

Abstract
Rotamer libraries are used in protein structure determination, structure prediction, and design. The backbone-dependent rotamer library consists of rotamer frequencies and their mean dihedral angles and variances as a function of the backbone dihedral angles ϕ and ψ. Previous versions of this rotamer library were not developed with smoothness in mind, although some structure prediction and protein design methods would strongly benefit from smoothing. A new version of the backbone-dependent rotamer library has been developed using adaptive kernel density estimates for the rotamer frequencies and adaptive kernel regression for the mean dihedral angles and variances. The formulation presented allows for evaluation of the rotamer probabilities, mean angles and variances at any ϕ, ψ point, i.e. as a continuous function of ϕ and ψ. Continuous probability density estimates for the non-rotameric degrees of freedom of amides, carboxylates, and aromatic side chains have been modeled as a function of the backbone dihedral angles and rotamers of the remaining degrees of freedom. New backbone-dependent rotamer libraries at varying levels of smoothing are available from http://dunbrack.fccc.edu.
INTRODUCTION
Rotamers are discrete conformations of organic molecules arising from large barriers to rotation about single bonds. Protein side-chain rotamer libraries, which contain frequencies, mean dihedral angles, and standard deviations of common conformations (Dunbrack and Cohen, 1997; Dunbrack and Karplus, 1993; Lovell et al., 2000) are used extensively in structure determination, structure prediction, and protein design. The subdivision of dihedral angle space into rotamers for the sp3-sp3 hybridized degrees of freedom enables fast enumeration over all possible conformers. In structure determination they are used as a search space in the process of fitting side-chain conformations to electron density (Adams et al., 2002; Headd et al., 2009) as well as in a number of structure validation methods (Davis et al., 2004). In structure prediction, they are used as a discrete search space of conformations (Desmet et al., 1992; Dunbrack and Karplus, 1993), and log rotamer probabilities are sometimes used as a term in scoring functions (Canutescu et al., 2003; Krivov et al., 2009; Liang and Grishin, 2002; Rohl et al., 2004b). In protein design, the sequence is altered by substituting in rotamers of different residue types and scoring these conformations in the environment of the side chain, including the rest of the protein and ligands and/or protein partners (Gordon et al., 2003; Kuhlman and Baker, 2004). Thus rotamer libraries form a critical element in much of computational structural biology, and their ongoing development remains an important task.
We have previously developed backbone-dependent rotamer libraries in which the rotamer frequencies and mean dihedral angles and their standard deviations are given on a 10°×10° grid of the backbone dihedral angles ϕ and ψ (Dunbrack, 2002; Dunbrack and Cohen, 1997). These libraries were developed using a Bayesian formalism by combining a prior estimate of the probabilities for each (ϕ, ψ) bin with raw counts of the rotamers in overlapping 20°×20° bins (Dunbrack and Cohen, 1997). The prior estimates came from modeling the observed (ϕ, ψ)-dependent frequencies as the product of ϕ and ψ dependencies. The mean dihedral angles and their variances were determined with a Bayesian normal model that combined separate ϕ- and ψ-dependent estimates with data points around each (ϕ, ψ) grid point.
In attempting to optimize the most recent version of this rotamer library (Dunbrack, 2002) in the program Rosetta (Rohl et al., 2004b) we found that both the rotamer probabilities and the mean dihedral angles and their standard deviations were quite bumpy in their variation with ϕ and ψ, a result of using raw counts in the probability estimates and calculation of simple averages. Rosetta uses the first derivatives of the rotamer probabilities, −∂log P(r|ϕ, ψ)/∂ϕ and −∂log P(r|ϕ, ψ)/∂ψ, in the local minimization of its scoring function (Leaver-Fay et al., 2011). The jaggedness of the rotamer library is likely to cause artifacts in any structure determination, prediction, or design program that models backbone flexibility and utilizes local minimization of scoring terms based on the backbone-dependent rotamer library. Backbone flexibility is increasingly incorporated into comparative modeling and protein design (Friedland et al., 2008; Smith and Kortemme, 2008).
Another shortcoming of the previous libraries was the treatment of non-rotameric degrees of freedom, in particular the amide, carboxylate and aromatic dihedral angle degrees of freedom (the terminal χ angles of Asn, Asp, Gln, Glu, Phe, Trp, His, Tyr). These degrees of freedom, connecting sp3 to sp2 hybridized groups, are difficult to describe as “rotamers” with mean dihedral angles and variances about these means. Instead, they are usually quite broadly distributed with asymmetric density distributions (Lovell et al., 1999). These distributions may vary with the backbone conformation, as the polar side chains interact electrostatically with the local backbone and the aromatic side chains encounter large steric clashes dependent on ϕ and ψ. It is therefore desirable to calculate a full density distribution of these dihedral angles for each (ϕ, ψ) grid point and χ1 rotamer (or χ1,χ2 rotamer for Gln and Glu). This is a complex estimation problem involving the regression of a density onto two angular degrees of freedom.
In this paper, our aims in deriving a new backbone-dependent rotamer library are several: 1) to take advantage of the much larger dataset that is available now than at the time of the last library (2002); 2) to use electron density calculations to remove highly dynamic side chains (or protein segments) that have uncertain conformations or coordinates (Shapovalov and Dunbrack, 2007); 3) to derive accurate and smooth density estimates of rotamer populations and their relative frequencies, including rare rotamers, as a continuous function of backbone dihedral angles; 4) to derive smooth estimates of the mean values and variances of rotameric side-chain dihedral angles; 5) to improve the treatment of non-rotameric degrees of freedom, i.e. those are not well described by the rotamer model; and 6) to employ methods producing meaningful estimates of rotamer frequencies, dihedral angles means and variances in the Ramachandran areas lacking experimental data.
In order to produce smooth and continuous estimates of the rotamer probabilities in this work, we use kernel density estimation. A kernel is a non-negative symmetric function, such as a Gaussian, that integrates to 1.0 and is centered on each data point. Density estimates at specific query points are determined by summing the values of the kernel functions centered on the data points. The smoothness of the density estimate is determined by the form of the kernel, in particular its bandwidth. Wider kernels produce smoother density estimates, while narrow kernels produce bumpier estimates. For each rotamer r of a given residue type we determine a probability density estimate, ρ(ϕ, ψ|r), essentially a Ramachandran distribution for each rotamer, and then use Bayes’ rule to invert this density to produce an estimate of the rotamer probability, P(r|ϕ, ψ):
| (1) |
where P(r) is the backbone-independent probability of rotamer r.
Density estimates for angles are more appropriately modeled using the von Mises probability density function as the kernel rather than Gaussian or other non-periodic kernels (Mardia and Jupp, 2000). The von Mises distribution has the form: ρ(x)= exp(κ cosx)/I0(κ), where x is an angle on the circle and I0 is the modified Bessel function of the first kind of order zero. The concentration parameter, κ, is inversely proportional to the squared width of the von Mises kernel, with larger values of κ producing narrower kernels. In order to deal with the large variation in density of data points on the Ramachandran map, we use adaptive kernel density estimation (Abramson, 1982; Breiman et al., 1977), in which the bandwidth is allowed to vary with the local density of data points. In this way, in sparse regions the kernels placed on each data point are wider, while in dense regions the kernels are narrower.
An important feature of our rotamer libraries is the ϕ, ψ-dependence of the means and variances of the dihedral angles for each rotamer, especially for χ1. Due to interactions with the local backbone, both steric and electrostatic, these average angles have strong and systematic variation with ϕ and ψ for each rotamer (Dunbrack and Cohen, 1997). For this purpose in the new rotamer library, we use adaptive kernel regression estimators (Brockmann et al., 1993) to determine χ̄|ϕ, ψ, r as smoothly varying functions of the backbone dihedrals. For the kernel regressions, we make the concentration parameters of all kernels, κ, adaptive to the same local density of data around the query point, rather than the data point as in the kernel density estimates. We also make the variance heteroscedastic, that is dependent on the backbone dihedral angles ϕ and ψ.
In our earlier libraries, all dihedral angle degrees of freedom were treated as “rotameric”. That is, the entire dihedral angle space was broken up into bins and conformations counted. For asparagine, for instance, in 1997 we divided χ2 into three bins, (−90°,−30°], (−30°,+30°], (+30°,+90°], by considering OD1 and ND2 atoms as indistinguishable. Later in 2002, we used the reduce program of Word et al. (Word et al., 1999) to orient OD1 and ND2 of Asn as well as possible, considering hydrogen bonding patterns. We then divided χ2 in the range [−180°,180°) into six bins, with different offsets depending on the χ1 rotamer. In each of these bins, we calculated mean dihedral angles and standard deviations. This is a poor model for the density, which is broadly distributed and asymmetric. In this work, we produce probability density estimates for the non-rotameric degrees of freedom: ρ(χn|r−n,ϕ, ψ), where r−n in this case represents the rotameric degrees of freedom. This is accomplished by combining the techniques of adaptive kernel density estimation and adaptive kernel regression. These probability distributions will be useful in minimizing the conformational energies of flexible degrees of freedom on smooth potential energy surfaces in the form of U = −logρ(χn|r−n,ϕ,ψ).
The rotamer libraries described here are evaluated on a 10°×10° grid of ϕ and ψ, but it should be noted that the use of kernels with support from −π to π allows us to develop functions that can be evaluated as continuous functions of ϕ and ψ, i.e. at any value of ϕ and ψ, not just those on a predefined grid. This is in contrast to our previous rotamer library formulation using multinomial functions, which required integer counts of each rotamer type within square bins of ϕ, ψ space.
RESULTS
Data set
The data set used in the new rotamer library was prepared through a series of steps. We first determined the full list of protein-containing PDB entries for which we could obtain electron densities from the Uppsala Electron Density Server (EDS) (Kleywegt et al., 2004). We have shown previously that side chains with sp3-sp3 hybridized bonds with non-rotameric dihedral angles, those far from the typical mean values for (60°, 180°, 300°), have much lower electron density than average (Shapovalov and Dunbrack, 2007). This list was then filtered by the PISCES server (Wang and Dunbrack, 2005) and run through the SIOCS program to flip Asn, Gln, and His terminal dihedral angles to account for hydrogen bonding. Finally, we obtained a list of 3,985 protein chains from 3,845 entries with resolution better than or equal to 1.8 Å, an R-factor cutoff of 0.22, and mutual sequence identity of the chains of 50% or less.
We distinguish between rotameric and non-rotameric degrees of freedom based on the hybridization state of the atoms involved in the dihedral angle. Dihedral degrees of freedom are centered on sp3-sp3 hybridized bonds and exhibit three narrow, approximately symmetric peaks in their probability density distributions. As an example, the χ1 density for methionine is shown in Figure 1A, with gauche+ (g+), trans (t), and gauche− (g−) peaks at approximately 60°, 180°, and 300° respectively. Non-rotameric degrees of freedom in protein side chains, by contrast, are centered on sp3-sp2 bonds, and exhibit broad and often asymmetric probability density distributions. As examples, the χ2 probability densities for asparagine and tryptophan are shown in Figure 1B and Figure 1C for each of the three χ1 rotamers of these residue types. The χ3 densities for Gln depend on both the χ1 and χ2 rotamers as shown in Figure 1D, E, and F.
Figure 1. Backbone-independent distribution of rotameric and non-rotameric χ.
