

Abstract
Functional gene clustering is a statistical approach for identifying the temporal patterns of gene expression measured at a series of time points. By integrating wavelet transformations, a power dimension-reduction technique, noisy gene expression data is smoothed and clustered allowing for new patterns of functional gene expression profiles to be identified. We implement the idea of wavelet dimension reduction into the mixture model for gene clustering, aimed to de-noise the data by transforming an inherently high-dimensional biological problem to its tractable low-dimensional representation. As a first attempt of its kind, we capitalize on the simplest Haar wavelet shrinkage technique to break an original signal down into its spectrum by taking its averages and differences and, subsequently, detect gene expression patterns that differ in the smooth coefficients extracted from noisy time series gene expression data. The method is shown to be effective on simulated data and and on recent time course gene expression data. Supplementary Material is available at www.liebertonline.com.
Key words: algorithms, statistics
1. Introduction
Although high-throughput technologies, such as DNA microarrays and proteomics platforms, have provided researchers with a set of unprecedented tools to ask and address various fundamental questions in developmental biology and biomedicine, the use of these technologies that generate enormous amounts of gene or protein data from biological entities relies critically on statistical analysis and modeling of the data.
The past decade has witnessed an astonishing development of statistical methods for cataloguing the patterns of gene expression and using these distinct patterns to assessing developmental functions and mechanisms of a biological phenomena (Eisen et al., 1998; Ramoni et al., 2002; Ghosh and Chinnaiyan, 2002; McLachlan et al., 2002; Zapala and Schork, 2006). More recently, there has been a considerable body of literature about the derivations of statistical methods for clustering time-dependent gene expression (Qian et al., 2001; Holter et al., 2001; Zhao et al., 2001; Park et al., 2003; Bar-Joseph et al., 2003; Luan and Li, 2003; Ernst et al., 2005; Storey et al., 2005; Ma et al., 2006; Ng et al., 2006; Inoue et al., 2007; Kim et al., 2008; Wang et al., 2009).
The central idea of functional gene clustering is to mathematically model the mean vectors for each gene pattern within the mixture model context incorporating the structure of covariance of the gene expressions measured at discrete time points. Such mathematical modeling has two major advantages. First, instead of estimating every mean at each time point and every element in the covariance matrix, functional clustering only needs to estimate a reduced number of mathematical parameters that model the mean-covariance structures. This provides greater power to detect significantly differentiated patterns during a time course. Second, gene expression profiles related to many biological processes have a certain pattern, which can be described robustly by mathematical functions. By estimating the parameters that determine mathematical functions, the genetic differentiation over time course can be estimated and tested. The results from these biologically justified models are, therefore, more closer to biological reality.
Despite its statistical and biological relevances, functional clustering has two significant limitations that may prevent its broad and deep uses in some particular situations. First, it does not allow the number of repeated measurements (defined as the dimensionality of observation) to unlimitedly increase for robust parameter estimation. While increased dimensionality possesses richer information, structural modeling of high-dimensional variances and correlations will be computationally expensive. With increasing dimension, the computation of inverse covariance matrix will tend to be unstable. Second, in practice, the sparsity of a data set increases exponentially with its dimensionality. Functional gene clustering based on a multivariate normal density function will be affected for high-dimensional data as measurement error will become increasingly problematic in parameter estimation of the classical mixture models.
An efficient treatment of high-dimensional microarray data is through dimensionality reduction, i.e., the transformation that brings data from a high- to low-order dimension. It has been shown that models with low dimension are not only computationally efficient, but also more robust than high dimensional models. Wavelet transforms that preserve signal pattern and yield better or comparative classification accuracy provide a powerful tool for dimensionality reduction (Donoho, 1995; Donoho and Johnstone, 1994).
In this article, we derive a wavelet-based de-noising method for functional clustering of time-dependent microarray gene expression data. By reducing the dimensionality of data, this method improves the accuracy and power of gene cluster detection in many situations.
2. Methods
2.1. Wavelet Transform
According to wavelet transform methodology, an original signal is divided into two sequences each with a length equal to a half of the original signal length (Mallat et al., 1989; Vidakovic, 1999; Jensen and la Cour-Harbo, 2001). The first sequence, denoted as the smooth coefficients (or approximation coefficients), corresponds to an approximation process of the original signal, whereas the second sequence, denoted as the detail coefficients, corresponds to the detail information (subtleties) that is complementary to the approximation process. We use c−r and d−r to denote the smooth and detail coefficients, respectively, where superscript −r indicates the resolution level at which the initial sequence is split into smooth and detail coefficients. Since detail coefficients are contaminated severely by random errors, shrinking them to zero will be helpful for reducing the overall noise level of the signal (Donoho, 1995; Donoho and Johnstone, 1994).
