

Abstract
Bottom-up fabrication of self-assembled nanomaterials requires control over forces and interactions between building blocks. We here report on the formation and architecture of supramolecular structures constructed from two different peptide amphiphiles. Inclusion of four alanines between a 16-mer peptide and a 16-carbon long aliphatic tail resulted in a secondary structure shift of the peptide headgroups from alpha helices to beta sheets. A concomitant shift in self-assembled morphology from nano-ribbons to core-shell wormlike micelles was observed by cryogenic transmission electron microscopy (cryo-TEM) and atomic force microscopy (AFM). In presence of divalent magnesium ions, these a priori formed supramolecular structures interacted in distinct manners, highlighting the importance of peptide amphiphile design in self-assembly.
Self-assembly is a powerful approach to construct advanced functional materials with control over dimensions on the nanoscale.1 Peptide-based building blocks have gained the attention of bioengineers because their natural origin provides a biocompatible and functional platform for construction of biomaterials.2 Moreover, advances in peptide synthesis enable such technologies for scale up to production levels. The challenge, however, remains to control the self-assembly process accurately at the molecular level in order to fabricate well-defined materials, whether these are nanosized colloids for drug delivery, macroscopic scaffolds for tissue engineering or coatings.2
Nature serves as inspiration for fabrication of new materials that rely on self-assembly and exhibit well-defined properties. Structural proteins have evolved as molecular building blocks that come together based on interactions, often mediated by small folded peptide segments. These peptides are potential candidates for design and fabrication of 3-dimensional structural systems. Moreover, they possess inherent biofunctionality able to guide cell behavior - or interfere with it - depending on the application in mind. However, short peptides isolated from the protein generally lose their capacity to form active, near-native secondary structures.
One approach to enable innate folding of synthetic peptides that emulate the folding in the whole protein and simultaneously induce the formation of nanosized, peptide-based materials is to modify them with a hydrophobic fatty acid tail.2,3 The resulting chimeric molecules, termed peptide amphiphiles (PAs), self-assemble due to hydrophobic interactions and stabilize secondary structure motifs on their surface as a result of peptide tethering and crowding.4 Apart from the predominant role of hydrophobic interactions of the tails, peptide headgroup interactions are important for controlling stability and the shape in the aggregated state.4-6 Solution ionic strength and/or pH has often been used as the trigger for self-assembly through screening of charges between neighboring peptides.7,8 The effect of ion type and valency has also been investigated, revealing the possibility of using it to tune inter-peptide forces.9,10 Overall, it has become evident that understanding of peptide-peptide interactions is a key in determining not only how peptides behave at the molecular level but additionally in controlling the macroscopic properties of the formed materials.4,10,11
We recently demonstrated that peptide folding of the short 16-mer peptide p5314-29 (derived from the MDM2-binding site of tumor suppressor protein p53) could be manipulated by the selection of appropriate linkers that covalently attached it to a palmitic tail.4 The resulting peptide amphiphiles self-assembled into elongated micelles that exhibited different type and extent of peptide secondary structure. Among the findings, we observed an interesting shift from a predominantly alpha helical fold to a beta-sheet structure when four alanines were introduced between the N-terminus of p5314-29 and the palmitic tail. Considering that both peptide structure and supramolecular assembly are critical to developing an efficient delivery system of this inhibitory peptide, we here examine in detail the supramolecular structure of these two peptide amphiphiles and study how these are influenced by high concentration of Mg2+ ions. Our results reveal striking differences in self-assembly stemming from the folding of the peptide into distinct secondary structure motifs, which have general implications for designing biomaterials as well as for studying peptide misfolding diseases.
Results & Discussion
Self-assembly of peptide amphiphiles
The chemical structures of the peptide amphiphiles (PAs) used in our study consist of an aliphatic palmitic tail and an oligopeptide fragment (Figure 1). The two PAs differ solely by four alanines interposed between the palmitic tail and the amino acid sequence LSQETFSDLWKLLPEN from peptide p5314-29. Hydrophobic forces predominantly drive the self-assembly of these PAs above the critical association concentration (CAC) of approximately 2 uM;4 the corresponding peptides do not self-associate at concentrations up to 0.5 mM. Despite the similarity in architecture and amino acid content, the inclusion of 4 alanines between p5314-29 and the palmitic tail had a striking effect on headgroup interactions and peptide folding as was demonstrated using circular dichroism (CD) spectroscopy (Figure 2).
Figure 1.
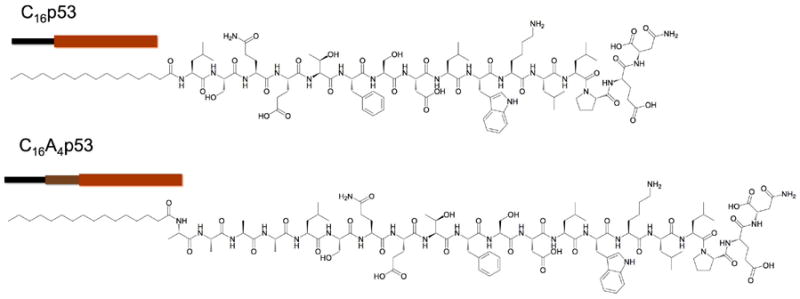
Chemical structure of peptide amphiphiles (PAs) used in this study
Figure 2.
