

Abstract
Breathing is controlled by a distributed network involving areas in the neocortex, cerebellum, pons, medulla, spinal cord, and various other subcortical regions. However, only one area seems to be essential and sufficient for generating the respiratory rhythm: the preBötzinger complex (preBötC). Lesioning this area abolishes breathing and following isolation in a brain slice the preBötC continues to generate different forms of respiratory activities. The use of slice preparations led to a thorough understanding of the cellular mechanisms that underlie the generation of inspiratory activity within this network. Two types of inward currents, the persistent sodium current (INaP) and the calcium-activated non-specific cation current (ICAN), play important roles in respiratory rhythm generation. These currents give rise to autonomous pacemaker activity within respiratory neurons, leading to the generation of intrinsic spiking and bursting activity. These membrane properties amplify as well as activate synaptic mechanisms that are critical for the initiation and maintenance of inspiratory activity. In this review, we describe the dynamic interplay between synaptic and intrinsic membrane properties in the generation of the respiratory rhythm and we relate these mechanisms to rhythm generating networks involved in other behaviors.
Keywords: Pacemaker, Respiration, Persistent sodium current, Calcium-activated non-specific cation current, preBötzinger complex, Rhythm generation, Bursting
Introduction
The ability of the nervous system to generate rhythmicity is almost ubiquitous, as the majority of brain regions are rhythmical under certain conditions. Rhythmically active networks are found for example in the neocortex, hippocampus, amygdala, cerebellum, basal ganglia, thalamus, locus coeruleus, ventral tegmentum area, medulla, and spinal cord. Rhythmic activity is associated with behaviors such as sleep, wakefulness, arousal, motivation, addiction, memory consolidation, cognition, fear, olfaction, locomotion, and breathing. In this review, we will focus on the neuronal network that gives rise to the breathing rhythm in mammals. Breathing behavior involves a larger network which is distributed throughout the nervous system and it includes areas such as the neocortex, cerebellum, amygdala, pons, medulla, and spinal cord [1–7]. However, only one region within this larger network seems to be essential for the generation of the respiratory rhythm itself, and this region is termed the preBötzinger complex [8]. The preBötC is located within the ventrolateral medulla. When lesioned, breathing ceases [9–12], and when isolated in a slice preparation, respiratory rhythm generation continues [8, 13]. Many of the characteristics of the fictive rhythmic activities generated within the preBötzinger complex have remarkable similarities with three activity patterns: normal respiratory activity (sometimes referred to as eupneic activity), sigh-activity, and gasping [14]. However, this similarity does not imply that in the intact nervous system the preBötC functions in isolation, nor does it imply that the rhythmic activities generated in the isolated network mirror the complexity, functionality, and plasticity of the entire network. Although we are far from a complete understanding of how different forms of breathing are generated in an intact animal, the ability to study this network in isolation has led to important insights into the mechanisms of respiratory rhythm generation. Many of these insights will be included in this review and they will be discussed in the context of mechanisms of rhythm generation in general.
Defining pacemaker neurons
The preBötC contains neurons that are capable of autonomously generating action potentials (spiking, Fig. 1b) and bursts of action potentials (bursting, Fig. 1c, cell 2) [8, 15]. These properties are not uncommon, since many types of neurons distributed throughout the nervous system are autonomously active. The key criterion to define these activities as “autonomous” or sometimes also “intrinsic” is that synaptic input is not required for their generation. Neurons capable of periodic spiking in the absence of synaptic input are referred to as autonomous spiking pacemakers [16]. Different types of neurons with this activity pattern are found in the subthalamic nucleus, nucleus basalis, globus pallidum, raphe nuclei, cerebellum, locus ceruleus, ventral tegmental area, and substantia nigra [16–20]. Depending on the modulatory conditions the same neuron can also generate bursting activity [17]. Following this nomenclature, the respiratory network contains both autonomous spiking pacemakers and bursting neurons [15, 21].
Fig. 1.

The PreBötzinger Complex contains rhythmically active neurons with different autonomous firing properties. a Schematic representation of the brainstem slice preparation containing the preBötzinger Complex (for space reasons abbreviated PBC in this particular schematic), nucleus ambiguus (NA), the spinal trigeminal nucleus (SP5), the inferior olive (IO), the vagus nucleus (X), and the hypoglossus nucleus (XII). Example of an integrated trace of an extracellular recording from the surface of the preBötC and a simultaneous intracellular recording of an inspiratory pacemaker with intrinsic bursting properties. During each population burst, this neuron receives synaptic input (shaded yellow) which activates bursting in this neuron. Injecting a depolarizing current (IM) into the neuron induces ectopic bursting and reveals the voltage dependency of this neuron. This leads to the conclusion that bursting arises autonomously within the neuron (shaded blue). Modified from Pena et al. [25]. b and c Neurons of the preBötC that are rhythmically active when embedded in the network (left traces) show distinct firing patterns following synaptic isolation (right traces). Cell 1 is an example of an autonomous spiking neuron, which spontaneously generates action potentials following synaptic isolation (right trace). Cell 2 is an example of a bursting neuron displaying high-frequency trains of action potentials (i.e., bursts) that result from both intrinsic and synaptic membrane properties when embedded in the network (left trace). Bursting continuous autonomously following synaptic isolation (right trace). Cell 3, simultaneously recorded with cell 2, represents a neuron that is not capable of autonomous activity. It becomes “silent” following synaptic isolation (right trace). Cells 2 and 3 are simultaneous recordings from two inspiratory neurons that were recorded before and after blockade of fast synaptic transmission, modified from Pena et al. [25]. Scale bars apply to all recordings
Thus, consistent with the nomenclature in other systems, we will use the term “pacemaker” for neurons that autonomously generate bursting as well as autonomously generate action potentials (or “spikes”). Spiking and bursting pacemakers do not necessarily represent distinct classes of neurons; instead, they often represent “different activity states” of the same neurons. Autonomous spiking in control can change to a bursting state in the presence of norepinephrine (Fig. 2a) [15], or from a bursting state into autonomous spiking following the blockade of endogenous 5-HT2A receptor activation (Fig. 2b) [18]. Some autonomously spiking neurons can burst during the rebound from a brief hyperpolarizing current pulse (Fig. 2c), or when activated in the rhythmically active network (Fig. 2c, right panel). The transition into the burst is not always abrupt. In many pacemakers, autonomously generated action potentials augment in a ramp-like manner during the transition into the intrinsically generated burst (Fig. 3a). The same neuron generates similar ramps as it transitions into the network burst (Fig. 3b).
Fig. 2.

Autonomous spiking and bursting in preBötC neurons represent “different activity states” within the same neuronal subtype and do not necessarily represent distinct classes of neurons. For example, neuromodulation can activate conditional burst properties in neurons of the preBötC. a Norepinephrine (NE) activates bursting (right trace) in an autonomous spiking neuron (left trace). Both traces were obtained in synaptic isolation. Modified from Viemari and Ramirez [15]. b Blockade of 5-HT2A receptor activation turns autonomous bursting (right trace) into autonomous spiking (left trace) in synaptic isolation. Modified from [92]. c An autonomous spiking neuron can generate rebound bursts following the injection of a brief hyperpolarizing current pulse (left panel). Although a similar effect is caused when embedded in the network, the same current injection evokes a much smaller membrane potential deflection (right panel). The injected hyperpolarizing pulse is schematically shown in blue below the trace (-DC)
Fig. 3.

The discharge pattern in many pacemakers is characterized by a an autonomously augmenting ramp which generates action potentials that transition into a burst. Intrinsic properties (shaded blue) create the augmenting ramp during autonomous bursting in synaptic isolation. b Embedded in the network, this pattern is similarly present within the same neuron. In this condition, both synaptic input (yellow) and intrinsic properties (blue) will likely contribute to the generation of the ramp pattern. The integrated population activity (red) from the preBötC occurs in phase with the bursting of the individual neuron
Cellular mechanisms of bursting in the respiratory network
Respiratory neurons that can autonomously generate spiking (Fig. 1b, cell 1) and bursting activity (Fig. 1c, cell 2) after blockade of fast synaptic transmission have been described under a variety of conditions, not only in in vitro slice preparations [22–26] but also in the so-called in situ preparation [27]. The activity pattern of autonomously active neurons can range from autonomously spiking to weak, irregular spiking and to regular bursting. The variety of activity patterns is explained by the ion channel diversity and the balance between inward and outward currents, a topic that has received considerable attention in invertebrate neurons [28–30]. Within the respiratory network two types of inward currents are particularly important in conferring autonomous activity patterns including bursting [26]. Some pacemakers continue to burst following the blockade of all voltage-dependent calcium channels using cadmium [CdCl2] as a general calcium channel antagonist (Fig. 4a). Neurons with these properties are often referred to as cadmium-insensitive pacemaker neurons (CI, Fig. 4a) [25, 26, 31, 32]. Among these CI pacemakers, a subset of neurons is capable of generating two types of bursts (Fig. 4a (2)) [33].
Fig. 4.

The population of autonomous bursting neurons can be classified as cadmium-sensitive and cadmium-insensitive pacemaker neurons. a Cadmium-insensitive (CI) pacemaker neurons are rhythmically bursting in synaptic isolation (1) and continue to burst in the presence of cadmium (Cd + + ) (2). In the same CI pacemaker neuron, two types of bursts can be generated successively (insets3a and 3b). Modified from Pena et al. [25]: simultaneous dual intracellular whole cell recordings of two CI pacemaker neurons recorded here in the presence of Cd + + as well as riluzole. While one neuron (top trace) becomes autonomously spiking in the presence of riluzole, the other neuron continues to generate bursts (lower trace) (5). b Cadmium-sensitive pacemaker (CS) neurons are rhythmically bursting in synaptic isolation, but become autonomously spiking or, as shown in (b), quiescent in the presence of Cd + + . c CI and CS pacemaker neurons display distinct voltage-dependent modulation of the bursting frequency. While CS pacemaker cells show in general a plateau of burst frequency near 1 Hz, CI pacemakers cells can have a burst frequency range over 2 Hz. Modified from [26]
The respiratory network also contains pacemakers that cease bursting in cadmium. These neurons are referred to as cadmium-sensitive (CS) pacemaker neurons [25, 26, 32]. The inward currents leading to cadmium-insensitive and cadmium-sensitive bursting have distinct voltage-dependent properties (Fig. 4c), play differential roles in respiratory rhythm generation, and are differentially modulated by neuromodulators. Increasing evidence indicates that bursting in cadmium-insensitive pacemaker neurons is mediated by the persistent sodium current [25, 34], while bursting in cadmium-sensitive pacemakers is mediated by the calcium-activated non-specific cation current (ICAN) [25, 32]. These two ionic mechanisms will be separately discussed in the subsequent sections.