As the simplest wavelet transform, discrete Haar transform calculates detail coefficients by subtracting successive values in the sequence (Walker, 1999). The data of expression profile for gene i measured at T time points can be expressed as
 |
(1) |
The smooth and detail coefficients of the original signal after the first-resolution Haar wavelet transform are arrayed by
 |
(2) |
where τ is the new sequence index used after applying the Haar wavelet transform. It can be seen that variation in detail coefficients at resolution 1 only reflects local fluctuations between the nearest neighbors in the sequence. Similarly, smooth coefficients at resolution 2 are obtained by summing pairs of resolution 1 smooth coefficients. At each resolution, the number of smooth and detail coefficients obtained drops by ½. The process can be repeated, each time reducing the dimension of the smooth and detail coefficients (Walker, 1999).
The pattern of the smooth coefficients in the wavelet space resembles the signal pattern in the time space. In Figure 1, c−0 represents a sample of repeated measurements of a pattern at 24 time points. The smooth coefficients of the first and second Haar wavelet transformation are plotted as c−1 and c−2, respectively. The pattern of c−1 and c−2 coefficients conform to the signal pattern although they are in two different resolution levels. Because of the similarity, it is reasonable to model the original pattern based on low-dimensional smooth coefficients.
FIG. 1.

The original periodic profile of a gene (c0), subject to Haar wavelet transform at the first (c−1) and second resolutions (c−2). The transformed data preserve most information of the original signal, although the lower-order resolution tends to be close to the original signal than does the higher-order resolution.
2.2. Thresholding
In wavelet transform, we need to find an approximation of the original signal which is smooth and can also adequately represent the input signal. Such an approximation can be detected by two thresholding approaches—the hard threshold filter (Hh) and the soft threshold filter (Hs). The hard threshold filter, also known as the “keep or kill” method (Aboufadel and Schlicker, 1999), removes coefficients below a threshold value determined by the estimated noise variance. The soft threshold shrinks large coefficients towards zero but also completely removes the smaller coefficients (Ghael et al., 1997). Since the de-noised signal is irreversibly different than the noisy signal, thresholding induces a loss of information.
The two thresholding approaches will produce different results. To make the resulting signal smoother, the soft threshold filter should be used, whereas, to make computation fasters, the hard threshold filter may be used. In practice, it is difficult to choose a threshold value because a small threshold value may not be able to remove a noise while a large threshold value introduces a bias. Many approaches have been available to determine an optimal threshold value. One universal method is to assign a threshold value given by
 |
(3) |
where  is the estimator of standard deviation of the noise, and T is the length of the input vector (Donoho and Johnstone, 1994). The hard thresholding rule is defined as
is the estimator of standard deviation of the noise, and T is the length of the input vector (Donoho and Johnstone, 1994). The hard thresholding rule is defined as
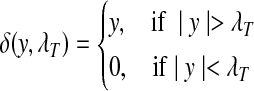 |
(4) |
As pointed out in Donoho and Johnstone (1994) and Johnstone and Silverman (1997), for a sequence of normal distributed random variables 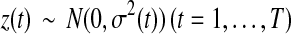 , we have
, we have
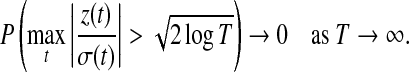 |
(5) |
Hence, if a detail coefficient is truly zero, then with a high probability it is estimated as zero in terms of the hard thresholding rule. The expected number of 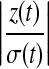 greater than the threshold tends to zero. For most applications, the hard thresholding rule only keeps those detail coefficients that are significantly greater than zero. Here, the hard thresholding rule is used to either keep or kill the whole level of detail coefficients.
greater than the threshold tends to zero. For most applications, the hard thresholding rule only keeps those detail coefficients that are significantly greater than zero. Here, the hard thresholding rule is used to either keep or kill the whole level of detail coefficients.
The following procedure is proposed to perform data dimensionality reduction through wavelet transforms:
(1) Select proper orthogonal wavelet filters;
(2) Calculate empirical variances for the detail coefficients;
(3) Apply the hard thresholding rule to the detail coefficients;
(4) Truncate the whole level of the detail coefficients if they are all set to zero by (3), and keep the whole level of the detail coefficients otherwise;
(5) Repeat procedures (1) to (4) for user-prescribed j times.
Different wavelet filters vary in filter length. A longer filter length wavelet tends to “average” over more signal points. The purpose of hard thresholding is to reduce the dimensionality of the data by truncating certain levels of detail coefficients. The variance estimator 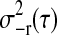 for each detail coefficient d−r(τ) is suggested in Donoho and Johnstone (1994) and Johnstone and Silverman (1997), i.e.,
for each detail coefficient d−r(τ) is suggested in Donoho and Johnstone (1994) and Johnstone and Silverman (1997), i.e.,
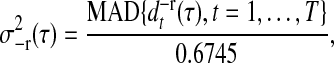 |
(6) |
where MAD denotes the median absolute deviation and 0.6745 is chosen to adjust for a normal distribution.
