

Abstract
We have obtained haplotypes from the autosomal glucocerebrosidase pseudogene (psGBA) for 100 human chromosomes from worldwide populations, as well as for four chimpanzee and four gorilla chromosomes. In humans, in a 5420-nucleotide stretch analyzed, variation comprises 17 substitutions, a 3-bp deletion, and a length polymorphism at a polyadenine tract. The substitution rate on the pseudogene (1.23 ± 0.22 × 10−9 per nucleotide and year) is within the range of previous estimates considering phylogenetic estimations. Recombination within the pseudogene was recognized, although the low variability of this locus prevented an accurate measure of recombination rates. At least 13% of the psGBA sequence could be attributed to gene conversion from the contiguous GBA gene, whereas the reciprocal event has been shown to lead to Gaucher disease. Human psGBA sequences showed a recent coalescence time (∼200,000 yr ago), and the most ancestral haplotype was found only in Africans; both observations are compatible with the replacement hypothesis of human origins. In a deeper timeframe, phylogenetic analysis showed that the duplication event that created psGBA could be dated at ∼27 million years ago, in agreement with previous estimates.
In the last few years, studies on human genetic variation have undertaken the complete ascertainment of nuclear genomic sequences. The difficulty in ascertaining haplotypes in a diploid region has led the field first toward the study of X chromosome-linked regions, because haplotypes can be obtained directly from the amplification of X chromosomes in males (Zietkiewicz et al. 1997, 1998; Nachman et al. 1998; Harris and Hey 1999; Kaessman et al. 1999). Several studies to date have analyzed autosomal sequences in worldwide samples (Harding et al. 1997; Clark et al. 1998; Rana et al. 1999; Fullerton et al. 2000). Nevertheless, to the best of our knowledge, pseudogene sequences in humans have not yet been analyzed in worldwide samples.
The term “pseudogene” comprises a wide group of nonfunctional loci with a marked diversity of characteristics. They have been described as dead genes, because they are homologous to their functional source gene but contain nucleotide changes that prevent the production of a functional genetic product. Most pseudogenes are created by one of two mechanisms: tandem duplication or retrotransposition from a functional gene; however, more complex cases have been described (for review, see Cooper 1999). Tandem duplication originates nonprocessed pseudogenes, which are usually linked to their source gene and retain the exon–intron structure of the functional gene. Many duplications include the promoter region, and this allows some pseudogenes to be transcribed.
One such tandem duplication originated the GBA pseudogene (psGBA), the nonfunctional duplicate of the GBA gene, which encodes for the glucocerebrosidase protein (Expasy, EC 3.2.1.45). Mutations on GBA produce Gaucher disease (GD) (OMIM 230800, 230900, and 231000 for Gaucher type 1, type 2, and type 3, respectively). More than 80,000 affected people in the world make GD the most prevalent lipid accumulation disorder.
GBA was mapped to 1q21 (Shafit-Zagardo et al. 1981; Devine et al. 1982; Ginns et al. 1985), and GBA cDNA was first cloned and sequenced from a fibroblast library (Sorge et al. 1985). The complete genomic sequences of GBA and psGBA were described some years later (GenBank J03059 and J03060, respectively; Horowitz et al. 1989). The GBA gene is 7.6 kb long, and it is divided into 11 exons and 10 introns. psGBA is located 16 kb downstream from GBA (Zimran et al. 1990; Winfield et al. 1997); it contains the same exon and intron number and structure as GBA, although its length is ∼5.7 kb (Fig. 1). GBA is longer than psGBA because of several Alu insertions in intronic tracts of GBA and a 55-bp deletion in exon 9 of psGBA (exon and intron notations in this report will follow the gene nomenclature). Despite the length difference, psGBA has maintained 96% sequence identity with the functional GBA gene. The high degree of sequence identity and the physical proximity between psGBA and GBA allows gene conversion events from psGBA to GBA (Hong et al. 1990; Latham et al. 1991), resulting in aberrant gene sequences that cause GD.
Figure 1.

Schematic representation of GBA and psGBA. Exons are depicted as boxes with identification numbers inside. Alu sequences are represented as black boxes. The PCR-amplified and sequenced areas are indicated. The transcripts for both regions are represented with smaller boxes under each region: (a) psGBA transcript described by Sorge et al. (1990); (b) psGBA transcript described by Imai et al. (1993). The length of all tracts is roughly proportional to their actual nucleotide length.
A rare trait of psGBA is that it is transcribed, because two TATA boxes and two CAT boxes in the GBA promoter area are preserved in psGBA, except for a substitution in the second CAT box (Horowitz et al. 1989). However, the activity of the psGBA promoter does not reach the levels of the GBA active promoter (Reiner and Horowitz 1988). Two psGBA transcripts have been described (Fig. 1; Sorge et al. 1990; Imai et al. 1993).
The duplication within 1q21 is present in rhesus monkeys, and thus it may have occurred before the divergence of the great apes and Old World monkeys, 25 million yr ago (Mya). The presence in the two duplication copies of an Alu sequence of the Sx family, which is 40 My old, places the age of duplication between 25 and 40 Mya (Winfield et al. 1997).
We analyzed sequence variation in psGBA in a sample of 100 worldwide chromosomes as a means to study variation in a human autosomal noncoding region. We have inferred the effects of recurrent mutation, recombination, and gene conversion on the phylogeny and polymorphism spectrum of this pseudogene. This is the first time that a wide spectrum of variability is reported for a completely noncoding autosomal tract, and that population genetic analyses are derived from a pseudogene.
RESULTS
Nucleotide and Haplotype Diversity
We ascertained 5420-bp psGBA haplotypes in 100 human, four chimpanzee, and four gorilla chromosomes (GenBank AF267177, AF272642, and AF272641, for the human, chimpanzee, and gorilla sequences, respectively). One variable position and two haplotypes were detected in the two chimpanzee samples analyzed. As for the two gorilla samples, seven variable positions and three haplotypes were detected.
Among 100 human chromosomes analyzed, 18 variable sites, all of them diallelic, were identified, which corresponds to an average density of one segregating site in every 301 bp. Eleven individuals were homozygous for the whole tract analyzed. Only one deletion, of 3 bp, was detected. The other 17 variants were single-nucleotide substitutions. Among those, transitions were more frequent than transversions: 15 transitions (88.2% of the single nucleotide substitutions) against two transversions (11.8%). In addition to the 18 segregating sites, a short polymorphic polyadenine tract was found in position 3109, corresponding to the Alu insertion in intron 7. The exact number of adenines could not be read with certainty in the diploid sequences, but it was clear in the haploid sequences, in which alleles containing 9–11 repeats were detected. This site has not been included in the haplotype determination and further analysis because it follows different evolutionary patterns (i.e., mutation mechanisms and rates) than the rest of the variable sites. Seven of the 18 total segregating sites (38.9%) are singletons (the rarer nucleotide variant appears once in the sample), and five (27.8%) are doubletons (the rarer nucleotide variant appears twice in the sample).
None of the GBA gene counterparts of the polymorphic psGBA sites has been shown to be variable, except for nucleotide 4291: We have found a frequency of 6% A and 94% G in psGBA, and in the GBA gene a frequency of 70% A and 30% G was reported (Beutler et al. 1992). And, vice versa, the remaining polymorphisms reported in GBA do not vary in psGBA in the present sample set. Site 4614, a C/G polymorphism in humans, is also variable in gorillas, where A/G alleles were observed.
In humans, the 18 segregating positions define 25 different haplotypes (Table 1). Haplotype diversity was 0.853. For the chimpanzee and gorilla alleles, only those positions that are polymorphic in the human sequence are shown in Table 1. Two major haplotypic groups are distinguishable in that table. Each group has a haplotype with high frequency (i.e., 3 and 17). Together, haplotypes 3 and 17 account for 52% of the total chromosomes.