The persistent sodium current in cadmium-insensitive pacemaker neurons
Voltage-dependent Na + currents are critical determinants of neuronal excitability. These currents can be discriminated based on their two distinct inactivation properties: (a) the fast transiently activated Na + currents (INaT) are responsible for the initial depolarization leading to action potentials, while (b) the non-inactivating low-voltage activated persistent Na + currents (INaP) contribute to the autonomous generation of action potentials and bursting. The persistent sodium current is a prominent contributor to the generation of intrinsic bursting and spontaneous action potential discharge in a variety of neurons [35–37]. It is typically activated at around − 60 mV and reaches a peak at − 40 to − 20 mV [22, 38–44]. Neurons that possess persistent sodium-dependent bursting properties are distributed throughout the central nervous system [40, 45–47]. This type of bursting is voltage dependent: the frequency of bursting increases with increasing depolarizing current injections (Fig. 4c) [26]. Despite its relatively small amplitude the persistent sodium current can also boost synaptic input as has been demonstrated in a variety of networks [48, 49]. In addition, the persistent sodium current provides a depolarizing drive towards the firing threshold of action potentials [50].
Riluzole or relatively low concentrations of TTX have been used to selectively block the persistent component of the sodium current, which in CI pacemakers abolishes bursting in the majority of neurons [25, 34]. However, in 29% of CI pacemaker neurons, bursting persists in relatively high concentrations of riluzole (20–50 μM) as well as cadmium (200 μM) [25]. Figure 4a (4) exemplifies two simultaneously recorded CI pacemaker neurons, one which ceased to burst (Fig. 4a (4), upper trace), and one which continued to burst in riluzole and cadmium (Fig. 4a (4), lower trace). One of the persisting bursts is shown at a faster time scale in Fig. 4a (5) to illustrate that the size of the action potentials was drastically reduced, indicating that the sodium current must have been partly blocked. This has important implications, because one cannot assume that bursting in all CI pacemakers is blocked when applying riluzole even at relatively high concentrations. Consequently, it is also unjustified to conclude that CI pacemakers are not essential for respiratory rhythm generation if network rhythmicity persists in riluzole, because almost 30% of these pacemaker neurons continue to burst. Among the CI pacemakers that ceased to burst, 59% continue to generate autonomous spiking after the application of riluzole (Fig. 4a (4), upper trace) [25] suggesting that spontaneous action potential generation may either involve other currents that are not blocked by these agents or, more likely, that this substance did not completely block the persistent sodium current [25].
One of the issues in blocking the persistent sodium current is that riluzole modulates both the transient and persistent sodium current to approximately the same degree [43]. However, riluzole has a greater affinity for the inactivated compared with the deinactivated (closed channel) state, thus leading to a relatively greater reduction in the persistent (INaP) versus transient component (INaT) of the sodium current [43]. Interestingly, Ptak et al. [43] found that the peak persistent sodium conductance, current density, and input resistance of interneurons isolated from the preBötC were greater than in the neighboring region of the rostral VRG. INaP density was also found to be greater in preBötC pacemaker versus nonpacemaker neurons [31], consistent with the notion that this current plays a critical role in conveying bursting properties to respiratory neurons. Again, this has important implications because any experiment that aims at abolishing selectively the persistent sodium current will also abolish to a certain degree the transient sodium current (see Fig. 4a (4 and 5)). Thus, a fine concentration balance determines whether these agents block only the persistent sodium component or, in addition, also the generation of action potentials. This limitation makes it experimentally impossible to achieve 100% selectivity. Moreover, selectivity is particularly difficult to achieve in a slice preparation in which any substance depends on diffusion to reach the neuronal target. Indeed Koizumi and Smith [34] demonstrated that pharmacological applications of TTX or riluzole cause spatially and temporally non-uniform pharmacological attenuation of INaP. This may also explain the simultaneous recording in Fig. 4a (4) in which bursting was blocked in one, but not in the other, CI pacemaker. These considerations explain why it has been very difficult to unambiguously assess the obligatory role of autonomous intrinsic activity in the generation of the respiratory rhythm [34].
Future approaches aimed at dissecting the relative contributions of the different forms of the sodium current may need to involve molecular tools. Voltage-gated Na + channels are composed of a pore-forming 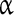 subunit and one or two associated
subunit and one or two associated 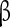 subunits [51, 52]. Nine functional types of
subunits [51, 52]. Nine functional types of 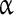 subunit mRNAs have been identified, of which Nav1.1, Nav1.2, Nav1.3, Nav1.5, and Nav1.6 are expressed within the central nervous system [53–55]. Mutations in the Nav1.1 gene (SCN1A) have been implicated in epilepsy [56–58], and Nav1.6 is thought to contribute to the generation of the persistent Na + current, but the contribution of Nav1.1 and 1.2 was also suggested following mutations in Nav1.6 [42, 59, 60]. The INaP in the preBötC seems to be determined by the Nav1.1 and Nav1.2
subunit mRNAs have been identified, of which Nav1.1, Nav1.2, Nav1.3, Nav1.5, and Nav1.6 are expressed within the central nervous system [53–55]. Mutations in the Nav1.1 gene (SCN1A) have been implicated in epilepsy [56–58], and Nav1.6 is thought to contribute to the generation of the persistent Na + current, but the contribution of Nav1.1 and 1.2 was also suggested following mutations in Nav1.6 [42, 59, 60]. The INaP in the preBötC seems to be determined by the Nav1.1 and Nav1.2 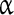 subunits [43].
subunits [43].
Four different types of the 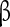 subunits have been described in a variety of systems [61, 62]. These subunits play diverse roles and are capable of modifying the biophysical and pharmacological properties of the
subunits have been described in a variety of systems [61, 62]. These subunits play diverse roles and are capable of modifying the biophysical and pharmacological properties of the 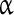 subunit [63, 64]. The
subunit [63, 64]. The 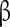 subunits have been implicated in conferring insensitivity to anti-epilepsy drug therapy [65] and mutation in the
subunits have been implicated in conferring insensitivity to anti-epilepsy drug therapy [65] and mutation in the 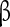 1 gene (SCNA1B) has been associated with epilepsy [66]. Particular importance for controlling the persistent sodium has been attributed to the
1 gene (SCNA1B) has been associated with epilepsy [66]. Particular importance for controlling the persistent sodium has been attributed to the 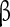 4 subunit. Detailed electrophysiological characterizations suggest that Nav
4 subunit. Detailed electrophysiological characterizations suggest that Nav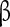 4 acts as an endogenous blocking protein that delays Na + channels from entering fast-inactivated states by rapid, unstable binding and unbinding at negative voltages [67, 68]. However, the role of the
4 acts as an endogenous blocking protein that delays Na + channels from entering fast-inactivated states by rapid, unstable binding and unbinding at negative voltages [67, 68]. However, the role of the 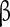 subunits in the respiratory network has, to the best of our knowledge, not been addressed.
subunits in the respiratory network has, to the best of our knowledge, not been addressed.
The calcium-activated nonselective cationic current in CS pacemaker neurons
In CS pacemakers, blockade of all voltage-activated calcium channels abolishes rhythmic bursting, suggesting that a calcium-dependent mechanism underlies their intrinsic bursting properties (Fig. 4b) [26, 32]. As first demonstrated by Pena et al. [25], bursting in most (82%) CS neurons is blocked by flufenamic acid (FFA), a blocker of the calcium-activated nonselective cationic current (ICAN). But FFA does also not block bursting in 100% of the CS pacemakers, an important caveat when assessing the obligatory role of this type of burst mechanism. This and subsequent studies confirmed that ICAN is critical for bursting in CS pacemaker neurons [25, 32]. The ICAN has been found in many non-neuronal [69, 70] and neuronal cells [71–73]. This current possesses several unique functional properties. Since ICAN is voltage independent and does not inactivate, it can maintain long-lasting depolarizations over a broad range of membrane potentials. Furthermore, it is activated and facilitated by increased concentrations of internal calcium [73, 74], which can be provided through voltage-gated calcium channels or by the release of calcium from internal stores.
The channels that mediate the ICAN have been suggested to belong to the transient receptor potential (TRP) family [75, 76]. These channels were first discovered in Drosophila [77] and consist of six transmembrane spanning segments (S1–S6) with a cation permeable pore (S5–S6). In general, this superfamily of channels is divided into seven subfamilies, of which six have been found to be functional in numerous mammalian tissues (i.e., the cardiovascular system, the immune system, or the kidney (for detailed review, see [78]) and are also expressed in the brain [79–81].
Most isoforms of the TRP channel family are widely distributed in the brain [82] and their important roles have already been defined for sensation of temperature (TRPV) [83], taste (TRPM5) [84], hearing and mechanosensation [85], neuronal outgrowth (TRPM7) [86], and cell survival (TRPC5) [79]. In addition, a number of studies showed that postsynaptic currents evoked by metabotropic glutamate receptors (mGluR1) are carried by TRPC1 channels, suggesting a role in synaptic plasticity [80, 87, 88].
In respiratory neurons, ICAN seems to depend on the activation of TRPM4 and/or TRPM5 [76]. These two isoforms are expressed in the region containing the preBötC and an intracellular analogue of phosphatidylinositol 4,5 (PIP2) leads to a FFA reversible augmentation of the inspiratory drive potential. This finding is consistent with the role of PIP2 in regulating calcium-activated TRPM4 channels [89, 90]. However, despite this circumstantial evidence, the molecular identity of the ICAN current remains unresolved. Ben-Mabrouk and Tryba [75] could not verify TRPM4/5 expression in the preBötC and proposed evidence that ICAN may instead depend on the TRPC3 or TRPC7 channels. In their experiments, the authors used the antagonist SKF-96365 to show that the effect of NK-1 receptor activation by substance P is reversible by blockade of the TRPC channels and that they could reverse the effects of substance P on CS pacemaker neurons. Thus, it is conceivable that different types of TRP channels contribute to the generation of ICAN within the respiratory network.