2.3. Wavelet-based functional clustering
2.3.1. Likelihood
Suppose there are n genes each measured at T equally-spaced time points. Let 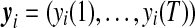 be the gene expression data for gene i. If these genes are clustered into J patterns, this means that any one of the genes (i) is assumed to arise from one (and only one) of the J possible expression patterns. Thus, the distribution of gene expression data is expressed as the J-component mixture probability density function, i.e.,
be the gene expression data for gene i. If these genes are clustered into J patterns, this means that any one of the genes (i) is assumed to arise from one (and only one) of the J possible expression patterns. Thus, the distribution of gene expression data is expressed as the J-component mixture probability density function, i.e.,
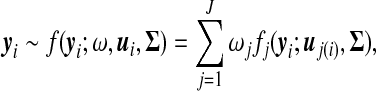 |
(7) |
where 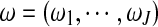 is the mixture proportions which are non-negative and sum to unity;
is the mixture proportions which are non-negative and sum to unity; 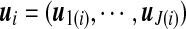 contains the component- (or pattern) specific mean vector for gene i; and Σ contains residual variances and covariances among the T time points for gene i which are common for all gene expression patterns. For a given gene i, the probability density function of the jth gene expression pattern or cluster, fj(yi; uj(i), Σ), is assumed to be multivariate normally distributed with mean vector
contains the component- (or pattern) specific mean vector for gene i; and Σ contains residual variances and covariances among the T time points for gene i which are common for all gene expression patterns. For a given gene i, the probability density function of the jth gene expression pattern or cluster, fj(yi; uj(i), Σ), is assumed to be multivariate normally distributed with mean vector
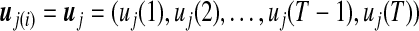 |
(8) |
and common covariance matrix Σ. For simplicity of notation, we drop the understood index i from the mean vector uj(i) as it is understood to depend on gene i.
As shown in equation (2), the original signal (1) is subject to wavelet transform at the first resolution. Correspondingly, the smooth and detail coefficients of gene pattern-specific mean signals uj after the first-resolution Haar wavelet transform can be arrayed as
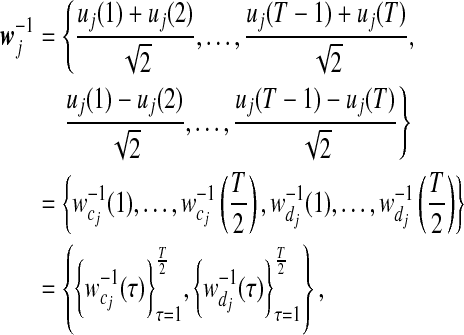 |
(9) |
Now, let z−r be the new variable with a reduced dimension T−r (T−r < T) transformed from the rth resolution Haar wavelet. The likelihood function based on a mixture model containing J gene expression patterns can be rewritten, in terms of z−r, as
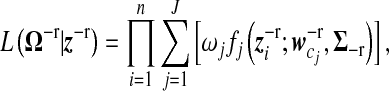 |
(10) |
where 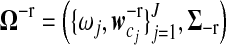 contains unknown parameters,
contains unknown parameters, 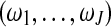 are the mixture proportions of J different gene expression patterns, as shown in equation (7), and
are the mixture proportions of J different gene expression patterns, as shown in equation (7), and 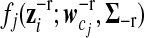 is the multivariate normal distribution of gene i that belongs to gene expression pattern j, in which
is the multivariate normal distribution of gene i that belongs to gene expression pattern j, in which 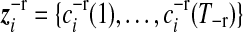 is a vector of smooth coefficients for gene i,
is a vector of smooth coefficients for gene i, 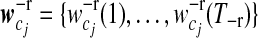 is a vector of expected smooth coefficients for gene expression pattern j and Σ−r is the (T−r × T−r) residual covariance matrix for the smooth coefficients.
is a vector of expected smooth coefficients for gene expression pattern j and Σ−r is the (T−r × T−r) residual covariance matrix for the smooth coefficients.
2.3.2. Modeling wavelet-based mean vectors
It is well known that the transcript levels of many DNA microarrays in terms of the amount of mRNAs vary with a particular pattern in time course. For example, the amount of mRNAs within the cell division cycle may change periodically (Spellman et al., 1998; De Lichtenberg et al., 2005). The regulation of these genes in a periodic manner coincident with the cell cycle may help maintain proper order during cell division and may also aid in conserving limited resources. The oscillation of cell cycle-regulated genes can be mathematically described by a simple periodic Fourier function expressed as a linear combination of cosine and sine waves. Thus, by estimating the parameters that define the periodic curves for individual genes, we can determine the differences in the temporal pattern of gene expression.
