

Abstract
The lipoglycopeptide antibiotic teicoplanin has proven efficacy against gram-positive pathogens. Teicoplanin is distinguished from the vancomycin-type glycopeptide antibiotics, by the presence of an additional cross-link between the aromatic amino acids 1 and 3 that is catalyzed by the cytochrome P450 monooxygenase Orf6* (CYP165D3). As a goal towards understanding the mechanism of this phenol-coupling reaction, we have characterized recombinant Orf6* and determined its crystal structure to 2.2 Å resolution. Although the structure of Orf6* reveals the core fold common to other P450 monooxygenases, there are subtle differences in the disposition of secondary structure elements near the active site cavity necessary to accommodate its complex heptapeptide substrate. Specifically, the orientation of the F and G helices in Orf6* results in a more closed active site than found in the vancomycin oxidative enzymes OxyB and OxyC. In addition, Met226 in the I helix replaces the more typical Gly/Ala residue that is positioned above the heme porphyrin ring, where it forms a hydrogen bond with a heme iron-bound water molecule. Sequence comparisons with other phenol-coupling P450 monooxygenases suggest that Met226 plays a role in determining the substrate regiospecificity of Orf6*. These features provide further insights into the mechanism of the cross-linking mechanisms that occur during glycopeptide antibiotics biosynthesis.
Keywords: Glycopeptide antibiotic, vancomycin, teicoplanin, cytochrome P450 monooxygenase
Introduction
Glycopeptide antibiotics exert their antimicrobial activity against gram-positive bacteria by targeting the N-acyl-D-Ala-D-Ala peptide termini of peptidoglycan precursors and inhibiting cell wall synthesis1. One representative of this class of antibacterials, vancomycin (compound 1 in Fig. 1), has been approved for clinical use and is used as the “last-resort” treatment against multiple-drug-resistant bacteria2, including methicillin-resistant Staphylococcus aureus (MRSA)3,4 and drug-resistant pneumococci5. However, a considerable number of vancomycin-resistant bacteria have emerged, including vancomycin-resistant enterococci (VRE)6 and vancomycin-intermediate and -resistant S. aureus (VISA and VRSA, respectively)7,8. The lipoglycopeptide antibiotic teicoplanin (compound 3 in Fig. 1) has proven efficacy against MRSA and VRSA9, and has shown superior pharmacokinetic profile in comparison to vanocymin10. The ability of teicoplanin to counter vancomycin resistant pathogens has prompted research efforts towards the design of lipoglycopeptide derivatives with increased antimicrobial properties relative to the parent compounds.
Figure 1.

Chemical structures of type I (1 vancomycin, 2 balhimycin), and type IV (3 teicoplanin, 4 A47934, and 5 A40926) glycopeptide antibiotics.
The general structure of glycopeptide and lipoglycopeptide antibiotics consists of a heptapeptide backbone containing aromatic amino acids whose side chains are oxidatively coupled via biaryl or biarylether bridges to generate an aglycon core11 (Fig. 1). These cross-links force the peptide backbone into a rigid conformation that is crucial to the primary antibiotic activity of glycopeptide antibiotics12. Previous studies have shown that linear or various mono-cyclic or bi-cyclic intermediates of the corresponding antibiotic have minimal antimicrobial activity13-17. The cross-linked aglycon undergoes further tailoring modifications including N-methylation, halogenation, glycosylation, and/or acylation18,19.
The elucidation of biosynthesis gene clusters of several glycopeptides has established the general route of the complex structure assembly20-23. The building blocks, which are mostly non-proteinogenic amino acids, are assembled by nonribosomal peptide synthetase (NRPS)1. The electron-rich aromatic side chains facilitate oxidative cross-linking reactions, which are carried out by a series of cytochrome P450 monooxygenases (P450s). For example, in the vancomycin-type (type I) glycopeptide antibiotic balhimycin (compound 2 in Figure 1) system, three heme-dependent P450s (OxyA, OxyB, OxyC) are responsible for the three cross-links in the heptapeptide substrate13,14. Genetic and chemical analyses have defined the regiospecificity and timing of the oxidative phenol-coupling reactions in the balhimycin cluster13,14,17. The first cross-linking occurs between residues 4 and 6 (C-O-D ring) and is catalyzed by OxyB13,14,17, the second is between residues 2 and 4 (D-O-E ring) and is catalyzed by OxyA, and the third is between residues 5 and 7 (AB ring) and is catalyzed by OxyC13,14,17. It has been suggested that the cross-linking reactions occur when the heptapeptide is still attached to the peptidyl carrier domain (PCD) as a thioester through its carboxy-terminus24. This is supported by the in vitro activity studies of OxyB25, using peptide substrates that are conjugated to a PCD domain.
A distinguishing feature of teicoplanin-type (type IV) glycopeptide antibiotics is an additional cross-link between residues 1 and 3 (F-O-G ring), which, in contrast to vancomycin-like glycopeptides, consists of non-proteinogenic aromatic amino acids26 (Fig. 1). Consequently, there are four P450s in biosynthetic systems of teicoplanin and related lipoglycopeptides12,21-23, namely orf5*, 6*, 7*, and 9* in the teicoplanin cluster 22. While the different oxidative enzymes from the same biosynthetic cluster show only modest sequence identity (i.e. less than 40%), orthologous enzymes from different species are highly similar. For example, Orf5*, 7*, and 9* from the tecioplanin gene cluster show strong sequence correspondence to OxyA, OxyB, and OxyC from the vancomycin cluster (similarity: 90%, 80%, 84%; identity: 78%, 72%, and 74%). This conservation has been used to infer function for the teicoplanin oxidative enzymes in catalyzing the cross-link between residues 2-4, 4-6, and 5-7, respectively22. The remaining P450, Orf6* (CYP165D3), is likely to be responsible for the additional cross-link between residue 1 and 3 that is unique to the teicoplanin aglycone22. Gene-knockout studies on the teicoplanin-type glycopeptide antibiotic A47934 (compound 4 in Figure 1) confirmed four P450 proteins that are responsible for the four cross-links in this type of compound and suggest a likely order of the four phenol-coupling reactions. Intriguingly, the additional 1-3 cross-link in A47934, catalyzed by the Orf6* orthologue StaG, is thought to be the second phenol-coupling reaction12.