Although ICAN and INaP are two inward currents that importantly contribute to autonomous bursting and action potential generation in respiratory neurons, it would be wrong to conclude that respiratory neurons exclusively contain either one or the other inward current. Neurons that cease to burst following blockade of ICAN will likely contain also INaP, yet in these neurons this current alone does not suffice to promote intrinsic bursting. CS pacemakers continue to autonomously generate action potentials (22% of control) after blockade of ICAN [25]. This remaining autonomous activity could involve INaP. Vice versa, neurons that cease to burst upon blockade of INaP are likely to also possess ICAN, yet this current alone does not suffice to promote intrinsic bursting in these neurons. Another important remark: neurons that possess ICAN and/or INaP, but do not burst in the absence of synaptic input, can potentially burst when synaptic excitatory inputs enable the activation of these inward currents [31]. Thus, autonomously bursting neurons are on one end of a spectrum of autonomously active pacemakers that generate autonomous activity ranging from spiking non-bursty, weakly bursty, to strong bursting activity. Moreover, this spectrum is not fixed. The propensity to burst can be altered by changes in the ratio of inward and outward currents (e.g., Fig. 2). An excellent example is the recent study by Zavala-Tecuapetla et al., demonstrating how the interactions between the KCa channels and various inward currents alter bursting in respiratory pacemaker neurons [91]. In addition, bursting is known to be affected by changes in the modulatory milieu, as well as long-term changes associated with homeostatic or other forms of long-term regulatory plasticity.
Pacemaker properties and their interaction with neuromodulators
Even in reduced conditions, i.e., when isolated in a slice preparation, respiratory pacemaker neurons are continuously modulated by biogenic amines and peptides that include norepinephrine (NE), serotonin (5-HT), acetylcholine (ACh), and substance P (SP) [15, 33, 92–98]. Their action is mediated via different receptors, allowing for highly specific and tightly regulated control of the neuronal discharge pattern. Hence, neuromodulators play important roles in the alteration of pacemaker properties.
In synaptically isolated conditions, NE has been shown to stimulate different types of inspiratory neurons contained in the preBötC in vitro [15]. This action presumably involves 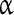 1,
1, 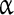 2, and
2, and 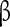 -noradrenergic mechanisms. NE induces ICAN-dependent bursting properties in spiking pacemaker neurons (Fig. 2a), and it depolarizes CI bursting pacemakers and increases their burst frequency. In CS pacemakers, NE increases the burst amplitude of the depolarizing potential and the number of action potentials during the burst. However, in contrast to the situation in CI pacemakers, NE does not affect the burst frequency in CS pacemakers [15]. This differential effect is preserved at the network level, since only the modulation of the burst amplitude but not the frequency depends on the activation of ICAN [15]. Therefore, by inducing bursting in pacemaker neurons, NE can change the balance between bursting and spiking pacemakers, which has very specific consequences at the network level.
-noradrenergic mechanisms. NE induces ICAN-dependent bursting properties in spiking pacemaker neurons (Fig. 2a), and it depolarizes CI bursting pacemakers and increases their burst frequency. In CS pacemakers, NE increases the burst amplitude of the depolarizing potential and the number of action potentials during the burst. However, in contrast to the situation in CI pacemakers, NE does not affect the burst frequency in CS pacemakers [15]. This differential effect is preserved at the network level, since only the modulation of the burst amplitude but not the frequency depends on the activation of ICAN [15]. Therefore, by inducing bursting in pacemaker neurons, NE can change the balance between bursting and spiking pacemakers, which has very specific consequences at the network level.
Substance P activates the inspiratory frequency at the network and behavioral level [9, 32, 93, 99–102] and also plays a critical role in the stability of the respiratory rhythm in vitro [75, 93, 103] and in vivo [99]. At the cellular level, SP slowly depolarizes spiking pacemaker neurons, leading to an increase in their intrinsic firing rate. SP activates bursting in CS pacemakers and, to a lesser extent, also in CI pacemakers, causing an increase in burst amplitude, frequency, and duration [93]. SP has been shown to activate neurokinin-1 receptors [100] that are coupled to Gq/11 proteins, that in turn activate phospholipase C. This intracellular cascade eventually induces ICAN, a mechanism that likely contributes to the effects of SP on burst amplitude. Recently, it has been suggested that this mechanism involves the activation of transient receptor protein canonical channels (TRPCs) [75], a mechanism which was also demonstrated in HEK293 cells [104]. SP-mediated effects on respiratory neurons are reversed by the application of an ICAN antagonist (flufenamic acid), but not by an INaP antagonist (riluzole) in vitro. Since TRPCs are non-selective cation channels permeable to monovalent and divalent ions [105], they might mediate ICAN in CS neurons [75]. Thus, similar to NE, SP modulates pacemaker properties in respiratory neurons via ICAN and it can alter the balance between autonomous spiking, bursting, and synaptic mechanisms.
Acetylcholine is a modulator that can alter the balance between two different types of CI-dependent bursting within the same individual neuron. As briefly mentioned above, a subset of CI pacemakers is capable of generating two types of bursts following synaptic isolation: relatively small-amplitude bursts (Fig. 4a (3a)) occurring at a fast frequency, and large-amplitude bursts (Fig. 4a (3b)) occurring at a slow frequency [33]. As demonstrated in the study by Tryba et al. [33], muscarinic activation alters the ratio of the number of autonomously generated low and large amplitude bursts in these neurons. The frequency of low amplitude bursts is inhibited, while the frequency of large amplitude bursts increased. It is very intriguing that this shift in the autonomous activity mirrors the activity of the network since muscarine inhibits the frequency of the small amplitude network bursts representing fictive eupneic activity, while the frequency of large amplitude sighs is significantly increased both in slices and the working heart-brainstem preparation [33]. This effect is likely mediated by M3 cholinergic receptors. It is important to emphasize that acetylcholine modulates the respiratory activity not only via muscarinic but also nicotinic receptors [33, 94–97].
Taken together, these results suggest that neuromodulators like NE, SP, or ACh can modulate the activity states of pacemakers from spiking to bursting, and they regulate the degree as well as types of bursting. Since neuromodulator levels change as a function of the sleep/wake state or the level of oxygenation, the contribution of different pacemaker properties to the rhythm generation is not fixed.
The synaptic determinants of respiratory rhythms
Much attention has been given to the role of synaptic transmission, and in particular the role of inhibitory mechanisms in respiratory rhythm generation [106, 107]. Synaptic inhibition is critical for the generation of the different phases of the respiratory rhythm. Moreover, in the intact network synaptic inhibition is important for establishing some of the characteristics of the eupneic rhythm [108–111]. Within the preBötC, synaptic inhibition also regulates pacemaker activity. In the rhythmically active network, bursting neurons are bombarded by synaptic inhibition not only during the expiratory phase, but also during inspiration [112]. As a consequence, bursting in some, but not all, bursting neurons, is suppressed during inspiration [113]. Thus, the degree of bursting within the respiratory network depends in part on synaptic inhibition, which is modulated by numerous conditions.
For example during hypoxic conditions, synaptic inhibition is reduced, which in turn reduces the suppressive synaptic influence on bursting. This reduction in synaptic inhibition will result in a greater contribution of CI bursting neurons during gasping [14, 25]. At the same time, however, hypoxia will lead to the inhibition of the majority of CS pacemakers [25]. Thus, the reconfiguration of the respiratory network during the transition from eupneic into gasping activity involves a complex change in the relative contribution of synaptic and autonomous bursting and spiking mechanisms [25].
The discovery that respiratory rhythmic activity persists following the blockade of synaptic inhibition in vitro [112, 114] and in situ [115] shifted attention towards better understanding the role of synaptic excitation in respiratory rhythm generation. Early on, AMPA receptors (AMPAr) were identified as the essential mechanisms for generating drive potentials in respiratory neurons [23, 90–93]. The blockade of AMPAr leads to the loss of population-level rhythmicity in the preBötC [26, 116–118]. However, AMPA is only one of several excitatory synaptic mechanisms that are important for respiratory rhythm generation within this network. This conclusion is well illustrated for the simultaneous generation of eupneic and sigh activity [14, 118, 119]. Fictive eupneic and fictive sigh activity are characterized by distinct integrated waveform patterns and population burst frequencies [14]. Fictive sigh burst generation is inhibited by antagonism of the P/Q-type Ca2 + channel or activation of the group III metabotropic glutamate receptor (mGluR), mGluR8 [119]. Moreover, while complete blockade of AMPAr inhibits fictive eupnea and sighs, incomplete blockade of AMPAr only abolishes fictive sighs. This dose-dependent inhibition of fictive sigh frequency by the antagonism of AMPAr suggests that the underlying mechanisms of fictive sigh burst frequency involves a degree of synaptic coupling similar to that proposed for the CA3 region of the hippocampus [95, 96].
