to be determined. Further, the transient species probed in this manner do not necessarily represent on-pathway intermediates during complex assembly. To understand the protein assembly pathway, it is crucial that on-pathway intermediates during protein assembly can be isolated. To this end, we chose the interaction between the Signal Recognition Particle (SRP) and the SRP receptor (SR) as a model system.
Rapid assembly of a stable SRP•SR complex is required to efficiently deliver cargo proteins to cellular membranes during cotranslational protein targeting, and it is essential for proper protein localization in all cells (18, 19). Formation of a stable SRP•SR complex is mediated by specific interactions between their NG domains (comprised of a GTPase, G domain, and a helical N domain) (Fig. 1A). However, free SRP and SR are not in the optimal conformation to bind one another, and extensive rearrangements must occur in both proteins to attain a stable complex (20). Previous kinetic studies showed that stable SRP•SR complex assembly begins with the formation of a transient “early” intermediate (Fig. 1A, step 1), which forms quickly (kon = 5.8 × 106 M-1 s-1) but is unstable (Kd ∼ 4–10 μM and koff ∼ 62 s-1) (21). This intermediate then slowly rearranges (k2 ∼ 1.5 s-1) to form the final stable complex, which is stabilized by a large, continuous interaction surface between the NG domains of both proteins (Fig. 1A, Left and step 2) (21). Importantly, complex assembly can be stalled at the early intermediate stage by leaving out guanosine 5′-triphosphate (GTP) while maintaining the kinetic competence of this intermediate (Fig. 1A) (21). This allowed us to isolate this intermediate and directly characterize its global structure, dynamics, and stability in this work. The results identified distinct interaction interfaces used by the early intermediate, and provide direct evidence for extensive conformational search in this intermediate and the importance of long-range electrostatic interactions in its stability. Further, the conformational distribution of the early intermediate is exquisitely sensitive to the biological cues of the SRP, providing potential mechanisms for biological regulation.
Fig. 1.

Mapping the interaction interface of the SRP•SR complexes using EPR spectroscopy. (A) (Left) Crystal structure of the SRP•SR NG-domain complex (1RJ9). (Right) A multistep mechanism for SRP•SR complex assembly involving formation of an early intermediate (step 1), and rearrangement to form the stable complex (step 2). Removal of GTP stalls the complex at the early intermediate stage. “T” denotes GTP. (B) Nitroxide spin probes labeled at specific SR residues change mobility upon formation of the early intermediate (blue), the stable complex (red), or both. The different classes of spin-probe mobility changes are defined in the text. Black denotes the apo-SR. (C) Interaction surface of the stable complex (Left) and early intermediate (Right) mapped by EPR. “N” and “G” denote the N and G domains of SRP and SR, respectively. The red line outlines the interaction surface in the cocrystal structure (20) of the stable complex.
Results
Map the Binding Interface of the Early Intermediate.
The interaction surface in the stable SRP•SR complex is formed primarily by close contacts between their G domains, with limited contacts between the N domains near the NG-domain interface contributing the remainder of the interface (Fig. 1A) (20, 22). We first asked whether the early intermediate forms the same or distinct interaction interface. To this end, electron paramagnetic resonance (EPR) spectroscopy was used to probe the interaction surface (23–25). Based on the cocrystal structure of the stable SRP•SR NG-domain complex, we selected residues in the vicinity of the interaction surface on SR for replacement by cysteine, which allowed site-directed spin labeling with the nitroxide probe (1-oxy-2,2,5,5-tetramethyl-3-pyrrolinyl-3-methyl) methanethiosulfonate. Only the sites where cysteine replacement and nitroxide labeling did not substantially affect the SRP–SR interaction were used for EPR measurements (Fig. S1 A and B). The residues on or near the dimer interface are likely to undergo significant changes in spin-probe mobility upon complex formation. These changes are measured by the linewidth of the central resonance (Fig. 1B and Fig. S1 C and D, ΔH0) and the overall breadth of the EPR spectra, especially the intensity of hyperfine splitting that arises from highly immobile populations of spin probes relative to the mobile population (Fig. 1B, dashed vs. solid arrows) (23).
To validate this approach, we first characterized the interaction surface of the stable complex formed with a nonhydrolyzable GTP analogue, 5′-guanylylimido-diphosphate. Twelve residues underwent significant EPR spectral changes upon complex formation (Fig. 1B and Fig. S1C, red vs. black). The majority of spin probes at these positions underwent changes in both central linewidth and the relative population of immobile species (Fig. 1B). For two of these residues, T356 and N426, changes in probe mobility was not obvious from the central linewidth but was detectable from changes in EPR spectral shape (Fig. 1B and Fig. S1C). Collectively, these data provided a view of the interaction surface in the stable complex that is consistent with the cocrystal structure (Fig. 1C, black vs. red outline) (20). This validated EPR as a powerful tool to probe the interaction surface of the complex.
