

Abstract
This work presents the initial characterization of transgenic mice with mutations in a primary zinc binding residue (H80), either alone or with a G93A mutation. H80G;G93A SOD1 transgenic mice developed paralysis with motor neuron loss, and ubquitin inclusion-type rather than mitochondrial vacuolar pathology. Unlike G93A SOD1 related disease, the course was not accelerated by over-expression of CCS. H80G SOD1 transgenic mice did not manifest disease at levels of SOD1 transgene expressed.
The H80G mutation altered certain biochemical parameters of both human wild type SOD1 and G93A SOD1. The H80G mutation does not substantially change the age dependent accumulation of G93A SOD1 aggregates and hydrophobic species in spinal cord. However, both H80G;G93A SOD1 and H80G SOD1 lack dismutase activity, the ability to form homodimers, and cooperativity with CCS, indicating that their dimerization interface is abnormal. The H80G mutation also made SOD1 susceptible to protease digestion.
The H80G mutation alters the redox properties of SOD1. G93A SOD1 exists in either reduced or oxidized form, whereas H80G;G93A SOD1 and H80G SOD1 exist only in a reduced state. The inability of SOD1 with an H80G mutation to take part in normal oxidation-reduction reactions has important ramifications for disease mechanisms and pathology in vivo.
Keywords: Amyotrophic lateral sclerosis, motor neuron, Superoxide dismutase, copper, mitochondria, inclusions
Mutant Cu, Zu superoxide dismutase (SOD1) causes one form of familial amyotrophic lateral sclerosis (fALS) by a toxic gain that may involve mitochondrial dysfunction. Transgenic mice expressing G37R or G93A SOD1 develop a progressive motor phenotype that is characterized pathologically by mitochondrial vacuolization (Dal Canto & Gurney 1994, Wong et al. 1995). Both wild type (WT) and fALS linked mutant SOD1s can be found in mitochondrial fractions isolated from spinal cord, suggesting a direct effect of SOD1 on mitochondrial function (Liu et al. 2004, Vijayvergiya et al. 2005, Higgins et al. 2002, Deng et al. 2006). In support of this hypothesis, over-expression of copper chaperone for SOD1 (CCS) in G93A or G37R SOD1 transgenic mice increases the mitochondrial localization of mutant SOD1 and greatly accelerates disease (Son et al. 2007, Son et al. 2009). This accelerated disease state is characterized by a marked enhancement of mitochondrial vacuolar pathology and by a selective deficiency of mitochondrial cytochrome c oxidase (COX) (Son et al. 2008, Son et al. 2009). These pathological and biochemical alterations provide not only strong evidence that G93A and G37R and SOD1 directly impact mitochondrial function, but also insight into underlying mechanisms.
The redox state of SOD1 is potentially a critical aspectof its mitochondrial toxicity. WT SOD1 can exist in either a reduced or oxidized form depending on the isomerization status of residues C57 and C146, although it is the oxidized form that is mature and enzymatically active. CCS plays a crucialrole in the maturation of SOD1 not only as a strict copper chaperone, but also as a facilitator of the oxidized form of wild type SOD1 in vivo (Culotta et al. 1997, Casareno et al. 1998, Schmidt et al. 2000, Furukawa & O’Halloran 2006, Seetharaman et al. 2009). Expression of CCS in wild type SOD1 transgenic mice results in a marked change to where virtually all SOD1 monomers exist in the oxidized form (Proescher et al. 2008). In contrast, over-expression of CCS in G93A or G37R SOD1 transgenic mice greatly favors the reduced SOD1 state (Proescher et al. 2008, Son et al. 2009). However, transgenic mice expressing SOD1 disease causing mutants, G86R or L126Z, which exist only in a reduced state, do not show mitochondrial vacuolar pathology nor can they be accelerated by the over-expression of CCS protein. These observations suggest a close correlation between SOD1 redox states, the ability to be accelerated by CCS, and mitochondrial pathology. To further test this hypothesis, we sought to generate transgenic mice over-expressing a SOD1 protein harboring a G93A mutation that would exist only in a reduced state, predicting that such mice might develop disease without mitochondrial pathology or susceptibilityto be accelerated by CCS.
There is little information available on the effect of primary zinc binding site SOD1 mutations in vivo. Only one case report exists of an individual developing sporadic ALS (no family history) and found to have a zinc binding site H80R SOD1 mutation (Alexander et al. 2002). However, whether this mutation is disease causing or a rare polymorphism is unclear. The lack of zinc binding site mutations associated with disease contrasts strongly to the pattern of multiple mutations observed in disease for other functional domains of SOD1, including the copper binding site. However, there have been familial SOD1 mutations in exon 5 at codon 124 which serve to alter the zinc electrostatic loop and disrupt zinc binding (Hosler et al. 1996). Human H80R and D124V SOD1 mutations have been expressed in yeast or inmouse motor neuron cell lines (Ferri et al. 2006, Seetharaman et al. 2010). These studies indicated that mutant SOD1 proteins are in reduced form and primarily monomeric with a weak affinity to the SOD1-like domain (domain II) of CCS. Transgenic mice expressing a glycine mutation in the joint copper/zinc binding residue (H63) in the context of other copper binding site mutations (Quad null) develop motor neuron disease, but the in vivo effects of a selective zinc binding site mutation remain unclear (Wang et al. 2003). We hypothesized that zinc binding site mutations might disrupt disulfide bond formation without mutating the copper binding residues or cysteine residues and consequently generated transgenic mice expressing H80G SOD1 or H80G;G93A SOD1.
Experimental procedures
Generation and characterization of H80GSOD1 and H80G; G93A SOD1 transgenic mice
The mutant SOD1 constructs were cloned into mouse prion promoter (Wang et al. 2005, Krishnan et al. 2006, Borchelt et al. 1996). The human H80G SOD1 cDNA or human H80G;G93A SOD1 cDNA were cloned into MoPrP Xho vector (12 kb) at the unique XhoI site between exons 2 and 3 (JHU-2; Johns Hopkins Special Collection distributed by American Type Culture Collection.) H80GSOD1 or H80G;G93ASOD1 transgenes were detected by PCR analysis on tail DNA using the following primers: sense primer 5′-GGC CCA GTG CAG GGC ATC ATC A-3′ and an antisense primer 5′-CAC ATC GGC CAC ACC ATC TTT G-3′. The expression levels of SOD1 transgenes in the positive lines were analyzed by Western Blotting on spinal cord extracts of four months old mice. The lines with highest expression levels were chosen for further analysis.
Transgenic mice expressing the low copy number human G93A SOD1 mutation (B6SJL-TgNSOD1-G93A SOD1; 1Gurdl JR2300) were originally obtained from The Jackson Laboratory (Bar Harbor, ME). This line develops motor deficits at about 6 months and has a mean survival of about 240 days (Puttaparthi et al. 2002). Transgenic mice expressing wild type (WT) human SOD1 (line N29, B6SJL-WT-SOD1) were also obtained from The Jackson Laboratory. Previously we have described the generation and characterization of CCS transgenic line 17 mice. When line 17 CCS transgenic mice were crossed with G93A SOD1 mice, the CCS/G93A SOD1 dual transgenic mice develop accelerated neurological deficits, with a mean survival of 36 days(Son et al. 2007). In the present experiments, line 17 CCS transgenic mice were crossed to either H80G SOD1 or H80G;G93A SOD1 mice to obtain CCS/H80G SOD1 or CCS/H80G;G93A SOD1 dual transgenic mice.
