

Abstract
Cooperative dysregulation of gene sequence and expression may contribute to cancer formation and progression. The Cancer Genome Atlas (TCGA) Network recently cataloged gene sequence and expression data for a collection of glioblastoma multiforme (GBM) tumors. We developed an automated, model-free method to rapidly and exhaustively examine the correlation among somatic mutation and gene expression and interrogated 149 GBM tumor samples from the TCGA. The method identified 41 genes whose mutation status is highly correlated with drastic changes in the expression (z-score ±2.0), across tumor samples, of other genes. Some of the 41 genes have been previously implicated in GBM pathogenesis (e.g., NF1, TP53, RB1 and IDH1) and others, while implicated in cancer, had not previously been highlighted in studies using TCGA data (e.g., SYNE1, KLF6, FGFR4, and EPHB4). The method also predicted that known oncogenes and tumor suppressors participate in GBM via drastic over-and under-expression, respectively. Additionally, the method identified a known synthetic lethal interaction between TP53 and PLK1, other potential synthetic lethal interactions with TP53, and correlations between IDH1 mutation status and the overexpression of known GBM survival genes.
Keywords: Glioblastoma, The Cancer Genome Atlas, Synthetic Lethal Interactions, TP53, IDH1
Introduction
Cancer is a complex disease arising from the concerted effect of multiple (epi) genetic changes that yield pathway dysregulation via alterations in gene copy number, DNA methylation, gene expression, and molecular function (1–3). Specific combinations of these alterations can vary, even in histologically similar cancers. Until recently, the limited scalability of genetic experiments forbade complete characterization of these complexities and variances; now, large-scale cancer genomics experiments can catalog alterations with up to full-exome coverage across 10’s to 100’s of samples (1, 2, 4). Bioinformatics techniques can interrogate this data and identify alterations that cooperatively drive cancer (5–10), even with patient specificity (6).
Alterations that affect gene expression levels (e.g., copy-number alteration (CNA) and DNA methylation) in cancer genomes have been used to identify driver genes and molecular subtypes of a particular cancer (11, 12). Such alterations have identified oncogenes activated via increased expression (as can occur with EGFR, for instance) or tumor suppressors deactivated via decreased expression (as can occur with RB1, for instance). Verhaak et al. showed that four clinically relevant glioblastoma (GBM) subtypes could be defined using a subset of The Cancer Genome Atlas (TCGA) GBM expression data (11). In that study, the authors also grouped mutation and CNA with the expression-defined GBM subtypes. Increased expression can further be used to identify cancer-specific essential genes, oncogene addiction, and synthetic lethality (13, 14).
Understanding subtype and patient-specific combinatorial patterns of (epi) genetic alterations in tumors has promise to inform therapeutic regimens. First, expression patterns common to a subtype may be informative with respect to the drugs most suitable for a group of patients. For example, the neural GBM subtype has a high rate of EGFR and ERBB2 over-expression, but patients with neural GBM that are not EGFR and/or ERBB2 positive may not benefit from receptor tyrosine kinase inhibitors. Second, alterations in off-target genes can modulate the efficacy of targeted therapies (i.e., drug resistance). For instance, EGFR-positive tumors respond to gefitinib, but amplification of the MET proto-oncogene can cause resistance (15). Tumors over-expressing ERBB2 respond to trastuzumab, but PI3K mutation can cause trastuzumab resistance (15). Finally, cancer-specific essential genes, oncogene addiction, and synthetic lethality can be druggable vulnerabilities in tumors (13, 14). Notably, while current methods for synthetic lethal screening can identify such vulnerabilities, some studies suggest that considering isolated pairwise interactions limits generalizability. For example, three groups screened unique KRAS-driven cancers for synthetic lethal interactions and recovered three unique lists of genes synthetic lethal with KRAS mutation (16); this suggests that the identified synthetic lethal interactions were a subset of larger, more complex networks (i.e., context specificity).
Many current bioinformatics approaches for assessing complex patterns of (epi) genetic aberrations in cancer rely on pre-existing knowledge of gene annotations, gene sets, protein-protein interactions, and curated pathways. Gene-set enrichment analysis is a widely-used method for interpreting differential gene expression levels, based on previously described functions and pathway memberships. Vaske et al. have used CNA and expression data to infer patient-specific pathway activities in TCGA GBM samples (6). In that report, the authors identified GBM subtypes using pathways inferred from the National Cancer Institute-Nature Pathway Interaction Database.
Here, we present a new approach to identify genes that tumors require for progression and survival, with patient-level specificity, by exhaustively and rapidly detecting correlations among gene expression and mutation. The method makes inferences directly from a collection of cancer genome samples and does not depend on pre-existing knowledge of gene function or interactions. We propose that this unbiased approach has utility to complement the findings of current gene-set and pathway-based methods. We apply the method to examine the correlation between expression and mutation in TCGA GBM tumor samples. Our results suggest that this approach can be useful for identifying genes that participate in cancer progression, networks of genes that promote cancer via combined genetic and transcriptome alterations, druggable cancer-specific genes, and synthetic lethal interactions.
Materials and Methods
We developed a novel computational method to identify genes potentially important in tumorigenesis and cancer-specific survival genes from correlations among somatic mutation and expression in cancer genomics data (see Figure 1). The algorithm compares the sample (patient)-specific mutation status of each gene with the expression level of each gene, across all tumor samples. Genes with drastic mutation-correlated differential expression, and the corresponding mutated genes, are returned for analysis. The algorithm also identifies statistically significant mutation-mutation coincidence and mutual exclusivity. Gene networks are constructed containing all significant correlations and automated literature searches are used to illuminate clinically relevant findings. Findings presented here were identified using all TCGA GBM samples for which expression and mutation data were available and have a p-value < 0.01 and a false discovery rate < 0.05.
Figure 1.

Algorithm
We begin by building two matrices, one expression and one mutation, which are gene (row) by sample (column) (Figure 1A). At this stage, the expression matrix is populated by the factored, three-platform data (see Data) and the mutation matrix is binary: 1 (true) if any mutation (see Data) occurs in a particular gene in a particular sample, otherwise the element is 0 (false).
Next, two-class, unpaired Significance Analysis of Microarrays (SAM) (17) is used to find genes that are differentially expressed with respect to the mutation status of a particular gene across all samples (i.e., the two classes are defined by the binary mutation vector for that particular gene from the mutation matrix) (Figure 1B). SAM was employed using a moderated t-statistic and the random seed was set to a constant (rand=123) for reproducibility. To correct for multiple testing, 100 random permutations of the class labels were made and a cutoff false discovery rate (FDR) of 0.05 applied. Genes with an FDR < 0.05 are considered to have significant mutation-correlated differential expression and are passed to the next stage of the algorithm.