Here we report the recombinant expression and characterization of Orf6* from the teicoplanin producer Actinoplanes teichomyceticus. We also present the 2.2 Å resolution crystal structure of Orf6*, which is the first structure for a P450 responsible for the additional 1-3 cross-link in the teicoplanin-type glycopeptide antibiotics. A comparison with the available structures of the phenol-coupling monooxygenases (OxyB and OxyC) in vancomycin system reveals a possible mechanism for Orf6* regiospecificity.
Materials and Methods
Cloning, Protein Expression, and Purification
Actinoplanes teichomyceticus was purchased from the American Tissue Culture Collection and used directly as a template for polymerase chain reaction amplifications without purification of the genomic DNA. The orf6* gene was amplified by polymerase chain reaction (PCR) using the following primers (restriction sites underlined): Orf6*_fwd, 5′-AAGCAGCCGCATATGGCACTTCCTCTGCCGCATCAGCGG-3′, Orf6*_rev, 5′-CTAGCTCGAGTCACCACTCAAGGGGAAGCTCCTC-3′. The PCR products were digested with NdeI and XhoI, and ligated into NdeI/XhoI digested pET28b (Novagen) to give the overexpression plasmid pET28-orf6* with an amino-terminal hexahistidine affinity tag.
For protein production, pET28-orf6* was transformed into the Escherichia coli Rosetta (DE3) strain. A pre-culture is grown overnight at 37° C in Terrific Broth (TB) with kanamycin (50 μg/ml) and chloramphenicol (34 μg/ml) and used to inoculate (4%, v/v) 400 ml of TB medium with the antibiotics and the resultant culture was grown at 37° C in shaker flasks. When the optical density at 600 nm (OD600) reached 0.5, δ-aminolevulinic acid was added to 0.15 mM. Protein production was induced at OD600 of 1.0 with the addition of 0.5 mM isopropyl-1-thio-β-D-galactopyranoside and growth was continued at 18° C. A second aliquot of δ-aminolevulinic acid was added (0.15 mM) 20 hours after induction. After another 24 hours the culture was harvested by centrifugation at 3,500 rpm for 25 minutes and the resultant pellet was resuspended in 20 mM Tris, pH 8.0, 1 M NaCl, 30 mM imidazole, and 10% (v/v) glycerol. Harvested cells were disrupted by four passes through an Avestin C5 Emulsiflex French Press and insoluble aggregates and cellular debris were removed by centrifugation at 15, 000 rpm for 1 hour.
Recombinant Orf6* from the above-clarified supernatant was applied to a 10 ml Talon resin (Clontech) column that was charged with nickel sulfate and pre-equilibrated with 20 mM Tris, pH 8.0, 1 M NaCl, 30 mM imidazole. After elution from the nickel affinity resin with 200 mM imidazole, the polyhistidine tag was removed with thrombin (1 unit/mg, MP Biomedicals). As for most bacterial P450s, and as evidenced by carbon monoxide difference spectrum, imidazole elution from the nickel affinity column does not affect the folding of the P450 protein. The protein was dialyzed into 20 mM Tris, pH 8.0, 50 mM NaCl and applied to a 5 ml HiTrap Q column (GE Healthcare), and then eluted with a linear gradient of NaCl from 50 mM to 1 M over 60 ml. Orf6* was further purified by size-exclusion chromatography (Superdex 75 16/60, GE Healthcare) prior to crystallization. Selenomethionine-incorporated Orf6* was produced by the method of van Duyne et al.27 and purified in the same manner as described above. Protein concentration was estimated by Bradford assay. The final yield for both native and selenomethionine-incorporated Orf6* was approximately 10 mg/L.
UV-visible spectroscopy
The UV-visible absorption spectra for Orf6* were measured with a Varian Cary 3 double-beam spectrophotometer. After thermal equilibration at 25 °C the baseline was zeroed between 200 and 900 nm and the UV-visible spectra of the substrate-free Orf6* (0.1 mg/ml in 20 mM HEPES, pH 7.5, 100 mM KCl) were recorded. The protein was reduced by addition of 2 mg of solid sodium dithionite and spectra were measured under the same condition. The reduced protein was divided into two tandem cuvettes (sample and reference) and CO was bubbled through the sample cuvette for 30 seconds, then the difference spectrum was measured. The protein-imidazole complex was generated by adding 100 mM imidazole to Orf6* in the same buffer and spectra were measured under the same condition.
Crystallization
Orf6* crystals were grown by the hanging drop vapor diffusion method. 1 μl (8 mg/ml in a solution containing 20 mM HEPES, pH 7.5, 100 mM KCl) protein was mixed with 1 μl precipitant solution containing 0.2 M sodium acetate, 0.1 M Tris, pH 8.5, and 20% (w/v) polyethylene glycol 4, 000. The mixture drop was equilibrated over a well containing the same precipitant solution at 8 °C, and crystals reached their maximum size after 3 days. Selenomethionine-incorporated Orf6* was crystallized under the same condition. The crystal was soaked in cryoprotectant solution containing the precipitant solution supplemented with 30% (v/v) glycerol anhydrous prior to vitrification in liquid nitrogen.
Phasing and Structure Determination
Crystals of selenomethionine-incorporated Orf6* (SeMet Orf6*) consistently diffracted to higher resolutions, and were subsequently used for data collection and structure refinement. Diffraction data were collected to 2.2 Å resolution at an insertion device line (LS-CAT-Sector 21 ID-D, Advanced Photon Source, Argonne, IL), and integrated and scaled using the HKL200028. SeMet Orf6* crystals display the symmetry of space group P31 with unit cell parameters a = 74.3 Å, b = 74.3 Å, c = 75.5 Å and contain one molecule per asymmetric unit. The 2.2 Å resolution data set had four-fold redundancy with an overall Rmerge 0.066, ad I/σ(I) = 4.8 in the highest resolution shell.
Selenium sites were identified using HySS29,30 and heavy atom parameters were further refined using PHENIX29,30 to yield an initial figure of merit of 0.390 to 2.2 Å resolution. Solvent flattening further improved the quality of the initial map permitting 80% of the main chain and 35% of the side chain residues to be automatically built by PHENIX. This initial model was further improved by automated building using ARP/wARP31 to yield a model with nearly all of the main chain and 70% of the side chains. The remainder of the model was fitted using XtalView32 and further improved by rounds of refinement with REFMAC533 and manual building. Multiple rounds of manual model building were interspersed with refinement using REFMAC5 to complete structure refinement. Cross-validation used 5% of the data in the calculation of the free R factor34. The stereochemistry of the model was routinely monitored throughout the course of refinement using PROCHECK35. Relevant data collection and refinement statistics are summarized on Table 1.
