

Abstract
In this study, we investigated on a systems level how complex protein interactions underlying cell polarity in yeast determine the dynamic association of proteins with the polar cortical domain (PCD) where they localize and perform morphogenetic functions. We constructed a network of physical interactions among >100 proteins localized to the PCD. This network was further divided into five robust modules correlating with distinct subprocesses associated with cell polarity. Based on this reconstructed network, we proposed a simple model that approximates a PCD protein's molecular residence time as the sum of the characteristic time constants of the functional modules with which it interacts, weighted by the number of edges forming these interactions. Regression analyses showed excellent fitting of the model with experimentally measured residence times for a large subset of the PCD proteins. The model is able to predict residence times using small training sets. Our analysis also revealed a scaffold protein that imposes a local constraint of dynamics for certain interacting proteins.
Keywords: budding yeast, modeling, network modularity
The study of many complex biological systems has reached a stage where much is known about the molecular components and their functional capacity and interactions. A challenge at hand is how to integrate this wealth of information to explain complex behaviors at the systems level. Cell polarity in the budding yeast Saccharomyces cerevisiae represents one such example (1–4). Functional analyses of the proteins involved have uncovered several subfunctions in the establishment and maintenance of cell polarity, including GTPase signaling, actomyosin-based transport, exocytic deposition of membrane components and cell wall materials, and endocytic recycling (1–4). Spatiotemporal coordination of these subfunctions leads to the morphogenesis of a bud required for cell division, or a shmoo-like projection required for mating.
Existing data suggest that the polar cortical domain (PCD) is a dynamic assembly of loosely interacting components (5–8). Hence, “protein dynamics” in this study refers to the cycle of association and dissociation of polarity proteins with the PCD. Detailed analysis of Cdc42, a key regulator of cell polarity, has shown that the stability and shape of the PCD require balanced molecular flux and appropriately specified parameters governing the rates of Cdc42 dynamics (6, 9). A more challenging question is how a system composed of hundreds of different protein molecules associating and dissociating at different rates could function coherently and bring about precise morphogenetic outcomes. A basic assumption that we make to investigate this problem on a systems level is that the dynamics of the peripherally associated PCD proteins are governed by complex interactions.
In this study, we first constructed a modularized network of physical interactions among all the known proteins localizing to and participating in the functions of the PCD. We then used an inverse fluorescence recovery after photobleaching (iFRAP) analysis to measure the dynamics of a subset of the PCD proteins in two of the key functional modules. Initially, we explored simple correlations between iFRAP time and diverse network parameters describing node properties. A lack of correlation in these analyses led us to construct and test a mathematically simple but conceptually rich model, which relates protein dynamics not only to protein interactions but also to the modular network structure and intrinsic kinetic parameters associated with module functions.
Results
Construction of a Modular Network of Interactions Among PCD Proteins.
We used the genome-wide GFP-tagged yeast protein localization database (10) to generate a comprehensive list of proteins that localize to the PCD at the tip of the nascent bud. The expected localization was confirmed for 111 proteins (SI Appendix, Table S1). We added several Rho and Rab family GTPases to this list, which are known to localize to the small bud tip (1, 2, 11). To reduce unnecessary complexity, subunits of two tightly associated stoichiometric complexes, the exocyst (12) and the Arp2/3 complex (13), were grouped together as single nodes as Exo and Arp, respectively. Two nodes were used to represent actin: actin cables (Act1C) serving transport function and actin patches (Act1P) mediating endocytosis.
We next used the BioGrid database (version 2.0.51) to construct a network of protein interactions containing 99 nodes and 302 linkages (SI Appendix, Fig. S1). Finally, we used a computational method designed to unravel modules in the network (14–16). Because protein interaction networks built from information in databases often contain false-positive or missing linkages, we designed a scheme that combined network reconstructions (17) with modularity maximization to control for error sensitivity in module identification (SI Appendix, Fig. S2 A–D). This analysis identified five consensus modules in the PCD network (SI Appendix, Fig. S3). Proteins classified in the same module typically have known functions within a common subprocess related to cell polarity. Four of the five modules correlate with functions known to be required for polarity and morphogenesis in yeast and are designated as Signaling, Transport, Endocytosis, and Exocytosis modules (Fig. 1A and SI Appendix, Table S2). The remaining module, named Mitotic Exit, includes proteins involved in the regulation of the exit from mitosis (18).
