

Abstract
Systems biology is an emerging discipline that combines high-content, multiplexed measurements with informatic and computational modeling methods to better understand biological function at various scales. Here we present a detailed review of the methods used to create computational models and conduct simulations of immune function, We provide descriptions of the key data gathering techniques employed to generate the quantitative and qualitative data required for such modeling and simulation and summarize the progress to date in applying these tools and techniques to questions of immunological interest, including infectious disease. We include comments on what insights modeling can provide that complement information obtained from the more familiar experimental discovery methods used by most investigators and why quantitative methods are needed to eventually produce a better understanding of immune system operation in health and disease.
Keywords: high-throughput analysis, modeling, genomics, proteomics, RNAi
Introduction
Immune responses are symphonies of molecular and cellular interactions, with each player doing its part to produce the composite behavior we see as effective host defense, or when dis-coordinated, as immunopathology or immunodeficiency. Just as the listening separately to the notes played by individual instruments fails to capture the ensemble effect achieved when an entire orchestra plays in unison, so too are we limited in our understanding of how the immune system operates when we focus only on the properties or actions of one or a few unconnected components.
In the 19th and early 20th century, biology was largely a study of physiology, the integrated basis for the functionality of an organism. However, with the advent of new instrumentation and technology in the late 1970’s, especially recombinant DNA methods, biology became progressively more reductionist, with a focus on individual cells and molecules, and immunology was no exception to this trend. The new knowledge acquired by the field through many such detailed studies has been enormously important in developing a parts list of the components involved in immune processes and in identifying some of the contributions of these molecular and cellular elements to the overall functioning of the system. Nonetheless, this information has still yielded only limited insights into the way these various elements integrate with each other to give rise to complex immunological behaviors, especially into how small quantitative changes in individual component function affect more global properties. This latter issue is of substantial importance in understanding how polymorphisms linked to disease by large-scale genetic studies influence immune function, as just one example.
At the same time as new tools were developed for and applied to ever finer dissection of the cell, genes, and proteins of the immune system, another set of technological advances increased the rate of data acquisition from a trickle to a stream to a river that has, with the commoditization of microarrays (1), the widespread use of deep sequencing methods (2), the advent of highly multiplexed flow cytometry (3), and the availability of high-throughput proteomics (4), turned into a torrent. Rather than exploring a single element in depth, these latter technologies are employed for broad probing of the state of biological systems (gene expression, protein identity, or substrate modification). This has led to a major change in how research is conducted in many laboratories – rather than experiments being designed based on pre-formed hypotheses derived from past training and knowledge of the literature, high-throughput methods are being used for unbiased exploration of the properties of a system to then generate novel hypotheses (5). It is up to the investigator to sort through the massive amount of new data flowing from the various multiplex technologies, a process that requires substantial ability in statistics and/or the capacity to properly use algorithms and software developed by experts in mathematics and computation. We have thus entered a new domain, in which the skills of ‘bioinformaticians’ are becoming essential elements in the research efforts of many laboratories. The technical capacity to generate these large-scale, in some cases global, datasets has in turn led to the emergence of the new discipline of ‘systems biology’, which in its simplest form is the old physiology recast in modern guise. It constitutes an attempt by the field to move from the very specific, from the detail, from the single molecule or gene, to a quantitative analysis of such elements as they operate together to give rise to behaviors not possessed by any single component alone – so-called ‘emergent properties,’ (6) the symphony rather than the notes of just the violin or oboe.
Many investigators consider bioinformatics synonymous with systems biology. But the truth is more complex. While statistical analysis of large datasets to look for trends, to cluster individual components into related groups, or to uncover connectivity among elements to produce large network maps are all essential to making use of such extensive information, these approaches alone fall short of moving the field from mere organization of knowledge to a deeper understanding of the principles underlying a system’s behavior or to an explanation of its mode of operation. The most common output from informatic manipulation of data elements is a non-parametric graph that shows qualitative interactions – often referred to as a ‘hairball’ or ‘ridiculome’1 because of the enormous complexity of such global depictions. In truth, these graphs are extremely useful for illustrating relationships between elements and for understanding the organization of components into operational modules, but they do not allow the investigator to predict how alterations in the concentration or efficiency of function of a particular element will influence the overall system’s activity or to discern why/how certain properties of the system arise from its elements. But in the end, this is just what we want from such a systemic analysis; the ability to fathom how higher-level function emerges from components that on their own lack the capacity in question and to predict how perturbations of individual elements will change this behavior, both for the basic insight this provides and for the potential clinical utility of such information.