Table 1.
psGBA Haplotypes and Their Frequencies in 100 Human Chromosomes
| Position | 184 | 234 | 308-10 | 2253 | 2266 | 2723 | 3617 | 3968 | 4008 | 4020 | 4274 | 4291 | 4419 | 4614 | 4635 | 4938 | 5001 | 5061 | GBA | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Haplotype | t | a | ctc | t | g | g | g | c | c | c | c | g | t | c | c | t | t | c | Freqa | |
| 3 | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | 29 | 7−/7+ |
| 8 | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | c | . | . | 5 | 4+ |
| 4 | . | . | . | . | . | . | . | . | . | . | . | . | c | . | . | . | . | . | 4 | 2+ |
| 20 | g | g | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | 2 | |
| 2 | . | . | . | . | . | . | . | . | . | . | . | . | . | . | t | . | . | . | 1 | 1+ |
| 21 | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | c | . | 1 | |
| 23 | . | . | . | . | . | . | a | t | . | . | . | . | . | . | . | . | . | . | 1 | |
| 29 | . | . | . | . | . | . | a | . | . | . | . | . | . | . | . | . | . | . | 1 | |
| 30 | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | t | 1 | 1+ |
| 1 | . | . | . | . | . | . | . | . | . | . | . | a | . | . | . | . | . | . | 1 | 1− |
| 5 | . | . | . | . | a | . | . | . | . | . | . | . | . | . | . | . | . | . | 1 | 1− |
| 6 | . | . | . | . | . | . | . | . | . | . | . | . | . | g | . | . | . | . | 1 | |
| 7 | . | . | . | c | . | . | . | . | . | . | . | . | . | g | . | . | . | . | 1 | 1+ |
| 25 | . | . | . | c | . | . | . | . | . | . | . | . | . | . | . | . | . | . | 3 | 2+ |
| 17 | . | . | . | c | a | . | . | . | . | t | . | . | . | . | . | c | . | . | 23 | 12− |
| 19 | . | . | . | c | a | . | . | . | . | t | . | a | . | . | . | c | . | . | 4 | 3− |
| 26 | . | . | . | c | . | . | . | . | . | t | . | . | . | . | . | c | . | . | 3 | 2− |
| 15 | . | . | . | c | a | . | . | . | . | . | . | a | . | . | . | c | . | . | 1 | |
| 13 | . | . | . | c | a | . | . | . | . | . | . | . | . | . | . | c | . | . | 3 | 1−/2+ |
| 16 | . | . | − | c | a | . | . | . | . | . | . | . | . | . | . | c | . | . | 1 | |
| 22 | . | . | . | c | a | . | . | . | . | . | . | . | . | . | . | . | . | . | 1 | |
| 24 | . | . | . | c | . | . | . | t | . | . | t | . | . | . | . | c | . | . | 1 | 1+ |
| 27 | . | . | . | c | . | a | . | . | . | . | . | . | . | . | . | c | . | . | 1 | |
| 10 | . | . | . | c | . | . | . | . | . | . | . | . | . | . | . | c | . | . | 9 | 1−/5+ |
| 11 | . | . | . | c | . | . | . | . | t | . | . | . | . | . | . | c | . | . | 1 | |
| Chimpanzee | . | . | . | c | . | . | . | t | . | . | . | a | c | g | . | c | . | . | 4 | |
| Gorilla | . | . | . | c | . | . | . | t | a | . | . | a | c | g/a | . | c | . | . | 3/1b | |
| Human GBA | . | . | ttc | . | a | . | . | t | a | t | . | g/a | c | g | . | . | . | . |
The states in chimpanzee, gorilla, and the human GBA gene (GenBank J03059) are shown for the human psGBA variable sites. Polymorphisms caused by a CpG dinucleotide are shown in bold. The nomenclature for the haplotypes is arbitrary. The association between GBA and psGBA haplotypes could be resolved for 54 of 100 chromosomes, and is indicated in the GBA column (the number indicates number of chromosomes).
Freq, haplotypic frequency.
Of the four gorilla alleles analyzed, three had a G in position 4614 and one presented an A.
No clear geographic structure is observed in the distribution of human haplotypes (Table 2). To evaluate the effect of the differences among populations on the general variation, we calculated Fst, which was 0.128 (P < 0.0001). This value is within the range for previous estimated Fst for mitochondrial DNA, Y-chromosome, and autosomal polymorphisms, which indicates that most of the human genetic variation is due to differences within, rather than among, populations (Barbujani et al. 1997; Jorde et al. 2000).
Table 2.
Population Haplotype Frequencies
| Hapa | BIAb | TAN | SAH | BAS | CAT | DRU | YAK | CHI | MAY | NAS | SWA-EU | SSAFR | Total |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 3 | 1 | 4 | 2 | 2 | 4 | 3 | 6 | 3 | 4 | 9 | 5 | 29 | |
| 17 | 2 | 4 | 4 | 2 | 3 | 2 | 3 | 3 | 9 | 2 | 23 | ||
| 10 | 4 | 1 | 1 | 1 | 2 | 1 | 5 | 9 | |||||
| 8 | 3 | 2 | 2 | 3 | 5 | ||||||||
| 4 | 4 | 4 | |||||||||||
| 19 | 3 | 1 | 3 | 4 | |||||||||
| 13 | 2 | 1 | 3 | ||||||||||
| 25 | 2 | 1 | 3 | ||||||||||
| 26 | 3 | 3 | |||||||||||
| 20 | 1 | 1 | 2 | 2 | |||||||||
| 5 | 1 | 1 | |||||||||||
| 1 | 1 | 1 | 1 | ||||||||||
| 6 | 1 | 1 | 1 | ||||||||||
| 7 | 1 | 1 | 1 | ||||||||||
| 2 | 1 | 1 | |||||||||||
| 11 | 1 | 1 | 1 | ||||||||||
| 15 | 1 | 1 | |||||||||||
| 16 | 1 | 1 | 1 | ||||||||||
| 21 | 1 | 1 | 1 | ||||||||||
| 22 | 1 | 1 | 1 | ||||||||||
| 23 | 1 | 1 | |||||||||||
| 24 | 1 | 1 | 1 | ||||||||||
| 27 | 1 | 1 | |||||||||||
| 29 | 1 | 1 | |||||||||||
| 30 | 1 | 1 | 1 | ||||||||||
| 2Nc | 10 | 10 | 10 | 10 | 10 | 10 | 10 | 10 | 10 | 10 | 30 | 20 | 100 |
| kd | 5 | 6 | 5 | 6 | 5 | 4 | 4 | 5 | 4 | 5 | 10 | 9 | 25 |
| Private Hape | 2 | 3 | 1 | 2 | 2 | 0 | 1 | 2 | 2 | 2 | 5 | 4 | — |
Hap, haplotype.
BIA, Biaka; TAN, Tanzanians; SAH, Saharawi; BAS, Basques; CAT, Catalans; DRU, Druze; YAK, Yakut; CHI, Chinese; MAY, Maya; NAS, Nasioi; SWA-EU, South West Asia and Europe (Basques, Catalans, and Druze); SSAFR, sub-Saharan Africans (Tanzanians and Biaka).
2N, sample size in number of chromosomes.
k, number of different haplotypes found in each population.
Private Hap, number of haplotypes found solely in that population.
Nucleotide diversity as the average heterozygosity, π, was 0.00044 (Nei and Li 1979). From π, the θ̂ estimator was 2.40, which corresponds to the mean number of pairwise differences. The variability observed for psGBA is low in comparison to the values observed for other nuclear loci (Table 3). The θ̂W statistic is a different estimator of the variability that can be computed from the number of segregating sites (S) in a given locus (Watterson 1975), and was estimated as 3.28 for psGBA.
Table 3.
Summary of Human Nuclear Sequence Variation Studies
| Authors | Year | Region | La | nb | Sc | kd | π |
|---|---|---|---|---|---|---|---|
| Hey | 1997 | PDHA1, X chre | 1.8 | 8 | 4 | 4 | 0.0015 |
| Zietkiewicz et al. | 1998 | Dystrophin, X chr | 7.6 | 250 | 35 | 36 | 0.0013 |
| Nachman et al. | 1998 | Introns from 7 loci, X chr | 11.4 | 10 | 20 | — | 0.0008 |
| Harris and Hey | 1999 | PDHA1, X chr | 4.2 | 35 | 25 | 11 | 0.0022 |
| Kaessman et al. | 1999 | Non coding region, X chr | 10.2 | 69 | 33 | 20 | 0.0005 |
| Jaruzelska et al. | 1999a | ZFX, X chr | 1.1 | 336 | 10 | 11 | 0.0011 |
| Dorit et al. | 1995 | ZFY, Y chr | 0.7 | 38 | 0 | 1 | 0 |
| Hammer | 1995 | YAP region, Y chr | 2.6 | 16 | 5 | 5 | 0.0004 |
| Whitfield et al. | 1995 | SRY, Y chr | 18.3 | 5 | 3 | 4 | 0.0003 |
| Jaruzelska et al. | 1999b | ZFY, Y chr | 0.7 | 205 | 1 | 2 | 0.00006 |
| Shen et al. | 2000 | SMCY, UTY1, DBY, DFFY, Y chr | 81 | 53 | 98 | — | 0.000052 |
| Li and Sadler | 1991 | 49 autosomal loci | 75 | 2 | — | — | 0.0011† |
| Fullerton et al. | 1994 | β-globin, chr 11 | 3.1 | 36 | 17 | 17 | 0.0014 |
| Harding et al. | 1997 | β-globin, chr 11 | 2.7 | 349 | 35 | 30 | 0.0018 |
| Clark et al. | 1998 | LPL, chr 8 | 9.7 | 142 | 88 | 88 | 0.0020 |
| Grimsley et al. | 1998 | HLA-H pseudogene, chr 6 | 0.3 | 34 | 15 | 11 | 0.0196 |
| Rieder et al. | 1999 | ACE, chr 17 | 24 | 22 | 78 | 13 | 0.0009 |
| Halushka et al. | 1999 | 75 genes | 190 | 149 | — | — | 0.0008 |
| Rana et al. | 1999 | MC1R, chr 16 | 0.95 | 242 | 6 | 6 | 0.0020 |
| Fullerton et al. | 2000 | ApoE, chr 19 | 5.5 | 192 | 22 | 31 | 0.0005 |
| This study | 2001 | psGBA, chr 1 | 5.4 | 100 | 18 | 25 | 0.0004 |
In the cases in which the X or the Y chromosomes were studied, the standardized π (nucleotide diversity) is shown (π values were multiplied by 4/3 or by 4, respectively, to make them comparable to autosomes).