Next, an expression matrix is created, this time only containing genes deemed to have significant mutation-correlated differential expression in the previous step. Then, the matrix is converted to two binary matrices (one for significant over-expression and one for significant under-expression) with the following calculation: 1) the z-score for each expression matrix element is calculated with respect to that element’s row (i.e., gene specific); this is repeated for each row (gene). 2) For the over-expressed binary matrix, any element with a z-score > 2.0 is 1 (true), otherwise the element is 0 (false); for the under-expressed binary matrix an element is 1 if the z-score < −2.0 and 0 otherwise (Figure 1C). Then, Fisher’s exact p-value is calculated for each gene in the expression matrix by populating a two-by-two contingency table with a binary expression vector (category one) and the mutation vector (category two); this process is repeated for each binary expression vector from the binary expression matrix (Figure 1D). This calculation allowed us to recover only genes that had drastic mutation-correlated over- and under-expression and to assign each correlation with an exact p-value. Mutation-correlated over- and under-expressed genes with a p-value < 0.01 (Fisher’s exact test) and an FDR < 0.05 (see Multiple Testing Correction) are hierarchically clustered using heatmap.2 from the R package, and mutations are plotted across the samples (Figure 1E). The entire process is repeated once for each mutated gene.
Pairwise mutation-mutation correlation
Two-by-two contingency tables were constructed for every pairwise mutation vector to find significant (p-value < 0.01, Fisher’s exact test) mutational co-occurrence and mutual exclusivity. Coincident pairwise mutation in at least three samples was additionally required to declare significant mutational co-occurrence.
Multiple testing correction
For both mutation-mutation correlation and mutation-correlated over-and under-expression, a potential discovery is declared when Fisher’s exact p-value is less than 0.01. For each potential discovery the algorithm makes 1000 random permutations of the columns (samples) and counts the correlations inferred from the permutated data (i.e., false discoveries). If the calculated FDR is greater than 0.05, the potential discovery is rejected as false. Every correlation presented in this paper has a Fisher’s exact p-value less than 0.01 with a FDR less than 0.05.
Data
We obtained expression data for GBM samples at the TCGA website (http://tcga-data.nci.nih.gov/docs/publications/gbm_exp/). This expression data was gathered on three individual microarray platforms, including Affymetrix Human Exon ST GeneChips, Affymetrix HT-HG-U133A GeneChips, and custom designed Agilent 244,000 feature gene expression microarrays (11). A single estimate of the relative expression for each gene in each sample was obtained using factor analysis (11). We removed any gene that had a missing value in any sample used for this study (reducing the total from 11,861 to 11,828).
We obtained Phase I GBM sequence data from the TCGA website (http://tcga-data.nci.nih.gov/tcga/). We obtained Phase II GBM sequence data from Baylor College of Medicine (personal communication, David A. Wheeler). All mutations labeled Validated and Nonsilent, and Somatic or LOH (loss of heterozygosity) were used. There were a total of 583 genes that met these criteria and 149 samples for which both expression and mutation data were available.
Literature mining
All literature mining used to highlight results (Table 1) was automated to increase efficiency and reduce user bias. Importantly, the literature mining was used only to interpret results, not as an input to the algorithm. To highlight potentially important genes that were identified, the algorithm searched 2,438,505 abstracts and titles for PubMed keyword “cancer” and the gene of interest; the same procedure was carried out for 16,237 GBM-specific articles. To determine if genes had been described in previous studies using TCGA GBM data, we downloaded references 5–12, and converted them to text for automated searching. All mutation correlated over-expressed genes were cross referenced with a list of known GBM survival genes (see Table 2) (18). We retrieved summaries from the Entrez Gene database; these summaries were used to determine if mutation-correlated over-expressed genes were oncogenes and if mutation-correlated under-expressed genes were tumor suppressors (Figure 1F and Table 3).
Table 1.
| Gene symbol | Mutations | Diff. expr. genes | Pubmed hits for cancer | Pubmed hits for glioblastoma | Previously highlighted in study using TCGA GBM data? |
|---|---|---|---|---|---|
| AKR1C3 | 16 | 5 | 82 | 4 | Yes |
|
| |||||
| ANK1 | 3 | 2 | 9 | 0 | No |
|
| |||||
| ATM | 2 | 165 | 2175 | 20 | Yes |
| KLF6 | 2 | 165 | 103 | 6 | No |
| LEMD3 | 2 | 165 | 5 | 0 | No |
|
| |||||
| CHL1 | 2 | 2 | 15 | 0 | No |
|
| |||||
| ConsReg523 | 7 | 10 | 0 | 0 | No |
|
| |||||
| DST (dystonin) | 12 | 14 | 14 | 0 | No |
|
| |||||
| EP300 | 2 | 8 | 155 | 0 | Yes |
| FGFR4 | 2 | 8 | 139 | 4 | No |
|
| |||||
| EP400 | 3 | 6 | 9 | 0 | No |
| FN1 | 3 | 6 | 58 | 2 | No |
|
| |||||
| EPHB4 | 3 | 149 | 104 | 0 | No |
|
| |||||
| FBXW7 | 2 | 259 | 109 | 4 | No |
| FURIN | 2 | 259 | 252 | 8 | Yes |
|
| |||||
| FGFR1 | 3 | 2 | 491 | 19 | Yes |
|
| |||||
| HPN (Hepsin) | 11 | 3 | 82 | 1 | No |
|
| |||||
| IDH1 | 11 | 1001 | 126 | 41 | Yes |
|
| |||||
| INHBC | 3 | 2 | 5 | 0 | No |
|
| |||||
| KCNG1 | 3 | 8 | 0 | 0 | No |
|
| |||||
| LGALS3BP | 3 | 4 | 54 | 1 | No |
|
| |||||
| LUM | 3 | 8 | 6 | 0 | No |
|
| |||||
| MADD | 10 | 15 | 18 | 0 | No |
|
| |||||
| MAPK9 | 2 | 396 | 2 | 0 | No |
|
| |||||
| MARK1 | 2 | 20 | 14 | 0 | No |
|
| |||||
| MET (c-met) | 3 | 10 | 1674 | 52 | Yes |
|
| |||||
| MKI67 | 27 | 2 | 7866 | 218 | No |
|
| |||||
| MSH6 | 4 | 3 | 320 | 9 | Yes |
|
| |||||
| MYST4 | 3 | 2 | 11 | 0 | No |
|
| |||||
| NF1 | 19 | 10 | 1916 | 41 | Yes |
|
| |||||
| NOS3 | 3 | 59 | 190 | 1 | No |
|
| |||||
| PI15 | 2 | 47 | 1 | 0 | No |
|
| |||||
| PTK2B | 2 | 55 | 38 | 0 | No |
|
| |||||
| RB1 | 9 | 12 | 1025 | 21 | Yes |
|
| |||||
| SRGAP1 | 2 | 5 | 5 | 0 | No |
|
| |||||
| STK36 | 3 | 2 | 4 | 0 | No |
|
| |||||
| SYNE1 | 10 | 543 | 5 | 0 | No |
|
| |||||
| TCF12 | 2 | 28 | 25 | 0 | Yes |
|
| |||||
| TP53 | 48 | 38 | 4011 | 189 | Yes |
|
| |||||
| TRPM3 | 4 | 318 | 1 | 0 | No |
|
| |||||
| WISP1 | 2 | 2 | 39 | 0 | No |
Table 2.