Excitatory synaptic mechanisms that contribute to the generation of fictive eupneic activity and, in particular, inspiratory drive potentials, also involve the NMDA receptor (NMDAr), mGluR1, and mGluR5 [118–120]. The transient Ca2 + influx resulting from the activation of these postsynaptic glutamatergic receptors in turn activates a signaling cascade that leads to the activation of ICAN, thus causing the amplification of the inspiratory drive potential [120]. Modeling studies linking ICAN activation with the AMPA receptor and mGluR1 suggest that rhythmicity can theoretically emerge from an interconnected network of glutamatergic neurons that do not exhibit intrinsic bursting [110]. These theoretical considerations have contributed to the formulation of the group pacemaker hypothesis as proposed by Rekling and others [2, 121]. In its extreme iteration this hypothesis questions the obligatory role of intrinsic voltage-dependent bursting properties. Clearly, it is undeniable that glutamatergic synaptic transmission is essential for the formation of the eupneic network bursts, as this mechanism can be pharmacologically tested. By contrast, there is currently no tool to unambiguously test the obligatory role of bursting pacemakers. As discussed in this review and illustrated in Fig. 4, no pharmacological approach can block all bursting neurons. While it is currently impossible to test the obligatory role of bursting neurons with the existing tools, it can be demonstrated experimentally that respiratory rhythm generation ceases in the presence of FFA and riluzole [25, 122]. Moreover, numerous studies demonstrate that various forms of autonomous pacemaker activity exist within the preBötC [23, 25, 26, 33, 113, 123]. Thus, aside from theoretical considerations of extreme conditions, it would almost be a miracle if the abundance of autonomous activity would simply go away during the emergence of the respiratory rhythm. The next paragraph will discuss the interactions that are known to exist between synaptic mechanisms and autonomously generated pacemaker activity.
Interactions between pacemaker properties and synaptic mechanisms
Respiratory neurons are experimentally identified as being autonomous pacemakers by synaptically isolating them from the network. If a neuron continues to burst it is identified as a “pacemaker” (Fig. 1a and c, cell 2) [23, 25, 27, 87]. However, synaptic isolation and testing its response to current injection (Fig. 1a) are only experimental tools to unambiguously assess a neuron’s ability to generate autonomous bursting or spiking activity. But, it cannot be assumed that a neuron’s discharge pattern in synaptic isolation mirrors the activity of the neuron when it is embedded in the functional network. As shown in many studies, bursting and other intrinsic membrane properties engage in a rich and dynamic interplay with synaptic excitation and inhibition, as well as a wide range of neuromodulatory processes. Although implicit in the word “interplay”, it must also be emphasized that similarly synaptic and modulatory mechanisms engage in a rich and dynamic interplay with intrinsic bursting and spiking properties. Figure 5 shows the activity of a bursting pacemaker neuron in synaptic isolation (Fig. 5a) and when embedded in the functional network (Fig. 5b). While autonomously generated activity will contribute to a synaptic amplification during the network burst and to the activity preceding the network burst, the discharge characteristics of the bursting neuron in the network are not the same as in synaptic isolation (Figs. 1b, 2, 3, and 5). Thus, any attempt to imply that either synaptic or intrinsic membrane properties are responsible for rhythm generation must assume that either one of these properties functions separately from the others. However, there is no evidence for a functional separation of synaptic and intrinsic membrane properties, and there is no evidence that such a functional separation would even be possible. In the functional network, the intrinsic generation of bursting and action potentials will inevitably impact and be impacted by synaptic interactions.
Fig. 5.

a Activity of a bursting pacemaker neuron in synaptic isolation. Activity is only driven by intrinsic membrane properties (blue). b Activity of the same neuron embedded in the functional network. Note that the discharge characteristics of bursting in the network are not the same as in synaptic isolation. The neuron’s activity is presumably dependent on both intrinsic properties (blue) and network activity (yellow)
Determinants of burst amplitude
In synaptically isolated neurons, the intrinsically generated burst amplitude is determined by the net inward current. As described above, two types of inward currents are important determinants of the intrinsically generated burst: INaP or ICAN (Fig. 4). However, this conclusion does not imply that these currents are the only or even the major determinants of the net inward current. As these currents are activated they will interact with other ion channels that will also contribute to the generation of the burst. Activation of ICAN or INaP will bring the membrane potential to the voltage range that will also activate other voltage-dependent ionic conductances and channels, including the INaT currents and the various voltage-dependent calcium channels (P/Q-, L-, and N-type calcium channels). The depolarization by these voltage-gated channels may suffice to remove the Mg2 + block leading to the voltage-dependent opening of the NMDA-receptor, thus further contributing to the amplitude of the burst [124]. The generation of the burst will become regenerative as any of these currents will elevate the membrane potential to a level that will open more voltage-activated ion channels, which in turn will raise the calcium levels which then will increase the activation of ICAN. Autonomous spiking will also contribute to the slowly increasing intrinsic depolarizing drive which, together with the increasing levels of intracellular calcium, will bring the neurons into the regenerative state that will result in the generation of the burst (Figs. 3 and 5).
In the functional network, the burst amplitude will be the sum of these net inward currents generated intrinsically and the net synaptic currents generated by the network. The relationship between intrinsic and synaptic currents is nonlinear, due to the voltage and calcium dependency of these currents. In the functional network, bursting properties will amplify synaptic input in a non-linear and time-dependent manner as demonstrated in other motor systems [125]. If the intrinsically generated burst is activated late into the population burst it will act as an amplifier, while if the intrinsically generated burst precedes the population burst this mechanism will contribute to the events that initiate the population burst, as will be discussed in the next paragraph.
Determinants of burst onset
In synaptically isolated bursting neurons the timing of burst onset is determined by various intrinsic membrane properties. In synaptically isolated CI pacemakers, burst generation has been elegantly studied, both experimentally and computationally [22, 23, 32, 126, 127]. According to these studies, INaP inactivation occurring during the burst contributes to the termination of the burst. At the same time this inactivation also sets the conditions for the onset of the subsequent burst, since the slow kinetics of the recovery from this inactivation determines the timing of burst onset. In the intact network, this slow voltage-dependent process will be influenced and partly determined by other intrinsic membrane properties and synaptic inputs. Synaptically evoked excitatory inputs will initially slow the recovery from inactivation, but upon reaching a certain voltage level they will trigger the activation of an INaP-dependent intrinsic burst. Thus, the amount of synaptic excitation needed to trigger a protracted burst will depend on the inactivation state of the channel. Immediately following a network burst, the threshold for activating the intrinsic burst will be high. But, this threshold will gradually decrease as the time interval following the network burst increases, up to the time point when an INaP-dependent burst will be activated intrinsically.
Bursts that are intrinsically generated prior to the population burst are often observed in bursting neurons and they are referred to as “ectopic” bursts (Fig. 1a, c, cell 2 and Fig. 5b) [2, 25, 33]. Assuming that bursting neurons are embedded in the respiratory network, these ectopic bursts may contribute to the initiation of the network burst [21]. It is important to emphasize that the threshold for synaptic or intrinsic activation of a burst will vary among individual neurons and individual respiratory cycles. The amount of activateable INaP, the inactivation status, the amount of synaptic inputs, and the location of ion channels and synaptic receptors on the dendrite and soma of the respiratory neurons will not be the same in any two given neurons at any given time. Thus, ectopic bursts will be generated in some but not all neurons during some but not all cycles. Bursting will act as a non-linear amplifier in some cycles, an initiating mechanism in other cycles, and the onset of each population burst will depend on the overall integration of all these intrinsic and synaptic factors at the cellular and network level.
The above considerations focused on the synaptic interactions of INaP-dependent bursting. Different rules apply to ICAN-dependent bursting. The onset of an ICAN burst will be determined by an increase in intracellular calcium, which will lead to the opening of TRP channels and thus an influx of Na + and Ca2 + . Because processes that raise intracellular calcium will open TRP channels, the voltage-dependent opening of calcium channels will also contribute to the activation of ICAN, which will render bursting in CS pacemakers voltage dependent (Fig. 4c). However, note the voltage-dependency is different than that observed in CI pacemakers (Fig. 4c) [119]. In the rhythmically active network, the synaptic volley that is derived from the population burst will generate action potentials that will create sufficient calcium influx to trigger the ICAN current, thereby leading to a boost of excitatory synaptic input. Besides AMPA receptors, any excitatory synaptic input involving the activation of the NMDAr, mGluR1, and mGluR5 will contribute to the activation of ICAN [120]. The transient Ca2 + influx derived from the activation of these postsynaptic glutamatergic receptors will activate a signaling cascade that activates ICAN and thus causes the amplification of the inspiratory drive potential [120]. However, ICAN activation does not depend only on synaptic properties. Following synaptic isolation, ICAN-dependent bursting persists in CS pacemaker neurons (Fig. 4b). Bursting in these pacemaker neurons is abolished when action potentials are blocked with TTX [26], suggesting that the intrinsically generated action potentials are an important source of intracellular calcium that is required to activate ICAN. This conclusion is consistent with calcium imaging studies that demonstrate substantial and widespread somatic Ca2 + influx, which is generated by TTX-sensitive action potentials [128]. Interestingly, NE induces ICAN-dependent bursting only in spiking pacemakers (Fig. 1b), but not in silent non-pacemakers [15] further supporting the conclusion that spiking activity plays a critical role in the generation of bursting.
An interesting property of pacemaker neurons is the presence of the background inward current [122, 129]. This background current presumably constitutes the INaP current, which continuously depolarizes pacemaker neurons rendering these neurons spontaneously active. This current keeps pacemaker neurons in the firing range, even if the extracellular potassium concentrations are altered [122, 129]. This important conclusion is often overlooked in computational modeling. Thus, we want to emphasize that this background current is an important mechanism that explains why pacemaker neurons burst not only at 8 mM extracellular potassium, but also at 3 mM extracellular potassium. In other words, raising the extracellular potassium concentration is not required to bring pacemakers into a bursting range, as is often assumed. The background current may also contribute to the generation of action potentials that intrinsically ramp up prior to the ICAN-dependent burst in many CS pacemakers (Fig. 3a). Assuming that at least 50% of bursting neurons are excitatory [130], this will lead to an intrinsically generated ramp-up of EPSPs that precedes the population burst. Indeed this synaptic ramp is also seen in nonpacemaker neurons (Fig. 1c, cell 3). Thus, autonomously generated spike activity in many respiratory neurons will inevitably contribute to the existing synaptic mechanisms.