We next used this approach to locate the interface of the early intermediate. Three classes of residues were identified that underwent distinct EPR spectral changes upon formation of the early or stable complex (Fig. 1B). Residues in class I, represented by V242 (Fig. 1B and Fig. S1C, purple), underwent similar reductions in spin-probe mobility upon formation of both the early and stable complexes, suggesting that they are involved in the interface of both complexes. Residues in class II, represented by S429 (Fig. 1B and Fig. S1C, brown), underwent substantial immobilization of the spin probe in the early intermediate, but these probes became more mobile in the stable complex, suggesting that they are engaged in stronger (in the cases of S429 and T451) or distinct patterns (in the cases of L433 and E487) of interactions in the early intermediate. Residues in class III, represented by I237 (Fig. 1B and Fig. S1C, green), exhibited substantial changes in spin-probe mobility only in the stable complex, suggesting that they are specifically involved in the formation of the stable complex.
Collectively, eight residues underwent substantial spectral changes upon formation of the early intermediate (Fig. 1B, classes I and II). Compared to the stable complex, these residues reside primarily in or near the N domain (Fig. 1C, Right), suggesting that the early intermediate has a detectable interaction surface that partly overlaps with but is distinct from that of the stable complex (Fig. 1C). In addition, some of the residues that changed mobility specifically in the stable complex (I237, Q425, N426; Fig. 1C and Fig. S2, green) were in or adjacent to the conserved “TAKGG” and “QLLIADV” motifs, which act as a hinge at the NG-domain interface to readjust the relative orientation of the G and N domains during stable complex formation (20). The absence of significant spectral changes at these positions in the early intermediate suggests that this crucial rearrangement has not taken place at the early intermediate stage.
To independently identify the interaction surface of the early intermediate, we introduced 24 mutations in SR, all of which map to the heterodimer interface in the stable complex (Fig. 2A). These mutations disrupt either the interactions at the dimer interface or the rearrangement at the NG-domain interface, and each impairs formation of the stable complex by 5- to 200-fold (22). Several of them were also near the residues engaged in the dimer interface of the early intermediate as identified by EPR (compare Fig. 2A vs. Fig. 1C, Right). We tested whether these mutations disrupted the stability of the early intermediate using fluorescence resonance energy transfer (FRET) between coumarin (DACM)-labeled SRP C235 and BODIPY-fluorescein (BODIPY-FL)–labeled FtsY C487 (21). To our surprise, most of these mutations did not disrupt the early intermediate (Fig. 2B, black bars). Only three mutations caused moderate reductions in the stability of the early intermediate (2- to 4-fold), and a combination of all three mutations destabilized the early intermediate by only 8-fold (Fig. 2B, gray bars and Fig. 2C).
Fig. 2.

Mutations that disrupt the stable complex did not significantly affect the early intermediate. (A) Positions of SR mutations (cyan and blue) studied herein are shown in the surface representation of the SR. The three moderately defective mutants are highlighted in blue. (B) The stability of the early intermediate is insensitive to many mutations that disrupt the stable complex. (C) The stabilities of the early intermediates formed by mutant SRs were determined by equilibrium titrations. Nonlinear fits gave Kd values of 4.1 μM for wild-type SR, 13.2 μM for SR (K306A), 17.3 μM for SR (L393W), and 31.3 μM for SR (K306A:L393W:A421W).
The mutational analyses provided independent support for important conclusions from the EPR experiments, including the paucity of G-domain interactions and the absence of conformational readjustments at the NG-domain interface in the early intermediate. On the other hand, these data raised additional questions, because they showed that the early intermediate was insensitive to many mutations near its putative interaction surface identified by EPR. Two models to reconcile these results were tested and verified in the experiments below. First, the major interactions that stabilize the early intermediate may lie further outside the G domain and its vicinity, where most of the mutations above were located. Second, the early intermediate may not have a defined structure but rather contains multiple conformations, each with a distinct interface. Mutations that disrupt a specific interface do not affect alternative conformations, and hence do not significantly affect the overall stability of the intermediate. In contrast, the stable complex has a more defined structure and hence is more susceptible to mutations that disrupt its interface.
Conformational Dynamics in the Early Intermediate.
