

Abstract
GAF domains are ubiquitous motifs present in cyclic GMP (cGMP)-regulated cyclic nucleotide phosphodiesterases, certain adenylyl cyclases, the bacterial transcription factor FhlA, and hundreds of other signaling and sensory proteins from all three kingdoms of life. The crystal structure of the Saccharomyces cerevisiae YKG9 protein was determined at 1.9 Å resolution. The structure revealed a fold that resembles the PAS domain, another ubiquitous signaling and sensory transducer. YKG9 does not bind cGMP, but the isolated first GAF domain of phosphodiesterase 5 binds with Kd = 650 nM. The cGMP binding site of the phosphodiesterase GAF domain was identified by homology modeling and site-directed mutagenesis, and consists of conserved Arg, Asn, Lys and Asp residues. The structural and binding studies taken together show that the cGMP binding GAF domains form a new class of cyclic nucleotide receptors distinct from the regulatory domains of cyclic nucleotide-regulated protein kinases and ion channels.
Keywords: cGMP/crystal structure/GAF domain/ GMP receptor/YKG9
Introduction
3′, 5′ cyclic guanosine monophosphate (cGMP) was one of the earliest second messenger molecules to be discovered, and has recently been the focus of intense and renewed fascination because of its critical role as the downstream transducer of the effects of nitric oxide (NO) (Moncada and Higgs, 1995; Hobbs and Ignarro, 1996; Murad, 1996). cGMP is also the intracellular messenger responsible for transducing atrial natriuretic factor (ANF) signaling (Garbers and Dubois, 1999), and it is the critical messenger molecule in the initiation of signaling in mammalian phototransduction (Stryer, 1991; Baylor, 1996). The most immediate cellular effect of NO and ANF is to allosterically promote the synthesis of cGMP by their receptors, which are themselves guanylyl cyclases. Conversely, the hydrolysis of cGMP by phosphodiesterase-6 (PDE6) is one of the pivotal early steps in mammalian phototransduction.
There are two main structural classes of cGMP receptor in mammalian cells. The first includes the regulatory domain of the cGMP-activated protein kinase (PKG; Weber et al., 1989; Shabb et al., 1990) and the gating domain of cGMP- and cNMP-gated ion channels (Altenhofen et al., 1991; Kumar and Weber, 1992). These domains are homologous to each other, and to the regulatory subunit of cAMP-dependent protein kinase (PKA), the cAMP-activated ras guanine nucleotide exchange factor Epac (de Rooij et al., 1998) and the bacterial catabolite activator protein (CAP). The crystal structures of the cAMP binding PKA regulatory subunit and of CAP are known (McKay and Steitz, 1981; Su et al., 1995). The structures of the cGMP binding PKG regulatory domain and the ion channel cNMP gating domain have been modeled on this basis (Weber et al., 1989; Shabb et al., 1990; Altenhofen et al., 1991; Kumar and Weber, 1992).
The second group of cGMP receptors are the cGMP-regulated families of cyclic nucleotide phosphodiesterases (PDEs; Corbin and Francis, 1999; Soderling and Beavo, 2000). This group comprises PDE2, PDE5 and PDE6. These enzymes have a dual role in cGMP metabolism. Not only are they allosterically regulated by cGMP at non-catalytic sites, they also hydrolyze cGMP to 5′-GMP. PDE5 and PDE6 are highly specific for cGMP as a substrate, while PDE2 hydrolyzes both cAMP and cGMP. The allosteric cGMP binding sites on these PDEs have been mapped to two homologous repeats in the N-terminal halves of the proteins. Two other bispecific PDE families, PDE10A and PDE11A, are homologous to PDEs 2, 5 and 6 in their N-termini, but appear to lack a high-affinity allosteric cGMP binding site (Soderling et al., 1999; Fawcett et al., 2000). The medical importance of these cGMP binding repeats for the normal function of these PDEs is highlighted by the naturally occurring mutation H258N in human PDE6B, which leads to congenital stationary night blindness in humans (Gal et al., 1994).
Motifs homologous to the cGMP binding repeats of the PDEs are found in a vast family (>440 proteins in the non-redundant database; Schultz et al., 1998) of signal transducing proteins, as revealed by sensitive sequence analysis techniques (Aravind and Ponting, 1997; Figure 1). These motifs are named ‘GAF domains’ for their presence in cGMP-regulated cyclic nucleotide PDEs, certain adenylyl cyclases and the bacterial transcription factor FhlA. The GAF domains of cGMP-regulated PDEs are set apart from most others by the presence of a NKXnD motif implicated in cGMP binding in PDE5 and PDE6 (Turko et al., 1996; Granovsky et al., 1998).

Fig. 1. Domain structure of mammalian GAF and PAS domain-containing phosphodiesterases and other representative GAF domain proteins. Domain structures were obtained from SMART (Schultz et al., 1998), Pfam (Bateman et al., 1999) and Soderling and Beavo (2000).
The presence of GAF domains in such a large and diverse family of proteins suggested the possibility of new and unexpected roles for cGMP signaling. It has been inferred that at least some GAF domains constitute independent cGMP binding modules, based on the known roles of these sequences in cGMP-regulated PDEs (Charbonneau et al., 1990; Aravind and Ponting, 1997). On the other hand, neither the cGMP binding properties nor any other characterization of isolated GAF domains have been reported to date. In this study, we sought to clarify the functions of GAF domains by first characterizing the cGMP binding properties of three recombinant GAF domains. We established that the first GAF domain of PDE5 (GAFa) bound cGMP, whereas neither the second domain of PDE5 (GAFb) nor the GAF protein encoded by the Saccharomyces cerevisiae open reading frame YKG9 bound cGMP. As we were unable to crystallize PDE5-GAFa, we proceeded to solve the crystal structure of the YKG9 protein, which we used as a template for the GAF domain fold. We have modeled the structure of the high-affinity cGMP binding PDE5-GAFa domain based on this template. We used the modeled structure, together with sequence alignments, existing structure–function data (Turko et al., 1996, 1998) and site-directed mutagenesis, to identify structural determinants for high-affinity cGMP binding.
Results
cGMP binding to GAF domains
In order to determine whether isolated GAF domains are capable of binding cGMP, both GAF domains of PDE5 were expressed as recombinant glutathione S-transferase (GST) fusions. Because of the low sequence similarity between different GAF domains, the optimal choices of boundaries for the recombinant constructs were uncertain. A longer and shorter form of each GAF domain was tested for expression. The longer forms of both failed to express soluble protein, hence the shorter constructs, together with the YKG9 protein, were assayed for binding to both cGMP and cAMP.
The first PDE5 GAF (GAFa) bound cGMP with a Kd of 0.65 µM, whereas the PDE5-GAFb and YKG9 did not bind cGMP (Figure 2). None of the proteins binds cAMP (data not shown). The finding of high-affinity binding to PDE5-GAFa is consistent with previous observations of cGMP binding to a PDE5 mutant (PDE5 D478A) whose GAFb domain was inactivated. This PDE5 mutant exhibited a Kd of 0.5 µM (McAllister-Lucas et al., 1995). Thus, the properties of isolated GAF domains from PDE5 appear to mirror their properties in the context of the intact enzyme. cGMP does not bind measurably to the isolated PDE5-GAFb, which is the low-affinity site in intact PDE5. On the other hand, the isolated PDE5-GAFa manifests the same high specificity for cGMP over cAMP as intact PDE5.