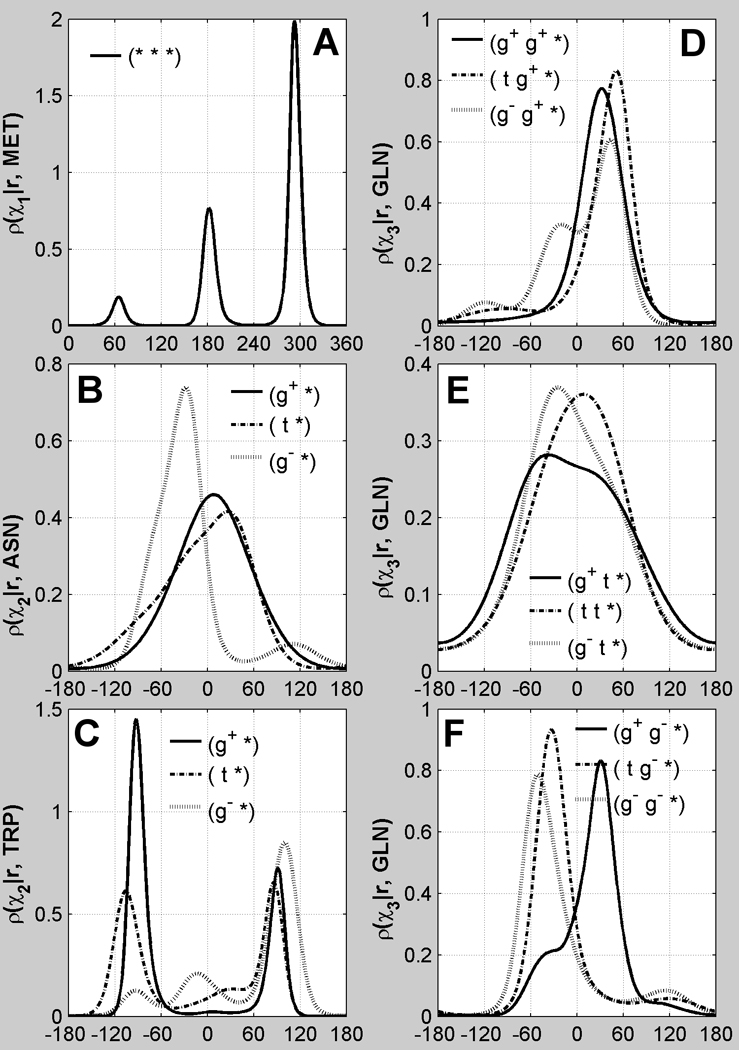
Panel A shows a probability density distribution of dihedral angles for a rotameric degree of freedom tightly and symmetrically clustered near the canonical values of 60°, 180° and 300° based on Met χ1 data regardless of χ1, χ2 or χ3 rotamer (*, *, *). Panels B and C depict distribution of the non-rotameric χ2 degree of freedom of Asn and Trp respectively for each of their χ1 rotamers: g+, t and g−. Panels D, E, and F show the backbone-independent distribution of non-rotameric χ3 of Gln for each of its (χ1, χ2) rotamers. Non-rotameric χ3 distributions for Gln are dependent on both the χ1 and χ2 rotamers. The distributions of the non-rotameric degrees of freedom are very broad and asymmetric and cannot be modeled with a rotameric model.
We calculated the electron density at the atom coordinates of 3,985 chains using methods described earlier (Shapovalov and Dunbrack, 2007) and calculated the geometric mean of the electron density at the atomic positions in each residue as a quality filter to remove disordered residues – those with electron densities in the bottom 25th percentile for each residue type. For the rotamer library calculations the resulting number of residues totaled 581,128 and their individual counts are given in Table S1 in the Supplementary Material along with the degrees of freedom defined for each side-chain type. We also accounted for incorrectly modeled leucine residues (see Figure S1 and Table S2), and we analyzed trans and cis proline separately, as well as disulfide-bonded and non-disulfide-bonded cysteines.
Deriving backbone-dependent rotamer probabilities from kernel density estimates
The challenging statistical problem that the backbone-dependent rotamer library presents is shown in Figure 2, a scatter plot of the nine leucine rotamer types on the Ramachandran map. The goal is to calculate P(r|ϕ, ψ), the probability of each rotamer as a function of the backbone dihedrals ϕ and ψ. The non-uniform distribution in ϕ,ψ and the large differences in overall populations and distributions of the different rotamers must all be accounted for. Our solution to this problem is to use adaptive kernel density estimates (AKDE) to obtain rotamer probability density functions, ρ(ϕ, ψ|r) from the input dataset {ϕi, ψi, ri} and to use Bayes’ rule to invert these densities to obtain the rotamer probabilities, P(r|ϕ, ψ).
Figure 2. The backbone-dependent rotamer library problem.
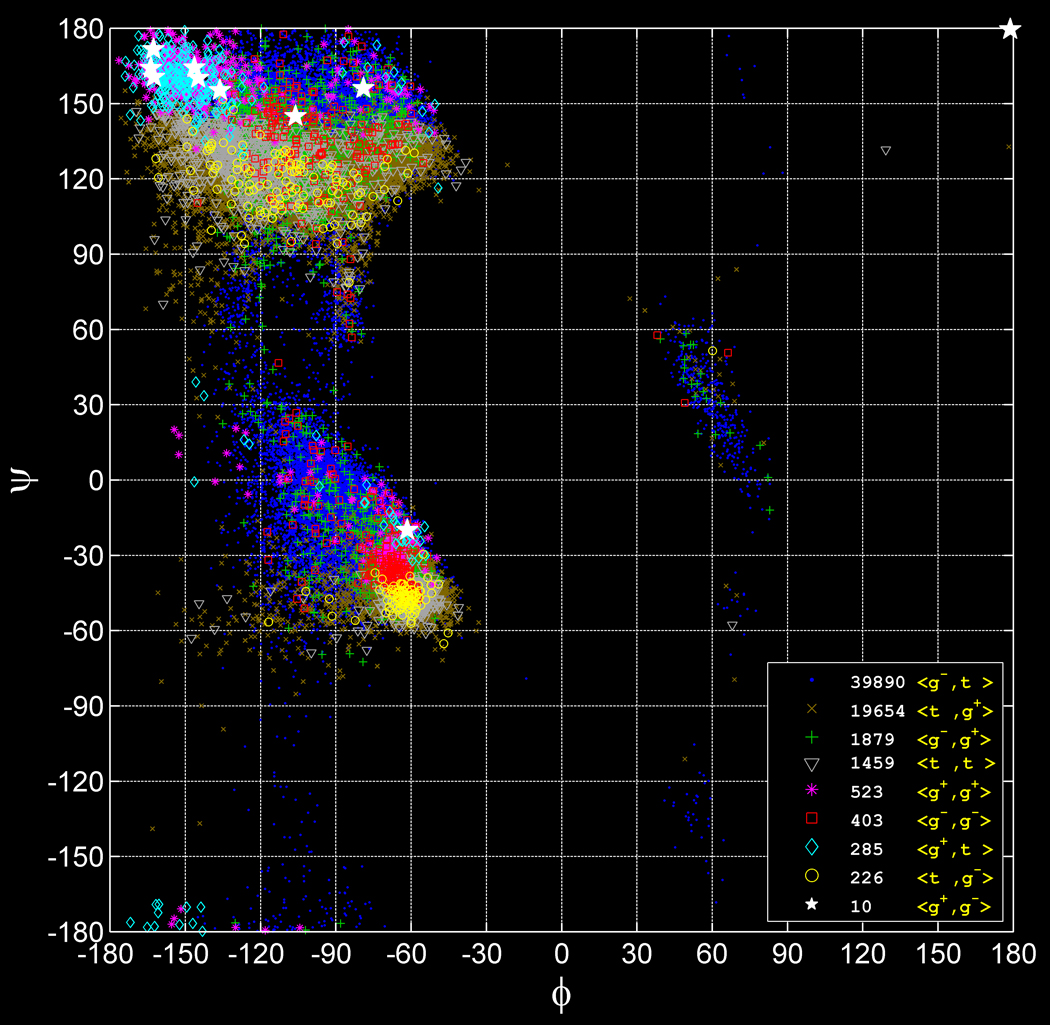
ϕ-ψ scatter plot of nine leucine rotamers and statistics of the total number of rotamers of each type. The scatter plot has larger and brighter markers for rare rotamers and smaller and darker markers – for abundant rotamers. The total number of rotamers differs significantly among the 9 types. The relative distributions of each rotamer depend strongly on backbone conformation.
As an example, in the top row of Figure 3 we show the probability density functions ρ(ϕ, ψ|r) of the g+, trans and g− rotamers of valine above their resulting backbone-dependent probabilities, P(r|ϕ, ψ), shown in the bottom row. The three rotamers have notably different ϕ, ψ probability densities which in turn produce quite different relative frequencies of the three rotamers as a function of ϕ and ψ. These estimates match conformational analysis of synpentane interactions (Wiberg and Murcko, 1988) of the side-chain Cγ1 and Cγ2 atoms with atoms of the backbone whose positions are dependent on ϕ and ψ (Dunbrack and Cohen, 1997; Dunbrack and Karplus, 1994).
Figure 3. Rotamer Ramachandran densities and their corresponding backbone-dependent rotamer probabilities from the new 2010 Rotamer Library.
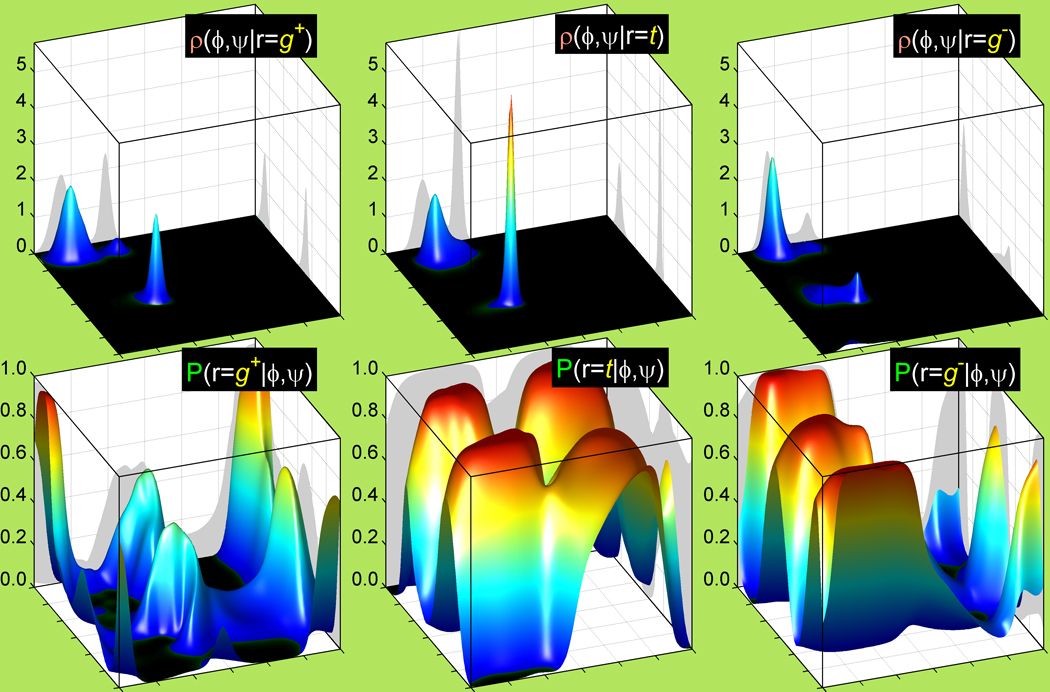
Top: Smoothed Ramachandran probability density functions (PDFs) of the backbone conformation (ϕ, ψ) for g+, trans and g− rotamers (left to right) of Val computed with Adaptive Kernel Density Estimation. ϕ and ψ are plotted along x-axis and y-axis respectively within their standard limits of (−180°, 180°). The PDFs are plotted along the z-axis and scaled in 1/radian2. For every rotamer the density integrates to 1 over the whole Ramachandran area. Bottom: Corresponding 2010 smooth backbone-dependent rotamer probabilities, calculated by inverting the Ramachandran densities in the top row with Bayes’ rule. The probabilities of all three g+, t and g− rotamers sum up to 1 for every (ϕ, ψ). The Val bandwidth radius is 5° and the concentration parameter, κ is 120. These values match the 5% stepdown from the optimal log-likelihood score for additional smoothness with the best SCWRL4 prediction rates (see Results).