To test whether the early intermediate samples a broad distribution of conformations, time-resolved FRET (TR-FRET) was used to measure the distance distribution between donor (DACM) and acceptor (BODIPY-FL) dyes labeled at specific sites on SRP and SR in different SRP•SR complexes. These measurements provided nanosecond snapshots of fluorescence decay of the donor dye (Fig. S3), from which donor–acceptor distance distributions of the respective complex could be derived (26). We analyzed the fluorescence decay curves using both the least-squares fitting (Fig. S4) and maximum entropy (Fig. 3) methods. These algorithms produce the narrowest and broadest distance distributions, respectively, that satisfy the experimental measurements, and the distance distributions in the ensemble of SRP•SR complex likely reside in between these two extreme representations (SI Methods). Given this, substantial caution was taken in the interpretation of the distance distributions such that the conclusions are largely independent of the method used to represent the data. Moreover, we focused on the changes in the distance distribution in the different SRP•SR complexes, which are less sensitive to biases introduced by different data representation.
Fig. 3.

Conformational distribution of the early intermediate is broad and is restricted by formation of the stable complex or the cargo. (Left) Positions of the G–G (A), NG–NG (B), and N–N (C) FRET pairs in the stable SRP•SR NG-domain complex. (Right) FRET distance distributions, P(r), for each FRET pair in the early intermediate (blue), stable complex (red), and the early intermediate bound to cargo (green), derived from maximal entropy analyses of the TR-FRET data as described in SI Methods.
Three pairs of residues were used to measure distance distributions between the G domains (Fig. 3A, G–G), the NG-domain interfaces (Fig. 3B, NG–NG), and the N domains (Fig. 3C, N–N) of both proteins. Anisotropy measurements confirmed that all the fluorophores are relatively free rotamers and hence suitable for distance measurements (SI Methods and Table S1). For all three pairs, the early intermediate exhibited broad distance distributions spanning approximately 25–60 Å without a single dominant population (Fig. 3 and Fig. S4, blue). In contrast, the distributions became significantly more restricted in the stable complex (Fig. 3 and Fig. S4, red). For the G–G and NG–NG pairs (Fig. 3 A and B and Fig. S4 A and B, red), a predominant population was observed in the stable complex with a distance in good agreement with the cocrystal structure (33 Å in crystal structure vs. 37 Å for the G–G pair, and 30 Å in crystal structure vs. 31 Å for the NG–NG pair) (20). In comparison, the N–N pair displayed a broader distribution in the stable complex, with a peak centered around 37 Å (Fig. 3C and Fig. S4C, red). This observation can be partially explained by the limited interactions between the N domains in the stable complex, which might allow these domains to have more flexibility (20, 27). Taken together, these results provided direct evidence that the early intermediate contained a large ensemble of conformations that are similar in stability, whereas the stable complex has a more specific structure, particularly at the G domains and NG-domain interfaces.
Comparison of these distance distributions also provided clues to the complex assembly process. A significant population of molecules with distances as short as approximately 25 Å was observed for the N–N pair in the early intermediate, but this population diminished in the stable complex (Fig. 3C and Fig. S4C). In contrast, a significant population of molecules exhibited long distances (45–60 Å) for the G–G pair in the early intermediate, which also diminished in the stable complex (Fig. 3A and Fig. S4A). This suggests that SRP•SR complex assembly initiates from close contacts between their N domains in the early intermediate, whereas the G domains are further apart.
Electrostatic Interactions Drive Formation and Stability of the Early Intermediate.
Consistent with the notion that complex assembly initiates with contacts between the N domains, adaptive Poisson–Boltzmann solver (APBS) calculation (28) revealed clusters of positively and negatively charged residues, respectively, on the surface of N domains of SRP and SR (Fig. 4A). Interactions between these electrostatically complementary surfaces were supported by their evolutionary conservation (Fig. S5A), and by molecular docking simulations using the ClusPro 2.0 program (29), which generated molecular models for the early intermediate. Two groups, each containing an ensemble of approximately 90 structures, scored significantly higher than all the alternative configurations (representatives in each group are shown in Fig. 4B). In the “N” group, the N domains of SRP and SR contact one another via the electrostatically complementary surfaces identified in the APBS calculation (Fig. 4B, Left); in the “G” group, the G domains of the proteins contact one another via an interface that is shifted away from the heterodimer interface in the stable complex (Fig. 4B, Right vs. Inset). The nucleotide-binding cavity of SRP and SR were exposed in both groups (Fig. 4B, Left and Right), explaining why formation of the early intermediate is a nucleotide-independent process.
