

Abstract
We have investigated the mechanism of binding single-stranded DNA (ssDNA) into the central channel of the ring-shaped T7 gp4A′ helicase–primase hexamer. Presteady-state kinetic studies show a facilitated five-step mechanism and provide understanding of how a ring-shaped helicase can be loaded on the DNA during the initiation of replication. The effect of a primase recognition sequence on the observed kinetics suggests that binding to the helicase DNA-binding site is facilitated by transient binding to the primase DNA-binding site, which is proposed to be a loading site. The proposed model involves the fast initial binding of the DNA to the primase site on the outside of the helicase ring, a fast conformational change, a ring-opening step, migration of the DNA into the central channel of the helicase ring, and ring closure. Although an intermediate protein–DNA complex is kinetically stable, only the last species in the five-step mechanism is poised to function as a helicase at the unwinding junction.
Keywords: DNA unwinding/fluorescence stopped-flow/kinetics/motor protein/protein–DNA interactions
Introduction
Helicases are motor proteins that unwind double-stranded DNA/RNA (dsDNA/RNA) using the free energy from NTP hydrolysis. Helicases from various organisms, including bacteriophages, bacteria, archaea, viruses and eukaryotes, have been shown to form ring-shaped hexamers (Matson and Kaiser-Rogers, 1990; Lohman and Bjornson, 1996; Patel and Picha, 2000). Most of these assemble into rings in the absence of DNA but many require Mg2+ and/or NTP for hexamer formation. Most of the hexameric helicases involved in DNA replication have been proposed to bind only one strand of the duplex DNA within the central channel while excluding the complementary strand (Bujalowski and Jezewska, 1995; Egelman et al., 1995; Yu et al., 1996; Smelkova and Borowiec, 1998; Fouts et al., 1999; Morris and Raney, 1999). The translocation of the helicase along the single-stranded DNA (ssDNA) passing through its central channel may play a major role in DNA unwinding. This mode of DNA binding topologically links the ring-shaped helicase and the DNA and contributes to the processivity of translocation and unwinding (Patel and Picha, 2000). The location of the ssDNA-binding site in the central channel of the ring however poses a problem. Free DNA cannot access the ssDNA-binding site within the central channel of the ring, suggesting that there must be a specific mechanism to load the helicase efficiently onto the DNA during initiation. Using presteady state kinetics, we have elucidated such a mechanism for T7 gp4A′ helicase–primase binding to ssDNA. The kinetic mechanism determined for gp4A′ has provided general insights into how hexameric helicases can be loaded on the DNA during initiation.
T7 gp4A′ contains both helicase and primase activities, and it does not require any accessory proteins to bind or unwind DNA. Although the two activities reside on separate domains of the protein, they are dependent on each other and the isolated domains are not fully active (Bird et al., 1997; Guo et al., 1999). The C-terminal domain is mostly responsible for the helicase function and the N-terminal domain for the primase (Patel et al., 1992; Frick et al., 1998). The gp4A′ interacts preferentially with ssDNA with a Kd <100 nM (Hingorani and Patel, 1993). This interaction requires hexamer formation and dTTP binding, but not hydrolysis (Picha and Patel, 1998; Picha et al., 2000). The interactions of the ssDNA with the primase domain are weak (Kd ∼10 µM) (Frick and Richardson, 1999). Thus, the tight binding of the ssDNA to the helicase occurs in the helicase DNA-binding site of the C-terminal domain. The role of the N-terminal domain in helicase function is unknown. The ssDNA-binding site on the helicase domain is located in the central channel of the ring (Egelman et al., 1995; Yu et al., 1996). In a recently reported crystal structure, it can be shown that the postulated ssDNA-binding motif, H4, within the central channel is not easily accessible to free DNA (Sawaya et al., 1999). The primase ssDNA-binding site, on the other hand, appears to be easily accessible to free DNA and is proposed to be located on the outside of the ring (Kusakabe et al., 1998).
In previous studies, we have dissected the kinetic pathway of 30mer ssDNA binding using stopped-flow fluorescence methods to understand how a preformed hexamer of gp4A′ loads onto a ssDNA during initiation. These studies have shown that the pathway of 30mer ssDNA binding to a preformed hexamer of gp4A′ consists of a minimum of four steps (Picha et al., 2000). We were able to show that ssDNA binding does not involve disassembly and re-assembly of hexamer subunits around the ssDNA, and ssDNA binding might occur by a ring-opening or a threading mechanism. In the study presented here, presteady state dTTPase-, helicase- and ssDNA-binding kinetics showed that ssDNA binds to gp4A′ by a five-step ring-opening mechanism. The initial encounter of the ssDNA with the hexamer occurs at a close to diffusion-limited rate constant, and our studies suggest that this interaction may be with the primase–ssDNA-binding site on gp4A′. Based on the kinetic stability of the intermediates, we propose that ring-opening and migration of the ssDNA into the central channel occur after the ssDNA is bound at the primase site. We show that the five-step mechanism is both necessary and sufficient, and only the last species in the pathway is proficient in carrying out the functions of a helicase.
Results
Previous stopped-flow ssDNA-binding studies (Picha et al., 2000) have shown that a preformed ring-shaped hexamer of gp4A′ protein binds a 30mer ssDNA by a four-step mechanism, shown below:
![]()
Where, E is gp4A′ hexamer, D is 30mer ssDNA and ED1 through ED4 are the various helicase–ssDNA complexes in the pathway of ssDNA binding. This four-step mechanism was elucidated by monitoring time-dependent fluorescence changes in the protein upon ssDNA binding, and the intrinsic rate constants were determined by kinetic simulation and global fitting. Because the ssDNA-binding site is in the central channel of the gp4A′ hexamer, we did not expect the kinetic pathway to consist of a simple bimolecular ssDNA-binding event. The four-step pathway is, however, complex and raises the following questions: are all steps in the ssDNA-binding pathway necessary, and do they lead to the formation of a competent helicase–ssDNA complex that is proficient in carrying out the functions of a helicase? A competent species of T7 gp4A′ would be able to unwind dsDNA and hydrolyze dTTP at the ssDNA-stimulated level. We therefore used these activities as criteria to assess the kinetic competence of the proposed helicase–ssDNA intermediates. The dTTPase and helicase activities were measured in the time period of ssDNA binding. These studies, described below, led to the proposal of a five-step ssDNA-binding mechanism and provided insights into the nature of the various helicase–ssDNA intermediates.
The presteady state kinetics of dTTP hydrolysis
The gp4A′ hexamer has an intrinsic dTTPase activity that is stimulated ∼10-fold by 30mer ssDNA and ∼100-fold by ssM13 DNA (Patel et al., 1992; Picha and Patel, 1998). We wished to determine which of the helicase–ssDNA species (ED1 through ED4) in the pathway were capable of hydrolyzing dTTP at the ssDNA-stimulated rate. We therefore measured the presteady state kinetics of dTTPase in the time period of ssDNA binding, from milliseconds to seconds after mixing the gp4A′ hexamer with the ssDNA. When the experiment was carried out with the 30mer ssDNA, a kinetic lag in the formation of dTDP was observed (Figure 1A). The initial dTTPase rate was close to the intrinsic dTTPase rate, and after several seconds the rate increased to the 30mer stimulated steady-state rate. The lag kinetics were fit to the 30mer binding mechanism (using the intrinsic rate constants shown in Table I) to determine which of the helicase–ssDNA species were capable of hydrolyzing dTTP at the 30mer ssDNA-stimulated rate. The results indicated that none of the species from ED1 to ED4 was capable of hydrolyzing dTTP at the ssDNA-stimulated rate. The dashed line in Figure 1A shows that the expected kinetics would have a much shorter lag if ED4 was hydrolyzing dTTP at the ssDNA-stimulated rate. The longer lag observed in our experiments indicated that ED4 was not competent in hydrolyzing dTTP at the ssDNA-stimulated rate and needed to be converted to ED5 to become competent. Thus, the data were fit to a five-step mechanism that invoked an additional step, from ED4 to ED5, at a conversion rate of 0.05 s–1. The presteady state dTTPase experiments therefore provided evidence for a fifth step in the 30mer binding mechanism. This last step was not observed previously in the stopped-flow kinetics (Picha et al., 2000). This may be because the fluorescence properties of ED4 and ED5 are identical. Alternatively, the signal may be very small and is lost in photo-bleaching that accompanies the measurements.