Fig. 1.

Modular organization of the yeast PCD protein interaction network. (A) Visualization of the modularized PCD protein interaction network. Five modules with distinct functions constitute this network: Signaling (red), Transport (brown), Endocytosis (blue), Exocytosis (green), and Mitosis Exit regulation (gray). The different node shapes represent different universal roles as indicated. Cdc42 protein is used as an example to explain the method for reduction of the local connectivity of each node to the global connectivity to the modules with assigned kinetic parameters. (B) Description of the interactions (colored lines) of Cdc42 with proteins in different modules (large circles). (C) Mathematical representation of PCD protein interaction network and dynamics. The global connectivity of Cdc42 is represented by a 5D integer vector composed of the number of Cdc42’s linkages with each of the five modules. Each module is also assigned a characteristic time constant, τi (τ1–5 for Signaling, Endocytosis, Transport, Mitotic Exit, and Exocytosis, respectively).
Measurement of PCD Protein Dynamics in Live Yeast Cells.
To assess patterns of protein dynamics across the system, we performed an analysis of the dynamics for most components (those that are taggable with GFP and are not transmembrane proteins) in the Signaling and Transport modules, which are critical for cell polarity establishment, taking advantage of the yeast ORF-GFP strain collection (10). We chose to use iFRAP (19, 20), wherein the dissociation of fluorescent protein molecules from the PCD was measured after photobleaching of the cytosolic pool (Fig. 2 A–C). The half-time (t1/2) of fluorescence loss approximates the protein's residence time on the polar cortex.
Fig. 2.

Experimental measurements of protein dynamics using iFRAP. (A–C) Images from iFRAP experiments and sample curves for proteins GFP-Rho3, GFP-Rho1, and Pea2-GFP. Red lines encircle bleached areas, and yellow lines encircle areas of fluorescence measurements. Red arrows indicate the time of iFRAP bleaching. The raw fluorescence measurements were subjected to monoexponential curve fitting (solid red curves). (Scale bar: 2 μm.) (D) Dynamics (measured as iFRAP t1/2) of 11 proteins in the Signaling module. Boxes show the range of 1 SEM from the mean, represented by small squares. Vertical bars show the range of 1 SD from the mean. The horizontal line in the boxes represents the median. (E) Dynamics (measured as t1/2) of 18 proteins in the Transport module represented as described in D. Note that the vertical time scale (0–50 s) for the 15 proteins on the left is different from that (0–180 s) for the 3 proteins on the right.
To ensure comparison at the same stage of polarization, cells with a nascent bud 1.45 ± 0.2 μm in diameter were chosen for iFRAP measurements. A C-terminal GFP tag was present directly at the native chromosomal locus of each of the tagged proteins in haploid yeast cells (10). For the Rho family proteins, GFP must be introduced at the N terminus and the tagged proteins are functional (SI Appendix, Fig. S4). The t1/2 was calculated for each protein after curve fitting using a monoexponential function (21), a necessary simplification for the purpose of this study.
Fig. 2 displays sample iFRAP curves (Fig. 2 A–C and Movies S1, S2, and S3), and all measured t1/2 values for 29 proteins in the Signaling and Transport modules (Fig. 2 D and E and SI Appendix, Tables S4 and S5). This result shows that most components of the Transport and Signaling modules are highly dynamic, with t1/2 in the range of 10–30 s. At the most dynamic end (smallest t1/2) lies Bem1 (t1/2 = 10.16 ± 1.56 s), which was postulated to be an adaptor protein that tethers complexes containing Cdc42, Cdc24, and Cla4 (22). At the slow end of the spectrum, Pea2 (23), a component of the “polarisome” (24), associates with the bud tip with the highest stability (t1/2 = 92.3 ± 12.7 s), whereas other polarisome proteins, such as Bni1, Bud6, and Spa2, exhibited varying t1/2 values considerably smaller than that of Pea2, suggesting that these proteins do not interact with the PCD as a single unit. An ANOVA test found that proteins in the Signaling module are, on average, more dynamic than those in the Transport module (P = 4.26E-16), with an average t1/2 of 18.5 ± 0.72 s (mean ± SE of mean) and 25.1 ± 1.8 s, respectively. The t1/2 values are also significantly different for proteins within modules (P = 5.50E-56), however. Further analysis shows that there is no clear separation of t1/2 between components of the two modules, aside from the few very slow components in the Transport module (e.g., Pea2, Rho1) (SI Appendix, Table S6). This result suggests that although the average dynamics of the two modules are significantly different, the dynamics of individual components of these modules cannot be predicted based only on their module assignment.