For periodically regulated genes, they can be approximated by Fourier series (Lasser, 1996). Fourier series approximation can assess periodicity. So, by applying a Fourier series approximation, we can identify the genes whose RNA levels varied periodically within the cell cycle and further find the associated amplitudes and phases. For a given gene expression pattern, let uj(t) denote the expected gene intensity ratio value at time point 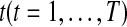 . Note that the ratio values are log transformed (base 2 for simplicity, so that log2 (Cy5/Cy3)) to treat inductions or repressions of identical magnitude as numerically equal but with opposite sign. The mean vector for a given gene expression pattern,
. Note that the ratio values are log transformed (base 2 for simplicity, so that log2 (Cy5/Cy3)) to treat inductions or repressions of identical magnitude as numerically equal but with opposite sign. The mean vector for a given gene expression pattern, 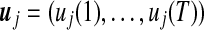 , can be modeled by a Fourier series approximation of order one. Thus, the log ratio gene expression value of gene expression pattern j at time point t can be expressed as
, can be modeled by a Fourier series approximation of order one. Thus, the log ratio gene expression value of gene expression pattern j at time point t can be expressed as
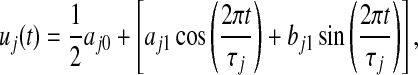 |
(11) |
where aj0 is the gene-specific fundamental frequency, aj1 and bj1 are the pattern-specific amplitude coefficients, which determine the times at which the gene achieves peak and trough expression levels, respectively, and τj is the gene-specific period of the cell cycle.
In general, the gene expression value of pattern j in time course can be mathematically fitted in form 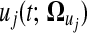 by a set of curve parameters
by a set of curve parameters 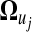 . The mean vector transformed at the first resolution transformation is expressed as
. The mean vector transformed at the first resolution transformation is expressed as
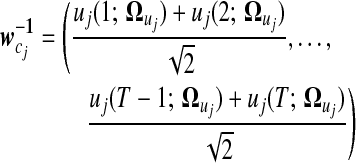 |
(12) |
Thus, by estimating 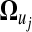 with transformed data at an appropriate transformation resolution −r, a gene expression curve in time course can be elucidated for individual patterns. Differences of the curves
with transformed data at an appropriate transformation resolution −r, a gene expression curve in time course can be elucidated for individual patterns. Differences of the curves 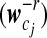 can be compared and tested for the statistical significance of time-dependent gene expression patterns.
can be compared and tested for the statistical significance of time-dependent gene expression patterns.
2.3.3. Modeling the covariance structure
It is not parsimonious to estimate all the elements in the covariance matrix among different time points because some structure exists for time-dependent variances and correlations. The covariance structure in the wavelet-domain can be modeled by a stationary first-order autoregressive (AR(1)) model (Diggle et al., 2002), expressed as
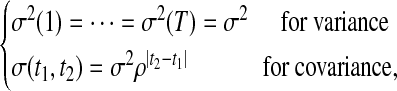 |
(13) |
where 0 < ρ < 1 is the proportion parameter with which the correlation decays with time lag. The parameters for the covariance structure are arrayed in Ωv = (ρ, σ2).
2.3.4. Estimation and tests
The standard EM algorithm is derived to estimate the parameters contained in the likelihood (10). Since the actual number of gene expression patterns is unknown, we will employ the commonly used model selection methods, AIC or BIC, to estimate the optimal number of components in the mixture model (10). After the optimal number of gene expression patterns is determined, a variety of biologically meaningful hypotheses can be formulated and tested. The most important hypothesis about overall differences in transcriptional expression profile among different patterns of microarray genes is formulated as
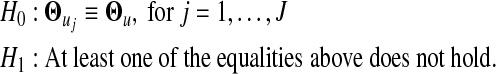 |
(14) |
The log-likelihood ratio (LR) test statistic is then calculated by
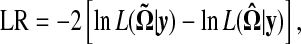 |
where the tildes and hats stand for the MLEs of the unknown parameters under the null and alternative hypotheses, respectively. The null hypothesis means that no different patterns of temporal expression exist among the genes studied, whereas the alternative hypothesis states that at least two different patterns can be identified. The critical threshold for claiming distinguishable expression patterns can be determined on the basis of simulation studies.
3. Implementation
3.1. Simulated data application
Time course gene expressions for 5000 genes were simulated over 40 equally spaced time points with mean expression profiles generated from one of the eight curves pictured in Figure 2. Residual error on the simulated series was generated from a stationary Gaussian autoregressive process with autocorrelation parameter ρ = 0.5 and standard deviation σ = 0.3. In the real data analysis performed below, we found the standard error of the clustered expression profiles to be around 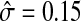 . Therefore the simulated data presents a smaller signal-to-noise ratio as compared to the real data analyzed below. Out of the 5000 simulated genes, 4000 were simulated from a flat signal with a constant value of one. The number of genes simulated under the other mean expression profiles are listed in Table 1.