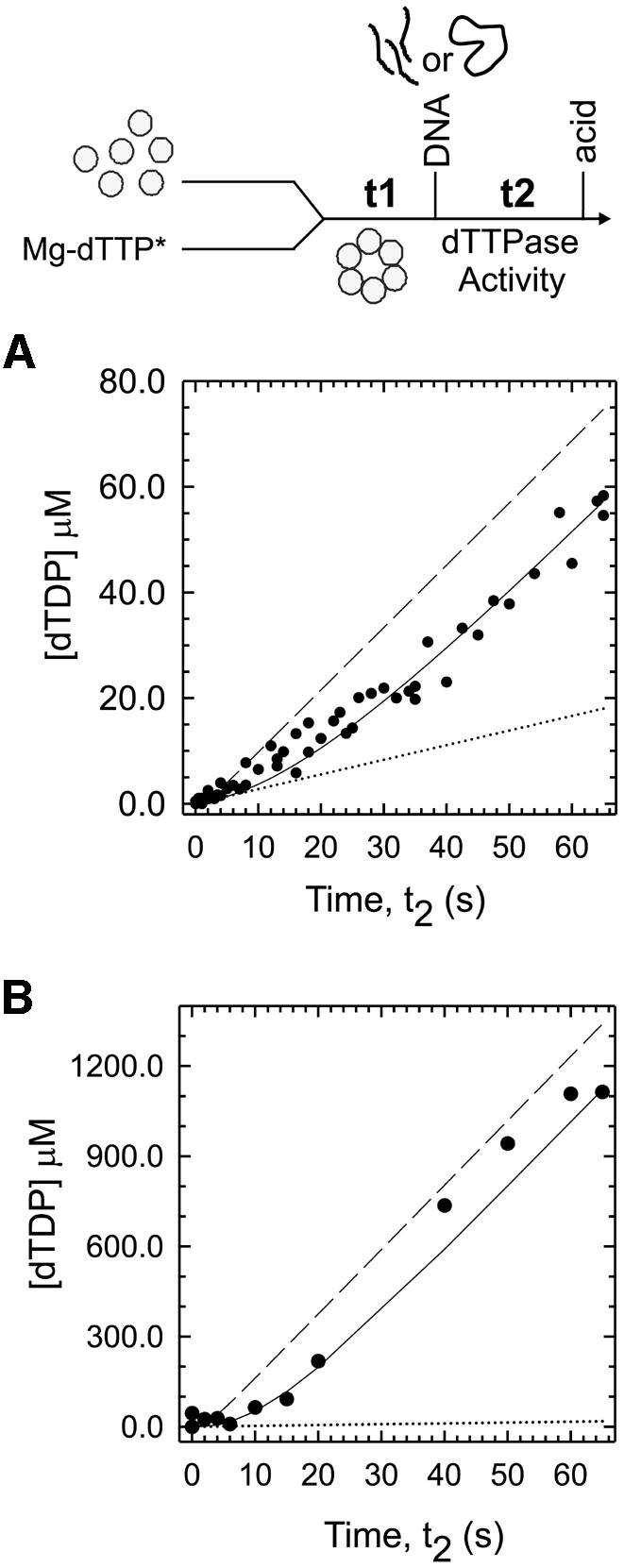
Fig. 1. The presteady state kinetics of dTTP hydrolysis in the presence of 30mer and ssM13 DNA. (A) Gp4A′ (1 µM hexamer, final concentration) preincubated with [α-32P]dTTP, Mg-dTTP (500 µM), and MgCl2 (7 mM) for t1 = 15 s was mixed for various times t2 with 30mer ssDNA (10 µM) in a rapid quenched flow instrument at 18°C. The formation of [α-32P]dTDP in the reaction time t2 was measured (circles). The data were fit to the 30mer binding mechanism (solid line) in Table I, with ED5 hydrolyzing dTTP at the 30mer stimulated rate (1.18 s–1), and the other species hydrolyzing at the intrinsic rate (0.277 s–1). The best fit provided a rate of 0.053 s–1 for the conversion of ED4 to ED5. The dashed line shows the predicted curve for a mechanism in which ED4 hydrolyzes dTTP at the 30mer-stimulated rate and the dotted line shows dTTP hydrolysis at the intrinsic rate (in the absence of DNA). (B) The presteady state kinetics of dTTP hydrolysis with ssM13 DNA (15 nM) was measured with 0.167 µM gp4A′ hexamer, [α-32P]dTTP, Mg-dTTP (10 mM) and MgCl2 (7 mM) as described for 30mer in (A). The lag kinetics (circles) were fit to the ssM13 DNA-binding mechanism shown in Table I (solid line) with ED5 hydrolyzing dTTP at the ssM13 DNA-stimulated rate (20.2 s–1) and the rest of the species hydrolyzing at the intrinsic rate (0.277 s–1). The dashed line shows the predicted curve for a mechanism in which ED4 hydrolyzes dTTP at the ssM13 DNA-stimulated rate and the dotted line shows the intrinsic dTTP hydrolysis.
Table I. DNA-binding mechanism with Mg-dTTP.

aRate constants k1–k4 were obtained from a previous study (Picha et al., 2000); k5 was obtained from the kinetics of dTTP hydrolysis (Figure 1) and DNA unwinding (Figure 2).
When the dTTPase kinetics were measured in the presence of a closed circular ssM13 DNA under presteady state conditions, a similar lag in dTDP formation was observed (Figure 1B) as with 30mer ssDNA, described above. The increase in the dTTPase rate from the intrinsic value (dotted line) to that of the ssM13-stimulated value took several seconds. Because the ssM13 DNA-stimulated rate is ∼100-fold higher than the intrinsic rate, the lag was more distinct. No lag in dTTPase was observed when the gp4A′ hexamer was preassembled on ssM13 DNA by adding dTTP without Mg2+ (data not shown). This indicates that the preassembled helicase–ssDNA complex is a competent complex or changes into one at a rate much faster than the steady state dTTPase rate. It also substantiates previous findings (Ahnert and Patel, 1997; Washington and Patel, 1998) that suggested that a helicase–ssDNA complex assembled in the absence of Mg2+ can initiate dsDNA unwinding without further rate-limiting steps. The observed lag in the formation of dTDP with ssM13 DNA was fit to the five-step ssM13 DNA-binding mechanism that was determined from stopped-flow fluorescence experiments (described below). The results were similar to those of the 30mer ssDNA, and showed that only ED5, but none of the intermediate species ED1–ED4, was capable of hydrolyzing dTTP at the ssM13 DNA-stimulated level.