All animal protocols were approved by our University’s Institutional Animal Care and Research Advisory Committee (University of Texas, Southwestern Medical Center, Dallas, TX) in compliance with National Institutes of Health guidelines.
Survival analysis
Animals unable to correct posture when placed on side were considered end stage and sacrificed. Survival analysis was performed using the Kaplan-Meier method.
Histology and Immunochemistry
Mice were overdosed with pentobarbital (250 mg/kg, i. p.) and perfused transcardially with PBS, followed by 4% paraformaldehyde in PBS. Spinal cords were dissected, post-fixed in 4% PFA, embedded in paraffin, sectioned, stained and viewed as previously described (Puttaparthi & Elliott 2005). In brief, sections of lumbar 5–6 spinal cord from paraffin-embedded tissue were stained with hematoxylin and eosin stain (H&E). For immuno-fluorescence, spinal cord sections were pretreated for 2hourswith 5% normal goat serum and 0.1% Triton X-100 and then incubated overnight at +4 °C with primary antibodies: mouse anti-COX1 subunit clone ID6E1A, 40 μg/ml (Molecular probes, Invitrogen) or rabbit anti-ubiquitin, 0.8μg/ml (Dako) or with rabbit anti-GFAP, 2.9μg/ml (Dako). The next day, the sections were washed with PBS and incubated for one hour at room temperature with secondary antibody: goat anti-mouse IgG labeled with Alexa Flour 488, 10 μg/ml, or with goat anti-rabbit IgG labeled with Alexa Fluor 555, 10 μg/ml (Molecular Probes, Invitrogen). Next, they were incubated for one minute with DAPI, 1/1000 dilution, (Molecular Probes, Invitrogen). The sections were washed and mounted with Gel/mount (Biomed Corp., Foster City, CA). Slides were viewed with an E800 fluorescent microscope (Nikon Instruments Inc) acquired by a cooled CCD camera (Roper Scientific) using Metamorph software.
Western blot analysis
Anesthetized mice were perfused transcardially with PBS. For Western blots, the dissected spinal cords were homogenized in 20 mM Tris- HCl, pH 7.5, 1 mM EDTA, 0.5% TritonX-100 (Sigma) with protease inhibitors. For western blot analysis homogenates or enriched mitochondrial fractions were derived from spinal cords of paralyzed 8 month G93A SOD1, paralyzed 1 month CCS/G3A SOD1, paralyzed 17–18 month H80G;G93A SOD1, paralyzed 17–18 month CCS/H80G;G93A SOD1, 18–20 month H80G SOD1, 18–20 month CCS/H80G SOD1, 7–8 month WT SOD1 transgenic mice or from 7–8 month non-transgenic mice. Mitochondria were isolated from dissected spinal cords using a mitochondrial isolation kit as suggested by the manufacturer (Sigma). The protein concentrations were determined by BCA protein assay (Thermo Scientific). Samples were resuspended in Laemmli sample buffer with β-mercaptoethanol, boiled and separated on Tris-glycine SDS gels and then transferred to polyvinylidene difluoride membranes (PVDF). The experiments were performed using the following primary antibodies: sheep anti-SOD1, 1/1600 dilution (Calbiochem, EMD Biosciences, Inc.), rabbit anti-SOD1, 0.2 μg/ml (Stressgen Biotechnologies, Inc.), rabbit anti-CCS, 0.1 μg/ml (FL-274 Santa Cruz, Biotechnology, Inc.), mouse anti-COX1 subunit, 1 μg/ml (clone 1D6) (Molecular Probes, Inc.; Invitrogen), mouse anti-Tim23, 1/2500 dilution (BD Biosciences), and rabbit anti-actin, 1 μg/ml (Sigma). Secondary antibodies were Alkaline Phosphatase conjugated polyclonal antibodies,0.08 μg/ml (Santa Cruz Biotechnology, Inc). The immuno-reactive signals were detected by Lumi-Phos WB substrate (Thermo Scientific).
Partially denaturing PAGE for the detection of dimers was modified from a published procedure (Tiwari et al. 2005). Spinal cords were homogenized in 20 mM Tris, pH 7.5, 0.5% Triton X-100with or without 1mM EDTA. The homogenates were briefly sonicated and spun at 17000xg for 20 minutes at +4C°. Supernatants were resuspended in sample loading buffer containing 0.2% SDS without any reducing agent, and instead of boiling, incubated at room temperature for 30 minutes. These partially denatured samples were separated on 18% Tris-Glycine SDS gels under non-reducing conditions. Control samples were separated on 4–20% Tris-Glycine-SDS gels under standard denaturing, reducing conditions. Prior to transfer onto PVDF membranes the proteins were subjected to in-gel reduction by incubating two times for 10 minutes each in transfer buffer containing 2% β-mercaptoethanol to increase antibody recognition of SOD1.
Dismutase activity analysis
Dismutase activity gels were performed as previously described with some modifications (Culotta et al. 1997, Krishnan et al. 2006). Mouse spinal cords were homogenized in 20 mM Tris, pH 7.5, and 0.5% Triton X-100, with or without 1mM EDTA, and without protease inhibitors. For dismutase activity analysis, 60 μg of total protein from spinal cord homogenates were resolved on 4–20% Tris-Glycine gels under non-denaturing, non-reducingconditions at 135 volts for 3.5 hours. Followingthe electrophoresis, gels were stained in a solution containing 50 mM potassium phosphate (pH 7.8), 300 μg/ml Nitro Blue Tetrazolium (NBT) (Sigma), 50 μg/ml riboflavin (Sigma) and 3.2 μl/ml TEMED (Sigma). After 45 minutesincubation in the dark, the blue NBT stain was developed by exposure to light for 5–10 minutes. Achromatic bands are regions of dismutase activity against a colored background.
Proteinase K treatment
Spinal cords were homogenized in PBS without protease inhibitors or detergents. The homogenates were briefly sonicated and spun at 17000xg for 30 minutes at +4C°. The supernatants were used for proteinase K digestions. The spinal cord samples derived from G93A SOD1 mice, were diluted 1/10 with spinal cord samples from non-transgenic mice in order to keep the SOD1 levels as well as the total protein levels comparable; 3μg of G93A SOD1 sample, mixed with 27 μg of non-transgenic sample, 30μg H80G;G93A SOD1 sample or 30μg of non-transgenic sample was digested with 100μg of proteinase K (Invitrogen) in 40μl total volume for 30 minutes at +37C° with constant gentle mixing. After digestion, proteinase K was inactivated by 5 mM PMSF (phenylmethylsulfonyl fluoride). Samples were separated under standard denaturing, reducing conditions on 4–20% Tris-Glycine SDS gels.