Table 1. Data collection, phasing and refinement statistics.
| SeMet Orf6* | |
|---|---|
| Data collection | |
| Space group | P31 |
| Cell dimensions | |
| a, b, c (Å) | 74.3, 74.3, 75.5 |
| Resolution (Å) | 50-2.2 (2.28-2.2)a |
| Rsym (%) | 6.6 (29.7) |
| I / σ(I) | 28.8 (4.8) |
| Completeness (%) | 96.2 (74.0) |
| Total reflections | 79,905 |
| Unique reflections | 22,760 |
| Redundancy | 3.5 (3.1) |
| Wilson B factor | 33.4 |
| Refinement | |
| Resolution (Å) | 25.0-2.2 |
| Number of reflections | 21,363 |
| Rwork / Rfreeb | 20.1%/25.7% |
| Number of atoms | |
| Protein | 2898 |
| Solvent | 184 |
| Heme | 43 |
| Average B value | |
| Protein | 30.8 |
| Solvent | 37.2 |
| Heme | 25.2 |
| R.m.s deviations | |
| Bond angles (Å) | 1.29 |
| Bond lengths (°) | 0.011 |
Highest resolution shell is shown in parenthesis.
R-factor = Σ(|Fobs|-k|Fcalc|)/Σ |Fobs|and R-free is the R value for a test set of reflections consisting of a random 5% of the diffraction data not used in refinement.
Results
UV-visible Spectroscopy
Amino-terminally hexahistidine-tagged Orf6* was heterologously expressed in recombinant E. coli Rosetta (DE3) and purified from the soluble fraction by Ni-NTA affinity chromatography, and was further purified by anion-exchange and size-exclusion chromatography after thrombolytic removal of the affinity tag. As shown in Fig. 2A, Orf6* gave characteristic UV-visible absorption spectra of P450 hemeprotein36. Specifically, the UV-visible absorption spectrum of substrate-free Orf6* has a Soret band at 419 nm, and β and α peaks at 536 and 570 nm, respectively, as is typical of oxidized P450s in the low-spin ferric state. Reduction of substrate-free Orf6* with sodium dithionite shifts the Soret band to 424 nm, and β and α peaks at 530 and 565 nm, respectively. The CO difference spectrum of reduced protein shows a prominent peak at 450 nm (Fig. 2B). These spectra show that the purified Orf6* is in the oxidized form, and can be reduced with sodium dithionite. Addition of imidazole to substrate-free Orf6* leads to a typical red shift in the Soret band to 429 nm, and β and α peaks to 543 and 565 nm indicating that imidazole binds to the heme iron (III) atom as an axial ligand37,38, a typical feature of P450 hemeproteins39.
Figure 2.
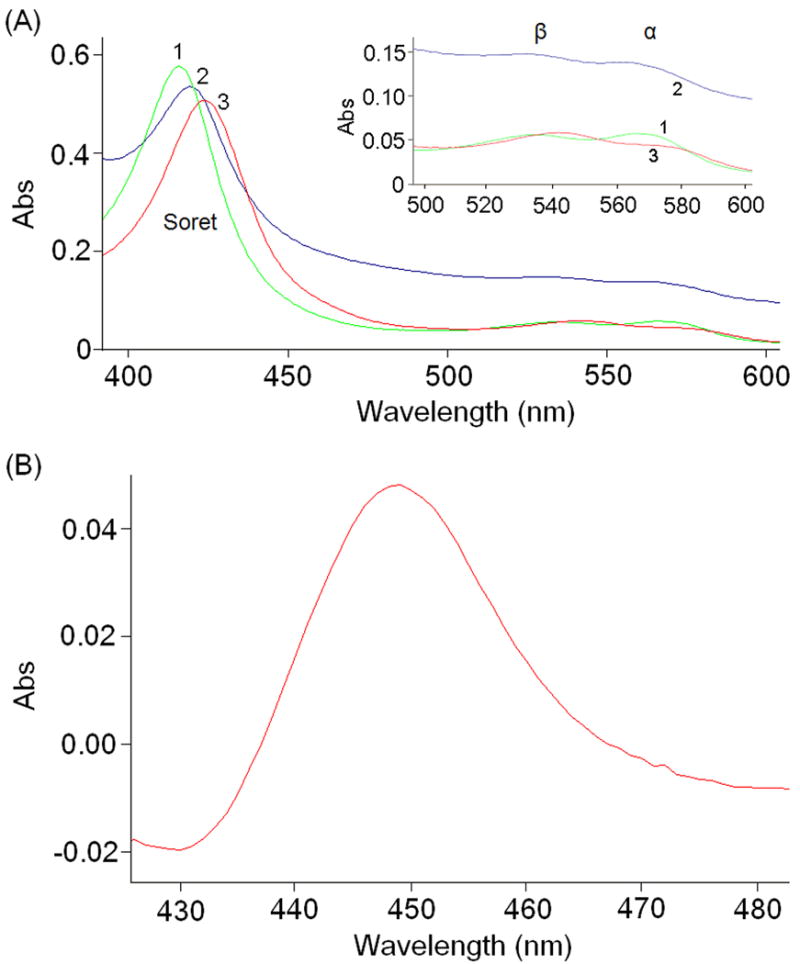
UV-visible absorption spectra of Orf6*. (A) Curve 1 (green) shows the spectrum of substrate-free Orf6*; Curve 2 (blue), after reduction with sodium dithionite; and Curve 3 (red), after addition of imidazole. (B) The CO-difference spectrum of reduced Orf6* showing the characteristic absorbance at 450 nm.
Overall Structure of Orf6*
The structure of Orf6* was determined to 2.2 Å using single-wavelength anomalous scattering from crystals grown with selenomethionine-incorporated protein (relevant data collection and refinement statistics are given in Table 1). As shown in Figs. 3A and B, Orf6* exhibits the typical overall structure and protein topography of most P450 heme-proteins40. The protein has a triangular prism shape with predominantly α-helical secondary structure (see Fig. 3B for a structure-based sequence alignment and secondary structure assignment) and the heme prosthetic group is embedded between the proximal L and distal I helices. The structure comprises 15 α-helices and 6 β-stands, and it can be roughly divided into α-rich and β-rich regions. When compared with the crystal structures of other P450s using DALI41, Orf6* shows the highest structural similarity to OxyB37, OxyC38, P450nor42, CYP105P143, and CYP105D644, among the substrate-free wild-type P450 structures (Table 2).