. Therefore the simulated data presents a smaller signal-to-noise ratio as compared to the real data analyzed below. Out of the 5000 simulated genes, 4000 were simulated from a flat signal with a constant value of one. The number of genes simulated under the other mean expression profiles are listed in Table 1.
FIG. 2.

True mean curves (eight in total) from which gene expression data was simulated from. Eighty percent of the expression profiles were simulated from a constant expression of one.
Table 1.
Original Number of Genes Allocated to the Eight Clusters Are Recorded Here Along with the Number of Genes that were Correctly and Incorrectly Classified; a Gene Is Said to be Classified to a Given Cluster if the Estimated Cluster Probability for the Corresponding Cluster Is at Least 90%
| Cluster | Original | Correctly classified | Incorrectly classified |
|---|---|---|---|
| Flat | 4000 | 3840 | 25 |
| Linear increase | 150 | 107 | 1 |
| Smile | 50 | 50 | 0 |
| Hat | 150 | 99 | 1 |
| Off-on-off | 200 | 163 | 1 |
| Abs(sin) | 100 | 85 | 3 |
| Curved increase | 150 | 150 | 0 |
| On-off | 200 | 198 | 0 |
| Total | 5000 | 4692 | 31 |
Without assuming the number of clusters is known (even though it is), we utilized the AIC and BIC empirically identify the optimal number of clusters. In Figure 3, the AIC and BIC values are graphed under three levels of Haar wavelet smoothing: no wavelet smoothing (r = 0), one level of smoothing (r = 1), and two levels of smoothing (r = 2). Without wavelet smoothing, AIC and BIC suggest five clusters, with one level of smoothing seven clusters are suggested, and eight clusters are suggested with two levels of smoothing. When two levels of smoothing are applied, a seemingly more robust number of clusters are selected by AIC/BIC, and the correct number of clusters (eight) were identified.
FIG. 3.

AIC (black) and BIC (red) values under three levels of wavelet smoothing.
Although the AIC/BIC values without wavelet smoothing are rather erratic, it is interesting to note that the overall minimum of AIC and BIC across the three levels of smoothing is obtained under no smoothing. Without wavelet smoothing, however, the AIC/BIC identified only five clusters, and even when eight clusters are carefully considered under no smoothing (refer to Appendix A in Supplementary Materials; for online Supplementary Material, see www.liebertonline.com), not all eight clusters are correctly identified.
Looking more carefully at the eight clusters identified by two levels of smoothing, the eight estimated mean curves are graphed in Figure 4 which are shown to closely follow the true cluster means displayed in Figure 2. The dimension reduction induced by the Haar wavelet transformation is evident in the wavelet means. These eight fitted clusters are individually analyzed in Figure 5.
FIG. 4.

Estimated wavelet mean curves of the eight clusters as selected by AIC and BIC under two levels of smoothing; all clusters were correctly identified.
FIG. 5.

Mean curves for each of the eight clusters identified by AIC and BIC under two levels of wavelet smoothing (r = 2) are individually graphed together with the time-course gene expressions for genes with greater than a 90% probability of belonging to the cluster. Gene expression profiles are colored according to their original cluster allowing inappropriately clustered genes to be easily identified. All of the clusters were correctly identified and true mean curves are graphed along with wavelet-estimated mean curves.
A gene will be classified to a specific cluster if it has at least a 90% estimated probability of belonging to that cluster. Some genes will not be classified to a specific cluster if the estimated cluster probabilities are all less than 90% (though the sum of estimated cluster probabilities is always 100%). For each identified cluster, the classified genes (based on the 90% threshold) are graphed in Figure 5, along with the mean wavelet curve and the true mean curve that corresponds to the cluster. Gene expression profiles are colored according to their original cluster allowing inappropriately clustered genes to be easily identified. The total number of correctly and incorrectly classified are included listed in Table 1. This simulation analysis indicates that incorporating wavelets into functional clustering can be a powerful tool for optimally identifying nonparametric signals and clusters within time-course data.
3.2. Real data application
This methodology is applied to time time course gene expression data published in Rustici et al. (2004). In their research, a total of 8 time-course experiments were performed with expression data collected at 18–22 times on 15-minute intervals. We analyzed data from one time-course experiment; the raw and processed datasets are accessible from ArrayExpress with accession number E-MEXP-54. After selecting genes with full time-course expression profiles, 2955 genes were used with 21 equally spaced time measurements taken for each gene.