L, length per locus is indicated as number of kilobases sequenced.
n, number of chromosomes.
S, number of segregating sites.
k, number of haplotypes observed.
chr, chromosome.
Nucleotide diversity for fourfold degenerate sites.
By the definition of a pseudogene, selection cannot act directly on it, but selective effects on neighboring genes can have a deep impact on sequence variability in pseudogenes. Thus, the neutral model of evolution cannot be assumed for psGBA. Under the neutral model, both θ̂π and θ̂W estimate 4Neμ, where Ne is the effective population size and μ is the mutation rate. To test whether psGBA evolves according to the neutral model, we estimated Tajima's D statistic (Tajima 1989), which compares both θ̂π and θ̂W estimators, and it was −0.76 (not significant, P > 0.10). The D* and F* statistics (Fu and Li 1993) can also be used to test whether mutations are selectively neutral. The D* statistic is based on the differences between the number of singletons and the total number of segregating positions, and it was −1.33 (not significant, P > 0.10). The F* statistic is based on the differences between the number of singletons and θ̂π, and it was found to be −1.34 for psGBA (not significant, P > 0.10). The results of these tests do not allow us to reject a neutral model to explain the results.
Polymorphism Patterns along psGBA: 5′ and 3′ Halves of the Pseudogene
The number of segregating positions and the nucleotide diversity along psGBA are represented in Figure 2. It can be seen that the segregating positions seem to concentrate toward the 3′ end of psGBA, and nucleotide diversity does not appear to be homogeneous along the pseudogene.
Figure 2.

Number of segregating positions (S), and nucleotide diversity (π) along psGBA. A window length of 100 nucleotides moved in steps of 25 nucleotides along the sequence was used.
In addition to the apparent nonuniform distribution of polymorphisms, a segment ∼2-kb long in the 5′ half (1946 nucleotides, 35.9 % of the total of nucleotides), from the CTC deletion in nucleotide 308 (exon 1) to the C/T polymorphism in nucleotide 2253 (exon 6), was found to lack any segregating site. Assuming that the distribution of polymorphic sites along the sequence is random, then the number of polymorphisms in a given segment would follow a Poisson distribution. The Poisson λ parameter according to the observed proportion of polymorphic sites in the remaining sequence (3474 bp) is λ = 1.83, and the probability of not observing any variable site in this 2-kb stretch is P = e−λ = 0.16. Therefore, the probability of absence of polymorphism in this region is not statistically significant and we cannot state that it evolves differently from the rest of the sequence. We can conclude, though, that if we had analyzed a shorter psGBA fragment, the results could have been biased.
The fact that 12 of the 17 segregating sites are placed in the 3′ half of the pseudogene made us check for possible differences in the variability between both halves of psGBA. We considered as the 5′ half the first 2710 bp and as the 3′ half the remaining 2710 bp. Six haplotypes and a nucleotide diversity of 0.00022 are observed in the 5′ half, against 16 haplotypes and a nucleotide diversity of 0.00066 in the 3′ half. The different number of segregating positions in both halves of psGBA was tested with a χ2 test (χ2 = 2.88, 1 d.f., P = 0.090), and no significant differences were found.
A possible explanation for the levels of variation in a given genomic region may be that they are a function of the frequency of hypermutable CpG dinucleotides. To check whether that was the case for the two halves of psGBA, the possible differences between (1) the total number of CpG dinucleotides (48 in the 5′ half, 32 in the 3′ half) and (2) the number of mutated CpGs in both halves of the pseudogene (one in the 5′ half, six in the 3′ half) were tested with a χ2 test. No significant differences were found concerning the number of CpGs between the two psGBA halves (χ2 = 3.20, 1 d.f., P = 0.07), but the number of mutated CpGs was significantly higher (χ2 = 6.68, 1 d.f., P = 0.009) in the 3′ half.
In summary, the apparent (but not statistically significant) difference in polymorphism between the 5′ and 3′ moieties of psGBA seems to be due to the higher mutability of CpG dinucleotides in the 3′ half, which is the only significant difference between both halves.
Substitution Rate
We have estimated the substitution rate as the number of differences over 2tL, L being the length of the segment compared and t the divergence time between species. We assumed a 7-My divergence time between gorillas and humans, and gorillas and chimpanzees, and a 5-My divergence time between chimpanzees and humans. Average substitution rates of 1.30 × 10−9, 1.43 × 10−9, and 1.014 × 10−9 per nucleotide and year were obtained for psGBA when comparing gorilla and human sequences (99 differences, 1.8% divergence), chimpanzee and human sequences (78 differences, 1.4% divergence), and gorilla and chimpanzee sequences (77 differences, 1.4% divergence), respectively. The mean weighted value for the substitution rate on psGBA is 1.23 ± 0.22 × 10−9.
The sequence of the GBA gene in chimpanzee was obtained (GenBank AF285236), and this allowed us to estimate the substitution rate for the GBA locus in the same way. As human GBA we used the sequence on GenBank J03059. Five small indels and 62 substitutions (36 transitions and 26 transversions) were detected between chimpanzee and human sequences along 7156 nucleotides from the GBA gene. The substitution rate for GBA between human and chimpanzee was estimated as 0.87 ± 0.11 × 10−9 per nucleotide and year. It should be noted that the confidence intervals for the substitution rates in the gene and in the pseudogene overlap slightly.
Recombination and Recurrent Mutation
psGBA is located in a centromeric area and, presumably, in a low-recombination genomic context. Nevertheless, some haplotypes present a pattern that could place the sequence in either of the two main groups observed among the haplotypes, that is, haplotypes 5, 8, and 25 and, to a lesser extent, 7, 22, and 1 (Table 1). This mixed pattern could be due to intragenic recombination, gene conversion, recurrent mutation, or back mutation events. In the absence of these processes, the maximum number of expected haplotypes for S diallelic segregating sites is S + 1 haplotypes. If psGBA has more alleles than would be expected from infinite-allele mutation alone, then at least one of these forces must have acted.
The absence of complete linkage disequilibrium can be also verified with the following rationale: If psGBA were in complete linkage disequilibrium, then we would expect that every haplotype constructed with the segregating sites only on the 3′ half of the locus would correspond to a single haplotype from the 5′ half of the locus. On the contrary, six haplotypes are observed for the 5′ half and 16 for the 3′ half, whereas when considering the whole segment 25 haplotypes appear.
To measure the relevance of intralocus recombination events on psGBA, the recombination parameter C = 4Nec, where Ne is the effective population size and c is the recombination rate per generation per base pair, was estimated. According to the estimation procedure suggested by Hudson (1987), in which the Ĉ estimator is based on the variance of the average number of nucleotide differences between pairs of sequences, Ĉ = 16.9. Nevertheless, this measure overestimates recombination if polymorphism is low and requires a long (more than 100-kb) segment, a large sample size, and high variability to be reliable (Hudson 1987). Even if the estimate was reliable, we should consider that possible recurrent mutational events are also counted as recombination events, and therefore recombination is overestimated. The γ estimator of recombination (Hey and Wakeley 1997) does not depend on the polymorphisms in the sample and is less biased by sample sizes and shorter DNA length than Hudson's estimator. The γ estimator was 1.271 for our sample set. From this estimate, the ratio of recombination events per mutation would be 0.39 (calculated as 4Neμ/4Nec = θ̂W/γ). Nonetheless, the γ estimator will yield an overestimate of recombination if there is homoplasy in the sample, as is probably the case for psGBA.
The minimum number of recombination events along psGBA necessary to explain the observed variability (Hudson and Kaplan 1985) was estimated as four, between sites 2253 and 2266, 2266 and 4020, 4020 and 4291, and 4291 and 4938. However, the scarce number of segregating positions should make us be extremely cautious when trying to identify possible recombinant chromosomes. In fact, only four segregating positions separate haplotype 3 from haplotype 17. In addition, two of the presumably most determinant positions in defining one or the other group (i.e., 2253, 2266, and 4938) are located on CpG dinucleotides (2253 and 4938), and therefore more than one mutational event could have originated the present observed variability at these positions. For example, it cannot be ascertained whether haplotype 8 was produced by a CpG recurrent mutation at 2253 or by a recombination event between haplotypes 3 and 10.