Fig. 2. Binding of cGMP to PDE5-GAFa and PDE5-GAFb, and YKG9.
Overall structure of the YKG9 GAF domain protein
The structure of YKG9 was determined to 1.9 Å resolution by multiwavelength anomalous dispersion (MAD) from non-covalently bound solvent Br– ions (Figure 3; Table I; Dauter and Dauter, 1999; Dauter et al., 2000). A YKG9 crystal was incubated for 45 s in cryoprotectant supplemented with 0.5 M NaBr. A three-wavelength MAD data set was collected at the Br K edge and at a high-energy remote point (Table I). The phasing power of the dispersive signal was limited, probably because the measured X-ray absorption spectrum reflects bulk Br– ions rather than those bound to the protein, and it is therefore difficult to locate the wavelength at which f′ of bound Br– ions is minimal. The phasing power of the f′′ component was stronger, and the density-modified map produced by solvent flipping (Abrahams and Leslie, 1996) yielded an experimental map into which all of the protein, except two N-terminal and one C-terminal residues, could be straightforwardly assigned. The structure determination represents the phasing of a 36 000 Da asymmetric unit from the anomalous signal of seven bound solvent Br– ions. The high quality of the electron density obtained for this reasonably large asymmetric unit suggests that MAD phasing using solvent Br– scatterers should prove to be widely applicable.

Fig. 3. (A) Electron density from solvent-flipped Br MAD map at 2.5 Å. (B) Overall structure of YKG9 dimer. (C and D) YKG9 monomer in two different views related by a 90° rotation about the y-axis.
Table I. Crystallographic data.
| Statistical parameters | |||
| wavelength (Å) | λ1 = 0.9199 Å | λ2 = 0.9196 Å | λ3 = 0.9150 Å |
| resolution (Å) | 1.9 | 1.9 | 1.9 |
| number of observed reflections | 898 200 | 897 099 | 901 143 |
| number of unique reflections | 36 219 | 36 234 | 36 203 |
| completeness of data (%) | 99.5 | 99.6 | 99.7 |
| overall I/σ (I) | 28.5 | 25.3 | 27.9 |
| completeness of outer shell (Å) | 98.6 (1.97–1.90) | 98.4 (1.97–1.90) | 99.5 (1.97–1.90) |
| I/σ (I) of outer shell (Å) | 2.78 (1.97–1.90) | 2.38 (1.97–1.90) | 2.73 (1.97–1.90) |
| Rsym (%) | 5.2 | 5.9 | 5.7 |
| Phasing and refinement statistics | |||
| λ | 0.9196 | 0.9199 | 0.9150 |
| RCullis (centric) | 0.89 | 0.83 | – |
| RCullis (ano) | 0.61 | 0.59 | 0.62 |
| phasing power (dispersive) | 0.76 | 0.94 | – |
| phasing power (ano) | 1.24 | 1.30 | 1.21 |
| resolution (Å) | 10–2.8 | ||
| figure of merit | 0.61 | ||
| Refinement statistics | |||
| resolution (Å) | 20–1.9 | ||
| unique data (F > 0) | 64 663 | ||
| R-factor (%) | 21.4 | r.m.s.d. from ideal bond length | 0.016 Å |
| Rfree (%) | 24.6 | r.m.s.d. from ideal bond angle | 1.73° |
| number of atoms | 3075 | r.m.s.d., B-factors of main chain | 1.6 Å2 |
| number of protein atoms | 2726 | r.m.s.d., B-factors of side chains | 2.4 Å2 |
| number of bromide ions | 9 |
The YKG9 structure is built around a central antiparallel six-stranded β-sheet with strand order 321654 (Figure 3C and D). The N- and C-terminal portions of the sequence form one outer layer of the structure, consisting of four helices: α1, α2, α3 and α5. The opposite outer layer of the structure is a mixture of loops and a short α-helix, α4. This layer does not fit into a standard class of supersecondary structure, so we refer to it as the ‘irregular’ layer. The N-terminus of intact YKG9 extends 41 residues before the N-terminal boundary of the GAF domain as defined by Aravind and Ponting (1997). The first three of these residues are disordered in one molecule (‘A’) in the dimer, and the first two are disordered in the other molecule. This 41 residue N-terminal extension to the GAF domain as defined previously (Aravind and Ponting, 1997) includes both α1 (residues 8–14) and α2 (residues 20–38). These α-helices, in particular α2, make up an integral part of the α-helical layer, suggesting that the stable folding unit for the GAF domain corresponds to residues 8–179 of YKG9. YKG9 dimerizes in solution as judged by gel filtration and dynamic light scattering, and it is found as a dimer of non-crystallographic symmetry within the crystal. The dimer interface buries 2200 Å2 of molecular surface area as compared with both monomers in isolation. Most of the dimer interface is made up of residues of the N-terminal tail, α3, β2, β3 and the β2–β3 loops (Figure 3B).
The YKG9 protein contains a buried cluster consisting of three acidic residues (Glu132, Asp149 and Asp151), His122 and three Cys residues (Cys91, Cys101 and Cys125). These residues face into a buried pocket sandwiched in between the central β-sheet and the irregular face (Figure 4A). The pocket has a calculated volume of 590 Å3 (Laskowski, 1995). Cys91 and Cys125 are disulfide bonded to each other in the structure. The disulfide is presumed to have formed in the crystal used in the structure solution because it was grown in the absence of reducing agent. The disulfide bond restricts the conformational freedom of the β2–β3 loops and β3–β4 loops containing these Cys residues. The presence of the disulfide linkage appears to stabilize YKG9 crystals, since crystals grown in high concentrations of dithiothreitol (DTT) are small and not of diffraction quality. Roughly 11–12 well-ordered solvent molecules are bound in the pocket, forming a solvent-filled tunnel that is contiguous with exterior solvent. His122 is hydrogen bonded to Glu132. Cys101 interacts closely with the side-chain of Asp149. The disulfide-bonded Cys pair is more distal to the other residues just noted. However, if Cys125 were reduced, a change in rotamer would bring it within 3.5 Å of the side-chain of Asp151. The three acidic residues form a tight cluster reminiscent of ‘hard’ divalent cation binding sites. One solvent molecule is coordinated by all three acidic side-chains in a manner suggesting that it is a cation rather than a water molecule. No divalent cations were present in the crystallization medium, however. Its refined B-factor, when treated as a water molecule, is 28 Å2. The B-factor is lower than the overall average for the protein, but very similar to that of protein residues and other bound solvent molecules within the pocket. It seems possible that this molecule is really an ammonium ion. Alternatively, one of the acidic side-chains might be protonated. The function of this cavity remains to be determined, although it appears to be a small molecule binding site.