To reach the results shown in Figure 3 for the new backbone-dependent rotamer library, we investigated and compared the results from a number of different methods. These are shown together in Figure 4 for the g+ rotamer of serine, P(r = g+|ϕ, ψ, aa = Ser). In the straightforward histogram approach (Figure 4A), the number of data points with a particular rotamer in every non-overlapping (ϕ, ψ) bin is counted and divided by the total number of data points of any rotamer type in the same bin. This approach produces crude estimates of the rotamer probabilities. The prevailing majority of the 10°×10° histogram bins have “unknown” values (set to 0 in the figure), produced by division of 0 points by 0 points. A large proportion of the bins have very spiky and extremely unreliable probability estimates.
Figure 4. Rotamer probability estimates produced by several methods and smoothing effect of adaptive kernel density with narrower or wider bandwidths.
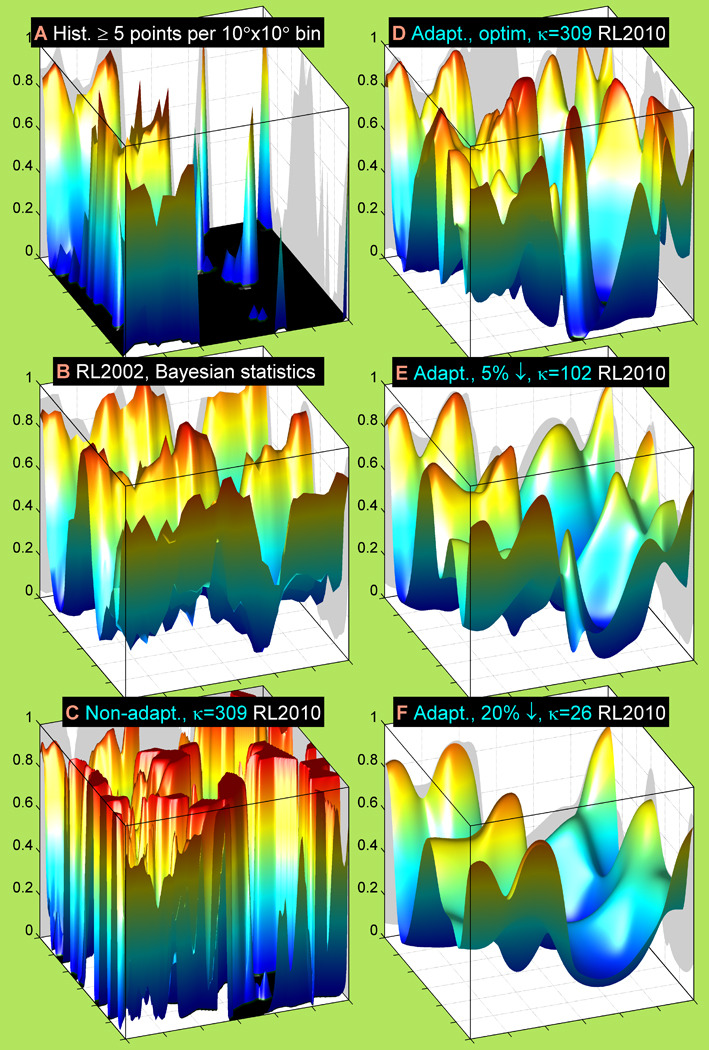
Non-overlapping 10°×10°-bin histogram (A), 2002 Bayesian (B), non-adaptive kernel density (C) and adaptive kernel density (D, E, F) estimates are shown for P(r = g+ | ϕ, ψ, aa = Ser). The histogram estimate (A) depicts only the bins with at least five points of any rotamer per bin. The non-adaptive kernel density estimate (C) has a fixed bandwidth (κ = 309, bandwidth radius, R = 3.3°), the same as for (D). The adaptive kernel density estimates with widening geometric-mean kernel bandwidth are ordered from (D) to (F). The maximum log-likelihood (κ = 309, R = 3.3°), 5%-stepdown (κ = 102, R = 6°) and 20%-stepdown (κ = 29, R = 11°) bandwidths are shown in panels (D), (E) and (F) respectively.
The Bayesian approach used in our 1997 and 2002 rotamer libraries used two-fold periodic kernels (although we did not call them as such at the time) to produce separate ϕ-dependent and ψ-dependent counts as a prior in the form of a Dirichlet function, which were combined with integer data counts in a multinomial likelihood to produce posterior estimates also in the form of Dirichlet functions (Dunbrack and Cohen, 1997). As shown in Figure 4B, this approach produced reasonable estimates for all values of ϕ and ψ but because of the integer counts in the Dirichlet function, the posterior estimates were very bumpy as a function of ϕ and ψ.
In this work, we are also using kernels to estimate the ϕ, ψ-dependent densities of each rotamer, but instead of combining them with data counts, we use the kernels directly to determine density estimates for each rotamer and Bayes’ rule to determine the rotamer probabilities. In our first attempt, we used kernel density estimates with fixed and constant kernel widths for all data points. The resulting rotamer probability for the serine g+ rotamer, calculated with a concentration parameter in the von Mises kernel function of κ = 309 (a bandwidth radius of 3.3°) is shown in Figure 4C. It reproduces the form of the Bayesian estimates but the transitions are rather sharp and it is very sensitive to outlier data in the ϕ, ψ space. A wider radius for the non-adaptive KDE data produces smoother estimates than shown in Figure 4C but such a radius flattens out the rotamer probabilities too much, leading to inaccurate probabilities even when data are plentiful (not shown).
To reduce the effect of outliers, we then employed adaptive kernel density estimates in which the kernel widths vary with the local density of data points. At higher densities, the kernels are narrower and at lower densities, such as in the vicinity of outliers, the kernels are wider, thus spreading out and minimizing their effect on the density estimates. The widths of the kernels are determined by a concentration parameter scaled with the square root of the local density of points, f̂(ϕ, ψ), obtained from some pilot estimate (in this case the non-adaptive kernel density). With the base kernel concentration parameter κ optimized to maximize the log-likelihood of P(r|ϕ, ψ)using 10-fold cross-validation, we calculated the rotamer probabilities shown in Figure 4D. The optimized value for serine is κ=309, so that the non-adaptive and adaptive rotamer probabilities in Figure 4C and D use the same value of κ. The adaptive version is much smoother than the non-adaptive version.
While eliminating the effects of the outliers, the changes in rotamer probability in Figure 4D may be sharper than optimal for programs like Rosetta that depend on the first derivatives of log P(r|ϕ, ψ). In order to increase the smoothness, we employed a penalized maximum likelihood procedure for optimizing the concentration parameter κ. This is a common procedure in density estimations (Eggermont and LaRiccia, 2001). The total log-likelihood expression can be modified in a number of ways. We use a simple approach that penalizes the average log likelihood by a fixed percentage of the range from its maximum value to its minimum value. In Figure 4E and Figure 4F, we show the g+ rotamer of serine calculated with concentration parameters such that the average log likelihood is 5% and 20% less than its full range shown in Figure 4D. Panels D, E, and F in Figure 4 thus illustrate the smoothing effect of the widening bandwidth radius (2°, 5° and 11°) of the adaptive KDEs on the rotamer probability estimates. The methods for choosing the optimized κ and the stepdown values of κ are illustrated in Figure S2. The optimized values of κ and the bandwidth radius and the same values for the 5% stepdown in the average log likelihood are given in Table S3. The appropriate choice of smoothing level may depend on the application for which the rotamer library is intended. We explore this further below.
For the rarer rotamers (those with less than 25 examples in the data set), we approximated the rotamer probability density ρ(ϕ, ψ|r) with rotamer data of the same side-chain type with one or more fewer degrees of freedom. In Figure 5, we present the rotamer probability estimates for the nine rotamers of leucine. For leucine, the {g+, g−} probability density was calculated with the {g+, X} data of leucine. The factor P(r) in Equation 1 is calculated based on the actual counts of the {g+, g−} rotamer, while ρ(ϕ, ψ|r) is calculated with the r=g+ data, producing a reasonable estimate of P(r = g+, g−|ϕ, ψ).
Figure 5. A complete set of backbone-dependent rotamer probabilities for leucine derived from adaptive kernel density estimates of the new 2010 rotamer library.
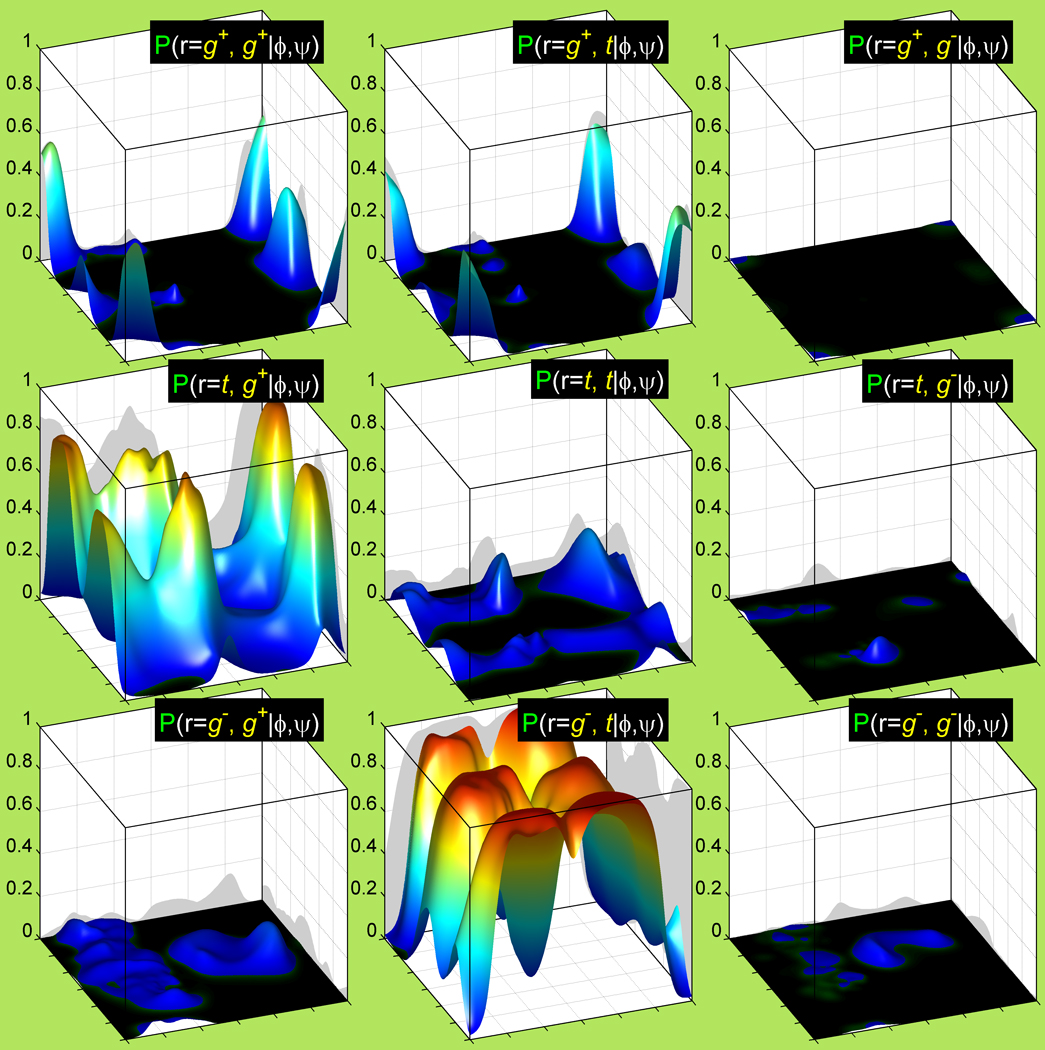
Leu demonstrates strong variation in its rotamer preferences both in the backbone-dependent and backbone-independent rotamer libraries. Some of its rotamers are restricted everywhere on the (ϕ, ψ) map, due to strong clashes of the side-chain conformations with its own backbone. The < g+, g− > rotamer has only 10 data points in our dataset while the total number of leucines is 64,329. The rare rotamer fix is used to calculate the Ramachandran probability density for the < g+, g− > rotamer using only the <g+> data.