Figure 3.

(A) Ribbon diagram showing the overall structure of Orf6*. Important secondary structural elements discussed in the text include F and G helices (colored in pink), I helix (colored in blue), and Cys loop (colored in red). The heme is shown in yellow sticks and the iron atom as a red sphere. Other secondary structural elements are colored in cyan. (B) Structure-based multiple sequence alignment of the type IV glycopeptide cross-linking cytochrome P450 monooxygenases Orf6* (teicoplanin), StaG (A47934) and Dbv13 (A40926) with those of the type I glycopeptide monooxygenases OxyB and OxyC (both from the vancomycin cluster). Residues that are unique to Orf6* and may be catalytically relevant are shown with a green arrowhead.
Table 2. Similarity of structure and sequence between Orf6* and other P450s.
| P450 namea | PDBb code | Z-score | RMSDc Å | Sequence identity % | FGd conformation relative to Orf6* |
|---|---|---|---|---|---|
| OxyB | 1lg9 | 44.9 | 2.2 | 38 | Open |
| CYP105 (MES) | 2z36 | 44.8 | 1.9 | 32 | Same |
| OxyC | 1ued | 44.7 | 2.1 | 37 | Open |
| CYP105P1 (FLI) | 3aba | 43.8 | 2.5 | 36 | Close |
| CYP105P1 (PIM) | 3e5k | 43.6 | 2.0 | 35 | Same |
| P450nor | 1rom | 43.4 | 2.0 | 26 | Same |
| CYP105P1 | 3e5j | 43 | 2.1 | 35 | Same |
| CYP105D6 | 3abb | 42.8 | 1.9 | 33 | Same |
P450s that show the highest structural similarity to Orf6*. Shown in parenthesis are bound substrates/ligands. MES: 2-(N-morpholino)-ethanesulfonic acid; FLI: Filipin I; PIM: 4-Phenyl-1H-imidazole.
Protein Data Bank.
Root mean square deviation.
F and G helices of the corresponding P450s.
Active Site Architecture
Orf6* shows the conserved four-helix bundle core, formed by D, E, I, and L helices, with the heme group confined between the I and L helices (Fig. 4A), and above the Cys-ligand loop adjacent to the amino-terminus of the L-helix. This Cys-ligand loop contains the P450 signature amino acid sequence FxxGxHxCxG, with the absolutely conserved Cys332 being the proximal ligand to the heme iron. The iron atom is in the heme plane and its distance to the thiolate sulfur of Cys332 is 2.3 Å. A solvent molecule, located 2.6 Å from the heme iron, completes the coordination shell (Fig. 4A), consistent with the low spin heme state of substrate-free Orf6* observed in the UV-visible spectra (Fig. 2).
Figure 4.
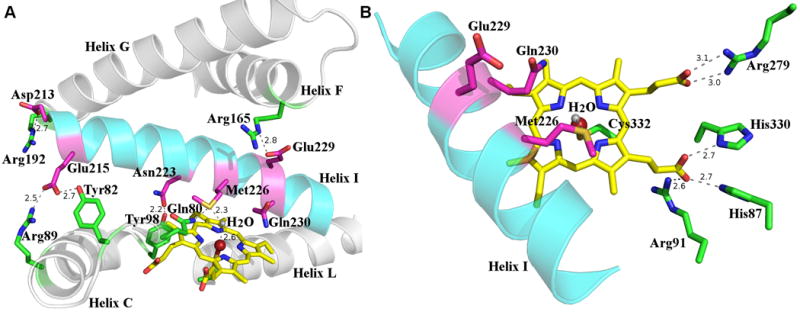
(A) Interactions between residues from I helix and those from other secondary structural elements. The I helix is shown in cyan and residues are colored in magenta, other helices are shown in gray and residues in these helices are colored in green. The heme is shown as a yellow stick, the iron shown as a maroon sphere, and the iron bound water as a gray sphere. Hydrogen bond interactions are shown as dash lines, next to which the distances are shown. (B) Active site of Orf6* showing critical catalytic residues and those interacting with the heme propionate groups. The I helix is shown in cyan, catalytically relevant residues conserved among type IV glycopeptide P450 monooxygenases are shown in magenta, Cys332 and other heme interacting residues shown in green. Hydrogen bond interactions are shown as dash lines, next to which the distances are shown.
The long I-helix spans the entire catalytic site and forms a wall above the heme pocket. In most P450s, two highly conserved and catalytically important residues, an acidic residue immediately followed by a threonine, are usually found in the I-helix over the pyrrole ring B of the heme group40,45. In contrast, in Orf6*, the acidic residue is Glu229 but the residue immediately to its carboxy-terminus is Gln230 (Fig. 4B). This variation is not unprecedented as the corresponding residues in OxyB are Asp239-Asn240, with the latter forming hydrogen bonds with two water molecules in the active site37. Although the side chain of Gln230 in Orf6* points to the heme group as in OxyB, there are no water molecules within hydrogen bonding distance to its backbone or side chain. This sequence variation is conserved at the corresponding place in the sequences of most of the phenol-coupling P450s involved in glycopeptide antibiotics biosynthesis (Fig. 3C). The Gln230 side chain might play a similar role to the hydroxyl group of the usual threonine in forming a proton delivery channel during catalysis45 or be involved in hydrogen bond interactions with the extended peptide substrate.
In striking contrast to the small neutral amino acid (glycine or alanine) typically found three residues before the conserved acidic residue in the I-helix of P450s37,38,42,46, Orf6* contains a methionine residue (Met226) (Fig. 4B) at this position. The side chain of Met226 points into the active site and the sulfur atom forms a hydrogen bond with the heme iron-coordinating water molecule. The function of Met226 is not yet clear, as a corresponding methionine residue is found in S. toyocaensis StaG but not in Nonomuraea sp. Dbv13 (Fig. 3C), both of which are P450s cross-linking the side chains of the aromatic residues 1 and 3 in the type IV glycopeptide antibiotics A47934 and A4092612,21, analogous to the Orf6* cross-linking reaction in teicoplanin biosynthesis22. All other putative phenol-coupling P450s in glycopeptide antibiotics biosynthesis have the more typical glycine or alanine at this position. The conservation of this methionine residue suggests that it plays a role in regiospecificity of the 1-3 cross-linking reaction. Attempts to carry out mutational analysis at Met226 are limited by the fact that attempts to produce the synthetic, singly cross-linked, heptapeptide substrate coupled to a PCD domain have thus far been unsuccessful.