Fig. 4. (A) The buried Cys/His/Asp/Glu pocket of YKG9 is compared with (B), the chromophore binding site of photoactive yellow protein. (C and D) The same images as (A) and (B) in a surface representation with basic residues colored blue, acidic red, hydrophobic orange and others gray. In (C) and (D) the surface is removed for a portion of the β3–α4 in YKG9 and the equivalent region of photoactive yellow protein in order to show the underlying cavities.
Unexpected structural similarities to PAS domains and profilin
Despite a lack of sequence homology to any previously determined structure, the YKG9 GAF domain revealed a number of unexpected similarities to other proteins in its three-dimensional structure. The non-redundant protein structure database was searched with VAST (Gibrat et al., 1996) and DALI (Holm and Sander, 1995). The YKG9 GAF domain structure has similarities to the actin binding protein profilin (PDB entries 3NUL and 1FIL), a penicillin binding domain (1PMD), d-Ala, d-Ala transpeptidase (3PTE) and the PAS domains of photoactive yellow protein (3PYP; Borgstahl et al., 1995) and the HERG potassium channel (1BYW; Cabral et al., 1998). The profilin and YKG9 GAF structures are superimposable over 95 residues in the central sheet and the helical layer, with a r.m.s.d. of 3.0 Å, despite having only 5% sequence identity (Figure 5A and B).

Fig. 5. Similarity between (A) YKG9 and (B) profilin, and (C) YKG9 and (D) photoactive yellow protein. Regions that can be superimposed within 3.0 Å are colored red.
The YKG9 structure also has strong similarity to the PAS domain of the oxygen-sensing histidine kinase FixL (1DRQ; Gong et al., 1998), although this recently released structure was not present in the version of the database that was searched. The similarities between the PAS and profilin folds have been noted (Borgstahl et al., 1995).
Photoactive yellow protein, which consists entirely of a single PAS domain, is closely superimposable over the 44 residues that comprise the β-sheet and the irregular layer (Figure 5C and D). The r.m.s.d. between YKG9 and photoactive yellow protein is the lowest for any of the matches at 1.6 Å. The GAF fold is a hybrid between the profilin fold, manifested in the helical and central layers, and the PAS fold, manifested in the central and irregular layers. The PAS fold contains a five-stranded β-sheet instead of a six-stranded one, and is lacking the GAF domain β3. The irregular layer of the GAF structure has considerable similarities to the corresponding region of photoactive yellow protein. Photoactive yellow protein contains a helix that corresponds topologically to the YKG9 α4, whereas profilin does not contain such a helix. The function of photoactive yellow protein as a photoreceptor depends on the presence of a cofactor, a p-hydroxycinnamoyl anion bound through a thioester linkage to Cys69. The most striking observation is that the cofactor binding site of photoactive yellow protein coincides in three dimensions with the Cys/His/Asp/Glu-containing pocket on the GAF domain. The aromatic ring of the chromophore overlays Cys125 of YKG9 in the superimposed structure (Figure 4). However, the side-chains of photoactive yellow protein that interact with the chromophore are not conserved in YKG9.
A model for the GAF domain of a cGMP-dependent phosphodiesterase
The structure of YKG9 provides an archetype for the larger family of GAF domains, including those of the cGMP PDEs. We used the crystal structure and a revised multiple sequence alignment derived by multiple threading (Figure 6) as the basis for constructing a three-dimensional model of PDE5-GAFa. The confidence level of this modeled structure is high in those regions that can be reliably aligned between PDE5 and YKG9. Conserved core secondary structures are most reliable, while the models for inserted loops are speculative. Three previously identified cGMP binding determinants, Asn276, Lys277 and Asp289 (McAllister-Lucas et al., 1995; Turko et al., 1996), are located on conserved core structures and can therefore be pinpointed reliably in the modeled structure of PDE5-GAFa.

Fig. 6. Threading and profile-based alignment of representative GAF domain sequences. Residues that are part of the aligned core, and can therefore be positioned in the three-dimensional homology models with good confidence, are shown with upper-case letters. The PDE5A sequence is bovine, the PDE2A and PDE6A sequences are human. Residues important for cGMP binding are highlighted in blue. Residues contributing directly to the YKG9 Cys/His/Asp/Glu cluster are marked in red. The PDE6 residue mutated in congenital night blindness is highlighted in white against a black background (top section). PYP, photoactive yellow protein, sequence aligned based on three-dimensional structure superposition.
In contrast to the relatively featureless molecular surface of YKG9, the PDE5-GAFa model reveals a molecular surface punctuated by a deep hydrophobic and acidic cavity adjacent to a smaller basic pocket (Figure 7A). These pockets lie over the C-terminus of α3, the N-terminus of α5, the start of β1 and the end of β6. Asn276 and Lys277 lie over the C-terminus of β6, and Asp289 is at the N-terminus of α5. An interaction of the N1 and N2 of the guanine ring with the Asp289 of the NKXnD motif would be consistent with the model proposed previously by Corbin and co-workers (Turko et al., 1996). In support of this model, selectivity for cGMP over cAMP is diminished at low pH, consistent with protonation of Asp289 (Turko et al., 1999). The pH dependence of nucleotide selectivity is almost completely abolished in the mutant D289N (Turko et al., 1999). An interaction between the guanine ring and the side-chain of Asn276 was suggested by previous mutagenic studies (Turko et al., 1996) and would also be consistent with the binding site identified here.
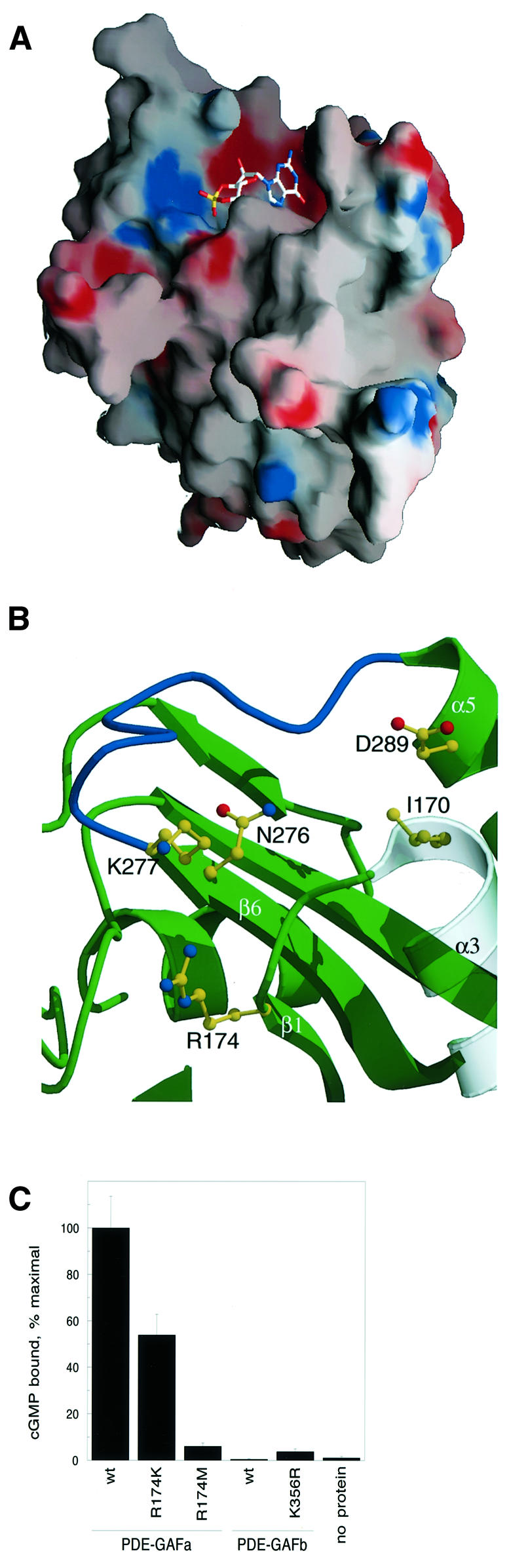
Fig. 7. (A) Surface representation of the PDE5-GAFa model and its cGMP binding site. A manually docked cGMP molecule is shown as a guide to the eye only. (B) Close-up view of the cGMP binding site of the PDE5-GAFa domain in the same orientation. The β6–α5 loop that potentially folds over bound cGMP is highlighted in blue. Ile170 is in the location corresponding to the PDE6 His residue mutated in congenital night blindness. The predicted guanine binding site is formed by Ile170, Asn276, Asp289 and their surroundings, while the predicted cyclic phosphate binding site is formed by Arg174, Lys277 and surrounding residues. (C) cGMP binding of Arg174 mutants in PDE5-GAFa, and the corresponding Lys356 mutant in PDE5-GAFb.
