

Abstract
African sleeping sickness is endemic in sub-Saharan Africa where the WHO estimates that 60 million people are at risk for the disease. Human African trypanosomiasis (HAT) is 100% fatal if untreated and the current drug therapies have significant limitations due to toxicity and difficult treatment regimes. No new chemical agents have been approved since eflornithine in 1990. The pentamidine analog DB289, which was in late stage clinical trials for the treatment of early stage HAT recently failed due to toxicity issues. A new protocol for the treatment of late-stage T. brucei gambiense that uses combination nifurtomox/eflornithine (NECT) was recently shown to have better safety and efficacy than eflornithine alone, while being easier to administer. This breakthrough represents the only new therapy for HAT since the approval of eflornithine. A number of research programs are on going to exploit the unusual biochemical pathways in the parasite to identify new targets for target based drug discovery programs. HTS efforts are also underway to discover new chemical entities through whole organism screening approaches. A number of inhibitors with anti-trypanosomal activity have been identified by both approaches, but none of the programs are yet at the stage of identifying a preclinical candidate. This dire situation underscores the need for continued effort to identify new chemical agents for the treatment of HAT.
African sleeping sickness – disease and impact
African sleeping sickness is a fatal vector-borne disease caused by the protozoal pathogen, Trypanosoma brucei [1–3]. It is endemic in sub-Saharan Africa where it is transmitted by the bite of the tsetse fly. The WHO estimates that as many as 60 million people are at risk to contract Human African trypanosomiasis (HAT), which is caused by the T. b. gambiense (West Africa) and T. b. rhodesiense (East Africa). T. b. gambiense accounts for greater than 90% of the disease, with tens of thousands of cases reported yearly [4, 5]. HAT is found primarily in rural communities and the primitive medical care in these regions likely results in significant under reporting of the disease burden. T.b. rhodesiense also infects both wild and domesticated animals. Cattle contribute significantly as a reservoir for human infection, and disease in these animals contributes to malnutrition. Several other subspecies of trypanosomes are also endemic in Africa but are limited to infection of animals (T.b. brucei, T. congolense and T. evansi). These species are unable to sustain human infection due to the presence of the trypanosome lytic factor (TLF) in human serum [6]. T.b. gambiense is inherently resistant to TLF, while T.b. rhodesiense, is protected from lysis through expression of a lytic resistance gene (serum resistance-associated (SRA)) protein.
Symptoms and diagnosis
The clinical manifestations of the HAT depend both on the subspecies of the parasite and on the stage of infection [2, 4]. T. brucei is an entirely extracellular parasite that in the early stages replicates in the blood, and in latter stages migrates into the central nervous system and is found in the cerebral spinal fluid (CSF). Early stage disease is manifest by fever, headache, malaise, weight loss, and arthralgia, with symptoms of fever, sometimes accompanied by rigor and vomiting, cycling over several day intervals. Skin legions or chancre may also be present. The underlying etiology of the cyclic symptoms is antigenic variation of the parasite surface coat (variant surface glycoprotein (VSG))[7]. Switching of the expressed VSG gene occurs at a sufficient frequency to render useless the host immune response. Late stage disease is marked by progressively worsening neurological symptoms including head-ache, sleep disorders, personality changes (e.g. anxiety, irritability, violence, delirium), motor weakness and visual impairments. T.b. rhoesiense causes a virulent, rapidly fatal disease with high parasitimia that kills patients within weeks to months. It progresses quickly to CNS involvement, and leads to multiple organ involvement, including significant cardiac symptoms [8, 9], endocrine and gastrointestinal problems. T.b. gambiense is characterized by low parasitimia and a slower time course before CNS involvement. Symptoms are manifest over many years leading to the classic neurological symptoms and progression to coma and death for which sleeping sickness has been named.
Disease diagnosis and stage assessment is primitive and relies on the microscopic identification of parasites in the blood, lymph or CSF, often requiring concentration techniques to increase sensitivity, particularly for patients infected with T. b. gambiense [10]. A card agglutination test is used to screen for possible T.b. gambiense infections with conformation relying on microscopic identification. Elevated white blood cells and IgM levels also suggest the presence of the parasite. PCR-based methods amenable to the field have been described but are not in wide use [11, 12].
Current state of drug therapy
Drugs are the only therapeutic option for the treatment of HAT as there is no vaccine and no prospects that one will be developed. HAT is managed with a combination of suramin (T.b. rhodesiense) and pentamidine (T.b. gambiense) for early stage disease prior to CNS involvement, and melarsoprol and eflornithine for late stage disease (Figure 1 and Table 1)[1]. It is likely that nifurtimox/eflornithine combination therapy (NECT) will supplant eflornithine alone as a treatment option. A just completed clinical trial led the WHO to place NECT on its Essential Medicines List and to the recommendation that it should be considered as the front line treatment for late stage T.b. gambiense by Government control programs[13]. Eflornithine is not effective against T.b. rhodesiense, thus melarsoprol remains the only option for late stage disease [14].
Figure 1.

Current clinically used drugs for the treatment of HAT.
Table 1.
Current Drugs for the treatment of HAT
| Drug | Use | Limitations | Mechanism of action |
Dosing |
|---|---|---|---|---|
| Suramin |
T. b. rhodesiense early stage only |
Does not cross the blood brain barrier; toxicity |
unknown | IV injection 100- 200 mg test dose then 1 g given on days 1, 3, 7, 14 and 21 |
| Pentamidine |
T. b. gambiense, early stage only |
Does not cross the blood brain barrier |
unknown | IM injection in single doses of 4.0 mg/kg per day for 7 days |
| Melarsoprol | Late Stage T. b rhodesiense; Late stage T. b. gambiense if eflornithine is unavailable |
Severe toxicity – causes reactive enceplapthy resulting in death in up to 6% of patients |
unknown | T.b. rhodesiense, 3 series of 3 daily doses IV with a 7 day rest period in between: 1.8, 2.7 and 3.6 mg/kg on days 1, 2, and 3 respectively with subsequent series at 3.6 mg/kg daily; T. b. gambiense, 2.2 mg/kg/day IV for 10 days |
| Eflornithine | Late state T. b gambiense |
Not effective against T. b rhodesiense; difficult dosing regime requiring prolong i.v. administration |
Mechanism based inhibitor of ornithine decarboxylase |
400 mg/kg/day in divided doses IV every 6 h for 14 days |
| Nifurtimox/ Eflornithine (NECT) |
Late stage T. b gambiense |
Not effective against T. b rhodesiense |
MOA of nifurtimox – activated by a NADH- dependent mitochondrial nitroreductase leading to the generation of intracellular free radicals[169] |
eflornithine 400 mg/kg/ day IV in divided doses every 12 h for 7 days and nifurtimox 15 mg/kg/day orally every 8 h for 10 days |
Early stage HAT. Suramin
Suramin has been available since 1920 and was developed as a follow-up to the observations that closely related dyes had anti-trypanosome activity (e.g. trypan blue) [1]. The mechanism of action is not known, though the fact that it is concentrated in the parasite via receptor-mediated endocytosis may account for its selective antityrpanosomal activity. Resistance has not occurred in the field, suggesting that it has multiple cellular targets. Suramin is effective against early stage infection by both T.b. rhodesiense and T.b. gambiense [1]. However its use is typically restricted to T.b. rhodesiense since pentamidine is available for treatment of T.b. gambiense. Suramin does not cross the blood brain barrier and is ineffective against late stage disease. A range of side effects have been reported and include nausea, vomiting, fatigue and shock followed by renal toxicity and neurological complications such as headache and peripheral neuropathy after several doses.
Early stage HAT. Pentamidine
Pentamidine is one of several diamidines that shows significant anti-trypanosomal activity [1, 15, 16]. The mechanism of action is not known for any of these, however pentamidine is significantly concentrated by the parasite (reaching millimolar levels) and this may be a key factor in its selective toxicity. Transport of pentamidine by T. brucei is energy dependent and requires the adenine/adeonsine P2 transporter in combination with a low-capacity high-affinity (HAPT) transporter and a high-capacity low affinity transporter (LAPT), explaining why resistance does not develop readily in the field [17]. A cell line containing a knockout of the P2 transporter was selected for pentamidine resistance, yielding a line that was 100-fold resistant to pentamidine as a result of the apparent loss of HAPT activity. This line is also highly resistant to melarsoprol suggesting that the loss of both the P2 and HAPT transporters is necessary to generate high-level pentamidine and melarsoprol resistance. In contrast, transport of other diamidines such as DB289 and diminazene aceturate occurs predominantly via the P2 transporter, allowing changes in a single protein to yield resistance.
Pentamidine is used for the treatment of early stage T.b. gambiense but not for T.b. rhodesiense where it has reduced activity. It has broad spectrum anti-parasitic [16] activity and has also been used for leishmaniasis [18]. Pentamidine is not effective against late stage HAT, and it had been presumed not to cross the blood brain barrier. Recent studies show that it enters the CNS but not in sufficient concentrations to be effective against late stage disease [19]. Pentamidine causes significant toxicity in at least half of the patients, with life threatening hypoglycemia being the most serious.
Late stage HAT. Melarsoprol
Melarsoprol is an organic arsenical that was discovered to have antitrypanosomal activity in 1949 and it is active against both stages of T.b. gambiense and T.b. rhodesiense [1]. Because of its extreme toxicity it is reserved for late stage disease, and is now only recommended for the treatment of late stage T.b. rhodesiense for which there are no other options. The molecular mechanism behind the trypanocidal activity of melarsoprol is not understood. A prodrug, it is rapidly converted to melarsin oxide after administration. It reacts with many biomolecules through reversible interaction with sulfhydryl groups in both the parasite and the host and its toxicity likely results from these interactions [1]. Increasing numbers of treatment failures are being reported [20] and field isolates that are ten-fold less sensitive to the drug have been identified. In the laboratory melarsoprol resistance has been associated with the P2 transporter[17]. However this appears to be only one of the factors leading to melarsopral treatment failure. Cells overexpressing the P-glycoprotein multidrug resistance pump are also less sensitive to melarsoprol [21]. Significantly, not all treatment failures result from drug resistance and other factors are thought to play a role [22].
Melarsoprol is reserved for the treatment of late stage T.b. rhodesiense and is recommended for T.b. gambiense only where eflornithine is unavailable. Historically the dosing schemes were empirically derived. For T.b. gambieinse a new 10-day dosage regiment was developed based on experimental assessment of pharmacokinetics and is now recommended [14]. Adaptation of the simplified dosing scheme for T.b. rhodesiense awaits clinical demonstration of its efficacy (C. Burri, personal communication). Melarsoprol is highly toxic resulting in a treatment related death rate reported to be 5.9% during the recent clinical trial of the 10-day treatment regime[14]. Reactive encephalopathy has an occurrence rate of 5–10% and half of those affected by this complication die. Beyond this, vomiting and abdominal cramping, peripheral neuropathy, hypertension and heart damage are also common side effects of the drug.
Late stage HAT. Eflornithine and nifurtimox/eflornithine combination (NECT)
Eflornithine, or D,L-α-difluoromethylornithine, is a mechanism-based inhibitor of the pyridoxal-5’-phosphate dependent enzyme, ornithine decarboxylase (ODC)[1]. ODC catalyzes the first committed step in the biosynthesis of polyamines (Figure 2), which are required for cell growth [23, 24]. Eflornithine originally was developed for cancer therapy though it was never registered for this use. It was discovered to have anti-trypanosomal activity in 1980, leading to its approval and registration for clinical use in 1990. Eflornithine is the only clinically utilized anti-trypanosomal agent with a known mechanism of action. X-ray structural data has captured the eflornithine complex with T. brucei ODC in a covalent bond with an active site cysteine [25] (Figure 3). Eflornithine is equally effective at inactivating both the trypanosomal and human enzymes, however important differences have been identified at the cellular level that are likely to contribute to the observed species selectivity and to the acceptable toxicological profile, including differences in intracellular turnover rate between the mamamalian and T. brucei enzymes.
Figure 2.

Polyamine biosynthetic pathway in T. brucei.
Figure 3.

Active site of ODC bound to eflornithine. Select residues within 4Å of eflornithine-PLP are displayed. Eflornithine binds in the dimer interface; subunit A (green), subunit B (pink), eflornithine (magenta) and the cofactor pyridoxal 5’-phosphate (yellow). Nitrogen atoms are blue, oxygen red, sulfur yellow and phosphate orange. The figure was generated in PyMol [170] using the pdb coordinate file 2TOD.
Eflornithine is recommended for treatment of late stage T.b. gambiense and has been clinically demonstrated to have a better safety profile than melarsoprol [26]. It is less effective against T.b. rhodesiense and is not used for this disease. The molecular basis for this species difference is not understood. Past issues with affordable supplies of the drug have been resolved clearing the way to more wide spread use of this drug. Large doses are required for effective treatment, which may be partially explained by the finding that eflornithine does not efficiently enter the CNS [27]. The treatment regiment is extremely challenging to administer in rural settings and represents the main limitation to its use.
A clinical trial of combination nifurtimox-eflornthine (NECT) using a shortened course of eflornithine has recently been completed [13]. The cure rate for NECT (96.5%) was higher than for eflornithine alone (91.6%), and fewer severe adverse events (grade 3–4) were recorded (14% vs 29%). Treatment related deaths were also lower for NECT (0.7%) versus eflornithine alone (2%), and both treatments led to substantially fewer treatment-related deaths than for melarsoprol (5.9%)[14]. The side effects for eflornithine alone include seizures, fever, infections, neutropenia, hypertension and diarrhea. These effects are generally reversible once drug is discontinued provided they are properly managed. Septic shock was identified as the cause of the eflornithine-related deaths in the study. For NECT fewer patients suffered from drug-related diarrhea, infections, fever, skin rash or hypertension, however more reported nausea, vomiting and tremors. NECT is significantly easier to administer than eflornithine alone, it requires less eflornthine, which while donated for HAT therapy is expensive to synthesize. Overall the data indicates that NECT should be considered the front line treatment for late-stage T.b. gambiense infection.