The active site conformation of Orf6* is stabilized by a number of hydrogen bonds between residues from I-helix and those from other secondary structure elements (Fig. 4A). Hydrogen bonds from the side chain carboxylate oxygens of the I-helix residues Glu229 and Asp213 to the backbone amides of the F-helix residue Arg165 and the G-helix residue Arg192, mediate the interaction of the I-helix with the F and G helices. The side chain carboxylate oxygens of Glu215 from the I-helix form hydrogen bonds with the guanidine nitrogen atom of Arg89 from the N-terminal portion of the C-helix, and the hydroxyl group of Tyr82 from the B′-C loop, respectively. The C-D loop forms interaction with the I-helix through the hydrogen bond between the hydroxyl group of Tyr98 and the side chain carbonyl oxygen of the I-helix residue Asn223.
Substrate Binding Pocket Comparison with Known P450s
A significant difference between the structures of Orf6* and other known P450s is in the orientation of F and G helices, which are believed to be important for substrate binding40,45,47. Although the F and G helices in Orf6* are similar to those in OxyB37 and OxyC38 in terms of length and fold, their relative orientations are significantly different. In particular, in Orf6* the F and G helices are rotated toward the active site, resulting in a much more closed substrate binding pocket (Figs. 5A, 5B, S1 and S2). This is unexpected because the Orf6* substrate, teicoplanin heptapeptide22, is nearly the same size as the vancomycin heptapeptide substrate of OxyB and OxyC16,20,37 (Fig. 5C). While the relative orientation of the F and G helices are similar in OxyB and OxyC (as would be expected given that they both work on the same substrate), the orientation of the F and G helices in Orf6* is much more similar to that of P450nor42, a nitric acid reductase whose substrate is substantially smaller compared to the teicoplanin aglycone substrate of Orf6*.
Figure 5.

(A) and (B) Orthogonal views of a least squared superposition of the crystal structure of Orf6* (in pink) with those of P450nor (in blue) and OxyB (in green). Note that the F and G helices of Orf6* and OxyB deviate significantly, despite the fact that both enzymes work on large, peptidic substrates. The F-G helices of Orf6* are in a closed conformation, similar to those observed in structures of P450s that work on small molecule substrates (e.g., P450nor). (C) A comparison of the substrates of Orf6*, OxyB and OxyC, which may provide a rationale for the positioning of the F and G helices in these enzymes. Existing cross-links in the substrate are shown as shaded circles and an arrow indicates the target site for each enzyme.
Prior analysis of the substrate binding pockets of OxyB and P450nor lead to the speculation that the larger size of the OxyB substrate dictated the need for the more open binding pocket37. However, our structure of Orf6* suggests that the binding pocket capped by the F and G helices may not only be determined by the substrate size, but also its orientation. Orf6* catalyzes the cross-linking at the amino-terminus of the teicoplanin heptapeptide22, while the target of OxyB is the bulky center of the vancomycin heptapeptide16,20,37. Consequently, the heptapeptide substrate must be positioned in the active site in substantially different orientations to afford the regiospecificities of each individual enzyme. While OxyC carries out the coupling reaction at the carboxy-terminus of the vancomycin heptapeptide, the open conformation of this enzyme can also be understood in the context of substrate orientation. Previous gene inactivation studies have suggested that the OxyC reaction is the last coupling reaction in vancomycin biosynthesis16,38, so its substrate, which is cross-linked between residues 2-4 and 4-6, has a rigid wide conformation that requires an open binding site (Fig. 5C). In contrast, genetic studies of A47934 biosynthesis12,21 imply that Orf6* catalyzes the second coupling reaction in teicoplanin biosynthesis. As a result, the Orf6* substrate is cross-linked only between residues 4-6 and the lack of a rigid structure in this substrate can be accommodated by a closed binding pocket in Orf6*.
In the Orf6* structure, the side chain of Glu229 in the I-helix forms a hydrogen bond with the backbone amide group of Arg165 from the F-helix (Figure 4A). This interaction likely stabilizes the inward conformation of F-helix, which in turn rotates the G-helix toward the active site. Such an interaction is not found in OxyB, OxyC, or P450nor but the Glu and Arg residues are conserved in both StaG and Dbv13 (Fig. 3C), both of which catalyze similar 1-3 coupling during the biosynthesis of the teicoplanin-type glycopeptide antibiotics A47934 and A40926, respectively. The F and G helices of Orf6* might undergo a further closure of the active site upon substrate binding, as has been observed in some structurally similar P450s (Table 2), such as CYP105P1 that binds the antifungal macrolide antibiotic filipin43,44.
Implications for Catalysis on PCD-coupled Substrate
Studies by Robinson and co-workers established that such oxidative coupling reaction only occurs on substrate peptides attached as thioesters to the NRPS module24. In the presence of a suitable electron source, purified recombinant OxyB can catalyze the 4-6 crosslink of a heptapeptide substrate conjugated to a peptide carrier domain (PCD) from the cognate NRPS through a thioester linkage25. As the Orf6* 1-3 cross-linking reaction occurs at the amino-terminus of the peptide substrate, and the PCD conjugation is through a carboxy-terminal linkage, the substrate can be correctly registered in the active site without requiring neither the PCD domain nor the phosphopantetheine arm to be substantially wedged into the binding pocket. In contrast, for the OxyB and OxyC reactions that occur at the center and carboxy-terminus of the peptide, their substrates are accommodated in an open binding pocket that is necessary to fit significant portions of both the heptapeptide substrate as well as the phosphopantetheine group (Fig. 5C).
