

Abstract
Defects in subunits of the conserved oligomeric Golgi (COG) complex represent a growing subset of congenital disorders of glycosylation (CDGs). In addition to altered protein glycosylation and vesicular trafficking, Cog-deficient patient fibroblasts exhibit a striking delay in the Golgi-disrupting effects of brefeldin A (BFA). Despite the diagnostic value of this BFA resistance, the molecular basis of this response is not known. To investigate potential mechanisms of resistance, we analyzed the localization of the large ARF-GEF, GBF1, in several Cog-deficient cell lines. Our results revealed mislocalization of GBF1 to non-Golgi compartments, in particular the ERGIC, within these cells. Biochemical analysis of GBF1 in control and BFA-treated fibroblasts demonstrated that the steady-state level and membrane recruitment is not substantially affected by COG deficiency, supporting a role for the COG complex in the localization but not membrane association of GBF1. We also showed that pretreatment of fibroblasts with bafilomycin resulted in a GBF1-independent BFA resistance that appears additive with the resistance associated with COG deficiency. These data provide new insight into the mechanism of BFA resistance in Cog-deficient cells by suggesting a role for impaired ARF-GEF localization.
Keywords: Golgi, retrograde traffic, Conserved Oligomeric Golgi complex, GBF1, BFA, Congenital Disorders of Glycosylation
INTRODUCTION
First discovered by Jaeken in 1980, congenital disorders of glycosylation (CDG) are a group of inherited disorders that typically involve defects in enzymes and proteins responsible for the biosynthesis of oligosaccharide precursors and glycoproteins [1–5]. Over the last several years, the molecular bases for CDGs have been expanded to include Golgi-localized proteins such as subunits of the V-type H+ ATPase (a proton transporter involved in vesicle acidification), and members of the COG complex (a multisubunit Golgi tethering complex that mediates vesicle transport steps within the secretory apparatus) [6–13]. These newly defined subtypes are characterized on a molecular and cellular level by alterations in protein glycosylation as well as defects in overall Golgi function and structure [14, 15]. Loss of COG complex integrity leads to impaired intra-Golgi retrograde transport of glycosyltransferases and nucleotide-sugar transporters within COPI-coated vesicles, mislocalization and/or decreased stability of these enzymes, and eventual protein and lipid glycosylation defects [16–22]. Studies in yeast have suggested that specific COG complex subunits are also involved in vesicle trafficking between the ER and Golgi although the exact function of COG at this interface is still unclear [23]. A role for the COG complex in ER-Golgi trafficking is further supported by the observation that Cog-deficient cells exhibit impaired recycling and stability of several Golgi and ERGIC proteins [15, 16].
In addition to changes in Golgi protein trafficking and stability, all Cog-deficient fibroblasts exhibit a delay in the Golgi-disrupting effect of the fungal metabolite, brefeldin A (BFA). We have previously shown that this delay appears to be caused by slower onset of BFA-induced tubule formation as opposed to impaired extension and fusion of these tubules with the ER [15]. Furthermore, certain Golgi proteins such as giantin and GPP130 were demonstrated to cluster around the microtubule-organizing center (MTOC) following prolonged exposure to BFA. The robust response of Cog-deficient fibroblasts to BFA has yielded a rapid and useful means of identifying novel COG defects among untyped CDG cases. BFA treatment has also become a general method for defining changes in Golgi function within other CDG subtypes, including the V-type H+ ATPase defects [11, 14]. Despite this important diagnostic value, the mechanisms underlying the unique effects of BFA in CDG fibroblasts have not been defined.
Delayed collapse of the Golgi upon treatment with various disrupting agents such as BFA can arise via multiple mechanisms. In light of the requirement for intact microtubules, chemical or genetic disruption of microtubule polymerization or integrity results in delayed BFA-induced Golgi collapse [24]. Another prerequisite for Golgi collapse in most cases is the release of COPI from the Golgi membrane, an event that occurs within minutes of BFA treatment due to the inactivation of ARFs [25]. Our earlier studies demonstrated that COPI release was not affected in the Cog7-deficient fibroblasts, indicating that the delayed response to BFA occurs via a COPI-independent mechanism [15]. Earlier work by several groups has also suggested that altered regulation and activity of large ARF-GEFs such as Golgi-specific brefeldin A resistant guanine nucleotide exchange factor 1 (GBF1) may confer resistance to BFA [26–30]. Indeed, overexpression of GBF1 has been demonstrated to protect cells from the Golgi-disrupting effects of BFA, possibly by sequestering the drug [31, 32]. GBF1, which actively cycles between the cytosol and membrane, is localized primarily at the Golgi but is also distributed in the ERGIC, ER and ER exit sites, suggesting that it is a cycling protein when membrane-bound [26, 28, 29, 33]. Upon BFA treatment, GBF1 becomes stabilized on the membrane and rapidly concentrated at the cis-Golgi [33]. This rapid concentration is followed by the onset of tubules that will eventually fuse with the ER and facilitate dissolution of the Golgi into a hybrid compartment [34, 35]. It is not clear whether this concentration of GBF1 at the Golgi is required for BFA-induced tubule formation or simply represents its enhanced stability on the membrane in the BFA-bound form [36]. GBF1 has been shown to interact with several Golgi and ERGIC proteins (such as p115), and it is well established that this protein plays a role in ER-Golgi transport steps and COPI recruitment [37, 38]. In light of these interactions and our earlier findings demonstrating altered protein recycling at the ER-Golgi boundary, we hypothesized that abnormal localization and dynamics of GBF1 may contribute to the delayed BFA-induced Golgi collapse in COG-deficient cells.
In this report, we provide evidence for the variable mislocalization of GBF1 in Cog7-deficient fibroblasts, Cog1-deficient ldlB CHO cells, and Cog knockdown HeLa cells. Biochemical analysis of GBF1 in control and BFA-treated fibroblasts demonstrated that the steady-state levels and membrane recruitment is not affected by COG deficiency, supporting a role for this complex in the localization but not membrane association of GBF1. The localization of the late Golgi ARF-GEF, brefeldin A-inhibited guanine nucleotide-exchange protein 1 (BIG1), was also altered in the COG-depleted HeLa cells, suggesting a common mechanism whereby the COG complex mediates the distribution of these proteins. Together, our findings uncover a potential mechanism of BFA resistance in Cog-deficient cells caused by mislocalization of ARF-GEFs and shed further light on the role of COG in vesicle trafficking at multiple locations within the secretory apparatus.