As only 21 time measurements are available for this dataset, only one level of Haar wavelet smoothing (r = 1) was incorporated. The initial values of the parameters in the EM algorithm were randomly selected from a normal distribution with mean 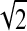 and variance 1. The AIC and BIC were used to identify the best number of clusters, and they both selected thirteen clusters. The AIC and BIC values are graphed in Figure 6.
and variance 1. The AIC and BIC were used to identify the best number of clusters, and they both selected thirteen clusters. The AIC and BIC values are graphed in Figure 6.
FIG. 6.

AIC (black) and BIC (red) values for functional clustering under one level of wavelet smoothing on the time course gene expression data considered in Rustici et al. (2004) indicates the optimal number of clusters to be thirteen.
The estimated mean curves for these clusters are graphed in Figure 7. As before in the simulated data analysis, a gene is classified to a given cluster if it has an estimated probability of belonging of at least 90%. For each of the identified clusters, the number of genes classified to that cluster are tabulated in Table 2. For instance, only four genes are seen to be strongly clustered in the first cluster whereas 447 genes indicate a strong classification to the last cluster. A total of 1540 genes (52%) were not strongly classified to one of the thirteen clusters. Each mean curve is individually graphed with the expression profiles of the genes belonging to the cluster in in Figure 8. Several interesting clusters were identified in this dataset. By plotting the individual mean curves with the classified genes separately, one can get a better sense of the true nature of each of the identified clusters. Some clusters were identified with only a few genes but with very unique profiles and other clusters indicate multiple gene with expressions in sync with a periodic signal. For biological relevance from this clustering application, the reader is can refer to the original application of this data in Rustici et al. (2004).
FIG. 7.

The thirteen estimated mean curves are graphed together.
Table 2.
The Number of Genes Classified to Each of the Thirteen Clusters Is Presented Here
| Cluster 1 | Cluster 2 | Cluster 3 | Cluster 4 | Cluster 5 | Cluster 6 | Cluster 7 | Cluster 8 | Cluster 9 | Cluster 10 | Cluster 11 | Cluster 12 | Cluster 13 | Unclassified |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 4 | 12 | 22 | 53 | 105 | 7 | 144 | 10 | 6 | 301 | 146 | 158 | 447 | 1540 |
FIG. 8.

Mean curves for each of the thirteen clusters identified are individually graphed along with individuals gene expressions for genes with at least 90% probability of belonging to such a cluster.
This dataset was also analyzed in Ning et al. (2010) utilizing a periodic Fourier series approximation in the mixture model to cluster the gene expression profiles. In this application, the AIC and BIC criteria selected nine distinct profiles, and these clusters are depicted in Figure 9. To quantitatively compare the 13 “wavelet clusters” with the 9 “Fourier clusters,” the proportion of overlap index was used. For each method, a gene was classified to a given cluster if it had greater than 90% posterior probability of belonging to the cluster. Given two clusters, the number of genes that were classified to both clusters forms the numerator of the index, and the total number of between the two clusters forms the denominator of the index. Therefore if two clusters perfectly match, the proportion of overlap is one, and if two clusters are disjoint, the proportion of overlap is zero. Some of the clusters matched up fairly closely, and matches and partial matches are provided in Table 3.
FIG. 9.

Mean curves for each of the nine clusters identified in Ning et al. (2010), which were produced by embedding a periodic Fourier series approximation into the mean expressions of the mixture components.
Table 3.
This Table Compares our Clustering Results that Utilize Wavelets with the Results Published in Ning et al. (2010)
| Wavelet cluster no. | Fourier cluster no. | Proportion of overlap |
|---|---|---|
| Cluster 3 | Cluster 5 | .870 |
| Cluster 6 | Cluster 6 | .857 |
| Cluster 9 | Cluster 3 | .600 |
| Cluster 7 | Cluster 7 | .467 |
| Cluster 2 | Cluster 1 | .348 |
| Cluster 13 | Cluster 9 | .203 |
| Cluster 10 | Cluster 9 | .109 |
A wavelet cluster is highly matches with a fourier cluster if it has a large proportion of overlap.
The computational requirements were not enormous. The computations were performed on a single desktop with a 4GHz (overclocked) Intel i7 quad-core processor. The R package multicore (Urbanek, 2009) was utilized to fully utilize the multiple cores. The source code to perform the real data analysis with corresponding dataset is available online at http://statgen.psu.edu.
4. Discussion
The studies of gene expression profiles in time course can help to understand the developmental machinery of gene regulation related to a biological process. A considerable body of literature has been available on statistical methods for characterizing different patterns of gene expression (Qian et al., 2001; Holter et al., 2001; Zhao et al., 2001; Park et al., 2003; Bar-Joseph et al., 2003; Luan and Li, 2003; Ernst et al., 2005; Storey et al., 2005; Ma et al., 2006; Ng et al., 2006; Inoue et al., 2007). When gene expression is measured at a long series of time points, it is possible that response data are contaminated by noises and, thus, the detection of patterns suffers from the so-called “curse of dimensionality.” As an increasingly popular means for data compression and de-noising in the context of signal and image processing (Donoho and Johnstone, 1994; Johnstone and Silverman, 1997), wavelet shrinkage has been used here to catalogue gene expression dynamics. This wavelet-based model projects higher dimensional data to a manageable lower dimensional subspace.