Fig. 4.

Electrostatic interactions between the N domains of SRP and SR stabilize the early intermediate and accelerate stable complex assembly. (A) The SRP and SR N domains contain complementarily charged surfaces. (B) Molecular docking simulation generated two groups of conformations (N and G) for the early intermediate. For comparison, the Inset in the middle shows the structure of the stable complex with the SRP NG domain aligned in the same orientation. The nucleotide bound to SRP is shown in CPK coloring. (C) The association rate constants predicted from Brownian dynamics calculations for formation of the early intermediate in the N or G group, and for the stable complex. The experimentally measured rate constants are in parentheses. (D) Charge complementarity between the N domains is critical to the stability of the early intermediate (black bars) and the kinetics of stable complex assembly (blue bars), determined using the FRET and GTPase assays, respectively, for the wild-type proteins (WT:WT), wild-type SRP and mutant SR (WT:RRLRR), mutant SRP and wild-type SR (RK3E:WT), and the charge reversal SRP and SR mutant pair (RK3E:RRLRR). The kinetic constants were derived from the data in Fig. S5D. FRET efficiency in the early intermediate was recorded at 5 μM FtsY, at which concentration the FRET value is most sensitive to changes in the stability of the early intermediate.
Both the N and G groups represent possible conformations within the ensemble of structures of the early intermediate, because all the residues that changed mobility in EPR measurements resided on the dimer interfaces of one or the other groups (Fig. S6A and B), and because both groups were needed to reproduce the experimentally observed broad distance distributions of FRET probes (compare Fig. S6C–E vs. Fig. 3, blue). Nevertheless, the following strongly suggests that the N group represents the major conformational ensemble. First, the residues that changed spin-probe mobility in the early intermediate are primarily in or near the N domain (Fig. 1C, Right). Second, most mutations in the G domain that could affect the G group did not abolish the stability of the early intermediate, suggesting that the conformers in the G group are less significantly populated in this intermediate. Third, in Brownian dynamics calculations (30), the association rate constant for the early intermediate estimated for the N group agreed well with the experimental value, whereas that for the G group was 30-fold slower (Fig. 4C).
What features in the N group make it the major conformation of the early intermediate? We reasoned that the complementarily charged surfaces on the N domains of SRP and SR could facilitate long-range electrostatic interactions that bring the two proteins into proximity (Fig. 4A). To test the contribution of these electrostatic interactions, we generated charge reversal mutants in which three basic residues (R35, R49, and K56) on the SRP N domain were mutated to glutamates (RK3E), and the glutamate residues in the EELEE motif on the SR N domain were mutated to arginines (RRLRR). Mutants SRP (RK3E) and SR (RRLRR) severely reduced the stability of the early intermediate (Fig. 4D and Fig. S5C); these mutants also caused 10- to 28-fold reductions in the association rate constant for stable SRP•SR complex assembly that correlated well with their reduced stabilities in the early intermediate (Fig. 4D, blue and Fig. S5D).
We further asked whether the SRP–SR interaction can be rescued by combining the charge reversal mutants of SRP and SR, which partially restores the electrostatic complementarity between their N domains (Fig. S5B). Indeed, the combination of the SRP (RK3E) and SR (RRLRR) mutants restored the stability of the early intermediate to within 3-fold of that of the wild-type protein (Fig. 4D, black bars and Fig. S5C). The kinetics of stable complex assembly was correspondingly rescued (Fig. 4D, blue bars and Fig. S5D). The incomplete rescue could be accounted for by the fact that, although the SRP (RK3E) mutation made the SRP N domain highly negatively charged, the SR (RRLRR) mutation rendered the SR N domain only moderately positively charged (Fig. S5B). Together, these results strongly support the notion that electrostatic interactions provide an important driving force to form and stabilize the early intermediate, which correspondingly enhances the kinetics of stable complex assembly.
Cargo Restricts Conformational Dynamics of the Early Intermediate.
The SRP–SR interaction is profoundly influenced by the cargos of SRP, the ribosome-nascent chain complexes (RNCs), which stabilize the early intermediate over 50-fold and accelerate stable complex assembly over 100-fold (18, 19). We speculated that the cargo could actively regulate the conformational dynamics of the early intermediate. To test this hypothesis, we used TR-FRET to measure the conformational distribution of the early intermediate in the presence of RNCFtsQ, which contains the first 74 amino acids of a known SRP substrate FtsQ. Notably, the cargo substantially altered the distance distribution of all the FRET pairs in the early intermediate, changing their broad distance distributions to more bimodal patterns (Fig. 3 and Fig. S4, green). Thus, the cargo restricts the dynamics of the early intermediate to a more limited conformational space, in which the successful selection of complementary structures might be enhanced. This could partly explain how the cargo enhances the kinetics of SRP•SR complex assembly and therefore effects efficient protein targeting (18, 19).