In order to gain further insights into the determinants of substrate recognition, we docked the co-crystal structure of the P450(Biol)-tetradecanoic acid-acyl carrier protein (ACP) complex48 onto that of Orf6* (Figs 6A, 6B). Despite the fact that P450(Biol) and Orf6* share less than 40% sequence identity, the ACP can be docked onto the structure of Orf6* without significant steric clashes. Although the PCD-peptide conjugate substrate of Orf6* is distinct from the tetradecanoic acid-ACP substrate of P450(Biol), this model yields a likely trajectory for the teicoplanin aglycone backbone. In this model, the unique Met226 present in Orf6* is located proximal to predicted locations of substrate residues 1 and 3, suggesting that this residue stabilizes the orientation of the substrate backbone to facilitate cross-linking (Fig. 6B). The sulfur atom Met226 may form hydrogen bonds with the phenol hydroxyls on either residues 1 and/or residue 3 of the substrate aglycone. Similarly, residue Gln230, which is conserved only among P450s that catalyze 1-3 cross-links on glycopeptides, may form hydrogen bonds with the phenol hydroxyls on residues 2 or 4 to stabilize the extended substrate prior to the coupling reaction.
Figure 6.

(A) Structure of the P450(Biol)-ACP-tetradecanoic acid complex near the vicinity of the active showing the F, G, and I helices (in pink), the ACP-bound substrate (in green) and catalytically important residues (in magenta). (B) A docking model of the putative Orf6* complex with the F, G, and I helices colored in cyan. This model suggests that Met226 and Gln230, which are conserved only among Orf6* orthologues, may play roles in steering and stabilizing the extended peptide substrate.
Discussion
In order to facilitate the development of novel glycopeptide antibiotics, we have focused on the cytochrome P450 monooxygenase Orf6* that is suggested to be the second coupling oxygenase in the biosynthesis of teicoplanin. Recombinant Orf6* yields a UV-visible absorption spectra characteristic of P450 heme-proteins, and indicates the existence of a low spin heme state for the substrate-free enzyme. The oxidized form of Orf6* can be reduced by sodium dithionite and bound via its heme iron (III) atom to imidazole. The 2.2 Å resolution crystal structure of Orf6* exhibits a typical P450-fold with a triangular prism shape. Among the substrate-free structures of known wildtype P450s, OxyB, OxyC, P450nor, and CYP105D6 show the highest similarity to Orf6*.
Sequence comparisons of all the phenol-coupling P450s involved in glycopeptide antibiotics biosynthesis suggest primary sequence conservations that may reflect substrate preferences of this subgroup of P450s. Among these, residues Arg165, and Gln230 are conserved only in Orf6*, Dbv13, and StaG, which catalyze the extra 1-3 cross-link found in type IV glycopeptides. While Met226 is also unique to Orf6* and StaG, this residue is a glycine in Dbv13, and the implications of this change in unclear. The conservations of these residues likely reflect their importance in stabilizing the extended peptide substrate of these particular P450 enzymes. The corresponding residues in OxyB, which catalyzes the 4-6 cross-link, are Leu174, and Asn240, and these residues are absolutely conserved among all 4-6 cross-linking monooxygenases in the biosynthetic clusters of teicoplanin (Orf7*), A47934 (StaH), and A40926 (Dbv12). These patterns of conservation imply the importance of these residues in determining the regiospecificity and timing of the phenol-coupling P450s. Further analysis of structure-function relationships will require reconstitution of in vitro activities using various synthetic peptide substrates that have been singly cross-linked and then coupled to the appropriate peptide carrier domains.
While this manuscript was under review, Schlichting and co-workers have independently reported the atomic coordinates for the structure of CYP165D3 (Orf6*) (PDB Code 3O1A). The 2.5 Å resolution structure of CPY165D3 is nearly identical to that presented here.
Supplementary Material
Acknowledgments
We thank Dr. Huimin Zhao for the help with the UV-visible absorption spectra experiments, and Dr. Stephen G. Sligar, Dr. Ilia G. Denisov, and Dr. David R. Nelson for insightful discussion. We also thank Vinayak Argarwal for data collection, and Joseph S. Brunzelle and staff at Life Sciences Collaborative Access Team (21-ID at Argonne National Labs, Advanced Photon Source) for facilitating data collection. This work is supported by a grant from NIGMS.
Footnotes
Accession numbers: Coordinates and structure factors have been deposited in the Protein Data Bank with accession number 3OO3.
References
- 1.Kahne D, Leimkuhler C, Lu W, Walsh C. Glycopeptide and lipoglycopeptide antibiotics. Chem Rev. 2005;105:425–448. doi: 10.1021/cr030103a. [DOI] [PubMed] [Google Scholar]
- 2.Moellering RC., Jr Vancomycin: a 50-year reassessment. Clin Infect Dis. 2006;42 1:S3–4. doi: 10.1086/491708. [DOI] [PubMed] [Google Scholar]
- 3.Anstead GM, Owens AD. Recent advances in the treatment of infections due to resistant Staphylococcus aureus. Curr Opin Infect Dis. 2004;17:549–555. doi: 10.1097/00001432-200412000-00007. [DOI] [PubMed] [Google Scholar]
- 4.Anstead GM, Quinones-Nazario G, Lewis JS., 2nd Treatment of infections caused by resistant Staphylococcus aureus. Methods Mol Biol. 2007;391:227–258. doi: 10.1007/978-1-59745-468-1_17. [DOI] [PubMed] [Google Scholar]