We have implemented the idea of wavelet dimension reduction into the mixture model for gene clustering, aimed to de-noise the data by transforming an inherently high-dimensional biological problem to its tractable low-dimensional representation. As a first attempt of its kind, we capitalize on the simplest Haar wavelet shrinkage technique to break an original signal down into spectrum by taking its averages and differences and, subsequently, to detect gene clusters that differ in the smooth coefficients extracting from noisy time series gene expression data. The wavelet thresholding approach that we utilized in this manuscript was constructed for equally spaced longitudinal data, however its extension to non-equally spaced data can be made possible through the development of second-generation wavelets (Pensky and Vidakovic, 2001; Jansen, 2003; Vanraes et al., 2002).
It is noted that the clusters identified with this method are meant to be exploratory. Identification of these clusters can help frame and guide biological investigations. Once data analysis has proceeded to the end point of our investigations, we are at a point where we can discuss mechanisms with biologists and hopefully work with them to understand the physical mechanisms.
This wavelet-based model will have many implications for addressing biologically meaningful hypotheses at the interplay between gene actions (or interactions) and developmental pathways in various complex biological processes or networks. Although our main application in this article is with time-course gene expression data, the techniques we have developed are generally applicable to other time-course datasets including applications to financial data where high-dimensional characteristics are ubiquitous. We hope that our method described within can provide a starting point for further exploration in the functional clustering of high-dimensional data.
Supplementary Material
Acknowledgments
This work was partially supported by NSF/NIH (joint grant DMS/NIGMS-0540745).
Disclosure Statement
No competing financial interests exist.
References
- Aboufadel E. Schlicker S. Discovering wavelets. Wiley; 1999. [Google Scholar]
- Bar-Joseph Z. Gerber G.K. Gifford D.K., et al. Continuous representations of time-series gene expression data. J. Comput. Biol. 2003;10:341–356. doi: 10.1089/10665270360688057. [DOI] [PubMed] [Google Scholar]
- De Lichtenberg U. Jensen L.J. Fausboll A., et al. Comparison of computational methods for the identification of cell cycle-regulated genes. Bioinformatics. 2005;21:1164. doi: 10.1093/bioinformatics/bti093. [DOI] [PubMed] [Google Scholar]
- Diggle P. Heagerty P. Liang K.Y., et al. Analysis of Longitudinal Data. Oxford University Press; New York: 2002. [Google Scholar]
- Donoho D.L. De-noising by soft-thresholding. IEEE Trans. Inform. Theory. 1995;41:613–627. [Google Scholar]
- Donoho D.L. Johnstone J.M. Ideal spatial adaptation by wavelet shrinkage. Biometrika. 1994;81:425. [Google Scholar]
- Eisen M.B. Spellman P.T. Brown P.O., et al. Cluster analysis and display of genome-wide expression patterns. Proc. Natl. Acad. Sci. USA. 1998;95:14863. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J. Nau G.J. Bar-Joseph Z. Clustering short time series gene expression data. Bioinformatics. 2005;21:159. doi: 10.1093/bioinformatics/bti1022. [DOI] [PubMed] [Google Scholar]
- Ghael S.P. Sayeed A.M. Baraniuk R.G. Improved wavelet denoising via empirical wiener filtering. Proc. SPIE. 1997;3169:389–399. [Google Scholar]
- Ghosh D. Chinnaiyan A.M. Mixture modelling of gene expression data from microarray experiments. Bioinformatics. 2002;18:275. doi: 10.1093/bioinformatics/18.2.275. [DOI] [PubMed] [Google Scholar]
- Holter N.S. Maritan A. Cieplak M., et al. Dynamic modeling of gene expression data. Proc. Natl. Acad. Sci. USA. 2001;98:1693. doi: 10.1073/pnas.98.4.1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue L.Y.T. Neira M. Nelson C., et al. Cluster-based network model for time-course gene expression data. Biostatistics. 2007;8:507–525. doi: 10.1093/biostatistics/kxl026. [DOI] [PubMed] [Google Scholar]
- Jansen M. Wavelet thresholding on non-equispaced data. Nonlinear Estimation Classification. 2003. p. 261.