Rotameric side-chain degrees of freedom: backbone-dependent kernel regression of χ means and variances
As with the backbone-dependent rotamer probabilities, we investigated a number of approaches in calculating the backbone-dependent means and standard deviations of side-chain dihedral angles for the rotameric degrees of freedom. In Figure 6 and Figure 7 respectively, we show the results of several different ways of calculating the μχ1 and σχ1 estimates for g+ rotamer of cysteine: μ(χ1|ϕ, ψ, r = g+) and σ(χ1|ϕ, ψ, r = g+). The simplest way is to average χ1 points and also calculate their standard deviation within non-overlapping 10°×10° bins. As with the histogram approach to rotamer probabilities described above, this method produces very crude and spiky estimates of μχ1 and its σχ1, as observed in Figure 6A and Figure 7A. In the bins with few data points, their means and deviations are statistically unreliable.
Figure 6. Rotameric χ mean estimates calculated with several methods and smoothing effects of query-adaptive kernel regressions.
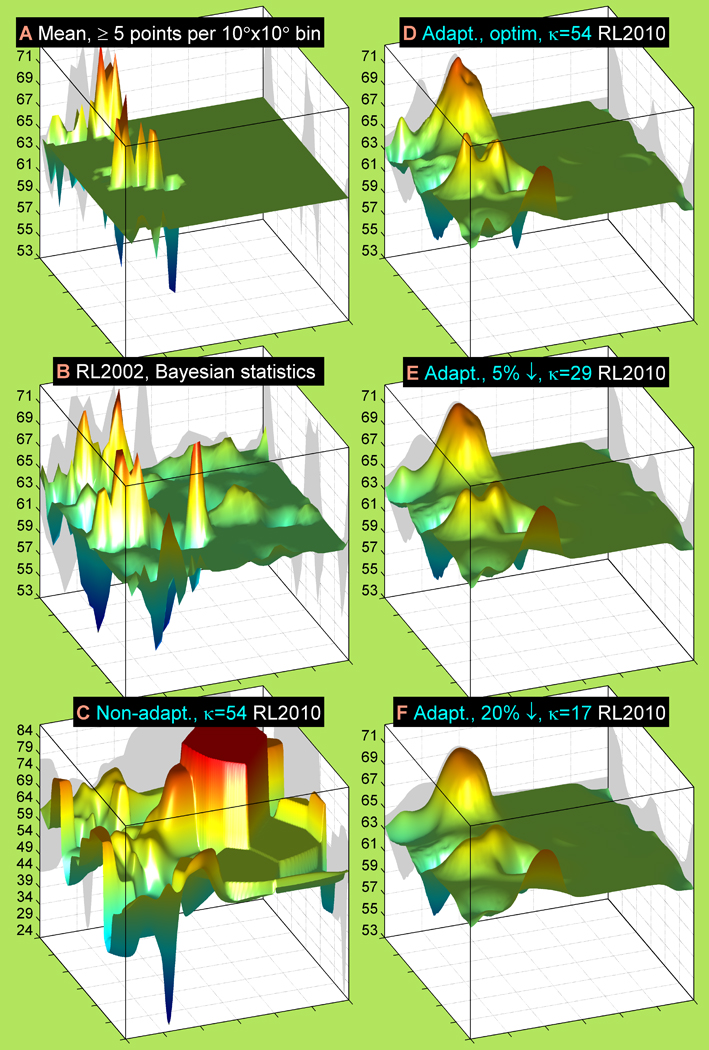
Non-overlapping 10°×10°-bin average (A), 2002 Bayesian (B), non-adaptive kernel regression (C) and query-adaptive kernel regression (D, E, F) estimates are shown for μ(χ | ϕ, ψ, r = g+, aa = Cys). The 10°×10°-bin average has only the bins with at least five g+ rotamers per bin. The non-adaptive kernel regression (C) has a fixed bandwidth (κ = 54, bandwidth radius, R = 8°), the same as for (D). The query-adaptive kernel regression estimates with widening geometric-mean kernel bandwidth are ordered from (D) to (F). The maximum log-likelihood (κ = 54, R = 8°), 5%-stepdown (κ = 29, R = 11°) and 20%-stepdown (κ = 17, R = 14°) bandwidths are in (D), (E) and (F) respectively.
Figure 7. Rotameric χ standard deviation estimates calculated with several methods and smoothing effects of query-adaptive kernel regressions.
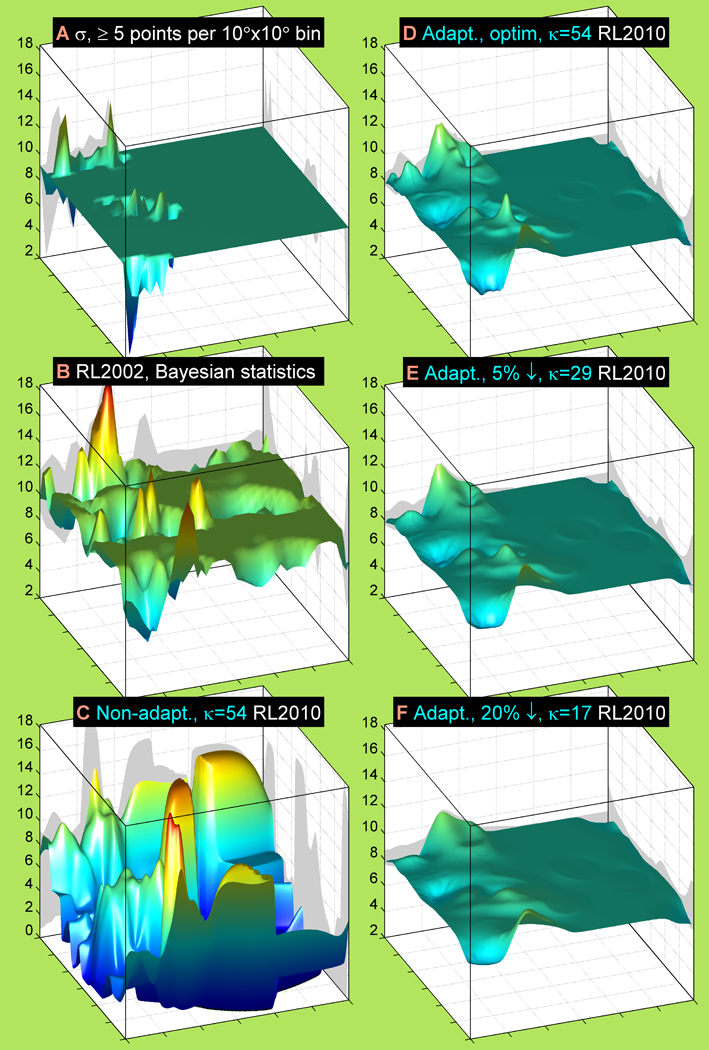
Non-overlapping 10°×10°-bin (A), 2002 Bayesian (B), non-adaptive kernel regression (C) and query-adaptive kernel regression (D, E, F) estimates are plotted for σ (χ | ϕ, ψ, r = g+, aa = Cys). The 10°×10°-bin estimate is shown only in the bins with at least five g+ rotamers per bin. Other information and parameters are the same as in Figure 6.
In the 1997 and 2002 rotamer libraries, we combined ϕ-dependent and ψ-dependent estimates of the mean angles and their variances with the data in overlapping 20°×20° bins in a Bayesian estimation procedure. The 2002 rotamer library estimates are shown in Figure 6B and Figure 7B. These estimates are extremely bumpy due to the large effect of a small number of side chains when the data are sparse. A non-adaptive kernel regression scheme also produces bumpy and extreme estimates, as shown for a bandwidth of 8° in Figure 6C and Figure 7C. This kernel captures very few data points at most query points and produces unreliable estimates of mean and standard deviation. The non-adaptive KR with a much wider bandwidth (not shown) is not as noisy but loses valuable features in the populated areas of (ϕ, ψ).
We thus moved to an adaptive scheme, applying query-adaptive kernel regression (KR) to estimate the rotameric χ means and their variances. The bandwidth varies as a function of the density local to the query point, rather than by the density around the data points, as used in the density estimates described earlier. We found that query-adaptive kernels provided regression curves and surfaces that more accurately modeled the observable variations in the χ angles as a function of ϕ and ψ than data-adaptive kernels.
For rotameric backbone-dependent χ mean and variance we utilized the sum of the squared residuals between the experimental χ points and the surface of the mean estimate as the objective function for minimization. The minimization was carried out for each χ angle of each rotamer separately. The optimal concentration parameters and their corresponding bandwidths used in the kernel regression can be found in Table S3.
As with the kernel density estimates, we also applied a simple form of penalized kernel regression, by stepping down the objective function by 2%, 5%, 10% and 20%. The values of κ that result from the 5% stepdown are also given in Table S3. Panels D, E, and F of Figure 6 and Figure 7 reveal the smoothing effect of the widening bandwidth radius (7°, 10° and 13°) of the query-adaptive KR of μχ1 and σχ1 respectively. Higher values of κ produce bumpier regression surfaces and lower κ produce flatter, smoother surfaces. As in the case with the rotamer probabilities, the appropriate level of smoothing may depend on the application.
For some (ϕ, ψ) values, clashes between the side-chain Xγ atom and backbone atoms whose positions are dependent on ϕ and ψ push the χ1 means away from their canonical values in order to relieve the clash (Dunbrack, 2002; Dunbrack and Cohen, 1997). For instance, the g+ rotamer shown in Figure 6 and Figure 7 has steric clashes with backbone atoms Oi and Ni+1 when ψ is near 120° and −60° respectively, and these interactions lead to a deviation in the χ1 dihedral angle means. In the unpopulated regions of the Ramachandran map, the query-dependent kernel regressions return to the backbone-independent mean value, which is a reasonable estimate since the angles do not usually vary more than about 15° from these values in any case. These are the flat areas in the μχ1 and σχ1 KR surfaces in Figure 6 and Figure 7. The σχ1 estimates are also larger when the side-chain and backbone atoms clash.
Non-rotameric side-chain degrees of freedom: backbone-dependent kernel regression of χ angle densities
The terminal dihedral angles of Asn, Asp, Gln, and Glu have very broad distributions, when considered independent of ϕ and ψ, as shown for Asn in Figure 1B and Gln in Figure 1D,E,F. The terminal dihedral angles of the aromatic amino acids have distributions broader than typical rotameric degrees of freedom and these are somewhat asymmetric as shown for Trp in Figure 1C. The normal model used for the rotameric degrees of freedom as for Met χ1 in Figure 1A (regression to a mean and standard deviation) is therefore inappropriate for these degrees of freedom, and therefore we refer to them as “non-rotameric.” The distributions of these non-rotameric angles vary significantly with ϕ and ψ. However, since they can not be modeled parametrically, they must be modeled with non-parametric density estimates. We therefore seek a method to determine a regression of the density of an angle onto the explanatory variables ϕ and ψ.