Resistance to eflornithine has not been observed in field, however it can be readily generated in the lab where reduced drug influx and/or increase putrescine uptake appear to account for drug resistance [1]. These data suggest that the increasing use of eflornithine as a single agent leaves it exposed to development of resistance, and to the worrying possibility that the only alternative drug to melarsoprol could be lost. NECT therapy has the added benefit that it may reduce the chance that resistance to eflornithine develops.
New drugs against HAT are needed
The management of HAT patients is complicated by the fact that 4 of 5 key drugs are administered parenetrally and significant issues of toxicity, difficulty treatment regimes or drug resistance are present. Beyond these issues the lack of a single agent that is effective against both species and both stages leads to the requirement that speciation and staging remain critical aspects of diagnosis, and this must be performed under the difficulty of rural community hospitals that lack modern medical equipment. There is a clear and pressing need for the development of safer more effective drugs for the treatment of HAT. T. brucei has a number of novel metabolic pathways and unusual biological features that are understudy for their potential in drug discovery programs. Below we summarize progress towards the identification of new-antitrypanosomal agents by exploring the biochemical pathways and systems that have been identified as potential drug targets through a review of both genetic and chemical data (Table 2).
Table 2.
Targets for the development of new drugs for HAT
| Pathway | Enzymes | Validation | Status |
|---|---|---|---|
| Polyamine biosynthesis |
ODC, AdoMetDC | Genetic and chemical |
AdoMetDC – Lead optimization |
| Trypanothione biosynthesis |
TryRed, TSHSyn | Genetic and chemical |
Hit to Lead |
| Energy Metabolism/glycolytic enzymes |
Hexokinase, Phosphoglucose isomerase, phosphofructokinase, fructose-1,6- bisphoshate aldolase; phosphoclycerate kinase; enolase |
Genetic and chemical |
PFK – Hit to Lead Aldolase – Hit identification |
| Purine and Pyrimidine metabolism |
DHFR-TS, pteridine reductase, cytidine triphosphate synthetase |
Genetic and chemical |
DHFR-TS – Hit to Lead |
| DNA modification | DNA topoisomerases | Genetic and chemical |
Topo – Hit identification |
| Fatty acid biosynthesis/utilization |
Elongases, N- myristoyltransferase |
Genetic and chemical |
NMT – Hit to Lead |
| Sphingolipid biosynthesis |
Sphingolipid synthase |
Genetic | |
| Protein modification | Prenylation | Genetic and chemical |
PFT – Lead optimization |
| Protein modification | Kinases | Genetic and chemical |
GSK3 – Hit to Lead |
| Protein modification | Proteases (TbCatB) | Genetic and chemical |
Hit identification and validation |
| tubulin | Tubulin assembly | Genetic and chemical |
Hit identification |
The genomes of the kinetoplastid protozoal pathogens (T. brucei, T. cruzi and Leishmania) are complete and a number of genetic tools are available to evaluate gene function and essentiality[3]. The trypanosomatids are diploids and knockouts of non-essential genes can be readily generated by homologous recombination. Regulated conditional knockout of essential genes in T. brucei is also feasible and is achieved by integration of a gene copy under the control of the tetracycline-inducible system prior to knockout of the endogenous alleles. RNAi is functional in T. brucei, but not in T. cruzi and most Leishmania species, making T. brucei the kinetoplastid of choice for genetic validation studies [28].
Polyamine and trpanothione biosynthesis
Polyamines are small carbon-based cations that are essential for cell growth in most organisms [23, 24]. They most likely mediate their cellular effects at the transcriptional and translational levels by interacting with nucleic acids. Further, spermidine is a required precursor for the essential covalent modification (hypusine modification) of eukaryotic initiation factor 5A. Polyamines are synthesized from the precursors L-ornithine and S-adenosylmethioine (AdoMet), with the first committed steps in the pathway catalyzed by ODC and S-adenosylmethioine decarbolyase (AdoMetDC) (Figure 2). These steps represent the main regulatory control points, and in mammalian cells the levels and activity of these enzymes are controlled at the transcriptional, translational and posttranslational level. The association of polyamines with cell growth and transformation led to significant interest in the pathway for the discovery of anit-prolifertaive agents. Significantly, HAT remains the only disease where targeting the polyamine pathway has led to a clinically useful drug. The success of eflornithine suggests that the polyamine pathway is a strong potential source for additional chemical species to treat HAT.
The polyamine biosynthetic pathway in trypanosomatids differs from the human host. First, spermidine is conjugated to glutathione to form a novel cofactor trypanothione that functions in place of glutathione in cellular redox reactions (Fig. 2) [29]. Much of the enzymatic machinery involved in redox metabolism has evolved to utilize trypanothione instead of glutathione. Trypanothione reductase (TrpRed) replaces glutathione reductase and trypanothione is used to provide reducing equivalents to tryparedoxin, which in turn is needed for detoxification of hydroperoxides and for the function of ribonucleotide reductase. Secondly, trypanosomatids do not make spermine nor do they encode the catabolic enzymes polyamine oxidase and spermidine/spermine N1-acetyltransferase, used by mammalian cells to interconvert the longer chain polyamines. Finally, the pathway is regulated by a novel mechanism. The active form of AdoMetDC is a heterodimer composed of the functional subunit and an inactive paralog, termed prozyme, which is present only in the genomes of the kinetoplastids, and functions as an allosteric activator of the functional subunit [30]. The expression levels of prozyme protein are regulated in response to depletion of AdoMetDC activity by RNAi-targeted gene silencing or chemical inhibition, providing a potential mechanism to control pathway flux [31].
Gene knock out strategies or RNAi-mediated gene silencing have demonstrated that the polyamine and trypanothione biosynthetic enzymes are essential. Of the enzymes in the pathway, only glutathione synthetase remains to be characterized. Depletion of ODC leads to loss of putrescine and trypanothione, correlating with cell growth arrest [32, 33]. The ODC knockout cell line was used to confirm that the mechanism of action of eflornthine is exclusively mediated through ODC. Knockdown of AdoMetDC, prozyme or spermidine synthase in blood form parasites leads to spermidine auxotropy, though interestingly spermidine levels are only partially reduced [31, 33, 34]. However knockdown of any of these enzymes also leads to complete depletion of trypanothione, which correlates with cell death. Finally genetic studies have also demonstrate that γ-glutamylcysteine syntehtase, TrpRed and TrpSyn are all also essential [35– 37].
In addition to the discovery of eflornithine, small molecule inhibitors of other enzyme in the pathway with cell activity have also been reported. The most actively targeted enzymes after ODC, are AdoMetDC, TrpRed and TrpSyn, with the data providing a strong case that both AdoMetDC and TrpSyn are “druggable” targets with good potential as targets in drug discovery programs.
AdoMetDC
Some of the most potent anti-trypanosomal agents that have been reported are inhibitors of AdoMetDC. Several inhibitors (e.g. MDL 73811) that were developed in anti-cancer programs are potent inhibitors of T. brucei growth and are also able to cure infections in mice. However despite the good activity MDL 73811 lacked metabolic stability and had poor brain penetration, thus it was not developed further for use in HAT. Recently, work to identify a stable analog of MDL 73811 identified Genz-644131, a potent irreversible inhibitor of the AdoMetDC/prozyme complex with excellent (EC50 = 0.1 nM) activity in vitro and the ability to cure infections in mice after i.p. dosing with 2 − 50 mg/kg QD or BID, depending on the parasite strain [38, 39](Figure 4). Genz-644131 has better metabolic stability than MDL 73811 and shows improved brain penetration, however it does not provide sterile cure against TREU 667, the CNS model strain of infection. Genz-644131 and MDL 73811 show good selectivity against the parasite over the host at the cellular level yet this difference is not explained by selectivity on the enzyme. T. brucei contains a novel AdoMetDC transporter that is absent in mammalian cells suggesting differential uptake of AdoMet analogs by the parasite may lead to selective toxicity [40]. The possibility that other inhibitor classes that show selectivity at the enzyme level can be identified is suggested by structural studies and kinetic analysis showing the active site of AdoMetDC is not conserved between species [41].
Figure 4.

Identified lead inhibitor series for AdoMetDC and TrpSyn
TrpRed and TrpSyn
TrpRed and TrpSyn both have the advantage of being novel enzymes in the parasite and for this reason extensive research efforts to identify inhibitors with anti-trypansomal activity have been undertaken. For TrpRed, HTS campaigns and structure-based methods have been employed to identify both reversible and mechanism-based inhibitors of the enzyme from an array of structural classes, including tricyclics, quinazolines, benzimidazoles, nitrobenzens, polyamine analogs and peptides ([42–47] and references therein). The common features of the various inhibitor classes include the requirement for positive charge and for a hydrophopic core. Despite these extensive efforts, inhibitors of TrpRed have not been amenable to lead optimization and no preclinical candidates have been identified that target the enzyme. Typically within a chemical series it has not been possible to demonstrate a correlation between enzyme and parasite efficacy, suggesting that off target effects contribute significantly to the observed anti-trypanosomal activity. TrpRed has a large active site that makes identifying drug like molecules that bind and fill the pocket challenging. For many inhibitors multiple binding modes and a tendency to bind multiple ligands simultaneously has been observed.
For TrpSyn, a recent HTS campaign identified a potent inhibitor of the enzyme (DDU 86439) that while only exhibiting micromolar activity against T. brucei, was demonstrated by genetic and biochemical studies to target TrpSyn within the cell [37](Figure 4). These studies provide strong validation of TrpSyn as a target.
Energy Metabolism
The T. brucei bloodstream form is solely dependent on glycolysis to provide ATP. As such, enzymes in this pathway are attractive targets for trypanocidal drugs [48, 49]. All of the enzymes in the glycolytic pathway have been described, isolated and purified, either through classical or recombinant means, allowing a comprehensive understanding of the kinetics and flux of the pathway to be developed (Figure 5). [50] As much of the detail around the pathway has been reviewed [49] only significant changes to understanding or attractiveness of the individual enzymes as drug targets will be covered here.
Figure 5.

Glycolysis pathway of T. brucei.
Hexokinase (TbHK1)
Hexokinase starts the glycolysis pathway, and transfers a phosphate from ATP to glucose. The T. brucei genome contains two nearly identical hexokinases (TbHK1 and TbHK2) that differ primarily in the C-terminus. This difference appears to be important in regulation of hexokinase activity through heterooligomer formation between TbHK1 and TbHK2 [51]. Myristate further regulates activity, apparently through modulation of oligomer formation [52]. Hexokinase was shown essential by RNAi [52] and an inhibitor of TbHK1, Lonidamine (Figure 6) is trypanocidal [53].
Figure 6.

Lonidamine, an inhibitor of TbHK1.
Phosphoglucose isomerase (TbPGI) and Phosphofructokinase (TbPFK)
Both enzymes have been expressed and their X-ray structures have been determined [54–56]. For TbPFK the crystal structures reveal unique features of the trypanosomal enzyme relative to other protozoal, bacterial and mammalian orthologs, and provide opportunities for design of species-specific inhibitors. The synthesis and evaluation of a series of 2,5-anydro-D-mannitol derivatives has been described (Table 3) [57].
Table 3.
2,5-anydro-D-mannitol Derivatives as TbPFK inhibitors.
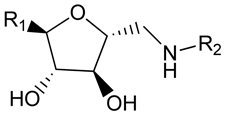 | |||
|---|---|---|---|
| R1 | R2 | TbPFK IC50 (µM) |
T. brucei S427 IC50 (µM) |
| -CH2OH | 3,4-dichlorobenzyl | 410 | 130 |
| -CH2OH | tetrahydronaphth-1-yl | >5000 | 830 |
| -CONH(3,4- dichlorobenzyl) |
3,4-dichlorobenzyl | 23 | 30 |
| CONH(cycloheptyl) | 3,4-dichlorobenzyl | 80 | 35 |
Fructose-1,6-Bisphosphate aldolase
The aldolase enzyme which effects reversible aldol cleavage of fructose 1,6-bisphosphate to dihydroxyacetone phosphate and D-glyceraldehyde-3-phosphate has been the target of a structure-based drug design program utilizing the crystal structure of the T. brucei enzyme [58]. Inhibitors based on the 1,6-dihydroxy-2-naphthaldehyde and 2,5-dihydroxybenzaldehyde scaffolds are selective for the T. brucei enzyme relative to the orthologous enzyme from rabbit muscle [59]. These phosphyorylated inhibitors did not exhibit activity against T. brucei, presumably due to poor membrane permeability. This issue was addressed through preparation of phosphate ester prodrugs, and modest activity in a whole cell assay has been observed (Figure 7)[60].
Figure 7.

Inhibitors of T. brucei fructose-1,6-bisphosphate aldolase.
Phosphglycerate Kinase and Enolase
Phosphoglycerate kinase, which converts 1,3-bisphosphoglycerate to 3-phosphoglycerate, is relatively less well characterized than other enzymes in the pathway. It has been reported, however, that the adenosine analog tubercidin, which is trypanocidal, inhibits TbPGKB [61]. The penultimate enzyme in the glycolytic pathway, Enolase, has been shown to have a flexible active site, based on the enzyme in complex with an inhibitor, 2-fluoro-2-phosphonoacetohydroxamate, which may allow for design of larger inhibitors [62].
Purine and Pyrimidine Metabolism
Purine metabolism
Like most obligate intracellular parasites, T. brucei has lost the capacity to synthesize purines de novo and depends on salvage from the host. Bloodstream T. brucei can take up different types of purines and interconvert them into essential cellular nucleotides. The parasite has developed a unique set of salvage transporters and enzymes, however no single protein is essential [63]. Programs to exploit parasite purine salvage rely on specific transport or activation of subversive drug substrates through enzymes and transporters that differ or are absent in the host [64]. For example, cordycepin, a 3'-deoxyadenosine targets T. brucei through incorporation into RNA, resulting in termination of synthesis [64, 65]. This antimetabolite pro-drug requires phosphorylation by adenosine kinase (AK) prior to incorporation into RNA. AK is not essential under normal T. brucei growth conditions in vitro, but down-regulation or inhibition leads to resistance [64, 66]. Cordycepin efficacy in murine models of HAT requires co-administration with the AK inhibitor coformycin to prevent deamination [65]. The combination demonstrates activity against a CNS infection in mice, suggesting permeation of brain parenchyma by both compounds. The potential toxicity of coformycin has led to suggestions of developing deamination resistant cordycepin or adenosine analogues [67].