To analyze further the origin of the possible recombinant alleles, we added data on three polymorphic sites analyzed for GBA (E. Mateu, F. Calafell, R. Martínez-Arias, A.Pérez-Lezaun, A. Andrés, J. Bertranpetit, unpubl.) to the haplotypic data for psGBA. Twelve polymorphisms in tight linkage disequilibrium define two main haplotypes for GBA, named + and −, respectively, with frequencies ∼70% and 30% in Africans and Asians and the reciprocal in Europeans (Beutler et al. 1992; Glenn et al. 1994; E. Mateu, F. Calafell, R. Martínez-Arias, A. Pérez-Lezaun, A. Andrés, J. Bertranpetit, unpubl.). Haplotype ascertainment from genotype data showed a significant linkage disequilibrium between haplotypes − and 17, and + and 3. Those haplotypes that were double haplotype homozygotes for GBA or psGBA allowed unambiguous phase resolution and determination of the joint GBA–psGBA haplotypes. From those, out of 14 chromosomes with psGBA haplotype 3, 7 were linked to GBA haplotype − and 7 to GBA haplotype +. The 12 resolvable psGBA haplotype 17 chromosomes were all linked to GBA haplotype −.
In the same way, haplotypes 25 and 7 were linked to GBA haplotype +. These two haplotypes have a C in position 2253 and therefore could be placed at first sight, erroneously, within haplogroup 17 (if position 2253 was taken as diagnostic). However, because GBA haplotype + seems not to be linked with haplogroup 17, it seems more likely that these are not recombinant haplotypes between haplotype group 17 and group 3 chromosomes, but rather that they have not yet lost 2253 C through repeated mutation at that CpG dinucleotide.
On the whole, it seems that recombination is low, although not absent, at psGBA and that other forces such as gene conversion and recurrent or back mutation may have had a prominent role in shaping the variability spectrum of psGBA.
Interlocus Gene Conversion
We aligned chimpanzee psGBA, human psGBA, and human GBA sequences (GenBank AF272642, AF267177, and J03059, respectively) to detect possible interlocus gene conversion events (gene conversion between different alleles at different loci) between psGBA and GBA and to assess their magnitude. Next, we added the sequence of the GBA gene in chimpanzee (GenBank AF285236) to detect the possible influence of gene conversion events in chimpanzees on those fragments where gene conversion in humans would be likely, and also to have an external reference for the GBA-specific sequence pattern. We would detect gene conversion as a string of nucleotide positions placed on a different haplotype background. We looked for gene-specific patterns in the pseudogenes, for pseudogene patterns in the genes, and also for nucleotide positions that interrupted those patterns (Fig. 3).
Figure 3.

Gene conversion tracts along psGBA. Positions at which both psGBA from chimpanzee and human differ and human GBA and psGBA are equal are considered possible gene conversion fragments from GBA to psGBA (light shading). The position following one of these blocks that was equal between both pseudogenes and different with the GBA gene (so that it interrupts the possible tract of gene conversion) is also indicated in the figure in lower case. Dark shadows indicate tracts where gene and pseudogene sequences are equal both in humans and in chimpanzees, but where orthologous sequences are different across species. Positions at which human psGBA, human GBA, and chimpanzee psGBA were different, or at which the GBA gene and psGBA in chimpanzee are equal and different from human psGBA, have not been taken into account. Variable sites found in the human pseudogene are represented in bold. (*) Similarity between genes and pseudogenes in position 1137 has to be taken with caution, because it is located after a single-nucleotide run of seven adenines, and the similarity could be a product of polymerase slippage and not of gene conversion. 1265i indicates the insertion after site 1265 in human psGBA.
When we compare the GBA and psGBA sequences in humans and chimpanzees, some tracts seem to be the clear result of gene conversion events from the human GBA gene to human psGBA. These are the fragments from 439 to 567, 1264 to 1265, 1628 to 1682, 1884 to 1982, 2105 to 2241, 4204 to 4261, and 4680 to 4910 (the numeration of human psGBA reported here is used [GenBank AF267177]). In these tracts, at least in two consecutive positions, human psGBA has the same pattern as human GBA, and it is different from psGBA in chimpanzee (to which it should be more similar). In addition, the gene pattern is the same in GBA from chimpanzee, while the characteristic pseudogene pattern would have been preserved only in psGBA in chimpanzee.
In other tracts, human GBA is the locus with a distinct sequence, and the sequence from chimpanzee GBA seems to have acquired the pattern from chimpanzee psGBA, such as in fragment 1295–1324. However, we have analyzed GBA in chimpanzee only for those positions that would allow us to recognize gene conversions in humans, and we cannot identify with certainty gene conversion tracts elsewhere in the chimpanzee sequences.
There is a third kind of tracts, in which psGBA and GBA sequences are equal within each species but different across species. These fragments are all located in introns, so that if gene conversion had occurred in both species, the direction could not be unequivocally established. Besides, recurrent or parallel mutations could have taken place, which could be mistaken for gene conversion.
Taking into account only the first type of fragments, in which gene conversion is more obvious, at least 709 bp of psGBA sequence (13% of the total length) is affected by this phenomenon. The high sequence similarity among the four sequences makes it difficult to pinpoint the extent of gene conversion, and therefore this is a minimum estimate, because gene conversion could extend longer and go unrecognized because of the high sequence similarity between gene and pseudogene. Gene conversion was random with respect to exon–intron distribution (χ2 = 1.216, 1 d.f., P = 0.27).
Recent gene conversion events may not have reached fixation in the population and remain polymorphic at psGBA. That could be the case for positions 2253, 2266, 4020, and 4938, because on them one allele corresponds to the state in psGBA in chimpanzee and the other variant to the state in the human GBA. Polymorphisms on sites 2253 and 4938 are located on a hypermutable CpG, which could as well account for the changes from G to A in this position.
Haplotype Phylogeny
A network with all possible phylogenetic links among haplotypes was constructed with the Network v, 2.0b software (Fig. 4). Reticulations in this median network can reveal homoplasy and possible recombination (Bandelt et al. 1995). In particular, three of the four estimated recombination events (between sites 2253 and 2266, 2266 and 4020, and 4020 and 4291) are reflected as reticulations in the network.
Figure 4.

Median network constructed from the 25 haplotypes in our sample. Haplotype number is shown inside the circles, and mutation positions are indicated on the branches linking two haplotypes. Circle areas are proportional to the frequency of the haplotypes. Branch lengths are proportional to the number of mutational events they represent.
Two major clades are observed, with centers in the two most frequent haplotypes, namely 3 (in 29 chromosomes) and 17 (in 23 chromosomes). The phylogenetic structure around haplotype 3 is clearly starlike, with 10 haplotypes radiating directly from it. No such starlike structure is observed around haplotype 17.
The extent of the phylogenetic separation between the haplotypes radiating from haplotypes 3 or 17 was ascertained by means of their pairwise difference distributions. Figure 5 shows the overall pairwise difference distribution, as well as the pairwise difference distributions within each haplotype group and between them. A slightly bimodal curve can be appreciated, which is caused by differences within (left-hand peak) and between (right-hand peak) haplotype groups 3 and 17, as shown by the separate pairwise distributions.
Figure 5.

Pairwise nucleotide difference distribution. The differences within haplogroup (hapg) 3, within haplogroup 17, and the differences resulting from the comparison between both haplogroups are shown. Haplogroup 3 comprised haplotypes 1, 2, 3, 4, 5, 6, 7, 8, 20, 21, 23, 25, 29, and 30, and haplogroup 17 comprised haplotypes 10, 11, 13, 15, 16, 17, 19, 22, 24, 26, and 27.
Time to the Most Recent Common Ancestor and Mutation Ages
To infer mutational ages and thus put the haplotype phylogeny in a historical frame, the method based on coalescence theory suggested by Griffiths and Tavaré (1994, 1998a, 1998b) was applied. Coalescence theory provides a mathematical tool for inferring backward in time the genealogy of genes or alleles sampled from a present population.
The ancestral state for the human psGBA was inferred as the state at each polymorphic site that would give the most parsimonious psGBA phylogeny, rooted with the chimpanzee psGBA. Human ancestral states for almost all polymorphic sites match those in the chimpanzee sequence, except for four positions (3968, 4291, 4419, and 4614), in which the nucleotide states present in chimpanzee are scattered on eight haplotypes located in external branches of the human network.