Hydrophobic interaction chromatography
Hydrophobic Interaction Chromatography (HIC) was performed as previously described with slight modifications (Zetterstrom et al. 2007). Mouse spinal cord samples were homogenized in 2ml of ice-cold phosphate buffer (10 mM potassium phosphate, pH 7.0, 0.15 M NaCl) containing EDTA-free protease inhibitors (Roche)and 20mM iodoacetamide (BIORAD) in glass-teflon homogenizer, sonicated with Sonic dismembrator (Fisher Scientific) briefly, and centrifuged at 17,000×g for 20 minutes at +4°C. The supernatants were subjected to HIC. The spinal cord samples derived from G93A SOD1 mice, were diluted 1/10 with spinal cord samples from non-transgenic mice in order to keep the SOD1 levels as well as the total protein levels comparable;25μg sample of G93A SOD1, mixed with 225μg of non-transgenic sample, 250 μg of H80G;G93A SOD1 or 250μg of H80G SOD1 sample was loaded on hydrophobic columns. One milliliter of Octyl-Sepharose CL-4B (Sigma) was packed in 0.8 × 10-cm columns and equilibrated withphosphate buffer. The columns were run stepwise at room temperature with gravity. 250μl sample was added, followed by 250 μl of phosphate buffer. The eluants were added back to the columns and incubated for 20 minutes. The columns were elutedtwice with 3 ml of phosphate buffer and after that twice with 3 ml of phosphate buffer diluted 1:10 with water. Finally, the columns were eluted with 4% SDS in 50 mM Tris HCl, pH 6.8. 0.25 ml fractions were collected and frozen at −80C°. Buffers contained 5mM freshly added iodoacetamide. The eluants and control samples were separated under standard, denaturing, reducing conditionson 10–20%Tris-Glycine SDS gels.
SOD1 disulfide analysis by iodoacetamide gel electrophoresis
SOD1 disulfide analysis was performed as previously described (Proescher et al. 2008). In brief, anesthetized mice were perfused transcardially with PBS containing 100mM iodoacetamide. Tissues were homogenized in buffer containing 50 mM Hepes, 2.5% SDS, 0.1 mM EDTA (pH 7.2) with 100 mM iodoacetamide, and homogenates were incubated at +37 °C for 30 minutes with gentle mixing. The protein concentrations were determined by BCA protein assay (Thermo Scientific). The homogenates were treated with Laemmli sample buffer without β-mercaptoethanol and separated on 15% Tris-Glycine SDS gels under non-reducing conditions. Prior to transfer onto PVDF membranes, the proteins were subjected to in-gel reduction by incubating two times for 10 min each in transfer buffer containing 2% β-mercaptoethanol to increase antibody recognition of SOD1.
Results
Characterization of mouse lines human H80G SOD1 or H80G;G93ASOD1
We selected the prion promoter to drive expression of the human SOD1 cDNAs, because prion promoter has been shown to express SOD1 transgenes within the CNS sufficiently high to produce a progressive motor phenotype (Wang et al. 2005). For the H80G SOD1 construct, six founding lines expressing higher levels of mutant SOD1 within spinal cord were established (Fig. 1a). H80G SOD1 transgenic mice were viable and fertile. These mice never exhibited abnormal motor reflexes or gait. Survival was not affected by expression of H80G SOD1 with these mice having a normal lifespan (>27 months). On pathological examination, the spinal cords of H80G SOD1 mice did not show a loss of motor neurons in the ventral horn (Fig. 1b and c). The degree of astrogliosis within spinal cord was comparable between aged H80G SOD1 mice and non-transgenic age matched controlsas detected byGFAP immuno-staining (Fig. 1d, e, f and g). Older H80G SOD1 mouse spinal cords demonstrate a normal pattern of ubiquitin immuno-reactivity comparable to age matched non-transgenic controls (Fig. 1d and e). Motor neurons in the spinal cords of H80G SOD1 mice did not exhibit ubiquitin positive cytoplasmic inclusions. Both H80G SOD1 mice and non-transgenic controls showed similar pattern and intensity of immuno-reactivity for the mitochondrially encoded subunit COX1 of complex IV within spinal cords (Fig. 1f and g). H80G SOD1 transgenic mice do not manifest a motor phenotype, spinal cord pathology or decreased survival. This indicates a lack of toxicity associated with this particular SOD1 zinc binding site mutation at the expression levels generated by our prion promoter construct.
Fig. 1.

Characterization of H80G SOD1 transgenic mice. (a) Western Blot analysis of the of SOD1 levels in spinal cord extracts of six original lines of four month old H80GSOD1 transgenic mice or non-transgenic (NTG) mouse. Line S was chosen for further analysis. (b and c) H & E stained sections from lumbar 5–6 ventral horn spinal cord of 23 month H80G SOD1 mouse and age-matched NTG mouse. (d-g) Immuno-staining of spinal cords of (e and g) the H80G SOD1 mouse or (d and f) age-matched NTG control; (d and e) stained for ubiquitin (red) and glial fibrillary acidic protein (GFAP, green); or (f and g) for complex IV subunit I (COX1, green) and GFAP (red). Magnification =200x(b-e); 400x (f and g).
For the H80G;G93A SOD1 construct, 12 founders were used for establishing lines. Western blot analysis showed that line H expressed the highest levels of H80G;G93A SOD1, and this line was used for further analysis (Fig. 2a). Transgenic mice expressing H80G;G93A SOD1 were also viable and fertile. Early development and motor function was normal. However, H80G;G93A SOD1 mice manifested a progressive motor phenotype characterized initially by hind limb paralysis. Median survival was 513 ± 14 days (Fig. 3). The ventral horn of weak H80G;G93A SOD1 mice showed marked neuronal loss and astrogliosis compared to wild type controls and equivalent to weak G93A SOD1 mice (Fig. 2b, c, d, g, j, and Fig. 1b). Pathological examination of spinal cord section showed prominent inclusion type pathology with numerous cytoplasmic ubiquitin inclusions which were not observed in control non-transgenic mice but are seen in weak G93A SOD1 mice (Fig. 2e, h, k). Vacuolar mitochondrial pathology was not observed, and no changes in COX1 subunit immuno-reactivity were noted in spinal motor neurons compared to controls (Fig. 2f, i, l). Thus, over-expression of H80G;G93A SOD1 in vivo leads to a progressive motor phenotype with decreased survival and an inclusion pathology, rather than a mitochondrial vacuolar pathology.
Fig. 2.

Characterization of H80G;G93A SOD1 SOD1 transgenic mice. (a) Western Blot analysis of SOD1 levels in spinal cord extracts of twelve lines of four month H80G;G93A SOD1 transgenic or NTG mice. (b and c) H & E stained section from lumbar 5–6 ventral horn spinal cord sections of (b) 14 month weak H80G;G93A SOD1 and (c) 8 month old weak G93A SOD1 mouse. (d-l) Immuno-staining of spinal cords (g, h, i) from H80G;G93A SOD1 mice,(d, e, f) age-matched NTG controls or (j, k, l) G93A SOD1 mice; (d, g, j) stained for GFAP( red), (e, h, k) stained for ubiquitin (red), and (f, i, l)stained for COX1 (green). Magnification=200x (b-e; g, h, j, k); 400x (f, i, l).
Fig. 3.

Over-expression of CCS does not affect survival of H80G or H80G;G93A SOD1 mice. Kaplan–Meier survival curve of H80G;G93A SOD1 transgenic mice (n=26), CCS/H80G;G93A SOD1 dual transgenic mice (n=27) and CCS/H80G SOD1 dual transgenic mice (n=10). The survival of H80G;G93A SOD1 mice does not significantly differ from the survival of CCS/H80G;G93A SOD1 mice. H80G SOD1 miceandCCS/H80G SOD1 mice have normal lifespan. Historical survival curves for G93A SOD1 and CCS/G93A SOD1 mice are also shown.