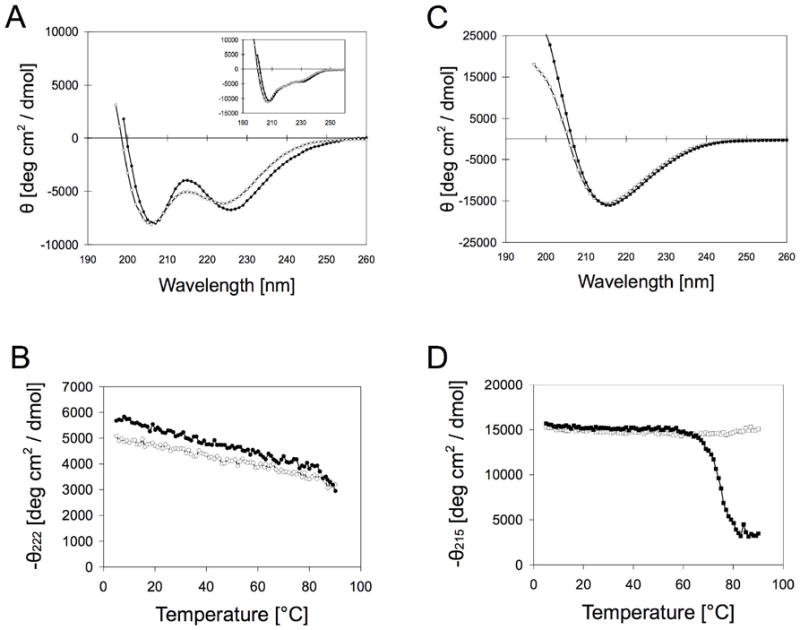
Circular dichroism spectroscopy of C16p53 and C16A4p53 in the presence and absence of 1 M MgCl2. (A) N-Bromosusuccinimide (NBS)-oxidized C16p53 in PBS folds into an alpha helix (open circles), which is slightly stabilized in presence of Mg2+ (filled circles). Inset shows original C16p53 CD spectra, which contain contributions from Trp23 stacking interactions (B) Melting curves for C16p53 confirm enhanced folding stability in the presence of Mg2+ (filled circles) compared to PBS (open circles). (C) C16A4p53, on the contrary, folds into beta sheets with comparable CD spectra in PBS (open squares) and PBS with 1 M MgCl2 (filled squares). (D) Melting curves of C16A4p53 in PBS (open squares) and PBS with 1 M MgCl2 (filled squares).
CD spectra in 10 mM PBS, well above the CAC of the PAs, showed that the peptide adopted a mostly alpha helical conformation in C16p53 assemblies, whereas it formed beta sheets in C16A4p53 assemblies (Figure 2A,C). In the presence of Mg2+ ions, the type of secondary structure was not altered and only a small stabilizing effect in folding was observed, particularly for C16p53. Peptide melting curves showed high folding stability of the peptide headgroups in micelles (Figure 2B,D). For C16A4p53, heating the peptide amphiphile solution above 60°C in the presence of divalent Mg2+ resulted in non-reversible precipitation of the micelles, hence the increase in molar ellipticity at 215 nm above that temperature.
When bound to MDM2, p5314-29 forms an amphipathic alpha helix, exposing a hydrophobic face containing Phe19, Trp23, and Leu26 side chains.12 The peptide in solution is mostly unstructured, but following PA assembly its conformational freedom is reduced and folding is energetically favored. Considering the above, we hypothesized that the peptide has a natural tendency to form an alpha helix, as is the case for C16p53. Interestingly, inclusion of alanines induces beta-sheet formation of the peptide headgroups. This result is non-trivial considering that alanines are frequently found in alpha helical segments of proteins and therefore are considered as alpha helix promoting amino acids.13 However, alanine-rich peptides forming beta-sheet have also been documented in nature, most notably in silk proteins14. We believe that the positioning of alanines close to the hydrophobic core of the PAs and the tendency to minimize the interfacial area between the core and the aqueous phase induces tight peptide packing and favors intermolecular hydrogen bonding. The small side chain of alanine, which is beneficial for formation of alpha helices, can also be viewed as advantageous for tightly packing beta-sheets. Indeed, a number of studies have utilized alanines as spacers in PAs to promote fiber formation through beta-sheet stacking.15-17