Another approach in targeting purine metabolism exploits transporters to accumulate toxic analogues within the parasite [68–71]. In one such study, melamine linked nitroheterocycles demonstrated good trypanocidal activity in vitro and in some cases in vivo [72]. The lack of correlation between affinity for P2 transporter and parasite killing suggested the involvement of other mechanisms or transporters such as HAPT1 and LAPT1 [73, 74]. In other studies, attempted delivery of melamine-linked eflornithine or fluoroquinolones was unsuccessful [75]. For Mannich bases, uptake into trypanosomes occurs efficiently in the presence or absence of the melamine moiety [71]. Exploitation of purine transporters for delivery of trypanocidal moieties requires further exploration.
Pyrimidine Metabolism
The pyrimidine metabolic pathway in T. brucei and other related parasites has received much less attention in comparison to pathways involved in the uptake and interconversion of purines. The presence of both the de novo biosynthetic and salvage pathways suggests redundant mechanisms to acquire pyrimidines, reducing the potential of this pathway in drug discovery. However, the suitability of dihydroorotate dehydrogenase (DHODH) and CTP synthetase as drug targets has been experimentally tested [76, 77]. DHODH is the fourth enzyme in the pyrimidine biosynthetic pathway. T. brucei has a cytoplasmic, fumarate-dependent class 1 DHODH, which has been characterized by X-ray structure analysis and RNAi knockdown, showing loss of this protein resulted in slowed growth but was not lethal [76]. The addition of 5- fluorouracil, an inhibitor of pyrimidine uptake, potentiated the effects of DHODH knockdown. Simultaneous inhibition of DHODH and pyrimidine uptake would be required to generate complete anti-trypanocidal effects.
Cytidine triphosphate synthetase (CTPS) is a key enzyme in nucleic acid and phospholipid biosynthesis and it is critical to T. brucei survival. Cultured T. brucei parasites have very low intracellular pools of CTP, attributed to the slow synthetic activity of CTPS [77, 78]. Drugs targeting T. brucei CTPS should be effective because unlike mammalian cells, the parasites can’t compensate for the loss of CTPS through salvage of cytidine. This idea has led to the evaluation and demonstration of effective activity of CTPS inhibitors 6-diazo-5-oxo-l-norleucine (DON) and alpha-amino-3-chloro-4,5-dihydro-5-isoxazoleacetic acid (acivicin) against T. brucei in vitro and in vivo [77, 78]. Avicin crosses the blood brain barrier, a requirement for treatment of stage 2 HAT. The potential toxicity of DON and avicin would require co-administered with purine bases to reduce such adverse reactions. This is likely to complicate the treatment regimen and potential outcome, thus limiting feasibility of this approach.
Pteridine Metabolism
Trypanosomatid protozoan parasites lack a de novo pathway for the synthesis of pteridines (folate and pterins) and rely on salvage from the host [79]. Pteridine salvage depends on transporters specific for folate and biopterin (FT1 and BT1, respectively) and at least two pteridine reductases. Bifunctional dihydrofolate reductase-thymidylate synthase (DHFR-TS) is specific for folate while pteridine reductase 1 (PTR1) reduces both folate and biopterin. Unlike humans and most other organisms that have monofunctional proteins, trypanosomatids express a bifunctional DHFR-TS in which a single polypeptide chain contains both catalytic activities [80]. DHFR-TS and PTR1, have been proposed as key drug targets in trypanosomatids because folates are essential for synthesis of thymidylate while pterins are implicated in parasites growth and oxidant resistance [81, 82].
Dihydrofolate reductase-thymidylate synthase
Inhibitors of DHFR have been successfully exploited for therapy of bacterial infections and other parasitic diseases such as malaria. However, classical inhibitors that target monofunctional DHFR are ineffective against trypansomatids. In Leishmania, this is due to drug resistance mechanisms, the most prominent of which is amplification of PTR1, which allows bypass of the DHFR block [83, 84]. Although this mechanism has not been demonstrated in T. brucei, the ability of PTR1 to catalyze the DHFR mediated reactions, suggests that a dual inhibitor or a combination of compounds targeting the two enzymes separately may be required to effectively exploit pteridine metabolism.
In T. brucei null mutants lacking DHFR-TS are resistant to antifolates and require thymidine supplemented media [85]. DHFR-TS knockouts of T. brucei could not be rescued with an enzymatically active TS on a plasmid, suggesting that both DHFR and TS are essential for thymidylate synthesis. Furthermore, DHFR-TS knockout T. brucei parasites cannot establish infection in mice, suggesting that thymidine concentrations in mouse blood are limiting. Although T. brucei DHFR studies have not identified a clear chemical scaffold for a focused lead discovery effort, validation data is sufficiently compelling to warrant efforts to exploit this target.
Pteridine reducatse-1
PTR1 and DHFR catalyze the same reactions, but utilize different catalytic mechanisms such that, if necessary to avoid PTR1 mediated bypass of DHFR, finding a common inhibitor could be challenging. However in T. brucei, PTR1 null mutants are not viable suggesting it maybe a target on its own [85]. The active site of T. brucei PTR1 is sufficiently distinct from that of DHFR to warrant a separate program to identify specific inhibitors. One such effort identified aminothiazole and aminobenzimidazole inhibitors from a fragment-based virtual screen. Structural determination of PTR1 bound to these inhibitors was used to inform a hit-expansion program to identify selective and potent inhibitors of the T. brucei enzyme [86]. Compounds from this effort were selective for T. brucei PTR1 over DHFR from the parasite or human, but a representative aminobenzimidazole with good potency for PTR1 (Kiapp = 7 nM) and favorable physicochemical properties for cellular permeation, showed limited ability to inhibit T. brucei growth in vitro. Apparently other chemical scaffolds have also failed to translate from potent PTR1 inhibition to tryapnocidal activity in culture [86]. Discovery of the basis for this limitation will be essential to any future efforts to progress inhibitors of PTR1 for HAT therapy.
DNA Topoisomerases
DNA topoisomerases catalyze the conversion of topological isomers of DNA and are essential for nucleic acid metabolism replication and transcription. Type I enzymes catalyze single strand breaks and type II make double stranded breaks on DNA [87]. Type I enzymes are further classified as IA or IB and the latter are key targets for the camptothecin class of anti-cancer compounds. Camptothecins act by trapping the enzyme in the complex with DNA substrate i.e. the cleavable complex. Trypanosomatids, have type IB topoisomerase that is sensitive to camptothecin [88, 89]. Unlike mammalian enzyme which is a single polypeptide, T. brucei topoisomerase is hetero-multimeric consisting of two separately encoded proteins. Both 90 kDa DNA binding and the 35 kDa catalytic subunits are essential in T. brucei [90]. A knockdown of one subunit results in decreased levels of protein from the second subunit and a concomitant reduction in synthesis of RNA and DNA, an expected consequence of interference with topoisomerases.
The potential of topoisomerase inhibitors as trypanocidal agents [91] has been demonstrated. Classical antibacterial fluoroquinolones such as KB5426, ofloxacin, and ciprofloxacin and camptothecin and non-camptothecin classes of topoisomerase inhibitors have shown activity against T. brucei [92, 93]. Recently, a series of indenoisoquinolines were shown to have trypanocidal activity in vitro and in vivo as shown by increased survival of mice challenged with a lethal infection [94]. Unfortunately none of the published topoisomerase inhibitors show a sufficient parasite versus host cell selectivity for development as safe and effective trypanocidal compounds. Future strategies for effective targeting of parasite topoisomerases should focus on defining structural differences between mammalian and T. brucei enzymes e.g. the unusual bi-subunit structure and association with both genomic and mitochondrial DNA.
Fatty Acid Biosynthesis
Like many eurokaryotes, biosynthesis of fatty acids is critical for the life cycle of T. brucei, yet the details of the biosynthetic pathways have only recently been elucidated [95, 96]. The uniqueness of these pathways provides the potential for selective intervention, with several candidate proteins now validated as targets. Particularly important to the parasite is generation of myristate, a key component in the glycosyl phosphatidylinositol (GPI) anchor of the variable surface glycoprotein (VSG) coat of the bloodstream form parasite. Though originally thought not to synthesize myristate, recent work has demonstrated that T. brucei uses a unique set of enzymes, elongases, to synthesize this important fatty acid. The essentiality of these enzymes has been validated using RNAi approaches[97]. This microsomal elongase system is responsible for synthesis of nearly all fatty acids in T. brucei, and uses butyryl-CoA as primer instead of acetyl-CoA as in other eukaryotes. Further exploration of fatty acid biosynthesis in the mitochondrion revealed a more classical type II fatty acid synthase and attendant carrier proteins, several of which were determined to be essential by RNAi [98–100].
A number of natural products (Figure 8) that inhibit fatty acid biosynthesis targets in other parasites (e.g. P. falciparum) have been shown to kill T. brucei in vitro [101–104].
Figure 8.

Natural product fatty acid biosynthesis inhibitors.
Fatty acid utilization
The enzyme responsible for attachment of myristate to the GPI anchor protein, N-myristoyltransferase (NMT), is essential for parasite viability based on RNAi [105]. Several inhibitors of fungal NMTs (Figure 9) inhibit the trypanosomal enzyme, and growth of T. brucei in vitro [106, 107]. Though these molecules are not drug-like, they have been employed for homology modeling using crystallographic information from fungal NMTs to suggest opportunities for rational design of new T. brucei selective inhibitors [108].
Figure 9.

Inhibitors of trypanosomal N-myristoyltransferase.
Sphingolipid biosynthesis
Connected to the fatty acid biosynthesis pathway, and critically important to viability of the trypanosome, is sphingolipid biosynthesis. The key enzyme in this pathway, sphingolipid synthase, has recently been identified and shown to be essential for T. brucei survival by RNAi and by chemical inhibition using aureobasidin A [109, 110].
Protein prenylation
Attachment of isoprenoids groups (e.g. farnesyl) to proteins is an important regulatory mechanism for signal transduction in trypanosomes. The enzyme responsible for this process, protein farnesyltransferase (PFT) has been targeted for screening efforts to identify leads for medicinal chemistry including a series of tetrahydroquinolines (Figure 10) active against both P. falciparum [111, 112] and T. brucei [113]. Notably, analogs of tipifarnib, an inhibitor of human PFT, with excellent activity against the related T. cruzi have also been described [114].
Figure 10.

Tetrahydroquinoline protein farnesyltransferase inhibitors.
Signal Transduction Pathways
Cell Cycle Kinases
The cell cycle of T. brucei is unusual in several regards, and as such may offer an effective approach to kill the parasite [115]. In particular, protein kinases involved in progression of trypanosomes through a number of cell cycle checkpoints have been suggested to be a attractive targets for drug discovery [116, 117]. This interest is based on several factors, including the growing understanding of the trypanosomatid cell cycle and availability of chemical starting points from orthologous mammalian targets [118, 119]. Genetic validation of numerous trypanosomal kinase targets through RNAi methods has been demonstrated, but very few have been validated chemically. Several groups, most notably the Drug Discovery Unit at Dundee University have initiated or planned HTS campaigns, with the hope of identifying leads to exploit the kinase targets [120].
One kinase target that has been chemically validated is TbGSK3. In mammalian cells, GSK3b has been shown to play a key role in multiple cellular processes including cell survival and death signaling, and can be inhibited by a multitude of ATP-site directed small molecules [121]. The T. brucei homolog (TbGSK3) can be inhibited by several commercial kinase inhibitors and these were shown to have anti-trypanosomal activity [122]. Additionally, a series of human GSK-3b inhibitors were evaluated in both assays, where good correlation between TbGSK3 inhibition and cell growth inhibition was reported (Table 4). A HTS campaign for inhibitors of TbGSK3 is planned, and may provide starting points for medicinal chemistry optimization of novel compounds. Critical to success of this effort, and generally for cell cycle kinases with mammalian homologs, will be identification of compounds that selectively inhibit the parasite enzyme.
Table 4.
Activity of GSK-3 focused inhibitors on TbGSK3 and T. brucei
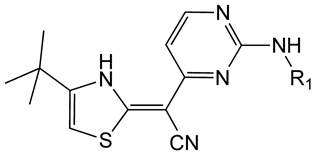 | ||
|---|---|---|
| R1 | TbGSK3 IC50 (nM) |
T.brucei EC50 (nM) |
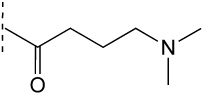 |
4 | 50 |
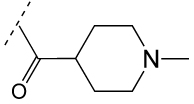 |
30 | 65 |
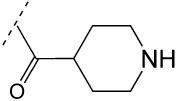 |
46 | 200 |
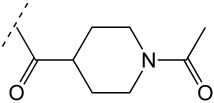 |
22 | 410 |
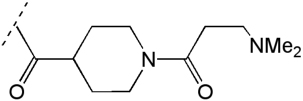 |
30 | 460 |
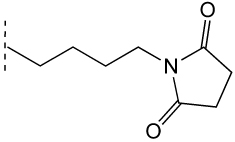 |
170 | 710 |
A second recently chemically validated target is TbAUK1, which is inhibited by hesperadin (Figure 11) in both biochemical phosphorylation and cell growth assays [123].
Figure 11.

Hesperadin
Proteases
Proteases have been extensively targeted for the development of chemotherapy for a number of proliferative diseases including cancer, viral pathogens and parasites. Proteases play a number of roles in disease progression and pathogenesis in both protozoal and worm pathogens [124]. In T. cruzi, a cysteine protease, cruzipain is involved in host cell invasion and inhibitors of this enzyme have reached late preclinical trials for Chagas’ disease [125]. The roles of proteases in T. brucei have been less well defined, however a number of recent studies have begun to explore the biology of proteases in T. brucei, and protease inhibitors with anti-trypanosomal activity have been reported.