|
Table 3.
| Mutated gene(s) | Under-expressed tumor suppressor | Over-expressed oncogene |
|---|---|---|
| FBXW7 FURRIN |
RARRES3 | RAF1 |
| IDH1 | RARRES3, DKK3, MCC | RAF1, MYCN, TET3, CDC25A |
| MAPK9 | RARRES3 | RAF1 |
| TRPM3 | RARRES3, DRAM | KRAS, CRKL |
| SYNE1 | MTUS1, ZFHX3, SPINT2 | RAF1 |
The algorithm developed for this study was written in Python (Figure 1). Entrez PubMed and Gene Summary database queries were made using Biopython. All calls to R and Bioconductor were made via the R interface for Python, RPy2 (http://rpy.sourceforge.net/rpy2.html). For the TCGA GBM dataset used here, total algorithm running time was less than five hours on a Linux workstation (two-core, 1.86 GHz processor and 4 GB of RAM).
Results and Discussion
Table 1 shows statistics for all genes where mutation status is significantly correlated with the drastic over- or under-expression, across tumor samples, of other genes. Our clustering scheme required genes be mutated in at least two samples, which reduced the total TCGA GBM set from 583 to 307. Forty-one of these mutated genes(~13%) were correlated with the drastic over- or under-expression of at least two of the 11,828 genes for which expression data was available. The low fraction of such correlations returned by our method partially reflects the stringency of the tests used to determine significance (see Materials and Methods). Comparing the numbers in columns 2 and 3 of Table 1 shows that there is no intrinsic bias of the algorithm to infer mutation-correlated over- or under-expression from frequency of mutation. For instance, HPN and IDH1 are each mutated in 11 samples, and IDH1 is correlated with the drastic over- or under-expression of 1001 genes, while HPN is only correlated with the drastic over- or under-expression of three genes. Low-frequency mutations also show a distribution of correlated expression. In the case of MAPK9, which was mutated in only two samples, there are 396 genes with correlated over-or under-expression. Conversely, CHL1 was mutated in two samples and only correlated with the differential expression of two genes.
If tumors select for genetic alterations that coordinate to promote cancer progression, then identifying coordinated genetic alterations could be useful to identify genes involved in tumorigenesis. Indeed, our approach identifies genes generally accepted to be involved in tumorigenesis (e.g., ATM, FGFR1, IDH1, MET, MSH6, NF1, RB1 and TP53). It is particularly difficult to assess the capacity of a genetic alteration to participate in cancer progression when that alteration is low frequency in the population; our approach identifies genes potentially involved in tumorigenesis that are mutated with low frequency in TCGA GBM tumor samples. For instance, ATM, KLF6, and LEMD3 are low-frequency mutations in TCGA GBM tumor samples, and have completely overlapping co-mutation (p-value 9 × 10−5 for each pairwise interaction, Fisher’s exact test). And, these low-frequency mutations are each highly correlated with the drastic over-or under-expression of 165 other genes. These observations suggest that ATM, KLF6, and LEMD3 may cooperatively promote tumorigenesis in some TCGA GBM samples.
EP300 and FGFR4, FBXW7 and FURIN, and EP400 and FN1 are also each exclusively co-mutated in TCGA GBM samples (Table 1). These four exclusively co-mutated sets of genes comprise 9 of the 41 mutated genes identified in this study (~22%), which may be unexpected. One potential explanation for this finding is that the mutant pairs have a specific epistatic relationship that is distinct from any of the mutations in isolation. A factor complicating the interpretation of the exclusively co-mutated sets is the occurrence of the of the so-called mutator phenotype. Each gene in the exclusively co-mutated sets is mutated in samples that are of the mutator phenotype, marked by higher-than-average mutation rates owing to mutation in mismatch repair genes. With the exception of LEMD3, EP400, and FN1, all genes in the co-mutated sets are well-studied cancer genes, and recurrence of mutations in these genes highlights them as potentially important in the progression of some gliomas. But, because these mutations were found in samples displaying the mutator phenotype, the possibility that some of them are passenger mutations has to be considered.
Columns 3–5 of Table 1 are derived from automated literature searches. While automated literature mining can be prone to false positives, large disparities among and within rows of Table 1 can highlight potentially important genes that can be investigated manually. For instance, KFL6 was found in the title or abstract of 103 papers on cancer and 6 specifically on GBM; however, KFL6 is a low-frequency mutation in TCGA GBM tumor samples and has never been highlighted in a study of TCGA GBM data. Manual investigation of PubMed ID’s returned by our method indicates that KFL6 is a well-studied cancer gene (19, 20). KLF6 is a putative tumor suppressor that mediates growth inhibition by over-expression of the cell cycle inhibitor CDKN1A (19). TCGA GBM samples contain at least one mutation in a previously reported KLF6 glioma mutation site (S77)(20). Similarly, EPHB4, FGFR4, FURIN, and NOS3 are all thought to be important in cancer progression; these genes are mutated with low-frequency in TCGA GBM tumor samples and not highlighted in previous studies using TCGA GBM data.
TP53 network
Figure 2 is a heatmap of genes whose over- or under-expression is significantly correlated with TP53 mutation. TP53 mutations clusters in two main groups on the right half the heatmap, with a few smaller clusters and outliers located among the samples. Aside from MDM2 (bottom of Figure 2), all genes in Figure 2 are over-expressed when TP53 is mutated. MDM2 is over-expressed when TP53 is wild type (i.e., MDM2 over-expression is mutually exclusive with TP53 mutation).
Figure 2.