According to the so called “group pacemaker” hypothesis [2], excitatory interconnections between preBötC neurons initiate positive feedback through recurrent excitation. These excitatory interconnections are the critical initiators of the respiratory rhythm. Although this hypothesis acknowledges that INaP and ICAN are present, the hypothesis proposes that these intrinsic mechanisms serve to amplify the synaptic depolarization rather then initiate the respiratory cycle. The group pacemaker hypothesis acknowledges also that bursting-pacemaker neurons participate in network activity but this hypothesis explicitly states that these neurons are not essential. According to this hypothesis, INaP and ICAN in most respiratory neurons are activated by excitatory synaptic input and are thus not “initiated” intrinsically. Yet, as shown in this and many other studies, INaP does not require excitatory synaptic input to be activated, because many respiratory neurons are autonomously active. The autonomously generated action potentials will drive ICAN in CS pacemaker neurons and INaP will drive CI pacemakers towards burst generation. If one considers how many respiratory neurons are capable of autonomously generating action potentials within the respiratory network, and that all bursting neurons are maintained in a depolarized membrane state through INaP current, the conclusion that synaptic drive is required to activate INaP seems unjustified. Thus, we propose that INaP and ICAN serve (a) to amplify the synaptic depolarization as well as (b) to initiate the synaptic events that contribute to the initiation of the respiratory rhythm.
Conclusion
The current debate on respiratory rhythm generation does not center on oversimplified concepts anymore. There is very little support for hypotheses that postulate that the respiratory rhythm emerges only through synaptic interactions or only through pacemaker properties. It seems that everybody agrees that synaptic and intrinsic membrane properties are involved in respiratory rhythm generation. However, it remains debated whether synaptic inputs or intrinsic membrane properties initiate the events that lead to the respiratory rhythm. The above considerations illustrate that the contribution of intrinsic and synaptic properties are inseparable in the functional network as both properties are intertwined in many ways in the rhythmically active network. Thus, arguing that rhythm generation is an “emergent synaptic property” seems as one-sided as arguing that rhythm generation is an “emergent bursting property”. Unless it is demonstrated that (a) a substantial release of excitatory synaptic transmitters can occur in the absence of intrinsic membrane properties such as INaP or ICAN, and (b) that this synaptic release suffices to initiate the events that lead to the activation of the respiratory cycle, it seems to us that the hypothesis that excitatory interconnections between preBötC neurons initiate the positive synaptic feedback which then generates the respiratory rhythm has little experimental support. Thus, while there is ample experimental evidence for the existence and contribution of synaptic as well as intrinsic mechanisms such as ICAN and INaP, there is little evidence that respiratory rhythm generation emerges through the interaction of either synaptic or intrinsic mechanisms alone. The question is whether it even makes sense to theoretically assume that such a condition exists. How (and why) would a network turn off all its rich intrinsic membrane properties to operate on synaptic mechanisms alone at any given moment?
Acknowledgements
This study was supported by the National Institutes of Health Grants R01 HL/NS-60120 and P01 HL-090554-01.
References
- 1.Alheid GF, Milsom WK, McCrimmon DR. Pontine influences on breathing: an overview. Respir. Physiol. Neurobiol. 2004;143(2–3):105–114. doi: 10.1016/j.resp.2004.06.016. [DOI] [PubMed] [Google Scholar]
- 2.Feldman JL, Negro CA. Looking for inspiration: new perspectives on respiratory rhythm. Nat. Rev. Neurosci. 2006;7(3):232–242. doi: 10.1038/nrn1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Davenport PW, Reep RL, Thompson FJ. Phrenic nerve afferent activation of neurons in the cat SI cerebral cortex. J. Physiol. 2010;588(Pt 5):873–886. doi: 10.1113/jphysiol.2009.181735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Harper RM. Sudden infant death syndrome: a failure of compensatory cerebellar mechanisms? Pediatr. Res. 2000;48(2):140–142. doi: 10.1203/00006450-200008000-00004. [DOI] [PubMed] [Google Scholar]
- 5.Harper RM, Woo MA, Alger JR. Visualization of sleep influences on cerebellar and brainstem cardiac and respiratory control mechanisms. Brain Res. Bull. 2000;53(1):125–131. doi: 10.1016/s0361-9230(00)00317-8. [DOI] [PubMed] [Google Scholar]
- 6.Subramanian HH, Holstege G. Periaqueductal gray control of breathing. Adv. Exp. Med. Biol. 2010;669:353–358. doi: 10.1007/978-1-4419-5692-7_72. [DOI] [PubMed] [Google Scholar]
- 7.Leupoldt A, Keil A, Chan PY, Bradley MM, Lang PJ, Davenport PW. Cortical sources of the respiratory-related evoked potential. Respir. Physiol. Neurobiol. 2010;170(2):198–201. doi: 10.1016/j.resp.2009.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Smith JC, Ellenberger HH, Ballanyi K, Richter DW, Feldman JL.Pre-Bötzinger complex: a brainstem region that may generate respiratory rhythm in mammals Science 1991254726–729.1991Sci...254..726S [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gray PA, Janczewski WA, Mellen N, McCrimmon DR, Feldman JL. Normal breathing requires preBötzinger complex neurokinin-1 receptor-expressing neurons. Nat. Neurosci. 2001;4(9):927–930. doi: 10.1038/nn0901-927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McKay LC, Janczewski WA, Feldman JL. Sleep-disordered breathing after targeted ablation of preBötzinger complex neurons. Nat. Neurosci. 2005;8(9):1142–1144. doi: 10.1038/nn1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ramirez JM, Schwarzacher SW, Pierrefiche O, Olivera BM, Richter DW. Selective lesioning of the cat pre-Bötzinger complex in vivo eliminates breathing but not gasping. J. Physiol. 1998;507(Pt 3):895–907. doi: 10.1111/j.1469-7793.1998.895bs.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tan W, Janczewski WA, Yang P, Shao XM, Callaway EM, Feldman JL. Silencing preBötzinger complex somatostatin-expressing neurons induces persistent apnea in awake rat. Nat. Neurosci. 2008;11:538–540. doi: 10.1038/nn.2104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ramirez JM, Quellmalz UJ, Richter DW. Postnatal changes in the mammalian respiratory network as revealed by the transverse brainstem slice of mice. J. Physiol. 1996;491(Pt 3):799–812. doi: 10.1113/jphysiol.1996.sp021258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lieske SP, Thoby-Brisson M, Telgkamp P, Ramirez JM. Reconfiguration of the neural network controlling multiple breathing patterns: eupnea, sighs and gasps. Nat. Neurosci. 2000;3(6):600–607. doi: 10.1038/75776. [DOI] [PubMed] [Google Scholar]
- 15.Viemari JC, Ramirez JM. Norepinephrine differentially modulates different types of respiratory pacemaker and nonpacemaker neurons. J. Neurophysiol. 2006;95(4):2070–2082. doi: 10.1152/jn.01308.2005. [DOI] [PubMed] [Google Scholar]
- 16.Surmeier DJ, Mercer JN, Chan CS. Autonomous pacemakers in the basal ganglia: who needs excitatory synapses anyway? Curr. Opin. Neurobiol. 2005;15(3):312–318. doi: 10.1016/j.conb.2005.05.007. [DOI] [PubMed] [Google Scholar]
- 17.Herrik KF, Christophersen P, Shepard PD. Pharmacological modulation of the gating properties of small conductance Ca2+-activated K+ channels alters the firing pattern of dopamine neurons in vivo. J. Neurophysiol. 2010;104(3):1726–1735. doi: 10.1152/jn.01126.2009. [DOI] [PubMed] [Google Scholar]
- 18.Ptak K, Yamanishi T, Aungst J, Milescu LS, Zhang R, Richerson GB, Smith JC. Raphe neurons stimulate respiratory circuit activity by multiple mechanisms via endogenously released serotonin and substance P. J. Neurosci. 2009;29(12):3720–3737. doi: 10.1523/JNEUROSCI.5271-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Raman IM, Gustafson AE, Padgett D. Ionic currents and spontaneous firing in neurons isolated from the cerebellar nuclei. J. Neurosci. 2000;20(24):9004–9016. doi: 10.1523/JNEUROSCI.20-24-09004.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Takahashi A, Shimamoto A, Boyson CO, DeBold JF, Miczek KA. GABA(B) receptor modulation of serotonin neurons in the dorsal raphe nucleus and escalation of aggression in mice. J. Neurosci. 2010;30(35):11771–11780. doi: 10.1523/JNEUROSCI.1814-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ramirez JM, Tryba AK, Pena F. Pacemaker neurons and neuronal networks: an integrative view. Curr. Opin. Neurobiol. 2004;14(6):665–674. doi: 10.1016/j.conb.2004.10.011. [DOI] [PubMed] [Google Scholar]
- 22.Negro CA, Koshiya N, Butera RJ, Smith JC. Persistent sodium current, membrane properties and bursting behavior of pre-Bötzinger complex inspiratory neurons in vitro. J. Neurophysiol. 2002;88(5):2242–2250. doi: 10.1152/jn.00081.2002. [DOI] [PubMed] [Google Scholar]