Discussion
Using the SRP–SR interaction as a model system, we analyzed the global structure, dynamics, and stability of an on-pathway intermediate during the assembly of a stable protein complex. The techniques used here would not provide atomic resolution information for the assembly intermediate; on the other hand, a combination of biochemical, biophysical, and theoretical approaches provided a set of complementary and self-consistent information that together revealed important global features of this intermediate and shed light on the association process of a relatively large and stable protein complex.
An intriguing finding of this work is that the interaction surface used by the early intermediate is quite distinct from that of the stable complex. Electrostatic interactions between complementarily charged surfaces on the N domains of SRP and SR provided the primary stabilizing force for the early intermediate (Movie S1). In contrast, more stereospecific interactions between the G domains, which provide most of the driving force for the stable complex, are rather weak at this stage of assembly. This explains the previous observations that formation of the early intermediate can occur independently of GTP and that nucleotides can rapidly exchange in this intermediate (21), and it is consistent with a recent cryo-EM analysis of an early cargo-SRP•SR targeting complex (31). The early intermediate studied herein is considerably more stable (Kd ∼ 4–10 μM and koff ∼ 62 s-1) than would be expected for encounter complexes and likely occurs at a stage later than simple diffusional encounter. The fact that this intermediate still has a distinct interaction surface than the final complex strongly suggests that productive protein–protein interactions can initiate at sites that are adjacent to, but quite distinct from, the final interaction surface.
Besides the electrostatic interaction between the N domains, a conserved electrostatic interaction between Lys399 in the G domain of SR and the GGAA tetraloop of the SRP RNA (the other component of SRP) also provides a crucial contact that stabilizes the early intermediate (approximately 12-fold) and accelerates stable complex assembly (32). Despite extensive mutagenesis, these two pairs of electrostatic interactions are the only ones that have been found thus far to contribute significantly to the stability of the early intermediate. Together, these results show that formation of the SRP•SR early intermediate is driven primarily by long-range electrostatic attractions. Consistent with this notion, the stability of the early intermediate and the rate of stable complex assembly have a strong dependence on ionic strength (32). Critical roles of electrostatic interactions in enhancing protein interaction kinetics have been predicted theoretically (33, 34) and demonstrated in multiple cases (35–38); our results further emphasize the role of such interactions in stabilizing assembly intermediates, which provides an effective way to accelerate the overall assembly process.
Another intriguing finding here is that TR-FRET measurements revealed a broad conformational distribution for the early intermediate (Fig. 5, blue; Movie S1). The broad conformational distribution is also supported by the observation that single mutations in the G domain do not significantly affect the stability of the early intermediate, whereas a combination of these mutations causes a substantial disruption of its stability. This provides direct evidence that a wide conformational space is explored by this intermediate, which could aid in the search and selection for the optimal structure conductive to forming the stable complex (39). Interestingly, the conformational space of the intermediate is actively regulated by the cargo of SRP (Fig. 5, green), which restricts the conformation of the early intermediate and produces a more bimodal pattern of distribution. These changes could potentially provide a mechanism to exert biological regulation (18, 19). Nevertheless, much more work will be needed to provide a molecular understanding of the conformational changes brought upon by the cargo and how these changes affect the complex assembly process.
Fig. 5.

Model of free energy landscapes for the protein assembly process. The conformational space is broad for the free proteins (gray) and the early intermediate (blue), but becomes more restricted in the steoreospecific stable complex (red) or when SRP is bound to the cargo (green).
Formation of the stable complex significantly restricts the distance distributions of both the G–G and NG–NG FRET pairs, consistent with the notion that a stable and stereospecific complex (Kd ∼ 16–30 nM) has a much more defined structure. In comparison, a broader distribution of FRET distances was exhibited by the N–N FRET probes, which might arise from a combination of the following factors. First, residual, albeit more restricted, conformational sampling still occurs in the stable complex (Fig. 5, red). Second, interactions in the stable complex primarily involve the G domains and the NG-domain interface, whereas contacts between the N domains are rather limited (Fig. 1A). Thus, the N domains are likely to have more flexibility than the G domains and can sample different configurations in the stable complex.