In the 1997 and 2002 rotamer libraries, non-rotameric χ angles were modeled in a manner very similar to the rotameric degrees of freedom despite the deficiencies of such modeling. This was accomplished by defining bins for each “rotamer,” establishing prior estimates formed from a product of individual ϕ-dependent and ψ-dependent distributions, and adding counts of χ2 in each bin from the neighborhood around each ϕ, ψ grid point. In the 2002 library, Asn had 6 χ2 bins for each χ1 rotamer over 360°, while Gln had 4 bins for χ3. Asp and Glu had 3 bins over 180°, while Phe and Tyr had 2 bins. His and Trp each had 3 bins of 120° each. For each bin, we calculated mean dihedral angles and their variances as well as relative populations. This is shown in the first row of Figure 8 for the {g+,t} rotamer of Gln for three different ϕ, ψ positions: near the α-helix region (−60°, −10°), near the β-sheet region (−150°, 180°) and near the polyproline-II region frequently occupied in loops (−80°, 180°). Each bar is located at the mean value of each bin and the horizontal bars indicate the standard deviation of the data in that bin, which is proportional to the bin widths.
Figure 8. Backbone-dependent treatment of non-rotameric side-chain χ: 2002 Rotamer Library, 2010 density model and 2010 discrete model.
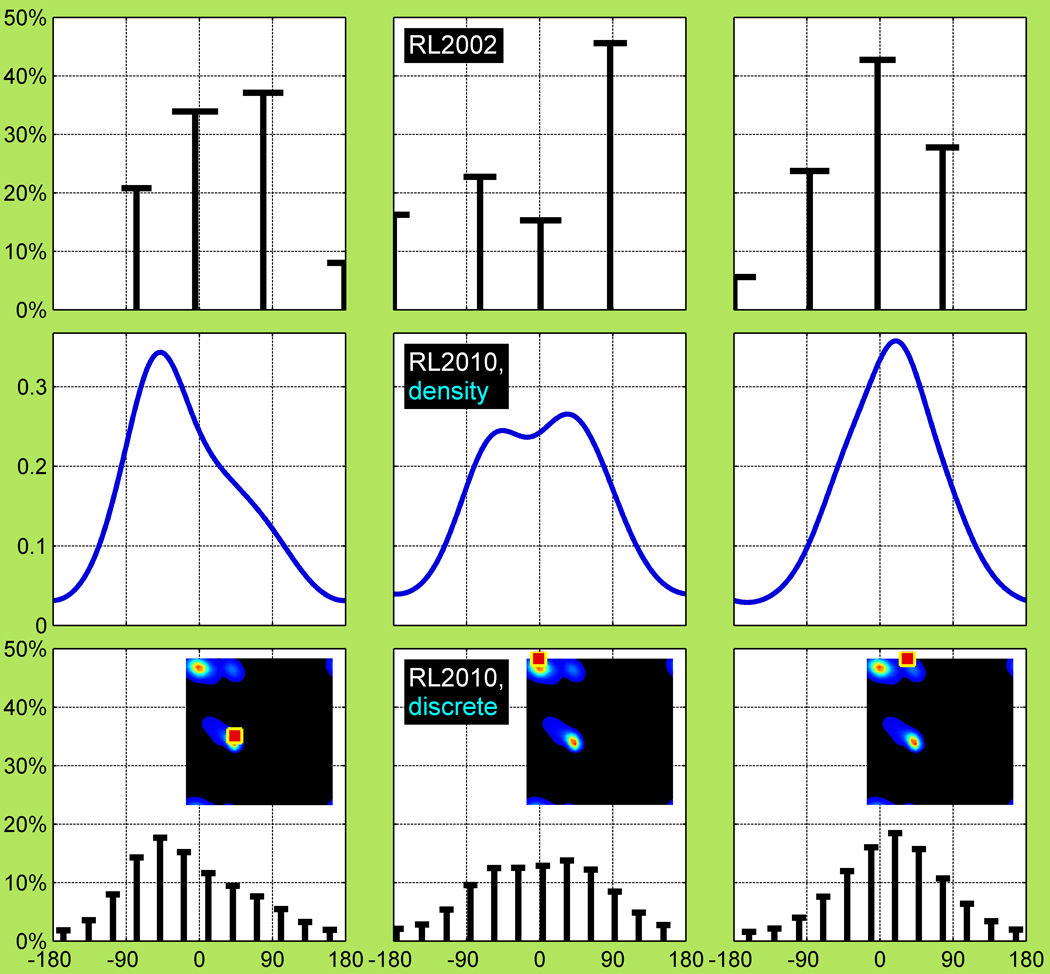
Backbone-dependent modeling of non-rotameric χ3 of χ1,χ2 rotamer = <g+,t> of Gln using Bayesian formalism of the 2002 rotamer library (top), 2010 query-adaptive KR of densities (middle) and 2010 binned “rotameric” model (bottom). These three models are provided at 3 different selected (ϕ, ψ) locations: (−60°, −10°), (−150°, 180°), and (−80°, 180°), indicated on the Ramachandran < g+, t > density insets in the bottom row. The top and bottom models are binned or “rotameric” while the middle model is continuous density. The “rotameric modeling” of the non-rotameric χ3 includes: r3 probabilities (heights of the bars), P(r3 | ϕ, ψ, r12 = < g+, t >) summing up to 1; χ3 means (positions of the bars), μ(χ3 | ϕ, ψ, r123); and χ3 standard deviations (lengths of the horizontal bars at the tip of the bars), σ(χ3 | ϕ, ψ, r123).
In the new 2010 rotamer library, we take a different approach and model the nonrotameric χ as continuous distributions as a function of (ϕ, ψ) for every rotamer combination of the rotameric degrees of freedom of the residue. For example, Gln has three side-chain degrees of freedom: rotameric χ1, χ2, and the terminal non-rotameric χ3. We therefore calculate backbone-dependent χ3 density distributions for each of the nine χ1,χ2 rotamers of Gln. We accomplish this by applying query-adaptive kernels to ϕ and ψ and data-adaptive kernels to the non-rotameric χn to estimate ρ(χn|ϕ, ψ, r−n), where n indicates the terminal dihedral angle and r−n indicates the rotamer of the non-terminal degrees of freedom. In the middle panel of Figure 8, the Gln χ3 density of the r−n = {g+, t} rotamer is evaluated every 1° for the same three (ϕ, ψ)’s as in the first row for the 2002 library. The distributions show that the modes are located at different positions for each ϕ,ψ point, the peaks are asymmetric, and in one case the distribution is bimodal. The curves roughly parallel the 2002 rotamer library, if the curves are integrated over 90° regions. For practical applications, we report backbone-dependent non-rotameric χn density every 10°.
To support existing applications such as SCWRL, which rely on our older 1997/2002 libraries and their format, for the new rotamer library we also create a new more detailed “rotameric” model for non-rotameric χ. To meet this goal and to accommodate a more complex distribution structure we increased the number of bins for the non-rotameric χ (Table S1). The rotamer bin width is decreased to 30°. The backbone-dependent probabilities are estimated by the product of the integrated continuous density over each bin and the corresponding backbone-dependent probabilities of r−n (see Eq. S39 and Eq. S40 in the Supplementary Material). The vertical bars are centered at the means and their horizontal bars specify the standard deviations of each of the 12 χ3 rotamers. These are estimated by integrating a product of the χ3 density and corresponding function over each of 12 bins (Eq. S39). Figure 8 thus illustrates binned and continuous models of non-rotameric χ angles and how the binned modeling has been changed since the 2002 analysis.
We also provide a movie of the probability density of χ2 for the g+ rotamer of Asn as a function of (ϕ, ψ) (Supplementary Movie S1). Additional figures and movies are available at http://dunbrack.fccc.edu.
Using the backbone-dependent rotamer library in structure prediction
The methods we have developed using kernel density estimates and kernel regressions have allowed us to develop smooth and statistically reliable backbone-dependent rotamer libraries. We can adjust the level of smoothing for different applications by adjusting the penalties in the objective functions for the rotamer probabilities and regressions. To choose a reasonable set of values, we tested a number of different libraries with our side-chain prediction program SCWRL4 (Krivov et al., 2009) and Rosetta (Rohl et al., 2004b). For SCWRL4 benchmarking we used the same testing set of 379 high-resolution protein monomers as in the original SCWRL4 work with a resolution cutoff of 1.8 Å and maximum mutual sequence identity of 30%. For Rosetta, we used a set of 50 monomeric, ligand-free proteins without disulfides and with resolution of 1.6 Å or better and less than 20% mutual sequence identity.
In the side-chain prediction literature, a side-chain torsion angle is considered correctly predicted if its value is within 40° from the experimental one. Using this traditional definition, in Table 1 we compare the best 2010 library vs. the older 2002 library in SCWRL4 prediction rates based on the flexible-rotamer-model (FRM) for each individual degree of freedom (χ1, χ2, χ3 and χ4) and the overall χ accuracy. The best 2010 rotamer library gives an overall increase of +0.67% in χ angle predictions on a test set of 379 proteins. This is a weighted average over χ1, χ1+2, χ1+2+3, and χ1+2+3+4 accuracies (see Eq. S42 and S43). While this is a modest increase, many highly populated side chain types are already at very high accuracies and cannot be improved much further. Except Pro (−0.3%) and Asn (−0.2%), the best 2010 library has performance better than 2002 for all residues types. Several dihedral angles have strong improvements in prediction rates, for example Trp χ2 +6%, Gln χ3 +4%, Phe χ2 +3%, Glu χ3 +3%, Ser χ1 +1%, Met χ3 +2%, Arg χ3 and χ4 +2%, Tyr χ2 +2% and Trp χ1 +1%.
Table 1.
2002 vs. best smooth 2010 rotamer libraries: benchmarking based on SCWRL4 side-chain conformation prediction accuracy.