- 23.Koshiya N, Smith JC.Neuronal pacemaker for breathing visualized in vitro Nature 19994006742360–363.1999Natur.400..360K [DOI] [PubMed] [Google Scholar]
- 24.Mekllen NM, Mishra D. Functional anatomical evidence for respiratory rhythmogenic function of endogenous bursters in rat medulla. J. Neurosci. 2010;30(25):8383–8392. doi: 10.1523/JNEUROSCI.5510-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pena F, Parkis MA, Tryba AK, Ramirez JM. Differential contribution of pacemaker properties to the generation of respiratory rhythms during normoxia and hypoxia. Neuron. 2004;43(1):105–117. doi: 10.1016/j.neuron.2004.06.023. [DOI] [PubMed] [Google Scholar]
- 26.Thoby-Brisson M, Ramirez JM. Identification of two types of inspiratory pacemaker neurons in the isolated respiratory neural network of mice. J. Neurophysiol. 2001;86(1):104–112. doi: 10.1152/jn.2001.86.1.104. [DOI] [PubMed] [Google Scholar]
- 27.St.-John WM, Stornetta RL, Guyenet PG, Paton JF. Location and properties of respiratory neurones with putative intrinsic bursting properties in the rat in situ. J. Physiol. 2009;587(Pt 13):3175–3188. doi: 10.1113/jphysiol.2009.170308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cymbalyuk GS, Gaudry Q, Masino MA, Calabrese RL. Bursting in leech heart interneurons: cell-autonomous and network-based mechanisms. J. Neurosci. 2002;22(24):10580–10592. doi: 10.1523/JNEUROSCI.22-24-10580.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Golowasch J, Buchholtz F, Epstein IR, Marder E. Contribution of individual ionic currents to activity of a model stomatogastric ganglion neuron. J. Neurophysiol. 1992;67(2):341–349. doi: 10.1152/jn.1992.67.2.341. [DOI] [PubMed] [Google Scholar]
- 30.Golowasch J, Casey M, Abbott LF, Marder E. Network stability from activity-dependent regulation of neuronal conductances. Neural Comput. 1999;11(5):1079–1096. doi: 10.1162/089976699300016359. [DOI] [PubMed] [Google Scholar]
- 31.Negro CA, Morgado-Valle C, Feldman JL. Respiratory rhythm: an emergent network property? Neuron. 2002;34(5):821–830. doi: 10.1016/s0896-6273(02)00712-2. [DOI] [PubMed] [Google Scholar]
- 32.Negro CA, Morgado-Valle C, Hayes JA, Mackay DD, Pace RW, Crowder EA, Feldman JL. Sodium and calcium current-mediated pacemaker neurons and respiratory rhythm generation. J. Neurosci. 2005;25(2):446–453. doi: 10.1523/JNEUROSCI.2237-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tryba AK, Pena F, Lieske SP, Viemari JC, Thoby-Brisson M, Ramirez JM. Differential modulation of neural network and pacemaker activity underlying eupnea and sigh-breathing activities. J. Neurophysiol. 2008;99(5):2114–2125. doi: 10.1152/jn.01192.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Koizumi H, Smith JC. Persistent Na + and K + -dominated leak currents contribute to respiratory rhythm generation in the pre-Bötzinger complex in vitro. J. Neurosci. 2008;28(7):1773–1785. doi: 10.1523/JNEUROSCI.3916-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brumberg JC, Nowak LG, McCormick DA. Ionic mechanisms underlying repetitive high-frequency burst firing in supragranular cortical neurons. J. Neurosci. 2000;20(13):4829–4843. doi: 10.1523/JNEUROSCI.20-13-04829.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Do MT, Bean BP. Subthreshold sodium currents and pacemaking of subthalamic neurons: modulation by slow inactivation. Neuron. 2003;39(1):109–120. doi: 10.1016/s0896-6273(03)00360-x. [DOI] [PubMed] [Google Scholar]
- 37.Taddese A, Bean BP. Subthreshold sodium current from rapidly inactivating sodium channels drives spontaneous firing of tuberomammillary neurons. Neuron. 2002;33(4):587–600. doi: 10.1016/s0896-6273(02)00574-3. [DOI] [PubMed] [Google Scholar]
- 38.Alzheimer C, Schwindt PC, Crill WE. Postnatal development of a persistent Na + current in pyramidal neurons from rat sensorimotor cortex. J. Neurophysiol. 1993;69(1):290–292. doi: 10.1152/jn.1993.69.1.290. [DOI] [PubMed] [Google Scholar]
- 39.Carr DB, Cooper DC, Ulrich SL, Spruston N, Surmeier DJ. Serotonin receptor activation inhibits sodium current and dendritic excitability in prefrontal cortex via a protein kinase C-dependent mechanism. J. Neurosci. 2002;22(16):6846–6855. doi: 10.1523/JNEUROSCI.22-16-06846.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kay AR, Sugimori M, Llinas R. Kinetic and stochastic properties of a persistent sodium current in mature guinea pig cerebellar Purkinje cells. J. Neurophysiol. 1998;80(3):1167–1179. doi: 10.1152/jn.1998.80.3.1167. [DOI] [PubMed] [Google Scholar]
- 41.Magistretti J, Alonso A. Biophysical properties and slow voltage-dependent inactivation of a sustained sodium current in entorhinal cortex layer-II principal neurons: a whole-cell and single-channel study. J. Gen. Physiol. 1999;114(4):491–509. doi: 10.1085/jgp.114.4.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Maurice N, Tkatch T, Meisler M, Sprunger LK, Surmeier DJ. D1/D5 dopamine receptor activation differentially modulates rapidly inactivating and persistent sodium currents in prefrontal cortex pyramidal neurons. J. Neurosci. 2001;21(7):2268–2277. doi: 10.1523/JNEUROSCI.21-07-02268.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ptak K, Zummo GG, Alheid GF, Tkatch T, Surmeier DJ, McCrimmon DR. Sodium currents in medullary neurons isolated from the pre-Bötzinger complex region. J. Neurosci. 2005;25(21):5159–5170. doi: 10.1523/JNEUROSCI.4238-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Drongelen W, Koch H, Elsen FP, Lee HC, Mrejeru A, Doren E, Marcuccilli CJ, Hereld M, Stevens RL, Ramirez JM. Role of persistent sodium current in bursting activity of mouse neocortical networks in vitro. J. Neurophysiol. 2006;96(5):2564–2577. doi: 10.1152/jn.00446.2006. [DOI] [PubMed] [Google Scholar]
- 45.Wu N, Enomoto A, Tanaka S, Hsiao CF, Nykamp DQ, Izhikevich E, Chandler SH. Persistent sodium currents in mesencephalic V neurons participate in burst generation and control of membrane excitability. J. Neurophysiol. 2005;93(5):2710–2722. doi: 10.1152/jn.00636.2004. [DOI] [PubMed] [Google Scholar]
- 46.Astman N, Gutnick MJ, Fleidervish IA. Persistent sodium current in layer 5 neocortical neurons is primarily generated in the proximal axon. J. Neurosci. 2006;26(13):3465–3473. doi: 10.1523/JNEUROSCI.4907-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fleidervish IA, Gutnick MJ. Kinetics of slow inactivation of persistent sodium current in layer V neurons of mouse neocortical slices. J. Neurophysiol. 1996;76(3):2125–2130. doi: 10.1152/jn.1996.76.3.2125. [DOI] [PubMed] [Google Scholar]
- 48.Schwindt P, Crill W. Mechanisms underlying burst and regular spiking evoked by dendritic depolarization in layer 5 cortical pyramidal neurons. J. Neurophysiol. 1999;81(3):1341–1354. doi: 10.1152/jn.1999.81.3.1341. [DOI] [PubMed] [Google Scholar]
- 49.Stuart G, Sakmann B. Amplification of EPSPs by axosomatic sodium channels in neocortical pyramidal neurons. Neuron. 1995;15(5):1065–1076. doi: 10.1016/0896-6273(95)90095-0. [DOI] [PubMed] [Google Scholar]
- 50.Pennartz CM, Bierlaagh MA, Geurtsen AM. Cellular mechanisms underlying spontaneous firing in rat suprachiasmatic nucleus: involvement of a slowly inactivating component of sodium current. J. Neurophysiol. 1997;78(4):1811–1825. doi: 10.1152/jn.1997.78.4.1811. [DOI] [PubMed] [Google Scholar]
- 51.Catterall WA. Molecular properties of brain sodium channels: an important target for anticonvulsant drugs. Adv. Neurol. 1999;79:441–456. [PubMed] [Google Scholar]
- 52.Catterall WA. From ionic currents to molecular mechanisms: the structure and function of voltage-gated sodium channels. Neuron. 2000;26(1):13–25. doi: 10.1016/s0896-6273(00)81133-2. [DOI] [PubMed] [Google Scholar]
- 53.Donahue LM, Coates PW, Lee VH, Ippensen DC, Arze SE, Poduslo SE. The cardiac sodium channel mRNA is expressed in the developing and adult rat and human brain. Brain Res. 2000;887(2):335–343. doi: 10.1016/s0006-8993(00)03033-x. [DOI] [PubMed] [Google Scholar]
- 54.Goldin AL. Resurgence of sodium channel research. Annu. Rev. Physiol. 2001;63:871–894. doi: 10.1146/annurev.physiol.63.1.871. [DOI] [PubMed] [Google Scholar]
- 55.Goldin AL, Barchi RL, Caldwell JH, Hofmann F, Howe JR, Hunter JC, Kallen RG, Mandel G, Meisler MH, Netter YB, Noda M, Tamkun MM, Waxman SG, Wood JN, Catterall WA. Nomenclature of voltage-gated sodium channels. Neuron. 2000;28(2):365–368. doi: 10.1016/s0896-6273(00)00116-1. [DOI] [PubMed] [Google Scholar]
- 56.Claes L, Del-Favero J, Ceulemans B, Lagae L, Broeckhoven C, Jonghe P. De novo mutations in the sodium-channel gene SCN1A cause severe myoclonic epilepsy of infancy. Am. J. Hum. Genet. 2001;68(6):1327–1332. doi: 10.1086/320609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Escayg A, MacDonald BT, Meisler MH, Baulac S, Huberfeld G, An-Gourfinkel I, Brice A, LeGuern E, Moulard B, Chaigne D, Buresi C, Malafosse A. Mutations of SCN1A, encoding a neuronal sodium channel, in two families with GEFS+2. Nat. Genet. 2000;24(4):343–345. doi: 10.1038/74159. [DOI] [PubMed] [Google Scholar]