The capacity of the peptide headgroup to adopt two different secondary structure motifs implies that folding of supramolecular structures is amenable to control. Several examples exist in the literature where a peptide switches between an alpha helix and a beta-sheet conformation either in a reversible or a non-reversible manner.5,18 Such transformations also bring in mind protein misfolding scenarios where a certain protein or peptide segment aggregates in beta-sheet fibrils. These amyloid fibers have been implicated with various pathologies.19 Here, the peptides studied adopt a controlled conformation in response to self-assembly. It is noteworthy that this occurs at particular conditions (concentration, pH, temperature etc.) in which the unmodified peptides show limited interactions. We therefore propose that peptide amphiphiles hold potential not only as building blocks of biomaterials but also as tools for studying peptide folding and peptide-peptide interactions.20
Imaging of supramolecular structures
The aforementioned differences in peptide headgroup structure, and therefore peptide-peptide interactions between C16p53 and C16A4p53, had a pronounced effect on supramolecular structure as observed using cryo-TEM (Figure 3). The C16p53 structures were scarce after sample preparation, presumably due to aggregate instability during the blotting procedure or adsorption of preferential binding of the PAs on the blotting paper. A couple of structures can be seen in figure 3A and several more in the supporting information (Figures S1 and S2). Careful examination of C16p53 structures revealed twisted ribbon morphology with a pitch that varied between 50 and 100 nm amongst different aggregates. The diameter of these aggregates oscillated between a minimum value of 5 nm and a maximum of 8 nm (Figure 3B and Figure S1). These dimensions excluded the possibility that the imaged structures are two intertwined core-shell micelles and suggest instead nano-ribbon morphology. Core-shell micelles twisted together would exhibit a width oscillating between a diameter and the double of that value. Moreover, individual micelles fraying at the ends of the supamolecular structures were not observed as previously seen in such aggregates.27 Taking into account that an alpha helix is formed (Figure 2A) and that Trp23 fluorescence indicates water shielding and a more hydrophobic environment,4 we propose the structure schematically presented in figure 4A. In this model the amphipathic alpha helix folds on itself, interacting with the acyl chain, while another PA symmetrically shields the acyl chain from water. The macroscopic association of C16p53 PAs is then preferentially linear and twisting occurs presumably due to accommodate close packing of alpha helices.
Figure 3.
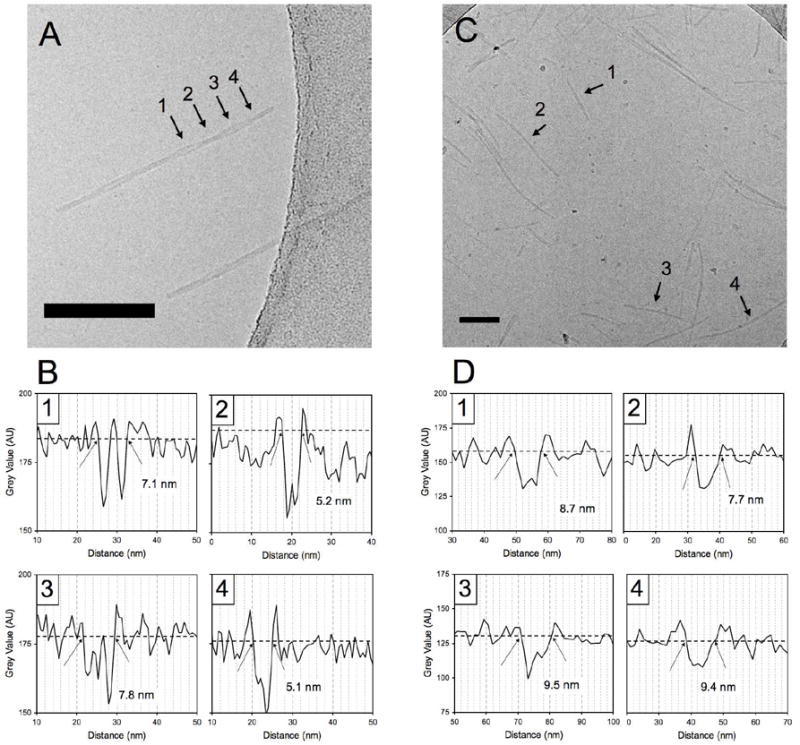
Cryo-TEM micrographs of C16p53 (A) and C16A4p53 (C) in PBS 10mM. C16p53 structures self-assembled into twisted ribbon structures, whereas C16A4p53 appeared as core-shell worm-like micelles. Pixel intensity profiles to determine aggregate width at positions indicated by the arrows in (A) and (C) are shown in (B) for C16p53 and (D) for C16A4p53, respectively. Scale bars: 100 nm
Figure 4.