| Categ | TRP | PHE | GLN | GLU | TYR | SER | ARG | HIS | LEU | MET | CYS | THR | ASP | ILE | VAL | LYS | ASN | PRO | ALL | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Best ’10 | χ1 | 94.1 | 98.1 | 85.0 | 81.0 | 97.1 | 75.4 | 83.1 | 93.5 | 96.4 | 90.2 | 93.2 | 94.3 | 90.5 | 98.5 | 96.9 | 82.8 | 91.7 | 87.1 | 90.15 |
| Old ’02 | 92.9 | 97.6 | 84.6 | 80.1 | 96.5 | 74.3 | 83.3 | 93.9 | 95.9 | 90.4 | 92.8 | 94.0 | 90.6 | 98.4 | 96.7 | 82.6 | 91.7 | 87.3 | 89.83 | |
| Δ(Best, Old) | +1.2 | +0.5 | +0.4 | +1.0 | +0.6 | +1.1 | −0.2 | −0.4 | +0.5 | −0.2 | +0.4 | +0.4 | −0.1 | +0.1 | +0.2 | +0.3 | 0.0 | −0.2 | +0.32 | |
| Best ’10 | χ1+2 | 84.6 | 96.6 | 71.1 | 68.0 | 94.8 | 72.9 | 66.4 | 91.9 | 81.9 | 84.7 | 91.0 | 72.3 | 76.7 | 83.9 | 81.73 | ||||
| Old ’02 | 78.9 | 93.7 | 71.0 | 67.5 | 92.9 | 72.5 | 64.6 | 91.2 | 81.9 | 83.8 | 90.6 | 72.5 | 77.0 | 84.3 | 81.01 | |||||
| Δ (Best, Old) | +5.7 | +2.9 | +0.1 | +0.6 | +1.9 | +0.5 | +1.8 | +0.8 | 0.0 | +0.9 | +0.4 | −0.2 | −0.3 | −0.4 | +0.72 | |||||
| Best ’10 | χ1+2+3 | 48.8 | 52.4 | 51.0 | 64.2 | 58.4 | 54.01 | |||||||||||||
| Old ’02 | 44.5 | 49.3 | 49.6 | 62.5 | 58.7 | 52.05 | ||||||||||||||
| Δ (Best,Old) | +4.2 | +3.1 | +1.5 | +1.7 | −0.3 | +1.96 | ||||||||||||||
| Best ’10 | χ1+2+3+4 | 38.1 | 39.9 | 38.99 | ||||||||||||||||
| Old ’02 | 36.3 | 39.6 | 38.01 | |||||||||||||||||
| Δ (Best,Old) | +1.8 | +0.2 | +0.98 | |||||||||||||||||
| Best ’10 | χall | 89.3 | 97.4 | 68.3 | 67.1 | 96.0 | 75.4 | 61.3 | 80.0 | 94.1 | 78.8 | 93.2 | 94.3 | 87.6 | 94.8 | 96.9 | 63.4 | 84.2 | 85.5 | 83.72 |
| Old ’02 | 85.9 | 95.7 | 66.7 | 65.6 | 94.7 | 74.3 | 60.4 | 79.2 | 93.5 | 78.3 | 92.8 | 94.0 | 87.2 | 94.5 | 96.7 | 63.4 | 84.3 | 85.8 | 83.04 | |
| Δ (Best,Old) | +3.4 | +1.7 | +1.6 | +1.5 | +1.2 | +1.1 | +0.9 | +0.7 | +0.6 | +0.5 | +0.4 | +0.4 | +0.4 | +0.3 | +0.2 | 0.0 | −0.2 | −0.3 | +0.67 | |
The performances of the new 2010 rotamer libraries were compared with the 2002 rotamer library. SCWRL4 was run on a set of 379 high-resolution proteins used previously (Krivov et al, 2009). The flexible rotamer model (FRM) of SCWRL4 was used, and crystal symmetry was used in the calculations (all side chains in all copies of the asymmetric unit were calculated simultaneously). Accuracy was evaluated on all side chains in the proteins excluding those with electron density in the bottom 25th percentile for each residue type. A predicted side-chain χ is considered correct if its value lies within 40° from its experimental value. For each residue type the 2002 and 2010 accuracies are provided for each individual χ angle. χall is an absolute average of all degrees of freedom for each residue (see Supplementary Material). ALL is an average accuracy among all 18 standard residue types.
To create smoother rotamer libraries from the 2010 data set, we determined lower κ’s (smoother functions) by finding the κ which had a lower value of the objective function by some percentage of its range (i.e., the maximum value minus the minimum value over all κ; see Figure S2 for an example). For SCWRL4, the best 2010 library is the one with the 5% stepdown in the objective functions from the optimal κ values. Increased smoothness (stepdowns of 10%, 20%, 25%) or reduced smoothness (2% or fully optimized) produce slightly lower prediction rates as shown in Table 1. For a more stringent definition of correct χ angles, within 10°, SCWRL4 demonstrates more improvement for 2010 vs. 2002, a total of +1.1% (data not shown).
Since the new rotamer libraries were developed in part to improve Rosetta performance when backbone flexibility is modeled, we tested Rosetta’s energy minimization protocols with the various rotamer libraries. After fitting the structures with standard bond lengths and bond angles, we separately ran two types of minimization tests on: FastRelax and ClassicRelax on the idealized structures generating 100 decoys for each. The FastRelax protocol (Tyka et al., 2011) consists of five rounds of the following: multiplying the repulsive van der Waals parameters by a scale factor C (0<C≤1), Monte Carlo simulated-annealing repacking of side chains using the rotamer library (replacing all side chains with random rotamers, several times over, with Metropolis criterion acceptance), and then continuous energy minimization of the backbone and side chains. The factor is ramped up from 0.02 to 1.0 over 4 steps in each round. The lowest energy structure when the scale factor is 1.0 is saved as a decoy. The ClassicRelax protocol (Bradley et al., 2005) consists of many rounds of small backbone perturbation moves (2°–3° in ϕ and ψ) and complete side-chain repacking, followed by backbone and side-chain continuous energy minimization. The FastRelax protocol is the one currently recommended for high-resolution refinement in Rosetta, but we decided to test the older protocol as well to see if it behaved differently. The results are shown in Table 2.
Table 2.
Effects of 2010 rotamer library smoothing in SCWRL4 and Rosetta
| 2002 | Optim | 2%↓ | 5%↓ | 10%↓ | 20%↓ | 25%↓ | 2009it10 | ||
|---|---|---|---|---|---|---|---|---|---|
| Side-chain prediction | |||||||||
| SCWRL4 | D(’10,’02), asymm. ED25–100% | 83.04% | +0.57% | +0.61% | +0.67% | +0.40% | +0.11% | −0.08% | N.D. |
| SCWRL4 | D(’10,’02), symm., ED0–100% | 79.33% | +0.55% | +0.59% | +0.64% | +0.39% | +0.11% | −0.11% | N.D. |
| Rosetta FastRelax | D(’10,’02), symm., ED0–100% | 72.82% | +0.48% | +0.21% | +0.10% | −0.09% | −1.04% | −1.44% | −1.45% |
| Rosetta ClassicRelax | D(’10,’02), symm., ED0–100% | 76.12% | +0.21% | +0.13% | 0.00% | −0.12% | −0.81% | −1.12% | −1.57% |
| RMSD differences | |||||||||
| Rosetta FastRelax: | D(’10,’02) / ’02 (backbone) | 1.112 | −2.23% | −0.37% | −0.19% | +0.04% | +0.67% | +1.36% | +0.63% |
| D(’10,’02) / ’02 (all atoms) | 1.596 | −1.83% | −0.49% | −0.58% | −0.19% | +0.76% | +1.22% | +1.01% | |
| Rosetta ClassicRelax: | D(’10,’02) / ’02 (backbone) | 1.081 | −1.21% | −0.67% | −2.06% | −1.35% | +0.64% | +2.41% | +0.18% |
| D(’10,’02) / ’02 (all atoms) | 1.517 | −0.02% | +0.28% | −0.76% | −0.21% | +1.36% | +2.36% | +1.91% | |
| TotalScoreMinusDun | |||||||||
| Rosetta FastRelax: | D(’10,’02) | −382.28 | 1.783 | −0.004 | −1.035 | −2.436 | −4.884 | −5.645 | −1.965 |
| Rosetta ClassicRelax: | D(’10,’02) | −379.00 | 0.871 | −0.692 | −1.548 | −2.685 | −5.068 | −5.504 | −1.970 |
2010 library names are listed in the first row. 2009it10 is a modified version of a developmental rotamer library created by using similar methods (with some important differences) in 2008. It is distributed with Rosetta3 and was recently described by Song et al. (2011). For side-chain accuracy, the absolute average percentage accuracy is given for the 2002 library, and the differences from those values are given for the other libraries (2010Library – 2002). For RMSD differences, the mean RMSD in (Å) is given for the 2002 library, and the percent differences from 2002 are given for the 2010 libraries. For TotalScoreMinusDun, the mean values are given for the 2002 library, and the differences (in Rosetta score units) are given for the 2010 libraries. For side-chain accuracy, “symm” indicates Asn, His, and Gln terminal dihedrals were treated as symmetric, while “asymm” indicates they are treated like other dihedral angles. ED25–100% indicates only side chains with electron density in the 25th to 100th percentile were included in the accuracy assessment. ED0–100% means all side chains were included. Better numbers are in bold type (higher side-chain accuracy, lower RMSDs), while worse numbers are in italic type. N.D.=not done. See also Figure S3.
The goal of these calculations is to perturb the backbone and side-chains from the native structure and to determine whether the energy function minimization is able to bring or keep the structure as close to native as possible, as measured by backbone and full-atom RMSDs. For Rosetta FastRelax we gained a 2.2% and 1.8% improvement for the optimized 2010 library relative to 2002 for the average backbone and full-atom RMSD’s respectively. For ClassicRelax, we achieved the best results with the smoother 5%-stepdown rotamer library. For this library, the backbone and full-atom RMSDs from native are 2.1% and 0.8% lower than the results with the 2002 rotamer library respectively.
The FastRelax decoys achieved the best side-chain accuracies with the optimized 2010 library compared to the 2010 libraries with additional smoothing. For cutoffs for correct predictions of 40° and 10°, the absolute average accuracies over all dihedral angles were 73.3% and 56.4% which is an improvement of 0.5% and 1.0% when comparing to the 2002 library respectively. The ClassicRelax decoys also achieve the best side-chain accuracies with the optimized 2010 library with average absolute accuracies of 76.3% (40°) and 58.9% (10°). SCWRL4 with crystal symmetry but without removing side chains in the bottom 25th percentile achieves an average absolute accuracy of 80.0% (40°) and 57.9% (10°) with the 5%-stepdown library. The crystal symmetry is responsible for about a 2% increase in average absolute accuracy (Krivov et al., 2009).
Note that in these calculations, neither SCWRL4 nor Rosetta has been optimized to work with the 2010 libraries. The SCWRL4 calculations used constant parameters for all residue types and all rotamer libraries. The distributed version of SCWRL4, by contrast, has optimized values for several parameters for each residue type. The Rosetta calculations used the standard “score12” scoring function, except for the different rotamer libraries. Song et al. have recently reported an optimization of Rosetta’s energy function for an earlier version of the rotamer libraries described here (Song et al., 2011). They modified the rotamer library to compensate for doubly-counted interactions such as side-chain/backbone hydrogen bonding and steric interactions. We tested one version of this rotamer library distributed with Rosetta3.1, “2009it10”; the results are shown in the last column of Table 2. Its side-chain and RMSD performances are worse than both the 2002 library and the optimized. 5% and 10% stepdown libraries presented here.
The backbone-dependent rotamer library is one component (designated “fa_dun” in Rosetta output) of several in the Rosetta scoring function, which includes repulsive and attractive van der Waals interactions, Ramachandran energies, solvation terms, and hydrogen bonding. We analyzed the scoring function values for the decoys generated with the two relax protocols and the various rotamer libraries, shown at the bottom of Table 2. As the smoothness is increased, the non-side-chain energy terms (“TotalScoreMinusDun”) optimized to lower values. This may be due to flatness of the smoother rotamer libraries, although the dynamic range of the smoother libraries is not significantly less than the fully optimized rotamer library.
One feature of the new rotamer libraries that improves the results of Rosetta is the nature of the non-rotameric degrees of freedom. For the 2002 library (“dun02” in Rosetta protocols), the non-rotameric degrees of freedom had between 2 (Phe, Tyr) and 6 (Asn) bins for rotamer probabilities, means, and standard deviations. In Rosetta, when the 2002 library is used, a harmonic energy term is applied to these mean values with a force constant inversely related to the standard deviation. When the developmental version of the rotamer libraries described here was implemented in Rosetta3 (“dun08” flag in Rosetta) (Leaver-Fay et al., 2011), Rosetta was modified to use the continuous probability estimates for the non-rotameric degrees of freedom. Thus these dihedral angles are free to change over a wide range in the smooth, backbone-dependent potentials, as shown for Gln in Figure 8. As a result, the output distributions of χ angles for these degrees of freedom are much closer to native structures than the results of the 2002 library, which are discretely distributed The distributions of χ2 for the decoys generated by FastRelax for the 2002 and optimized 2010 libraries are shown in Figure S3. The results may be compared to the backbone-independent χ2 distributions for Asn shown in Figure 1B.