Mutation ages were estimated using the maximum-likelihood estimate of θ, θ̂ML (4.45), which is the θ value that would yield the most likely coalescent tree. θ̂ML was calculated using the GENETREE program. Effective population size was estimated as Ne = 8345 from θ = 4Neμ, using the θ̂ML estimate and the previously calculated substitution rate per sequence and generation, μ = 13.33 × 10−5. Under these conditions, and assuming neutrality, the infinite-sites mutation model (haplotypes presumably affected by recurrent mutation or recombination were omitted from the analysis), random mating, no population substructure, constant population size, and a generation time of 20 yr, the coalescent time, or time to the most recent common ancestor (TMRCA) was estimated at 199,000 ± 58,600 yr (Fig. 6). Diversity data were estimated for the haplotypes compatible with the infinite-sites mutation model and were not significantly different from those estimated from the complete set of haplotypes, so that no major biases were introduced when computing the mutation ages. The age of the mutations in the gene genealogy range from 163,800 to 5170 yr (Table 4).
Figure 6.

Coalescent tree of the psGBA haplotypes, for the psGBA haplotypes compatible with the infinite-sites mutation model. The tree was computed using 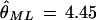 , Ne = 8345, μ = 13.33 × 10−5 per generation and year, and a 20-yr generation time, through 2 × 107 iterations. Numbers along branches represent mutational events. The continents where the haplotypes are present are indicated. (SWA-E) Southwest Asia and Europe.
, Ne = 8345, μ = 13.33 × 10−5 per generation and year, and a 20-yr generation time, through 2 × 107 iterations. Numbers along branches represent mutational events. The continents where the haplotypes are present are indicated. (SWA-E) Southwest Asia and Europe.
Table 4.
Time to Most Recent Common Ancestor (TMRCA) and Ages of the Deepest Mutations in the psGBA Phylogeny
| Mutation and TMRCA | Age, constant population | SD, constant population | Age, exponential population growth | SD, exponential population growth |
|---|---|---|---|---|
| TMRCA | 199,000 | 58,600 | 91,000 | 17,000 |
| 3968 | 163,800 | 48,400 | 81,600 | 13,900 |
| 4274 | 99,460 | 67,500 | 44,800 | 24,400 |
| 2253 | 95,000 | 32,700 | 58,000 | 11,400 |
| 4938 | 74,400 | 26,600 | 49,900 | 10,100 |
| 2266 | 74,000 | 28,300 | 53,200 | 12,000 |
Times have been computed under constant population size and exponential growth models. Ages are given in years and have been calculated with a generation time of 20 yr, a population size of 7330 individuals under the constant population model, and a population size of 14,170 individuals under the exponential population growth model.
Mutation 4938 would lead to haplotype 3 and the group of haplotypes radiating from it, and it has an estimated age of ∼74,400 ± 26,600 yr. To assess the reliability of the estimated ages, an independent estimate of the age of haplogroup 3 was calculated according to a Poisson distribution of mutations, as explained in Methods section. Haplotype 3 was considered to be the ancestral sequence of haplogroup 3, which was designed as those haplotypes that could be derived unequivocally from haplotype 3, that is, 2, 4, 6, 20, 21, 29, 30, and haplotype 3 itself. Age of haplogroup 3 can be inferred as 43,000 ± 11,900 yr, which overlaps with the previous estimate.
We also computed the TMRCA assuming a population growth model instead of a population with constant size. Growth parameter β̂ML and θ̂ML under an exponential growth model were estimated simultaneously with the GENETREE program. We assumed the same growth rate for all populations. We obtained a value of β̂ML of 11.4 and of θ̂ML of 8.6. From this θ̂ML value we obtain an effective population size of 16,125 individuals. TMRCA and mutation-age estimates obtained under a population growth model were markedly lower than those obtained under a model of constant population size (Table 4). The general pattern of the gene tree obtained under a growth model is the same as under a constant population model, except that branches have been shortened between mutations 2266 and 3968 and between 4938 and 3968. The difference in the likelihood of the trees under constant and growth models was not statistically significant.
Phylogenetic Relations among Orthologous and Paralogous Sequences
The neighbor-joining phylogenetic algorithm (Saitou and Nei 1987) was applied to distances between pairs of sequences estimated with the Kimura 2-parameter method. To assess the reliability of branching, 1000 bootstrap replicates were performed. Trees were constructed among the human psGBA haplotypes, among all the pseudogene sequences (human, chimpanzee, and gorilla), and among those and the GBA gene sequences in human (GenBank J03059) and chimpanzee (GenBank AF285236) (Fig. 7).
Figure 7.

Neighbor-joining trees representing GBA gene and psGBA in humans, chimpanzees, and gorillas. Tree robustness was assessed by 1000 bootstrap iterations, and the percentage of trees with a given node present in the bootstrap tree is shown in the figures (italic numbers on the branches). For the lower tree, only bootstrap fractions equal or larger than 50% are shown. Arrows indicate outgroup rooting with human GBA (in the middle tree, which links human and chimpanzee psGBA sequences) and with chimpanzee psGBA (in the lower tree).
It is clear from Figure 7 that the duplication event that created psGBA preceded hominoid speciation, because the GBA and psGBA sequences cluster clearly by homology rather than by species. The time of duplication for psGBA was calculated from the differences between GBA and psGBA in humans as number of differences over Lμ, L being the length compared between two sequences, and μ the sum of the estimated substitution rates for GBA and psGBA, because the divergence in the two branches after the duplication, for GBA and for psGBA, has to be considered. The estimate was 23.4 My with the human data and 23.2 My with the chimpanzee data.
DISCUSSION
We have sequenced an ∼5.5-kb stretch containing pseudogene psGBA in 100 chromosomes distributed among all major world geographic areas and found that psGBA has the lowest nucleotide diversity observed for an autosomal locus. On average, two randomly chosen sequences of nearly 5.5 kb will be different at about two nucleotide sites. This low value was not expected for a noncoding region such as a pseudogene, which would be seemingly free to accumulate variation unchecked by purifying selection. Next, we discuss the main evolutionary forces that may have acted on psGBA to create and shape genetic variation, as well as the inferences that can be drawn from that variation both on the evolutionary history of the region and on human evolution.
Genomic Forces Acting on psGBA: Mutation, Recombination, Gene Conversion
The substitution rate we have found in psGBA is not higher than the substitution rates described for functional genes. Substitution rate values for psGBA and GBA are indeed close. This fact might be taken into account when considering pseudogenes to estimate the rate of spontaneous mutation. The present results indicate a large heterogeneity in pseudogene mutation rates, as lower values than those considered “neutral” have been found in a clearly nonfunctional genomic region. Previous estimates of substitution rates should be taken with caution because they were estimated from a limited number of pseudogenes that have not been proven not to be under selective constraints (Li et al. 1981).
As shown by the different number of CpG dinucleotides that are found to be polymorphic in the 5′ and 3′ moieties of psGBA, mutation does not seem to have a homogeneous action along the pseudogene. Moreover, a phylogenetic network of psGBA haplotypes showed instances of repeated mutations at some sites, although most of psGBA is fixed.
The nonsignificance of Tajima and Fu and Li tests does not allow us to directly reject neutrality for psGBA. However, it does not imply absence of selection, either. It might be that to detect the effect of selection on psGBA these tests would require a larger sample size (Simonsen et al. 1995). Besides, the variability observed for psGBA (π = 0.00044) is low in comparison to the values observed for other autosomal loci, with nucleotide diversity values that range from 0.0196 for the HLA-H pseudogene (Grimsley et al. 1998) to 0.0005 for the Apolipoprotein E gene (Fullerton et al. 2000). The nucleotide diversity of psGBA is lower than for all autosomal coding regions. Although the standard tests for neutral evolution were not statistically significant, this low diversity could indicate selection having an effect on psGBA.
Gene conversion seems to have played an important role in the evolution of GBA and psGBA, because sequences at several tracts are probably due to this mechanism. Gene conversion is a homogenizing mechanism between homologous loci in the genome. It consists of a nonreciprocal transfer of information: An allele (information acceptor) is modified by a second allele (information donor) that remains unchanged. The length of the DNA segment converted can vary from a few base pairs to several hundreds. Gene conversion cannot be proved without ambiguity because its result is not distinguishable from a double crossing-over event. However, the probability of a double crossing-over event in a tract shorter than several hundred kilobases is extremely low (Broman and Weber 2000). Thirteen percent of the psGBA sequence probably has its origins on GBA. These ancient gene conversion tracts are fixed on psGBA; they may have happened well before the TMRCA of the current haplotypes, and therefore they do not have any effect on the observed variability. The fact that the gene conversion tracts detected are random with respect to exon–intron distribution was expected, because the transference of any DNA fragment to psGBA does not have functional implications. We cannot discard recent gene conversions as the cause for some of the segregating positions, that is, 2253, 2266, 4020, and 4938. Recurrent mutation due to the hypermutability of CpG dinucleotides could as well account for the mutations 2253 and 4938.