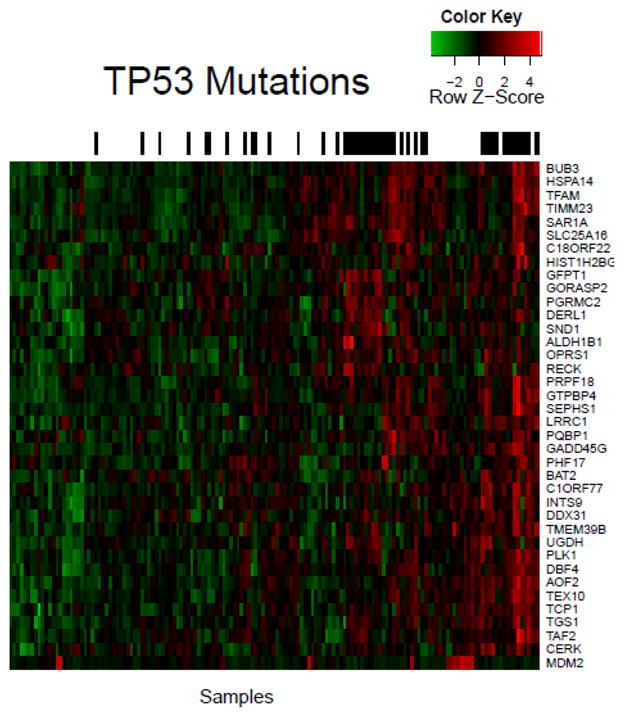
TP53 is a well-studied cancer gene, therefore method efficacy can be considered based on ability to capture known correlations. For instance, MDM2 is a negative regulator of tumor suppressor TP53, therefore MDM2 over-expression and TP53 mutation can have a redundant phenotype and can be mutually exclusive (21); our method recovers this mutual exclusivity (p-value 0.0075, Fisher’s exact test).
The observation that PLK1 over-expression occurs in cancer cells harboring TP53 mutation led some groups to speculate that inhibition of PLK1 may specifically killTP53 mutant cells(22, 23). Indeed, PLK1 inhibitors specifically kill cells harboring TP53 mutation (22, 23), suggesting TP53 and PLK1 may constitute a synthetic lethal interaction. We find a similar relation between TP53 and PLK1 in human TCGA GBM tumor samples (p-value 0.0099, Fisher’s exact test). Our method does not find significant correlation between PLK1 over-expression and the mutation status of any gene other than TP53.
DBF4 over-expression has been specifically linked to TP53 status (24). RNA-mediated interference of DBF4 was shown to specifically slow growth and reduce survival of melanoma cells (25). Our method found that DBF4 over-expression is correlated with TP53 mutation in TCGA GBM tumor samples (p-value 0.0099, Fisher’s exact test). TP53 and DBF4 may constitute a synthetic lethal pair, and DBF4 drugging might specifically kill cells harboring TP53 mutation. Our method does not find significant correlation between DBF4 over-expression and the mutation status of any gene other thanTP53.
Small-interfering RNA knockdown of the TP53-associated TCP1 gene resulted in slowed growth in a ovarian-carcinoma cell line (26). In a study using 186 breast cancer tumors, TCP1 subunit over-expression was shown to be correlated with TP53 mutation (27). We find significant correlation (p-value 0.0099, Fisher’s exact test) between TCP1 over-expression and TP53 mutation in TCGA GBM tumor samples. TP53 mutation may create a dependence on the over-expression of TCP1 and TCP1 may present a therapeutic vulnerability in some TP53-driven cancers.
BUB3 (28), HSPA14 (HSP60) (29), TFAM (30), GFTP1 (GFAT) (31), DERL1 (32), SND1 (33), ALDH1B1 (34), RECK (35), UGHD (36), AOF2 (LSD1) (37), GADD45G (38) and CERK (ceramide kinase) (39) have also been central genes in at least one cancer study where each was found to be over-expressed in certain cancers. DERL1 over-expression inspired Ran et al. (32) to target DERL1 with anti-DERL1 antibodies, which resulted in tumor growth suppression in mice. The UGDH inhibitors gallic acid and quercetin have strong antiproliferative effects in breast cancers over-expressing UGDH (36). Small-interfering-RNA mediated knockdown of AOF2 (LSD1) slows neuroblastoma cell growth in cells over-expressing AOF2 (37). Repression of CERK (ceramide kinase) in a human adenocarcinoma cell line over-expressing CERK induced apoptosis (40). HSPA14 (HSP60) inhibition can selectively induce apoptosis in tumor cells over-expressing HSPA14 (29). To the best of our knowledge, this is the first time the over-expression of these genes has been linked to TP53 mutation. Because inhibition of these genes induces effects specific to cancer cells, they may be druggable targets in cancers mutated in TP53.
Subclusters in Figure 2 arise from tumor samples sharing genes with similar TP53-mutation-correlated expression. For example, PLK1, DBF4, AOF2, TCP1, and CERK (defined here as cluster A) cluster together because they have similar expression across all samples. Over-expression of each cluster-A gene is associated with a druggable dependence in cancer cells (22, 23, 25, 26, 37, 40), and PLK1 (22), DBF4 (24), and TCP1 (27) are known to be over-expressed specifically in the context of TP53 mutation. Our method identifies groups of tumors that may have a dependence on multiple druggable targets. Tumor dependence on multiple over-expressed druggable genes may be of therapeutic relevance because low-concentration inhibitor cocktails could replace single-agent targeted therapies, resulting in increased therapeutic index(41).
Mutation-correlated differential expression among subclusters may also inform therapeutic regimens. For instance, GFPT1, GORASP2, PGRMC2, DERL1, SND1, ALDH1B1, OPRS1, and RECK (defined here as cluster B) are over-expressed in tumors distinct from those with cluster-A gene over-expression. Because cluster-A gene over-expression is a signature of cluster-A gene dependence, cluster-A gene inhibitors might inhibit tumors over-expressing cluster-A genes more than tumors lacking cluster-A gene over-expression. In that case, patients with cluster-A signatures and patients with cluster-B signatures may benefit from different drugging protocols, which our method highlights.
IDH1/SYNE1 networks
In a landmark study, Parsons et al. discovered a novel, high-frequency driver mutation in IDH1, highlighting the utility of unbiased genomics experiments(4). Focused studies by many groups confirmed the importance of IDH1 mutation in GBM. Of the 41 mutated genes returned by our method, IDH1 is one of the most studied genes in GBM (Table 1, column 5), which is striking considering its importance in GBM is recently discovered. Our method finds 1001 genes have drastic over-or under-expression associated with IDH1 mutation status; thisIDH1 network is by far the largest network returned by our method (Table 1, column 3). This suggests that IDH1 mutation is associated with an unique GBM (epi)genotype. Indeed, IDH1 mutation is a defining characteristic of the proneural GBM subtype (11) and the glioma CpG-island methylator phenotype(12).
Figure 3 is a graph representation of all IDH1 nearest and second-nearest neighbors. In this graph, nodes represent mutated genes, over-expressed oncogenes or GBM survival genes, or under-expressed tumor suppressors returned by our method. These types of coordinated (de)activation can drive cancer, and second nearest neighbors highlight networks connected by common genes.