The most active efforts to identify protease inhibitors with activity against T. brucei have focused on the cysteine proteases. T. brucei contains two Clan CA cysteine proteases, a cathepsin L-like protease, brucipain, and a cathepsin B protease, TbCatB, the latter of which is upregulated in bloodstream parasites. In vitro and in vivo RNAi studies have shown that TbCatB is essential for the growth, while brucipain is not [126, 127]. Data suggests that TbCatB plays a role in degradation of host transferrin for iron acquisition [126]. Purine-derived nitrile inhibitors of TbcatB with low micromolar inhibition against the enzyme show anti-trypanosomal activity, though no clear correlation between enzyme and cell-based activity emerged in these studies [128]. Thiosemicarbazone inhibitors of both rhodesain and TbcatB have also been reported, though these compounds are more potent on rhodesain and again no correlation between enzyme inhibition and cell-based activity was observed [129].
The roles of several other protease classes in the biology of the parasite have recently been described. A bacterial-like HsIVU protease that has ATP-dependent protease activity is localized to the mictochondria and is involved in replication of the kinetoplstid DNA, playing a role that is novel to the parasite [130]. Knockdown of this gene caused a significant growth defect. A family of surface metalloproteases have been identified with a role in cleavage of VSG from the parasite surface during transformation from blood form to procyclic form, though none appear essential on their own [131], and a family of metacaspases was identified to associate with RAB11-endosomes, though they have no role in programmed cell death and individually are not essential [132].
Tubulin
Disruption of microtubule assembly in T. brucei through interference with tubulin polymerization by benzimidazoles and dinitroaniline herbicides has been shown to inhibit parasite growth [133]. Due to the ubiquitous nature of tubulin in both the parasite and the mammalian host, selectivity for inhibition of parasite microtubule assembly is key to successful progression of compounds working by this mechanism. One class of molecules that has been extensively explored and appears to meet this requirement is the 3,5-dinitrosulfanilamides. Starting with the known herbicides trifluralin and oryzalin, Werbovetz and co-workers developed a series of dinitrosulfanilimides that demonstrated high potency versus T. brucei and low cytotoxicity (Table 5) [134, 135, 136]. As expected from their mode of action, these compounds demonstrated antimitotic effects on cultured T. brucei as assessed by flow cytometry, but the most potent compounds were not active in a HAT murine model.
Table 5.
Dinitrosulfanilimide SAR.
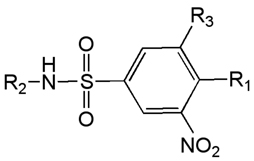 | |||||
|---|---|---|---|---|---|
| R1 | R2 | R3 |
T. b. brucei EC50 (µM) |
J774 cytotox EC50 (µM) |
Selectivity Index |
| N(nPr)2 | H | NO2 | 6.6 | 41 | 6.2 |
| N(nBu)2 | H | NO2 | 1.9 | 9.4 | 4.9 |
| N(nPr)2 | Ph | NO2 | 0.73 | 29 | 40 |
| N(nBu)2 | Ph | NO2 | 0.12 | 12 | 100 |
| NEt2 | Ph | NO2 | 2.8 | 48 | 17 |
| N(nPr)2 | 4-ClPh | NO2 | 13 | 10 | 0.8 |
| N(nPr)2 | iPr | NO2 | 4.1 | ND | NC |
| N(nPr)2 | Ph | H | 20 | 31 | 1.4 |
Metabolic instability was hypothesized as a reason for the lack of in vivo activity [137], and more recent efforts are directed at finding metabolically stable analogs, with only limited success [138]. A second area of concern, the potential mutagenicity of dinitro aromatics, has also been addressed, with preliminary results suggesting that the two nitro groups can be replaced by cyano with retention of anti-trypanosomal activity [139]. Finally, a related, but independently discovered series of 2,4-dinitro-6-trifluoromethyl aniline derivatives has been described which exhibit selective binding to trypanosomal α-tubulin relative to the mammalian protein [140].
Identification of Trypanocidal Compounds Through Whole Cell Assays
A complimentary and useful approach to the discovery of effective trypanocidal compounds that does not depend upon knowledge of the biochemical target has been evaluation of compounds by whole cell growth inhibition assay. The most commonly employed variant of this assay uses the fluorescent oxidation-reduction reagent Alamar Blue as an indicator of trypanosome viability [141]. Alternatively, an ATP-bioluminescence approach which correlates ATP release from trypanosomes with parasite viability has been reported [142]. Recent advances in liquid handling robotics have facilitated development of high-throughput assays in 96, 384 and 1536-well plate formats in both modes, which has enabled screening of large compounds libraries for starting points for chemical optimization.
Diamidines
Aromatic diamidines related to the clinically important drug for Stage 1 HAT, pentamidine, have been extensively explored for potential new compounds that might exhibit superior potency and lowered toxicity, and SAR for members of this series has been reviewed [16, 143]. Emerging from these efforts was the candidate drug DB289, which was progressed to clinical trials based on efficacy in both murine and non-human models of HAT [15, 144]. Unfortunately, clinical development of DB289 was terminated in 2008 due to safety concerns.
Numerous variants of the core region in the diamidine series have been reported in the past several years that seek to reduce the cytotoxicity observed in earlier candidates including phenoxymethylaromatic [145], 2-phenylbenzofuran [146], triaryl [147], imidazopyridine [148], thiophene [149], bis-benzofuran [150]and 3,5-diphenyloxazole [151] analogs. One strategy that was somewhat effective in this regard was the introduction of an N-isopropyl amidine, which although it reduced potency against T. brucei, reduced cytotoxicity to a greater extent (Figure 12A). A second area of focus in the diamidine series has been on preparation of analogs and potential pro-drugs that could provide orally active compounds. This work has continued to focus on utilization of O-methylamidoximes (Figure 12B) [148, 152].
Figure 12.

A. Effect of N-isopropyl amidines on Selectivity Index. B. O-Methylamidoxime prodrugs of Diamidines.
Lipophilic Amines
Several chemotypes have been identified, and SAR developed based on whole cell assays, for polycyclic amine scaffolds (Figure 13A)[153–157]. The aminoadamantane derivatives originated from the observation that rimantidine exhibited activity against bloodstream form of T. brucei in vitro. Early SAR studies in this series demonstrated some improvements in activity in vitro, but compounds evaluated in vivo were able only to suppress the rate of T. brucei infection and did not cure. More recently, analogs which sought to capitalize on the relationship between lipophilicity and potency have been described [156, 158].
Figure 13.

A. Trypanocidal polycyclic amines. B. Trypanocidal bicyclo[3.2.2] nonanes.
The 4-aminobicyclo[2.2.2]octan-2-one and 4-aminobicyclo[2.2.2]octan-2-ol derivatives were synthesized, evaluated against a range of parasites (L. donovani, P. falciparum, T. cruzi and T. brucei) and were found to be active against the T. b. rhodesiense STIB 900 strain [159]. Subsequent publications have explored the SAR of the series, particularly ester and ether analogs of the bicyclo[2.2.2]octanols [160, 161]. A general dependence of activity on lipophilicity was noted, but this improvement in potency was accompanied by a loss of selectivity. Additionally, dialkylamino-substituted esters of the bicyclo[2.2.2]octanols have demonstrated good in vitro potency and low cytotoxicity [153, 162]. (Table 6)
Table 6.
Bicyclo[2.2.2]octanol derivatives.
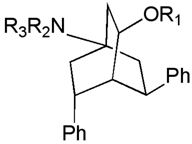 | ||||
|---|---|---|---|---|
| R1 | NR2R3 | T. b. rhod. EC50 (µM) |
L6 cytotox EC50 (µM) |
Selectivity Index |
| H | NMe2 | 3.0 | 130 | 45 |
| MeCO- | NMe2 | 4.75 | NT | NC |
| tBuCO- | NMe2 | 0.62 | 5.1 | 8.1 |
| 4-MeOC6H4CO- | NMe2 | 1.5 | 7.3 | 4.8 |
| C6H5CH2- | pyrrolidinyl | 1.3 | 7.1 | 5.4 |
| Et2NCH2CO- | NMe2 | 0.61 | 30 | 49 |
| 2-pyrrolidinylacetyl- | NMe2 | 0.21 | 33 | 160 |
| 2-pyrrolidinylacetyl- | pyrrolidinyl | 0.23 | 43 | 190 |
| 2-(4- piperazinyl)acetyl- |
NMe2 | 0.13 | 40 | 310 |
| 3- pyrrolidinylpropionyl- |
NMe2 | 0.076 | 26 | 340 |
| 3- pyrrolidinylpropionyl- |
pyrrolidinyl | 0.12 | 22 | 180 |
A variety of semithiocarbazone [160], oxime [163] and amino derivatives[164] at the 2-position of the bicyclo[2.2.2]octanones have also been evaluated. Most interesting amongst these are the 2-amino analogs, which exhibit good potency (IC50 < 1 µM) and selectivity (S.I.> 100). The third related class of polycyclic amines are the 2-azabicyclo[3.2.2]nonanes (Figure 13B). This ring-expanded variant of the bicyclo[2.2.2] octanes was synthesized by Beckmann rearrangement of the oxime derivatives described earlier. While the lactam derivatives obtained from this rearrangement were only weakly active, reduction to the diamines afforded compounds with improved potency and selectivity [165]. Substitution on the 2-aza nitrogen in the bicyclo[3.2.2]nonane generally resulted in loss of potency, which prompted exploration of substituted aryl rings as a means to further improve potency (Table 7)[154]. These efforts have culminated in the identification of several 4-substituted derivatives with excellent in vitro potency and selectivity. Representative compounds from the 4-aminobicyclo[2.2.2]octan-2-ol, 4-aminobicyclo[2.2.2]oct-2-ylamines and 2-azabicyclo[3.2.2]nonanes have been evaluated in mice, where they demonstrated prolongation of survival, but have not cure of T. brucei infection [157].
Table 7.
2-Azabicyclo[3.2.2]nonane derivatives.
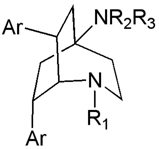 | |||||
|---|---|---|---|---|---|
| R1 | NR2R3 | Ar | T. b. rhod. EC50 (µM) |
L6 cytotox EC50 (µM) |
Selectivity Index |
| H | NMe2 | Ph | 0.60 | 110 | 180 |
| H | pyrrolidinyl | Ph | 1.2 | 120 | 100 |
| 2-(4- piperazinyl)acetyl- |
NMe2 | Ph | 46 | 200 | 4.3 |
| H | NMe2 | 4-ClPh | 0.061 | 8.7 | 140 |
| H | pyrrolidinyl | 4-ClPh | 0.066 | 10 | 150 |
| -CH2CO2Et | NMe2 | 4-ClPh | 1.8 | 5.1 | 2.9 |
| -CH2CONH2 | NMe2 | 4-ClPh | 0.91 | 26 | 28 |
| -CH2CH2NH2 | NMe2 | 4-ClPh | 0.50 | 10 | 20 |
| H | NMe2 | 4- CF3Ph |
0.08 | 9.4 | 120 |
Miscellaneous Natural and Synthetic Compounds
Using whole cell screening approaches, a number of natural products and synthetic compounds have been identified in the past several years as potential starting points for discovery of anti-trypanosomal agents. For example, synthesis of a library of 2-aryloxy anthra- and naphtha-quinones designed to incorporate features of the natural product lapachol and biocide triclosan [166] afforded a number of analogs with high potency and modest selectivity (Table 8) [167]. From the same laboratory, a series of 3,5-disubstituted-2H–pyrazoles and isoxazoles were synthesized and found to be active vs. T. b. rhodesiense (Table 9) [168].
Table 8.
Anti-trypanosomal 2-aryloxy-1,4-anthraquinones and 1,4-napthoquinones.
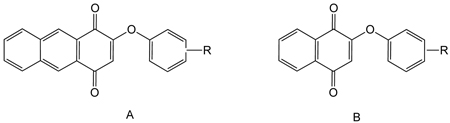 | ||||
|---|---|---|---|---|
| Template | R | T. b. rhod. EC50 (µM) |
L6 Cytotox EC50 (µM) |
Selectivity Index |
| A | 2,4-Cl2 | 0.065 | 0.49 | 31 |
| A | 2-Br, 4-F | 0.08 | 1.1 | 14 |
| A | H | 0.05 | 1.0 | 20 |
| B | 2,4-Cl2 | 0.29 | 5.2 | 18 |
| B | 2,4- (CH3)2 |
0.22 | 5.3 | 24 |
| B | 2,4-Br2 | 0.51 | 29 | 74 |
Table 9.
3,5-disubstituted azoles.