It might be worth noting that we have detected gene conversion from GBA (under selective pressure) to psGBA (nonfunctional, and therefore presumably without purifying selection), but not the other way around. Gene conversions from psGBA to GBA happen indeed and have been detected, because the individuals carrying those converted alleles are affected with GD (Koprivica et al. 2000; Stone et al. 2000). However, these individuals have a low fitness (GBA alleles with psGBA tracts interrupting the reading frame are lethal in homozygosity), and these GBA alleles are either not passed on to the next generation or are lost slowly over time because of purifying selection. Thus, the detailed knowledge of sequence variation at psGBA may be crucial for recognizing psGBA to GBA gene conversion events in GD chromosomes.
The GBA Region Phylogeny
When psGBA and GBA sequences from human and chimpanzee were compared, it was clear that the homologous human and chimpanzee pseudogenes were much more related to each other than to their paralogous genes. However, by bringing sequences from GBA to psGBA, gene conversion events would have partly homogenized the GBA–psGBA tract. We estimated that at least 13% of the human psGBA sequence was, in fact, GBA sequence transported by gene conversion. These tracts were fixed in all chromosomes in the sample, indicating that the gene conversion events that generated them preceded the MRCA of human variation. The homogenizing effect of gene conversion should be taken into account when estimating duplication times from the differences between psGBA and GBA. The calculated estimate for the duplication time at 23.4 My ago would be solely due to, at most, the 87% of the psGBA sequence not affected by gene conversion. Thus, the time estimate can be corrected for the length of the homogenized region, and a 26.9-Mya date is obtained. Besides the likely underestimate of the extent of gene conversion, this figure may have an additional downward bias. Since the duplication event and before one of the GBA copies was inactivated, both copies may have evolved under the same constraints and at the same slow rate, which would have later increased for the copy that became psGBA. Because we have assumed that the substitution rate for psGBA is constant after the duplication event, we may have underestimated the duplication date. Our estimate is at the low end for the age range from 25 to 40 My suggested previously (Winfield et al. 1997).
psGBA and Human Evolution
The haplotype that is closest to the human MRCA sequence is, according to the haplotype phylogeny, haplotype 24, which has been found only in the Biaka. The two most frequent haplotypes, which, as all other haplotypes, are more derived than 24, are found in Africans as well as in non-Africans. This pattern, in which the most ancestral sequences are found only in Africa, has been observed repeatedly in sequences in mitochondrial DNA (Vigilant et al. 1991), autosomes (Harding et al. 1997; Clark et al. 1998), the X chromosome (Hey 1997; Zietkiewicz et al. 1997), and the Y chromosome (Shen et al. 2000), as well as in Y-chromosome polymorphisms (Hammer et al. 1995; Underhill et al. 1997). This set of observations, among others, shows coalescence times younger than 1 My, and that genetic diversity in non-Africans is a phylogenetic subset of that in Africans, and, therefore, it is compatible with a common, recent origin of anatomically modern humans in Africa.
The structure of the haplotype phylogeny contains two haplotype groups that radiate, respectively, from haplotypes 3 and 17. The former haplotype group has a clearer starlike structure and is in a looser linkage disequilibrium with polymorphisms at the GBA gene. Both features indicate an older age for haplogroup 3 than for 17, which is confirmed by the ages estimated for the most derived mutations that define haplotypes 3 and 17, which are, respectively, 74,000 ± 27,300 yr ago for 4938 and 37,500 ± 16,000 yr ago for 4020.
The TMRCA estimated for psGBA, ∼199,000 yr ago under the constant population model and ∼91,000 yr ago under the growth model, is the most recent found to date for autosomal loci. Previous estimates from autosomal loci locate the TMRCA around 1 Mya (Harding et al. 1997; Clark et al. 1998). Our estimate would be closer to the age estimated recently for the Y chromosome (50,000 yr ago; Thomson et al. 2000), correcting for the fourfold lower population size of the Y chromosome, and for the Apolipoprotein E gene (300,000 yr ago; Fullerton et al. 2000). Nevertheless, one should be cautious when making inferences from genomic data to population history, because it might be that the ages we obtain are influenced more by genomic than by population events. Different genomic regions may have different evolutive histories. For instance, selection could have had an influence on shaping the psGBA variability pattern. This would shorten the psGBA gene genealogy observed currently. What is clear from the data on psGBA is that it is possible to obtain such recent ages for autosomal loci. Different coalescence ages are being obtained from different human loci. Thus, perhaps the distribution of coalescence times over a number of loci is more informative than any single-locus estimate.
In summary, we have shown the interplay of a number of forces, such as recombination, recurrent mutation, and gene conversion, in shaping the phylogeny and polymorphism of a human autosomal pseudogene. Both aspects of the dynamics of the genome region, genomic and population-based factors, have been uncovered in a complex but meaningful analysis.
METHODS
To have a global representation of the variability pattern on psGBA, five individuals from 10 populations representing all major world geographic areas were analyzed: Biaka Pygmies (from the Central African Republic) and Tanzanians (from the region of Morogoro in the South East of Tanzania), both from sub-Saharan Africa; Saharawi from Western Sahara, in North Africa; Druze from Northern Israel, in the Middle East; Basques and Catalans, both from the Iberian Peninsula, in Europe; Yakut (from Siberia) and Han Chinese, both from East Asia; Mayan from Yucatan, America; Nasioi from Bougainville Island in Melanesia, Pacific. Informed consent was obtained from all individuals included in this study. DNA from Basque, Catalan, Tanzanian, and Saharawi samples was extracted from fresh blood using a standard phenol–chloroform extraction method after digestion with proteinase K. DNA from Biaka, Mayan, Yakut, Chinese, Druze, and Nasioi was obtained from lymphoblastoid cell lines maintained in Kenneth and Judy Kidd's laboratory at Yale University. Two unrelated chimpanzees (Pan troglodytes) and two unrelated gorillas (Gorilla gorilla) were included in the sample to perform phylogenetic comparisons. In total, the precise sequence of 108 psGBA alleles has been determined, and almost 600 kb sequenced.
A 5.7-kb DNA segment encompassing the psGBA region was amplified using psGBA-specific primers (forward primer sequence: 5′-acatcacggtagcctcagcatgttgtg-3′; reverse primer sequence: 5′-ccccaagactggtttttctactctcatgac-3′). PCR conditions were as follows: 0.24 mM dNTPs, 6 × 10−5 mM each primer, 200–400 ng genomic DNA, 1.5 mM MgCl2 buffer, 3.5 units of High Fidelity enzyme mix in 50 μL final volume. The PCR profile starts with a denaturation step of 2 min at 94°C, followed by 10 cycles of 15 sec at 94°C, 30 sec at 60°C, 4 min at 68°C, 20 cycles with the same conditions but with 20 sec additional elongation per cycle, and a final elongation step for 8 min at 72°C. The PCR amplicon was sequenced directly on an automated sequencer (ABI PRISM 377, PE Biosystems), using the ABI PRISM dRhodamine Terminator Cycle Sequencing Ready Reaction kit with Ampli Taq Polymerase (PE Biosystems). DNA sequencing was performed using a battery of primers that yield sequencing fragments that overlap between successive reactions (details of the primers used are available on request). The chromatograms were imported into the Seqman II software (Lasergene package, DNASTAR Inc.), assembled, and analyzed. A visual screening was also performed to detect any suspected heterozygous site. Heterozygous sites were detected and the genotypes for all the individuals were obtained. A 5420-bp tract could be ascertained unambiguously for all the individuals of our sample set. PCR-amplified and sequenced regions are indicated in Figure 1. Position 1 was defined as the first nucleotide in this stretch, which corresponds to nucleotide 280 in the pseudogene sequence in Horowitz et al. (1989) (GenBank J03060). The same primer pair and PCR conditions were used to amplify psGBA from human, chimpanzee, and gorilla samples. The whole amplified segment was cloned for those samples for which haplotype assignment was not direct, that is, those with more than one heterozygous site. To discern the phase among them, tracts with heterozygous sites were resequenced in one clone from each cloned sample. The sequence of the other allele was inferred, and the phase was reconfirmed by sequencing a different clone.
The GBA sequence was obtained for one chimpanzee. PCR GBA-specific primers were used (forward: 981–1006, reverse: 8203–8224, according to GenBank J03059 for the human GBA sequence). PCR conditions were the same as those used for psGBA, except that the annealing temperature was decreased from 60°C to 58°C, and elongation time was increased from 4 to 8 min. Additional inner primers, located at positions 2436–2453, 4018–4001, and 3038–3057 (numeration from 5′ to 3′ according to GenBank J03059), were used for sequencing.