Figure 3.

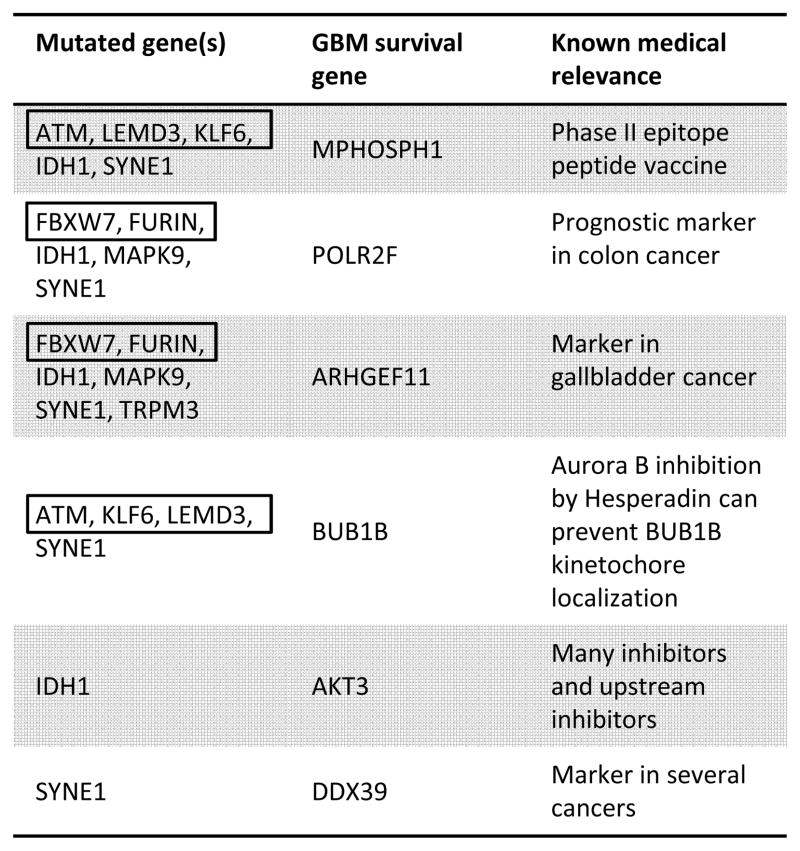
We find TCGA GBM tumors with IDH1 mutation are significantly correlated with the drastic over-expression of several known GBM survival genes (Figure 3 and Table 2) (18): MPHOSPH1, POLR2F, ARHGEF11, and AKT3 (p-value 4.1×10−5, 2.8×10−3, 2.8×10−3 and 2.8×10−3, respectively). Because IDH1 mutation is a defining characteristic of specific GBM (epi)genotypes, druggable dependencies associated with IDH1 mutation status could be clinically relevant. M-phase phosphoprotein 1 (MPHOSPH1) is known to be over-expressed in some bladder cancers (42). Recently, phase I/II trials using MPHOSPH1 peptide epitopes were shown to induce specific cytotoxic T lymphocytes against bladder cancers over-expressing MPHOSPH1 (42). Our results suggest that similar approaches may benefit some gliomas mutated in IDH1. AKT3 over-expression was found in a significant fraction of breast and prostate cancers (43), and has been reported as a possible oncogene and a potential glioma survival gene (18). Indeed, oncogene addiction could be considered a type of survival-gene dependence. Importantly, AKT3 is a well-studied cancer gene, and inhibitors of AKT3 and other genes in the AKT3 pathway exist (43). Here, we find that drastic over-expression of AKT3 is exclusively and significantly associated with IDH1 mutation. Over-expression of the GBM survival genes POLR2F and ARHGEF11 are known markers in colon (44) and gallbladder (45) cancer, respectively; unfortunately, we know of no drugs that target these genes. All known GBM survival genes whose drastic over-expression is correlated with mutation status of another gene, and the corresponding mutated genes, are shown in Table 2.
Our method identified several mutation-correlated over-expressed oncogenes and under-expressed tumor suppressors, the majority being significantly associated with IDH1 mutation status(Figure 3 and Table 3). The under-expression of tumor suppressors RARRES3, DKK3, and MCC was significantly correlated with IDH1 mutation (p-value 8.0×10−5, 9.0×10−3, and 2.8×10−3, respectively); DKK3 and MCC under-expression was exclusively associated with IDH1 mutation. Over-expression of the oncogenes RAF1, MYCN, TET3, and CDC25A was significantly correlated with IDH1 mutation (p-value 2.8×10−3, 9.0×10−3, 1.2×10−3 and 1.2×10−3, respectively); MYCN, TET3, and CDC25A over-expression was exclusively associated with IDH1 mutation. All known oncogenes whose drastic over-expression, and tumor suppressors whose drastic under-expression is correlated with mutation status of another gene, and the corresponding mutated genes, are shown in Table 3.
Our method finds 543 genes have drastic over- or under-expression associated with SYNE1 mutation status; this SYNE1 network is the second largest network returned by our method (Table 1, column 3). Similarly, SYNE1 participates in significant mutational co-occurrence more than any other gene; there are 12 mutation-mutation interactions involving SYNE1 (Figure 3). Also, SYNE1 is the only gene with which MSH6 and MLH1 have complete mutational overlap (1.1×10−5 and 2.2×10−4, respectively); MSH6 and MLH1 are mismatch repair genes whose mutation is known to cause the so-called mutator phenotype in GBM(46).
SYNE1 mutation is high-frequency in TCGA GBM tumor samples, but has not been highlighted in previous studies using TCGA GBM data (Table 1). Similarly, our method does not find any previous correlation between GBM and SYNE1 mutation in the literature (Table 1). SYNE1 mutation is known to influence cerebellar ataxia, and has recently been associated with lung, ovarian, and colorectal cancers (47). Our results suggest SYNE1 mutation is important in TCGA GBM tumor samples, and may be important in some glioblastomas in general.
We find SYNE1 mutation is significantly correlated with the over-expression of several known GBM survival genes (Table 2). BUB1B is a chromosome instability gene known to be involved in cancer (48). The aurora-B inhibitor hesperadin can prevent kinetochore localization of BUB1B and arrest cell cycle progression (49); hesperadin has not yet been proven effective in cancer clinical trials. We find this known GBM survival and chromosome instability gene to be over-expressed in the presence of SYNE1 mutation (p-value 8.6×10−4), suggesting BUB1B as a potential therapeutic target in some SYNE1 mutated gliomas. DDX39 is known to be over-expressed in several cancer types (50) and is a known GBM survival gene (18). We suggest possible a connection between these results, in thatDDX39 dependency may present as DDX39 over-expression. And, this dependency is significantly correlated with the mutation status of SYNE1 in TCGA GBM samples (p-value 8.6×10−4). Other survival genes having over-expression significantly correlated with SYNE1 mutation status include MPHOSPH1 and POLR2F (p-value 6.5×10−4 and 2.1×10−3, respectively), and were described above. Our method also finds that the under-expression of the MTUS1, ZFHX3, and SPINT2 tumor suppressors is significantly and exclusively correlated with the mutation status of SYNE1(p-value 2.1×10−3, 2.1×10−3, and 4.1×10−3, respectively). RAF1 oncogene over-expression is significantly correlated with SYNE1 mutation status (p-value 2.1×10−3).