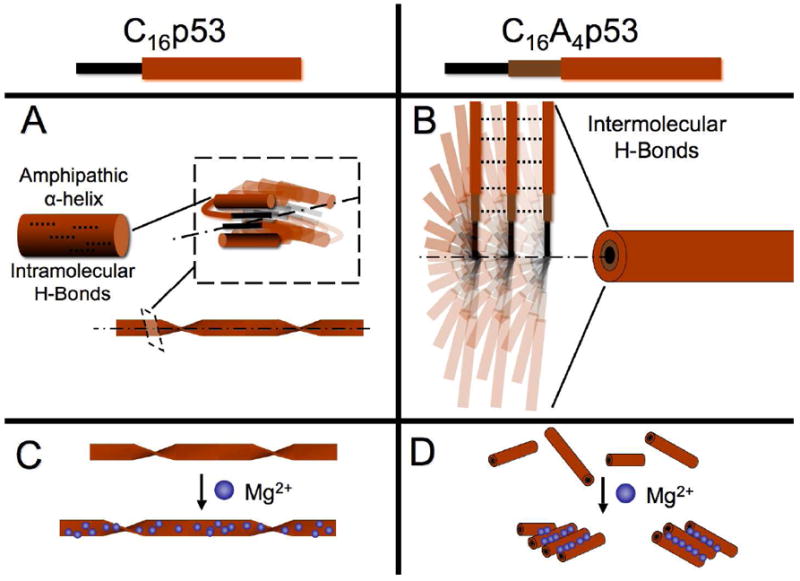
Schematic representation of supramolecular assemblies formed by C16p53 (A) and C16A4p53 (B) in PBS 10 mM. Presence of magnesium ions increases the pitch of C16p53 twisted structures (C) whereas it causes lateral aggregation of C16A4p53 worm-like micelles (D). The dotted lines in (A) and (B) represent H-bonds and the dot-dash lines the longitudinal axis of the supramolecular structures
In contrast to C16p53, C16A4p53 aggregates were readily imaged even at lower concentrations (Figure 3C). In this case, structures that resemble worm-like micelles of uniform thickness and finite length were imaged. Figure 3D presents diameter measurement of 4 different micelles. The average diameter obtained from averaging 40 such micelles from 6 different TEM micrographs was found to be 8.5 ± 0.9 nm (average ± standard deviation). This corresponds well with core-shell micelle morphology, with the core composed of the hydrocarbon chains and the shell made up of extended peptide headgroups (Figure 4B). This fibrillar structure is similar to those reported by analogous peptide amphiphiles rich in alanines immediately after the acyl chain (C16Ax-peptide) reported in literature.16,21,22 Hydrogen bonding occurs between adjacent PAs and runs axially along the micelles. It is not clear at this point whether there is a twist in the beta-sheets or they run parallel to the central axis.11,23,24 However, the minimum in the CD spectrum at 214 nm suggests linear PA stacking.23 Independent of the extent of disorder in beta-sheet formation along the micelle long-axis, inter-peptide hydrogen bonding in C16A4p53 is expected to increase stability of the supramolecular structures, compared to C16p53 where hydrogen bonding occurs intramolecularly.
The differences in supramolecular structure observed between the two PAs shed light on our previously reported findings in respect to their stiffness, obtained by dynamic light scattering (DLS) measurements.4 For C16p53, we determined that the persistence length exceeded 50 nm, while C16A4p53 micelles had a persistence length of less than 20 nm, despite both PAs having similar hydrodynamic diameters. Here, we show that the cause of this difference is the qualitatively different morphology of the supramolecular structure.
We next performed AFM in semi-wet conditions and observed the twisted ribbon morphology of C16p53 (Figure S3). The absence of excess water and/or the deposition process could have altered the structure morphology while inclusion of buffers was not possible due to their crystallization upon drying. Although AFM in liquid overcomes these issues, at physiological pH the net surface charge of the micelle is negative and they are repelled by the negatively charged mica surface, making imaging challenging. Addition of divalent ions addressed this problem and will be discussed below.
Effect of Mg2+ addition in PA solutions
Interactions between the building blocks in PA supramolecular assemblies are dependent on the presence of ions between these blocks at physiological pH. Divalent ions are often employed to form ionic bridges between neighboring PAs in fibrillar aggregates with the aim of stabilizing the structure.9,10 We studied the effect of magnesium ions at a high concentration after the PAs had already assembled in PBS. Moreover an excess of Mg2+ ions induced formation of cross-links between the PA structures and a mica surface facilitating AFM imaging in liquid.
Visual examination following addition of Mg2+ at a concentration of 1 M revealed major differences between C16p53 and C16A4p53 PA micelle suspensions: C16p53 aqueous suspensions remained clear whereas C16A4p53 suspensions rapidly turned turbid (Figure 5A-inset). We performed light scattering measurements to gain a better understanding of the process. Kinetics of scattering intensity at 90°, measured as counts of scattering events detected per second, showed that aggregation of C16A4p53 micelles occurred within seconds following addition of Mg2+ (Figure 5A). Instead, scattering intensity of C16p53 dropped slightly because of dilution with the MgCl2 solution and remained constant over at least 30 minutes. Diffusion coefficients before Mg2+ addition were 2.08×10-8 cm2/s and 3.52×10-8 cm2/s for C16p53 and C16A4p53 structures, respectively, corresponding to hydrodynamic diameters of 236 nm and 139 nm. In the case of C16A4p53, the jump in scattering intensity was concurrent with a rapid increase in diffusion coefficient as evidenced by the autocorrelation curves measured immediately after mixing with MgCl2. Interestingly, DLS measurements over the next 48 hours did not indicate a shift in autocorrelation curves, suggesting formation of stable aggregates (Figure 5B-bottom). Furthermore, in this time period, no precipitation was observed. On the contrary, the autocorrelation curves for C16p53 structures in presence of Mg2+ gradually shifted towards longer decay times, corresponding to higher diffusion coefficients (Figure 5B-top) while the solution remained transparent. We would like to note that the observed increases in diffusion coefficient could originate from viscosity increases parallel to structure modification, and therefore we have refrained from reporting data on hydrodynamic size.
Figure 5.