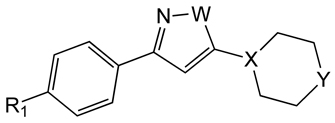 | ||||||
|---|---|---|---|---|---|---|
| R | W | X | Y | T. b. rhod. IC50 (µM) |
L6 Cytotox IC50 (µM) |
Selectivity Index |
| PhO- | NH | CH | O | 3.2 | 90 | 28 |
| PhO- | NH | N | O | 1.0 | 62 | 62 |
| PhO- | NH | N | N-Boc | 2.9 | 30 | 10 |
| PhO- | O | N | O | 5.9 | 170 | 29 |
| 4- NH2C6H4O |
NH | N | O | 18 | >270 | 15 |
| PhO- | NH | N | NH | 1.1 | 18 | 16 |
References
- 1.Barrett MP, Boykin DW, Brun R, Tidwell RR. Human African trypanosomiasis: pharmacological re-engagement with a neglected disease. Br J Pharmacol. 2007;152(8):1155–1171. doi: 10.1038/sj.bjp.0707354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kennedy PG. The continuing problem of human African trypanosomiasis (sleeping sickness) Ann Neurol. 2008;64(2):116–126. doi: 10.1002/ana.21429. [DOI] [PubMed] [Google Scholar]
- 3.Stuart K, Brun R, Croft S, Fairlamb A, Gurtler RE, McKerrow J, Reed S, Tarleton R. Kinetoplastids: related protozoan pathogens, different diseases. J Clin Invest. 2008;118(4):1301–1310. doi: 10.1172/JCI33945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fevre EM, Wissmann BV, Welburn SC, Lutumba P. The burden of human african trypanosomiasis. PLoS Negl Trop Dis. 2008;2(12):e333. doi: 10.1371/journal.pntd.0000333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Simarro PP, Jannin J, Cattand P. Eliminating human African trypanosomiasis: where do we stand and what comes next? PLoS Med. 2008;5(2):e55. doi: 10.1371/journal.pmed.0050055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pays E, Vanhollebeke B, Vanhamme L, Paturiaux-Hanocq F, Nolan DP, Perez-Morga D. The trypanolytic factor of human serum. Nat Rev Microbiol. 2006;4(6):477–486. doi: 10.1038/nrmicro1428. [DOI] [PubMed] [Google Scholar]
- 7.Stockdale C, Swiderski MR, Barry JD, McCulloch R. Antigenic variation in Trypanosoma brucei: joining the DOTs. PLoS Biol. 2008;6(7):e185. doi: 10.1371/journal.pbio.0060185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blum JA, Zellweger MJ, Burri C, Hatz C. Cardiac involvement in African and American trypanosomiasis. Lancet Infect Dis. 2008;8(10):631–641. doi: 10.1016/S1473-3099(08)70230-5. [DOI] [PubMed] [Google Scholar]
- 9.Blum JA, Schmid C, Burri C, Hatz C, Olson C, Fungula B, Kazumba L, Mangoni P, Mbo F, Deo K, Mpanya A, Dala A, Franco JR, Pohlig G, Zellweger MJ. Cardiac Alterations in Human African Trypanosomiasis (T.b. gambiense) with Respect to the Disease Stage and Antiparasitic Treatment. PLoS Negl Trop Dis. 2009;3(2):e383. doi: 10.1371/journal.pntd.0000383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chappuis F, Loutan L, Simarro P, Lejon V, Buscher P. Options for field diagnosis of human african trypanosomiasis. Clin Microbiol Rev. 2005;18(1):133–146. doi: 10.1128/CMR.18.1.133-146.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mugasa CM, Laurent T, Schoone GJ, Kager PA, Lubega GW, Schallig HD. Nucleic acid sequence-based amplification with oligochromatography for detection of Trypanosoma brucei in clinical samples. J Clin Microbiol. 2009;47(3):630–635. doi: 10.1128/JCM.01430-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Njiru ZK, Mikosza AS, Armstrong T, Enyaru JC, Ndung'u JM, Thompson AR. Loop-Mediated Isothermal Amplification (LAMP) Method for Rapid Detection of Trypanosoma brucei rhodesiense. PLoS Negl Trop Dis. 2008;2(1):e147. doi: 10.1371/journal.pntd.0000147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Priotto G, Kasparian S, Mutombo W, Ngouama D, Ghorashian S, Arnold U, Ghabri S, Baudin E, Buard V, Kazadi-Kyanza S, Ilunga M, Mutangala W, Pohlig G, Schmid C, Karunakara U, Torreele E, Kande V. Nifurtimox-eflornithine combination therapy for second-stage African Trypanosoma brucei gambiense trypanosomiasis: a multicentre, randomised, phase III, non-inferiority trial. Lancet. 2009;374(9683):56–64. doi: 10.1016/S0140-6736(09)61117-X. [DOI] [PubMed] [Google Scholar]
- 14.Schmid C, Richer M, Bilenge CM, Josenando T, Chappuis F, Manthelot CR, Nangouma A, Doua F, Asumu PN, Simarro PP, Burri C. Effectiveness of a 10-day melarsoprol schedule for the treatment of late-stage human African trypanosomiasis: confirmation from a multinational study (IMPAMEL II) J Infect Dis. 2005;191(11):1922–1931. doi: 10.1086/429929. [DOI] [PubMed] [Google Scholar]
- 15.Mdachi RE, Thuita JK, Kagira JM, Ngotho JM, Murilla GA, Ndung'u JM, Tidwell RR, Hall JE, Brun R. Efficacy of the novel diamidine compound 2,5-Bis(4-amidinophenyl)- furan-bis-O-Methlylamidoxime (Pafuramidine, DB289) against Trypanosoma brucei rhodesiense infection in vervet monkeys after oral administration. Antimicrob Agents Chemother. 2009;53(3):953–957. doi: 10.1128/AAC.00831-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bakunova SM, Bakunov SA, Patrick DA, Kumar EV, Ohemeng KA, Bridges AS, Wenzler T, Barszcz T, Jones SK, Werbovetz KA, Brun R, Tidwell RR. Structure-activity study of pentamidine analogues as antiprotozoal agents. J Med Chem. 2009;52(7):2016–2035. doi: 10.1021/jm801547t. [DOI] [PubMed] [Google Scholar]
- 17.de Koning HP. Ever-increasing complexities of diamidine and arsenical crossresistance in African trypanosomes. Trends Parasitol. 2008;24(8):345–349. doi: 10.1016/j.pt.2008.04.006. [DOI] [PubMed] [Google Scholar]
- 18.Croft SL. Kinetoplastida: new therapeutic strategies. Parasite. 2008;15(3):522–527. doi: 10.1051/parasite/2008153522. [DOI] [PubMed] [Google Scholar]
- 19.Sanderson L, Dogruel M, Rodgers J, De Koning HP, Thomas SA. Pentamidine movement across the murine blood-brain and blood-cerebrospinal fluid barriers: effect of trypanosome infection, combination therapy, P-glycoprotein, and multidrug resistance-associated protein. J Pharmacol Exp Ther. 2009;329(3):967–977. doi: 10.1124/jpet.108.149872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Robays J, Nyamowala G, Sese C, Betu Ku Mesu Kande V, Lutumba P, Van der Veken W, Boelaert M. High failure rates of melarsoprol for sleeping sickness, Democratic Republic of Congo. Emerg Infect Dis. 2008;14(6):966–967. doi: 10.3201/eid1406.71266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shahi SK, Krauth-Siegel RL, Clayton CE. Overexpression of the putative thiol conjugate transporter TbMRPA causes melarsoprol resistance in Trypanosoma brucei. Mol Microbiol. 2002;43(5):1129–1138. doi: 10.1046/j.1365-2958.2002.02831.x. [DOI] [PubMed] [Google Scholar]
- 22.Maina N, Maina KJ, Maser P, Brun R. Genotypic and phenotypic characterization of Trypanosoma brucei gambiense isolates from Ibba, South Sudan, an area of high melarsoprol treatment failure rate. Acta Trop. 2007;104(2–3):84–90. doi: 10.1016/j.actatropica.2007.07.007. [DOI] [PubMed] [Google Scholar]
- 23.Pegg AE, Feith DJ. Polyamines and neoplastic growth. Biochemical Society transactions. 2007;35(Pt 2):295–299. doi: 10.1042/BST0350295. [DOI] [PubMed] [Google Scholar]
- 24.Casero RA, Jr, Marton LJ. Targeting polyamine metabolism and function in cancer and other hyperproliferative diseases. Nature reviews. 2007;6(5):373–390. doi: 10.1038/nrd2243. [DOI] [PubMed] [Google Scholar]
- 25.Grishin NV, Osterman AL, Brooks HB, Phillips MA, Goldsmith EJ. X-ray structure of ornithine decarboxylase from Trypanosoma brucei: the native structure and the structure in complex with alpha-difluoromethylornithine. Biochemistry. 1999;38(46):15174–15184. doi: 10.1021/bi9915115. [DOI] [PubMed] [Google Scholar]
- 26.Balasegaram M, Young H, Chappuis F, Priotto G, Raguenaud ME, Checchi F. Effectiveness of melarsoprol and eflornithine as first-line regimens for gambiense sleeping sickness in nine Medecins Sans Frontieres programmes. Trans R Soc Trop Med Hyg. 2009;103(3):280–290. doi: 10.1016/j.trstmh.2008.09.005. [DOI] [PubMed] [Google Scholar]
- 27.Sanderson L, Dogruel M, Rodgers J, Bradley B, Thomas SA. The blood-brain barrier significantly limits eflornithine entry into Trypanosoma brucei brucei infected mouse brain. J Neurochem. 2008;107(4):1136–1146. doi: 10.1111/j.1471-4159.2008.05706.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ullu E, Tschudi C, Chakraborty T. RNA interference in protozoan parasites. Cell Microbiol. 2004;6(6):509–519. doi: 10.1111/j.1462-5822.2004.00399.x. [DOI] [PubMed] [Google Scholar]
- 29.Krauth-Siegel RL, Comini MA. Redox control in trypanosomatids, parasitic protozoa with trypanothione-based thiol metabolism. Biochim Biophys Acta. 2008;1780(11):1236–1248. doi: 10.1016/j.bbagen.2008.03.006. [DOI] [PubMed] [Google Scholar]
- 30.Willert EK, Fitzpatrick R, Phillips MA. Allosteric regulation of an essential trypanosome polyamine biosynthetic enzyme by a catalytically dead homolog. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(20):8275–8280. doi: 10.1073/pnas.0701111104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Willert EK, Phillips MA. Regulated expression of an essential allosteric activator of polyamine biosynthesis in African trypanosomes. PLoS Pathog. 2008;4(10):e1000183. doi: 10.1371/journal.ppat.1000183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li F, Hua S, Wang C, Gottesdiener K. Trypanosoma brucei brucei: characterization of an ODC null bloodstream form mutant the action of alpha-difluoromethylornithine. Exp. Parasitiol. 1998;88:255–257. doi: 10.1006/expr.1998.4237. [DOI] [PubMed] [Google Scholar]
- 33.Xiao Y, McCloskey DE, Phillips MA. RNA interference-mediated silencing of ornithine decarboxylase and spermidine synthase genes in Trypanosoma brucei provides insight into regulation of polyamine biosynthesis. Eukaryot Cell. 2009;8(5):747–755. doi: 10.1128/EC.00047-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Taylor MC, Kaur H, Blessington B, Kelly JM, Wilkinson SR. Validation of spermidine synthase as a drug target in African trypanosomes. Biochem J. 2008;409(2):563–569. doi: 10.1042/BJ20071185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huynh TT, Huynh VT, Harmon MA, Phillips MA. Gene knockdown of g-glutamylcysteine synthetase by RNAi in the parasitic protozoa Trypanosoma brucei demonstrates that it is an essential enzyme. J. Biol. Chem. 2003;278:39794–39800. doi: 10.1074/jbc.M306306200. [DOI] [PubMed] [Google Scholar]
- 36.Krieger S, Schwarz W, Ariyanayagam MR, Fairlamb AH, Krauth-Siegel RL, Clayton C. Trypanosomes lacking trypanothione reductase are avirulent and show increased sensitivity to oxidative stress. Molecular microbiology. 2000;35:542–552. doi: 10.1046/j.1365-2958.2000.01721.x. [DOI] [PubMed] [Google Scholar]
- 37.Wyllie S, Oza SL, Patterson S, Spinks D, Thompson S, Fairlamb AH. Dissecting the essentiality of the bifunctional trypanothione synthetase-amidase in Trypanosoma brucei using chemical and genetic methods. Mol Microbiol. 2009 doi: 10.1111/j.1365-2958.2009.06761.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Barker RH, Jr, Liu H, Hirth B, Celatka CA, Fitzpatrick R, Xiang Y, Willert EK, Phillips MA, Kaiser M, Bacchi CJ, Rodriguez A, Yarlett N, Klinger JD, Sybertz E. Novel S-adenosylmethionine decarboxylase inhibitors for the treatment of human African trypanosomiasis. Antimicrob Agents Chemother. 2009;53(5):2052–2058. doi: 10.1128/AAC.01674-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bacchi CJ, Barker RH, Jr, Rodriguez A, Hirth B, Rattendi D, Yarlett N, Hendrick CL, Sybertz E. Trypanocidal activity of 8-methyl-5'-{[(Z)-4-aminobut-2-enyl]-(methylamino)}adenosine (Genz-644131), an adenosylmethionine decarboxylase inhibitor. Antimicrob Agents Chemother. 2009;53(8):3269–3272. doi: 10.1128/AAC.00076-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Goldberg B, Rattendi D, Lloyd D, Sufrin JR, Bacchi CJ. In situ kinetic characterization of methylthioadenosine transport by the adenosine transporter (P2) of the African Trypanosoma brucei brucei and Trypanosoma brucei rhodesiense. Biochemical Pharmacol. 2001;61:449–457. doi: 10.1016/s0006-2952(00)00560-8. [DOI] [PubMed] [Google Scholar]
- 41.Beswick T, Willert E, Phillips M. Mechanisms of allosteric regulation of Trypanosoma cruzi S-adenosylmethionine decarboxylase. Biochemistry. 2006;45(25):7797–7807. doi: 10.1021/bi0603975. [DOI] [PubMed] [Google Scholar]