Other Considerations
One important distinction to make, when considering alteration co-occurrence in cancer, is whether identified interactions have true cellular dependence or if they are correlated for an unidentified reason. For instance, our method recovered several GBM survival genes whose over-expression was correlated with IDH1 mutation status. But, IDH1 mutation was found to be associated with a broadly altered (epi)genotype in this and other GBM studies. Therefore, IDH1 mutation and survival gene over-expression could be selected for by similar or overlapping hubs from the IDH1 network, but not by each other. In that scenario, the complex networks could vary among patients and cancer types reducing the generalizability of drugging protocols. Furthermore, any inhibitor targeting an over-expressed gene will be limited in efficacy to scenarios where that gene is significantly over-expressed in the patients tumor relative to their healthy tissue.
Elucidating true interaction dependence could also be informative. For instance, if mutation in hypothetical gene A created a strict cellular dependence on the over-expression of hypothetical gene B, then by definition there would be a requirement for gene B over-expression to precede gene A mutation during cancer progression. This temporal ordering would be required because cells harboring mutation in gene A, but not over-expressing gene B, would be eliminated from the population. In cancer genomics data this would manifest as significant correlation between gene A mutation and gene B over-expression, and on average, a greater number of samples over-expressing gene B, compared with those harboring gene A mutation. The requirement for such temporal ordering could be exploited for prognosis as well as provide an obvious therapeutic target.
Conclusion
In this report we developed an intuitive and unbiased method to exhaustively interrogate cancer genomics data to identify genes that tumors require for progression and survival. The method identified many genes known to promote GBM pathogenesis and highlighted several genes not previously associated with GBM as potentially important in GBM pathogenesis. Additionally, the algorithm identified known druggable cancer-specific dependencies, survival genes, and potential synthetic lethal interactions. And, all observations were identified with patient specificity, which could increase clinical utility.
This algorithm should be a useful complement to existing methods. Because it is exhaustive, and unbiased in that all genes are tested regardless of prior association to disease, our new algorithm may identify novel correlations that add to the existing/emerging picture of gliomas and caner in general. Furthermore, development of model-free approaches, such as that developed in this study, may be applicable to a wide range of genes and pathways as they do not rely on previously curated pathway or interaction databases.
A useful addition to our algorithm might be to consider site- or domain-specific mutation. While this is expected to be noisy for most genes, genes with multiple domain-specific functions may influence distinct, mutation-specific regulatory changes. One difficulty in implementing such a strategy would be distinguishing protein functional regions in an automated fashion.
One improvement to our method would be the ability to automatically return known inhibitors for inferred therapeutic vulnerabilities. It is not immediately obvious how this improvement could be implemented, owing to a lack of systematic annotation in the literature; however, assembling the correct drug databases might be one approach. If successful, clinical cancer genomics data would be algorithmic input, and the output could consist of therapeutic vulnerabilities ranked by known druggability.
Important open questions include the origin of drug resistance and the generalizability of synthetic lethal interactions. Most inhibitors targeting a specific driver gene have only modest success, often owing to off-target alterations. Similarly, synthetic lethal killing of tumor cells with generalizability has yet to be demonstrated, suggesting the potential existence of a synthetic lethal network. Therefore, a comprehensive list of compensatory alterations that cause drug resistance or facilitate viability in the presence of targeted synthetic lethality may be useful. The information imparted from such a compendium could allow clinicians to cut cancer off at the pass. To that end, the combined effort of high-throughput cancer (epi)genomics experiments and complementary bioinformatics approaches is indispensible.
Supplementary Material
Acknowledgments
We thank Dr. Bert Vogelstein for his critical reading of the manuscript. This work was funded by NIH NCI grant CA135877 and NSF DBI CAREER award 0845275 to RK.
Footnotes
Supporting material for this manuscript includes six supplementary files: SYNE1 and IDH1 survival analysis and occurrence of mutations highlighted in this study in the COSMIC database (Supplementary Figure 1 and Table 1); individual heatmaps for each of the 41 genes whose mutation status is significantly correlated with the over- and under-expression of other genes (Supplementary Heatmaps); the raw data, including p-values, for every correlation returned by our algorithm (Supplementary Mutation-expression correlation and Supplementary Mutation-mutation correlation); the sample-specific mutation type (e.g., non-sense, splice site, frame shift) for each mutated gene highlighted in the study, the zygosity, and the CHASM predictions for each missense mutation (Supplementary Mutation type, zygosity, and score); legends for all spreadsheets (Supplementary Spreadsheet legends).