The cGMP binding site contains a potential cyclic phosphate binding site that includes Arg174 and Lys277. Previous mutagenesis implicated Lys277 in cGMP binding (Turko et al., 1996), but the role of Arg174 has not been examined before. Arg174 from the N-terminus of β1 joins Lys277 to form the basic pocket. To test the model implicating Arg174 in cGMP binding to PDE-GAFa, mutants were constructed replacing this residue with Lys or Met. Replacement of Arg174 with Met drastically reduced cGMP binding to PDE-GAFa, indicating that a positive charge in this position is critical for function (Figure 7C). The Arg→Lys mutant was much less deleterious, indicating that the presence of a positive charge is the most important factor at this site. Mutation of the analogous position in PDE5-GAFb (Lys356) to Arg resulted in little improvement in cGMP binding, consistent with the only modest reduction in cyclic nucleotide binding to R174K PDE-GAFa.
Like YKG9, GAFa is a dimer in solution as judged by gel filtration, and it seems likely that it dimerizes through a similar interface to that observed in YKG9. The dimerization observed here is consistent with previous reports that intact PDE2 and PDE5, as well as proteolytic fragments comprising their regulatory domains, are dimers (Thomas et al., 1990; Stroop and Beavo, 1991). When GAFa is modeled as a monomer, the region corresponding to the YKG9 dimer interface contains more exposed hydrophobic residues than other parts of the structure, including Leu164, Leu194, Phe195 and Val197, consistent with a role in dimer formation or possibly other types of protein–protein interaction.
Discussion
GAF and PAS: an extended superfamily of phototransducing and signaling domains
PAS domains, named for Per, ARNT and Sim, are a ubiquitous class of sensory transduction domains (Ponting and Aravind, 1997; Pellequer et al., 1998; Reppert, 1998; Sassone-Corsi, 1998). The striking similarity in the structures of the GAF and PAS domains reveals a clear evolutionary relationship between the two. These two domain families taken separately each represent an exceptionally large number of proteins implicated in sensory and signaling pathways. A total of 877 PAS domains are found in 574 different proteins in the non-redundant sequence database, whereas 497 GAF domains are found in 446 different proteins (Schultz et al., 1998). Both domain families include members that have direct roles as photon receptors. The photoactive yellow protein is an archetypal PAS domain photoreceptor (Pellequer et al., 1998). It is intriguing that the Cys/His/Asp/Glu pocket of the YKG9 protein corresponds in three dimensions to the p-hydroxycinnamoyl binding site of photoactive yellow protein (Figure 4; Borgstahl et al., 1995; Genick et al., 1998) and the heme binding pocket of FixL (Gong et al., 1998). The observation of a close similarity in both topology and binding site location between the GAF and PAS domains provides for the first time a direct link between two of the largest structural classes of soluble sensory transducers.
The phytochromes are a major class of signal transducing photoreceptors in plants (Quail et al., 1995), and one of the largest groups of GAF domain proteins (Figure 1). Photons are absorbed by a tetrapyrrole chromo phore that is covalently attached to a Cys residue in the GAF domain of the phytochrome. This Cys residue is found in an ∼25 amino acid segment inserted between β4 and β5, placing it over the irregular layer of the GAF domain. This region is absent from YKG9 and most other GAF domains. Model building positions the inserted region directly above the unusual Cys/His/Asp/Glu pocket of YKG9. A homology model (not shown) of phytochrome E of Arabidopsis thaliana reveals that this predominantly hydrophobic pocket is maintained in this protein. It therefore appears that the tetrapyrrole binding pocket of the phytochromes is formed at essentially the same site as the Cys/His/Asp/Glu pocket of YKG9 and the heme and chromophore binding sites of the FixL and photoactive yellow protein PAS domains, respectively. The new relationship between PAS domain-containing photoreceptors and phytochromes extends to other soluble GAF domain-containing photoreceptors as well, as exemplified by the cyanobacterial chromatic adaptation sensor (Kehoe and Grossman, 1996). Circadian regulation in cyanobacteria depends on a GAF domain-containing protein, the circadian input kinase (CikA; Schmitz et al., 2000), in contrast to the PAS domain-containing circadian regulatory proteins of eukaryotes. The similarity established between GAF and PAS domains is consistent with an evolutionary connection between circadian regulation in eukaryotes and cyanobacteria.
The connection between PAS and GAF domains sheds new light on the domain structure of PDEs. Of the 11 PDE families, PDEs 2, 5, 6, 10 and 11 contain GAF domains in their N-terminal regulatory regions, and one, PDE8, contains a PAS domain. The function of the single N-terminal PAS domain of PDE8 is not known and the presence of a PAS domain in a PDE was unexpected at the time of its discovery (Soderling et al., 1998). The similarity between the GAF and PAS domains makes the occurrence of a PAS domain in a PDE seem much less surprising and suggests that PDE8 should be grouped, at least in a structural sense, along with PDEs 2, 5, 6, 10 and 11.
cGMP recognition by GAF domains
To date, just one structural class of cyclic nucleotide receptors has been characterized, that comprises the bacterial CAP (McKay and Steitz, 1981), the cyclic nucleotide-regulated protein kinases PKG and PKA (Weber et al., 1989; Su et al., 1995) and the cyclic nucleotide-gated ion channels (Altenhofen et al., 1991; Kumar and Weber, 1992). This class has been referred to as the ‘cNMP’ domain family (Schultz et al., 1998). The GAF domains of the cGMP-regulated PDEs represented a potentially different class of cyclic nucleotide receptors, since they lacked any sequence homology to the cNMP motif. The structure of the YKG9 protein shows no similarity to the cNMP domain and thus establishes beyond any doubt that there are at least two entirely different structural classes of cyclic nucleotide receptors.