Diversity parameters were calculated with the DnaSP software (DNA Sequence Polymorphism version 3.14; Rozas and Rozas 1999). Only single-nucleotide substitutions were considered in the calculations of the diversity parameters. Fst and haplotype ascertainment between psGBA and GBA polymorphisms were estimated with the Arlequin version 2.000 package (Schneider et al. 2000). Network 2.0b analysis software was used to establish median-joining networks among the haplotypes of our sample set (Bandelt et al. 1995). Neighbor-joining trees (Saitou and Nei 1987) were built from a sequence distance matrix computed with the DNADIST program, in the Phylip 3.5c package (Felsenstein 1989). The SITES program (http://heylab.rutgers.edu/) was used to compute the γ recombination parameter. The GENETREE program was used to estimate coalescence times and the age of mutations (Griffiths and Tavaré 1994; 1998a; 1998b). In addition, we calculated an independent estimate of the age of haplogroup 3; considering a constant-rate neutral mutation process, the number of mutations that would have accumulated in a given sample of sequences springing from a common ancestral sequence follows a Poisson distribution with mean λ = μ t, where μ is the mutation rate per segment and per year and t is the time elapsed since the coalescence of all the sequences. We can estimate λ as an average number of differences from the ancestral sequence in haplogroup 3 (Bertranpetit and Calafell 1996). The standard error of λ was estimated as (λ/n)1/2, where n is the number of chromosomes considered (Rando et al. 1998).
Acknowledgments
We are indebted to K.M. Weiss and A. Buchanan for technical assistance and helpful comments on an earlier version of the manuscript. We thank Kenneth K. Kidd, Judith R. Kidd, and B. Bonné-Tamir for sharing DNA samples. This research was supported by Dirección General de Investigación Científica y Técnica (Spanish Government) grant PB98–1064, and by Generalitat de Catalunya, Grup de Recerca Consolidat 1998SGR00009. R.M.-A. received a fellowship from the Spanish Ministry of Education and Culture (AP96).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL jaume.bertranpetit@cexs.upf.es; FAX 34–93–542 28 02.
Article published on-line before print: Genome Res., 10.1101/gr.167701.
Article and publication are at www.genome.org/cgi/doi/10.1101/gr.167701.
REFERENCES
- Bandelt H, Forster P, Sykes BC, Richards MB. Mitochondrial portraits of human populations using median networks. Genetics. 1995;141:743–753. doi: 10.1093/genetics/141.2.743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbujani G, Magagni A, Minch E, Cavalli-Sforza L. An appointment of human DNA diversity. Proc Natl Acad Sci. 1997;94:4516–4519. doi: 10.1073/pnas.94.9.4516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertranpetit J, Calafell F. Genetic and geographical variability in cystic fibrosis: Evolutionary considerations. In: Cardew G, editor. Variation in the human genome. Chichester: Wiley; 1996. pp. 97–118. (Ciba Foundation Symposium 197). [DOI] [PubMed] [Google Scholar]
- Beutler E, West C, Gelbart T. Polymorphisms in the human glucocerebrosidase gene. Genomics. 1992;12:795–800. doi: 10.1016/0888-7543(92)90311-f. [DOI] [PubMed] [Google Scholar]
- Broman KW, Weber JL. Characterization of human crossover interference. Am J Hum Genet. 2000;66:1911–1926. doi: 10.1086/302923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark GA, Weiss KM, Nickerson DA, Taylor SL, Buchanan A, Stengard J, Salomaa V, Vartiainen E, Perola M, Boerwinkle E, et al. Haplotype structure and population genetic inferences from nucleotide-sequence variation in human lipoprotein lipase. Am J Hum Genet. 1998;63:595–612. doi: 10.1086/301977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper DN. Pseudogenes and their formation. In: Cooper DN, editor. Human Gene Evolution. Oxford: BIOS Scientifics Publishers Ltd.; 1999. pp. 265–296. [Google Scholar]
- Devine EA, Smith M, Arredongo-Vega FX, Shafit-Zagardo B, Desnick RJ. Regional assignment of the structural gene for human acid β-glucosidase to q42→qter on chromosome 1. Cytogenet Cell Genet. 1982;33:340–344. doi: 10.1159/000131781. [DOI] [PubMed] [Google Scholar]
- Dorit RL, Akashi H, Gilbert W. Absence of polymorphisms at the ZFY locus on the human Y chromosome. Science. 1995;268:1183–1185. doi: 10.1126/science.7761836. [DOI] [PubMed] [Google Scholar]
- Felsenstein J. PHYLIP-phylogeny inference package (version 3.2) Cladistics. 1989;5:164–166. [Google Scholar]
- Fu YX, Li WH. Statistical tests of neutrality of mutations. Genetics. 1993;133:693–709. doi: 10.1093/genetics/133.3.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fullerton SM, Harding RM, Boyce AJ, Clegg JB. Molecular and population genetic analysis of allelic sequence diversity at the human β-globin locus. Proc Natl Acad Sci. 1994;91:1805–1809. doi: 10.1073/pnas.91.5.1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fullerton SM, Clark AG, Weiss KM, Nickerson DA, Taylor ST, Stengård JH, Salomaa V, Vartiainen E, Perola M, Boerwinkle E, et al. Apolipoprotein E variation at the sequence haplotype level: Implications for the origin and maintenance of a major human polymorphism. Am J Hum Genet. 2000;67:881–900. doi: 10.1086/303070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginns E, Choudary PV, Tsuji S, Martin B, Stubblefield B, Sawyer J, Hozier J, Barranger J. Gene mapping and leader polypeptide sequence of human glucocerebrosidase: Implications for Gaucher disease. Proc Natl Acad Sci. 1985;82:7101–7105. doi: 10.1073/pnas.82.20.7101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glenn D, Gelbart T, Beutler E. Tight linkage of pyruvate kinase (PKLR) and glucocerebrosidase (GBA) genes. Hum Genet. 1994;93:635–638. doi: 10.1007/BF00201562. [DOI] [PubMed] [Google Scholar]
- Griffiths RC, Tavaré S. Ancestral inference in population genetics. Stat Sci. 1994;9:307–319. [Google Scholar]
- ————— The age of a mutation in a general coalescent tree. Commun Stat. 1998a;14:273–295. [Google Scholar]
- ————— The ages of mutations in gene trees. Ann Appl Prob. 1998b;9:567–590. [Google Scholar]
- Grimsley C, Mather KA, Ober C. HLA-H: A pseudogene with increased variation due to balancing selection at neighboring loci. Mol Biol Evol. 1998;15:1581–1588. doi: 10.1093/oxfordjournals.molbev.a025886. [DOI] [PubMed] [Google Scholar]
- Halushka MK, Fan J-B, Bentley K, Hsie L, Shen N, Weder A, Cooper R, Lipshutz R, Chakravarti A. Patterns of single-nucleotide polymorphisms in candidate genes for blood-pressure homeostasis. Nat Genet. 1999;22:239–247. doi: 10.1038/10297. [DOI] [PubMed] [Google Scholar]
- Hammer MF. A recent common ancestry for human Y chromosomes. Nature. 1995;378:376–378. doi: 10.1038/378376a0. [DOI] [PubMed] [Google Scholar]
- Harding RM, Fullerton SM, Griffiths RC, Bond J, Cox MJ, Schneider JA, Moulin DS, Clegg JB. Archaic African and Asian lineages in the genetic ancestry of modern humans. Am J Hum Genet. 1997;60:772–789. [PMC free article] [PubMed] [Google Scholar]
- Harris EE, Hey J. X chromosome evidence for ancient human histories. Proc Natl Acad Sci. 1999;96:3320–3324. doi: 10.1073/pnas.96.6.3320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hey J. Mitochondrial and nuclear genes present conflicting portraits of human origins. Mol Biol Evol. 1997;14:166–172. doi: 10.1093/oxfordjournals.molbev.a025749. [DOI] [PubMed] [Google Scholar]
- Hey J, Wakeley J. A coalescent estimator of the population recombination rate. Genetics. 1997;145:833–846. doi: 10.1093/genetics/145.3.833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong CM, Ohashi T, Yu XY, Weiler S, Barranger JA. Sequence of two alleles responsible for Gaucher disease. DNA Cell Biol. 1990;9:233–241. doi: 10.1089/dna.1990.9.233. [DOI] [PubMed] [Google Scholar]
- Horowitz M, Wilder S, Horowitz Z, Reiner O, Gelbart T, Beutler E. The human glucocerebrosidase gene and pseudogene: Structure and evolution. Genomics. 1989;4:87–96. doi: 10.1016/0888-7543(89)90319-4. [DOI] [PubMed] [Google Scholar]
- Hudson RR. Estimating the recombination parameter of a finite population model without selection. Genet Res. 1987;50:245–250. doi: 10.1017/s0016672300023776. [DOI] [PubMed] [Google Scholar]