CCS over-expression does not accelerate disease in H80G;G93A SOD1 mice
Because over-expression of CCS protein in G93ASOD1 mice results in a nearly ten-fold acceleration of disease severity with marked enhancement of mitochondrial toxicity, we wished to determine whether H80G;G93A SOD1 mice would also show disease acceleration in the setting of CCS over-expression (Son et al. 2007). We therefore crossed our CCS high expressing mouse line with H80G;G93A SOD1 transgenic mice to obtain offspring that would express H80G;G93A SOD1 alone or with CCS over-expression. The survival curves for these genotypes are shown in Fig. 3. The median survival for H80G;G93A SOD1 mice over-expressing CCS is 505 days, which is not statistically different form the median survival seen in H80G;G93A SOD1 mice. The pathology in H80G;G93A SOD1 mice over-expressing CCS also appeared unchanged with ubiquitin inclusion type pathology, rather than mitochondrial vacuolation. Because isolated COX deficiency is a biochemical hallmark of accelerated disease in G93A SOD1 mice over-expressing CCS, we therefore assessed COX1 subunit steady state levels in mitochondrial fractions obtained from spinal cords of the various transgenic mouse lines (Fig. 4). COX1 levels are markedly reduced in CCS/G93A SOD1 mice compared to non-transgenic and G93A SOD1 mice as has been previously reported (Son et al. 2008). In contrast, H80G;G93A SOD1 mice over-expressing CCS express similar steady state levels of COX1 compared to H80G;G93A SOD1 mice within spinal cord. We also crossed H80G SOD1 line with CCS transgenic mice to obtain H80G SOD1 over-expressing CCS. These CCS/H80G SOD1 dual mice did not manifest motor symptoms and exhibited a normal lifespan (Fig. 3). These results indicate that unlike G93A SOD1 mice thatshow marked acceleration of disease with CCS over-expression, CCS over-expression has no effect on survival or pathology in H80G;G93A SOD1 transgenic mice.
Fig. 4.
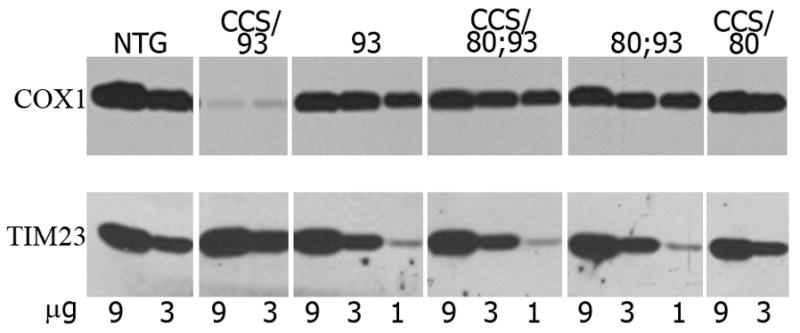
Over-expression of CCS does not cause a reduction in COX1 levels in mitochondria in H80G;G93A SOD1 or H80G SOD1 mice. COX1 levels are significantly reduced only in mitochondria of CCS/G93A SOD1 mice. Western blot analysis of complex IV structural subunit COX1 levels in mitochondrally enriched fractions derived from mouse spinal cords. 9, 3, or 1μg of each sample was used for western blots, which were probed with anti-COX1 or withanti-TIM23, used as an indicator for mitochondrial protein loading. Abbreviationsfor Figs. 4–7: non-transgenic=NTG; G93A SOD1=93, CCS/G93A SOD1=CCS/93, H80G;G93A SOD1=80;93, CCS/H80G;G93A SOD1=CCS/80;93, CCS/H80G SOD1=CCS/80, human wild type SOD1=WT SOD1. Transgenic human SOD1=h, and endogenousmouse SOD1=m.
Effects of the H80G mutation on biochemical properties of G93A SOD1 protein
Our results indicate that although both H80G;G93A SOD1 and G93A SOD1 transgenic mice develop progressive motor neuron disease, the type of pathology and susceptibility to acceleration by CCS over-expression are quite disparate. These differences suggest that H80G mutation leads to a fundamental alteration in the biochemical properties of wild type SOD1 and G93A SOD1. In order to test this hypothesis, we compared various aspects of SOD1 biochemistry including dismutase activity, ability to form SOD1 homodimers, ability to react with CCS, propensity for aggregation, protease resistance, hydrophobicity, and redox state in H80G;G93A, H80G and G93A SOD1 obtained from spinal cord extracts.
The dismutase activity of the various SOD1 mutations is shown in Fig. 5a. A dismutase activity gel assay shows that G93A SOD1has strong activity, agreeing with previously published results (Gurney et al. 1994). In contrast to G93A SOD1, both H80G and H80G;G93A SOD1 moieties do not possess dismutase activity, and exhibit only a single band thatis identical to non-transgenics, which corresponds to endogenous murine SOD1. The presence of CCS over-expression does not appear to enhance dismutase activity in either the H80G or H80G;G93A SOD1 mutants. The absence of EDTA in the homogenization buffer did not influence results of the dismutase assay (Fig. 5b).
Fig. 5.

(a and b). Dismutase activity gel assay. Zinc binding site mutations H80G;G93A SOD1 and H80G SOD1 result in a loss of dismutase activity. (c, d, e). Detection of SOD1 dimers. H80G;G93A SOD1 and H80G SOD1 proteins exits only in monomeric form, whereas wild type SOD1 and G93A SOD1 can dimerize. (c and e) Spinal cord homogenates separated under partially denaturing, non-reducing conditions, (d) control samples analyzed under standard reducing conditions. In (b) and (e) the homogenates were prepared in buffer containing or lacking 1 mM EDTA. Western blots probed with anti-SOD1. Under non-reducing conditions, the signal of wild type SOD1 is weaker than that of G93ASOD1, and the signal of endogenous mouse SOD1 is below detectable levels. In order to have comparable levelsof SOD1 protein, 5–30 μg of total protein was loaded per lane.
Both human wild type and G93A SOD1 proteins are known to form homodimers. We thereforeasked whether the addition of an H80G mutation at the zinc binding site would alter the ability of these SOD1 proteins to homodimerize. Under partially denaturing and non-reducing western blot conditions, human wild type SOD1 and G93A SOD1 derived from spinal cord homogenates of transgenic mice show both monomeric and dimeric forms (Fig. 5c). The dimeric forms can easily be reduced to monomers with the addition of a reducing agent (Fig. 5d). In contrast, human H80G and H80G;G93A SOD1 proteins exist only as monomers, indicating that H80G mutation interferes with SOD1 homodimerization (Fig. 5c and d). The absence of EDTA in the homogenization buffer did not influence results of the dimerization assay (Fig. 5e).
Earlier in vitro work had suggested that zinc binding site mutations would prevent the accumulation of insoluble SOD1 positive high molecular weight complexes (HMWCs) (Krishnan et al. 2006). We wished to assess this potential within spinal cord from in vivo specimens (Fig. 6a). Western blot analysis under standard denaturing and reducing conditions shows that SOD1 positive HMWCs accumulate in the spinal cords of weak H80G;G93A SOD1 mice in a pattern resembling that seen in weak G93A SOD1 mice whereas such complexes cannot be detected in non-transgenic or H80G SOD1 mice. These results indicate that the ability of G93A SOD1 to form HMWCs is not altered by the addition of an H80G mutation.
Fig. 6.