Dynamic light scattering was used to study the effect of Mg2+ on PA self-assembly. (A) Scattering intensity at 90° was monitored with time following mixing (t=2 min) of 200 uM PA solution with MgCl2 solution (C16p53: circles, C16A4p53: squares). Inset shows PA solutions immediately following MgCl2 addition (final Mg2+ concentration: 1 M). (B) Normalized autocorrelation curves obtained by DLS for C16p53 (top) and C16A4p53 (bottom).
In order to understand the effect of magnesium ions on the two PAs, we probed their structure following MgCl2 addition using both AFM and cryo-TEM. AFM of the PAs in the presence of divalent Mg2+ ions allowed visualization of C16p53 aggregates (Figure 6), which were again scarce in samples prepared for cryo-TEM (Figure 7). C16p53 exhibited the same twisted ribbon morphology as in the absence of magnesium ions. In this case, however, structures seemed to be longer, a finding that agrees well with DLS results. Aggregate height oscillated from 2.5 nm to 5.0 nm with a periodicity ranging from 100 and 200 nm (Figure 6). The height of the aggregates was lower than the width measured using cryo-TEM (5-10 nm; Figure 7). This difference could originate from compression of the structures on the mica surface by the AFM tip. Alternatively, flattening of the aggregates on the surface of mica could occur. The measured pitch was similar between the two techniques and higher than that observed in the absence of Mg2+.
Figure 6.

Atomic force micrograph of C16p53 structures associated with a mica surface in the presence of 1 M MgCl2 (A). Measurements from one such structure (B) revealed a regular pitch of approximately 180 nm and a height of 5 nm at the higher points (C).
Figure 7.
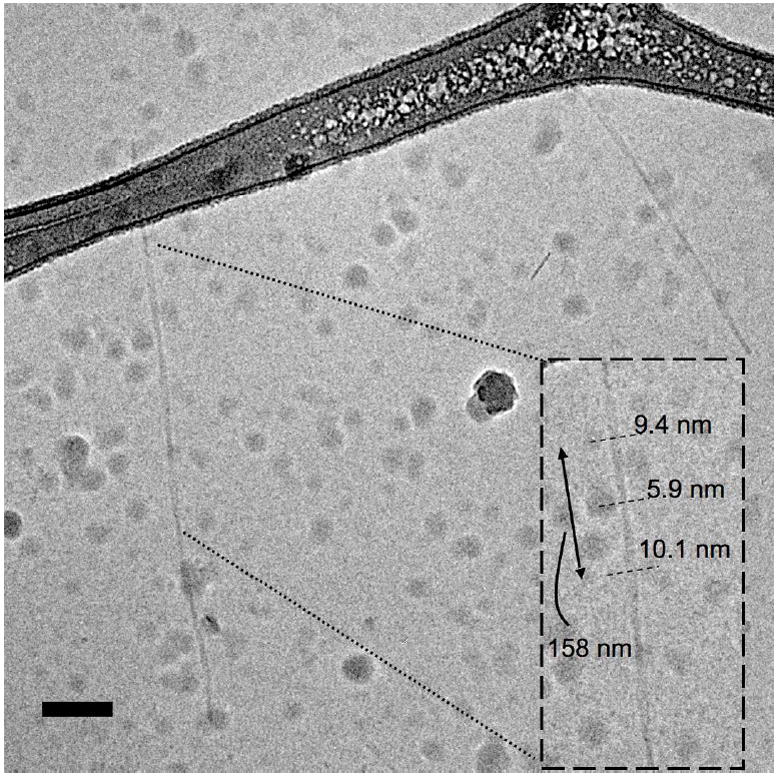
CryoTEM image of C16p53 PAs in the presence of 1 M MgCl2 forming twisted ribbons. Measurements of structure width and periodicity are shown in the inset. The globular structures present in the image are surface ice contamination. Scale bar: 100 nm.
C16A4p53 imaged by AFM under liquid in the presence of MgCl2 showed a strikingly different image compared to C16p53 (Figure 8). Aggregates of micelles with lengths similar to those observed by cryo-TEM in absence of Mg2+ (Figure 3B) were visualized. Association of the micelles appeared to be along the long axis of the micelles (Figure S4). Micelle diameter was uniform and a height of approximately 10 nm was measured, which corresponds well with the width obtained by cryo-TEM. The bundling of individidual micelles was also imaged using cryo-TEM (Figure 9) and negative stain TEM (Figure S5).
Figure 8.