The YKG9 structure provides a three-dimensional template for modeling other GAF domains, including those of the PDEs. The use of multiple threading alignment based on the solved structure makes it possible to generate reasonably reliable homology models of GAF domains from PDEs and other proteins based on the YKG9 crystal structure. The accumulated data on residues involved in cGMP binding (McAllister-Lucas et al., 1995; Turko et al., 1996) made it possible to identify the cGMP binding site unambiguously. The cGMP binding site is formed by parts of α3, α5, β1 and β6. This is on the opposite side of the central β-sheet from the Cys/His/Asp/Glu cavity formed between the irregular layer and the β-sheet, and the site has no counterpart in the PAS domain. The cGMP site is as near the general region of the fold as the poly-l-proline- and actin-binding sites of profilin (Schutt et al., 1993; Thorn et al., 1997), but no precise alignment of the sites can be derived.
The model of the binding site strongly suggested Arg174 as a key ligand for the cyclic phosphate moiety. The correct prediction of a role for Arg174 in cGMP binding lends considerable confidence to the identification of the cGMP binding site. In PDE5-GAFb, which binds cGMP with much lower affinity than PDE5-GAFa, the position corresponding to Arg174 is a Lys. The mutational analysis shows that while the R174K mutation reduces binding measurably, this change alone cannot account for all of the difference in affinity between PDE5-GAFa and PDE5-GAFb. Indeed, the reverse mutation, K356R in PDE5-GAFb does not restore binding. This is not necessarily surprising, since there are several other sequence differences between PDE5-GAFa and PDE-GAFb in the α3–β1 junction and the β6–α5 loop, both in the vicinity of the binding site. Aside from the absolute conservation of the Asn, Lys and Asp surrounding the guanine base, and the usually conserved presence of either an Arg or Lys near the phosphate, no other PDE GAF domain residues are completely conserved. Aside from the PDEs, few GAF domains contain the full (R/K)XmNKXnD motif required for cGMP binding. However, two of the three adenylyl cyclases of Anabaena contain GAF domains that fit this pattern, suggesting that these enzymes could be regulated by cGMP.
The cGMP binding site structure (Figure 7B) is consistent with previous proposals for the mechanism of selectivity for cGMP over cAMP (McAllister-Lucas et al., 1995; Turko et al., 1996, 1999). Interactions between Asp289 and the guanine N1 and N2 offer at least a partial explanation for binding of cGMP in preference to cAMP (McAllister-Lucas et al., 1995; Turko et al., 1996, 1999). The binding site suggests a possible explanation for the effects of the H258N mutation in PDE6B, which causes congenital stationary night blindness in humans (Gal et al., 1994). In the PDE5-GAFa–cGMP complex model, the counterpart of this residue, Ile170, is located very near the cGMP binding site, making it a good candidate for a direct interaction with cGMP.
The cGMP complex model suggests a possible mechanism for ligand-induced conformational changes in PDE regulation. There is evidence for such allosteric regulation in several PDEs. cGMP binding allosterically stimulates hydrolysis of both cAMP and cGMP by the catalytic domain of PDE2 (Stroop and Beavo, 1991). cGMP binding to allosteric sites on PDE5 does not directly regulate the catalytic rate but it is required for the subsequent phosphorylation of Ser92 by PKA and PKG (Turko et al., 1998). In the case of PDE6, cGMP binding to the α and β catalytic subunits dramatically enhances their interaction with the regulatory γ subunit, and in turn, with transducin (D’Amours and Cote, 1999; Mou et al., 1999). The β6–α5 loop is predicted to be highly exposed and possibly disordered in the absence of cGMP. The loop is poised to form a lid over the cGMP upon binding (Figure 7B). Among a number of PDE GAF domains, this loop contains a Gly–Lys sequence that could interact with the phosphate group. Such an interaction seems likely because, in its absence, interactions with the Arg174 and Lys277 alone would leave the cyclic phosphate substantially exposed to solvent. A conformational change such as a disorder→ order transition in this loop could serve to transmit the cGMP binding signal to other domains or subunits of the enzyme. The β2–β3 loop also abuts the cGMP site, and could potentially change conformation upon cGMP binding. This offers one model for the allosteric effects of cGMP in PDE regulation.
Conclusion
The crystal structure of a GAF domain has revealed substantial new insights into the evolution and function of a very large class of biological signaling and sensory transducers. The GAF domain proteins are now clearly linked in their evolution to the equally ubiquitous PAS domain-containing class of signaling and sensory proteins. Indeed, there is considerable overlap between these two groups since a substantial number of proteins contain both PAS and GAF domains. The common theme among both classes is the binding, either covalent or non-covalent, of a remarkably diverse set of regulatory small molecules. The putative ligands for many of the PAS and GAF domains remain unidentified, suggesting that there still exists a wealth of yet-to-be-discovered small molecule signaling and sensory pathways.
The analysis of cGMP binding to the GAF domains of PDE5 revealed conclusively that these domains form a novel class of cyclic nucleotide receptors dissimilar to the ‘cNMP’ motif of CAP, PKA, PKG and the cyclic nucleotide-gated ion channels. A combination of theoretical and mutational analysis revealed determinants for specific recognition of cGMP. The sequence determinants for cGMP binding have been expanded, defining a (R/K)XmNKXnD motif. This motif is rarely found in GAF domains other than those of the phosphodiesterases, suggesting that the cGMP binding function of these GAF domains is specialized and evolutionarily recent.
Materials and methods
Protein expression and purification
The S.cerevisiae open reading frame coding for the hypothetical protein YKG9 was amplified by PCR from the DNA template (ORF YKL69W; Research Genetics) and subcloned into the NcoI and StuI sites of the pGEX-2T-parallel (Sheffield et al., 1999) modified to code for a rTEV protease cleavage site instead of a thrombin cleavage site. The GST fusion protein was expressed in BL21(DE3) cells (Novagen). Cells were grown to an OD600 of 0.6–0.7 and induced with 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG). After induction, the cells were grown for an additional 3–4 h and harvested. The harvested cells were resuspended in lysis buffer containing 100 mM NaCl, 50 mM Tris pH 8.0 and 2 mM DTT. The resuspended cells were subjected to three passes in a French press (750 p.s.i.). The lysate was centrifuged at 18 000 r.p.m. for 1 h. The supernatant was incubated with glutathione–Sepharose 4B (Pharmacia) matrix for 1 h. After incubation, the beads were washed with 10 column volumes of lysis buffer. The wash was followed by rTEV protease cleavage in lysis buffer for ≥4 h at room temperature. The proteolytic cleavage product was purified on a Superdex 200 gel filtration column (Pharmacia) equilibrated with 20 mM Tris pH 7.4 and 2 mM DTT on an FPLC system. The yield was ∼6.5 mg purified YKG9 protein from an 8 l culture.