The features of the early intermediate during protein assembly bear intriguing analogies to molten globules during protein folding, in that both are relatively resistant to many mutations and have a broad free energy landscape that allows the protein(s) to sample multiple configurations (40). Also analogous to the protein folding process, the energy landscape of protein assembly appears to be funnel shaped and becomes narrower as the free proteins transition through the intermediate and progress toward the steoreospecific complex (Fig. 5), as predicted by theoretical work (7, 10–12, 33, 41). These findings could represent general features of transient intermediates during assembly of stable protein complexes and provide a framework to understand their roles in enhancing protein interactions and biological regulation.
Methods
EPR.
EPR spectra were acquired with a 9.4-GHz (X-band) Bruker EMX EPR spectrometer with an ER 4119HS cavity at 20–23 °C. 40% glycerol was present in all samples to eliminate the global tumbling motion of proteins. All scans were carried out using a microwave power of 5 mW, a modulation amplitude of 2 G and a magnetic field sweep width of 100 G. The central linewidth of EPR spectra was the same at microwave powers of 0.2–5 mW. Averaged spectra were obtained from 32–64 scans, and background signals were subtracted. For specific conditions in the EPR measurements, see SI Methods.
Steady-State Fluorescence.
All measurements were carried out at 25 °C in SRP buffer [50 mM KHEPES, pH 7.5, 150 mM KOAc, 2 mM Mg(OAc)2, 2 mM DTT, 0.01% Nikkol] on a Fluorolog-3-22 spectrofluorometer (Jobin Yvon), using an excitation wavelength of 380 nm and an emission wavelength of 470 nm. FRET efficiency was calculated as described (21). To compare the relative equilibrium stabilities of the early intermediates formed by different SR mutants, 4 μM BODIPY-FL–labeled SR were incubated with 1 μM DACM-labeled SRP in the absence of GTP. Because formation of the early intermediate is rapid but has a high Kd (4–10 μM), the FRET value at the subsaturating SR concentration provided a sensitive measure of the changes in its stability. For representative mutants, equilibrium titrations were carried out. The data were fit to Eq. 1,
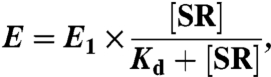 |
[1] |
in which E1 is the FRET end point with saturating SR, and Kd is the equilibrium dissociation constant of the early intermediate.
TR-FRET Measurements.
TR fluorescence decay measurements were carried out in SRP buffer with a picosecond streak camera (C5680; Hamamatsu Photonics) in the photon-counting mode (42), using an excitation wavelength of 355 nm generated from a third harmonic of a regeneratively amplified mode-locked Nd-YAG laser (pulsewidth is approximately 15 ps) (Vanguard, Spectra-Physics). A band-pass filter of 450 ± 5 nm was used as the emission filter. There was no observable fluorescence from buffer or unlabeled protein. DACM fluorescence decay kinetics was measured in both short (5-ns) and long (20-ns) timescale, with time resolutions of approximately 10 ps and approximately 40 ps, respectively. For specific conditions during the TR-FRET measurements and data analyses, see SI Methods.
Supplementary Material
Acknowledgments.
We thank B.S.P. Araujo for modeling the effect of fluorophore linkers on distant measurements; H.B. Gray and P.E. Wright for insightful discussions; and D.C. Rees, T.F. Miller III, and members of the Shan laboratory for comments on the manuscript. This work was supported by National Institutes of Health Grants GM078024 to S.-o.S. and GM068041 to J.R.W., DARPA Protein Design Processes to S.L.M., and career awards from the Burroughs Welcome Foundation, the Henry and Camille Dreyfus Foundation, the Arnold and Mabel Beckman Foundation, and the David and Lucile Packard Foundation to S.-o.S.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1019051108/-/DCSupplemental.