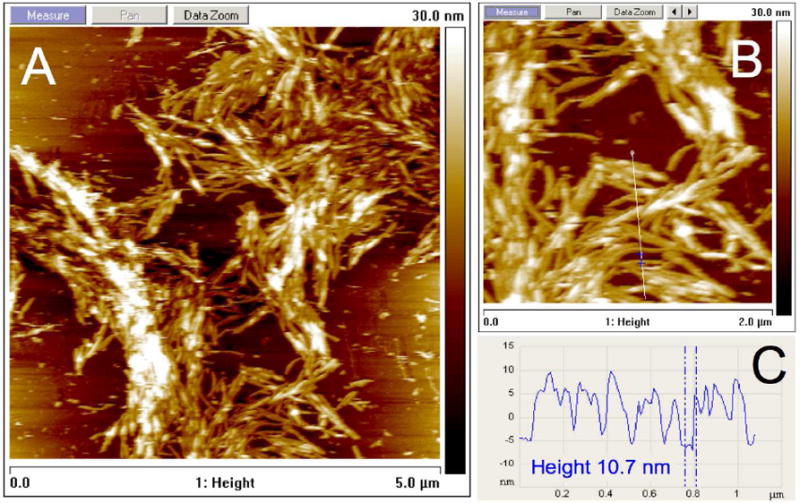
Atomic force micrograph of C16A4p53 structures associated with a mica surface in the presence of 1 M MgCl2 revealing extensive association of PA micelles (A). The micelle height was between 10 and 11 nm (B, C).
Figure 9.

Cryo-TEM image of C16A4p53 PAs in the presence of 1 M MgCl2. Association along the long axis of individual micelles was evident. Scale bar: 100 nm.
Our results highlight major differences upon Mg2+ ion addition between the two different peptide amphiphiles. These differences are not likely to be a result from direct binding or interactions of magnesium ions with alanine side chains as these are non-polar and hydrophobic. Instead, the differences are expected to originate from the different peptide secondary structures induced. The two different folded patterns not only possess different overall morphology but also dictate the positioning of the anionic residues within the supramolecular structure. In the case of C16A4p53, p5314-29 is extended with Glu28 on the outer surface of the corona. Magnesium ions could therefore act as crosslinks between adjacent micelles (Figure 4D) and result in the observations of TEM (Figure 9) and AFM (Figure 8). For C16p53, elongation of the twisted ribbons may result from a stabilizing effect of ionic crosslinking between adjacent PAs in the structure. Lateral aggregation of the ribbons is unlikely considering their geometry and the contact points between them;25 energetically costly untwisting would be necessary to induce a similar effect. Our results were obtained with magnesium as the divalent ion as this is the prevalent divalent ion in intracellular matrix, present in numerous protein and RNA structures. Studies of peptide amphiphile systems have sometimes revealed significant changes in interactions depending on the ion type.8-10,26 It would be of interest to test the effect of divalent ion size, especially on C16p53 structures where presumably ions interact with less accessible, glutamic acid anionic side chains.
In contrast to previous reports of structures with similar morphologies where the self-assembled structure was stabilized by beta-sheets20,27-31 the peptide here adopts a mostly alpha-helical conformation. Pashuck and Stupp recently reported on the formation of twisted ribbons, which upon aging, formed helical ribbons.29 The effect of aging on C16p53 structures is under investigation; however, during preparation of samples reported here a heating step of 1 hour at 60°C was performed, which makes formation of kinetically trapped structures unlikely. Concentration of the sample has also been implicated in determining supramolecular structure morphology28 and merits further investigation. In contrast to the twisted ribbon morphology, the fibrillar, core-shell morphology observed for C16A4p53 has often been observed in peptide amphiphile systems.7,11
In summary, we have presented in detail the process of self-assembly into supramolecular structures of two p5314-29 peptide amphiphiles differing solely by a four-alanine ‘linker’. The presence of this ‘linker’ had a dramatic effect on peptide secondary structure in the self-assembled state, shifting the energy balance from favoring formation of an alpha-helix in the case of C16p53 to formation of beta-sheets for C16A4p53. This shift in turn determined the morphology of the supramolecular structures as well as their assembly in the presence of a divalent cation (Mg2+). Such differences are important for the design of p5314-29 peptide delivery systems based on PAs and have implications for a number of similar systems encountered in literature. Previously, we have shown that C16p53 PAs readily enter cells following disassembly into isolated PAs;32 altering interactions between PAs through incorporation of linkers constitutes a potential control mechanism over stability and delivery. Furthermore, our results revealed promise for use of peptide amphiphiles to study peptide-peptide interactions and their aggregation processes. Intermolecular interactions are amplified in PAs due to condensation of the peptide on the surface of the supramolecular structures. Even though contributions of the alkyl chains are always present, the effective concentration required to observe peptide-peptide interactions is greatly reduced and energy contributions can be extracted through systematic selection of spacers and conditions.
Methods
Materials
Peptide amphiphiles (PAs) were synthesized as previously described.33 Their identity was verified by electrospray ionization mass spectrometry and purity was determined using analytical HPLC on a reverse-phase C4 column (Vydac). Peptide amphiphiles with purity greater than 95% were used. In order to prepare micelles, PAs were dissolved in a 1:1 mixture of chloroform and methanol. The organic solvents were then evaporated under N2 flow eventually forming a film on a glass vial wall. The film was dried in vacuum and subsequently hydrated in water or buffer at 60 °C for 1 h. All samples were stored at 4°C and used within 1 week of preparation.
Spectroscopy
Circular dichroism (CD) spectroscopy was performed on a Jasco J-815 spectropolarimiter. Peptide amphiphile solutions (100 uM) were analyzed using 1 mm pathlength cuvettes, while temperature was controlled within 0.1 °C using a Peltier device.