It is the domain of ‘modelers’ to move from informatic analyses into this more functional realm. Mathematical or computational modeling is not a new endeavor, especially in immunology, but it is a less widely employed and appreciated aspect of the emerging discipline of systems biology as compared to bioinformatic analysis of data. But we believe that the two are complementary and indeed, each cannot reach its full potential without the other. Computational simulation is only effective if the modeler has in hand the properly processed and analyzed data necessary to instantiate a model close to biological reality (in terms of element identity, organization, and quantitative parameters). At any level of resolution, from molecules, to cells, to tissues, to a complete organism, the modeler needs the contributions of informaticians to develop a realistic and valid model structure for further computational processing. On the other hand, without modeling, the mere organization of data does not add the necessary insight into global system performance sought by biologists.
From the perspective of the practicing immunologist, what does systems biology in all its guises have to offer? Isn’t experimentation - not mathematical twiddling - really the essential activity involved in gaining new understanding of how the immune system functions? And yes, informatics is useful for handling large datasets, but isn’t its major value in discovery of new interesting molecules or genes so one can go back into the lab to study these in detail using comfortable experimental tools and techniques? In this review we argue that these existing paradigms are changing - that the value of traditional experimental studies will increase dramatically if more quantitative tools are introduced into mainstream immunology research in the form of analytic measurements, formalized model generation, simulations, and computer predictions, built on a foundation involving systematic measurements organized and parsed by informatics approaches.
What is the basis for this view? We suggest that too often, interpretation of experimental data is limited by a failure to intuit the complex, non-linear behaviors typical of highly connected systems with large numbers of feedback connections (7; 8), which of course perfectly describes the immune system. Add to this the exponential increase and decrease in lymphocyte cell numbers during adaptive immune responses, properties that markedly amplify the influence of very small differences in the activity of molecular circuits or cells (9; 10), and the need for more formal representations and quantitative analyses of immune function becomes even more evident.
We are not talking here about the type of ad hoc ‘theoretical immunobiology’ that has acquired a questionable name in the past. Rather, we are referring to combining rich experimental datasets and existing knowledge in the field with newer efforts to obtain more quantitative measurements of biochemical or cellular parameters suitable for computer modeling and simulation. Predictions of system behavior from such simulations obtained under defined conditions corresponding to experimentally testable situations amount to ‘in silico experimentation’ (11). These predicted outcomes must then be put to the test at the bench, to examine the strength of the underlying computational model. Through iterative cycles of such model building, simulation, prediction, experiment, and model refinement (when experimental results and prediction disagree as they inevitably will), one can develop much more complete and informative models of immunological processes than those we formulate purely in our imagination or represent as simplified cartoons in reviews.
What about the scale of such models? Many investigators bemoan the presumed need for ‘completeness’ in order to achieve a useful model and despair of obtaining the data needed to reach this ultimate goal. Although for bacteria or yeast it is possible to undertake truly ‘system-level’ studies involving the measurement all gene transcripts or proteins expressed by a cell under various conditions and the systematic perturbation of each of these elements through mutation, this is clearly impractical or impossible for more complex organisms. However, useful models that represent emergent behaviors need not involve the entire system – the complex properties of subcircuits or modules that form key parts of larger networks are valuable to investigate and simulate on their own (7; 12) even if the eventual goal must be to stitch such incomplete models together into a grander scheme that more truly reflects overall physiology. To conduct studies at the ‘systems’ level, it is merely necessary to at least move up the scale from individual component dissection to a consideration of integrated behavior of sets of connected components (molecules, cells, even disparate tissues). Such efforts help us organize our thinking about the aspects of the subnetwork’s structure that give rise to its specific properties [amplification, noise suppression, time-gated function, and so on (13)], and can assist in understanding the underlying control circuits that regulate behaviors like switching between tolerant and immune states (8), the antigen thresholds required for induction of responses, original antigenic sin, the choice of CD4 effector fate, and many others. In concert with the critical efforts already underway to systematically obtain data on gene expression in immune cells (14) and to quantify aspects of immune function previously examined in a more qualitative manner, we can begin to generate a body of models for many such modules of immune function. These in turn can each be refined by contributions from many investigators in the field, hopefully over time approaching the underlying reality more and more closely, and leading eventually to the generation of an integrated ‘supermodel’ as these smaller pieces prove their worth through rigorous experimental testing. It is an opportunity for all immunologists to contribute to and receive back from a group undertaking that ultimately supports their own specific research interests while advancing the entire field. Rather than being concerned about ‘big science’ in thinking about systems approaches, we hope that immunologists will view systems immunology as the new immunophysiology, with opportunities for all to participate and to benefit.