References
- 1.Alberts B. The cell as a collection of protein machines: Preparing the next generation of molecular biologists. Cell. 1998;92:291–294. doi: 10.1016/s0092-8674(00)80922-8. [DOI] [PubMed] [Google Scholar]
- 2.Janin J. The kinetics of protein-protein recognition. Proteins. 1997;28:153–161. doi: 10.1002/(sici)1097-0134(199706)28:2<153::aid-prot4>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 3.Schreiber G. Kinetic studies of protein-protein interactions. Curr Opin Struct Biol. 2002;12:41–47. doi: 10.1016/s0959-440x(02)00287-7. [DOI] [PubMed] [Google Scholar]
- 4.Schreiber G, Haran G, Zhou HX. Fundamental aspects of protein-protein association kinetics. Chem Rev. 2009 doi: 10.1021/cr800373w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ubbink M. The courtship of proteins: Understanding the encounter complex. FEBS Lett. 2009;583:1060–1066. doi: 10.1016/j.febslet.2009.02.046. [DOI] [PubMed] [Google Scholar]
- 6.Koshland DE. Application of a theory of enzyme specificity to protein synthesis. Proc Natl Acad Sci USA. 1958;44:98–104. doi: 10.1073/pnas.44.2.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ma B, Kumar S, Tsai CJ, Nussinov R. Folding funnels and binding mechanisms. Protein Eng. 1999;12:713–720. doi: 10.1093/protein/12.9.713. [DOI] [PubMed] [Google Scholar]
- 8.Schreiber G, Shaul Y, Gottschalk KE. Electrostatic design of protein-protein association rates. Methods Mol Biol. 2006;340:235–249. doi: 10.1385/1-59745-116-9:235. [DOI] [PubMed] [Google Scholar]
- 9.Boehr DD, Nussinov R, Wright PE. The role of dynamic conformational ensembles in biomolecular recognition. Nat Chem Biol. 2009;5:789–796. doi: 10.1038/nchembio.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shoemaker BA, Portman JJ, Wolynes PG. Speeding molecular recognition by using the folding funnel: The fly-casting mechanism. Proc Natl Acad Sci USA. 2000;97:8868–8873. doi: 10.1073/pnas.160259697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Papoian GA, Ulander J, Wolynes PG. Role of water mediated interactions in protein-protein recognition landscapes. J Am Chem Soc. 2003;125:9170–9178. doi: 10.1021/ja034729u. [DOI] [PubMed] [Google Scholar]
- 12.Papoian GA, Wolynes PG. The physics and bioinformatics of binding and folding-an energy landscape perspective. Biopolymers. 2003;68:333–349. doi: 10.1002/bip.10286. [DOI] [PubMed] [Google Scholar]
- 13.Tang C, Iwahara J, Clore GM. Visualization of transient encounter complexes in protein-protein association. Nature. 2006;444:383–386. doi: 10.1038/nature05201. [DOI] [PubMed] [Google Scholar]
- 14.Volkov AN, Worrall JA, Holtzmann E, Ubbink M. Solution structure and dynamics of the complex between cytochrome c and cytochrome c peroxidase determined by paramagnetic NMR. Proc Natl Acad Sci USA. 2006;103:18945–18950. doi: 10.1073/pnas.0603551103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fawzi NL, Doucleff M, Suh JY, Clore GM. Mechanistic details of a protein-protein association pathway revealed by paramagnetic relaxation enhancement titration measurements. Proc Natl Acad Sci USA. 2010;107:1379–1384. doi: 10.1073/pnas.0909370107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Crowley PB, Ubbink M. Close encounters of the transient kind: Protein interactions in the photosynthetic redox chain investigated by NMR spectroscopy. Acc Chem Res. 2003;36:723–730. doi: 10.1021/ar0200955. [DOI] [PubMed] [Google Scholar]
- 17.Xu X, et al. Dynamics in a pure encounter complex of two proteins studied by solution scattering and paramagnetic NMR spectroscopy. J Am Chem Soc. 2008;130:6395–6403. doi: 10.1021/ja7101357. [DOI] [PubMed] [Google Scholar]
- 18.Zhang X, Schaffitzel C, Ban N, Shan SO. Multiple conformational switches in a GTPase complex control co-translational protein targeting. Proc Natl Acad Sci USA. 2009;106:1754–1759. doi: 10.1073/pnas.0808573106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang X, Rashid R, Wang K, Shan SO. Sequential checkpoints govern substrate selection during cotranslational protein targeting. Science. 2010;328:757–760. doi: 10.1126/science.1186743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Egea PF, et al. Substrate twinning activates the signal recognition particle and its receptor. Nature. 2004;427:215–221. doi: 10.1038/nature02250. [DOI] [PubMed] [Google Scholar]