Further testing is needed of the different rotamer libraries in various protocols (ab initio structure prediction, comparative modeling, docking, protein design, etc.) to determine which is most suitable for each application. On our website, http://dunbrack.fccc.edu we provide access to the full range of rotamer libraries described here, as well as images and movies of the distributions. For most purposes, the 5%-stepdown library may be most appropriate, since it provides a good tradeoff between appropriate details and smoothness of the probability distributions.
DISCUSSION
The backbone-dependent rotamer libraries we have developed previously have found uses in many different applications in protein structure prediction (Andrusier et al., 2007; Bower et al., 1997; Hartmann et al., 2007; Krieger et al., 2009; Krivov et al., 2009; Liang and Grishin, 2002; Mendes et al., 2001; Rohl et al., 2004a; Smith et al., 2007; Zhang et al., 2004) and protein design (Calhoun et al., 2003; Dahiyat and Mayo, 1997; Kuhlman and Baker, 2000; Pokala and Handel, 2005; Saraf et al., 2006; Stiebritz and Muller, 2006). In these applications, both the backbone-dependent probabilities and the backbone-dependent dihedral angles have made important contributions. We therefore have taken great care in producing a new backbone-dependent rotamer library, testing many different ways of estimating the probabilities and regression functions that make up the library.
A number of different technical obstacles have been overcome in developing the new rotamer library. In our previous libraries, we did not use methods that reliably produced smoothly varying estimates of the rotamer probabilities and dihedral angles with backbone ϕ and ψ. The kernel density estimates and regressions used here coupled with the penalized maximum likelihood optimization of the smoothing parameters have produced smooth, reliable estimates of the library values. Filtering by electron density and adaptive kernel density estimates and regressions reduced the effects of outliers in Ramachandran space.
An important innovation in this rotamer library is the treatment of non-rotameric degrees of freedom. The previous model of a small number of χ angle bins for these dihedrals sometimes resulted in likely artifacts in structure prediction and design. For instance, Rosetta previously placed harmonic energy functions on each of the “rotamers” of χn, which for the amides and carboxylates in particular created potential functions with 4 or 6 minima with large energy barriers in between. However, these degrees of freedom do not fit a rotamer model of discrete side-chain conformations with relatively small dihedral angle variances. Instead, they have widely distributed densities, and especially in the case of Asp and Asn, strong backbone-dependence. In the new rotamer library, smooth densities are achieved with a novel combination of query-dependent adaptive kernels on ϕ, ψ and data-dependent adaptive kernels on the χ angles, effectively the regression of an angular density onto two angular explanatory variables.
Two other studies have presented analyses similar to that of the backbone-dependent rotamer library. Amir et al. used the data from our 2002 library (850 proteins) and cubic splines to produce both joint and conditional probability distributions of ϕ, ψ, and χ angles (Amir et al., 2008). Such an analysis does emphasize smoothness of the probability distributions. Harder et al. have recently developed a generative model of protein side-chain conformations called BASILISK (Harder et al., 2010). It generates samples of side-chain dihedral angles for given input backbone dihedral angles. It is also capable of returning a log-likelihood value for any query side-chain conformation (χ1, χ2, χ3, χ4) given a backbone conformation. Because it ties together χ angle probabilities of different residue types, it does have incorrect ordering of rotamer probabilities such as serine for which the g-χ1 rotamer is not the most common.
Neither of these programs use the rotamer model and thus may not be easily incorporated into programs that utilize such models to enumerate all possible rotamers in structure prediction and design. It should be noted that our methods for determining the non-rotameric χ angle densities can be used for any of the side-chain degrees of freedom, not just the non-rotameric ones. So for instance, it is possible to create estimates (including multidimensional estimates) for ρ(χ|ϕ, ψ) for rotameric degrees of freedom independent of rotamer state. Such a model would include changes in probability of the rotamers, the positions of modes in the density, as well as covariance of the dihedral angles with respect to each other and the backbone dihedral angles. We are currently exploring the utility of such probability density estimates.
We believe the new backbone-dependent rotamer library has a number of useful characteristics that will make it useful in a variety of applications in protein structure determination, prediction, and design.
METHODS
The full methods are given in the Supplementary Material.
Deriving backbone-dependent rotamer probabilities from Ramachandran densities of each rotamer from adaptive kernel density estimates
We want to determine the rotamer probabilities, P(r|ϕ, ψ, aa), for each amino acid type, aa, and each rotamer r, so that:
| (2) |
for any values of (ϕ, ψ). Using Bayes’ rule (see Equation 1), these probabilities can be derived from the Ramachandran probability density functions of each rotamer, ρ(ϕ, ψ|r,aa) and the backbone-independent frequencies of each rotamer, P(r|aa). The sum in the denominator of Equation 1 is over all rotamers of a given residue type. P(r|aa) can be calculated easily from the observed frequencies of each rotamer in the dataset. However, to calculate accurate and smooth estimates of P(r|ϕ, ψ, aa), we require accurate and smooth estimates of ρ(ϕ, ψ|r, aa). We drop “aa” from the formulas below. Also we denote probabilities with P and probability densities with ρ.
Smooth estimates of ρ(ϕ, ψ|r) can be calculated from kernel density estimates. A kernel is a non-negative function that integrates to 1. In one dimension, a kernel density estimate may be written:
| (3) |
where K is the kernel function, N is the number of data points, and h is the kernel bandwidth. For instance, if the kernel is Gaussian, h is the square root of the variance, or σ.
Since Ramachandran probability density is defined for the backbone torsion angles ϕ and ψ as two arguments, we use a two-dimensional kernel density estimate using the von Mises distribution as the kernel. The non-adaptive or fixed-bandwidth KDE in two dimensions for Ramachandran data can be written as the sum over products of ϕ- and ψ- von Mises kernels for Nr data points of rotamer type, r:
| (4) |
In this case defines a radius of the two-dimensional hump covering 67% of the kernel density. I0 is the Bessel function of the first kind of order 0; it normalizes the kernels to 1. For simplicity we do not place a caret on top of kernel density or kernel regression estimates.
To reduce the effect of outliers, we use adaptive kernel density estimates (AKDE) in which the bandwidth parameter (κ) varies across the sample data points, depending on the local density of the data (Abramson, 1982; Breiman et al., 1977). For the Ramachandran density, the AKDE is:
| (5) |
The adaptive parameters λi are based on a pilot estimate of the Ramachandran density for the residue type as a whole:
| (6) |
For the pilot estimate, we use the non-adaptive kernel density estimate given in Eq. 4. The factor g is simply the geometric mean of the pilot density estimates at the N data points. We use α=1/2, a value which is commonly used to regulate the magnitude of how much sample points from the sparsely populated regions have their bandwidths expanded and how much those in the populated regions have their bandwidths shrunk relative to the geometric mean sample point (Abramson, 1982; Silverman, 1986).
We chose the parameter κ for each residue type using cross validation of the average log likelihood of the rotamers as described in the Supplementary Material.
Adaptive kernel regression (KR) for the rotameric χ angles and variances
The second major component of the rotamer library is the backbone-dependent population means, μ and standard deviations, σ of the available side-chain dihedral angles (χ1, χ2, χ3 and χ4). We model the regression relation between the response variable, χ and the explanatory variables (ϕ, ψ):
| (7) |
where m(ϕi, ψi|r) is the unknown regression function, ν(ϕi, ψi) is the variance, and εi are random observation errors normally distributed with a mean of zero and variance 1. Given that side chains in backbone-constrained conformations experience greater uncertainty in their χ angles, we assume the standard deviation of the observation errors vary as a function of ϕ and ψ, that is, the model is heteroscedastic. In this case the regression function is the conditional expectation or population mean of χ given the backbone conformation:
| (8) |
| (9) |
Since we do not expect μ(χ|ϕ, ψ, r) and σ2(χ|ϕ, ψ, r) to vary rapidly with ϕ and ψ, we use the Nadaraya-Watson or local constant kernel regression (KR) estimator to model them. It corresponds to a local constant or zero-order polynomial, kernel-weighted least squares fit:
| (10) |
The appropriate adaptive kernel for regression onto the angles ϕ and ψ is again a symmetric two-dimensional von Mises kernel:
| (11) |
However, in this case, we use a kernel that is adaptive based on the query point rather than the data point:
| (12) |
This estimator can adapt to the density of sample points, taking a larger bandwidth where points are sparse. It can adapt to changes in residual variance in case of heteroscedacity, smoothing more where residual variance is high. The estimator can adapt to the structure of the regression function, smoothing more in flat parts of the surface and less in steeper parts. This leads to improved smoothness that is one of our goals of better side-chain modeling.
Backbone-dependent modeling of non-rotameric degrees of freedom
The terminal dihedral angle for certain side chain types is not well described as a rotamer. These include the terminal degrees of freedom of Asn, Asp, Glu, and Gln. The aromatic residues, Phe, Tyr, His, and Trp, also have more broadly distributed χ2 angles than rotameric degrees of freedom, although not to the same extent as the amide and carboxylate groups. We model the terminal dihedral angle of side chains with non-rotameric degrees of freedom, χn, as continuous probability density functions as a function of the backbone conformation, (ϕ, ψ), ρ(χn|ϕ, ψ, r−n), where r−n denotes the rotamer of the rotameric degrees of freedom (χ1 for Asn, Asp, and the aromatics; χ1, χ2 for Gln and Glu), such that:
| (13) |
With ρ(χn|ϕ, ψ, r−n) in hand on a fine grid of χn values, we can calculate binned probabilities at any desired resolution, 5°, 10°, or 30° for instance.
Modeling ρ(χn|ϕ, ψ, r−n) is effectively the regression of a probability density function (PDF) onto the explanatory variables ϕ, ψ; that is, we want a separate ρ(χn) for any ϕ, ψ. We have calculated Ramachandran map PDFs with data-point adaptive kernels, while we have found that regressions were better produced using query-point adaptive kernels. We achieve the backbone-dependent non-rotameric χn density modeling by computing the backbone-dependent KR of the χn densities, each of which is based on an individual χn data point taken from the input sample:
| (14) |
where χi are the data points of χn and Kϕ, ψ(ϕ − ϕi, ψ − ψi) is the query-adaptive kernel with the same expression as in Eq. 11 and its κ is the von Mises concentration parameter in the (ϕ, ψ) space. We take the kernels on χ to be one-dimensional von Mises functions (Eq. 6) centered on χi taken from the data sample:
| (15) |
The concentration parameter, κ1d sets the overall bandwidth in the χn space and is chosen independently from its counterpart, the (ϕ, ψ)-space κ. λi are the scaling parameters calculated in the data-adaptive fashion in accordance with the one-dimensional χi backbone-independent density:
| (16) |
where f̂χ(χn|r−n) is a χn pilot density estimate and α=1/2. The pilot density is modeled with a non-adaptive KDE with the same concentration parameter, κ1d:
| (17) |
The χn concentration parameters, κ1d/λi (Eq. 15) are data-adaptive in order to produce a true PDF that integrates to 1. If κ1d/λi is query-adaptive, the resulting function would not integrated to 1 and would not meet the definition of a PDF (Sain, 1994).
Note that κ and κ1d have different and specific values for each rotamer, r−n. It is also worth pointing out that in very empty parts of the (ϕ, ψ) map where κ/λϕψ → 0, the KR of the χn densities defaults to the backbone-independent density:
| (18) |
Further details on optimizing the bandwidths and converting non-rotameric density into rotamer probabilities for the non-rotameric degrees of freedom are given in the Supplementary Material.
Availability
The 2010 rotamer libraries are available from http://dunbrack.fccc.edu. The website also presents additional images of the backbone-dependent probabilities, dihedral angle means, and movies of the non-rotameric probability densities.