References
- 1.Jones S, Zhang X, Parsons DW, Lin JC-H, Leary RJ, Angenendt P, et al. Core Signaling Pathways in Human Pancreatic Cancers Revealed by Global Genomic Analyses. Science. 2008;321:1801–6. doi: 10.1126/science.1164368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McLendon R, Friedman A, Bigner D, Van Meir E, Brat D, Mastrogianakis G, et al. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–8. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yeang C-H, McCormick F, Levine A. Combinatorial patterns of somatic gene mutations in cancer. The FASEB Journal. 2008;22:2605–22. doi: 10.1096/fj.08-108985. [DOI] [PubMed] [Google Scholar]
- 4.Parsons DW, Jones S, Zhang X, Lin JC-H, Leary RJ, Angenendt P, et al. An Integrated Genomic Analysis of Human Glioblastoma Multiforme. Science. 2008;321:1807–12. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gaire RK, Bailey J, Bearfoot J, Campbell IG, Stuckey PJ, Haviv I. MIRAGAA—a methodology for finding coordinated effects of microRNA expression changes and genome aberrations in cancer. Bioinformatics. 2010;26:161–7. doi: 10.1093/bioinformatics/btp654. [DOI] [PubMed] [Google Scholar]
- 6.Vaske CJ, Benz SC, Sanborn JZ, Earl D, Szeto C, Zhu J, et al. Inference of patient-specific pathway activities from multi-dimensional cancer genomics data using PARADIGM. Bioinformatics. 2010;26:237–45. doi: 10.1093/bioinformatics/btq182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brennan C, Momota H, Hambardzumyan D, Ozawa T, Tandon A, Pedraza A, et al. Glioblastoma Subclasses Can Be Defined by Activity among Signal Transduction Pathways and Associated Genomic Alterations. PLoS ONE. 2009;4:e7752. doi: 10.1371/journal.pone.0007752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Freire P, Vilela M, Deus H, Kim Y-W, Koul D, Colman H, et al. Exploratory Analysis of the Copy Number Alterations in Glioblastoma Multiforme. PLoS ONE. 2008;3:e4076. doi: 10.1371/journal.pone.0004076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cerami E, Demir E, Schultz N, Taylor BS, Sander C. Automated Network Analysis Identifies Core Pathways in Glioblastoma. PLoS ONE. 2010;5:e8918. doi: 10.1371/journal.pone.0008918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bredel M, Scholtens DM, Harsh GR, Bredel C, Chandler JP, Renfrow JJ, et al. A Network Model of a Cooperative Genetic Landscape in Brain Tumors. JAMA. 2009;302:261–75. doi: 10.1001/jama.2009.997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Verhaak RGW, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, et al. Integrated Genomic Analysis Identifies Clinically Relevant Subtypes of Glioblastoma Characterized by Abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17:98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Noushmehr H, Weisenberger DJ, Diefes K, Phillips HS, Pujara K, Berman BP, et al. Identification of a CpG Island Methylator Phenotype that Defines a Distinct Subgroup of Glioma. Cancer Cell. 2010;17:510–22. doi: 10.1016/j.ccr.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Luo J, Solimini NL, Elledge SJ. Principles of Cancer Therapy: Oncogene and Non-oncogene Addiction. Cell. 2009;136:823–37. doi: 10.1016/j.cell.2009.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McManus KJ, Barrett IJ, Nouhi Y, Hieter P. Specific synthetic lethal killing of RAD54B-deficient human colorectal cancer cells by FEN1 silencing. Proceedings of the National Academy of Sciences. 2009;106:3276–81. doi: 10.1073/pnas.0813414106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ikediobi ON. Somatic pharmacogenomics in cancer. Pharmacogenomics J. 2008;8:305–14. doi: 10.1038/tpj.2008.8. [DOI] [PubMed] [Google Scholar]
- 16.Singh A, Settleman J. Oncogenic K-ras “addiction” and synthetic lethality. Cell Cycle. 2009;8:2676–8. doi: 10.4161/cc.8.17.9336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proceedings of the National Academy of Sciences. 2001;98:5116–21. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thaker NG, Zhang F, McDonald PR, Shun TY, Lewen MD, Pollack IF, et al. Identification of Survival Genes in Human Glioblastoma Cells by Small Interfering RNA Screening. Molecular Pharmacology. 2009;76:1246–55. doi: 10.1124/mol.109.058024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Narla G, Heath KE, Reeves HL, Li D, Giono LE, Kimmelman AC, et al. KLF6, a Candidate Tumor Suppressor Gene Mutated in Prostate Cancer. Science. 2001;294:2563–6. doi: 10.1126/science.1066326. [DOI] [PubMed] [Google Scholar]
- 20.Jeng Y-M, Hsu H-C. KLF6, a putative tumor suppressor gene, is mutated in astocytic gliomas. International Journal of Cancer. 2003;105:625–9. doi: 10.1002/ijc.11123. [DOI] [PubMed] [Google Scholar]
- 21.Ichimura K, Bolin MB, Goike HM, Schmidt EE, Moshref A, Collins VP. Deregulation of the p14ARF/MDM2/p53 Pathway Is a Prerequisite for Human Astrocytic Gliomas with G1-S Transition Control Gene Abnormalities. Cancer Research. 2000;60:417–24. [PubMed] [Google Scholar]
- 22.Degenhardt Y, Greshock J, Laquerre S, Gilmartin AG, Jing J, Richter M, et al. Sensitivity of Cancer Cells to Plk1 Inhibitor GSK461364A Is Associated with Loss of p53 Function and Chromosome Instability. Molecular Cancer Therapeutics. 2010;9:2079–89. doi: 10.1158/1535-7163.MCT-10-0095. [DOI] [PubMed] [Google Scholar]
- 23.Sur S, Pagliarini R, Bunz F, Rago C, Diaz LA, Kinzler KW, et al. A panel of isogenic human cancer cells suggests a therapeutic approach for cancers with inactivated p53. Proceedings of the National Academy of Sciences. 2009;106:3964–9. doi: 10.1073/pnas.0813333106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bonte D, Lindvall C, Liu H, Dykema K, Furge K, Weinreich M. Cdc7-Dbf4 kinase overexpression in multiple cancers and tumor cell lines is correlated with p53 inactivation. Neoplasia (New York, NY) 2008;10:920. doi: 10.1593/neo.08216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nambiar S, Mirmohammadsadegh A, Hassan M, Mota R, Marini A, Alaoui A, et al. Identification and functional characterization of ASK/Dbf4, a novel cell survival gene in cutaneous melanoma with prognostic relevance. Carcinogenesis. 2007;28:2501–10. doi: 10.1093/carcin/bgm197. [DOI] [PubMed] [Google Scholar]
- 26.Macleod K, Mullen P, Sewell J, Rabiasz G, Lawrie S, Miller E, et al. Altered ErbB Receptor Signaling and Gene Expression in Cisplatin-Resistant Ovarian Cancer. Cancer Research. 2005;65:6789–800. doi: 10.1158/0008-5472.CAN-04-2684. [DOI] [PubMed] [Google Scholar]