- Hudson R, Kaplan NL. Statistical properties of the number of recombination events in the history of a sample of DNA sequences. Genetics. 1985;111:147–164. doi: 10.1093/genetics/111.1.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai K, Nakamura M, Yamada M, Asano A, Yokoyama S, Tsuji S, Ginns E. A novel transcript from a pseudogene for human glucocerebrosidase in non-Gaucher disease cells. Gene. 1993;136:365–368. doi: 10.1016/0378-1119(93)90497-q. [DOI] [PubMed] [Google Scholar]
- Jaruzelska J, Zietkiewicz E, Batzer M, Cole DEC, Moisan JP, Scozzari R, Tavaré S, Labuda D. Spatial and temporal distribution of the neutral polymorphisms in the last ZFX intron: Analysis of the haplotype structure and genealogy. Genetics. 1999a;152:1091–1101. doi: 10.1093/genetics/152.3.1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaruzelska J, Zietkiewicz E, Labuda D. Is selection responsible for the low level of variation in the last intron of the ZFY locus? Mol Biol Evol. 1999b;16:1633–1640. doi: 10.1093/oxfordjournals.molbev.a026076. [DOI] [PubMed] [Google Scholar]
- Jorde LB, Watkins WS, Bamshad MJ, Dixon ME, Ricker CE, Seielstad MT, Batzer MA. The distribution of human genetic diversity: A comparison of mitochondrial, autosomal, and Y-chromosome data. Am J Hum Genet. 2000;66:979–988. doi: 10.1086/302825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaessman H, Heiβig F, Haeseler A, Pääbo S. DNA sequence variation in a non-coding region of low recombination on the human X chromosome. Nat Genet. 1999;22:78–81. doi: 10.1038/8785. [DOI] [PubMed] [Google Scholar]
- Koprivica V, Stone DL, Park JK, Callahan M, Frisch A, Cohen IJ, Tayebi N, Sidransky E. Analysis and classification of 304 mutant alleles in patients with type 1 and type 3 Gaucher disease. Am J Hum Genet. 2000;66:1777–1786. doi: 10.1086/302925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latham TE, Theophilus BDM, Grabowski GA, Smith FI. Heterogeneity of mutations in the acid β-glucosidase gene of Gaucher disease patients. DNA Cell Biol. 1991;10:15–21. doi: 10.1089/dna.1991.10.15. [DOI] [PubMed] [Google Scholar]
- Li W-H, Sadler LA. Low nucleotide diversity in man. Genetics. 1991;129:513–523. doi: 10.1093/genetics/129.2.513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W-H, Gojobori T, Nei M. Pseudogenes as a paradigm of neutral evolution. Nature. 1981;292:237–239. doi: 10.1038/292237a0. [DOI] [PubMed] [Google Scholar]
- Nachman MW, Bauer VL, Crowell SL, Charles FA. DNA variability and recombination rates at X-linked loci in humans. Genetics. 1998;150:1133–1141. doi: 10.1093/genetics/150.3.1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nei M, Li W-H. Mathematical model for studying genetic variation in terms of restriction endonucleases. Genetics. 1979;76:5369–5273. doi: 10.1073/pnas.76.10.5269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rana BK, Hewett-Emmett D, Jin L, Chang BH, Sambuughin N, Lin M, Watkins S, Bamshad M, Jorde LB, Ramsay M, et al. High polymorphism at the human melanocortin 1 receptor locus. Genetics. 1999;151:1547–1557. doi: 10.1093/genetics/151.4.1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rando JC, Pinto F, González AM, Hernández M, Larruga JM, Cabrera VM, Bandelt H-J. Mitochondrial DNA analysis of Northwest African populations reveals genetic exchange with European, Near-Eastern, and sub-Saharan populations. Ann Hum Genet. 1998;62:531–550. doi: 10.1046/j.1469-1809.1998.6260531.x. [DOI] [PubMed] [Google Scholar]
- Reiner O, Horowitz M. Differential expression of the human glucocerebrosidase-coding gene. Gene. 1988;17:469–478. doi: 10.1016/0378-1119(88)90511-2. [DOI] [PubMed] [Google Scholar]
- Rieder MJ, Taylor SL, Clark AG, Nickerson NA. Sequence variation in the human angiotensin converting enzyme. Nat Genet. 1999;22:59–62. doi: 10.1038/8760. [DOI] [PubMed] [Google Scholar]
- Rozas J, Rozas R. DnaSP version 3: An integrated program for molecular population genetics and molecular evolution analysis. Bioinformatics. 1999;15:174–175. doi: 10.1093/bioinformatics/15.2.174. [DOI] [PubMed] [Google Scholar]
- Saitou N, Nei M. The neighbour-joining method: A new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4:406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- Schneider S, Kueffer JM, Roessli D, Excoffier L. Arlequin ver.2000: A software environment for the analysis of population genetics data. Switzerland: Geneva, Genetics and Biometry Laboratory, University of Geneva; 2000. [Google Scholar]
- Shafit-Zagardo B, Devine EA, Smith M, Arredongo-Vega F, Desnick RJ. Assignment of the gene for acid β-glucosidase to human chromosome 1. Am J Hum Genet. 1981;33:564–575. [PMC free article] [PubMed] [Google Scholar]
- Shen P, Wang F, Underhill PA, Franco C, Yang W-H, Roxas A, Sung R, Lin AA, Hyman RW, Vollrath D, et al. Population genetic implications from sequence variation in four Y chromosome genes. Proc Natl Acad Sci. 2000;97:7354–7359. doi: 10.1073/pnas.97.13.7354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonsen KL, Churchill GA, Aquadro CF. Properties of statistical tests of neutrality for DNA polymorphism data. Genetics. 1995;141:413–429. doi: 10.1093/genetics/141.1.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorge J, West C, Westwood B, Beutler E. Molecular cloning and nucleotide sequence of human glucocerebrosidase cDNA. Proc Natl Acad Sci. 1985;82:7289–7293. doi: 10.1073/pnas.82.21.7289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorge J, Gross E, West C, Beutler B. High level transcription of the glucocerebrosidase pseudogene in normal subjects and patients with Gaucher disease. J Clin Invest. 1990;86:1137–1141. doi: 10.1172/JCI114818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone DL, Tayebi N, Orvisky E, Stubblefield B, Madike V, Sidransky E. Glucocerebrosidase gene mutations in patients with type 2 Gaucher disease. Hum Mutat. 2000;15:181–188. doi: 10.1002/(SICI)1098-1004(200002)15:2<181::AID-HUMU7>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- Tajima F. Statistical method for testing the neutral mutation hypothesis. Genetics. 1989;123:585–595. doi: 10.1093/genetics/123.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson R, Pritchard JK, Shen P, Oefner PJ, Feldman MW. Recent common ancestry of human Y chromosome: Evidence from DNA sequence data. Proc Natl Acad Sci. 2000;97:7360–7365. doi: 10.1073/pnas.97.13.7360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Underhill PA, Jin L, Lin AA, Mehdi SQ, Jenkins T, Vollrath D, Davis RW, Cavalli-Sforza LL, Oefner PJ. Detection of numerous Y chromosome biallelic polymorphisms by denaturing high-performance liquid chromatography. Genome Res. 1997;7:996–1005. doi: 10.1101/gr.7.10.996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigilant L, Stoneking M, Harpending H, Hawkes K, Wilson AC. African populations and the evolution of human mitochondrial DNA. Science. 1991;253:1503–1507. doi: 10.1126/science.1840702. [DOI] [PubMed] [Google Scholar]
- Watterson GA. On the number of segregating sites in genetical models without recombination. Theor Popul Biol. 1975;7:256–276. doi: 10.1016/0040-5809(75)90020-9. [DOI] [PubMed] [Google Scholar]
- Whitfield LS, Sulston JE, Goodfellow PN. Sequence variation of the human Y chromosome. Nature. 1995;378:379–380. doi: 10.1038/378379a0. [DOI] [PubMed] [Google Scholar]
- Winfield SL, Tayebi N, Martin BM, Ginns EI, Sidransky E. Identification of three additional genes contiguous to the glucocerebrosidase locus on chromosome 1q21: Implications for Gaucher disease. Genome Res. 1997;7:1020–1026. doi: 10.1101/gr.7.10.1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zietkiewicz E, Yotova V, Jarnik M, Korab-Laskowska M, Kidd KK, Modiano D, Scozzari R, Stoneking M, Tishkoff S, Batzer M, et al. Nuclear DNA diversity in worldwide distributed human populations. Gene. 1997;205:161–171. doi: 10.1016/s0378-1119(97)00408-3. [DOI] [PubMed] [Google Scholar]
- ————— Genetic structure of the ancestral population of modern humans. J Mol Evol. 1998;47:146–155. doi: 10.1007/pl00006371. [DOI] [PubMed] [Google Scholar]
- Zimran A, Sorge J, Gross E, Kubitz M, West C, Beutler E. A glucocerebrosidase fusion gene in Gaucher disease. J Clin Invest. 1990;85:219–222. doi: 10.1172/JCI114415. [DOI] [PMC free article] [PubMed] [Google Scholar]