- 5.Cunha BA. Antimicrobial therapy of multidrug-resistant Streptococcus pneumoniae, vancomycin-resistant enterococci, and methicillin-resistant Staphylococcus aureus. Med Clin North Am. 2006;90:1165–1182. doi: 10.1016/j.mcna.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 6.Murray BE. Vancomycin-resistant enterococcal infections. N Engl J Med. 2000;342:710–721. doi: 10.1056/NEJM200003093421007. [DOI] [PubMed] [Google Scholar]
- 7.Hiramatsu K, Hanaki H, Ino T, Yabuta K, Oguri T, Tenover FC. Methicillin-resistant Staphylococcus aureus clinical strain with reduced vancomycin susceptibility. J Antimicrob Chemother. 1997;40:135–136. doi: 10.1093/jac/40.1.135. [DOI] [PubMed] [Google Scholar]
- 8.Sieradzki K, Roberts RB, Haber SW, Tomasz A. The development of vancomycin resistance in a patient with methicillin-resistant Staphylococcus aureus infection. N Engl J Med. 1999;340:517–523. doi: 10.1056/NEJM199902183400704. [DOI] [PubMed] [Google Scholar]
- 9.Van Bambeke F. Glycopeptides and glycodepsipeptides in clinical development: a comparative review of their antibacterial spectrum, pharmacokinetics and clinical efficacy. Curr Opin Investig Drugs. 2006;7:740–749. [PubMed] [Google Scholar]
- 10.Malabarba A, Goldstein BP. Origin, structure, and activity in vitro and in vivo of dalbavancin. J Antimicrob Chemother. 2005;55 2:ii15–20. doi: 10.1093/jac/dki005. [DOI] [PubMed] [Google Scholar]
- 11.Donadio S, Sosio M, Stegmann E, Weber T, Wohlleben W. Comparative analysis and insights into the evolution of gene clusters for glycopeptide antibiotic biosynthesis. Mol Genet Genomics. 2005;274:40–50. doi: 10.1007/s00438-005-1156-3. [DOI] [PubMed] [Google Scholar]
- 12.Hadatsch B, Butz D, Schmiederer T, Steudle J, Wohlleben W, Sussmuth R, Stegmann E. The biosynthesis of teicoplanin-type glycopeptide antibiotics: assignment of p450 mono-oxygenases to side chain cyclizations of glycopeptide a47934. Chem Biol. 2007;14:1078–1089. doi: 10.1016/j.chembiol.2007.08.014. [DOI] [PubMed] [Google Scholar]
- 13.Bischoff D, Pelzer S, Bister B, Nicholson GJ, Stockert S, Schirle M, Wohlleben W, Jung G, Sussmuth RD. The Biosynthesis of Vancomycin-Type Glycopeptide Antibiotics-The Order of the Cyclization Steps. Angew Chem Int Ed Engl. 2001;40:4688–4691. doi: 10.1002/1521-3773(20011217)40:24<4688::aid-anie4688>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 14.Bischoff D, Pelzer S, Holtzel A, Nicholson GJ, Stockert S, Wohlleben W, Jung G, Sussmuth RD. The Biosynthesis of Vancomycin-Type Glycopeptide Antibiotics-New Insights into the Cyclization Steps Angew Chem Int Ed Engl. 2001;40:1693–1696. doi: 10.1002/1521-3773(20010504)40:9<1693::aid-anie16930>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 15.Pelzer S, Reichert W, Huppert M, Heckmann D, Wohlleben W. Cloning and analysis of a peptide synthetase gene of the balhimycin producer Amycolatopsis mediterranei DSM5908 and development of a gene disruption/replacement system. J Biotechnol. 1997;56:115–128. doi: 10.1016/s0168-1656(97)00082-5. [DOI] [PubMed] [Google Scholar]
- 16.Stegmann E, Pelzer S, Bischoff D, Puk O, Stockert S, Butz D, Zerbe K, Robinson J, Sussmuth RD, Wohlleben W. Genetic analysis of the balhimycin (vancomycin-type) oxygenase genes. J Biotechnol. 2006;124:640–653. doi: 10.1016/j.jbiotec.2006.04.009. [DOI] [PubMed] [Google Scholar]
- 17.Süssmuth RD, Pelzer S, Nicholson G, Walk T, Wohlleben W, Jung G. New Advances in the Biosynthesis of Glycopeptide Antibiotics of the Vancomycin Type from Amycolatopsis mediterranei. Angew Chem Int Ed Engl. 1999;38:1976–1979. doi: 10.1002/(SICI)1521-3773(19990712)38:13/14<1976::AID-ANIE1976>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 18.Sussmuth RD, Wohlleben W. The biosynthesis of glycopeptide antibiotics--a model for complex, non-ribosomally synthesized, peptidic secondary metabolites. Appl Microbiol Biotechnol. 2004;63:344–350. doi: 10.1007/s00253-003-1443-z. [DOI] [PubMed] [Google Scholar]
- 19.Hubbard BK, Walsh CT. Vancomycin assembly: nature's way. Angew Chem Int Ed Engl. 2003;42:730–765. doi: 10.1002/anie.200390202. [DOI] [PubMed] [Google Scholar]
- 20.Pelzer S, Sussmuth R, Heckmann D, Recktenwald J, Huber P, Jung G, Wohlleben W. Identification and analysis of the balhimycin biosynthetic gene cluster and its use for manipulating glycopeptide biosynthesis in Amycolatopsis mediterranei DSM5908. Antimicrob Agents Chemother. 1999;43:1565–1573. doi: 10.1128/aac.43.7.1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pootoolal J, Thomas MG, Marshall CG, Neu JM, Hubbard BK, Walsh CT, Wright GD. Assembling the glycopeptide antibiotic scaffold: The biosynthesis of A47934 from Streptomyces toyocaensis NRRL15009. Proc Natl Acad Sci U S A. 2002;99:8962–8967. doi: 10.1073/pnas.102285099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li TL, Huang F, Haydock SF, Mironenko T, Leadlay PF, Spencer JB. Biosynthetic gene cluster of the glycopeptide antibiotic teicoplanin: characterization of two glycosyltransferases and the key acyltransferase. Chem Biol. 2004;11:107–119. doi: 10.1016/j.chembiol.2004.01.001. [DOI] [PubMed] [Google Scholar]
- 23.Sosio M, Stinchi S, Beltrametti F, Lazzarini A, Donadio S. The gene cluster for the biosynthesis of the glycopeptide antibiotic A40926 by nonomuraea species. Chem Biol. 2003;10:541–549. doi: 10.1016/s1074-5521(03)00120-0. [DOI] [PubMed] [Google Scholar]
- 24.Puk O, Huber P, Bischoff D, Recktenwald J, Jung G, Sussmuth RD, van Pee KH, Wohlleben W, Pelzer S. Glycopeptide biosynthesis in Amycolatopsis mediterranei DSM5908: function of a halogenase and a haloperoxidase/perhydrolase. Chem Biol. 2002;9:225–235. doi: 10.1016/s1074-5521(02)00101-1. [DOI] [PubMed] [Google Scholar]