MATERIALS AND METHODS
Reagents, Antibodies and Cell Lines
Reagents and sources were as follows: BFA was from Calbiochem, Golgicide A was a gift from Dr. David Haslam (Washington University School of Medicine, Saint Louis, MO) and AG1478 was purchased from Sigma. Anti-GBF1 monoclonal antibodies were purchased from BD Biosciences, anti-BIG1 polyclonal antibodies were purchased from Santa Cruz, anti-giantin rabbit polyclonal antisera was from Covance and anti-human ERp29 rabbit polyclonal antisera was purchased from Thermo Scientific. Sec31 polyclonal antibodies were kindly provided by Dr. Fred Gorelick (Yale University). All fluorophore-conjugated secondary antibodies were obtained from Invitrogen.
Wild type and Cog7-deficient patient fibroblasts were maintained in DMEM supplemented with 10% fetal bovine serum and 100 U/mL penicillin and 100 U/mL streptomycin. Wild type and ldlB CHO cells were grown in RPMI supplemented with 10% fetal bovine serum and 100 U/mL penicillin and 100 U/mL streptomycin.
HeLa cells stably expressing GalNAcT2-GFP were obtained from Dr. Brian Storrie (UAMS). These cells were cultured in DMEM/F-12 50/50 medium supplemented with 15 mM HEPES, 2.5 mM L-glutamine, 10% FBS, and 0.4 mg/ml G418 sulfate. Cells were grown at 37°C and 5% CO2 in a 90% humidified incubator.
siRNA-induced knockdowns
For the knockdowns of stably-expressing GalNAcT2-GFP HeLa cells, siRNA duplexes were obtained from Dharmacon: human Cog3 (AGACTTGTGCAGTTTAACA; [18]) and human Cog6 (AGATATGACAAGTCGCCTA; [39]). Transfection was performed using Lipofectamine RNAiMAX (Invitrogen), following a protocol recommended by the manufacturer. One cycle of transfection (100 nM siRNA each) was performed and cells were analyzed 72 h after transfection.
Immunofluorescence and Confocal Microscopy
For immunofluorescence experiments, cells were seeded on coverslips and cultured overnight prior to drug treatments. Following treatments, cells were washed with phosphate-buffered saline, fixed in 3.7% formaldehyde in PBS and stained as described previously [15]. The standard dilution buffer for both primary and secondary antibody incubations contained PBS, pH 7.4, 0.1% Triton X-100 and 1mg/mL bovine serum albumin. Confocal images were acquired using an Olympus FV1000 laser scanning microscope equipped a 60x oil immersion (numerical aperture 1.4) objective. Stacks of 0.25 μm (based on calculated optimums) optical sections were collected in the z-dimension, and subsequently collapsed into a single image (maximum intensity or z-projection) unless otherwise noted.
Treatment with Golgi-Disrupting Agents and Quantitation of Effects
Patient fibroblasts were treated with 5 μg/mL BFA for various times (5–20 min) and GalNAcT2-GFP HeLa cells for 5, 10, 20, 30, and 60 min to induce Golgi collapse. For the Golgicide A (GCA) and AG1478 experiments, cells were treated with these compounds in complete growth media with either 10 μM GCA or 20 μM AG1478 for various times and analyzed as described above. Quantitation of the percentage of cells with an ER staining pattern was scored by eye in at least three independent experiments.
The percentage of GBF1 localized on the Golgi apparatus in GalNAcT2-GFP HeLa cells was analyzed using Image J software (NIH). The integrated density of the GBF1 that colocalized with GalNAcT2-GFP was calculated after subtraction of the background. The integrated density of the GBF1 signal for the entire cell was also analyzed. The percent Golgi localization was calculated by dividing the Golgi integrated density by the total integrated density. Three separate experiments were done, with the number of cells for each experiment ranging between 45–55.
Subcellular fractionation of endogenous GBF1
Fibroblasts (2 ×107) treated with or without 5 μg/mL BFA for 5 min were placed on ice and collected in cold PBS by scraping. The pelleted cells were disrupted in 400 μL homogenization buffer (50mM HEPES-KOH, pH 7.5, 100mM KCl, 1mM MgCl2, 1mM DTT) by passage through a 27-gauge needle three times. The homogenate was centrifuged for 10 min at 1000 × g at 4°C to remove debris and nuclei. The post-nuclear supernatant was then subjected to high-speed centrifugation (100,000 × g) for 45 min at 4°C using a Beckman TLA 100.2 rotor. The resulting supernatant was reserved and designated the cytosolic fraction. The remaining pellet was washed with buffer, solubilized in PBS containing 1% Triton X-100, 0.1% SDS and designated the membrane fraction. Fractions containing the same volume of original cells were analyzed by 8% SDS-PAGE. After SDS-PAGE, proteins were transferred to nitrocellulose membrane and blotted with a monoclonal antibody against GBF1. The blots were scanned and the density of signal of each fraction was quantitated using Image J software (NIH).
RESULTS
GBF1 is localized primarily to the ERGIC compartment in Cog1-deficient ldlB CHO cells
One possible mechanism for the delayed response of Cog-deficient cells to BFA is altered dynamics of the ARF-GTP exchange factor, GBF1. To explore this possibility, we first assessed the localization of this protein in WT and Cog-deficient CHO cells. Wild type and Cog1-deficient ldlB CHO cells were co-stained with antisera against GBF1 and the Golgi marker giantin and visualized by confocal microscopy (Figure 1). Membrane-associated GBF1 was detected primarily on the Golgi in wild type CHO cells, as evidenced by the high degree of co-localization between GBF1 and giantin. In contrast, very little GBF1 staining on the Golgi was noted in untreated ldlB cells, where it was localized instead to evenly distributed, punctate structures throughout the cytoplasm. Although giantin is a Cog-sensitive protein (or GEAR) in ldlB cells, we do not believe that the failure of GBF1 and giantin to co-localize reflects the mislocalization of giantin in these cells. Indeed, little or no co-localization of GBF1 with other Golgi markers including GM130 was observed (data not shown). In order to better define the peripheral localization of GBF1 in WT and ldlB cells, we co-stained with markers of the ERGIC (ERGIC-53), ER exit sites (Sec31) and ER (ERp29). Although there was significant overlap between GBF1 and Sec31 in WT cells, minimal co-localization with the other marker proteins was detected. In contrast, we found a high degree of overlap between ERGIC-53 and GBF1 in the ldlB cells, but little co-localization with Sec31 and ERp29 (see Supplemental Figure 1 for separation of GBF1 and ERp29 images). These results indicate GBF1 predominantly resides in the ERGIC compartment in Cog-deficient CHO cells.
Figure 1. GBF1 is mislocalized primarily to the ERGIC in Cog1-deficient ldlB CHO cells.