We first searched for simple correlations between the measured protein residence times and parameters describing node properties, such as degree, the number of edges (or interactions) connected with a node. No significant linear correlation was found between degree and t1/2 values (SI Appendix, Fig. S5A). Likewise, no significant linear correlation was found between t1/2 values and any of the several commonly used parameters, such as betweenness (SI Appendix, Fig. S5B), participation coefficient (SI Appendix, Fig. S5C), and others (SI Appendix, Table S7). We also investigated if proteins that interact with each other tended to have similar dynamics on a global level by testing if a protein's t1/2 is closer to its interaction partners than its noninteraction partners for all 29 proteins. Analysis showed no differences for 18 proteins (SI Appendix, Table S8). For the other 11 proteins, 4 (yellow) were shown to have more similar dynamics to the noninteraction partners, with 5 being more similar to the interaction partners and 2 (green) being equally different from both noninteraction and interaction partners (SI Appendix, Table S8). These results suggest no simple correlations between dynamics and a protein's interactive properties.
Modeling Protein Dynamics Based on a Modular Network Structure.
Our failure in finding simple correlations between iFRAP times and node properties might be explained by a lack of consideration for the modular architecture of the network as well as some intrinsic kinetic properties in module functions. To construct a model incorporating inputs from these more global properties, we first reduced the modular network to a simpler structure: the interactions of a given PCD protein p with other proteins are replaced by the interactions between the protein and each of the modules that harbors the interacting proteins. Thus, the protein p is described by the integer 5D vector Np = {Np,1, Np,2, Np,3, Np,4, Np,5}, where Np,i denotes the number of interactions of the protein p with proteins belonging to the ith module (Fig. 1 B and C, using the protein Cdc42 as an example). To introduce dynamics into this system, we assigned a single characteristic kinetic parameter, τi, to the ith module. τi has the dimension of time but has no absolute biological meaning. Thus, the network's dynamic properties are described by another 5D vector, τ = {τ1, τ2, τ3, τ4, τ5}. The main model equation (Eq. 1):
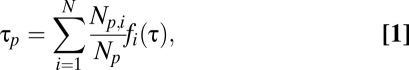 |
where
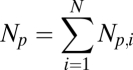 |
relates the reduced network protein vector, Np, to the protein's dynamic parameter, τp. This formula means that τp is a weighted average of functions fi (τ) that depend on some or all parameters τi. The choice of the functions fi is limited only by a restriction that it should have the dimension of time. In the simplest case, the dynamics of any protein interacting with the ith module are determined solely by this module [i.e., fi (τ) = τi], given by Eq. 2:
 |
We scale τp as the residence time of protein p on the polar cortex.
Validation of the Model and the Model's Predictive Ability.
To evaluate if the model (Eq. 2) fits well with the experimentally observed protein dynamics, we utilized a multivariate regression analysis using the normalized number of interactions that each of the 29 proteins has with each module of the network as the predictor variables and the experimentally determined t1/2 values as the variables to be predicted (Materials and Methods). The resulting Pearson coefficient between predicted and measured t1/2 values was 0.66 (P = 0.0001), suggesting that the model performs well in describing the experimental measurements.