The presteady state kinetics of dsDNA unwinding
The competency of the helicase–ssDNA intermediates was also tested by measuring the helicase activity in the time-period of ssDNA binding. The presteady state dTTPase experiments indicated that only ED5 was capable of hydrolyzing dTTP at the ssDNA-stimulated rate. Therefore, ED5 was expected to be the earliest complex capable of unwinding dsDNA. To investigate this, and to determine whether the five-step mechanism was sufficient in describing the formation of a complex capable of dsDNA unwinding, the single-turnover unwinding kinetics were measured using a synthetic fork-DNA with a 50 bp dsDNA region. When the gp4A′ hexamer was rapidly mixed with the fork-DNA to start the reaction (condition A), the observed unwinding rate was 4 bp/s (Figure 2, triangles). When the gp4A′ hexamer was preassembled on the fork-DNA and the reaction was started by adding Mg2+ (condition B), the unwinding rate was ∼10-fold faster, ∼30 bp/s (Figure 2, circles). These results demonstrate that the slow assembly of the hexamer on the fork-DNA limits unwinding. To determine which of the helicase–ssDNA species in the 30mer binding pathway were competent in unwinding dsDNA, the unwinding kinetics under condition A were fit to the 30mer binding mechanism (Table I) followed by steps of dsDNA unwinding. The results showed that none of the species from ED1 through ED4 was competent, but ED5 was competent in unwinding dsDNA at the rate of 30 bp/s. The dashed curve in Figure 2 shows the predicted unwinding kinetics if ED4 was capable of unwinding dsDNA at 30 bp/s. The fit of the observed data provided an ED4 to ED5 conversion rate of 0.08–1, which is very similar to the rate obtained from the dTTPase experiments. This showed that the formation of ED5 from ED4 is the rate limiting step for DNA unwinding under condition A. Therefore, the presteady state dTTPase and helicase kinetics provided evidence for the existence of the species ED5 and indicated that it was formed from ED4 at a rate of 0.05–0.08–1.
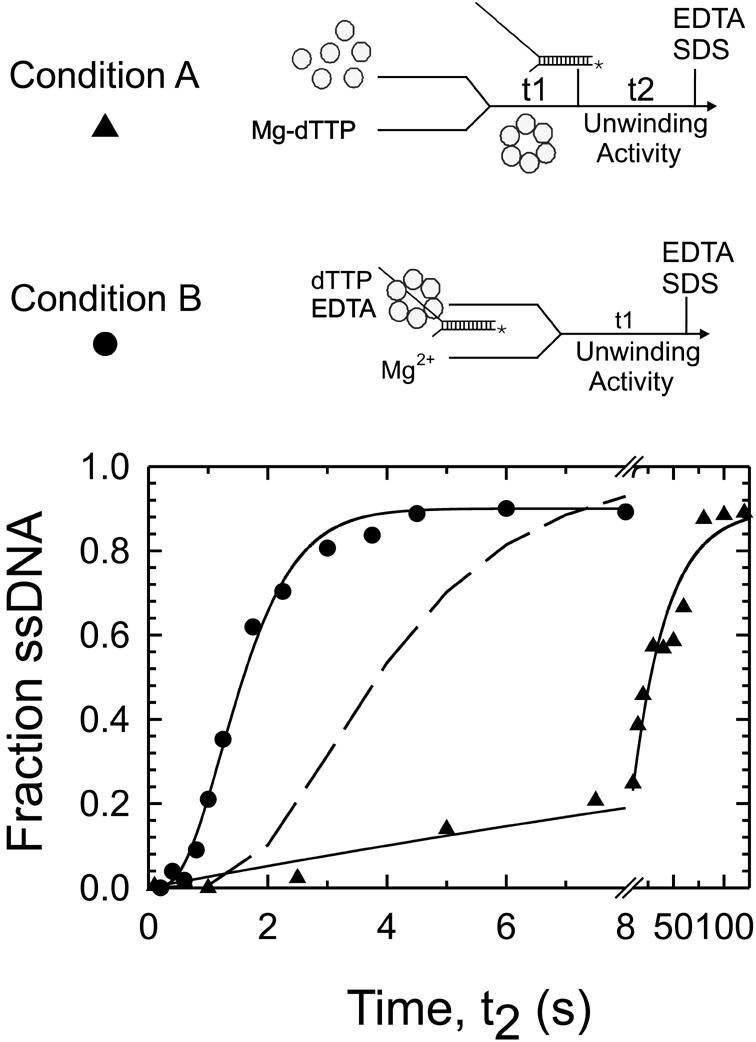
Fig. 2. The presteady state kinetics of dsDNA unwinding. The unwinding of a 50 bp fork-DNA was measured under single turnover conditions in a rapid quench–flow instrument at 18°C. Under condition A (triangles), gp4A′ (0.33 µM hexamer, final concentration) was preincubated with Mg-dTTP (4 mM) and MgCl2 (7 mM), and then added to radiolabeled fork-DNA (5 nM) to start the reaction. The ssDNA formed after varying reaction times was determined. Under condition B (circles), gp4A′ (0.33 µM hexamer) was pre-assembled on the fork-DNA (5 nM) in the presence of dTTP (4 mM) and EDTA (1.5 mM) and the unwinding reaction was initiated by adding MgCl2 (11 mM). Under condition A, unwinding occurred at an observed rate of 2.65 bp/s and under condition B at 30 bp/s. The dashed line shows the predicted curve for unwinding under condition A if ED4 was competent and unwound at a rate of 30 bp/s.
The mechanism of ssM13 DNA binding
The ssM13 DNA-binding mechanism was determined from stopped-flow protein fluorescence experiments, similar to those described for the 30mer ssDNA (Picha et al., 2000). A gp4A′ hexamer, preformed in the presence of Mg-dTTP, was mixed with ssM13 DNA and the resulting changes in the protein fluorescence were monitored. Four kinetic phases were observed, and these were monitored at increasing concentrations of ssM13 DNA (Figure 3). The rate of the first phase increased linearly with the ssM13 DNA concentration and represented the bimolecular ssDNA-binding step. The rate of the second phase increased hyperbolically, and the rates of the third and fourth phases remained constant with increasing ssDNA. These kinetics were fit to a linear multi-step ssDNA-binding mechanism, and the intrinsic rate constants of each step were determined by numerical integration and global fitting, as described for the 30mer ssDNA (Picha et al., 2000).

Fig. 3. Stopped-flow kinetics of ssM13 DNA binding. (A) The time courses (shown in log-scale) of protein fluorescence changes resulting from mixing a preincubated solution of gp4A′ (142 nM hexamer, final concentration), Mg-dTTP (2 mM) and MgCl2 (7 mM free) with various concentrations of ssM13 DNA (15–60 nM) are shown. The solid black lines show the fit of the kinetic data to the ssM13 DNA-binding mechanism shown in Table I. (B) The time courses were fit to the sum of four exponentials, and the rates were plotted against the ssDNA concentrations (5–60 nM). The rate of the first phase increased linearly with increasing ssDNA with a slope of 0.2 nM–1s–1 and a y-intercept of 5.4 s–1. (C) The rate of the second phase increased hyperbolically and reached a maximum rate of 5.2 s–1 (y-intercept = 0). (D) The rate of the third phase remained constant at ∼0.5 s–1. (E) The rate of the fourth phase remained constant at ∼0.02 s–1.