(a) SOD1 positive HMWPCs accumulating in the spinal cords of paralyzed G93A SOD1 mice can also be detected in paralyzed H80G;G93A SOD1 mice in western blot analysis. In order to have comparable amounts of SOD1 protein, 5–30 μg of spinal cords homogenates was loaded per lane. (a) High exposure was used for the detection of HMWPCs. A lower exposure of the same samples for the detection of corresponding monomers is shown below. (b) G93A SOD1 is more protease resistant than H80G;G93A SOD1. 5 μg protein from controls (PK −) and digested samples (PK +) was loaded per lane. (c) Both G93A SOD1 and H80G;G93A SOD1 proteins reveal hydrophobic properties. Spinal cord samples were used for hydrophobic column chromatography. 20ul (1/12.5 of the volume) of the main eluants (upper panel) and 10μg of controls (lower panel) were loaded per lane. The endogenous mouse SOD1 cannot be detected in eluants. The samples were analyzed on western gels under standard denaturing, reducing conditions and probed with anti-SOD1.
Wild type SOD1 as well as several SOD1 mutants, including G93A SOD1, has a high degree of protease resistance (Ratovitski et al. 1999). In order to assess whether the addition of H80G mutation would alter protease resistance of G93A SOD1 protein, we digested G93A SOD1 and H80G;G93A SOD1 spinal cord extractswith ProteinaseK (Fig. 6b). H80G;G93A SOD1 was degraded after 30 minutes Proteinase K treatment at +37C°, whereas most of the G93A SOD1 remains intact. Wild type endogenous mouse SOD1 is protease resistant under these conditions. Our data suggest that additional H80G mutation has caused changes in the G93A SOD1 protein conformation, making it more sensitive to protease digestion.
The hydrophobic nature of G93A SOD1 protein has been linked to aggregation and disease toxicity. We therefore assessed whether the addition of the H80G mutation to G93A SOD1 would alter its hydrophobicity in spinal cord extracts from paralyzed mice (Fig. 6c). Using hydrophobic interaction chromatography, we observe greatdifferences in binding to the hydrophobic column between G93A SOD1 and WT SOD1, agreeing with previously published results (Zetterstrom et al. 2007). A fraction of G93A SOD1 is firmly bound to the column and can be eluted only with the appropriate reagents, in contrast to WT SOD1, which is unable to bind the column. For H80G;G93A SOD1 we also observe a substantial fraction binding to the hydrophobic column comparable to G93A SOD1 when controlled for both total protein and SOD1 loading. Thus, the hydrophobic properties of G93A SOD1 are preservedin the context of the additional H80G zinc binding site mutation. The SOD1 polypeptide with H80G SOD1 mutation also revealed hydrophobic properties.
Wild type SOD1 and G93A SOD1 can exist in either an oxidized or reduced form in vivo as related to intra-molecular disulfide bonds at residues C57 or C146. Previous published work has suggested that the redox state of G93A SOD1 is crucial for its mitochondrial toxicity, which is enhanced by CCS over-expression (Proescher et al. 2008). Because H80G;G93A SOD1 transgenic mice manifest very little mitochondrial pathology and their disease was not accelerated by CCS over-expression, we hypothesized that the addition of the H80G mutation to G93A SOD1 would alter its redox state. We therefore compared redox states of G93A SOD1 and H80G;G93A SOD1 as well as H80G SOD1proteins isolated from spinal cord extracts by using an iodoacetamide based assay. In this assay disulfide oxidized and disulfide reduced SOD1 are differentiated by denaturing, non-reducing gel electrophoresis following thiol protection by iodoacetamide (Fig. 7). In the reduced forms, the free cysteine residues are modified by iodoacetamide and therefore have a slower mobilityin non-reducing SDS-PAGEanalysis.G93A SOD1 monomers form two bands, with the higher band representing a reduced form and the lower band representing an oxidized form (Fig. 7, upper panel). The ratio of reduced to oxidized form of G93A SOD1 mutantis increased in the setting of CCS over-expression as we have shown previously (Son et al. 2009). In contrast, H80G;G93A SOD1 exists in only a single state which does not change with CCS over-expression. This single band represents the reduced form in standard reducing SDS-PAGE analysis (Fig. 7, lower panel). Similarly, H80G SOD1 exhibits only one band corresponding to the reduced form, which is not altered by CCS over-expression. These results indicate that the addition of the H80G mutation to SOD1 alters its redox potential and only allows for the presence of a reduced form of the SOD1 protein in vivo. The H80G mutation renders SOD1 more comparable to the G86R and L126Z in terms of its altered redox state. In agreement, H80G;G93A SOD1 transgenic mice have pathology similar toG86R SOD1 and L126Z SOD1 transgenic mice. They sharean inclusion type rather than mitochondrial pathology, maintainnormal levels of COX1, and are unaffected by over-expression of CCS (Son et al. 2009). The biochemical properties of G93A SOD1, H80G; G93A SOD1, H80G SOD1, and WTSOD1 proteins are summarized in Table 1.
Fig. 7.
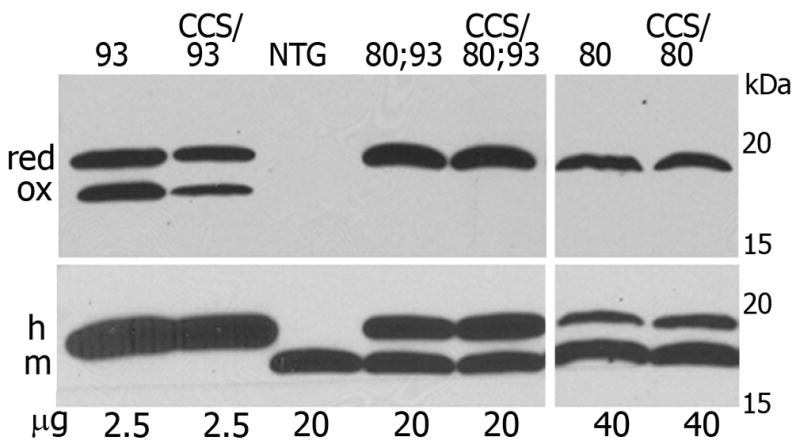
H80G;G93A SOD1 and H80GSOD1 proteins exist only in reduced form, in contrast to G93A SOD1 that can be in oxidized or reduced form. Iodoacetamide treated spinal cord homogenates were separated under non-reducing conditions (upper panel) or under reducing conditions (lower panel) in SDS gels. Oxidized (ox) and reduced (red) forms of SOD1 can be detected in non-reducing gels. The SOD1 positive bands are not quantitative due to the modification of SOD1 with iodoacetamide. Endogenous mouse SOD1 (m) migrates slightly faster than human transgenic SOD1 (h). It can be detected only in reduced form, in samples containing larger amounts of protein.