- 42.Richardson JL, Nett IR, Jones DC, Abdille MH, Gilbert IH, Fairlamb AH. Improved Tricyclic Inhibitors of Trypanothione Reductase by Screening and Chemical Synthesis. ChemMedChem. 2009;4(8):1333–1340. doi: 10.1002/cmdc.200900097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Perez-Pineiro R, Burgos A, Jones DC, Andrew LC, Rodriguez H, Suarez M, Fairlamb AH, Wishart DS. Development of a novel virtual screening cascade protocol to identify potential trypanothione reductase inhibitors. J Med Chem. 2009;52(6):1670–1680. doi: 10.1021/jm801306g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Patterson S, Jones DC, Shanks EJ, Frearson JA, Gilbert IH, Wyatt PG, Fairlamb AH. Synthesis and Evaluation of 1-(1-(Benzo[b]thiophen-2-yl)cyclohexyl)piperidine (BTCP) Analogues as Inhibitors of Trypanothione Reductase. ChemMedChem. 2009;4(8):1341–1353. doi: 10.1002/cmdc.200900098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Holloway GA, Charman WN, Fairlamb AH, Brun R, Kaiser M, Kostewicz E, Novello PM, Parisot JP, Richardson J, Street IP, Watson KG, Baell JB. Trypanothione reductase high-throughput screening campaign identifies novel classes of inhibitors with anti-parasitic activity. Antimicrob Agents Chemother. 2009 doi: 10.1128/AAC.01568-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stump B, Eberle C, Schweizer WB, Kaiser M, Brun R, Krauth-Siegel RL, Lentz D, Diederich F. Pentafluorosulfanyl as a novel building block for enzyme inhibitors: trypanothione reductase inhibition and antiprotozoal activities of diarylamines. Chembiochem. 2009;10(1):79–83. doi: 10.1002/cbic.200800565. [DOI] [PubMed] [Google Scholar]
- 47.Cavalli A, Lizzi F, Bongarzone S, Brun R, Krauth-Siegel RL, Bolognesi ML. Privileged structure-guided synthesis of quinazoline derivatives as inhibitors of trypanothione reductase. Biorganic and Medicinal Chemistry Letters. 2009;19:3031–3035. doi: 10.1016/j.bmcl.2009.04.060. [DOI] [PubMed] [Google Scholar]
- 48.Verlinde CLMJ, Hannaert V, Blonski C, Willson M, Périé JJ, Fothergill-Gilmore LA, Opperdoes FR, Gelb MH, Hol WGJ, Michels PAM. Glycolysis as a target for the design of new anti-trypanosome drugs. Drug Resistance Updates. 2001;4(1):50–65. doi: 10.1054/drup.2000.0177. [DOI] [PubMed] [Google Scholar]
- 49.Lakhdar-Ghazal F, Blonski C, Willson M, Michels P, Perie J. Glycolysis and Proteases as Targets for the Design of New Anti- Trypanosome Drugs. Current Topics in Medicinal Chemistry. 2002;2(5):439. doi: 10.2174/1568026024607472. [DOI] [PubMed] [Google Scholar]
- 50.Albert MA, Haanstra JR, Hannaert V, Van Roy J, Opperdoes FR, Bakker BM, Michels PA. Experimental and in silico analyses of glycolytic flux control in bloodstream form Trypanosoma brucei. J Biol Chem. 2005;280(31):28306–28315. doi: 10.1074/jbc.M502403200. [DOI] [PubMed] [Google Scholar]
- 51.Morris MT, DeBruin C, Yang Z, Chambers JW, Smith KS, Morris JC. Activity of a second Trypanosoma brucei hexokinase is controlled by an 18-amino-acid C-terminal tail. Eukaryot Cell. 2006;5(12):2014–2023. doi: 10.1128/EC.00146-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chambers JW, Kearns MT, Morris MT, Morris JC. Assembly of Heterohexameric Trypanosome Hexokinases Reveals That Hexokinase 2 Is a Regulable Enzyme. J. Biol. Chem. 2008;283(22):14963–14970. doi: 10.1074/jbc.M802124200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chambers JW, Fowler ML, Morris MT, Morris JC. The anti-trypanosomal agent lonidamine inhibits Trypanosoma brucei hexokinase 1. Molecular and Biochemical Parasitology. 2008;158(2):202–207. doi: 10.1016/j.molbiopara.2007.12.013. [DOI] [PubMed] [Google Scholar]
- 54.Arsenieva D, Appavu BL, Mazock GH, Jeffery CJ. Crystal structure of phosphoglucose isomerase from Trypanosoma brucei complexed with glucose-6-phosphate at 1.6 A resolution. Proteins. 2009;74(1):72–80. doi: 10.1002/prot.22133. [DOI] [PubMed] [Google Scholar]
- 55.Abell L, Schineller J, Keck P, Villafranca J. Effect of metal-ligand mutations on phosphoryl transfer reactions catalyzed by E. coli glutamine synthetase. Biochemistry. 1995;34:16695–16702. doi: 10.1021/bi00051a018. [DOI] [PubMed] [Google Scholar]
- 56.McNae IW, Martinez-Oyanedel J, Keillor JW, Michels PA, Fothergill-Gilmore LA, Walkinshaw MD. The crystal structure of ATP-bound phosphofructokinase from Trypanosoma brucei reveals conformational transitions different from those of other phosphofructokinases. J Mol Biol. 2009;385(5):1519–1533. doi: 10.1016/j.jmb.2008.11.047. [DOI] [PubMed] [Google Scholar]
- 57.Nowicki MW, Tulloch LB, Worralll L, McNae IW, Hannaert V, Michels PA, Fothergill-Gilmore LA, Walkinshaw MD, Turner NJ. Design, synthesis and trypanocidal activity of lead compounds based on inhibitors of parasite glycolysis. Bioorg Med Chem. 2008;16(9):5050–5061. doi: 10.1016/j.bmc.2008.03.045. [DOI] [PubMed] [Google Scholar]
- 58.Chudzik DM, Michels PA, de Walque S, Hol WG. Structures of type 2 peroxisomal targeting signals in two trypanosomatid aldolases. J Mol Biol. 2000;300(4):697–707. doi: 10.1006/jmbi.2000.3910. [DOI] [PubMed] [Google Scholar]
- 59.Dax C, Duffieux F, Chabot N, Coincon M, Sygusch J, Michels PAM, Blonski C. Selective Irreversible Inhibition of Fructose 1,6-Bisphosphate Aldolase from Trypanosoma brucei. Journal of Medicinal Chemistry. 2006;49(5):1499–1502. doi: 10.1021/jm050237b. [DOI] [PubMed] [Google Scholar]
- 60.Azema L, Lherbet C, Baudoin C, Blonski C. Cell permeation of a Trypanosoma brucei aldolase inhibitor: evaluation of different enzyme-labile phosphate protecting groups. Bioorg Med Chem Lett. 2006;16(13):3440–3443. doi: 10.1016/j.bmcl.2006.04.010. [DOI] [PubMed] [Google Scholar]
- 61.Drew ME, Morris JC, Wang Z, Wells L, Sanchez M, Landfear SM, Englund PT. The Adenosine Analog Tubercidin Inhibits Glycolysis in Trypanosoma brucei as Revealed by an RNA Interference Library. J. Biol. Chem. 2003;278(47):46596–46600. doi: 10.1074/jbc.M309320200. [DOI] [PubMed] [Google Scholar]
- 62.de ASNMV, Gomes Dias SM, Mello LV, da Silva Giotto MT, Gavalda S, Blonski C, Garratt RC, Rigden DJ. Structural flexibility in Trypanosoma brucei enolase revealed by X-ray crystallography and molecular dynamics. FEBS J. 2007;274(19):5077–5089. doi: 10.1111/j.1742-4658.2007.06027.x. [DOI] [PubMed] [Google Scholar]
- 63.el Kouni H. Potential chemotherapeutic targets in the purine metabolism of parasites. Pharmacology and Therapeutics. 2003;99:283–309. doi: 10.1016/s0163-7258(03)00071-8. [DOI] [PubMed] [Google Scholar]
- 64.Luscher A, Onal P, Schweingruber AM, Maser P. Adenosine kinase of Trypanosoma brucei and its role in susceptibility to adenosine antimetabolites. Antimicrob Agents Chemother. 2007;51(11):3895–3901. doi: 10.1128/AAC.00458-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rottenberg MEMW, Ferella M, Petitto-Assis F, Goto H, Kristensson K, McCaffrey R, Wigzell H. Treatment of African trypanosomiasis with cordycepin and adenosine deaminase inhibitors in a mouse model. Journal of Infectious Diseases. 2005;192(9):1658–1665. doi: 10.1086/496896. [DOI] [PubMed] [Google Scholar]
- 66.Vodnala M, Fijolek A, Rofougaran R, Mosimann M, Maser P, Hofer A. Adenosine kinase mediates high affinity adenosine salvage in Trypanosoma brucei. J Biol Chem. 2008;283(9):5380–5388. doi: 10.1074/jbc.M705603200. [DOI] [PubMed] [Google Scholar]
- 67.Pettitt A. Mechanism of action of purine analogues in chronic lymphocytic leukaemia. British Journal of Hematology. 2003;121 doi: 10.1046/j.1365-2141.2003.04336.x. 692-70. [DOI] [PubMed] [Google Scholar]
- 68.Baliani A, Peal V, Gros L, Brun R, Kaiser M, Barrett MP, Gilbert IH. Novel functionalized melamine-based nitroheterocycles: synthesis and activity against trypanosomatid parasites. Org Biomol Chem. 2009;7(6):1154–1166. doi: 10.1039/b813394h. [DOI] [PubMed] [Google Scholar]
- 69.Barrett MP, Gilbert IH. Targeting of toxic compounds to the trypanosome's interior. Adv Parasitol. 2006;63:125–183. doi: 10.1016/S0065-308X(06)63002-9. [DOI] [PubMed] [Google Scholar]
- 70.Chollet CBA, Wong PE, Barrett MP, Gilbert IH. Targeted delivery of compounds to Trypanosoma brucei using the melamine motif. Bioorganic and Medicinal Chemistry Letters. 2009;17(6):2512–2523. doi: 10.1016/j.bmc.2009.01.058. [DOI] [PubMed] [Google Scholar]
- 71.Wenzel IN, Wong PE, Maes L, Muller TJ, Krauth-Siegel RL, Barrett MP, Davioud-Charvet E. Unsaturated Mannich bases active against multidrug-resistant Trypanosoma brucei brucei strains. ChemMedChem. 2009;4(3):339–351. doi: 10.1002/cmdc.200800360. [DOI] [PubMed] [Google Scholar]
- 72.Baliani A, Bueno GJ, Stewart ML, Yardley V, Brun R, Barrett MP, Gilbert IH. Design and synthesis of a series of melamine-based nitroheterocycles with activity against Trypanosomatid parasites. J Med Chem. 2005;48(17):5570–5579. doi: 10.1021/jm050177+. [DOI] [PubMed] [Google Scholar]
- 73.Landfear SM. Drugs and transporters in kinetoplastid protozoa. Adv Exp Med Biol. 2008;625:22–32. doi: 10.1007/978-0-387-77570-8_3. [DOI] [PubMed] [Google Scholar]
- 74.Lanteri CA, Stewart ML, Brock JM, Alibu VP, Meshnick SR, Tidwell RR, Barrett MP. Roles for the Trypanosoma brucei P2 transporter in DB75 uptake and resistance. Mol Pharmacol. 2006;70(5):1585–1592. doi: 10.1124/mol.106.024653. [DOI] [PubMed] [Google Scholar]
- 75.Chollet C, Baliani A, Wong PE, Barrett MP, Gilbert IH. Targeted delivery of compounds to Trypanosoma brucei using the melamine motif. Bioorg Med Chem. 2009;17(6):2512–2523. doi: 10.1016/j.bmc.2009.01.058. [DOI] [PubMed] [Google Scholar]
- 76.Arakaki TL, Buckner FS, Gillespie JR, Malmquist NA, Phillips MA, Kalyuzhniy O, Luft JR, Detitta GT, Verlinde CL, Van Voorhis WC, Hol WG, Merritt EA. Characterization of Trypanosoma brucei dihydroorotate dehydrogenase as a possible drug target; structural, kinetic and RNAi studies. Mol Microbiol. 2008;68(1):37–50. doi: 10.1111/j.1365-2958.2008.06131.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hofer A, Steverding D, Chabes A, Brun R, Thelander L. Trypanosoma brucei CTP synthetase: a target for the treatment of African sleeping sickness. Proc Natl Acad Sci U S A. 2001;98(11):6412–6416. doi: 10.1073/pnas.111139498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Fijolek AHA, Thelander L. Expression, purification, characterization, and in vivo targeting of trypanosome CTP synthetase for treatment of African sleeping sickness. Journal of Biological Chemistry. 2007;282(16):11858–18865. doi: 10.1074/jbc.M611580200. [DOI] [PubMed] [Google Scholar]
- 79.Beck JT. U.B. Nutritional requirements of wild-type and folate transport-deficient Leishmania donovani for pterins and folates. Molecular and Biochemical Parasitology. 1990;43(2):221–230. doi: 10.1016/0166-6851(90)90147-e. [DOI] [PubMed] [Google Scholar]
- 80.Gamarro F, Yu PL, Zhao J, Edman U, Greene PJ, Santi D. Addendum to "Trypanosoma brucei dihydrofolate reductase-thymidylate synthase: gene isolation expression characterization of the enzyme". Mol Biochem Parasitol. 1995;75(1):127. doi: 10.1016/0166-6851(95)00059-a. [DOI] [PubMed] [Google Scholar]
- 81.Moreira W, Leblanc E, Ouellette M. The role of reduced pterins in resistance to reactive oxygen and nitrogen intermediates in the protozoan parasite Leishmania. Free Radic Biol Med. 2009;46(3):367–375. doi: 10.1016/j.freeradbiomed.2008.10.034. [DOI] [PubMed] [Google Scholar]
- 82.Nare B, Garraway LA, Vickers TJ, Beverley SM. PTR1-dependent synthesis of tetrahydrobiopterin contributes to oxidant susceptibility in the trypanosomatid protozoan parasite Leishmania major. Curr Genet. 2009;55(3):287–299. doi: 10.1007/s00294-009-0244-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bello AR, Nare B, Freedman D, Hardy L, Beverley SM. PTR1: a reductase mediating salvage of oxidized pteridines and methotrexate resistance in the protozoan parasite Leishmania major. Proc Natl Acad Sci U S A. 1994;91(24):11442–11446. doi: 10.1073/pnas.91.24.11442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.El Fadili A, Kundig C, Roy G, Ouellette M. Inactivation of the Leishmania tarentolae pterin transporter (BT1) and reductase (PTR1) genes leads to viable parasites with changes in folate metabolism and hypersensitivity to the antifolate methotrexate. J Biol Chem. 2004;279(18):18575–18582. doi: 10.1074/jbc.M400652200. [DOI] [PubMed] [Google Scholar]