- 58.Wallace RH, Scheffer IE, Barnett S, Richards M, Dibbens L, Desai RR, Lerman-Sagie T, Lev D, Mazarib A, Brand N, Ben-Zeev B, Goikhman I, Singh R, Kremmidiotis G, Gardner A, Sutherland GR, George AL, Mulley JC, Berkovic SF. Neuronal sodium-channel alpha1-subunit mutations in generalized epilepsy with febrile seizures plus. Am. J. Hum. Genet. 2001;68(4):859–865. doi: 10.1086/319516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Raman IM, Sprunger LK, Meisler MH, Bean BP. Altered subthreshold sodium currents and disrupted firing patterns in Purkinje neurons of Scn8a mutant mice. Neuron. 1997;19(4):881–891. doi: 10.1016/s0896-6273(00)80969-1. [DOI] [PubMed] [Google Scholar]
- 60.Vega-Saenz de Miera EC, Rudy B, Sugimori M, Llinas R.Molecular characterization of the sodium channel subunits expressed in mammalian cerebellar Purkinje cells Proc. Natl. Acad. Sci. USA 199794137059–7064.1997PNAS...94.7059D [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yu FH, Catterall WA. Overview of the voltage-gated sodium channel family. Genome Biol. 2003;4(3):207. doi: 10.1186/gb-2003-4-3-207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yu FH, Westenbroek RE, Silos-Santiago I, McCormick KA, Lawson D, Ge P, Ferriera H, Lilly J, DiStefano PS, Catterall WA, Scheuer T, Curtis R. Sodium channel beta4, a new disulfide-linked auxiliary subunit with similarity to beta2. J. Neurosci. 2003;23(20):7577–7585. doi: 10.1523/JNEUROSCI.23-20-07577.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Brackenbury WJ, Isom LL. Voltage-gated Na + channels: potential for beta subunits as therapeutic targets. Expert Opin. Ther. Targets. 2008;12(9):1191–1203. doi: 10.1517/14728222.12.9.1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Isom LL. Sodium channel beta subunits: anything but auxiliary. Neuroscientist. 2001;7(1):42–54. doi: 10.1177/107385840100700108. [DOI] [PubMed] [Google Scholar]
- 65.Uebachs M, Opitz T, Royeck M, Dickhof G, Horstmann MT, Isom LL, Beck H. Efficacy loss of the anticonvulsant carbamazepine in mice lacking sodium channel beta subunits via paradoxical effects on persistent sodium currents. J. Neurosci. 2010;30(25):8489–8501. doi: 10.1523/JNEUROSCI.1534-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wallace RH, Wang DW, Singh R, Scheffer IE, George AL, Phillips HA, Saar K, Reis A, Johnson EW, Sutherland GR, Berkovic SF, Mulley JC. Febrile seizures and generalized epilepsy associated with a mutation in the Na + -channel beta1 subunit gene SCN1B. Nat. Genet. 1998;19(4):366–370. doi: 10.1038/1252. [DOI] [PubMed] [Google Scholar]
- 67.Aman TK, Raman IM. Inwardly permeating Na ions generate the voltage dependence of resurgent Na current in cerebellar Purkinje neurons. J. Neurosci. 2010;30(16):5629–5634. doi: 10.1523/JNEUROSCI.0376-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bant JS, Raman IM.Control of transient, resurgent, and persistent current by open-channel block by Na channel beta4 in cultured cerebellar granule neurons Proc. Natl. Acad. Sci. USA 20101072712357–12362.2010PNAS..10712357B [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Colquhoun D, Neher E, Reuter H, Stevens CF.Inward current channels activated by intracellular Ca in cultured cardiac cells Nature 19812945843752–754.1981Natur.294..752C [DOI] [PubMed] [Google Scholar]
- 70.Maruyama Y, Peterson OH.Single-channel currents in isolated patches of plasma membrane from basal surface of pancreatic acini Nature 19822995879159–161.1982Natur.299..159M [DOI] [PubMed] [Google Scholar]
- 71.Congar P, Leinekugel X, Ben-Ari Y, Crepel V. A long-lasting calcium-activated nonselective cationic current is generated by synaptic stimulation or exogenous activation of group I metabotropic glutamate receptors in CA1 pyramidal neurons. J. Neurosci. 1997;17(14):5366–5379. doi: 10.1523/JNEUROSCI.17-14-05366.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Prisco GV, Pearlstein E, Ray D, Robitaille R, Dubuc R. A cellular mechanism for the transformation of a sensory input into a motor command. J. Neurosci. 2000;20(21):8169–8176. doi: 10.1523/JNEUROSCI.20-21-08169.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Partridge LD, Valenzuela CF. Ca2 + store-dependent potentiation of Ca2 + -activated non-selective cation channels in rat hippocampal neurones in vitro. J. Physiol. 1999;521(Pt 3):617–627. doi: 10.1111/j.1469-7793.1999.00617.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Launay P, Fleig A, Perraud AL, Scharenberg AM, Penner R, Kinet JP. TRPM4 is a Ca2 + -activated nonselective cation channel mediating cell membrane depolarization. Cell. 2002;109(3):397–407. doi: 10.1016/s0092-8674(02)00719-5. [DOI] [PubMed] [Google Scholar]
- 75.Ben-Mabrouk F, Tryba AK. Substance P modulation of TRPC3/7 channels improves respiratory rhythm regularity and ICAN-dependent pacemaker activity. Eur. J. Neurosci. 2010;31(7):1219–1232. doi: 10.1111/j.1460-9568.2010.07156.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Crowder EA, Saha MS, Pace RW, Zhang H, Prestwich GD, Negro CA. Phosphatidylinositol 4,5-bisphosphate regulates inspiratory burst activity in the neonatal mouse preBötzinger complex. J. Physiol. 2007;582(Pt 3):1047–1058. doi: 10.1113/jphysiol.2007.134577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Minke B. Drosophila mutant with a transducer defect. Biophys. Struct. Mech. 1977;3(1):59–64. doi: 10.1007/BF00536455. [DOI] [PubMed] [Google Scholar]
- 78.Nilius B, Owsianik G, Voets T, Peters JA. Transient receptor potential cation channels in disease. Physiol. Rev. 2007;87(1):165–217. doi: 10.1152/physrev.00021.2006. [DOI] [PubMed] [Google Scholar]
- 79.Greka A, Navarro B, Oancea E, Duggan A, Clapham DE. TRPC5 is a regulator of hippocampal neurite length and growth cone morphology. Nat. Neurosci. 2003;6(8):837–845. doi: 10.1038/nn1092. [DOI] [PubMed] [Google Scholar]
- 80.Kim SJ, Kim YS, Yuan JP, Petralia RS, Worley PF, Linden DJ.Activation of the TRPC1 cation channel by metabotropic glutamate receptor mGluR1 Nature 20034266964285–291.2003Natur.426..285K [DOI] [PubMed] [Google Scholar]
- 81.Moran MM, Xu H, Clapham DE. TRP ion channels in the nervous system. Curr. Opin. Neurobiol. 2004;14(3):362–369. doi: 10.1016/j.conb.2004.05.003. [DOI] [PubMed] [Google Scholar]
- 82.Mori Y, Takada N, Okada T, Wakamori M, Imoto K, Wanifuchi H, Oka H, Oba A, Ikenaka K, Kurosaki T. Differential distribution of TRP Ca2 + channel isoforms in mouse brain. NeuroReport. 1998;9(3):507–515. [PubMed] [Google Scholar]
- 83.Caterina MJ, Leffler A, Malmberg AB, Martin WJ, Trafton J, Petersen-Zeitz KR, Koltzenburg M, Basbaum AI, Julius D.Impaired nociception and pain sensation in mice lacking the capsaicin receptor Science 20002885464306–313.2000Sci...288..306C [DOI] [PubMed] [Google Scholar]
- 84.Zhang Y, Hoon MA, Chandrashekar J, Mueller KL, Cook B, Wu D, Zuker CS, Ryba NJ. Coding of sweet, bitter, and umami tastes: different receptor cells sharing similar signaling pathways. Cell. 2003;112(3):293–301. doi: 10.1016/s0092-8674(03)00071-0. [DOI] [PubMed] [Google Scholar]
- 85.Corey DP. New TRP channels in hearing and mechanosensation. Neuron. 2003;39(4):585–588. doi: 10.1016/s0896-6273(03)00505-1. [DOI] [PubMed] [Google Scholar]
- 86.Aarts M, Iihara K, Wei WL, Xiong ZG, Arundine M, Cerwinski W, MacDonald JF, Tymianski M. A key role for TRPM7 channels in anoxic neuronal death. Cell. 2003;115(7):863–877. doi: 10.1016/s0092-8674(03)01017-1. [DOI] [PubMed] [Google Scholar]
- 87.Bengtson CP, Tozzi A, Bernardi G, Mercuri NB. Transient receptor potential-like channels mediate metabotropic glutamate receptor EPSCs in rat dopamine neurones. J. Physiol. 2004;555(Pt 2):323–330. doi: 10.1113/jphysiol.2003.060061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Tozzi A, Bengtson CP, Longone P, Carignani C, Fusco FR, Bernardi G, Mercuri NB. Involvement of transient receptor potential-like channels in responses to mGluR-I activation in midbrain dopamine neurons. Eur. J. Neurosci. 2003;18(8):2133–2145. doi: 10.1046/j.1460-9568.2003.02936.x. [DOI] [PubMed] [Google Scholar]
- 89.Karashima Y, Prenen J, Meseguer V, Owsianik G, Voets T, Nilius B. Modulation of the transient receptor potential channel TRPA1 by phosphatidylinositol 4,5-biphosphate manipulators. Pflügers Arch. 2008;457(1):77–89. doi: 10.1007/s00424-008-0493-6. [DOI] [PubMed] [Google Scholar]
- 90.Nilius B, Mahieu F, Prenen J, Janssens A, Owsianik G, Vennekens R, Voets T. The Ca2 + -activated cation channel TRPM4 is regulated by phosphatidylinositol 4,5-biphosphate. EMBO J. 2006;25(3):467–478. doi: 10.1038/sj.emboj.7600963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zavala-Tecuapetla C, Aguileta MA, Lopez-Guerrero JJ, Gonzalez-Marin MC, Pena F. Calcium-activated potassium currents differentially modulate respiratory rhythm generation. Eur. J. Neurosci. 2008;27(11):2871–2884. doi: 10.1111/j.1460-9568.2008.06214.x. [DOI] [PubMed] [Google Scholar]