Dynamic light scattering (DLS)
DLS was performed on a system (Brookhaven Instruments) that consisted of an avalanche photodiode detector to measure scattering intensity from a 632.8 nm HeNe laser (Melles Griot) as a function of delay time. Temperature was maintained at 25.0 ± 0.1°C using a circulating water bath. The first-order autocorrelation function was measured at 90° (q = 0.0189 nm-1). The autocorrelation function was then fit using a second-order cumulant to extract the average decay rate, Γ1. The quantity Γ1/q2 was then taken as the apparent diffusion coefficient, Dapp. Scattering intensity was measured as scattering events per s (counts/s) at 90°.
Atomic force microscopy
Typical AFM experiment follows the deposition of 50uL of PA solution on freshly cleaved V1 grade mica surface (from Ted Pella) for 10min in a closed humid chamber. After deposition, samples were washed with appropriate buffer. Samples were either imaged under buffer conditions in AFM liquid chamber or dried in the stream of nitrogen and imaged under air. The AFM images were recorded in the amplitude modulated tapping mode using MultiMode microscope equipped with E scanner controlled by Nanoscope IIIA (Veeco, Santa Barbara, CA). For experiments performed under liquid, silicon nitride probes (MLCT-AUHM) with resonance frequency ~8 kHz and spring constant k~0.03 N/m (Veeco Probes, Santa Barbara, CA) were used. Images in air were collected using silicon probes (NSC12/without Al coating) with resonance frequency ~160-200 kHz and spring constant k~4.5-6.5 N/m (MicroMasch, Wilsonville, OR). Images were processed and analyzed using NanoScope® software (Veeco, Santa Barbara, CA) and leveled by a first order plane fit in order to correct the sample tilt.
Transmission electron microscopy
All PA samples used for TEM imaging were prepared at a concentration between 1.0-2.5 mg/mL and were imaged at 200kV with a JEOL JEM2010F (Figures 3 & S1) and an FEI Technai G2 Sphera microscope (Figures 7, 9 & S5). Images were recorded digitally with a Gatan Ultrascan 4000 CCD camera on JEM2010F and 1000 CCD camera on Technai microscopes. Images were analyzed using the Gatan Digital Micrograph software.
Negative stain samples were prepared by placing a 3 μL droplet of PA solution onto a glow discharged formvar coated copper grid for 5 minutes. The excess liquid was wicked away by filter paper. A 3 μL droplet of 2% (w/v) aqueous phosphotungstic acid was placed on the grid and left for 5 minutes before similarly being wicked away. The samples were left to dry overnight before imaging.
Cryogenic transmission electron microscopy (cryo-TEM) samples were prepared using the environmentally controlled FEI Vitrobot Mark IV (24°C, 100% humidity). A 3.5 μL droplet of the sample was pipetted onto a glow discharged Quantifoil or lacey carbon coated copper grid. The sample was blotted once with filter paper for 2 seconds before being plunged into liquid nitrogen cooled liquid ethane. The samples were placed in a Gatan cryo-holder and were kept below -170°C throughout imaging. Samples were imaged using low-dose mode.
Supplementary Material
Acknowledgments
We thank B. Lin for critical comments on the manuscript and T. Neumann for help with AFM in air. This work was supported by National Heart, Lung and Blood Institute grant 5 U54 CA119335-04, the MRSEC Program of the National Science Foundation under award DMR05-20415 and NIH grant P41RR02250.
Footnotes
SUPPORTING INFORMATION PARAGRAPH Additional cryo-TEM (Figures S1, S2), AFM (Figure S3) images of C16p53 PAs and AFM (Figure S4) and negative stain TEM images of C16A4p53 (Figure S5) are presented. This material is available free of charge via the Internet at http://pubs.acs.org.
Contributor Information
Dimitris Missirlis, Email: missirlis@uni-heidelberg.de.
Arkadiusz Chworos, Email: achworos@cbmm.lodz.pl.
Caroline J. Fu, Email: cjfu@bcm.edu.
Htet A. Khant, Email: htet.khant@bcm.edu.
Daniel V. Krogstad, Email: dkrogstad@engineering.ucsb.edu.
Matthew Tirrell, Email: mvtirrell@berkeley.edu.