- 27.Ooe A, Kato K, Noguchi S. Possible involvement of CCT5, RGS3, and YKT6 genes up-regulated in p53-mutated tumors in resistance to docetaxel in human breast cancers. Breast Cancer Research and Treatment. 2007;101:305–15. doi: 10.1007/s10549-006-9293-x. [DOI] [PubMed] [Google Scholar]
- 28.Yuan B, Xu Y, Woo J-H, Wang Y, Bae YK, Yoon D-S, et al. Increased Expression of Mitotic Checkpoint Genes in Breast Cancer Cells with Chromosomal Instability. Clinical Cancer Research. 2006;12:405–10. doi: 10.1158/1078-0432.CCR-05-0903. [DOI] [PubMed] [Google Scholar]
- 29.Ghosh JC, Dohi T, Kang BH, Altieri DC. Hsp60 Regulation of Tumor Cell Apoptosis. Journal of Biological Chemistry. 2008;283:5188–94. doi: 10.1074/jbc.M705904200. [DOI] [PubMed] [Google Scholar]
- 30.Cormio A, Guerra F, Cormio G, Pesce V, Fracasso F, Loizzi V, et al. The PGC-1[alpha]-dependent pathway of mitochondrial biogenesis is upregulated in type I endometrial cancer. Biochemical and Biophysical Research Communications. 2009;390:1182–5. doi: 10.1016/j.bbrc.2009.10.114. [DOI] [PubMed] [Google Scholar]
- 31.Paterson A, Kudlow J. Regulation of glutamine:fructose-6-phosphate amidotransferase gene transcription by epidermal growth factor and glucose. Endocrinology. 1995;136:2809–16. doi: 10.1210/endo.136.7.7789306. [DOI] [PubMed] [Google Scholar]
- 32.Ran Y, Hu H, Hu D, Zhou Z, Sun Y, Yu L, et al. Derlin-1 Is Overexpressed on the Tumor Cell Surface and Enables Antibody-Mediated Tumor Targeting Therapy. Clinical Cancer Research. 2008;14:6538–45. doi: 10.1158/1078-0432.CCR-08-0476. [DOI] [PubMed] [Google Scholar]
- 33.Ho J, Kong J-W-F, Choong L-Y, Loh M-C-S, Toy W, Chong P-K, et al. Novel Breast Cancer Metastasis-Associated Proteins. Journal of Proteome Research. 2008;8:583–94. doi: 10.1021/pr8007368. [DOI] [PubMed] [Google Scholar]
- 34.The Gene Expression Profiles of Medulloblastoma Cell Lines Resistant to Preactivated Cyclophosphamide. Current Cancer Drug Targets. 2008;8:172–9. doi: 10.2174/156800908784293631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kitajima S, Miki T, Takegami Y, Kido Y, Noda M, Hara E, et al. Reversion-inducing cysteine-rich protein with Kazal motifs interferes with epidermal growth factor receptor signaling. Oncogene. 2010 doi: 10.1038/onc.2010.448. [DOI] [PubMed] [Google Scholar]
- 36.Hwang EY, Huh J-W, Choi M-M, Choi SY, Hong H-N, Cho S-W. Inhibitory effects of gallic acid and quercetin on UDP-glucose dehydrogenase activity. FEBS Letters. 2008;582:3793–7. doi: 10.1016/j.febslet.2008.10.010. [DOI] [PubMed] [Google Scholar]
- 37.Schulte JH, Lim S, Schramm A, Friedrichs N, Koster J, Versteeg R, et al. Lysine-Specific Demethylase 1 Is Strongly Expressed in Poorly Differentiated Neuroblastoma: Implications for Therapy. Cancer Research. 2009;69:2065–71. doi: 10.1158/0008-5472.CAN-08-1735. [DOI] [PubMed] [Google Scholar]
- 38.Flores O, Burnstein KL. GADD45{gamma}: a New Vitamin D-Regulated Gene that Is Antiproliferative in Prostate Cancer Cells. Endocrinology. 2010;151:4654–64. doi: 10.1210/en.2010-0434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ruckhäberle E, Karn T, Rody A, Hanker L, Gätje R, Metzler D, et al. Gene expression of ceramide kinase, galactosyl ceramide synthase and ganglioside GD3 synthase is associated with prognosis in breast cancer. Journal of Cancer Research and Clinical Oncology. 2009;135:1005–13. doi: 10.1007/s00432-008-0536-6. [DOI] [PubMed] [Google Scholar]
- 40.Mitra P, Maceyka M, Payne SG, Lamour N, Milstien S, Chalfant CE, et al. Ceramide kinase regulates growth and survival of A549 human lung adenocarcinoma cells. FEBS Letters. 2007;581:735–40. doi: 10.1016/j.febslet.2007.01.041. [DOI] [PubMed] [Google Scholar]
- 41.Teicher BA. Combinations of PARP, hedgehog and HDAC inhibitors with standard drugs. Current Opinion in Pharmacology. 2010;10:397–404. doi: 10.1016/j.coph.2010.04.014. [DOI] [PubMed] [Google Scholar]
- 42.Obara W, Tsunoda T, Yoshida K, Kanehira M, Takata R, Katagiri T, et al. Phase I/II study of novel HLA-A24 restricted DEPDC1 and MPHOSPH1 peptide vaccine for bladder cancer. J Clin Oncol (Meeting Abstracts) 2010;28:e13122. [Google Scholar]
- 43.Lindsley C, Barnett S, Layton M, Bilodeau M. The PI3K/Akt pathway: recent progress in the development of ATP-competitive and allosteric Akt kinase inhibitors. Current Cancer Drug Targets. 2008;8:7–18. doi: 10.2174/156800908783497096. [DOI] [PubMed] [Google Scholar]
- 44.Antonacopoulou AG, Grivas PD, Skarlas L, Kalofonos M, Scopa CD, Kalofonos HP. POLR2F, ATP6V0A1 and PRNP Expression in Colorectal Cancer: New Molecules with Prognostic Significance? Anticancer Research. 2008;28:1221–7. [PubMed] [Google Scholar]
- 45.Kim J, Kim H, Lee K, Lee J, Choi S, Paik S, et al. Gene expression profiles in gallbladder cancer: the close genetic similarity seen for early and advanced gallbladder cancers may explain the poor prognosis. Tumor Biology. 2008;29:41–9. doi: 10.1159/000132570. [DOI] [PubMed] [Google Scholar]
- 46.Purow B, Schiff D. Advances in the genetics of glioblastoma: are we reaching critical mass? Nat Rev Neurol. 2009;5:419–26. doi: 10.1038/nrneurol.2009.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Doherty JA, Rossing MA, Cushing-Haugen KL, Chen C, Van Den Berg DJ, Wu AH, et al. ESR1/SYNE1 Polymorphism and Invasive Epithelial Ovarian Cancer Risk: An Ovarian Cancer Association Consortium Study. Cancer Epidemiology Biomarkers & Prevention. 2010;19:245–50. doi: 10.1158/1055-9965.EPI-09-0729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ricke RM, van Ree JH, van Deursen JM. Whole chromosome instability and cancer: a complex relationship. Trends in Genetics. 2008;24:457–66. doi: 10.1016/j.tig.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hauf S, Cole RW, LaTerra S, Zimmer C, Schnapp G, Walter R, et al. The small molecule Hesperadin reveals a role for Aurora B in correcting kinetochore–microtubule attachment and in maintaining the spindle assembly checkpoint. The Journal of Cell Biology. 2003;161:281–94. doi: 10.1083/jcb.200208092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sugiura T, Nagano Y, Noguchi Y. DDX39, upregulated in lung squamous cell cancer, displays RNA helicase activities and promotes cancer cell growth. Cancer biology & therapy. 2007;6:957. doi: 10.4161/cbt.6.6.4192. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.