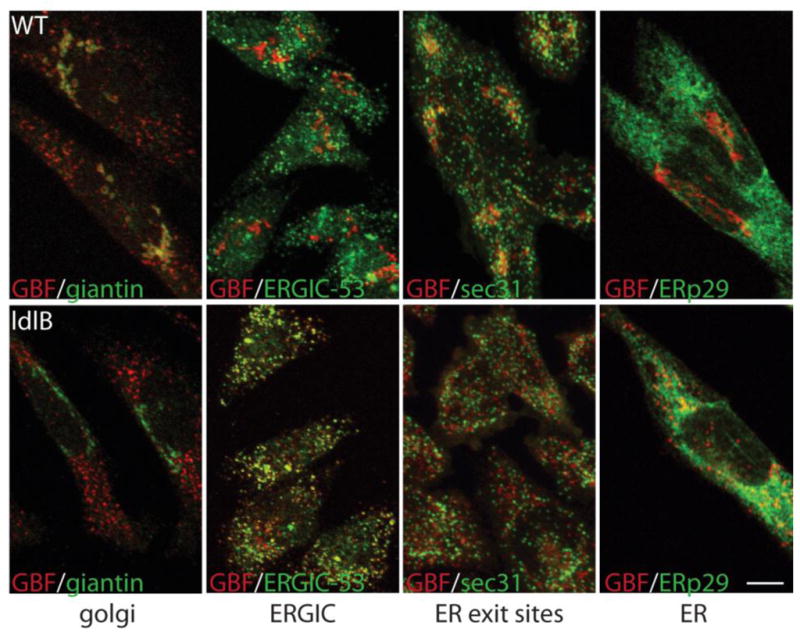
WT and ldlB CHO cells were co-stained with GBF1 and markers of the Golgi (giantin), ERGIC (ERGIC-53), ER exit sites (Sec31) and ER (ERp29) and visualized by confocal microscopy. Bar = 10 μm.
ldlB CHO cells are resistant to the Golgi-disrupting effects of BFA
Since mislocalization of GBF1 (the molecular target of BFA) to the ERGIC in ldlB cells might correspond with BFA resistance, we monitored the localization of GBF1 and giantin in BFA-treated WT and ldlB cells. In line with previous studies [28,29,32–34], GBF1 shifts from the Golgi to a localization consistent with ER and/or ER exit sites in wild type CHO cells following treatment (Figure 2A,B). A similar redistribution of GBF1 was noted in BFA treated ldlB cells but this effect was less pronounced, likely due to the fact that the majority of the protein was already redistributed and/or restricted to these non-Golgi compartments (Figure 2C,D). Unlike WT cells, giantin remained largely localized to the Golgi region in ldlB cells even after prolonged BFA exposure, indicating that these cells exhibit the same delayed effect of BFA as noted previously in Cog-deficient human fibroblasts (Figure 2B,D).
Figure 2. ldlB CHO cells are resistant to the Golgi-disrupting effects of BFA.

Wild type and COG1-deficient ldlB CHO cells were cultured in the absence (A,C) or presence (B,D) of 5 μg/mL for 40 min, co-stained with giantin and GBF1 antibodies and analyzed by confocal microscopy. Although GBF1 is readily detected on the Golgi in WT cells (A), very little Golgi staining of GBF1 was observed in the ldlB cells (C). Collapse of the Golgi marker giantin is observed in WT cells following BFA treatment (B) but the ldlB cells are resistant (D). Arrowheads denote cells that are enlarged in the far right panels. Bar = 10 μm.
Both GBF1 and BIG1 are mislocalized in COG knockdown HeLa cells
GBF1 and BIG1 localization was assessed in wild type and Cog3- and Cog6-knockdown HeLa cells to look at the effects of acute Cog depletion (Figure 3). GBF1 was again readily detected on the Golgi in WT cells as determined by its co-localization with GalNAcT2-GFP. Cog3 knockdown cells, however, exhibited a high degree of peripheral GBF1 staining. The majority of GBF1 in both Cog3 KD (87 ± 4%) and COG6 KD (78 ± 10%) cells was peripheral/cytosolic and not co-localized with the GalNAcT2-GFP-positive Golgi membranes (Figure 3). Interestingly, the late Golgi ARF-GEF BIG1 was strikingly redistributed in the Cog3 KD cells, suggesting that the Golgi association of other large ARF-GEFs is sensitive to COG deficiency. To address whether acute knockdown of Cog subunits leads to BFA resistance, we treated Cog3 and Cog6 KD cells with BFA for various times and monitored the extent of Golgi collapse using GFP-GalNAcT2 as a marker (Supplemental Figure 2). Our results demonstrated that Cog3 KD, and to a lesser extent, Cog6 KD cells exhibited delayed collapse of the Golgi into the ER, demonstrating that acute knockdown of Cog subunits also leads to BFA resistance.
Figure 3. Both GBF1 and BIG1 are mislocalized in COG knockdown HeLa cells.

GalNAcT2-GFP HeLa cells were mock transfected or transfected with Cog3 and Cog6 siRNA. After 72 hours, cells were fixed with 4% paraformaldehyde, permeabilized with Triton X-100, treated with anti-GBF1 and anti-BIG1 antibodies, and developed using HyLite-555-conjugated anti-mouse and HyLite-647-conjugated anti-rabbit antibodies. Samples were viewed by dual-laser confocal microscopy (63x oil objective). Control cells (A), Cog3 KD cells (B) and Cog6 KD cells (C). Bar = 10 μm.
Delayed Golgi collapse in Cog-deficient cells is specific to agents that bind GBF1
Before investigating the possible involvement of GBF1 localization on Cog-deficient fibroblasts, we first wanted to determine whether the delayed Golgi collapse induced by BFA in these cells is specific to this drug. To do so, we incubated wild type and Cog7-deficient fibroblasts with two additional Golgi-disrupting agents, Golgicide A (GCA) and tryphostin (AG1478). GCA and AG1478, both identified in screens for compounds that regulate intracellular Golgi trafficking, have been previously shown to specifically impact GBF1 function but not other large ARF-GEFs such as BIG1 and BIG2 [40, 41]. These compounds induce rapid Golgi collapse via tubules in a manner that is highly similar to BFA. Compared to the complete collapse of the Golgi seen in treated WT cells (Figure 4A-C), treatment of Cog7-deficient cells with both compounds resulted in a significant delay in the redistribution of cis-Golgi localized giantin to the ER (Figure 4D-F). The percentage of total cells that exhibit complete GCA-induced collapse of the Golgi is much greater in WT cells when compared to Cog7-deficient cells (64.1% in WT vs. 1.1% in Cog7 after 17 min treatment). Interestingly, these cells were almost completely resistant to the effects of AG1478, with only minimal Golgi collapse noted after 30 min treatment. Together, these findings support a role for GBF1 in the delayed Golgi collapse associated with Cog deficiency.
Figure 4. Delayed Golgi collapse in Cog-deficient cells is specific to agents that bind GBF1.