PDE5-GAFa and PDE5-GAFb domains were subcloned from the full-length bovine PDE5A cDNA. Primer pairs containing homology to DNA coding for residues 132–335 and 147–321 were used in PCRs to generate longer and shorter versions, respectively, of PDE5-GAFa. Longer and shorter versions corresponded to residues 316–526 and 332–512 of PDE5-GAFb. Gel-purified PCR products were cleaved with EcoRI and SpeI and ligated into the same sites of pGEX-2T-parallel. Expression in BL21(DE3) cells and purification were the same as for YKG9.
cGMP binding assays
Binding of cyclic nucleotides was assayed using a modification of a previously described protocol (McAllister-Lucas et al., 1995). Briefly, 10 pmol of protein were incubated for 1 h at 4°C in the presence of 10 mM sodium phosphate buffer pH 7.0, 1 mM EDTA, 2.5 mM DTT and various concentrations (0.2–24 µM) of cGMP or cAMP (with trace [3H]cyclic nucleotide). Samples were applied to pre-moistened HAWP filters (0.45 µm, Millipore) and rinsed with 10 mM potassium phosphate buffer pH 6.8 containing 1 mM EDTA. Dry filters were counted for 3H using liquid (Biosafe II) scintillation counting. Background levels of nucleotide binding to filters were determined by excluding protein from the assay.
Site-directed mutagenesis
Mutants of PDE5-GAFa were generated using the Quik-Change site-directed mutagenesis kit (Stratagene) with the PDE5-GAFa-pGEX-2T wild-type construct serving as template. Primers for incorporating the mutations R174K and R174M were CTCATCTCCGCCGACAAA TACTCCTTATTCCTC and CTCATCTCCGCCGACATGTACTCC TTATTCCTC, respectively, along with their complements (mutated amino acids are in bold). The K356R mutant of PDE5-GAFb was constructed using the primer TTCATGCAGGTGCAGAGATG CACCATTTTCATA and its complement with the GAFb-pGEX-2T wild-type construct as template. Expression and purification were as described for YKG9 and PDE5 GAFs.
Protein crystallization
YKG9 protein was concentrated to 20 mg/ml in 50 mM Tris pH 8.0, 2 mM DTT, and used for crystallization. Crystals were obtained by the hanging drop diffusion method at room temperature using the Wizard II sparse matrix screen (Emerald Biostructures). Protein and well solution were mixed at a ratio of 1:1. Tetragonal crystals appeared among heavy precipitate after 48 h of vapor diffusion against a well solution of 0.1 M Tris–HCl pH 7.0, 0.2 M Li2SO4 and 2.0 M (NH4)2SO4. Crystallization conditions were then optimized and better crystals (0.8 × 0.3 × 0.3 mm) were obtained using 0.1 M Tris–HCl pH 7.8, 0.1 M Li2SO4 and 1.8 M (NH4)2SO4. For data collection at 100°K, crystals were transferred to a solution containing 2.5 M (NH4)2SO4, 0.1 M Tris–HCl pH 8.0, 0.05 M Li2SO4, 10% (v/v) glycerol and 30% (w/v) sucrose. Crystals were frozen in a stream of cold nitrogen gas at 100°K. A native crystal diffracted to 2.2 Å using a laboratory rotating anode (Rigaku RU-100) source X-rays and 1.9 Å at beamline X9B, NSLS, Brookhaven National Laboratory. The crystals belong to space group P43212 with a = b = 73.64 Å, c = 162.48 Å and two YKG9 molecules per asymmetric unit.
Crystallographic data collection, phasing and refinement
The phase problem was solved using MAD from non-covalently bound Br– ions (Dauter and Dauter, 1999; Dauter et al., 2000). Various concentrations of NaBr, between 0.5 and 1.0 M, and soaking times were tested before arriving at an appropriate combination. Increasing the NaBr concentration above 0.5 M or the soaking time above 45 s resulted in increased mosaicity of the crystal and reduced diffracting power. A single crystal was used for collecting data at three wavelengths near the Br K edge at beamline X9B at the NSLS. Data were collected from a single crystal mounted at an arbitrary orientation. To maximize the anomalous signal needed, inverse beam data were collected in 1° oscillations for 60° with an exposure of 30 s. Data were collected on an ADSC Quantum 4 CCD detector. All three data sets were >98% complete. The intensities were integrated and scaled using the HKL package (Otwinowski and Minor, 1997).
The positions of the bromide ions were independently located using SHELXS (Sheldrick, 1986) and Shake and Bake (SnB; Miller et al., 1994) using the anomalous differences with data from the peak wavelength where the maximum Δf′′ contribution was expected. SHELX found a total of 20 sites while SnB found 19 sites. The phasing power, figure of merit and RCullis were evaluated using the top 3, 7 and 19 sites from SnB, ranked by peak height. The calculations were carried out in space group P41212 with MLPHARE (Otwinowski and Minor, 1997), and the statistics were modestly superior for the seven-site solution as compared with the others. Phases for the seven-site solution were recalculated for the enantiomeric solution in P43212, and both phase sets calculated at 2.8 Å resolution were input into Solomon (Abrahams and Leslie, 1996) for density modification and phase extension to 2.6 Å. The P43212 phase set converged with a figure of merit of 0.76 and a correlation coefficient of 0.61, whereas the P41212 phase set failed to converge. The electron density map was skeletonized using the BONES option in MAPMAN and the model was built into the density with O (Jones et al., 1991). No non-crystallographic symmetry averaging was carried out since the map was completely interpretable without averaging. Refinement was carried out using CNS (Brünger et al., 1998) using torsional dynamics and the maximum likelihood target function. After each cycle of refinement, the Fo – Fc and 2Fo – Fc maps were generated for manual model building. Water molecules were automatically added using options available in CNS. Only those waters that were more than a specified distance cut-off (2.5 Å) from a neighboring atom and >2.5 σ in peak height were used.
Homology modeling
The multiple sequence alignment of GAF domains in the SMART database (Schultz et al., 1998; http://smart.embl-heidelberg.de/smart) was recalculated using the combined threading and sequence profile method of Bryant and co-workers (Panchenko et al., 1999, 2000). The modified alignment was used to generate a homology model of PDE5-GAFa using the program LOOK version 3.5.2 (C.Lee, T.Tversky and P.Gentry, Molecular Applications Group, http://www.bioinformatics.ucla.edu/genemine).
Coordinates
Coordinates have been deposited in the protein databank at the RCSB with accession codes 1F5M (YKG9 crystal structure) and 1FL4 (PDE5-GAFa homology model).
Acknowledgments
Acknowledgements
We thank Z.Dauter for advice on the Br– MAD technique and for assistance with data collection at beamline X9B, NSLS, Brookhaven National Laboratory, S.Bryant, A.R.Panchenko and A.Marchler-Bauer for the multiple threading alignment, J.Corbin for discussions and for providing the PDE5 cDNA, and A.Hickman for comments on the manuscript. Correspondence and requests for materials should be addressed to J.H.H. at jh8e@nih.gov.