Table 1.
| G93A SOD1 | 80G;G93A SOD1 | H80G SOD1 | WT SOD1 | |
|---|---|---|---|---|
| Normal properties: | ||||
| Reactions with CCS | yes | no | no | yes |
| Dimerization | yes | no | no | yes |
| Dismutase activity | yes | no | no | yes |
| Oxidation of disulfide bridges | yes | no | no | yes |
| Abnormal properties: | ||||
| Protease resistance | yes | no | no | yes |
| Aggregation | yes | yes | no | no |
| Hydrophobicity | yes | yes | yes | no |
| Pathogenicity | yes | yes | no* | no |
at the achieved expression levels
Discussion
This work explores the biochemical properties and in vivo effects of the human SOD1 primary zinc binding site mutation H80G. Many experiments addressing the effect of zinc on SOD1 protein have been done by using zinc depleted SOD1 originally derived from purified holo-SOD1(Sahawneh et al. 2010, Roberts et al. 2007). In those cases, holo-SOD1 has already attained its native conformational state that is likely perturbed by stripping zinc. However, when zinc-binding sites are altered in vivo during SOD1 synthesis and initial folding, the effect of zinc on SOD1 structure and function can be assessed with more biological relevance.
In this work, we present the initial characterization of transgenic mice with mutations in a primary zinc binding residue (H80), either alone or in the context of a G93A mutation. Over-expression of H80G SOD1 did not yield a motor phenotype or spinal cord pathology, at least at the levels of the SOD1 transgene we were able to achieve in our mouse lines. In contrast, over-expression of H80G;G93A SOD1 did result in a progressive motor phenotype characterized by severe motor neuron loss, ubiquitin positive inclusions and gliosis. Analysis of SOD1 expressed in spinal cord extracts confirmed that the H80G mutation fundamentally altered certain biochemical parameters of both human wild type SOD1 and G93A SOD1. SOD1 proteins harboring H80G mutation are vulnerable to proteases. They lack dismutase activity, are unable to form SOD1 homodimers, cannot normally react with CCS, and fail to undergo normal redox cycle. G93A SOD1 and H80G;G93A SOD1 share certain characteristics, including hydrophobicityandsusceptibilityto form HMWPCs.
Hydrophobic column chromatography analysis can be used to study hydrophobic regions on the surface of proteins. Wild type SOD1, which is mainly a cytosolic protein, is hydrophilic and does not bind to hydrophobic columns. Mutations might increase hydrophobicity by causing metal deficiency, decreasing the stability of SOD1 polypeptide and exposing hydrophobic patches on its surface due to conformational changes. The hydrophobic forms of several SOD1 mutants were found to be zinc deficient, disulfide reduced and mostly monomeric (Tiwari et al. 2009, Tiwari et al. 2005, Zetterstrom et al. 2007). Furthermore, analysis of the hydrophobic and aggregation properties of various human SOD1 mutations indicate that there is a correlation between exposure of hydrophobic surfaces and aggregation (Munch & Bertolotti 2010). In the spinal cords of G93A SOD1 transgenic mice, there is an age-dependent increase of misfolded hydrophobic SOD1 subfractions (Zetterstrom et al. 2007). Similarly, there is an age-dependent accumulation of aggregates and HMWPCs in G93A SOD1 spinal cords (Johnston et al. 2000, Puttaparthi et al. 2003, Karch et al. 2009, Trumbull & Beckman 2009). The G93A mutation has been found to selectively destabilize the remote metal binding region in SOD1 and that might contribute to these properties (Museth et al. 2009). The zinc binding site mutation H80G does not appear to fundamentally alter these properties in G93A SOD1 in vivo, as H80G;G93A SOD1 displays both hydrophobicity and propensity to aggregate.
However, H80G and H80G;G93A mutants do exhibit changes in other biochemical properties compared to wild type SOD1 or G93A SOD1. G93A SOD1 retains dismutase activity, whereas both H80G;G93A and H80G mutants have lost their activity (Krishnan et al. 2006). H80G;G93A and H80G mutants have also lost their ability to normally interactwith CCS and to form SOD1 homodimers, indicating that the their dimerization interface has undergone structural changes. This finding agrees with studies where zinc deficient human D83S SOD1 expressed in E. Coli was found to be unstable (Sahawneh et al. 2010). In addition, human H80R and D124V SOD1 expressed in yeast are more prone to monomerization, and lose their ability to interact normally with CCS (Seetharaman et al. 2010, Sahawneh et al. 2010). Similarly, Proteinase K digestion experiments confirm structural differences between G93A SOD1 and H80G;G93A SOD1 based on susceptibility to protease digestion (Ratovitski et al. 1999).
The addition of a zinc binding site mutation to G93A does appear to radically alter the redox state of the protein in vivo, which has important ramifications for disease mechanisms and pathology. Human SOD1 has four cysteine residues at positions 6, 57, 111, and 146. Cys 57 and Cys 146 form intra-subunit disulfide bond, important for copper metallation, SOD1 maturation and its interaction with CCS (Furukawa & O’Halloran 2006, Seetharaman et al. 2009). In human H80R and D124V SOD1 expressed in yeast, up to four cysteine residues can be reduced and remain in the reduced state even in the presence of CCS (Seetharaman et al. 2010). These findings indicate that the conformation of these metal deficient H80R and D124V mutants is probably distorted due to reduction of intra-molecular disulfide bonds. These mutants cannot co-operate normally with CCS and mature, and are considered to be folding intermediates prone to oligomerization. The SOD1 disulfide analysis of spinal cord samples demonstrate that H80G;G93A SOD1 also exists only in a reduced state in vivo, unlike G93A SOD1 that can exist in either reduced or oxidized form and therefore can undergo the redox cycle. We have previously shown in vivo that CCS over-expression favors the oxidized form of wild type SOD1 but promotes the reduced from of G93A or G37R SOD1 (Proescher et al. 2008, Son et al. 2009). In contrast, CCS over-expression does not have any effect on H80G;G93A SOD1 or H80G SOD1, which already exist only in the reduced state. This pattern closely resembles what is observed for other SOD1 mutants such as G86R and L126Z (Son et al. 2009).
The ability of SOD1 mutant polypeptides to undergo redox cycles provide a possible explanation for the vacuolar mitochondrial pathology observed in certain SOD1 mutant transgeniclines but not in others. The import of proteins into the intermembrane space (IMS) of mitochondria is dependent on coupled oxidation-reduction reactions of cysteine rich proteins, including CCS (Mesecke et al. 2005). The Mia 40-Erv1 disulfide relay system drives the import of these proteins into the IMS of mitochondria in reduced, unfolded state, where they are trapped after being oxidatively folded (Bihlmaier et al. 2008, Reddehase et al. 2009, Hell 2008). Since CCS facilitates the entry of SOD1 into mitochondria, the mitochondrial localization of SOD1 is mainly dependent on its ability to productively co-operate with to CCS. Both in vivo and in vitro experiments have shown that increase in CCS levels markedly increase the mitochondrial load of SOD1 (Kawamata & Manfredi 2008, Son et al. 2007). Even a potential secondary mitochondrial localization and maturation pathways for SOD1 via glutathione are dependent on oxidation–reduction reactions (Carroll et al. 2004). The accumulation of mutant SOD1 within mitochondria appears to correlate with mitochondrial toxicity, vacuolation, and COX deficiency in vivo. G93A SOD1, with the addition zinc binding site mutation H80G, loses its ability to interact with CCS and undergo maturation, remaining fixed in the reduced form. Its inability to take part of normal oxidation-reduction reactions would prevent its localization in mitochondrial inter-membrane space where it might exert direct mitochondrial toxicity. In support of this idea, H80G;G93A SOD1 transgenic mice do not have mitochondrial vacuolar pathology or COX deficiency, and their disease is not accelerated by over-expression of CCS. These results favor the hypothesis that the ability of SOD1 mutants to produce mitochondrial vacuolization is dependent on its redox properties.