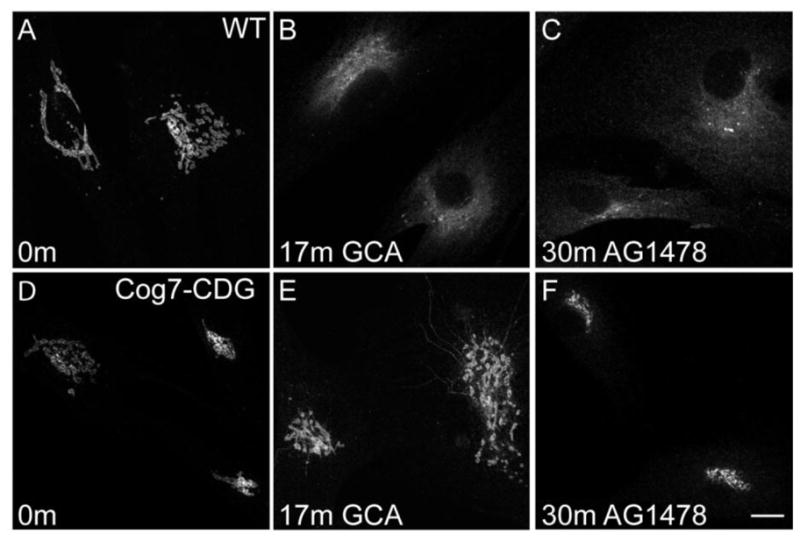
WT (A-C) and Cog7-deficient (D-F) fibroblasts were treated with 10 μM golgicide A or 20 μM AG1478 for the times shown and stained with antibodies against giantin. Bar = 10 μm.
The redistribution of GBF1 to the ERGIC in Cog7-deficient fibroblasts may be masked by the juxtaposition of ERGIC and Golgi membranes
Cog7-deficient cells exhibit a profound delay in the Golgi-disrupting effects of BFA. We investigated whether altered GBF1 distribution might underlie this effect by analyzing the steady-state localization of GBF1 and ERGIC in WT and Cog7-deficient human fibroblasts. As in the other cell lines studied, GBF1 is localized primarily to the Golgi region and showed some overlap with ERGIC-53 in WT fibroblasts, in particular in areas that are juxtaposed to this region (Figure 5A). In Cog7-deficient fibroblasts, we observed a greater peripheral distribution of GBF1. Minimal overlap between ERp29 and GBF1 was observed, however, suggesting that this peripheral staining does not reside in the ER compartment (data not shown). While still detected within the Golgi region in these cells, the boundary of GBF1 staining was less defined compared to WT fibroblasts and exhibited greater overlap with ERGIC-53. Since markers of the ERGIC compartment were previously shown to localize closer to the cis-Golgi membrane in Cog7-deficient cells, we next performed co-localization experiments with giantin and ERGIC-53 in these cells (Figure 5B). As shown, the juxtaposition of these markers and the Golgi-like staining pattern of ERGIC-53 (see arrow) are clearly evident in Cog7-deficient cells. In contrast, a more differentiated boundary of ERGIC-53 and giantin is observed in WT cells. Thus, it is possible that the majority of GBF1 is in fact mislocalized to the ERGIC compartment in the Cog7-deficient fibroblasts but this redistribution is masked in light of the shift of the ERGIC membranes closer to the cis-Golgi face.
Figure 5. The redistribution of GBF1 to the ERGIC in Cog7-deficient fibroblasts may be masked by the juxtaposition of ERGIC and Golgi membranes.

WT and Cog7-deficient fibroblasts were co-stained with antibodies against ERGIC-53 and GBF1 (panel A) and giantin (panel B). Compared to wild type cells, more peripheral GBF1 staining is observed in the Cog7-deficient fibroblasts as well as a shift of the GBF1-positive ERGIC membranes closer to the cis-Golgi face. Bar = 10 μm.
The steady-state levels and membrane recruitment of GBF1 are not significantly affected in Cog-deficient fibroblasts
Although we are able to visualize GBF1 staining on the Golgi in Cog7-deficient cells, it is possible that impaired recruitment at the Golgi or overall steady-state level of GBF1 in these fibroblasts might be altered, accounting for the delayed effects on BFA. To explore this possibility, we performed Western blot experiments and membrane recruitment assays in WT and Cog7-deficient fibroblasts and CHO cells. As shown in Figure 6A, there were no statistically significant differences in the steady-state levels of GBF1 in WT and Cog7 (111 ± 18% of WT) fibroblasts or WT and ldlB (88 ± 19% of WT) CHO cells. For the recruitment assays, fibroblasts were subjected to cytosol/membrane fractionation before and after BFA treatment. Total membrane and cytosolic pools were resolved by SDS-PAGE and GBF1 detected by Western blot analysis (Figure 6B). BFA treatment results in rapid and nearly complete shift of GBF1 from the cytosol to the membrane in both wild type and Cog-deficient cells. Quantitation of the fraction of GBF1 found in the membrane and cytosolic pools before and after BFA treatment (Figure 6C) revealed a consistently greater fraction of GBF1 within the membrane-bound fraction in the Cog-deficient cells (24% in WT vs. 41% in Cog7-deficient fibroblasts). Based on these findings, we conclude that overall levels and membrane association of GBF1 is not altered by defects in the Cog7 subunit. It is noteworthy that a larger fraction of GBF1 is associated with the membrane fraction in Cog-deficient cells under steady-state conditions which may reflect enhanced membrane residence/stability of this protein within pre-Golgi compartments.
Figure 6. Steady-state levels and BFA-induced recruitment of GBF1 onto membranes is not affected in Cog7-deficient fibroblasts.
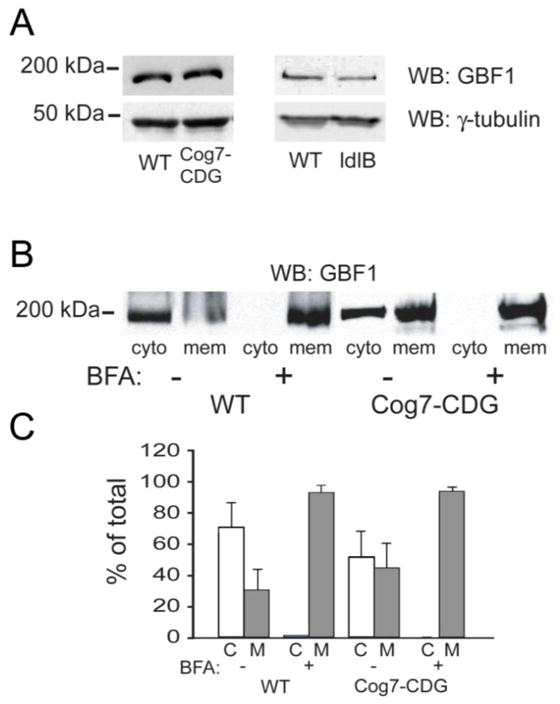
(A) Western blot analysis of GBF1 levels in WT and Cog7-deficient fibroblasts and WT and ldlB CHO cells. (B) Wild type and COG7-deficient fibroblasts were treated with BFA for 5 min. followed by disruption of the cells and cytosol/membrane fractionation. Total membrane and cytosolic pools were resolved by SDS-PAGE and GBF1 detected by Western blot analysis. (C) The percent of total GBF1 in each pool was quantified and plotted. The average from three independent experiments and the standard deviations is shown.