Table I shows the final ssDNA-binding mechanism for the ssM13 and 30mer ssDNAs in the presence of Mg-dTTP. The overall mechanism for ssDNA binding is very similar for both ssDNAs. Because multiple gp4A′ hexamers can bind to a single molecule of ssM13 DNA, we cannot make a direct comparison of the bimolecular rate constants of the two ssDNAs. Nevertheless, both ssDNAs were found to bind at very fast rates resulting in ED1 (2×107M–1s–1 with the 30mer ssDNA, 4×108M–1s–1 with the ssM13 DNA). ED1 was converted to ED2 at a fast rate of 132 s–1 in the 30mer binding pathway. This rate could not be accurately determined for the ssM13 DNA, because high concentrations of ssM13 DNA could not be used in the stopped-flow experiments. However, it is estimated to be >18 s–1, since that was the observed rate at the highest concentration of ssDNA used. A series of slower conformational changes followed the fast initial steps. ED2 was converted to ED3 at 5.0 s–1 in the presence of ssM13 DNA and at 3.4 s–1 in the presence of 30mer. The ED3 to ED4 conversion rate was 0.5 s–1 for both ssDNAs. The conversion of ED4 to ED5 occurred at 0.02 s–1 with ssM13 DNA and could not be measured in fluorescence experiments with the 30mer. However, the dTTPase stimulation experiments with both DNAs showed a rate of 0.05–0.08 s–1 for the conversion of ED4 to ED5. Thus, considering the different lengths of the substrate ssDNAs, the overall mechanisms of ssDNA binding as determined from the stopped-flow and presteady state ssDNA-stimulated dTTPase experiments are very similar for both 30mer and ssM13 DNAs.
The kinetic data presented above established the requirement of a five-step ssDNA-binding mechanism for the formation of a competent helicase. The nature of the intermediate species in this mechanism is unknown, but the final species is well characterized. Studies in the presence of Mg-dTMP-PCP (deoxythymidine [β, γ, methylene] triphosphate) have shown that ssDNA is stably bound within the central channel at equilibrium (Egelman et al., 1995; Yu et al., 1996), and such a species has a half-life of dissociation >60 min (data not shown; Matson and Richardson, 1985). We therefore used kinetic stability as a criterion to determine when the ssDNA migrates into the central channel during the ssDNA-binding process. The 30mer binding mechanism (Table II) shows that the first irreversible step is the conversion of ED3 to ED4, suggesting that ED4 is the first stable helicase–ssDNA complex. This was tested experimentally by the following chase filter-binding experiments.
Table II. DNA-binding mechanism with Mg-dTMP-PCP.

The measurement of 30mer ssDNA binding by the chase filter-binding assay
The rationale for this experiment was to generate the various helicase–ssDNA intermediates as a function of time with a radiolabeled 30mer ssDNA. Then excess nonradiolabeled ssDNA was added as a chase, and the amount of radiolabeled ssDNA bound to the protein determined. These experiments were carried out with Mg-dTMP-PCP rather than Mg-dTTP, because a stable helicase–ssDNA complex is formed only with the nonhydrolyzable analog, and this complex can be quantified by the nitrocellulose-DEAE membrane binding assay (Patel and Hingorani, 1995; Hingorani and Patel, 1996). The stopped-flow kinetic studies have shown that the helicase–ssDNA intermediates are formed in the millisecond time-scale (Picha et al., 2000). Therefore, the gp4A′ hexamer was mixed with radiolabeled 30mer ssDNA in a rapid quenched-flow instrument from 5 ms to several seconds. An excess of chase was added, and after 25 s the radioactive ssDNA that remained bound to the hexamer was quantified by the membrane-binding assay.
The resulting 30mer binding kinetics, determined by this chase filter-binding assay, were biphasic (Figure 4). The kinetics were analyzed as described in the Materials and methods to determine when a kinetically stable helicase–ssDNA complex was formed (a species that does not dissociate in the presence of the chase). The models in which ED2 or ED3 were assumed to be kinetically stable did not fit the data (short-dashed and dashed lines, Figure 4B and C). The observed kinetics fit well when we assumed that ED4 was a kinetically stable species. In fact, the chase data could be described with essentially the same intrinsic rate constants as determined from the stopped-flow experiments (Table II). Only the rate constant for the conversion of ED3 to ED4 varied somewhat. The chase data at 400 nM and 1.1 µM 30mer ssDNA were described best with rates of 0.09 and 0.23 s–1, respectively, in comparison with 0.21 s–1 as determined in the stopped-flow experiments (Picha et al., 2000). These experiments, in addition to verifying the kinetic mechanism of 30mer ssDNA binding obtained by fluorescence measurements, indicated that ED4 is the first species in the ssDNA-binding pathway, where the ssDNA is stably bound to the gp4A′ hexamer.

Fig. 4. The kinetics of 30mer ssDNA binding measured by chase-filter DNA-binding assay. (A) Gp4A′ (142 nM hexamer, final concentration), Mg-dTMP-PCP (2 mM), and MgCl2 (7 mM free) was mixed with 5′-[32P]30mer ssDNA (400 nM or 1.1 µM) for varying times in a rapid quench–flow instrument at 18°C. The amount of radiolabeled 30mer bound to gp4A′ hexamer, after 25 s of chase addition (4 or 11 µM non-radiolabeled 30mer ssDNA, in buffer H with 2 mM Mg-dTMP-PCP, 7 mM MgCl2) was determined. (B) and (C) show the chase kinetics with 400 nM and 1.1 µM ssDNA, respectively. The kinetics of ssDNA binding fit to the sum of two exponentials (Equation 1). The amplitudes and rates at 400 nM 30mer were 51.9 nM and 11.7 s–1 for the first phase and 90.4 nM and 0.094 s–1 for the second phase, respectively. The amplitudes and rate at 1.1 µM 30mer were 65.4 nM and >30 s–1 for the first phase and 61.3 nM and 0.4 s–1 for the second phase, respectively. The solid black lines are predicted chase kinetics curves from the 30mer ssDNA-binding mechanism, shown in Table II (formation of ED4 is irreversible). The short- and long-dashed lines represent the predicted formation ED2 or ED3, respectively, assuming that they are stable protein–DNA complexes.
The above experiments raise the next question. Where is the ssDNA bound in the ED1, ED2 and ED3 complexes? Both ED1 and ED2 are formed at very fast rates compared with the rest of the species. ED1 is formed at a close to diffusion-limited rate and its conversion to ED2 is fast. We reasoned that the fast ssDNA-binding event can not occur at the ssDNA-binding site located within the central channel because that site would be inaccessible to free ssDNA. We propose that the fast encounter of the ssDNA occurs at a site that is readily accessible and located on the outside of the ring. A known ssDNA-binding site that was proposed to be accessible and located on the outside, is the primase ssDNA-binding site present on the N-terminal domain of gp4A′ (Kusakabe et al., 1998). The interactions of the ssDNA with the primase ssDNA-binding site have been measured to be weak with a Kd in the range of 10 µM (Frick and Richardson, 1999). In fact, the Kd for ED1 is weak as well (2.5 µM). To investigate the hypothesis that the initial interactions of the ssDNA occur at the primase ssDNA-binding site of gp4A′, we have designed the following stopped-flow kinetic experiments.
The effect of the primase recognition sequence on the kinetics of ssDNA binding
The primase site of T7 gp4A′ recognizes sequences such as 3′-CTGGT on ssDNA, which it uses as a template to synthesize RNA primers when ATP or ApC and CTP are provided (Scherzinger et al., 1977; Tabor and Richardson, 1981). It has been shown that the primase domain interacts more tightly with the ssDNA that contains a primase recognition sequence (Frick and Richardson, 1999). Therefore, if the first interaction between the helicase and the ssDNA occurred at the primase ssDNA-binding site, then the presence of a primase recognition sequence (and the nucleotide substrates for primer synthesis) may affect the overall kinetics of ssDNA binding. On the other hand, if the initial encounter were to occur with the helicase ssDNA-binding site directly, then having a primase sequence in the ssDNA should have no effect on the kinetics of ssDNA binding. A primase ssDNA recognition sequence (3′-CTGGT) was introduced by changing only one base in the 30mer ssDNA at position 19 from the 5′ end (A to G change). This minimal change allowed a direct comparison to be made between the kinetic mechanisms of binding with the 30mer ssDNA (without the primase sequence) and the pri30mer ssDNA (with the primase sequence).