Next, we tested the predictive ability of the model by selecting 10 proteins (Bni1, Cdc42, Cdc24, Cla4, Bem1, Myo2, Spa2, Ste20, Rho3, Mkk1) t1/2 values as the training set to calculate the module characteristic times (τ). We reasoned that the choice of this training set must be such that it is most informative about all modules of the network, and should therefore include proteins with maximal numbers of nonzero elements of the integer vector Np. Using the regression analysis, the t1/2 measurements from this training set predicted characteristic times, τ, to be 12.85, 27.27, 20.89, 82.20 and 3.86 s for the Signaling, Endocytosis, Transport, Mitotic Exit, and Exocytosis modules, respectively. These τ values were then used to predict the t1/2 values for the other 19 proteins using Eq. 2. The resulting Pearson correlation coefficient between the predicted and measured values was 0.56 (P = 0.0092). Furthermore, because the large τ calculated for the Mitotic Exit module predicts relatively large t1/2 values for the mitotic exit proteins exhibiting mostly intramodule interactions (e.g., 73.0 and 82.2 s for Kel1 and Lte1), we performed iFRAP analysis on Kel1 and Lte1, and yielded average t1/2 values of 87.4 and 49.8 s, respectively (SI Appendix, Fig. S6 and SI Appendix, Table S9). The overall correlation between predicted values and experimental measurement for the 21 proteins not used for the training set was 0.57 (P = 0.0071) (Fig. 3A).
Fig. 3.

Correlation of measured vs. model-predicted residence times and training set analysis. (A) Correlation of mean measured residence times with the values predicted by the model, utilizing the characteristic module times (τi) calculated using the 10-component training set, for the proteins in the Signaling and Transport modules (black dots) and 2 proteins in the Mitotic Exit module (blue dots). The three outliers (Pea2, Rho1, and Lrg1) are represented by brown dots. (Inset) Correlation for the training set components (green dots). (B) Performance of qualified training sets randomly drawn (black dots) from the measured t1/2 values of 29 proteins in Signaling and Transport modules in predicting the residence times for the 19 remaining proteins in these modules. The y axis represents the errors (as square root of error mean square) obtained by the least-squares multivariate regression method using each training set (Materials and Methods). The x axis indicates the correlation coefficient between the measured t1/2 values and predicted residence values. The red point indicates the performance of our chosen training set. (C) Same analysis and representation as in B, except for predicting residence times for 21 proteins (19 as in B plus the 2 proteins in the Mitotic Exit module).
To test if the training set that we chose represented the best training set, 1,000 randomly drawn sets of 10 proteins t1/2 values were used to build a linear regression model and predict residence time for either the 19 remaining Signaling and Transport module proteins (Fig. 3B) or 21 proteins (adding Lte1 and Kel1) (Fig. 3C). Random sets not connected to all five modules (thus unable to make predictions globally) or predicted negative τ values were removed from the analysis. This analysis found that many different training sets are capable of significant predictions and that our chosen set was among the best performers (Fig. 3 B and C).
We next tested whether modularity is indeed required for the model by investigating the scenario wherein the entire network is considered as a single module. The integer 5D vector Np was reduced to a single integer of the node, and in this scenario, the dynamics of the system would be characterized by a single τ. The model predicts a single residence time for all network components approximated by τ, which can be estimated as the average of the measured t1/2. Such predicted residence times do not correlate with the experimentally measured t1/2 values (Pearson correlation coefficient = 0), suggesting that network modularity is a critical requirement for our model to predict protein dynamics.
Local Scaffolding Effect of Pea2.
The dynamics of the three slow proteins of the Transport module (Pea2, Rho1, and Lrg1) were not well predicted by the global model (Fig. 3A). We investigated the possibility that the slowest protein (Pea2), postulated to be a scaffold protein (24), may have a strong effect on the dynamics of proteins that directly interact with it. A physical interaction between Rho1 and Pea2 was not reported in the literature, but in vivo evidence of this interaction can be shown by FRET in live cells expressing Pea2-mCherry and GFP-Rho1 under their native promoter (SI Appendix, Fig. S7). Supporting a strong effect of Pea2 on Rho1 dynamics, deletion of PEA2 rendered Rho1 significantly more dynamic (Fig. 4 and SI Appendix, Fig. S8). A further examination of proteins having a close connection with Pea2 in the network revealed a trend of increasing dynamics (t1/2), along a hierarchy of interactions descending from Pea2 (Fig. 4A). Like Rho1, the t1/2 for two of the first-tier Pea2 interactors, Spa2 and Bud6, were significantly reduced in pea2Δ (Fig. 4B; SI Appendix, Fig. S8; and SI Appendix, Table S10). The localization of the other two, Msb3 and Msb4, was drastically diminished in the mutant, which prevented dynamics measurement by iFRAP. In contrast, pea2Δ did not consistently or as drastically alter the dynamics of proteins further down the hierarchy from Pea2 (Fig. 4, SI Appendix, Fig. S8; and SI Appendix, Table S10).