Bafilomycin treatment renders cells resistant to BFA and augments the delayed BFA response in Cog7-deficient cells in a GBF1-independent manner
In addition to Cog-CDG cells, variable BFA resistance has been observed in other CDG patient fibroblasts including those with defects in the proton transporter ATP6VOA2 [11]. To investigate whether the delayed response to BFA in these cells occurs in a GBF1-dependent manner, we inhibited vacuolar proton transport in wild type human fibroblasts using bafilomycin and then monitored the dynamics of giantin following BFA treatment. As shown in Figure 7, bafilomycin treatment results in a delay in the effects of BFA treatment. Although the vast majority of cells (~95%) exhibit an ER staining pattern after 20 min. exposure to BFA, pretreatment with bafilomycin significantly delays this collapse (Figure 7A). Complete BFA-induced collapse of giantin into the ER is noted in both control and bafilomycin-pretreated cells by 40 min. exposure. Importantly, we did not observe any obvious mislocalization of GBF1 to pre-Golgi compartments in bafilomycin-treated fibroblasts, suggesting that the delayed response to BFA is a GBF1-independent phenomenon (Figure 7C). Furthermore, the effects of bafilomycin on BFA resistance were not mediated by delayed COPI release since the kinetics of β-COP dissociation from the Golgi membrane was comparable to untreated cells (data not shown). Since COG deficiency and inhibition of vacuolar proton transport appear to result in BFA resistance via unique mechanisms, we investigated whether bafilomycin treatment would augment the delayed response to BFA in Cog7-deficient fibroblasts. As shown in Figure 7A and B, pretreatment of Cog7-deficient fibroblasts with 50 nM bafilomycin resulted in an additive delay in the BFA-induced Golgi collapse compared to untreated Cog7-deficient cells and bafilomycin pretreatment alone. This additive effect is particularly apparent at 40 min. BFA treatment, where bafilomycin pretreatment resulted in a 75% decrease in the percentage of cells with an ER staining only (80% with BFA alone vs. 20% with BFA and bafilomycin). Qualitative analysis of the bafilomycin-treated wild type cells revealed that the onset of BFA-induced Golgi collapse was not substantially altered since similar numbers of giantin-positive tubules were observed in both untreated and treated cells after 12 min of BFA exposure. These tubules, however, persisted much longer in the bafilomycin-treated cells compared to wild type, suggesting that their fusion with the ER was delayed (Figure 7A). This finding is consistent with studies indicating that certain membrane fusion events require proper acidification. Collectively, these results demonstrate that the effects of Cog depletion and Vo-type ATPase proton transport blockade are additive.
Figure 7. Bafilomycin treatment renders wild type cells resistant to BFA and augments the delayed BFA response in Cog-deficient cells in a GBF1-independent manner.

(Panel A) Wild type and Cog7-deficient fibroblasts were pretreated for 4 hr with 50 nM bafilomycin followed by BFA treatment (5 μg/mL) for either 20 or 40 min. The cells were stained for GBF1 and giantin and visualized by confocal microscopy. (Panel B) Quantitation of the percentage of cells with an ER staining pattern is shown. (Panel C) WT fibroblasts treated with and without 50 nM bafilomycin for 3 hr were stained for GBF1. Bar = 10 μm.
DISCUSSION
GBF1, normally present on the Golgi as well as the ERGIC and ER exit sites, was mislocalized in all of the Cog-deficient cells studied, although to varying degrees. Unlike other Cog-sensitive proteins (or GEARs), however, the stability of GBF1 is not affected by its mislocalization, since comparable levels of this protein were found in all the Cog-deficient cells (Figure 6). The most striking redistribution was observed in the ldlB CHO cells, where GBF1 was almost completely absent (but still detectable) from the Golgi membrane. It is noteworthy that COPI remains localized to the Golgi in these cells [42], suggesting that these low levels of Golgi-associated GBF1 are still sufficient to sustain COPI recruitment. Indeed, Lowe and coworkers made similar observations with regard to GBF1 and COPI localization within mitotic cells [43]. Although Golgi staining was clearly observed in the Cog-deficient fibroblasts and HeLa cells, a greater fraction of GBF1 appeared to be found in peripheral, non-Golgi sites. The basis for the variable localization of GBF1 in the different Cog-deficient cells is not clear but may be related to the organization of pre-Golgi compartments in these cell types (see Figure 5), differences in the localization of the unidentified GBF1 membrane receptor or even the degree of knockdown and specificity of the individual COG subunits.
A role for GBF1 mislocalization in the BFA resistance of Cog-deficient cells is suggested but not proven by these findings. Earlier work by several groups has demonstrated that alterations in GBF1 levels can influence sensitivity to BFA treatment. More specifically, the effect of GBF1 overexpression appears to be mediated by the ability of excess cytosolic GBF1 to bind BFA and reduce its action on endogenous pools. In the Cog-deficient cells, a role for GBF1 in BFA resistance would not be attributed to altered levels of the protein but rather the mislocalization of GBF1 (or its membrane receptor) to non-Golgi compartments. It is possible, however, that other direct or indirect consequences of Cog deficiency, or molecular compensation in response to loss of Cog complex integrity, are responsible for the BFA resistance in these cells. Recent work has demonstrated differences in the lipid content of the Golgi in Cog-deficient cells [22]. Such changes might alter the mixing of lipids following fusion of these tubules with the ER. Moreover, lipids such as PI4P play an important role the recruitment of GBF1 to the membrane [44]. Thus, effects on proteins and enzymes involved in phosphatidylinositol metabolism in Cog-deficient cells might also underlie GBF1 mislocalization. While our data does not support impaired recruitment of GBF1 to the membrane, it is equally possible that this change in Golgi lipids alone alters the rate of tubule formation in a manner that is independent of GBF1. We believe the finding that acute knockdown of Cog subunits also resulted in BFA resistance (Supplemental Figure 2) would argue against molecular compensation as the underlying cause of BFA resisitance but cannot rule out that this compensation occurs rapidly upon Cog depletion in order to maintain Golgi function.