- 25.Woithe K, Geib N, Zerbe K, Li DB, Heck M, Fournier-Rousset S, Meyer O, Vitali F, Matoba N, Abou-Hadeed K, Robinson JA. Oxidative phenol coupling reactions catalyzed by OxyB: a cytochrome P450 from the vancomycin producing organism. implications for vancomycin biosynthesis J Am Chem Soc. 2007;129:6887–6895. doi: 10.1021/ja071038f. [DOI] [PubMed] [Google Scholar]
- 26.Malabarba A, Ferrari P, Gallo GG, Kettenring J, Cavalleri B. Teicoplanin, antibiotics from Actinoplanes teichomyceticus nov. sp. VII. Preparation and NMR characteristics of the aglycone of teicoplanin. J Antibiot (Tokyo) 1986;39:1430–1442. doi: 10.7164/antibiotics.39.1430. [DOI] [PubMed] [Google Scholar]
- 27.Van Duyne GD, Standaert RF, Karplus PA, Schreiber SL, Clardy J. Atomic structures of the human immunophilin FKBP-12 complexes with FK506 and rapamycin. J Mol Biol. 1993;229:105–124. doi: 10.1006/jmbi.1993.1012. [DOI] [PubMed] [Google Scholar]
- 28.Otwinowski Z, Borek D, Majewski W, Minor W. Multiparametric scaling of diffraction intensities. Acta Crystallogr A. 2003;59:228–234. doi: 10.1107/s0108767303005488. [DOI] [PubMed] [Google Scholar]
- 29.Grosse-Kunstleve RW, Adams PD. Substructure search procedures for macromolecular structures. Acta Crystallogr D Biol Crystallogr. 2003;59:1966–1973. doi: 10.1107/s0907444903018043. [DOI] [PubMed] [Google Scholar]
- 30.Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr D Biol Crystallogr. 2002;58:1948–1954. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- 31.Perrakis A, Morris R, Lamzin VS. Automated protein model building combined with iterative structure refinement. Nat Struct Biol. 1999;6:458–463. doi: 10.1038/8263. [DOI] [PubMed] [Google Scholar]
- 32.McRee DE. XtalView/Xfit--A versatile program for manipulating atomic coordinates and electron density. J Struct Biol. 1999;125:156–165. doi: 10.1006/jsbi.1999.4094. [DOI] [PubMed] [Google Scholar]
- 33.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 34.Kleywegt GJ, Brunger AT. Checking your imagination: applications of the free R value. Structure. 1996;4:897–904. doi: 10.1016/s0969-2126(96)00097-4. [DOI] [PubMed] [Google Scholar]
- 35.Laskowski RA, Rullmannn JA, MacArthur MW, Kaptein R, Thornton JM. AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J Biomol NMR. 1996;8:477–486. doi: 10.1007/BF00228148. [DOI] [PubMed] [Google Scholar]
- 36.Hill HAO, Röder A, Williams RJP. Cytochrome P-450 suggestions as to the structure and mechanism of action. Naturwissenschaften. 1970;57:69–72. [Google Scholar]
- 37.Zerbe K, Pylypenko O, Vitali F, Zhang W, Rouset S, Heck M, Vrijbloed JW, Bischoff D, Bister B, Sussmuth RD, Pelzer S, Wohlleben W, Robinson JA, Schlichting I. Crystal structure of OxyB, a cytochrome P450 implicated in an oxidative phenol coupling reaction during vancomycin biosynthesis. J Biol Chem. 2002;277:47476–47485. doi: 10.1074/jbc.M206342200. [DOI] [PubMed] [Google Scholar]
- 38.Pylypenko O, Vitali F, Zerbe K, Robinson JA, Schlichting I. Crystal structure of OxyC, a cytochrome P450 implicated in an oxidative C-C coupling reaction during vancomycin biosynthesis. J Biol Chem. 2003;278:46727–46733. doi: 10.1074/jbc.M306486200. [DOI] [PubMed] [Google Scholar]
- 39.Jefcoate CR. Measurement of substrate and inhibitor binding to microsomal cytochrome P-450 by optical-difference spectroscopy. Methods Enzymol. 1978;52:258–279. doi: 10.1016/s0076-6879(78)52029-6. [DOI] [PubMed] [Google Scholar]
- 40.Graham SE, Peterson JA. How Similar Are P450s and What Can Their Differences Teach Us? Arch Biochem Biophys. 1999;369:24–29. doi: 10.1006/abbi.1999.1350. [DOI] [PubMed] [Google Scholar]
- 41.Holm L, Rosenstrom P. Dali server: conservation mapping in 3D. Nucleic Acids Res. 2010;38(Suppl):W545–549. doi: 10.1093/nar/gkq366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Park SY, Shimizu H, Adachi S, Nakagawa A, Tanaka I, Nakahara K, Shoun H, Obayashi E, Nakamura H, Iizuka T, Shiro Y. Crystal structure of nitric oxide reductase from denitrifying fungus Fusarium oxysporum. Nat Struct Biol. 1997;4:827–832. doi: 10.1038/nsb1097-827. [DOI] [PubMed] [Google Scholar]
- 43.Xu LH, Fushinobu S, Ikeda H, Wakagi T, Shoun H. Crystal structures of cytochrome P450 105P1 from Streptomyces avermitilis: conformational flexibility and histidine ligation state. J Bacteriol. 2009;191:1211–1219. doi: 10.1128/JB.01276-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xu LH, Fushinobu S, Takamatsu S, Wakagi T, Ikeda H, Shoun H. Regio- and stereospecificity of filipin hydroxylation sites revealed by crystal structures of cytochrome P450 105P1 and 105D6 from Streptomyces avermitilis. J Biol Chem. 2010;285:16844–16853. doi: 10.1074/jbc.M109.092460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Denisov IG, Makris TM, Sligar SG, Schlichting I. Structure and chemistry of cytochrome P450. Chem Rev. 2005;105:2253–2277. doi: 10.1021/cr0307143. [DOI] [PubMed] [Google Scholar]
- 46.Oshima R, Fushinobu S, Su F, Zhang L, Takaya N, Shoun H. Structural evidence for direct hydride transfer from NADH to cytochrome P450nor. J Mol Biol. 2004;342:207–217. doi: 10.1016/j.jmb.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 47.Gotoh O. Substrate recognition sites in cytochrome P450 family 2 (CYP2) proteins inferred from comparative analyses of amino acid and coding nucleotide sequences. J Biol Chem. 1992;267:83–90. [PubMed] [Google Scholar]
- 48.Cryle MJ, Schlichting I. Structural insights from a P450 Carrier Protein complex reveal how specificity is achieved in the P450(BioI) ACP complex. Proc Natl Acad Sci U S A. 2008;105:15696–15701. doi: 10.1073/pnas.0805983105. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.