- 85.Sienkiewicz N, Jaroslawski S, Wyllie S, Fairlamb AH. Chemical and genetic validation of dihydrofolate reductase-thymidylate synthase as a drug target in African trypanosomes. Mol Microbiol. 2008;69(2):520–533. doi: 10.1111/j.1365-2958.2008.06305.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Mpamhanga CP, Spinks D, Tulloch LB, Shanks EJ, Robinson DA, Collie IT, Fairlamb AH, Wyatt PG, Frearson JA, Hunter WN, Gilbert IH, Brenk R. One scaffold, three binding modes: novel and selective pteridine reductase 1 inhibitors derived from fragment hits discovered by virtual screening. J Med Chem. 2009;52(14):4454–4465. doi: 10.1021/jm900414x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Chowdhury AR MH. DNA topoisomerases in life and death: implications in kinetoplastid protozoa. Current Molecular Medicine. 2004;4(6):711–722. doi: 10.2174/1566524043360302. [DOI] [PubMed] [Google Scholar]
- 88.Balaña-Fouce RRC, Pérez-Pertejo Y, Díaz-González R, Reguera RM. Targeting atypical trypanosomatid DNA topoisomerase I. Drug Discovery Today. 2006;11(15):733–740. doi: 10.1016/j.drudis.2006.06.014. [DOI] [PubMed] [Google Scholar]
- 89.Deterding A DF, Thompson KA, Steverding D. Anti-trypanosomal activities of DNA topoisomerase inhibitors. Acta Tropica. 2005;93(3):311–316. doi: 10.1016/j.actatropica.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 90.Bakshi RP. S.T. RNA interference of Trypanosoma brucei topoisomerase IB: both subunits are essential. Molecular and Biochemical Parasitology. 2004;136(2):249–255. doi: 10.1016/j.molbiopara.2004.04.006. [DOI] [PubMed] [Google Scholar]
- 91.Steverding D. W.X. Evaluation of anti-sleeping-sickness drugs and topoisomerase inhibitors in combination on Trypanosoma brucei. Jouurnal of Antimicrobial Chemotherapy. 2009;63(6):1293–1295. doi: 10.1093/jac/dkp120. [DOI] [PubMed] [Google Scholar]
- 92.Nenortas EBC, Shapiro TA. Antitrypanosomal activity of fluoroquinolones. Antimicrobial Agents and Chemotherapy. 1999;43(8):2066–2068. doi: 10.1128/aac.43.8.2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Nenortas EKT, Burri C, Shapiro TA. Antitrypanosomal activities of fluoroquinolones with pyrrolidinyl substitutions. Antimicrobial Agents and Chemotherapy. 2003;47(9) doi: 10.1128/AAC.47.9.3015-3017.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bakshi RPSD, Morrell A, Cushman M, Shapiro TA. Activity of indenoisoquinolines against African trypanosomes. Antimicrobial Agents and Chemotherapy. 2009;53(1):123–128. doi: 10.1128/AAC.00650-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lee SH, Stephens JL, Englund PT. A fatty-acid synthesis mechanism specialized for parasitism. Nat Rev Microbiol. 2007;5(4):287–297. doi: 10.1038/nrmicro1617. [DOI] [PubMed] [Google Scholar]
- 96.van Hellemond JJ, Tielens AG. Adaptations in the lipid metabolism of the protozoan parasite Trypanosoma brucei. FEBS Lett. 2006;580(23):5552–5558. doi: 10.1016/j.febslet.2006.07.056. [DOI] [PubMed] [Google Scholar]
- 97.Lee SH, Stephens JL, Paul KS, Englund PT. Fatty acid synthesis by elongases in trypanosomes. Cell. 2006;126(4):691–699. doi: 10.1016/j.cell.2006.06.045. [DOI] [PubMed] [Google Scholar]
- 98.Stephens JL, Lee SH, Paul KS, Englund PT. Mitochondrial fatty acid synthesis in Trypanosoma brucei. J Biol Chem. 2007;282(7):4427–4436. doi: 10.1074/jbc.M609037200. [DOI] [PubMed] [Google Scholar]
- 99.Autio KJ, Guler JL, Kastaniotis AJ, Englund PT, Hiltunen JK. The 3-hydroxyacyl-ACP dehydratase of mitochondrial fatty acid synthesis in Trypanosoma brucei. FEBS Lett. 2008;582(5):729–733. doi: 10.1016/j.febslet.2008.01.051. [DOI] [PubMed] [Google Scholar]
- 100.Guler JL, Kriegova E, Smith TK, Lukes J, Englund PT. Mitochondrial fatty acid synthesis is required for normal mitochondrial morphology and function in Trypanosoma brucei. Mol Microbiol. 2008;67(5):1125–1142. doi: 10.1111/j.1365-2958.2008.06112.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Tasdemir D, Guner ND, Perozzo R, Brun R, Donmez AA, Calis I, Ruedi P. Anti-protozoal and plasmodial FabI enzyme inhibiting metabolites of Scrophularia lepidota roots. Phytochemistry. 2005;66(3):355–362. doi: 10.1016/j.phytochem.2004.11.013. [DOI] [PubMed] [Google Scholar]
- 102.Karioti A, Skaltsa H, Linden A, Perozzo R, Brun R, Tasdemir D. Anthecularin: a novel sesquiterpene lactone from Anthemis auriculata with antiprotozoal activity. J Org Chem. 2007;72(21):8103–8106. doi: 10.1021/jo701751w. [DOI] [PubMed] [Google Scholar]
- 103.Tasdemir D, Topaloglu B, Perozzo R, Brun R, O'Neill R, Carballeira NM, Zhang X, Tonge PJ, Linden A, Ruedi P. Marine natural products from the Turkish sponge Agelas oroides that inhibit the enoyl reductases from Plasmodium falciparum, Mycobacterium tuberculosis and Escherichia coli. Bioorg Med Chem. 2007;15(21):6834–6845. doi: 10.1016/j.bmc.2007.07.032. [DOI] [PubMed] [Google Scholar]
- 104.Giddens AC, Nielsen L, Boshoff HI, Tasdemir D, Perozzo R, Kaiser M, Wang F, Sacchettini JC, Copp BR. Natural product inhibitors of fatty acid biosynthesis: synthesis of the marine microbial metabolites pseudopyronines A and B and evaluation of their anti-infective activities. Tetrahedron. 2008;64(7):1242–1249. [Google Scholar]
- 105.Price HP, Menon MR, Panethymitaki C, Goulding D, McKean PG, Smith DF. Myristoyl-CoA:protein N-myristoyltransferase, an essential enzyme and potential drug target in kinetoplastid parasites. J Biol Chem. 2003;278(9):7206–7214. doi: 10.1074/jbc.M211391200. [DOI] [PubMed] [Google Scholar]
- 106.Panethymitaki C, Bowyer PW, Price HP, Leatherbarrow RJ, Brown KA, Smith DF. Characterization and selective inhibition of myristoyl-CoA:protein N-myristoyltransferase from Trypanosoma brucei and Leishmania major. Biochem J. 2006;396(2):277–285. doi: 10.1042/BJ20051886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Bowyer PW, Tate EW, Leatherbarrow RJ, Holder AA, Smith DF, Brown KA. N-myristoyltransferase: a prospective drug target for protozoan parasites. ChemMedChem. 2008;3(3):402–408. doi: 10.1002/cmdc.200700301. [DOI] [PubMed] [Google Scholar]
- 108.Sheng C, Ji H, Miao Z, Che X, Yao J, Wang W, Dong G, Guo W, Lu J, Zhang W. Homology modeling and molecular dynamics simulation of N-myristoyltransferase from protozoan parasites: active site characterization and insights into rational inhibitor design. J Comput Aided Mol Des. 2009;23(6):375–389. doi: 10.1007/s10822-009-9267-2. [DOI] [PubMed] [Google Scholar]
- 109.Sutterwala SS, Hsu FF, Sevova ES, Schwartz KJ, Zhang K, Key P, Turk J, Beverley SM, Bangs JD. Developmentally regulated sphingolipid synthesis in African trypanosomes. Mol Microbiol. 2008;70(2):281–296. doi: 10.1111/j.1365-2958.2008.06393.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Mina JG, Pan SY, Wansadhipathi NK, Bruce CR, Shams-Eldin H, Schwarz RT, Steel PG, Denny PW. The Trypanosoma brucei sphingolipid synthase, an essential enzyme and drug target. Mol Biochem Parasitol. 2009 doi: 10.1016/j.molbiopara.2009.06.002. [DOI] [PubMed] [Google Scholar]
- 111.Olepu S, Suryadevara PK, Rivas K, Yokoyama K, Verlinde CL, Chakrabarti D, Van Voorhis WC, Gelb MH. 2-Oxo-tetrahydro-1,8-naphthyridines as selective inhibitors of malarial protein farnesyltransferase and as anti-malarials. Bioorg Med Chem Lett. 2008;18(2):494–497. doi: 10.1016/j.bmcl.2007.11.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Bendale P, Olepu S, Suryadevara PK, Bulbule V, Rivas K, Nallan L, Smart B, Yokoyama K, Ankala S, Pendyala PR, Floyd D, Lombardo LJ, Williams DK, Buckner FS, Chakrabarti D, Verlinde CL, Van Voorhis WC, Gelb MH. Second generation tetrahydroquinoline-based protein farnesyltransferase inhibitors as antimalarials. J Med Chem. 2007;50(19):4585–4605. doi: 10.1021/jm0703340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Eastman RT, Buckner FS, Yokoyama K, Gelb MH, Van Voorhis WC. Thematic review series: lipid posttranslational modifications. Fighting parasitic disease by blocking protein farnesylation. J Lipid Res. 2006;47(2):233–240. doi: 10.1194/jlr.R500016-JLR200. [DOI] [PubMed] [Google Scholar]
- 114.Kraus JM, Verlinde CL, Karimi M, Lepesheva GI, Gelb MH, Buckner FS. Rational modification of a candidate cancer drug for use against Chagas disease. J Med Chem. 2009;52(6):1639–1647. doi: 10.1021/jm801313t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Hammarton TC. Cell cycle regulation in Trypanosoma brucei. Mol Biochem Parasitol. 2007;153(1):1–8. doi: 10.1016/j.molbiopara.2007.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Doerig C. Protein kinases as targets for anti-parasitic chemotherapy. Biochim Biophys Acta. 2004;1697(1–2):155–168. doi: 10.1016/j.bbapap.2003.11.021. [DOI] [PubMed] [Google Scholar]
- 117.Naula C, Parsons M, Mottram JC. Protein kinases as drug targets in trypanosomes and Leishmania. Biochim Biophys Acta. 2005;1754(1–2):151–159. doi: 10.1016/j.bbapap.2005.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Hammarton TC, Kramer S, Tetley L, Boshart M, Mottram JC. Trypanosoma brucei Polo-like kinase is essential for basal body duplication, kDNA segregation and cytokinesis. Mol Microbiol. 2007;65(5):1229–1248. doi: 10.1111/j.1365-2958.2007.05866.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Parsons M, Worthey EA, Ward PN, Mottram JC. Comparative analysis of the kinomes of three pathogenic trypanosomatids: Leishmania major, Trypanosoma brucei and Trypanosoma cruzi. BMC Genomics. 2005;6:127. doi: 10.1186/1471-2164-6-127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Brenk R, Schipani A, James D, Krasowski A, Gilbert IH, Frearson J, Wyatt PG. Lessons learnt from assembling screening libraries for drug discovery for neglected diseases. ChemMedChem. 2008;3(3):435–444. doi: 10.1002/cmdc.200700139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Meijer L, Flajolet M, Greengard P. Pharmacological inhibitors of glycogen synthase kinase 3. Trends in Pharmacological Sciences. 2004;25(9):471–480. doi: 10.1016/j.tips.2004.07.006. [DOI] [PubMed] [Google Scholar]
- 122.Ojo KK, Gillespie JR, Riechers AJ, Napuli AJ, Verlinde CL, Buckner FS, Gelb MH, Domostoj MM, Wells SJ, Scheer A, Wells TN, Van Voorhis WC. Glycogen synthase kinase 3 is a potential drug target for African trypanosomiasis therapy. Antimicrob Agents Chemother. 2008;52(10):3710–3717. doi: 10.1128/AAC.00364-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Jetton N, Rothberg KG, Hubbard JG, Wise J, Li Y, Ball HL, Ruben L. The cell cycle as a therapeutic target against Trypanosoma brucei: Hesperadin inhibits Aurora kinase-1 and blocks mitotic progression in bloodstream forms. Mol Microbiol. 2009;72(2):442–458. doi: 10.1111/j.1365-2958.2009.06657.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.McKerrow JH, Rosenthal PJ, Swenerton R, Doyle P. Development of protease inhibitors for protozoan infections. Curr Opin Infect Dis. 2008;21(6):668–672. doi: 10.1097/QCO.0b013e328315cca9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Doyle PS, Zhou YM, Engel JC, McKerrow JH. A cysteine protease inhibitor cures Chagas' disease in an immunodeficient-mouse model of infection. Antimicrob Agents Chemother. 2007;51(11):3932–3939. doi: 10.1128/AAC.00436-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.O'Brien TC, Mackey ZB, Fetter RD, Choe Y, O'Donoghue AJ, Zhou M, Craik CS, Caffrey CR, McKerrow JH. A parasite cysteine protease is key to host protein degradation and iron acquisition. J Biol Chem. 2008;283(43):28934–28943. doi: 10.1074/jbc.M805824200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Abdulla MH, O'Brien T, Mackey ZB, Sajid M, Grab DJ, McKerrow JH. RNA Interference of Trypanosoma brucei Cathepsin B and L Affects Disease Progression in a Mouse Model. PLoS Negl Trop Dis. 2008;2(9):e298. doi: 10.1371/journal.pntd.0000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Mallari JP, Shelat AA, Obrien T, Caffrey CR, Kosinski A, Connelly M, Harbut M, Greenbaum D, McKerrow JH, Guy RK. Development of potent purine-derived nitrile inhibitors of the trypanosomal protease TbcatB. J Med Chem. 2008;51(3):545–552. doi: 10.1021/jm070760l. [DOI] [PubMed] [Google Scholar]