The COG complex is generally believed to regulate intra-Golgi retrograde transport via the tethering COPI-coated vesicles. Thus, the failure of GBF1 to localize to the Golgi in the ldlB cells is therefore of interest since it suggests that the anterograde recycling of this protein between the ERGIC and cis-Golgi is affected. A shift in the distribution of GBF1 to the ERGIC and ER exit sites has also been noted in cells that express a GTP-restricted mutant of Rab1b and those grown at 15°C [32, 45]. Therefore, loss of COG complex integrity and the subsequent mislocalization of GBF1 likely impact the formation and movement of selected transport intermediates between the ER, ERGIC and cis-Golgi. Along with our earlier findings, the current data suggest that actively recycling proteins such as ERGIC-53 or GBF1 exhibit particular sensitivity to loss of COG complex function. Since COPI has been implicated in ERGIC-to-Golgi transport events [46–48], it is possible that altered Cog-dependent tethering of COPI-coated transport intermediates from the cis-Golgi face to pre-Golgi compartments (or vice versa), in a manner analogous to its function in intra Golgi transport, is responsible for the mislocalization and impaired recycling of these proteins. Importantly, though, impaired COG function does not appear to substantially disrupt the constitutive flow of secreted proteins through the ER-Golgi interface. Further investigation of the mechanisms (direct or indirect) whereby the COG complex selectively mediates protein recycling between the ER and Golgi is warranted.
In addition to Cog-deficient cells, a delayed response to BFA has been noted in fibroblasts from patients with defects in the a2 subunit of the V-type ATPase transporter [11]. Our present experiments suggest that this delayed response likely occurs independently of GBF1. While treatment of normal fibroblasts with bafilomycin, an inhibitor of the V-type ATPase transporter, renders these cells resistant to the effects of BFA, we did not observe any GBF1 mislocalization upon treatment. In contrast to Cog-deficient cells, the most sensitive step in BFA-induced Golgi collapse in the bafilomycin-treated cells appears to be extension and fusion of tubules with the ER, not the initial formation of these tubules. This is supported by the fact that sustained tubulation of the Golgi is readily observed in bafilomycin- and BFA-treated cells (Figure 6A). Recent studies have indicated a function for vesicle acidification in the membrane fusion events [49, 50]. Thus, the delayed Golgi collapse in response to bafilomycin treatment may be a consequence of impaired fusion of BFA-induced tubules with the ER as opposed to altered concentration of GBF1 at the Golgi membrane. This scenario is supported by observations that treatment of cells with monensin or nigericin counteracts the effects of BFA by blocking tubule fusion with the ER [51]. Our observation that bafilomycin treatment augments the delayed response of COG7-deficient fibroblasts to BFA is consistent with two separate mechanisms of action. However, in light of the influence of V-ATPase-dependent intra-endosomal acidification on the recruitment of ARF6 and the small ARF-GEF ARNO, it is possible that these mechanisms might also intersect, in particular with the trans-Golgi network, to confer resistance to Golgi-disrupting agents such as BFA. We are currently exploring the possibility that the severe mislocalization of BIG1 in the Cog3 KD cells results from altered vesicle acidification.
In conclusion, our findings reveal mechanisms for unique response of Cog-deficient cells to Golgi-disrupting agents such as BFA and expand the list of proteins whose localization and movement at the ER-Golgi boundary is sensitive to loss of COG function. In the ongoing search for novel CDG defects, treatment with specific Golgi disrupting agents should continue to find a useful application in identifying sensitive transport events within the secretory apparatus and assessing overall Golgi dynamics.
Supplementary Material
Acknowledgments
We wish to thank Monty Krieger for providing us with the ldlB cells for this study, David Haslam for his generous gift of Golgicide A and Fred Gorelick for providing the Sec31 antisera. This work was supported by grants from the American Heart Association (R.S. 0535026N), National Science Foundation (V.L. MCB-0645163) and NIH (V.L. 1R01GM083144).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Jaeken J. Congenital disorders of glycosylation (CDG): it’s (nearly) all in it! Journal of Inherited Metabolic Disease. 2011 doi: 10.1007/s10545-011-9299-3. [DOI] [PubMed] [Google Scholar]
- 2.Jaeken J, Stibler H, Hagberg B. The carbohydrate-deficient glycoprotein syndrome. A new inherited multisystemic disease with severe nervous system involvement. Acta Paediatr Scand Suppl. 1991;375:1–71. [PubMed] [Google Scholar]
- 3.Jaeken J, van Eijk HG, van der Heul C, Corbeel L, Eeckels R, Eggermont E. Sialic acid-deficient serum and cerebrospinal fluid transferrin in a newly reCognized genetic syndrome. Clin Chim Acta. 1984;144:245–247. doi: 10.1016/0009-8981(84)90059-7. [DOI] [PubMed] [Google Scholar]
- 4.Haeuptle MA, Hennet T. Congenital disorders of glycosylation: an update on defects affecting the biosynthesis of dolichol-linked oligosaccharides. Human Mutation. 2009;30:1628–1641. doi: 10.1002/humu.21126. [DOI] [PubMed] [Google Scholar]
- 5.Eklund EA, Freeze HH. The congenital disorders of glycosylation: a multifaceted group of syndromes. NeuroRx. 2006;3:254–263. doi: 10.1016/j.nurx.2006.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Foulquier F, Ungar D, Reynders E, Zeevaert R, Mills P, Garcia-Silva MT, Briones P, Winchester B, Morelle W, Krieger M, Annaert W, Matthijs G. A new inborn error of glycosylation due to a Cog8 deficiency reveals a critical role for the Cog1-Cog8 interaction in COG complex formation. Human Molecular Genetics. 2007;16:717–730. doi: 10.1093/hmg/ddl476. [DOI] [PubMed] [Google Scholar]