- 21.Zhang X, Kung S, Shan SO. Demonstration of a multistep mechanism for assembly of the SRP•SRP receptor complex: Implications for the catalytic role of SRP RNA. J Mol Biol. 2008;381:581–593. doi: 10.1016/j.jmb.2008.05.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shan SO, Stroud RM, Walter P. Mechanism of association and reciprocal activation of two GTPases. PLoS Biol. 2004;2:1572–1581. doi: 10.1371/journal.pbio.0020320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Crane JM, Lilly AA, Randall LL. Characterization of interactions between proteins using site-directed spin labeling and electron paramagnetic resonance spectroscopy. Methods Mol Biol. 2010;619:173–190. doi: 10.1007/978-1-60327-412-8_11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McHaourab HS, Lietzow MA, Hideg K, Hubbell WL. Motion of spin-labeled side chains in T4 lysozyme. Correlation with protein structure and dynamics. Biochemistry. 1996;35:7692–7704. doi: 10.1021/bi960482k. [DOI] [PubMed] [Google Scholar]
- 25.Zhang X, Lee SW, Zhao L, Xia T, Qin PZ. Conformational distributions at the N-peptide/boxB RNA interface studied using site-directed spin labeling. RNA. 2010;16:2474–2483. doi: 10.1261/rna.2360610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kimura T, Lee JC, Gray HB, Winkler JR. Folding energy landscape of cytochrome cb562. Proc Natl Acad Sci USA. 2009;106:7834–7839. doi: 10.1073/pnas.0902562106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Focia PJ, Gawronski-Salerno J, Coon JS, Freymann DM. Structure of a GDP∶AlF4 complex of the SRP GTPases Ffh and FtsY, and identification of peripheral nucleotide interaction site. J Mol Biol. 2006;360:631–643. doi: 10.1016/j.jmb.2006.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc Natl Acad Sci USA. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Comeau SR, et al. ClusPro: Performance in CAPRI rounds 6–11 and the new server. Proteins. 2007;69:781–785. doi: 10.1002/prot.21795. [DOI] [PubMed] [Google Scholar]
- 30.Ermak DL, McCammon JA. Brownian dynamics with hydrodynamic interactions. J Chem Phys. 1978;69:1352–1360. [Google Scholar]
- 31.Estrozi LF, Boehringer D, Shan SO, Ban N, Schaffitzel C. Cryo-EM structure of the E. coli translating ribosome in complex with SRP and its receptor. Nat Struct Mol Biol. 2011;18:88–90. doi: 10.1038/nsmb.1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shen K, Shan SO. Transient tether between the SRP RNA and SRP receptor ensures efficient cargo delivery during cotranslational protein targeting. Proc Natl Acad Sci USA. 2010;107:7698–7703. doi: 10.1073/pnas.1002968107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Levy Y, Onuchic JN, Wolynes PG. Fly-casting in protein-DNA binding: Frustration between protein folding and electrostatics facilitates target recognition. J Am Chem Soc. 2007;129:738–739. doi: 10.1021/ja065531n. [DOI] [PubMed] [Google Scholar]
- 34.Miyashita O, Onuchic JN, Okamura MY. Transition state and encounter complex for fast association of cytochrome c2 with bacterial reaction center. Proc Natl Acad Sci USA. 2004;101:16174–16179. doi: 10.1073/pnas.0405745101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jones S, Thornton JM. Principles of protein–protein interactions. Proc Natl Acad Sci USA. 1996;93:13–20. doi: 10.1073/pnas.93.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schreiber G, Fersht AR. Rapid, electrostatically assisted association of proteins. Nat Struct Biol. 1996;3:427–431. doi: 10.1038/nsb0596-427. [DOI] [PubMed] [Google Scholar]
- 37.Vijayakumar M, et al. Electrostatic enhancement of diffusion-controlled protein-protein association: Comparison of theory and experiment on barnase and barstar. J Mol Biol. 1998;278:1015–1024. doi: 10.1006/jmbi.1998.1747. [DOI] [PubMed] [Google Scholar]
- 38.Kiel C, Selzer T, Shaul Y, Schreiber G, Herrmann C. Electrostatically optimized Ras-binding Ral guanine dissociation stimulator mutants increase the rate of association by stabilizing the encounter complex. Proc Natl Acad Sci USA. 2004;101:9223–9228. doi: 10.1073/pnas.0401160101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Harel M, Spaar A, Schreiber G. Fruitful and futile encounters along the association reaction between proteins. Biophys J. 2009;96:4237–4248. doi: 10.1016/j.bpj.2009.02.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pande VS, Rokhsar DS. Is the molten globule a third phase of proteins? Proc Natl Acad Sci USA. 1998;95:1490–1494. doi: 10.1073/pnas.95.4.1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wolynes PG, Onuchic JN, Thirumalai D. Navigating the folding routes. Science. 1995;267:1619–1620. doi: 10.1126/science.7886447. [DOI] [PubMed] [Google Scholar]
- 42.Lee JC, Engman KC, Tezcan FA, Gray HB, Winkler JR. Structural features of cytochrome c' folding intermediates revealed by fluorescence energy-transfer kinetics. Proc Natl Acad Sci USA. 2002;99:14778–14782. doi: 10.1073/pnas.192574099. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.