The 30mer and the pri30mer ssDNA-binding kinetics were determined in the presence of dTMP-PCP by monitoring the transient changes in gp4A′ fluorescence in a stopped-flow instrument. Three phases were observed, and their rates were dependent on the ssDNA concentrations (data not shown). The rate of the first phase increased linearly with increasing ssDNA, the rate of the second phase increased in a hyperbolic manner, and the rate of the third phase remained constant with increasing ssDNA. These dependencies indicated that the ssDNA-binding mechanism was essentially unaltered even though the ssDNA contained a primase recognition sequence. However, the intrinsic rate constants were affected (Table II), and these were determined by the method of numerical integration and global fitting, as described previously (Picha et al., 2000). To visualize how the altered intrinsic rate constants affect the overall kinetics of ssDNA binding, we simulated the time-dependent formation and decay of the helicase–ssDNA species for the two ssDNAs, using the respective intrinsic rate constants (Table II). This analysis showed that the ED2 was converted more slowly to ED3 in the pri30mer binding pathway (Figure 5A). This effect was more pronounced when the nucleotide substrates for RNA synthesis, ApC and CTP, were present. Similarly, the formation of ED4 was slower when the ssDNA contained a primase sequence (Figure 5B). The same experiments were carried out with a second set of ssDNAs. These ssDNAs were of a different sequence from the ones used in the above experiments. Again, in this second set, a primase recognition sequence was introduced into one of the two ssDNAs by a single nucleotide change. The same trends in the accumulation of ED2 and ED4 were observed for this second set of 30mer ssDNAs (data not shown). These results support the notion that the ssDNA first binds to the primase ssDNA-binding site to form ED1 and then ED2. It appears that when the ssDNA contains a primase recognition sequence and when the substrates for RNA synthesis are present, then the ssDNA is retained longer at the primase ssDNA-binding site and migrates into the central channel at a lower rate.

Fig. 5. Kinetics of formation and decay of the various helicase–ssDNA species in the 30mer ssDNA-binding pathways. The formation and decay of ED2 (A) and ED4 (B) in the 30mer ssDNA-binding pathways was simulated using the mechanism derived from the stopped-flow experiments (Table II). The concentrations of gp4A′ hexamer and ssDNA, in the simulations, were 142 nM and 1 µM, respectively. The curves for 30mer (solid line), pri30mer (dashed line) and pri30mer + ApC + CTP (short-dashed line) are overlaid for comparison.
Discussion
A number of hexameric helicases are involved in dsDNA replication, where their strand separation activity provides the ssDNA templates necessary for the replication machinery. The replicative helicases have to start unwinding dsDNA at specific sites in the genome, but it is unclear how they are loaded at the origin of replication. Many of these ring-shaped hexameric helicases bind preferentially to ssDNA regions of the fork-DNA and the DNA is bound within the central channel of the ring, which poses a problem during initiation. How does the helicase load onto the DNA efficiently if the ssDNA-binding site is located in the central channel and inaccessible to free DNA? We propose three general kinetic pathways by which a DNA can bind into the central channel of a preformed ring-shaped hexamer (Scheme 1). These are categorized as the ring-opening, threading and disassembly models. In the ring-opening model, the disruption of one subunit interface results in an open ring, which exposes the DNA-binding site and allows the DNA to bind in the central channel. In the threading model, none of the subunit interfaces is disrupted, and a ring-opening step is not required for DNA binding. Instead, a free end of the DNA threads into the hole and the DNA binds directly into the central channel of a closed ring. In the disassembly model, two or more of the subunit interfaces are disrupted, resulting in hexamer disassembly. The subunits then reassemble into a hexamer around the DNA.

To distinguish between the models shown in Scheme 1, we investigated the ssDNA-binding mechanism of T7 gp4A′. We have eliminated the disassembly pathway as a possible mechanism because the hexamer subunits do not disassemble before binding DNA and the 30mer binding kinetics were independent of protein concentration (Patel et al., 1994; Picha et al., 2000). To distinguish between the threading and ring-opening mechanisms, we compared the kinetics of 30mer and ssM13 DNA binding. The ssM13 DNA is circular and can not bind to a preformed ring of gp4A′ by the threading pathway. The combination of stopped-flow ssDNA binding and presteady state dTTPase and helicase kinetic studies revealed that both ssDNAs bind by essentially the same mechanism. The results are therefore consistent with a five-step ring-opening mechanism for ssDNA binding to a preformed hexamer of gp4A′, shown in Scheme 2.

In a simple ring-opening mechanism, shown in Scheme 1, DNA binding requires collision of the DNA within the central channel of an open ring. Thus, the ring-opening step has to coincide with the DNA collision step within the central channel of a transiently opened ring. For this reason, many of the encounters between the ssDNA and the helicase are predicted to be nonproductive, and the observed rates for ssDNA binding directly into the central channel are expected to be lower than diffusion-limited. Such a low rate was observed with the Escherichia coli DnaB helicase, which appears to use a simple ring-opening mechanism (Bujalowski and Jezewska, 2000). However, with gp4A′ the initial encounter of the ssDNA was nearly diffusion limited. To explain the fast binding of the ssDNA, we propose that the ssDNA first interacts with a readily accessible site on gp4A′, possibly on the outside of the helicase ring, before it migrates into the central channel as shown in Scheme 2. It has been proposed that the primase–ssDNA-binding site on the N-terminal domain of gp4A′ is accessible and located on the outside of the gp4A′ ring (Kusakabe et al., 1998). The stopped-flow studies reported here support the idea that the ssDNA is bound to the primase site in the ED1 and ED2 complexes (Scheme 2). The primase site interacts weakly with the ssDNA with a Kd close to 10 µM (Frick and Richardson, 1999) and indeed the Kd of ED1 is 2.5 µM (50 s–1/2×107 M–1s–1), as measured by stopped-flow studies. Our studies suggest that the primase ssDNA-binding site acts as a loading site, and the binding of the ssDNA to the primase site either triggers the ring-opening process or simply increases the local concentration of ssDNA, thereby increasing the probability of ssDNA binding in the central channel. Therefore the mechanism of DNA binding to the helicase DNA-binding site of T7 gp4A′ occurs by a facilitated mechanism.
To determine when the ring-opening and closing steps occur and when the ssDNA migrates into the central channel, we characterized the kinetic stability and the enzymatic activities of the helicase–ssDNA intermediates using presteady state dTTPase and helicase studies. The chase filter DNA-binding experiments indicated that ED4 was the first species that was kinetically stable. In addition, the kinetics of ssDNA binding by the chase filter binding method were entirely consistent with the mechanism determined from protein fluorescence changes. We propose that the ssDNA migrates into the central channel upon formation of ED4, as shown in Scheme 2. Interestingly, when we analyzed the dTTPase and the helicase activities of the intermediates, we found that none of the species from ED1 to ED4 was competent in hydrolyzing dTTP at the ssDNA-stimulated rate or unwinding dsDNA; only ED5 was competent. We therefore propose that ED5 is a final closed ring complex with the ssDNA bound in the central channel that is proficient in dsDNA unwinding. It is not clear exactly when the ring opens. Since the ssDNA is bound within the central channel only in the ED4 complex, the preceding steps ED1 to ED2 or ED2 to ED3 are possible candidates for the ring-opening steps. Because the ring-opening step involves disruption of a subunit–subunit interface, we reasoned that the ring-opening step is likely to be slow. We therefore assigned the slower ED2 to ED3 transition as the ring-opening step, as shown in Scheme 2.