- 7.Foulquier F, Vasile E, Schollen E, Callewaert N, Raemaekers T, Quelhas D, Jaeken J, Mills P, Winchester B, Krieger M, Annaert W, Matthijs G. Conserved oligomeric Golgi complex subunit 1 deficiency reveals a previously uncharacterized congenital disorder of glycosylation type II. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:3764–3769. doi: 10.1073/pnas.0507685103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Paesold-Burda P, Maag C, Troxler H, Foulquier F, Kleinert P, Schnabel S, Baumgartner M, Hennet T. Deficiency in COG5 causes a moderate form of congenital disorders of glycosylation. Human Molecular Genetics. 2009;18:4350–4356. doi: 10.1093/hmg/ddp389. [DOI] [PubMed] [Google Scholar]
- 9.Reynders E, Foulquier F, Leao Teles E, Quelhas D, Morelle W, Rabouille C, Annaert W, Matthijs G. Golgi function and dysfunction in the first COG4-deficient CDG type II patient. Human Molecular Genetics. 2009;18:3244–3256. doi: 10.1093/hmg/ddp262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu X, Steet RA, Bohorov O, Bakker J, Newell J, Krieger M, Spaapen L, Kornfeld S, Freeze HH. Mutation of the COG complex subunit gene COG7 causes a lethal congenital disorder. Nature Medicine. 2004;10:518–523. doi: 10.1038/nm1041. [DOI] [PubMed] [Google Scholar]
- 11.Kornak U, Reynders E, Dimopoulou A, van Reeuwijk J, Fischer B, Rajab A, Budde B, Nurnberg P, Foulquier F, Lefeber D, Urban Z, Gruenewald S, Annaert W, Brunner HG, van Bokhoven H, Wevers R, Morava E, Matthijs G, Van Maldergem L, Mundlos S. Impaired glycosylation and cutis laxa caused by mutations in the vesicular H+-ATPase subunit ATP6V0A2. Nature Genetics. 2008;40:32–34. doi: 10.1038/ng.2007.45. [DOI] [PubMed] [Google Scholar]
- 12.Kranz C, Ng BG, Sun L, Sharma V, Eklund EA, Miura Y, Ungar D, Lupashin V, Winkel RD, Cipollo JF, Costello CE, Loh E, Hong W, Freeze HH. COG8 deficiency causes new congenital disorder of glycosylation type IIh. Human Molecular Genetics. 2007;16:731–741. doi: 10.1093/hmg/ddm028. [DOI] [PubMed] [Google Scholar]
- 13.Zeevaert R, Foulquier F, Jaeken J, Matthijs G. Deficiencies in subunits of the Conserved Oligomeric Golgi (COG) complex define a novel group of Congenital Disorders of Glycosylation. Molecular Genetics and Metabolism. 2008;93:15–21. doi: 10.1016/j.ymgme.2007.08.118. [DOI] [PubMed] [Google Scholar]
- 14.Hucthagowder V, Morava E, Kornak U, Lefeber DJ, Fischer B, Dimopoulou A, Aldinger A, Choi J, Davis EC, Abuelo DN, Adamowicz M, Al-Aama J, Basel-Vanagaite L, Fernandez B, Greally MT, Gillessen-Kaesbach G, Kayserili H, Lemyre E, Tekin M, Turkmen S, Tuysuz B, Yuksel-Konuk B, Mundlos S, Van Maldergem L, Wevers RA, Urban Z. Loss-of-function mutations in ATP6V0A2 impair vesicular trafficking, tropoelastin secretion and cell survival. Human Molecular Genetics. 2009;18:2149–2165. doi: 10.1093/hmg/ddp148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Steet R, Kornfeld S. COG-7-deficient Human Fibroblasts Exhibit Altered Recycling of Golgi Proteins. Molecular Biology of the Cell. 2006;17:2312–2321. doi: 10.1091/mbc.E05-08-0822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Oka T, Ungar D, Hughson FM, Krieger M. The COG and COPI complexes interact to control the abundance of GEARs, a subset of Golgi integral membrane proteins. Molecular Biology of the Cell. 2004;15:2423–2435. doi: 10.1091/mbc.E03-09-0699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shestakova A, Zolov S, Lupashin V. COG complex-mediated recycling of Golgi glycosyltransferases is essential for normal protein glycosylation. Traffic (Copenhagen, Denmark) 2006;7:191–204. doi: 10.1111/j.1600-0854.2005.00376.x. [DOI] [PubMed] [Google Scholar]
- 18.Zolov SN, Lupashin VV. Cog3p depletion blocks vesicle-mediated Golgi retrograde trafficking in HeLa cells. Journal of Cell Biology. 2005;168:747–759. doi: 10.1083/jcb.200412003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Peanne R, Legrand D, Duvet S, Mir AM, Matthijs G, Rorher J, Foulquier F. Differential Effects of Lobe a and Lobe B of the Cog Complex on the Stability of B4galt1 and St6gal1. Glycobiology. 2010 doi: 10.1093/glycob/cwq176. [DOI] [PubMed] [Google Scholar]
- 20.Pokrovskaya ID, Willett R, Smith RD, Morelle W, Kudlyk T, Lupashin VV. COG complex specifically regulates the maintenance of Golgi glycosylation machinery. Glycobiology. 2011 doi: 10.1093/glycob/cwr028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Spessott W, Uliana A, Maccioni HJ. Defective GM3 synthesis in Cog2 null mutant CHO cells associates to mislocalization of lactosylceramide sialyltransferase in the Golgi complex. Neurochem Res. 2010;35:2161–2167. doi: 10.1007/s11064-010-0319-8. [DOI] [PubMed] [Google Scholar]
- 22.Spessott W, Uliana A, Maccioni HJ. Cog2 null mutant CHO cells show defective sphingomyelin synthesis. Journal of Biological Chemistry. 2010;285:41472–41482. doi: 10.1074/jbc.M110.150011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Morsomme P, Prescianotto-Baschong C, Riezman H. The ER v-SNAREs are required for GPI-anchored protein sorting from other secretory proteins upon exit from the ER. Journal of Cell Biology. 2003;162:403–412. doi: 10.1083/jcb.200212101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Duran JM, Valderrama F, Castel S, Magdalena J, Tomas M, Hosoya H, Renau-Piqueras J, Malhotra V, Egea G. Myosin motors and not actin comets are mediators of the actin-based Golgi-to-endoplasmic reticulum protein transport. Molecular Biology of the Cell. 2003;14:445–459. doi: 10.1091/mbc.E02-04-0214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yan JP, Colon ME, Beebe LA, Melancon P. Isolation and characterization of mutant CHO cell lines with compartment-specific resistance to brefeldin A. Journal of Cell Biology. 1994;126:65–75. doi: 10.1083/jcb.126.1.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Garcia-Mata R, Szul T, Alvarez C, Sztul E. ADP-ribosylation factor/COPI-dependent events at the endoplasmic reticulum-Golgi interface are regulated by the guanine nucleotide exchange factor GBF1. Molecular Biology of the Cell. 2003;14:2250–2261. doi: 10.1091/mbc.E02-11-0730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Niu TK, Pfeifer AC, Lippincott-Schwartz J, Jackson CL. Dynamics of GBF1, a Brefeldin A-sensitive Arf1 exchange factor at the Golgi. Molecular Biology of the Cell. 2005;16:1213–1222. doi: 10.1091/mbc.E04-07-0599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Szul T, Garcia-Mata R, Brandon E, Shestopal S, Alvarez C, Sztul E. Dissection of membrane dynamics of the ARF-guanine nucleotide exchange factor GBF1. Traffic (Copenhagen, Denmark) 2005;6:374–385. doi: 10.1111/j.1600-0854.2005.00282.x. [DOI] [PubMed] [Google Scholar]