Fig. 4.

Effects of the scaffold protein Pea2 on the dynamics of its interacting proteins. (A) Local hierarchical organization of protein dynamics descending from Pea2 (green). It can be observed that proteins (red) more closely interacting with Pea2, generally exhibit longer t1/2s than those (blue) further away from Pea2 in the hierarchy (the only exception is Yor304c-a). Shown are those components for which residence times were determined by iFRAP analysis. (B) Effects of pea2Δ on the dynamics of a subset of proteins in the hierarchy depicted in A examined by iFRAP. Shown are % changes of t1/2 in pea2Δ compared with those in the WT. Red bars represent proteins in the higher hierarchy that directly interact with Pea2, and blue bars represent those in the lower hierarchy shown in A.
To determine what role Pea2 might play in Rho1 polarization, we performed time-lapse microscopy to compare the kinetics of GFP-Rho1 polarization to the PCD in WT and pea2Δ cells. Interestingly, not only did Rho1 polarize robustly in the mutant cells, but, in fact, polarization occurred at a significantly higher rate in the mutant than in WT cells (Fig. 5 A and B; SI Appendix, Fig. S9; and Movies S4 and S5). Because Rho1 is a key regulator of cell wall glucan synthesis and remodeling (25), we measured the rate of cell wall growth in buds of WT and pea2Δ cells. Fluorescently labeled Con A was used to identify the regions of cell wall growth (26), and the rate of cell wall surface area increase was quantified over time (Fig. 5 C and D). Following the same trend as the effect on Rho1 polarization, the rate of cell wall growth in pea2Δ cells was also significantly higher than in the WT (P = 0.0020). These results suggest that Pea2 is not required for the establishment or maintenance of cell polarity but modulates the rates of Rho1 polarization and polarized cell surface growth.
Fig. 5.

Effects of Pea2 on polarization kinetics and the rate of polarized growth. (A) Observation of GFP-Rho1 polarization in WT (black) and pea2Δ cells (green) by time-lapse movies (Movies S4 and S5). The plots show representative time-dependent changes in the average fluorescence intensity of GFP-Rho1 in the PCD, normalized against the average intensity in the whole cell. (B) Quantification of the rates of Rho1 polarization in time-lapse movies as described in A. The polarization rate was defined as the slope at the inflection point of the sigmoid curve used for fitting. Statistical representation is the same as described in Fig. 2D. (C) Example cell images for cell wall growth during budding in WT and pea2Δ cells by fluorescent Con A staining. Regions staining only with Texas Red Con A but not FITC Con A (red-only zones in merged images) represent a recent area of cell wall growth. (Scale bar: 2 μm.) (D) Quantification of cell wall growth during budding in WT and pea2Δ cells by fluorescent Con A staining. Plots show the increase in the area of growth over time. Averages of measurements from 15 to 25 cells per time point per strain are shown. Error bars reflect SEM.
Discussion
The analysis presented above provides a systems level account of how a network of protein interactions explains dynamic properties of proteins required for polarity and morphogenesis in budding yeast. The use of a computational algorithm, based purely on maximization of a mathematically defined modularity function, identified a modular organization of the interaction network among cell polarity proteins that is strikingly consistent with known functional pathways involved in cell polarization. This result strongly supports the notion that modularity underlies the evolution of complex functions in biological systems (27–29).