References
- Abrahams J.P. and Leslie,A.G.W. (1996) Methods used in the structure determination of bovine F-1 ATPase. Acta Crystallogr. D, 52, 30–42. [DOI] [PubMed] [Google Scholar]
- Altenhofen W., Ludwig,J., Eismann,E., Kraus,W., Bonigk,W. and Kaupp,U.W. (1991) Control of ligand specificity in cyclic nucleotide-gated channels from rod photoreceptors and olfactory epithelium. Proc. Natl Acad. Sci. USA, 88, 9868–9872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aravind L. and Ponting,C.P. (1997) The GAF domain: an evolutionary link between diverse phototransducing proteins. Trends Biochem. Sci., 22, 458–459. [DOI] [PubMed] [Google Scholar]
- Bateman A., Birney,E., Durbin,R., Eddy,S.R., Finn,R.D. and Sonnhammer,E.L. (1999) Pfam 3.1: 1313 multiple alignments match the majority of proteins. Nucleic Acids Res., 27, 260–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baylor D. (1996) How photons start vision. Proc. Natl Acad. Sci. USA, 93, 560–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borgstahl G.E.O., Williams,D.R. and Getzoff,E.D. (1995) 1.4 Å structure of photoactive yellow protein, a cytosolic photoreceptor: unusual fold, active site and chromophore. Biochemistry, 34, 6278–6287. [DOI] [PubMed] [Google Scholar]
- Brünger A.T. et al. (1998) Crystallography and NMR system (CNS): a new software system for macromolecular structure determination. Acta Crystallogr. D, 54, 905–921. [DOI] [PubMed] [Google Scholar]
- Cabral J.H.M., Lee,A., Cohen,S.L., Chait,B.T., Li,M. and MacKinnon,R. (1998) Crystal structure and functional analysis of the HERG potassium channel N terminus: A eukaryotic PAS domain. Cell, 95, 649–655. [DOI] [PubMed] [Google Scholar]
- Charbonneau H., Prusti,R.K., Letron,H., Sonnenburg,W.K., Mullaney,P.J., Walsh,K.A. and Beavo,J.A. (1990) Identification of a noncatalytic cGMP-binding domain in both the cGMP-stimulated and photoreceptor cyclic-nucleotide phosphodiesterases. Proc. Natl Acad. Sci. USA, 87, 288–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbin J.D. and Francis,S.H. (1999) Cyclic GMP phosphodiesterase-5: target of sildenafil. J. Biol. Chem., 274, 13729–13732. [DOI] [PubMed] [Google Scholar]
- D’Amours M.R. and Cote,R.H. (1999) Regulation of photoreceptor phosphodiesterase catalysis by its non-catalytic cGMP-binding sites. Biochem. J., 340, 863–869. [PMC free article] [PubMed] [Google Scholar]
- Dauter Z. and Dauter,M. (1999) Anomalous signal of solvent bromides used for phasing of lysozyme. J. Mol. Biol., 289, 93–101. [DOI] [PubMed] [Google Scholar]
- Dauter Z., Dauter,M. and Rajashankar,K.R. (2000) Novel approach to phasing proteins: derivatization by short cryo-soaking with halides. Acta Crystallogr. D, 56, 232–237. [DOI] [PubMed] [Google Scholar]
- de Rooij J., Zwartkruis,F.J.T., Verheijen,M.H.G., Cool,R.H., Nijman,S.M.B., Wittinghofer,A. and Bos,J.L. (1998) Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature, 396, 474–477. [DOI] [PubMed] [Google Scholar]
- Fawcett L., Baxendale,R., Stacey,P., McGrouther,C., Harrow,I., Soderling,S., Hetman,J., Beavo,J.A. and Phillips,S.C. (2000) Molecular cloning and characterization of a distinct human phosphodiesterase gene family: PDE11A. Proc. Natl Acad. Sci. USA, 97, 3702–3707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gal A., Orth,U., Baehr,W., Schwinger,E. and Rosenberg,T. (1994) Heterozygous missense mutation in the rod cGMP phosphodiesterase β-subunit gene in autosomal dominant stationary night blindness. Nature Genet., 7, 64–67. [DOI] [PubMed] [Google Scholar]
- Garbers D.L. and Dubois,S.K. (1999) The molecular basis of hypertension. Annu. Rev. Biochem., 68, 127–155. [DOI] [PubMed] [Google Scholar]
- Genick U.K., Soltis,S.M., Kuhn,P., Canestrelli,I.L. and Getzoff,E.D. (1998) Structure at 0.85 Å resolution of an early protein photocycle intermediate. Nature, 392, 206–209. [DOI] [PubMed] [Google Scholar]
- Gibrat J.-F., Madej,T. and Bryant,S.H. (1996) Surprising similarities in structure comparison. Curr. Opin. Struct. Biol., 6, 377–385. [DOI] [PubMed] [Google Scholar]
- Gong W., Hao,B., Mansy,S.S., Gonzalez,G., Gilles-Gonzales,M.A. and Chan,M.K. (1998) Structure of a biological oxygen sensor: A new mechanism for heme-driven signal transduction. Proc. Natl Acad. Sci. USA, 95, 15177–15182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granovsky A.E., Natochin,M., McEntaffer,R.L., Haik,T.L., Francis,S.H., Corbin,J.D. and Artemyev,N.O. (1998) Probing domain functions of chimeric PDE6α′/PDE5 cGMP-phosphodiesterase. J. Biol. Chem., 273, 24485–24490. [DOI] [PubMed] [Google Scholar]
- Hobbs A.J. and Ignarro,L.J. (1996) Nitric oxide cyclic GMP signal transduction system. Methods Enzymol., 269, 134–148. [DOI] [PubMed] [Google Scholar]
- Holm L. and Sander,C. (1995) DALI—a network tool for protein structure comparison. Trends Biochem. Sci., 20, 478–480. [DOI] [PubMed] [Google Scholar]
- Jones T.A., Zou,J.Y., Cowan,S.W. and Kjeldgaard,M. (1991) Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A, 47, 110–119. [DOI] [PubMed] [Google Scholar]
- Kehoe D.M. and Grossman,A.R. (1996) Similarity of a chromatic adaptation sensor to phytochrome and ethylene receptors. Science, 273, 1409–1412. [DOI] [PubMed] [Google Scholar]
- Kumar V.D. and Weber,I.T. (1992) Molecular model of the cyclic GMP-binding domain of the cyclic GMP-gated ion channel. Biochemistry, 31, 4643–4649. [DOI] [PubMed] [Google Scholar]
- Laskowski R.A. (1995) SURFNET: a program for visualizing molecular surfaces, cavities and intermolecular interactions. J. Mol. Graph., 13, 323–330. [DOI] [PubMed] [Google Scholar]
- McAllister-Lucas L.M., Haik,T.L., Colbran,J.L., Sonnenburg,W.K., Seger,D., Turko,I.V., Beavo,J.A., Frances,S.H. and Corbin,J.D. (1995) An essential aspartic acid at each of two allosteric cGMP-binding sites of a cGMP-specific phosphodiesterase. J. Biol. Chem., 270, 30671–30679. [DOI] [PubMed] [Google Scholar]
- McKay D.B. and Steitz,T.A. (1981) Structure of catabolite gene activator protein at 2.9 Å resolution suggests binding to left-handed B-DNA. Nature, 290, 744–749. [DOI] [PubMed] [Google Scholar]