- 29.Szul T, Grabski R, Lyons S, Morohashi Y, Shestopal S, Lowe M, Sztul E. Dissecting the role of the ARF guanine nucleotide exchange factor GBF1 in Golgi biogenesis and protein trafficking. Journal of Cell Science. 2007;120:3929–3940. doi: 10.1242/jcs.010769. [DOI] [PubMed] [Google Scholar]
- 30.Turi TG, Webster P, Rose JK. Brefeldin A sensitivity and resistance in Schizosaccharomyces pombe. Isolation of multiple genes conferring resistance. Journal of Biological Chemistry. 1994;269:24229–24236. [PubMed] [Google Scholar]
- 31.Claude A, Zhao BP, Kuziemsky CE, Dahan S, Berger SJ, Yan JP, Armold AD, Sullivan EM, Melancon P. GBF1: A novel Golgi-associated BFA-resistant guanine nucleotide exchange factor that displays specificity for ADP-ribosylation factor 5. Journal of Cell Biology. 1999;146:71–84. [PMC free article] [PubMed] [Google Scholar]
- 32.Kawamoto K, Yoshida Y, Tamaki H, Torii S, Shinotsuka C, Yamashina S, Nakayama K. GBF1, a guanine nucleotide exchange factor for ADP-ribosylation factors, is localized to the cis-Golgi and involved in membrane association of the COPI coat. Traffic (Copenhagen, Denmark) 2002;3:483–495. doi: 10.1034/j.1600-0854.2002.30705.x. [DOI] [PubMed] [Google Scholar]
- 33.Zhao X, Lasell TK, Melancon P. Localization of large ADP-ribosylation factor-guanine nucleotide exchange factors to different Golgi compartments: evidence for distinct functions in protein traffic. Molecular Biology of the Cell. 2002;13:119–133. doi: 10.1091/mbc.01-08-0420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sciaky N, Presley J, Smith C, Zaal KJ, Cole N, Moreira JE, Terasaki M, Siggia E, Lippincott-Schwartz J. Golgi tubule traffic and the effects of brefeldin A visualized in living cells. Journal of Cell Biology. 1997;139:1137–1155. doi: 10.1083/jcb.139.5.1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lippincott-Schwartz J. Membrane cycling between the ER and Golgi apparatus and its role in biosynthetic transport. Subcellular Biochemistry. 1993;21:95–119. doi: 10.1007/978-1-4615-2912-5_5. [DOI] [PubMed] [Google Scholar]
- 36.Cherfils J, Melancon P. On the action of Brefeldin A on Sec7-stimulated membrane-recruitment and GDP/GTP exchange of Arf proteins. Biochemical Society Transactions. 2005;33:635–638. doi: 10.1042/BST0330635. [DOI] [PubMed] [Google Scholar]
- 37.Garcia-Mata R, Sztul E. The membrane-tethering protein p115 interacts with GBF1, an ARF guanine-nucleotide-exchange factor. EMBO Reports. 2003;4:320–325. doi: 10.1038/sj.embor.embor762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Monetta P, Slavin I, Romero N, Alvarez C. Rab1b interacts with GBF1 and modulates both ARF1 dynamics and COPI association. Molecular Biology of the Cell. 2007;18:2400–2410. doi: 10.1091/mbc.E06-11-1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Smith RD, Willett R, Kudlyk T, Pokrovskaya I, Paton AW, Paton JC, Lupashin VV. The COG complex, Rab6 and COPI define a novel Golgi retrograde trafficking pathway that is exploited by SubAB toxin. Traffic (Copenhagen, Denmark) 2009;10:1502–1517. doi: 10.1111/j.1600-0854.2009.00965.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pan H, Yu J, Zhang L, Carpenter A, Zhu H, Li L, Ma D, Yuan J. A novel small molecule regulator of guanine nucleotide exchange activity of the ADP-ribosylation factor and golgi membrane trafficking. Journal of Biological Chemistry. 2008;283:31087–31096. doi: 10.1074/jbc.M806592200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Saenz JB, Sun WJ, Chang JW, Li J, Bursulaya B, Gray NS, Haslam DB. Golgicide A reveals essential roles for GBF1 in Golgi assembly and function. Nature Chemical Biology. 2009;5:157–165. doi: 10.1038/nchembio.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Podos SD, Reddy P, Ashkenas J, Krieger M. LDLC encodes a brefeldin A-sensitive, peripheral Golgi protein required for normal Golgi function. Journal of Cell Biology. 1994;127:679–691. doi: 10.1083/jcb.127.3.679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Morohashi Y, Balklava Z, Ball M, Hughes H, Lowe M. Phosphorylation and membrane dissociation of the ARF exchange factor GBF1 in mitosis. Biochemical Journal. 2010;427:401–12. doi: 10.1042/BJ20091681. [DOI] [PubMed] [Google Scholar]
- 44.Dumaresq-Doiron K, Savard MF, Akam S, Costantino S, Lefrancois S. The phosphatidylinositol 4-kinase PI4KIIIalpha is required for the recruitment of GBF1 to Golgi membranes. Journal of Cell Science. 2010;123:2273–2280. doi: 10.1242/jcs.055798. [DOI] [PubMed] [Google Scholar]
- 45.Alvarez C, Garcia-Mata R, Brandon E, Sztul E. COPI recruitment is modulated by a Rab1b-dependent mechanism. Molecular Biology of the Cell. 2003;14:2116–2127. doi: 10.1091/mbc.E02-09-0625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lippincott-Schwartz J, Cole NB, Donaldson JG. Building a secretory apparatus: role of ARF1/COPI in Golgi biogenesis and maintenance. Histochemistry and Cell Biology. 1998;109:449–462. doi: 10.1007/s004180050247. [DOI] [PubMed] [Google Scholar]
- 47.Aridor M, Bannykh SI, Rowe T, Balch WE. Sequential coupling between COPII and COPI vesicle coats in endoplasmic reticulum to Golgi transport. Journal of Cell Biology. 1995;131:875–893. doi: 10.1083/jcb.131.4.875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rowe T, Aridor M, McCaffery JM, Plutner H, Nuoffer C, Balch WE. COPII vesicles derived from mammalian endoplasmic reticulum microsomes recruit COPI. Journal of Cell Biology. 1996;135:895–911. doi: 10.1083/jcb.135.4.895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Galli T, McPherson PS, De Camilli P. The V0 sector of the V-ATPase, synaptobrevin, and synaptophysin are associated on synaptic vesicles in a Triton X-100-resistant, freeze-thawing sensitive, complex. Journal of Biological Chemistry. 1996;271:2193–2198. doi: 10.1074/jbc.271.4.2193. [DOI] [PubMed] [Google Scholar]
- 50.Wada Y, Sun-Wada GH, Tabata H, Kawamura N. Vacuolar-type proton ATPase as regulator of membrane dynamics in multicellular organisms. Journal of Bioenergetics and Biomembranes. 2008;40:53–57. doi: 10.1007/s10863-008-9128-z. [DOI] [PubMed] [Google Scholar]
- 51.Barzilay E, Ben-Califa N, Hirschberg K, Neumann D. Uncoupling of brefeldin a-mediated coatomer protein complex-I dissociation from Golgi redistribution. Traffic (Copenhagen, Denmark) 2005;6:794–802. doi: 10.1111/j.1600-0854.2005.00317.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.