The most important result of our analysis is that the dynamics of most of the PCD proteins can be approximated by a simple mathematical model accounting for their connectivity to network modules and intrinsic kinetic parameters of module functions. Our model implies that if a protein interacts mainly with components of the module to which it belongs, its residence time should approximate the module's characteristic time. If a protein also interacts with other modules of the network, however, its residence time will deviate from the characteristic time of its assigned module. The physical meaning of this model is that proteins interacting with a given module are dynamically coupled with the function performed by this module. A well-known classic theory on dynamic coupling is the Kuramoto model of oscillator coupling, which deals with unimodular distribution of oscillator frequencies (30). With five modules, our system is more complex, and unlike the classic theory, which relies on the assumption of uniform interaction between oscillator units, degree varies considerably from node to node in the PCD network. The mathematical relationship between our model and the Kuramoto model was explored, and the findings are described in SI Appendix.
Although previous studies from us and others have suggested that some of the PCD proteins are dynamically localized (5, 6, 31, 32), others have raised the potential importance of molecules that are more stably anchored and may serve as structural scaffolds during polarity establishment and maintenance (33, 34). Our large-scale iFRAP data suggest that the PCD can be generally characterized as a highly dynamic molecular ensemble, possibly a design feature suitable for maintaining association with the rapidly growing membrane at the tip of the bud. Our analysis did reveal a scaffold protein, Pea2, that exhibits a large residence time and suppresses the dynamics of Rho1 and other interacting proteins. Unexpectedly, deletion of Pea2 did not prevent cell polarization or polarized growth but, instead, led to increased rates of these processes. A possible explanation is that a higher rate of Rho1 GTPase recycling from the cortical membrane may enable more rapid protein redistribution during symmetry breaking and faster cycles of glucan synthase activation.
Materials and Methods
Yeast Strains, Cell Culture, Growth Conditions, and Plasmids.
Techniques for yeast cell culture and genetics were essentially as described previously (35). Most of the GFP-tagged strains were from the library of GFP-tagged proteins in budding yeast (10). All yeast strains are isogenic to S288C and listed in SI Appendix, Table S11. The functionality of the GFP-Rho1– and GFP-Rho3–tagged proteins was assessed in rho1Δ and rho3Δ backgrounds (SI Appendix, Fig. S4).
Network Module Analysis.
Given a network, for a certain partition, P, of the nodes into modules, the modularity, M(P), is defined as follows:
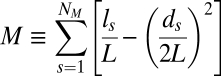 |
where NM is the number of modules, L is the number of links in the network, ls is the number of links between nodes in module s, and ds is the sum of the connectivity (degrees) of the nodes in module s. Modules (and the optimal number of modules) are typically identified by selecting the partition, P*, that maximizes M(P) (14). Further details about robust module identification are provided in SI Appendix.
Time-Lapse Imaging, iFRAP, and FRET Measurements.
All time-lapse images were acquired with an inverted Zeiss 200m outfitted with a spinning-disk confocal system (Yokagawa), a Plan-Apochromat 100×, a 1.4-N.A. oil objective, and an EM-CCD (C9100; Hamamatsu). Image acquisition and analysis were performed with Metamorph acquisition software (version 6.0; Molecular Devices).
Details about imaging experiments and data analysis are provided in SI Appendix.
Regression Analysis and Statistics.
Regression analysis was performed using SAS 9.2 (SAS Institute). For the analysis comparing average t1/2 values for two modules, we fitted a model that included module effect and protein effect, such that the protein effect was considered as nested within the module. We also fitted a model without considering the module effect to compare the difference between all proteins. The Tukey method was used for pairwise comparison. A two-way ANOVA model was used for the analysis comparing cell wall growth and the analysis comparing the average t1/2 values between a protein itself and its directly interacting proteins or its indirectly interacting proteins (SI Appendix, Table S8).
Supplementary Material
Acknowledgments
We thank X. Fan for constructing GFP-Rho1 and GFP-Rho3 plasmids; L. A. N. Amaral for discussion of network analysis; S. Ramachandran for input on genetic analysis; and B. Slaughter, J. Schwartz, W. Marshall, E. Glynn, and J. Unruh for help in optimizing microscopy experiments. This work was supported by National Institutes of Health Grant R01GM057063 (to R.L.), Spanish Ministerio de Ciencia e Innovación (MICINN) Grant FIS2010-18639 (to R.G. and M. S.-P.), James S. McDonnell Foundation Research Award (to R.G. and M. S.-P.), European Union Grant PIRG-GA-2010-277166 (to R.G.), and European Union Grant PIRG-GA-2010-268342 (to M. S.-P.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1017567108/-/DCSupplemental.