The ring-opening mechanism is likely to be a preferred pathway for loading hexameric helicases on the DNA even under cellular conditions. Under cellular conditions, free ends of the DNA are not available; hence DNA binding by the threading mechanism is not possible. Similarly, if the helicase assembles into a stable hexamer with Mg2+ and/or NTP immediately after protein synthesis, before it encounters the target DNA, then disassembly and re-assembly of hexamer subunits around the DNA are unlikely to occur. The ring-opening is therefore the only reasonable mechanism for loading a stable hexameric helicase on the DNA under cellular conditions. The facilitated ring-opening ssDNA-binding mechanism elucidated for the T7 gp4A′ hexamer provides an efficient mechanism for loading a hexameric helicase on the DNA. As discussed above, a simple ring-opening pathway is inefficient because the steps of ring-opening and DNA binding have to coincide. In the facilitated ring-opening mechanism, the DNA is already bound to the helicase at a loading site and hence the DNA can be easily transferred into the central channel upon ring-opening. Such a mechanism also provides a way to achieve sequence-specific loading of helicase on the DNA. Helicases such as SV40 large T antigen and BPV E1 have distinct origin-binding sites that load these helicases at specific origins of replication (Dean et al., 1987; Deb and Tegtmeyer, 1987; Borowiec and Hurwitz, 1988; Ustav et al., 1991; Wilson and Ludes-Meyers, 1991). Other hexameric helicases, such as DnaB, do not appear to have a loading site; thus, E.coli DnaB binds ssDNA very slowly, ∼1000-fold slower than a diffusion-limited rate (Bujalowski and Jezewska, 2000). The DnaB helicase, however, interacts with the DnaC or the λP protein, each of which bind specifically to initiator complexes that form at the origins of chromosomal DNA replication (Wahle et al., 1989; Mallory et al., 1990; Allen and Kornberg, 1991). Bacillus subtilis phage SPP1 G40P helicase loads efficiently on the DNA in the presence of phage G38P and G39P proteins that bind at the origin (Ayora et al., 1999). Phage T4 gp41 helicase associates with gp59 that helps load the helicase on T4 gp32 coated DNA (Tarumi and Yonesaki, 1995). The MCM helicase proteins from eukaryotes and archaea associate with ORC and cdc6p proteins that help load these helicases at the origin of replication (Donovan et al., 1997; Dutta and Bell, 1997; Tanaka et al., 1997; Zou and Stillman, 1998). These accessory proteins may therefore provide the loading sites necessary to increase the efficiency of helicase binding to the DNA. The common theme is a preassociation step that brings the DNA in the vicinity of the helicase before the DNA is transferred into the central channel of the ring-shaped hexamer. Such a mechanism appears to be built into the T7 gp4A′ protein by the association of the primase and helicase functions.
Materials and methods
Enzymes
The gp4A′ protein is an M64L mutant of gp4A, which was expressed and purified as described previously (Patel et al., 1992; Hingorani and Patel, 1996). The T4 polynucleotide kinase for 5′-end labeling of ssDNA was purchased from Gibco-BRL.
Nucleotides, buffers and other reagents
dTTP was purchased from Sigma, and dTMP-PCP was purchased from USB. [α-32P]dTTP and [γ-32P]ATP were purchased from Amersham Pharmacia Biotech. Nitrocellulose membranes (NC BA-S) were purchased from Schleicher & Schuell, DEAE membranes (DE81) from Whatman and Bio-Gel P30 resin from Bio-Rad. Buffer H contains 50 mM NaCl, 20 mM Tris–Cl pH 7.5, and 10% glycerol.
DNA
Oligodeoxyribonucleotides were purchased from Integrated DNA Technologies (30mer: ACAGTACTCTAGTTACTGATCTGAGAT CAG; pri30mer: ACAGTACTCTAGTTACTGGTCTGAGATCAG). The sequences were designed to avoid the formation of hairpins and dimers. The ssDNAs were purified and their concentrations were determined as described previously (Picha et al., 2000). The extinction coefficients were: 30mer ε = 326 760 M–1cm–1 and pri30mer ε = 323 570 M–1cm–1. The 30mer ssDNA was radiolabeled as described previously (Picha and Patel, 1998). The ssM13mp18 DNA was purified as described previously (Sambrook et al., 1989).
Presteady state kinetics of dTTP hydrolysis in the presence of 30mer and ssM13 DNAs
The presteady state kinetics of dTTP hydrolysis were measured in a rapid quench–flow apparatus in the three-syringe mode. gp4A′ was incubated with [α-32P]dTTP and Mg-dTTP and free MgCl2 for 15 s (t1). Subsequently, ssDNA was added, and the reaction mixture was incubated for varying times t2 (0.1–65 s). The reactions were stopped with 0.5 M formic acid, and were separated by PEI Cellulose F thin layer chromatography (EM Science, Gibbstown, NJ) in 0.4 M KPO4 (pH 3.4). The radioactivity on the plates was quantitated using a PhosphorImager (Molecular Dynamics). The molar amount of dTDP formed was determined and plotted versus the reaction time, t2. The experiment with the 30mer ssDNA was conducted multiple times, and the data from eight separate t2-series were combined. The intrinsic dTTP hydrolysis rate of gp4A′ was determined separately in similar experiments without ssDNA.
To determine the kinetics of dTTP hydrolysis under conditions where gp4A′ hexamer was preassembled on ssM13 DNA, a two-syringe quench–flow set-up was used. The mixture of gp4A′, ssM13 DNA, EDTA and dTTP in buffer H was preincubated for ∼1 h, and then mixed with MgCl2 and a small amount of [α-32P]dTTP in buffer H in a quench–flow instrument. The reactions were stopped after 0.1–65 s with 0.5 M formic acid (∼90 µl). The separation of the reaction products and their analysis were carried out as described above.
Presteady state kinetics of dsDNA unwinding
The dsDNA unwinding experiments were carried out using a fork-DNA substrate, 36/15(50), containing a 36 nt 5′-tail and a noncomplementary 15 nt 3′-tail, on the same side of a 50 bp dsDNA region (Ahnert and Patel, 1997). The sequence of the 5′-tail strand was: 5′-TACGATCTAGCC TCAATACCAGGGTCAGGTTCGTTAGAGCGGATTACTATACTA CATTAGAATTCAGACTGTAGAGATTCGGTAAG-3′ and that of the 3′-tail strand was: 5′-CTTACCGAATCTCTACAGTCTGAATTCTAA TGTAGTATAGTAATCCGCTCATTGCTTGTATGGTC-3′. The underlined portions form the dsDNA region.
The unwinding experiments were carried out in a rapid quench–flow apparatus in the three-syringe mode at 18°C. Under condition A, gp4A′ was mixed with Mg-dTTP and free MgCl2 for 15 s. The radiolabeled fork-DNA was added to the protein to start the reaction. After 0.02–330 s, the reaction was stopped using 100 mM EDTA, 1% SDS, 500 nM unlabeled 5′-tail strand and bromophenol blue. The reaction products were separated and quantitated as described previously (Ahnert and Patel, 1997). The unwinding experiments under condition B were carried out in the quench–flow apparatus in the two-syringe mode. gp4A′, radiolabeled fork-DNA, dTTP and EDTA in buffer H were incubated for 1 h. The unwinding reactions were started by rapidly mixing the gp4A′ solution with MgCl2. After 0.1–8 s the reactions were stopped, and the unwinding time course was determined as described above.