Supplementary Material
ACKNOWLEDGMENTS
We thank Georgy Krivov, Benjamin Blum and Prof. Michael I. Jordan (University of California Berkeley) for helpful discussions. Brian Weitzner provided invaluable help in setting up the Rosetta calculations. This work was funded under NIH grants P20 GM76222 and R01 GM84453 to R.L.D.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Abramson IS. On bandwidth variation in kernel estimates - a square root law. Ann. Statist. 1982;10:1217–1223. [Google Scholar]
- Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr D Biol Crystallogr. 2002;58:1948–1954. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- Amir ED, Kalisman N, Keasar C. Differentiable, multi-dimensional, knowledge-based energy terms for torsion angle probabilities and propensities. Proteins. 2008;72:62–73. doi: 10.1002/prot.21896. [DOI] [PubMed] [Google Scholar]
- Andrusier N, Nussinov R, Wolfson HJ. FireDock: fast interaction refinement in molecular docking. Proteins. 2007;69:139–159. doi: 10.1002/prot.21495. [DOI] [PubMed] [Google Scholar]
- Bower MJ, Cohen FE, Dunbrack RL., Jr Prediction of protein side-chain rotamers from a backbone-dependent rotamer library: a new homology modeling tool. J Mol Biol. 1997;267:1268–1282. doi: 10.1006/jmbi.1997.0926. [DOI] [PubMed] [Google Scholar]
- Bradley P, Misura KM, Baker D. Toward high-resolution de novo structure prediction for small proteins. Science. 2005;309:1868–1871. doi: 10.1126/science.1113801. [DOI] [PubMed] [Google Scholar]
- Breiman L, Friedman JH, Purcell E. Variable kernel estimates of multivariate densities. Technometrics. 1977;19:135–144. [Google Scholar]
- Brockmann M, Gasser T, Herrmann E. Locally Adaptive Bandwidth Choice for Kernel Regression-Estimators. J. Am. Stat. Assoc. 1993;88:1302–1309. [Google Scholar]
- Calhoun JR, Kono H, Lahr S, Wang W, DeGrado WF, Saven JG. Computational design and characterization of a monomeric helical dinuclear metalloprotein. J Mol Biol. 2003;334:1101–1115. doi: 10.1016/j.jmb.2003.10.004. [DOI] [PubMed] [Google Scholar]
- Canutescu AA, Shelenkov AA, Dunbrack RL., Jr A graph-theory algorithm for rapid protein side-chain prediction. Protein Sci. 2003;12:2001–2014. doi: 10.1110/ps.03154503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahiyat BI, Mayo SL. De novo protein design: fully automated sequence selection. Science. 1997;278:82–87. doi: 10.1126/science.278.5335.82. [DOI] [PubMed] [Google Scholar]
- Davis IW, Murray LW, Richardson JS, Richardson DC. MOLPROBITY: structure validation and all-atom contact analysis for nucleic acids and their complexes. Nucleic Acids Res. 2004;32:W615–W619. doi: 10.1093/nar/gkh398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desmet J, De Maeyer M, Hazes B, Lasters I. The dead-end elimination theorem and its use in protein sidechain positioning. Nature. 1992;356:539–542. doi: 10.1038/356539a0. [DOI] [PubMed] [Google Scholar]
- Dunbrack RL., Jr Rotamer libraries in the 21st century. Curr Opin Struct Biol. 2002;12:431–440. doi: 10.1016/s0959-440x(02)00344-5. [DOI] [PubMed] [Google Scholar]
- Dunbrack RL, Jr, Cohen FE. Bayesian statistical analysis of protein side-chain rotamer preferences. Protein Sci. 1997;6:1661–1681. doi: 10.1002/pro.5560060807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunbrack RL, Jr, Karplus M. Backbone-dependent rotamer library for proteins. Application to side-chain prediction. J Mol Biol. 1993;230:543–574. doi: 10.1006/jmbi.1993.1170. [DOI] [PubMed] [Google Scholar]
- Dunbrack RL, Jr, Karplus M. Conformational analysis of the backbone-dependent rotamer preferences of protein sidechains. Nat Struct Biol. 1994;1:334–340. doi: 10.1038/nsb0594-334. [DOI] [PubMed] [Google Scholar]
- Eggermont PPB, LaRiccia VN. Maximum Penalized Likelihood Estimation: Volume I: Density Estimation. New York: Springer-Verlag; 2001. [Google Scholar]
- Friedland GD, Linares AJ, Smith CA, Kortemme T. A simple model of backbone flexibility improves modeling of side-chain conformational variability. J Mol Biol. 2008;380:757–774. doi: 10.1016/j.jmb.2008.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon DB, Hom GK, Mayo SL, Pierce NA. Exact rotamer optimization for protein design. J Comput Chem. 2003;24:232–243. doi: 10.1002/jcc.10121. [DOI] [PubMed] [Google Scholar]
- Harder T, Boomsma W, Paluszewski M, Frellsen J, Johansson KE, Hamelryck T. Beyond rotamers: a generative, probabilistic model of side chains in proteins. BMC Bioinformatics. 2010;11:306. doi: 10.1186/1471-2105-11-306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann C, Antes I, Lengauer T. IRECS: a new algorithm for the selection of most probable ensembles of side-chain conformations in protein models. Protein Sci. 2007;16:1294–1307. doi: 10.1110/ps.062658307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Headd JJ, Immormino RM, Keedy DA, Emsley P, Richardson DC, Richardson JS. Autofix for backward-fit sidechains: using MolProbity and real-space refinement to put misfits in their place. J Struct Funct Genomics. 2009;10:83–93. doi: 10.1007/s10969-008-9045-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleywegt GJ, Harris MR, Zou JY, Taylor TC, Wahlby A, Jones TA. The Uppsala Electron-Density Server. Acta Crystallogr D Biol Crystallogr. 2004;60:2240–2249. doi: 10.1107/S0907444904013253. [DOI] [PubMed] [Google Scholar]
- Krieger E, Joo K, Lee J, Lee J, Raman S, Thompson J, Tyka M, Baker D, Karplus K. Improving physical realism, stereochemistry, and side-chain accuracy in homology modeling: Four approaches that performed well in CASP8. Proteins. 2009;77 Suppl 9:114–122. doi: 10.1002/prot.22570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krivov GG, Shapovalov MV, Dunbrack RL., Jr Improved prediction of protein side-chain conformations with SCWRL4. Proteins. 2009;77:778–795. doi: 10.1002/prot.22488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhlman B, Baker D. Native protein sequences are close to optimal for their structures. Proc Natl Acad Sci U S A. 2000;97:10383–10388. doi: 10.1073/pnas.97.19.10383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhlman B, Baker D. Exploring folding free energy landscapes using computational protein design. Curr Opin Struct Biol. 2004;14:89–95. doi: 10.1016/j.sbi.2004.01.002. [DOI] [PubMed] [Google Scholar]
- Leaver-Fay A, Tyka M, Lewis SM, Lange OF, Thompson J, Jacak R, Kaufman K, Renfrew PD, Smith CA, Sheffler W, et al. ROSETTA3: an object-oriented software suite for the simulation and design of macromolecules. Methods Enzymol. 2011;487:545–574. doi: 10.1016/B978-0-12-381270-4.00019-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang S, Grishin NV. Side-chain modeling with an optimized scoring function. Protein Sci. 2002;11:322–331. doi: 10.1110/ps.24902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovell SC, Word JM, Richardson JS, Richardson DC. Asparagine and glutamine rotamers: B-factor cutoff and correction of amide flips yield distinct clustering. Proc Natl Acad Sci U S A. 1999;96:400–405. doi: 10.1073/pnas.96.2.400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovell SC, Word JM, Richardson JS, Richardson DC. The penultimate rotamer library. Proteins. 2000;40:389–408. [PubMed] [Google Scholar]
- Mardia KV, Jupp PE. Directional Statistics. London: Wiley; 2000. [Google Scholar]
- Mendes J, Nagarajaram HA, Soares CM, Blundell TL, Carrondo MA. Incorporating knowledge-based biases into an energy-based side-chain modeling method: application to comparative modeling of protein structure. Biopolymers. 2001;59:72–86. doi: 10.1002/1097-0282(200108)59:2<72::AID-BIP1007>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- Pokala N, Handel TM. Energy functions for protein design: adjustment with protein-protein complex affinities, models for the unfolded state, and negative design of solubility and specificity. J Mol Biol. 2005;347:203–227. doi: 10.1016/j.jmb.2004.12.019. [DOI] [PubMed] [Google Scholar]
- Rohl CA, Strauss CE, Chivian D, Baker D. Modeling structurally variable regions in homologous proteins with rosetta. Proteins. 2004a;55:656–677. doi: 10.1002/prot.10629. [DOI] [PubMed] [Google Scholar]
- Rohl CA, Strauss CE, Misura KM, Baker D. Protein structure prediction using Rosetta. Methods Enzymol. 2004b;383:66–93. doi: 10.1016/S0076-6879(04)83004-0. [DOI] [PubMed] [Google Scholar]
- Sain SR. Dept. of Statistics. Houston, Texas: Rice University; 1994. Adaptive kernel density estimation. [Google Scholar]
- Saraf MC, Moore GL, Goodey NM, Cao VY, Benkovic SJ, Maranas CD. IPRO: an iterative computational protein library redesign and optimization procedure. Biophys J. 2006;90:4167–4180. doi: 10.1529/biophysj.105.079277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapovalov MV, Dunbrack RL., Jr Statistical and conformational analysis of the electron density of protein side chains. Proteins. 2007;66:279–303. doi: 10.1002/prot.21150. [DOI] [PubMed] [Google Scholar]
- Silverman BW. Density Estimation for Statistics and Data Analysis. New York: Chapman & Hall; 1986. [Google Scholar]
- Smith CA, Kortemme T. Backrub-like backbone simulation recapitulates natural protein conformational variability and improves mutant side-chain prediction. J Mol Biol. 2008;380:742–756. doi: 10.1016/j.jmb.2008.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith RE, Lovell SC, Burke DF, Montalvao RW, Blundell TL. Andante: reducing side-chain rotamer search space during comparative modeling using environment-specific substitution probabilities. Bioinformatics. 2007;23:1099–1105. doi: 10.1093/bioinformatics/btm073. [DOI] [PubMed] [Google Scholar]
- Song Y, Tyka M, Leaver-Fay A, Thompson J, Baker D. Structure-guided forcefield optimization. Proteins. 2011 doi: 10.1002/prot.23013. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiebritz MT, Muller YA. MUMBO: a protein-design approach to crystallographic model building and refinement. Acta Crystallogr D Biol Crystallogr. 2006;62:648–658. doi: 10.1107/S0907444906013333. [DOI] [PubMed] [Google Scholar]
- Tyka MD, Keedy DA, Andre I, Dimaio F, Song Y, Richardson DC, Richardson JS, Baker D. Alternate states of proteins revealed by detailed energy landscape mapping. J Mol Biol. 2011;405:607–618. doi: 10.1016/j.jmb.2010.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Dunbrack RL., Jr PISCES: recent improvements to a PDB sequence culling server. Nucleic Acids Res. 2005;33:W94–W98. doi: 10.1093/nar/gki402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiberg KB, Murcko MA. Rotational barriers. 2. Energies of alkane rotamers. An examination of gauche interactions. J. Am. Chem. Soc. 1988;110:8029–8038. [Google Scholar]
- Word JM, Lovell SC, Richardson JS, Richardson DC. Asparagine and glutamine: using hydrogen atom contacts in the choice of side-chain amide orientation. J. Mol. Biol. 1999;285:1735–1747. doi: 10.1006/jmbi.1998.2401. [DOI] [PubMed] [Google Scholar]
- Zhang C, Liu S, Zhou Y. Accurate and efficient loop selections by the DFIRE-based all-atom statistical potential. Protein Sci. 2004;13:391–399. doi: 10.1110/ps.03411904. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.