References
- 1.Pruyne D, Bretscher A. Polarization of cell growth in yeast. I. Establishment and maintenance of polarity states. J Cell Sci. 2000;113:365–375. doi: 10.1242/jcs.113.3.365. [DOI] [PubMed] [Google Scholar]
- 2.Park HO, Bi E. Central roles of small GTPases in the development of cell polarity in yeast and beyond. Microbiol Mol Biol Rev. 2007;71:48–96. doi: 10.1128/MMBR.00028-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Casamayor A, Snyder M. Bud-site selection and cell polarity in budding yeast. Curr Opin Microbiol. 2002;5:179–186. doi: 10.1016/s1369-5274(02)00300-4. [DOI] [PubMed] [Google Scholar]
- 4.Wedlich-Soldner R, Li R. Yeast and fungal morphogenesis from an evolutionary perspective. Semin Cell Dev Biol. 2008;19:224–233. doi: 10.1016/j.semcdb.2008.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wedlich-Soldner R, Wai SC, Schmidt T, Li R. Robust cell polarity is a dynamic state established by coupling transport and GTPase signaling. J Cell Biol. 2004;166:889–900. doi: 10.1083/jcb.200405061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Slaughter BD, Das A, Schwartz JW, Rubinstein B, Li R. Dual modes of cdc42 recycling fine-tune polarized morphogenesis. Dev Cell. 2009;17:823–835. doi: 10.1016/j.devcel.2009.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.van Drogen F, Peter M. Spa2p functions as a scaffold-like protein to recruit the Mpk1p MAP kinase module to sites of polarized growth. Curr Biol. 2002;12:1698–1703. doi: 10.1016/s0960-9822(02)01186-7. [DOI] [PubMed] [Google Scholar]
- 8.Boyd C, Hughes T, Pypaert M, Novick P. Vesicles carry most exocyst subunits to exocytic sites marked by the remaining two subunits, Sec3p and Exo70p. J Cell Biol. 2004;167:889–901. doi: 10.1083/jcb.200408124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marco E, Wedlich-Soldner R, Li R, Altschuler SJ, Wu LF. Endocytosis optimizes the dynamic localization of membrane proteins that regulate cortical polarity. Cell. 2007;129:411–422. doi: 10.1016/j.cell.2007.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huh WK, et al. Global analysis of protein localization in budding yeast. Nature. 2003;425:686–691. doi: 10.1038/nature02026. [DOI] [PubMed] [Google Scholar]
- 11.Pruyne D, Bretscher A. Polarization of cell growth in yeast. J Cell Sci. 2000;113:571–585. doi: 10.1242/jcs.113.4.571. [DOI] [PubMed] [Google Scholar]
- 12.TerBush DR, Novick P. Sec6, Sec8, and Sec15 are components of a multisubunit complex which localizes to small bud tips in Saccharomyces cerevisiae. J Cell Biol. 1995;130:299–312. doi: 10.1083/jcb.130.2.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Machesky LM, Atkinson SJ, Ampe C, Vandekerckhove J, Pollard TD. Purification of a cortical complex containing two unconventional actins from Acanthamoeba by affinity chromatography on profilin-agarose. J Cell Biol. 1994;127:107–115. doi: 10.1083/jcb.127.1.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guimerà R, Amaral LA. Cartography of complex networks: Modules and universal roles. J Stat Mech. 2005;2005(P02001):P02001-1–P02001-13. doi: 10.1088/1742-5468/2005/02/P02001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guimerà R, Nunes Amaral LA. Functional cartography of complex metabolic networks. Nature. 2005;433:895–900. doi: 10.1038/nature03288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sales-Pardo M, Guimerà R, Moreira AA, Amaral LA. Extracting the hierarchical organization of complex systems. Proc Natl Acad Sci USA. 2007;104:15224–15229. doi: 10.1073/pnas.0703740104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guimerà R, Sales-Pardo M. Missing and spurious interactions and the reconstruction of complex networks. Proc Natl Acad Sci USA. 2009;106:22073–22078. doi: 10.1073/pnas.0908366106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bosl WJ, Li R. Mitotic-exit control as an evolved complex system. Cell. 2005;121:325–333. doi: 10.1016/j.cell.2005.04.006. [DOI] [PubMed] [Google Scholar]