Stopped-flow kinetics of 30mer and ssM13 DNA binding
The kinetic mechanisms of ssDNA binding with the 30mer ssDNAs (30mer, pri30mer and pri30mer + ApC + CTP) and ssM13 DNA were determined by stopped-flow fluorescence experiments, kinetic simulation and global fitting, as described previously (Picha et al., 2000). The case of ssM13 DNA posed a problem since many gp4A′ hexamers can bind to a single ssM13 DNA. Therefore, the concentration of ssDNA-binding sites on the ssM13 DNA does not change appreciably during the ssDNA-binding process. To describe this situation in the global fitting procedure, the differential equation describing the decrease of the ssM13 DNA concentration was excluded. In addition, the large size of the ssM13 DNA results in a non-ideal diffusion behavior, which along with the large number of protein binding sites, makes it difficult to compare the obtained bimolecular on-rates for the 30mer and ssM13 DNAs.
Rapid quench–flow chase and NC/DEAE ssDNA-binding assay
The experiment was carried out in a rapid quench–flow instrument (KinTek Quench–flow Model-3, KinTek Corp., State College, PA). A mixture of gp4A′, Mg-dTMP-PCP and MgCl2 in buffer H was mixed with a solution of 5′-[32P]30mer ssDNA, Mg-dTMP-PCP, and MgCl2 in buffer H at 18°C. After varying times (t1 = 0.015–0.3 s), a 10-fold molar excess of non-radiolabeled 30mer ssDNA was added as a chase. After 25 s chase time, a 20 µl aliquot was filtered through a NC/DEAE membrane assembly, and the molar amount of helicase–ssDNA complex was determined as described previously (Hingorani and Patel, 1993; Patel and Hingorani, 1995). To account for the nonspecific ssDNA binding to the membranes and the effectiveness of the chase, the following experiment was carried out. The radiolabeled ssDNA was first mixed with the chase ssDNA and then gp4A′ was added. The reaction was filtered and analyzed as above. About 0.2% of the radiolabeled ssDNA was bound to the protein under these conditions, and this was subtracted from the data. The ssDNA-binding kinetics best fit to the sum of two exponentials (Equation 1).
[Bound DNA] = a1 × (1 – e–k1 × t ) + a2 × (1 – e–k2 × t ) + C (1)
[Bound ssDNA] is the amount of ssDNA bound to the protein and retained on the NC filter, a1 and a2 are the amplitudes, and k1 and k2 are the rate constants of the two phases. C is a constant.
Kinetic simulations
The ssDNA-binding kinetics, measured by the chase filter binding assay, were computer simulated with the software ‘Scientist’ (MicroMath Research, L.C.) as described previously (Picha et al., 2000). The simulations employed the 30mer ssDNA-binding mechanism and the rate constants in Table II (30mer). The concentration of each species, from E to ED4, after mixing the hexamer with the ssDNA, was determined by numerical integration of the differential equations describing the mechanism shown in Table II. E represents gp4A′ hexamer, D represents ssDNA and ED1—ED5 are the various helicase–ssDNA complexes in the pathway of ssDNA binding. To calculate the amount of each species that was converted to ED4 during the chase time of 25 s, we used the method of net rate constants (Cleland, 1975) and arrived at Equation 2.
![]()
![]()
ED4chase is the amount of ED4 containing radiolabeled ssDNA after the 25 s chase time. ED2(t1) through ED4(t1) are the amounts of the respective species bound to radiolabeled ssDNA at the time of chase addition. The time course of ED4 formation was plotted and compared with the observed chase data.
The presteady state dTTPase experiments with the 30mer and ssM13 DNAs were simulated using the respective ssDNA-binding mechanisms (Table I) with the software ‘Scientist’. The differential equations describing each of the species from E to ED5 in the ssDNA-binding mechanisms were used to describe the amount of each species at any given time throughout the reaction. The kinetics of dTDP formation were then determined by assigning a hydrolysis rate to each species. For example, when ED5 was hydrolyzing at the ssDNA-stimulated rate (ks) and the rest of the species at the intrinsic rate (ki), the formation of dTDP at any given time was described by H where:
dH/dt = ki × (E + ED1 + ED2 + ED3 + ED4) + ks × (ED5).
Acknowledgments
Acknowledgements
We thank Mikhail Levin for his artistic help in preparing the figures. This work was supported by National Institutes of Health Grant GM55310.
References
- Ahnert P. and Patel,S.S. (1997) Asymmetric interactions of hexameric bacteriophage T7 DNA helicase with the 5′- and 3′-tails of the forked DNA substrate. J. Biol. Chem., 272, 32267–32273. [DOI] [PubMed] [Google Scholar]
- Allen G.C.J. and Kornberg,A. (1991) Fine balance in the regulation of DnaB helicase by DnaC protein in replication in Escherichia coli.J. Biol. Chem., 266, 22096–22101. [PubMed] [Google Scholar]
- Ayora S., Stasiak,A. and Alonso,J.C. (1999) The Bacillus subtilis bacteriophage SPP1 G39P delivers and activates the G40P DNA helicase upon interacting with the G38P-bound replication origin. J. Mol. Biol., 288, 71–85. [DOI] [PubMed] [Google Scholar]
- Bird L.E., Hakansson,K., Pan,H. and Wigley,D.B. (1997) Characterization and crystallization of the helicase domain of bacteriophage T7 gene 4 protein. Nucleic Acids Res., 25, 2620–2626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borowiec J.A. and Hurwitz,J. (1988) ATP stimulates the binding of simian virus 40 (SV40) large tumor antigen to the SV40 origin of replication. Proc. Natl Acad. Sci. USA, 85, 64–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bujalowski W. and Jezewska,M.J. (1995) Interactions of Escherichia coli primary replicative helicase DnaB protein with single-stranded DNA. The nucleic acid does not wrap around the protein hexamer. Biochemistry, 34, 8513–8519. [DOI] [PubMed] [Google Scholar]
- Bujalowski W. and Jezewska,M.J. (2000) Kinetic mechanism of the single-stranded DNA recognition by Escherichia coli replicative helicase DnaB protein. Application of the matrix projection operator technique to analyze stopped-flow kinetics. J. Mol. Biol., 295, 831–852. [DOI] [PubMed] [Google Scholar]
- Cleland W.W. (1975) Partition analysis and the concept of net rate constants as tools in enzyme kinetics. Biochemistry, 14, 3220–3224. [DOI] [PubMed] [Google Scholar]
- Dean F.B., Dodson,M., Echols,H. and Hurwitz,J. (1987) ATP-dependent formation of a specialized nucleoprotein structure by simian virus 40 (SV40) large tumor antigen at the SV40 replication origin. Proc. Natl Acad. Sci. USA, 84, 8981–8985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deb S.P. and Tegtmeyer,P. (1987) ATP enhances the binding of simian virus 40 large T antigen to the origin of replication. J. Virol., 61, 3649–3654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donovan S., Harwood,J., Drury,L.S. and Diffley,J.F. (1997) Cdc6p-dependent loading of Mcm proteins onto pre-replicative chromatin in budding yeast. Proc. Natl Acad. Sci. USA, 94, 5611–5616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta A. and Bell,S.P. (1997) Initiation of DNA replication in eukaryotic cells. Ann. Rev. Cell. Dev. Biol., 13, 293–332. [DOI] [PubMed] [Google Scholar]