References
- 1.Zhang S. Nat Biotechnol. 2003;21:1171–1178. doi: 10.1038/nbt874. [DOI] [PubMed] [Google Scholar]
- 2.Zhao X, Pan F, Xu H, Yaseen M, Shan H, Hauser CA, Zhang S, Lu JR. Chem Soc Rev. 2010;39:3480–3498. doi: 10.1039/b915923c. [DOI] [PubMed] [Google Scholar]
- 3.Versluis F, Marsden HR, Kros A. Chem Soc Rev. 2010;39:3434–3444. doi: 10.1039/b919446k. [DOI] [PubMed] [Google Scholar]
- 4.Missirlis D, Farine M, Kastantin M, Ananthanarayanan B, Neumann T, Tirrell M. Bioconjug Chem. 2010;21:465–475. doi: 10.1021/bc900383m. [DOI] [PubMed] [Google Scholar]
- 5.Shimada T, Lee S, Bates FS, Hotta A, Tirrell M. J Phys Chem B. 2009;13:13711–13714. doi: 10.1021/jp901727q. [DOI] [PubMed] [Google Scholar]
- 6.Niece KL, Hartgerink JD, Donners JJ, Stupp SI. J Am Chem Soc. 2003;125:7146–7147. doi: 10.1021/ja028215r. [DOI] [PubMed] [Google Scholar]
- 7.Hartgerink JD, Beniash E, Stupp SI. Proc Natl Acad Sci U S A. 2002;99:5133–5138. doi: 10.1073/pnas.072699999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Anderson JM, Andukuri A, Lim DJ, Jun HW. ACS Nano. 2009;3:3447–3454. doi: 10.1021/nn900884n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stendahl JC, Rao MS, Guler MO, Stupp SI. Adv Funct Mater. 2006;16:499–508. [Google Scholar]
- 10.Greenfield MA, Hoffman JR, de la Cruz MO, Stupp SI. Langmuir. 2010;26:3641–3647. doi: 10.1021/la9030969. [DOI] [PubMed] [Google Scholar]
- 11.Paramonov SE, Jun HW, Hartgerink JD. J Am Chem Soc. 2006;128:7291–7298. doi: 10.1021/ja060573x. [DOI] [PubMed] [Google Scholar]
- 12.Kussie PH, Gorina S, Marechal V, Elenbaas B, Moreau J, Levine AJ, Pavletich NP. Science. 1996;274:948–953. doi: 10.1126/science.274.5289.948. [DOI] [PubMed] [Google Scholar]
- 13.Blaber M, Zhang XJ, Matthews BW. Science. 1993;260:1637–1640. doi: 10.1126/science.8503008. [DOI] [PubMed] [Google Scholar]
- 14.Numata K, Kaplan DL. Adv Drug Deliv Rev. 2010;62:1497–1508. doi: 10.1016/j.addr.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rajangam K, Behanna HA, Hui MJ, Han X, Hulvat JF, Lomasney JW, Stupp SI. Nano Lett. 2006;6:2086–2090. doi: 10.1021/nl0613555. [DOI] [PubMed] [Google Scholar]
- 16.Tovar JD, Claussen RC, Stupp SI. J Am Chem Soc. 2005;127:7337–7345. doi: 10.1021/ja043764d. [DOI] [PubMed] [Google Scholar]
- 17.Beniash E, Hartgerink JD, Storrie H, Stendahl JC, Stupp SI. Acta Biomater. 2005;1:387–397. doi: 10.1016/j.actbio.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 18.Altman M, Lee P, Rich A, Zhang S. Protein Sci. 2000;9:1095–1105. doi: 10.1110/ps.9.6.1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chiti F, Dobson CM. Annu Rev Biochem. 2006;75:333–366. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]
- 20.Meijer JT, Roeters M, Viola V, Lowik DWPM, Vriend G, van Hest JCM. Langmuir. 2007;23:2058–2063. doi: 10.1021/la0625345. [DOI] [PubMed] [Google Scholar]
- 21.Hung AM, Stupp SI. Langmuir. 2009;25:7084–7089. doi: 10.1021/la900149v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cui H, Pashuck ET, Velichko YS, Weigand SJ, Cheetham AG, Newcomb CJ, Stupp SI. Science. 2010;327:555–559. doi: 10.1126/science.1182340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pashuck ET, Cui H, Stupp SI. J Am Chem Soc. 2010;132:6041–6046. doi: 10.1021/ja908560n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jiang HZ, Guler MO, Stupp SI. Soft Matter. 2007;3:454–462. doi: 10.1039/b614426h. [DOI] [PubMed] [Google Scholar]
- 25.Nyrkova IA, Semenov AN, Aggeli A, Boden N. Eur Phys J B. 2000;17:481–497. [Google Scholar]
- 26.Dai Q, Dong M, Liu Z, Prorok M, Castellino FJ. J Inorg Biochem. 2011;105:52–57. doi: 10.1016/j.jinorgbio.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Muraoka T, Cui H, Stupp SI. J Am Chem Soc. 2008;130:2946–2947. doi: 10.1021/ja711213s. [DOI] [PubMed] [Google Scholar]
- 28.Cui H, Muraoka T, Cheetham AG, Stupp SI. Nano Lett. 2009;9:945–951. doi: 10.1021/nl802813f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pashuck ET, Stupp SI. J Am Chem Soc. 2010;132:8819–8821. doi: 10.1021/ja100613w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Castelletto V, Hamley IW, Hule RA, Pochan D. Angew Chem Int Ed. 2009;48:2317–2320. doi: 10.1002/anie.200805500. [DOI] [PubMed] [Google Scholar]
- 31.Aggeli A, Nyrkova IA, Bell M, Harding R, Carrick L, McLeish TC, Semenov AN, Boden N. Proc Natl Acad Sci U S A. 2001;98:11857–11862. doi: 10.1073/pnas.191250198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Missirlis D, Khant H, Tirrell M. Biochemistry. 2009;48:3304–3314. doi: 10.1021/bi802356k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Berndt P, Fields GB, Tirrell M. J Am Chem Soc. 1995;117:9515–9522. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.