Acknowledgments
This work was supported by a grant from the NINDS (R01 NS055315) and the Muscular Dystrophy Association (MDA.
Abreviations
- ALS
Amyotrophic Lateral Sclerosis
- CCS
copper chaperone for SOD1
- COX
cytochrome c oxidase
- GFAP
glial fibrillary acidic protein
- H & E
hematoxylin and eosin
- HMWPCs
high molecular weight protein complexes
- HIC
Hydrophobic Interaction Chromatography
- IMS
intermembrane space
- NBT
Nitro Blue Tetrazolium
- NTG
non-transgenic
- PFA
paraformaldehyde
- PMSF
phenylmethylsulfonyl fluoride
- PBS
phosphate buffered saline
- PVDF
polyvinylidene difluoride
- SDS-PAGE
sodium dodecyl sulfate polyacrylamide gel electrophoresis
- SOD1
superoxide dismutase 1
- TEMED
N,N,N′,N′-Tetramethylethylenediamine
- Tris
tris(hydroxymethyl) aminomethane
- Ubiq
ubquitin
- WT
wild type
Footnotes
The authors report no financial disclosures or conflicts.
References
- Alexander MD, Traynor BJ, Miller N, Corr B, Frost E, McQuaid S, Brett FM, Green A, Hardiman O. “True” sporadic ALS associated with a novel SOD-1 mutation. Ann Neurol. 2002;52:680–683. doi: 10.1002/ana.10369. [DOI] [PubMed] [Google Scholar]
- Bihlmaier K, Mesecke N, Kloeppel C, Herrmann JM. The disulfide relay of the intermembrane space of mitochondria: an oxygen-sensing system? Ann N Y Acad Sci. 2008;1147:293–302. doi: 10.1196/annals.1427.005. [DOI] [PubMed] [Google Scholar]
- Borchelt DR, Davis J, Fischer M, et al. A vector for expressing foreign genes in the brains and hearts of transgenic mice. Genet Anal. 1996;13:159–163. doi: 10.1016/s1050-3862(96)00167-2. [DOI] [PubMed] [Google Scholar]
- Carroll MC, Girouard JB, Ulloa JL, Subramaniam JR, Wong PC, Valentine JS, Culotta VC. Mechanisms for activating Cu- and Zn-containing superoxide dismutase in the absence of the CCS Cu chaperone. Proc Natl Acad Sci U S A. 2004;101:5964–5969. doi: 10.1073/pnas.0308298101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casareno RL, Waggoner D, Gitlin JD. The copper chaperone CCS directly interacts with copper/zinc superoxide dismutase. J Biol Chem. 1998;273:23625–23628. doi: 10.1074/jbc.273.37.23625. [DOI] [PubMed] [Google Scholar]
- Culotta VC, Klomp LW, Strain J, Casareno RL, Krems B, Gitlin JD. The copper chaperone for superoxide dismutase. J Biol Chem. 1997;272:23469–23472. doi: 10.1074/jbc.272.38.23469. [DOI] [PubMed] [Google Scholar]
- Dal Canto MC, Gurney ME. Development of central nervous system pathology in a murine transgenic model of human amyotrophic lateral sclerosis. Am J Pathol. 1994;145:1271–1279. [PMC free article] [PubMed] [Google Scholar]
- Deng HX, Shi Y, Furukawa Y, et al. Conversion to the amyotrophic lateral sclerosis phenotype is associated with intermolecular linked insoluble aggregates of SOD1 in mitochondria. Proc Natl Acad Sci U S A. 2006;103:7142–7147. doi: 10.1073/pnas.0602046103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferri A, Cozzolino M, Crosio C, Nencini M, Casciati A, Gralla EB, Rotilio G, Valentine JS, Carri MT. Familial ALS-superoxide dismutases associate with mitochondria and shift their redox potentials. Proc Natl Acad Sci U S A. 2006;103:13860–13865. doi: 10.1073/pnas.0605814103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukawa Y, O’Halloran TV. Posttranslational modifications in Cu, Zn-superoxide dismutase and mutations associated with amyotrophic lateral sclerosis. Antioxid Redox Signal. 2006;8:847–867. doi: 10.1089/ars.2006.8.847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurney ME, Pu H, Chiu AY, et al. Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science. 1994;264:1772–1775. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- Hell K. The Erv1-Mia40 disulfide relay system in the intermembrane space of mitochondria. Biochim Biophys Acta. 2008;1783:601–609. doi: 10.1016/j.bbamcr.2007.12.005. [DOI] [PubMed] [Google Scholar]
- Higgins CM, Jung C, Ding H, Xu Z. Mutant Cu, Zn superoxide dismutase that causes motoneuron degeneration is present in mitochondria in the CNS. J Neurosci. 2002;22:RC215. doi: 10.1523/JNEUROSCI.22-06-j0001.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosler BA, Nicholson GA, Sapp PC, et al. Three novel mutations and two variants in the gene for Cu/Zn superoxide dismutase in familial amyotrophic lateral sclerosis. Neuromuscul Disord. 1996;6:361–366. doi: 10.1016/0960-8966(96)00353-7. [DOI] [PubMed] [Google Scholar]
- Johnston JA, Dalton MJ, Gurney ME, Kopito RR. Formation of high molecular weight complexes of mutantCu, Zn-superoxide dismutase in a mouse model for familial amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. 2000;97:12571–12576. doi: 10.1073/pnas.220417997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karch CM, Prudencio M, Winkler DD, Hart PJ, Borchelt DR. Role of mutant SOD1 disulfide oxidationand aggregation in the pathogenesis of familial ALS. Proc Natl Acad Sci U S A. 2009;106:7774–7779. doi: 10.1073/pnas.0902505106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamata H, Manfredi G. Different regulation of wild-type and mutant Cu, Zn superoxide dismutase localization in mammalian mitochondria. Hum Mol Genet. 2008;17:3303–3317. doi: 10.1093/hmg/ddn226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan U, Son M, Rajendran B, Elliott JL. Novel mutations that enhance or repress the aggregation potential of SOD1. Mol Cell Biochem. 2006;287:201–211. doi: 10.1007/s11010-005-9112-4. [DOI] [PubMed] [Google Scholar]
- Liu J, Lillo C, Jonsson PA, et al. Toxicity of familial ALS-linked SOD1 mutants from selective recruitment to spinal mitochondria. Neuron. 2004;43:5–17. doi: 10.1016/j.neuron.2004.06.016. [DOI] [PubMed] [Google Scholar]
- Mesecke N, Terziyska N, Kozany C, Baumann F, Neupert W, Hell K, Herrmann JM. A disulfide relay system in the intermembrane space of mitochondria that mediates protein import. Cell. 2005;121:1059–1069. doi: 10.1016/j.cell.2005.04.011. [DOI] [PubMed] [Google Scholar]
- Munch C, Bertolotti A. Exposure of hydrophobic surfaces initiates aggregation of diverse ALS-causing superoxide dismutase-1 mutants. J Mol Biol. 2010;399:512–525. doi: 10.1016/j.jmb.2010.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Museth AK, Brorsson AC, Lundqvist M, Tibell LA, Jonsson BH. The ALS-associated mutation G93A in human copper-zinc superoxide dismutase selectively destabilizes the remote metal binding region. Biochemistry. 2009;48:8817–8829. doi: 10.1021/bi900703v. [DOI] [PubMed] [Google Scholar]