- Miller R., Gallo,S.M., Khalak,H.G. and Weeks,C.M. (1994) SnB—crystal structure determination via Shake-and-Bake. J. Appl. Crystallogr., 27, 613–621. [Google Scholar]
- Moncada S. and Higgs,E.A. (1995) Molecular mechanisms and therapeutic strategies related to nitric oxide. FASEB J., 9, 1319–1330. [PubMed] [Google Scholar]
- Mou H., Grazio,H.J.,III, Cook,T.A., Beavo,J.A. and Cote,R.H. (1999) cGMP binding to noncatalytic sites on mammalian rod photoreceptor phosphodiesterase is regulated by binding of its γ and δ subunits. J. Biol. Chem., 274, 18813–18820. [DOI] [PubMed] [Google Scholar]
- Murad F. (1996) Signal transduction using nitric oxide and cyclic guanosine monophosphate. J. Am. Med. Assoc., 276, 1189–1192. [PubMed] [Google Scholar]
- Otwinowski Z. and Minor,W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol., 276, 307–326. [DOI] [PubMed] [Google Scholar]
- Panchenko A., Marchler-Bauer,A. and Bryant,S.H. (1999) Threading with explicit models for evolutionary conservation of structure and sequence. Proteins Suppl., 3, 133–140. [DOI] [PubMed] [Google Scholar]
- Panchenko A.R., Marchler-Bauer,A. and Bryant,S.H. (2000) Combina tion of threading potentials and sequence profiles improves fold recognition. J. Mol. Biol., 296, 1319–1331. [DOI] [PubMed] [Google Scholar]
- Pellequer J.L., Wager-Smith,K.A., Kay,S.A. and Getzoff,E.D. (1998) Photoactive yellow protein: A structural prototype for the three-dimensional fold of the PAS domain superfamily. Proc. Natl Acad. Sci. USA, 95, 5884–5890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponting C.P. and Aravind,L. (1997) PAS: a multifunctional domain family comes to light. Curr. Biol., 7, R674–R677. [DOI] [PubMed] [Google Scholar]
- Quail P.H., Boylan,M.T., Parks,B.M., Short,T.W., Xu,Y. and Wagner,D. (1995) Phytochromes: Photosensory perception and signal transduction. Science, 268, 675–680. [DOI] [PubMed] [Google Scholar]
- Reppert S.M. (1998) A clockwork explosion! Neuron, 21, 1–4. [DOI] [PubMed] [Google Scholar]
- Sassone-Corsi P. (1998) Molecular clocks: mastering time by gene regulation. Nature, 392, 871–874. [DOI] [PubMed] [Google Scholar]
- Schmitz O., Katayama,M., Williams,S.B., Kondo,T. and Golden,S.S. (2000) CikA, a bacteriophytochrome that resets the cyanobacterial circadian clock. Science, 289, 765–768. [DOI] [PubMed] [Google Scholar]
- Schultz J., Milpetz,F., Bork,P. and Ponting,C.P. (1998) SMART, a simple modular architecture research tool: Identification of signaling domains. Proc. Natl Acad. Sci. USA, 95, 5857–5864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schutt C.E., Myslik,J.C., Roxycki,M.D., Goonesekere,N.C.W. and Lindberg,U. (1993) The structure of crystalline profilin-β-actin. Nature, 365, 810–816. [DOI] [PubMed] [Google Scholar]
- Shabb J.B., Ng,L. and Corbin,J.D. (1990) One amino acid change produces a high-affinity cGMP binding site in cAMP-dependent protein kinase. J. Biol. Chem., 265, 16031–16034. [PubMed] [Google Scholar]
- Sheffield P., Garrard,S. and Derewenda,Z. (1999) Overcoming expression and purification problems of RhoGDI using a family of parallel expression vectors. Protein Expr. Purif., 15, 34–39. [DOI] [PubMed] [Google Scholar]
- Sheldrick G.M. (1986) SHELXS86—program for crystal structure solution. University of Gottingen, Gottingen, Germany.
- Soderling S.H. and Beavo,J.A. (2000) Regulation of cAMP and cGMP signaling: new phosphodiesterases and new functions. Curr. Opin. Cell Biol., 12, 174–179. [DOI] [PubMed] [Google Scholar]
- Soderling S.H., Bayuga,S.J. and Beavo,J.A. (1998) Cloning and characterization of a cAMP-specific cyclic nucleotide phospho diesterase. Proc. Natl Acad. Sci. USA, 95, 8991–8996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soderling S.H., Bayuga,S.J. and Beavo,J.A. (1999) Isolation and characterization of a dual-substrate phosphodiesterase gene family: PDE10A. Proc. Natl Acad. Sci. USA, 96, 7071–7076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stroop S.D. and Beavo,J.A. (1991) Structure and function studies of the cGMP-stimulated phosphodiesterase. J. Biol. Chem., 266, 23802–23809. [PubMed] [Google Scholar]
- Stryer L. (1991) Visual excitation and recovery. J. Biol. Chem., 266, 10711–10714. [PubMed] [Google Scholar]
- Su Y., Dostmann,W.R.G., Herberg,F.W., Durick,K., Xuong,N.H., Ten-Eyck,L. Taylor,S.S. and Varughese,K.I. (1995) Regulatory subunit of protein kinase-A—structure of a deletion mutant with cAMP binding domains. Science, 269, 807–813. [DOI] [PubMed] [Google Scholar]
- Thomas M.K., Francis,S.H. and Corbin,J.D. (1990) Characterization of a purified bovine lung cGMP-binding cGMP phosphodiesterase. J. Biol. Chem., 265, 14964–14970. [PubMed] [Google Scholar]
- Thorn K.S., Christensen,H.E.M., Shigeta,R., Huddler,D., Shalaby,L., Lindberg,U., Chua,N.H. and Schutt,C.E. (1997) The crystal structure of a major allergen from plants. Structure, 5, 19–32. [DOI] [PubMed] [Google Scholar]
- Turko I.V., Haik,T.L., McAllister-Lucas,L.M., Burns,F., Francis,S.H. and Corbin,J.D. (1996) Identification of key amino acids in a conserved cGMP-binding site of cGMP-binding phosphodiester ases—a putative NKX(n)D motif for cGMP binding. J. Biol. Chem., 271, 22240–22244. [DOI] [PubMed] [Google Scholar]
- Turko I.V., Francis,S.H. and Corbin,J.D. (1998) Binding of cGMP to both allosteric sites of cGMP-binding cGMP-specific phospho diesterase (PDE5) is required for its phosphorylation. Biochem. J., 329, 505–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turko I.V., Francis,S.H. and Corbin,J.D. (1999) Studies of the molecular mechanism of discrimination between cGMP and cAMP in the allosteric sites of the cGMP-binding cGMP-specific phosphodiesterase (PDE5). J. Biol. Chem., 274, 29038–29041. [DOI] [PubMed] [Google Scholar]
- Weber I.T., Shabb,J.B. and Corbin,J.D. (1989) Predicted structures of the cGMP binding domains of the cGMP-dependent protein kinase—a key alanine threonine difference in evolutionary divergence of cAMP and cGMP binding sites. Biochemistry, 28, 6122–6127. [DOI] [PubMed] [Google Scholar]