- 19.Lippincott-Schwartz J, Altan-Bonnet N, Patterson GH. Photobleaching and photoactivation: Following protein dynamics in living cells. Nat Cell Biol. 2003;5(Suppl):S7–S14. [PubMed] [Google Scholar]
- 20.Rabut G, Doye V, Ellenberg J. Mapping the dynamic organization of the nuclear pore complex inside single living cells. Nat Cell Biol. 2004;6:1114–1121. doi: 10.1038/ncb1184. [DOI] [PubMed] [Google Scholar]
- 21.Spector DL, Goldman RD. Live Cell Imaging: A Laboratory Manual. Plainview, NY: Cold Spring Harbor Laboratory Press; 2005. [Google Scholar]
- 22.Gulli MP, et al. Phosphorylation of the Cdc42 exchange factor Cdc24 by the PAK-like kinase Cla4 may regulate polarized growth in yeast. Mol Cell. 2000;6:1155–1167. doi: 10.1016/s1097-2765(00)00113-1. [DOI] [PubMed] [Google Scholar]
- 23.Chenevert J, Valtz N, Herskowitz I. Identification of genes required for normal pheromone-induced cell polarization in Saccharomyces cerevisiae. Genetics. 1994;136:1287–1296. doi: 10.1093/genetics/136.4.1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sheu YJ, Santos B, Fortin N, Costigan C, Snyder M. Spa2p interacts with cell polarity proteins and signaling components involved in yeast cell morphogenesis. Mol Cell Biol. 1998;18:4053–4069. doi: 10.1128/mcb.18.7.4053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Madden K, Snyder M. Cell polarity and morphogenesis in budding yeast. Annu Rev Microbiol. 1998;52:687–744. doi: 10.1146/annurev.micro.52.1.687. [DOI] [PubMed] [Google Scholar]
- 26.Nern A, Arkowitz RA. G proteins mediate changes in cell shape by stabilizing the axis of polarity. Mol Cell. 2000;5:853–864. doi: 10.1016/s1097-2765(00)80325-1. [DOI] [PubMed] [Google Scholar]
- 27.Callebaut W, Rasskin-Gutman D. Modularity: Understanding the Development and Evolution of Natural Complex Systems (Vienna Series in Theoretical Biology) Cambridge, MA: MIT Press; 2005. [Google Scholar]
- 28.Kirschner M, Gerhart J. Evolvability. Proc Natl Acad Sci USA. 1998;95:8420–8427. doi: 10.1073/pnas.95.15.8420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wagner A. Robustness and Evolvability in Living Systems. Princeton: Princeton Univ Press; 2007. [Google Scholar]
- 30.Kuramoto Y. Chemical Oscillations, Waves, and Turbulence. New York: Dover Publications; 1984. [Google Scholar]
- 31.Maddox PS, Bloom KS, Salmon ED. The polarity and dynamics of microtubule assembly in the budding yeast Saccharomyces cerevisiae. Nat Cell Biol. 2000;2:36–41. doi: 10.1038/71357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Valdez-Taubas J, Pelham HR. Slow diffusion of proteins in the yeast plasma membrane allows polarity to be maintained by endocytic cycling. Curr Biol. 2003;13:1636–1640. doi: 10.1016/j.cub.2003.09.001. [DOI] [PubMed] [Google Scholar]
- 33.Blumer KJ, Cooper JA. Go ahead, break my symmetry! Nat Cell Biol. 2003;5:1048–1049. doi: 10.1038/ncb1203-1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Irazoqui JE, Gladfelter AS, Lew DJ. Scaffold-mediated symmetry breaking by Cdc42p. Nat Cell Biol. 2003;5:1062–1070. doi: 10.1038/ncb1068. [DOI] [PubMed] [Google Scholar]
- 35.Sherman F, Fink GR, Hicks JB. Yeast Genetics. Plainview, NY: Cold Spring Harbor Laboratory Press; 1987. Methods in. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.