- Proescher JB, Son M, Elliott JL, Culotta VC. Biological effects of CCS in the absence of SOD1 enzyme activation: implications for disease in a mouse model for ALS. Hum Mol Genet. 2008;17:1728–1737. doi: 10.1093/hmg/ddn063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puttaparthi K, Elliott JL. Non-neuronal induction of immunoproteasome subunits in an ALS model: possible mediation by cytokines. Exp Neurol. 2005;196:441–451. doi: 10.1016/j.expneurol.2005.08.027. [DOI] [PubMed] [Google Scholar]
- Puttaparthi K, Gitomer WL, Krishnan U, Son M, Rajendran B, Elliott JL. Disease progression in a transgenic model of familial amyotrophic lateral sclerosis is dependent on both neuronal and non-neuronal zinc binding proteins. J Neurosci. 2002;22:8790–8796. doi: 10.1523/JNEUROSCI.22-20-08790.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puttaparthi K, Wojcik C, Rajendran B, DeMartino GN, Elliott JL. Aggregate formation in the spinal cord of mutant SOD1 transgenic mice is reversible and mediated by proteasomes. J Neurochem. 2003;87:851–860. doi: 10.1046/j.1471-4159.2003.02028.x. [DOI] [PubMed] [Google Scholar]
- Ratovitski T, Corson LB, Strain J, Wong P, Cleveland DW, Culotta VC, Borchelt DR. Variation in the biochemical/biophysical properties of mutant superoxide dismutase 1 enzymes and the rate of disease progression in familial amyotrophic lateral sclerosis kindreds. Hum Mol Genet. 1999;8:1451–1460. doi: 10.1093/hmg/8.8.1451. [DOI] [PubMed] [Google Scholar]
- Reddehase S, Grumbt B, Neupert W, Hell K. The disulfide relay system of mitochondria is required for the biogenesis of mitochondrial Ccs1 and Sod1. J Mol Biol. 2009;385:331–338. doi: 10.1016/j.jmb.2008.10.088. [DOI] [PubMed] [Google Scholar]
- Roberts BR, Tainer JA, Getzoff ED, Malencik DA, Anderson SR, Bomben VC, Meyers KR, Karplus PA, Beckman JS. Structural characterization of zinc-deficient human superoxide dismutase and implications for ALS. J Mol Biol. 2007;373:877–890. doi: 10.1016/j.jmb.2007.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahawneh MA, Ricart KC, Roberts BR, et al. Cu, Zn-superoxide dismutase increases toxicity of mutant and zinc-deficient superoxide dismutase by enhancing protein stability. J Biol Chem. 2010;285:33885–33897. doi: 10.1074/jbc.M110.118901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt PJ, Kunst C, Culotta VC. Copper activation of superoxide dismutase 1 (SOD1) in vivo. Role for protein-protein interactions with the copper chaperone for SOD1. J Biol Chem. 2000;275:33771–33776. doi: 10.1074/jbc.M006254200. [DOI] [PubMed] [Google Scholar]
- Seetharaman SV, Prudencio M, Karch C, Holloway SP, Borchelt DR, Hart PJ. Immature copper-zinc superoxide dismutase and familial amyotrophic lateral sclerosis. Exp Biol Med (Maywood) 2009;234:1140–1154. doi: 10.3181/0903-MR-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seetharaman SV, Winkler DD, Taylor AB, et al. Disrupted zinc-binding sites in structures of pathogenic SOD1 variants D124V and H80R. Biochemistry. 2010;49:5714–5725. doi: 10.1021/bi100314n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son M, Fu Q, Puttaparthi K, Matthews CM, Elliott JL. Redox susceptibility of SOD1 mutants is associated with the differential response to CCS over-expression in vivo. Neurobiol Dis. 2009;34:155–162. doi: 10.1016/j.nbd.2009.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son M, Leary SC, Romain N, Pierrel F, Winge DR, Haller RG, Elliott JL. Isolated cytochrome c oxidase deficiency in G93A SOD1 mice overexpressing CCS protein. J Biol Chem. 2008;283:12267–12275. doi: 10.1074/jbc.M708523200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son M, Puttaparthi K, Kawamata H, Rajendran B, Boyer PJ, Manfredi G, Elliott JL. Overexpression of CCS in G93A-SOD1 mice leads to accelerated neurological deficits with severe mitochondrial pathology. Proc Natl Acad Sci U S A. 2007;104:6072–6077. doi: 10.1073/pnas.0610923104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiwari A, Liba A, Sohn SH, Seetharaman SV, Bilsel O, Matthews CR, Hart PJ, Valentine JS, Hayward LJ. Metal deficiency increases aberrant hydrophobicity of mutant superoxide dismutases that cause amyotrophic lateral sclerosis. J Biol Chem. 2009;284:27746–27758. doi: 10.1074/jbc.M109.043729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiwari A, Xu Z, Hayward LJ. Aberrantly increased hydrophobicity shared by mutants of Cu, Zn-superoxide dismutase in familial amyotrophic lateral sclerosis. J Biol Chem. 2005;280:29771–29779. doi: 10.1074/jbc.M504039200. [DOI] [PubMed] [Google Scholar]
- Trumbull KA, Beckman JS. A role for copper in the toxicity of zinc-deficient superoxide dismutase to motor neurons in amyotrophic lateral sclerosis. Antioxid Redox Signal. 2009;11:1627–1639. doi: 10.1089/ars.2009.2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijayvergiya C, Beal MF, Buck J, Manfredi G. Mutant superoxide dismutase 1 forms aggregates in the brain mitochondrial matrix of amyotrophic lateral sclerosis mice. J Neurosci. 2005;25:2463–2470. doi: 10.1523/JNEUROSCI.4385-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Slunt H, Gonzales V, Fromholt D, Coonfield M, Copeland NG, Jenkins NA, Borchelt DR. Copper-binding-site-null SOD1 causes ALS in transgenic mice: aggregates of non-native SOD1 delineate a common feature. Hum Mol Genet. 2003;12:2753–2764. doi: 10.1093/hmg/ddg312. [DOI] [PubMed] [Google Scholar]
- Wang J, Xu G, Slunt HH, Gonzales V, Coonfield M, Fromholt D, Copeland NG, Jenkins NA, Borchelt DR. Coincident thresholds of mutant protein for paralytic disease and protein aggregation caused by restrictively expressed superoxide dismutase cDNA. Neurobiol Dis. 2005;20:943–952. doi: 10.1016/j.nbd.2005.06.005. [DOI] [PubMed] [Google Scholar]
- Wong PC, Pardo CA, Borchelt DR, Lee MK, Copeland NG, Jenkins NA, Sisodia SS, Cleveland DW, Price DL. An adverse property of a familial ALS-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron. 1995;14:1105–1116. doi: 10.1016/0896-6273(95)90259-7. [DOI] [PubMed] [Google Scholar]
- Zetterstrom P, Stewart HG, Bergemalm D, Jonsson PA, Graffmo KS, Andersen PM, Brannstrom T, Oliveberg M, Marklund SL. Soluble misfolded subfractions of mutant superoxide dismutase-1s are enriched in spinal cords throughout life in murine ALS models. Proc Natl Acad Sci U S A. 2007;104:14157–14162. doi: 10.1073/pnas.0700477104. [DOI] [PMC free article] [PubMed] [Google Scholar]