- 129.Mallari JP, Shelat A, Kosinski A, Caffrey CR, Connelly M, Zhu F, McKerrow JH, Guy RK. Discovery of trypanocidal thiosemicarbazone inhibitors of rhodesain and TbcatB. Bioorg Med Chem Lett. 2008;18(9):2883–2885. doi: 10.1016/j.bmcl.2008.03.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Li Z, Lindsay ME, Motyka SA, Englund PT, Wang CC. Identification of a bacterial-like HslVU protease in the mitochondria of Trypanosoma brucei and its role in mitochondrial DNA replication. PLoS Pathog. 2008;4(4):e1000048. doi: 10.1371/journal.ppat.1000048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Grandgenett PM, Otsu K, Wilson HR, Wilson ME, Donelson JE. A function for a specific zinc metalloprotease of African trypanosomes. PLoS Pathog. 2007;3(10):1432–1445. doi: 10.1371/journal.ppat.0030150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Helms MJ, Ambit A, Appleton P, Tetley L, Coombs GH, Mottram JC. Bloodstream form Trypanosoma brucei depend upon multiple metacaspases associated with RAB11-positive endosomes. J Cell Sci. 2006;119(Pt 6):1105–1117. doi: 10.1242/jcs.02809. [DOI] [PubMed] [Google Scholar]
- 133.Ochola DO, Prichard RK, Lubega GW. Classical ligands bind tubulin of trypanosomes and inhibit their growth in vitro. J Parasitol. 2002;88(3):600–604. doi: 10.1645/0022-3395(2002)088[0600:CLBTOT]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 134.Bhattacharya G, Salem MM, Werbovetz KA. Antileishmanial dinitroaniline sulfonamides with activity against parasite tubulin. Bioorg Med Chem Lett. 2002;12(17):2395–2398. doi: 10.1016/s0960-894x(02)00465-1. [DOI] [PubMed] [Google Scholar]
- 135.Werbovetz KA, Sackett DL, Delfin D, Bhattacharya G, Salem M, Obrzut T, Rattendi D, Bacchi C. Selective antimicrotubule activity of N1-phenyl-3,5-dinitro-N4,N4-di-n-propylsulfanilamide (GB-II-5) against kinetoplastid parasites. Mol Pharmacol. 2003;64(6):1325–1333. doi: 10.1124/mol.64.6.1325. [DOI] [PubMed] [Google Scholar]
- 136.Bhattacharya G, Herman J, Delfin D, Salem MM, Barszcz T, Mollet M, Riccio G, Brun R, Werbovetz KA. Synthesis and antitubulin activity of N1- and N4-substituted 3,5-dinitro sulfanilamides against African trypanosomes and Leishmania. J Med Chem. 2004;47(7):1823–1832. doi: 10.1021/jm0304461. [DOI] [PubMed] [Google Scholar]
- 137.Wu D, George TG, Hurh E, Werbovetz KA, Dalton JT. Pre-systemic metabolism prevents in vivo antikinetoplastid activity of N1,N4-substituted 3,5-dinitro sulfanilamide, GB-II-150. Life Sci. 2006;79(11):1081–1093. doi: 10.1016/j.lfs.2006.03.028. [DOI] [PubMed] [Google Scholar]
- 138.George TG, Johnsamuel J, Delfin DA, Yakovich A, Mukherjee M, Phelps MA, Dalton JT, Sackett DL, Kaiser M, Brun R, Werbovetz KA. Antikinetoplastid antimitotic activity and metabolic stability of dinitroaniline sulfonamides and benzamides. Bioorg Med Chem. 2006;14(16):5699–5710. doi: 10.1016/j.bmc.2006.04.017. [DOI] [PubMed] [Google Scholar]
- 139.George TG, Endeshaw MM, Morgan RE, Mahasenan KV, Delfin DA, Mukherjee MS, Yakovich AJ, Fotie J, Li C, Werbovetz KA. Synthesis, biological evaluation, and molecular modeling of 3,5-substituted-N1-phenyl-N4,N4-di-n-butylsulfanilamides as antikinetoplastid antimicrotubule agents. Bioorg Med Chem. 2007;15(18):6071–6079. doi: 10.1016/j.bmc.2007.06.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Giles NL, Armson A, Reid SA. Characterization of trifluralin binding with recombinant tubulin from Trypanosoma brucei. Parasitol Res. 2009;104(4):893–903. doi: 10.1007/s00436-008-1271-2. [DOI] [PubMed] [Google Scholar]
- 141.Räz B, Iten M, Grether-Bühler Y, Kaminsky R, Brun R. The Alamar Blue® assay to determine drug sensitivity of African trypanosomes (T.b. rhodesiense and T.b. gambiense) in vitro. Acta Tropica. 1997;68(2):139–147. doi: 10.1016/s0001-706x(97)00079-x. [DOI] [PubMed] [Google Scholar]
- 142.Mackey ZB, Baca AM, Mallari JP, Apsel B, Shelat A, Hansell EJ, Chiang PK, Wolff B, Guy KR, Williams J, McKerrow JH. Discovery of trypanocidal compounds by whole cell HTS of Trypanosoma brucei. Chem Biol Drug Des. 2006;67(5):355–363. doi: 10.1111/j.1747-0285.2006.00389.x. [DOI] [PubMed] [Google Scholar]
- 143.Werbovetz K. Diamidines as antitrypanosomal, antileishmanial and antimalarial agents. Curr Opin Investig Drugs. 2006;7(2):147–157. [PubMed] [Google Scholar]
- 144.Thuita JK, Karanja SM, Wenzler T, Mdachi RE, Ngotho JM, Kagira JM, Tidwell R, Brun R. Efficacy of the diamidine DB75 and its prodrug DB289, against murine models of human African trypanosomiasis. Acta Trop. 2008;108(1):6–10. doi: 10.1016/j.actatropica.2008.07.006. [DOI] [PubMed] [Google Scholar]
- 145.Patrick DA, Bakunov SA, Bakunova SM, Suresh Kumar EV, Chen H, Jones SK, Wenzler T, Barzcz T, Werbovetz KA, Brun R, Tidwell RR. Synthesis and antiprotozoal activities of dicationic bis(phenoxymethyl)benzenes, bis(phenoxymethyl)naphthalenes, and bis(benzyloxy)naphthalenes. Eur J Med Chem. 2009 doi: 10.1016/j.ejmech.2009.03.014. [DOI] [PubMed] [Google Scholar]
- 146.Bakunov SA, Bakunova SM, Wenzler T, Barszcz T, Werbovetz KA, Brun R, Tidwell RR. Synthesis and antiprotozoal activity of cationic 2-phenylbenzofurans. J Med Chem. 2008;51(21):6927–6944. doi: 10.1021/jm800918v. [DOI] [PubMed] [Google Scholar]
- 147.Arafa RK, Ismail MA, Munde M, Wilson WD, Wenzler T, Brun R, Boykin DW. Novel linear triaryl guanidines, N-substituted guanidines and potential prodrugs as antiprotozoal agents. Eur J Med Chem. 2008;43(12):2901–2908. doi: 10.1016/j.ejmech.2008.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Ismail MA, Arafa RK, Wenzler T, Brun R, Tanious FA, Wilson WD, Boykin DW. Synthesis and antiprotozoal activity of novel bis-benzamidino imidazo[1,2-a]pyridines and 5,6,7,8-tetrahydro-imidazo[1,2-a]pyridines. Bioorg Med Chem. 2008;16(2):683–691. doi: 10.1016/j.bmc.2007.10.042. [DOI] [PubMed] [Google Scholar]
- 149.Gonzalez JL, Stephens CE, Wenzler T, Brun R, Tanious FA, Wilson WD, Barszcz T, Werbovetz KA, Boykin DW. Synthesis and antiparasitic evaluation of bis-2,5-[4-guanidinophenyl]thiophenes. Eur J Med Chem. 2007;42(4):552–557. doi: 10.1016/j.ejmech.2006.11.006. [DOI] [PubMed] [Google Scholar]
- 150.Bakunova SM, Bakunov SA, Wenzler T, Barszcz T, Werbovetz KA, Brun R, Hall JE, Tidwell RR. Synthesis and in vitro antiprotozoal activity of bisbenzofuran cations. J Med Chem. 2007;50(23):5807–5823. doi: 10.1021/jm0708634. [DOI] [PubMed] [Google Scholar]
- 151.Patrick DA, Bakunov SA, Bakunova SM, Kumar EV, Lombardy RJ, Jones SK, Bridges AS, Zhirnov O, Hall JE, Wenzler T, Brun R, Tidwell RR. Synthesis and in vitro antiprotozoal activities of dicationic 3,5-diphenylisoxazoles. J Med Chem. 2007;50(10):2468–2485. doi: 10.1021/jm0612867. [DOI] [PubMed] [Google Scholar]
- 152.Ansede JH, Voyksner RD, Ismail MA, Boykin DW, Tidwell RR, Hall JE. In vitro metabolism of an orally active O-methyl amidoxime prodrug for the treatment of CNS trypanosomiasis. Xenobiotica. 2005;35(3):211–226. doi: 10.1080/00498250500087671. [DOI] [PubMed] [Google Scholar]
- 153.Faist J, Seebacher W, Schlapper C, Kaiser M, Brun R, Saf R, Weis R. Antiplasmodial and antitrypanosomal activity of bicyclic amides and esters of dialkylamino acids. Bioorg Med Chem. 2009;17(10):3595–3603. doi: 10.1016/j.bmc.2009.04.002. [DOI] [PubMed] [Google Scholar]
- 154.Berger H, Weis R, Kaiser M, Brun R, Saf R, Seebacher W. Novel azabicyclo[3.2.2]nonane derivatives and their activities against Plasmodium falciparum K1 and Trypanosoma brucei rhodesiense. Bioorg Med Chem. 2008;16(12):6371–6378. doi: 10.1016/j.bmc.2008.05.007. [DOI] [PubMed] [Google Scholar]
- 155.Schlapper C, Seebacher W, Kaiser M, Brun R, Saf R, Weis R. Bicyclo[2.2.2]octyl esters of dialkylamino acids as antiprotozoals. Bioorg Med Chem. 2007;15(16):5543–5550. doi: 10.1016/j.bmc.2007.05.042. [DOI] [PubMed] [Google Scholar]
- 156.Papanastasiou I, Tsotinis A, Kolocouris N, Prathalingam SR, Kelly JM. Design, synthesis, and trypanocidal activity of new aminoadamantane derivatives. J Med Chem. 2008;51(5):1496–1500. doi: 10.1021/jm7014292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Weis R, Seebacher W. New bicyclic amines: synthesis and SARs of their action against the causative organisms of malaria and sleeping sickness. Curr Med Chem. 2009;16(11):1426–1441. doi: 10.2174/092986709787846479. [DOI] [PubMed] [Google Scholar]
- 158.Kolocouris N, Zoidis G, Foscolos GB, Fytas G, Prathalingham SR, Kelly JM, Naesens L, De Clercq E. Design and synthesis of bioactive adamantane spiro heterocycles. Bioorg Med Chem Lett. 2007;17(15):4358–4362. doi: 10.1016/j.bmcl.2007.04.108. [DOI] [PubMed] [Google Scholar]
- 159.Weis R, Brun R, Saf R, Seebacher W. 4-Aminobicyclo[2.2.2]octanone Derivatives with Antiprotozoal Activities. Monatshefte für Chemie / Chemical Monthly. 2003;134(7):1019–1026. [Google Scholar]
- 160.Seebacher W, Brun R, Kaiser M, Saf R, Weis R. Synthesis and evaluation of the antitrypanosomal and antiplasmodial activities of new 4-aminobicyclo[2.2.2]octane derivatives. Eur J Med Chem. 2005;40(9):888–896. doi: 10.1016/j.ejmech.2005.03.018. [DOI] [PubMed] [Google Scholar]
- 161.Weis R, Schlapper C, Brun R, Kaiser M, Seebacher W. Antiplasmodial and antitrypanosomal activity of new esters and ethers of 4-dialkylaminobicyclo[2.2.2]octan-2-ols. Eur J Pharm Sci. 2006;28(5):361–368. doi: 10.1016/j.ejps.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 162.Schlapper C, Seebacher W, Faist J, Kaiser M, Brun R, Saf R, Weis R. Antiplasmodial and antitrypanosomal activities of aminobicyclo[2.2.2]octyl omega-aminoalkanoates. Eur J Med Chem. 2009;44(2):736–744. doi: 10.1016/j.ejmech.2008.05.002. [DOI] [PubMed] [Google Scholar]
- 163.Seebacher W, Schlapper C, Brun R, Kaiser M, Saf R, Weis R. Synthesis of new esters and oximes with 4-aminobicyclo[2.2.2]octane structure and evaluation of their antitrypanosomal and antiplasmodial activities. Eur J Med Chem. 2006;41(8):970–977. doi: 10.1016/j.ejmech.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 164.Weis R, Berger H, Kaiser M, Brun R, Saf R, Seebacher W. Synthesis of bicyclic amines and their activities against Trypanosoma brucei rhodesiense and Plasmodium falciparum K1. Arch Pharm Res. 2008;31(6):688–697. doi: 10.1007/s12272-001-1214-5. [DOI] [PubMed] [Google Scholar]
- 165.Seebacher W, Weis R, Kaiser M, Brun R, Saf R. Synthesis of 2-azabicyclo[3.2.2]nonanes from bicyclo[2.2.2]octan-2-ones and their activities against Trypanosoma brucei rhodesiense and Plasmodium falciparum K1. J Pharm Pharm Sci. 2005;8(3):578–585. [PubMed] [Google Scholar]
- 166.Paul KS, Bacchi CJ, Englund PT. Multiple triclosan targets in Trypanosoma brucei. Eukaryot Cell. 2004;3(4):855–861. doi: 10.1128/EC.3.4.855-861.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Bolognesi ML, Lizzi F, Perozzo R, Brun R, Cavalli A. Synthesis of a small library of 2-phenoxy-1,4-naphthoquinone and 2-phenoxy-1,4-anthraquinone derivatives bearing anti-trypanosomal and anti-leishmanial activity. Bioorg Med Chem Lett. 2008;18(7):2272–2276. doi: 10.1016/j.bmcl.2008.03.009. [DOI] [PubMed] [Google Scholar]
- 168.Kuettel S, Zambon A, Kaiser M, Brun R, Scapozza L, Perozzo R. Synthesis and evaluation of antiparasitic activities of new 4-[5-(4-phenoxyphenyl)-2H–pyrazol-3-yl]morpholine derivatives. J Med Chem. 2007;50(23):5833–5839. doi: 10.1021/jm700938n. [DOI] [PubMed] [Google Scholar]
- 169.Wilkinson SR, Taylor MC, Horn D, Kelly JM, Cheeseman I. A mechanism for cross-resistance to nifurtimox and benznidazole in trypanosomes. Proc Natl Acad Sci U S A. 2008;105(13):5022–5027. doi: 10.1073/pnas.0711014105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.DeLano WL. The PyMOL Molecular Graphics System. http://www.pymol.org. 2002 [Google Scholar]