- 92.Pena F, Ramirez JM. Endogenous activation of serotonin-2A receptors is required for respiratory rhythm generation in vitro. J. Neurosci. 2002;22(24):11055–11064. doi: 10.1523/JNEUROSCI.22-24-11055.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Pena F, Ramirez JM. Substance P-mediated modulation of pacemaker properties in the mammalian respiratory network. J. Neurosci. 2004;24(34):7549–7556. doi: 10.1523/JNEUROSCI.1871-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Shao XM, Feldman JL. Acetylcholine modulates respiratory pattern: effects mediated by M3-like receptors in preBötzinger complex inspiratory neurons. J. Neurophysiol. 2000;83(3):1243–1252. doi: 10.1152/jn.2000.83.3.1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Shao XM, Feldman JL. Mechanisms underlying regulation of respiratory pattern by nicotine in preBötzinger complex. J. Neurophysiol. 2001;85(6):2461–2467. doi: 10.1152/jn.2001.85.6.2461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Shao XM, Feldman JL. Pharmacology of nicotinic receptors in preBötzinger complex that mediate modulation of respiratory pattern. J. Neurophysiol. 2002;88(4):1851–1858. doi: 10.1152/jn.2002.88.4.1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Shao XM, Feldman JL. Cholinergic neurotransmission in the preBötzinger complex modulates excitability of inspiratory neurons and regulates respiratory rhythm. Neuroscience. 2005;130(4):1069–1081. doi: 10.1016/j.neuroscience.2004.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Tryba AK, Pena F, Ramirez JM. Gasping activity in vitro: a rhythm dependent on 5-HT2A receptors. J. Neurosci. 2006;26(10):2623–2634. doi: 10.1523/JNEUROSCI.4186-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Doi A, Ramirez JM. State-dependent interactions between excitatory neuromodulators in the control of breathing. J. Neurosci. 2010;30:8251–8262. doi: 10.1523/JNEUROSCI.5361-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Gray PA, Rekling JC, Bocchiaro CM, Feldman JL. Modulation of respiratory frequency by peptidergic input to rhythmogenic neurons in the preBötzinger complex. Science. 1999;286(5444):1566–1568. doi: 10.1126/science.286.5444.1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Shvarev YN, Lagercrantz H, Yamamoto Y. Biphasic effects of substance P on respiratory activity and respiration-related neurones in ventrolateral medulla in the neonatal rat brainstem in vitro. Acta Physiol. Scand. 2002;174(1):67–84. doi: 10.1046/j.1365-201x.2002.00926.x. [DOI] [PubMed] [Google Scholar]
- 102.Thoby-Brisson M, Trinh JB, Champagnat J, Fortin G. Emergence of the pre-Bötzinger respiratory rhythm generator in the mouse embryo. J. Neurosci. 2005;25(17):4307–4318. doi: 10.1523/JNEUROSCI.0551-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Telgkamp P, Cao YQ, Basbaum AI, Ramirez JM. Long-term deprivation of substance P in PPT-A mutant mice alters the anoxic response of the isolated respiratory network. J. Neurophysiol. 2002;88(1):206–213. doi: 10.1152/jn.2002.88.1.206. [DOI] [PubMed] [Google Scholar]
- 104.Oh EJ, Gover TD, Cordoba-Rodriguez R, Weinreich D. Substance P evokes cation currents through TRP channels in HEK293 cells. J. Neurophysiol. 2003;90(3):2069–2073. doi: 10.1152/jn.00026.2003. [DOI] [PubMed] [Google Scholar]
- 105.Clapham DE, Montell C, Schultz G, Julius D. International Union of Pharmacology. XLIII. Compendium of voltage-gated ion channels: transient receptor potential channels. Pharmacol. Rev. 2003;55(4):591–596. doi: 10.1124/pr.55.4.6. [DOI] [PubMed] [Google Scholar]
- 106.Ballantyne D, Richter DW. Post-synaptic inhibition of bulbar inspiratory neurones in the cat. J. Physiol. 1984;348:67–87. doi: 10.1113/jphysiol.1984.sp015100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ogilvie MD, Gottschalk A, Anders K, Richter DW, Pack AI. A network model of respiratory rhythmogenesis. Am. J. Physiol. 1992;263(4 Pt 2):R962–R975. doi: 10.1152/ajpregu.1992.263.4.R962. [DOI] [PubMed] [Google Scholar]
- 108.Abdala AP, Rybak IA, Smith JC, Zoccal DB, Machado BH, St.-John WM, Paton JF. Multiple pontomedullary mechanisms of respiratory rhythmogenesis. Respir. Physiol. Neurobiol. 2009;168(1–2):19–25. doi: 10.1016/j.resp.2009.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Baekey DM, Molkov YI, Paton JF, Rybak IA, Dick TE. Effect of baroreceptor stimulation on the respiratory pattern: insights into respiratory-sympathetic interactions. Respir. Physiol. Neurobiol. 2010;174:135–145. doi: 10.1016/j.resp.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Rubin JE, Hayes JA, Mendenhall JL, Negro CA.Calcium-activated nonspecific cation current and synaptic depression promote network-dependent burst oscillations Proc. Natl. Acad. Sci USA 200910682939–2944.2009PNAS..106.2939R [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Smith JC, Abdala AP, Rybak IA, Paton JF. Structural and functional architecture of respiratory networks in the mammalian brainstem. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2009;364(1529):2577–2587. doi: 10.1098/rstb.2009.0081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ramirez JM, Telgkamp P, Elsen FP, Quellmalz UJ, Richter DW. Respiratory rhythm generation in mammals. Synaptic and membrane properties. Respir. Physiol. 1997;110(2–3):71–85. doi: 10.1016/s0034-5687(97)00074-1. [DOI] [PubMed] [Google Scholar]
- 113.Tryba AK, Pena F, Ramirez JM. Stabilization of bursting in respiratory pacemaker neurons. J. Neurosci. 2003;23(8):3538–3546. doi: 10.1523/JNEUROSCI.23-08-03538.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Shao XM, Feldman JL. Respiratory rhythm generation and synaptic inhibition of expiratory neurons in pre-Bötzinger complex: differential roles of glycinergic and GABAergic neural transmission. J. Neurophysiol. 1997;77(4):1853–1860. doi: 10.1152/jn.1997.77.4.1853. [DOI] [PubMed] [Google Scholar]
- 115.Ramirez JM, Viemari JC. Determinants of inspiratory activity. Respir. Physiol. Neurobiol. 2005;147(2–3):145–157. doi: 10.1016/j.resp.2005.05.003. [DOI] [PubMed] [Google Scholar]
- 116.Funk GD, Smith JC, Feldman JL. Generation and transmission of respiratory oscillations in medullary slices: role of excitatory amino acids. J. Neurophysiol. 1993;70(4):1497–1515. doi: 10.1152/jn.1993.70.4.1497. [DOI] [PubMed] [Google Scholar]
- 117.Greer JJ, Smith JC, Feldman JL. Role of excitatory amino acids in the generation and transmission of respiratory drive in neonatal rat. J. Physiol. 1991;437:727–749. doi: 10.1113/jphysiol.1991.sp018622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Lieske SP, Ramirez JM. Pattern-specific synaptic mechanisms in a multifunctional network. I. Effects of alterations in synapse strength. J. Neurophysiol. 2006;95(3):1323–1333. doi: 10.1152/jn.00505.2004. [DOI] [PubMed] [Google Scholar]
- 119.Lieske SP, Ramirez JM. Pattern-specific synaptic mechanisms in a multifunctional network. II. Intrinsic modulation by metabotropic glutamate receptors. J. Neurophysiol. 2006;95(3):1334–1344. doi: 10.1152/jn.00506.2004. [DOI] [PubMed] [Google Scholar]
- 120.Pace RW, Mackay DD, Feldman JL, Negro CA. Role of persistent sodium current in mouse preBötzinger complex neurons and respiratory rhythm generation. J. Physiol. 2007;580:485–496. doi: 10.1113/jphysiol.2006.124602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Rekling JC, Feldman JL. PreBötzinger complex and pacemaker neurons. Hypothesized site and kernel for respiratory rhythm generation. Annu. Rev. Physiol. 1998;60:385–405. doi: 10.1146/annurev.physiol.60.1.385. [DOI] [PubMed] [Google Scholar]
- 122.Tryba AK, Ramirez JM. Background sodium current stabilizes bursting in respiratory pacemaker neurons. J. Neurobiol. 2004;60(4):481–489. doi: 10.1002/neu.20050. [DOI] [PubMed] [Google Scholar]
- 123.Johnson SM, Smith JC, Funk GD, Feldman JL. Pacemaker behavior of respiratory neurons in medullary slices from neonatal rat. J. Neurophysiol. 1994;72(6):2598–2608. doi: 10.1152/jn.1994.72.6.2598. [DOI] [PubMed] [Google Scholar]
- 124.Ramirez JM, Richter DW. The neuronal mechanisms of respiratory rhythm generation. Curr. Opin. Neurobiol. 1996;6(6):817–825. doi: 10.1016/s0959-4388(96)80033-x. [DOI] [PubMed] [Google Scholar]
- 125.Ramirez JM, Pearson KG. Octopamine induces bursting and plateau potentials in insect neurones. Brain Res. 1991;549(2):332–337. doi: 10.1016/0006-8993(91)90477-d. [DOI] [PubMed] [Google Scholar]
- 126.Butera RJ, Rinzel J, Smith JC. Models of respiratory rhythm generation in the pre-Bötzinger complex. I. Bursting pacemaker neurons. J. Neurophysiol. 1999;82(1):382–397. doi: 10.1152/jn.1999.82.1.382. [DOI] [PubMed] [Google Scholar]
- 127.Negro CA, Wilson CG, Butera RJ, Rigatto H, Smith JC. Periodicity, mixed-mode oscillations, and quasiperiodicity in a rhythm-generating neural network. Biophys. J. 2002;82(1 Pt 1):206–214. doi: 10.1016/S0006-3495(02)75387-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Morgado-Valle C, Beltran-Parrazal L, DiFranco M, Vergara JL, Feldman JL. Somatic Ca2 + transients do not contribute to inspiratory drive in preBötzinger complex neurons. J. Physiol. 2008;586(18):4531–4540. doi: 10.1113/jphysiol.2008.154765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Chevalier M, Ben-Mabrouk F, Tryba AK. Background sodium current underlying respiratory rhythm regularity. Eur. J. Neurosci. 2008;28(12):2423–2433. doi: 10.1111/j.1460-9568.2008.06537.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Morgado-Valle C, Baca SM, Feldman JL. Glycinergic pacemaker neurons in preBötzinger complex of neonatal mouse. J. Neurosci. 2010;30(10):3634–3639. doi: 10.1523/JNEUROSCI.3040-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]