- Egelman E.H., Yu,X., Wild,R., Hingorani,M.M. and Patel,S.S. (1995) Bacteriophage T7 helicase/primase proteins form rings around single-stranded DNA that suggest a general structure for hexameric helicases. Proc. Natl Acad. Sci. USA, 92, 3869–3873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fouts E.T., Yu,X., Egelman,E.H. and Botchan,M.R. (1999) Biochemical and electron microscopic image analysis of the hexameric E1 helicase. J. Biol. Chem., 274, 4447–4458. [DOI] [PubMed] [Google Scholar]
- Frick D.N. and Richardson,C.C. (1999) Interaction of bacteriophage T7 gene 4 primase with its template recognition site. J. Biol. Chem., 274, 35889–35898. [DOI] [PubMed] [Google Scholar]
- Frick D.N., Baradaran,K. and Richardson,C.C. (1998) An N-terminal fragment of the gene 4 helicase/primase of bacteriophage T7 retains primase activity in the absence of helicase activity. Proc. Natl Acad. Sci. USA, 95, 7957–7962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo S., Tabor,S. and Richardson,C.C. (1999) The linker region between the helicase and primase domains of the bacteriophage T7 gene 4 protein is critical for hexamer formation. J. Biol. Chem., 274, 30303–30309. [DOI] [PubMed] [Google Scholar]
- Hingorani M.M. and Patel,S.S. (1993) Interactions of bacteriophage T7 DNA primase/helicase protein with single-stranded and double-stranded DNAs. Biochemistry, 32, 12478–12487. [DOI] [PubMed] [Google Scholar]
- Hingorani M.M. and Patel,S.S. (1996) Cooperative interactions of nucleotide ligands are linked to oligomerization and DNA binding in bacteriophage T7 gene 4 helicases. Biochemistry, 35, 2218–2228. [DOI] [PubMed] [Google Scholar]
- Kusakabe T., Baradaran,K., Lee,J. and Richardson,C.C. (1998) Roles of the helicase and primase domain of the gene 4 protein of bacteriophage T7 in accessing the primase recognition site. EMBO J., 17, 1542–1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohman T.M. and Bjornson,K.P. (1996) Mechanisms of helicase-catalyzed DNA unwinding. Annu. Rev. Biochem., 65, 169–214. [DOI] [PubMed] [Google Scholar]
- Mallory J.B., Alfano,C. and McMacken,R. (1990) Host virus interactions in the initiation of bacteriophage lambda DNA replication. Recruitment of Escherichia coli DnaB helicase by lambda P replication protein. J. Biol. Chem., 265, 13297–13307. [PubMed] [Google Scholar]
- Matson S.W. and Richardson,C.C. (1985) Nucleotide-dependent binding of the gene 4 protein of bacteriophage T7 to single-stranded DNA. J. Biol. Chem., 260, 2281–2287. [PubMed] [Google Scholar]
- Matson S.W. and Kaiser-Rogers,K.A. (1990) DNA helicases. Annu. Rev. Biochem., 59, 289–329. [DOI] [PubMed] [Google Scholar]
- Morris P.D. and Raney,K.D. (1999) DNA helicases displace streptavidin from biotin-labeled oligonucleotides. Biochemistry, 38, 5164–5171. [DOI] [PubMed] [Google Scholar]
- Patel S.S. and Hingorani,M.M. (1995) Nucleotide binding studies of bacteriophage T7 DNA helicase–primase protein. Biophys. J., 68, 186S–189S. [PMC free article] [PubMed] [Google Scholar]
- Patel S.S. and Picha,K.M. (2000) Structure and function of hexameric helicases. Annu. Rev. Biochem., in press. [DOI] [PubMed] [Google Scholar]
- Patel S.S., Rosenberg,A.H., Studier,F.W. and Johnson,K.A. (1992) Large scale purification and biochemical characterization of T7 primase/helicase proteins. Evidence for homodimer and heterodimer formation. J. Biol. Chem., 267, 15013–15021. [PubMed] [Google Scholar]
- Patel S.S., Hingorani,M.M. and Ng,W.M. (1994) The K318A mutant of bacteriophage T7 DNA primase–helicase protein is deficient in helicase but not primase activity and inhibits primase–helicase protein wild-type activities by heterooligomer formation. Biochemistry, 33, 7857–7868. [DOI] [PubMed] [Google Scholar]
- Picha K.M. and Patel,S.S. (1998) Bacteriophage T7 DNA helicase binds dTTP, forms hexamers and binds DNA in the absence of Mg2+. The presence of dTTP is sufficient for hexamer formation and DNA binding. J. Biol. Chem., 273, 27315–27319. [DOI] [PubMed] [Google Scholar]
- Picha K.M., Ahnert,P. and Patel,S.S. (2000) DNA binding in the central channel of bacteriophage T7 helicase–primase is a multistep process. Nucleotide hydrolysis is not required. Biochemistry, 39, 6401–6409. [DOI] [PubMed] [Google Scholar]
- Sambrook J., Fritsch,E.F. and Maniatis,T. (1989) Single-stranded, filamentous bacteriophage vectors. In Ford,N., Nolan,C. and Ferguson,M. (eds), Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp. 4.1–4.54. [Google Scholar]
- Sawaya M.R., Guo,S., Tabor,S., Richardson,C.C. and Ellenberger,T. (1999) Crystal structure of the helicase domain from the replicative helicase–primase of bacteriophage T7. Cell, 99, 167–177. [DOI] [PubMed] [Google Scholar]
- Scherzinger E., Lanka,E. and Hillenbrand,G. (1977) Role of bacteriophage T7 DNA primase in the initiation of DNA strand synthesis. Nucleic Acids Res., 4, 4151–4163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smelkova N.V. and Borowiec,J.A. (1998) Synthetic DNA replication bubbles bound and unwound with twofold symmetry by a simian virus 40 T-antigen double hexamer. J. Virol., 72, 8676–8681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabor S. and Richardson,C.C. (1981) Template recognition sequence for RNA primer synthesis by gene 4 protein of bacteriophage T7. Proc. Natl Acad. Sci. USA, 78, 205–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka T., Knapp,D. and Nasmyth,K. (1997) Loading of an Mcm protein onto DNA replication origins is regulated by Cdc6p and CDKs. Cell, 90, 649–660. [DOI] [PubMed] [Google Scholar]
- Tarumi K. and Yonesaki,T. (1995) Functional interactions of gene, 32, 41 and 59 proteins of bacteriophage T4. J. Biol. Chem., 270, 2614–2619. [DOI] [PubMed] [Google Scholar]
- Ustav M., Ustav,E., Szymanski,P. and Stenlund,A. (1991) Identification of the origin of replication of bovine papillomavirus and characterization of the viral origin recognition factor E1. EMBO J., 10, 4321–4329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahle E., Lasken,R.S. and Kornberg,A. (1989) The dnaB–dnaC replication protein complex of Escherichia coli. I. Formation and properties. J. Biol. Chem., 264, 2463–2468. [PubMed] [Google Scholar]
- Washington M.T. and Patel,S.S. (1998) Increased DNA unwinding efficiency of bacteriophage T7 DNA helicase mutant protein 4A′/E348K. J. Biol. Chem., 273, 7880–7887. [DOI] [PubMed] [Google Scholar]
- Wilson V.G. and Ludes-Meyers,J. (1991) A bovine papillomavirus E1-related protein binds specifically to bovine papillomavirus DNA. J. Virol., 65, 5314–5322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X., Hingorani,M.M., Patel,S.S. and Egelman,E.H. (1996) DNA is bound within the central hole to one or two of the six subunits of the T7 DNA helicase. Nature Struct. Biol., 3, 740–743. [DOI] [PubMed] [Google Scholar]
- Zou L. and Stillman,B. (1998) Formation of a preinitiation complex by S-phase cyclin CDK-dependent loading of Cdc45p onto chromatin. Science, 280, 593–596. [DOI] [PubMed] [Google Scholar]