- Jensen A. la Cour-Harbo A. Ripples in Mathematics: The Discrete Wavelet Transform. Springer Verlag; New York: 2001. [Google Scholar]
- Johnstone I.M. Silverman B.W. Wavelet threshold estimators for data with correlated noise. J. R. Statist. Soc. Ser. B. 1997;59:319–351. [Google Scholar]
- Kim B.R. Zhang L. Berg A., et al. A computational approach to the functional clustering of periodic gene expression profiles. Genetics. 2008;180:821–834. doi: 10.1534/genetics.108.093690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasser R. Introduction to Fourier Series. CRC; Boca Raton, FL: 1996. [Google Scholar]
- Luan Y. Li H. Clustering of time-course gene expression data using a mixed-effects model with b-splines. Bioinformatics. 2003;19:474. doi: 10.1093/bioinformatics/btg014. [DOI] [PubMed] [Google Scholar]
- Ma P. Castillo-Davis C.I. Zhong W., et al. A data-driven clustering method for time course gene expression data. Nucleic Acids Res. 2006;34:1261. doi: 10.1093/nar/gkl013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallat S.G., et al. A theory for multiresolution signal decomposition: the wavelet representation. IEEE Trans. Pattern Anal. Mach. Intell. 1989;11:674–693. [Google Scholar]
- McLachlan G.J. Bean R.W. Peel D. A mixture model-based approach to the clustering of microarray expression data. Bioinformatics. 2002;18:413. doi: 10.1093/bioinformatics/18.3.413. [DOI] [PubMed] [Google Scholar]
- Ng S.K. McLachlan G.J. Wang K., et al. A mixture model with random-effects components for clustering correlated gene-expression profiles. Bioinformatics. 2006;22:1745. doi: 10.1093/bioinformatics/btl165. [DOI] [PubMed] [Google Scholar]
- Ning L. McMurry T. Berg A., et al. Functional clustering of periodic transcriptional profiles through arma(p,q) PLoS ONE. 2010. (in press). [DOI] [PMC free article] [PubMed]
- Park T. Yi S.G. Lee S., et al. Statistical tests for identifying differentially expressed genes in time-course microarray experiments. Bioinformatics. 2003;19:694. doi: 10.1093/bioinformatics/btg068. [DOI] [PubMed] [Google Scholar]
- Pensky M. Vidakovic B. On non-equally spaced wavelet regression. Ann. Instit. Statist. Math. 2001;53:681–690. [Google Scholar]
- Qian J. Stenger B. Wilson C.A., et al. Partslist: a web-based system for dynamically ranking protein folds based on disparate attributes, including whole-genome expression and interaction information. Nucleic Acids Res. 2001;29:1750. doi: 10.1093/nar/29.8.1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramoni M.F. Sebastiani P. Kohane I.S. Cluster analysis of gene expression dynamics. Proc. Natl. Acad. Sci. USA. 2002;99:9121. doi: 10.1073/pnas.132656399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rustici G. Mata J. Kivinen K., et al. Periodic gene expression program of the fission yeast cell cycle. Nat. Genet. 2004;36:809–817. doi: 10.1038/ng1377. [DOI] [PubMed] [Google Scholar]
- Spellman P.T. Sherlock G. Zhang M.Q., et al. Comprehensive identification of cell cycle-regulated genes of the yeast Saccharomyces cerevisiae by microarray hybridization. Mol. Biol. cell. 1998;9:3273. doi: 10.1091/mbc.9.12.3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storey J.D. Xiao W. Leek J.T., et al. Significance analysis of time course microarray experiments. Proc. Natl. Acad. Sci. USA. 2005;102:12837. doi: 10.1073/pnas.0504609102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbanek S. multicore: Parallel processing of R code on machines with multiple cores or CPUs. R package version 0.1–3. 2009. www.rforge.net/multicore/ [Jun 1;2010 ]. www.rforge.net/multicore/
- Vanraes E. Jansen M. Bultheel A. Stabilised wavelet transforms for non-equispaced data smoothing. Signal Process. 2002;82:1979–1990. [Google Scholar]
- Vidakovic B. Statistical Modeling by Wavelets. Wiley; New York: 1999. [Google Scholar]
- Walker J.S. A Primer on Wavelets and Their Scientific Applications. CRC Press; Boca Raton, FL: 1999. [Google Scholar]
- Wang L. Chen X. Wolfinger R.D., et al. A unified mixed effects model for gene set analysis of time course microarray experiments. Statist. Appl. Genet. Mol. Biol. 2009;8:47. doi: 10.2202/1544-6115.1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zapala M.A. Schork N.J. Multivariate regression analysis of distance matrices for testing associations between gene expression patterns and related variables. Proc. Natl. Acad. Sci. USA. 2006;103:19430. doi: 10.1073/pnas.0609333103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L.P. Prentice R. Breeden L. Statistical modeling of large microarray data sets to identify stimulus-response profiles. Proc. Natl. Acad. Sci. USA. 2001;98:5631. doi: 10.1073/pnas.101013198. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.