As discussed above, there are two major threads in systems biology, informatics and modeling. Each has become such a large enterprise that we cannot do justice to both here, and so have opted to focus on the less commonly used modeling and simulation aspect. In the body of this review, we discuss the computational approaches and tools available for translating data into models suitable for simulation and prediction of biological behavior, the key technologies for data acquisition that contribute to effective computational modeling, and the limitations that must be overcome for their more effective use in supporting these endeavors. In each section, we provide examples of how these technologies and tools have already begun to contribute to a better understanding of the immune system. We end with a perspective on what can be expected in the next few years in this rapidly changing arena.
Modeling and Simulation Scales, Paradigms, Techniques, and Integrated Approaches
Systems biology puts a strong emphasis on quantitative data and strategies for comprehensive measurements of biological parameters. To understand why the cycle of hypothesis building, data acquisition, dynamical modeling and subsequent refinement of the hypothesis demands this emphasis, it is best to begin with a summary of the different questions that can be addressed by modeling and simulation, the various types of computational models that can be built, and the tools available or still needed for construction and simulation of these various models (Table 1).
TABLE 1.
Computational approaches and tools
| Modeling approach | Typical applications | Limitations | Tools |
|---|---|---|---|
| Individual particle-based stochastic | Small subcellular signaling processes, apects of bacterial biochemistry | Limited to small systems (in terms of space and chemical complexity) | MCell (32), Smoldyn (315), ChemCell (316), GetBonNie (non- spatial) (49) |
| Particle number stochastic | Signaling processes with important stochastic aspects (due to small system size or high sensitivity) | Limited to small systems (in terms of space and chemical complexity), less detail than individual particle simulation | MesoRD (35), SmartCell (33), GetBonNie (non- spatial) |
| Concentration-based spatial, non-stochastic | Cellular signaling processes with important spatial aspects | Either high spatial resolution or biochemical complexity, No stochasticity | Virtual Cell (37), Simmune (36) |
| Concentration-based, non-spatial, non-stochastic | Cellular signaling processes without spatial aspects | Assumption of global biochemical homogeneity in the simulated system | Copasi (46), E-cell (44), Cellware (45), Systems Biology Workbench (47), GetBonNie |
Modeling and Simulation at the Molecular Scale
Biologists study diverse questions that range from the molecular (vibrational modes that affect protein folding or molecular binding) to the organismal. Although substantial progress has been made in simulation techniques and in the development of specialized computer hardware that permit ab-initio molecular dynamics simulations of large peptide folding processes (15; 16), the most fundamental scale of computational models widely used in immunology is the scale describing the dynamics of molecular interactions in terms of rates of association, dissociation, and post-translational modification. Based on such molecular event rates, dynamic models can be developed that cast the time evolution of a particular part of cellular biochemistry into mathematical expressions that describe the rate of change per unit time of the number of its molecular species. Depending on the specific question at hand, the focus of such mathematical descriptions may be on the formation of individual multi-molecular signaling complexes (‘clusters’), on the biochemistry of subcellular compartments, or on the biochemical behavior of signaling pathways within entire cells. In most cases, the equations are too complicated to be solved ‘on a piece of paper’ and are instead used in computer simulations that iteratively ‘solve’ them for small time steps, to calculate the evolution of the system over biological time periods of seconds to hours or days.
Immunologists have long recognized the importance of early receptor-receptor ligation events in regulating cell function and differentiation state (17–19) and there is increasing evidence for a crucial role of the biochemical and conformational properties of the membrane and proximal actin and myosin fiber structures in guiding such events during lymphocyte interactions (20–25). These insights have prompted numerous theoretical studies focusing on the small-scale spatial aspects of cellular signaling. Stochastic spatially-resolved simulations (26) explore the early kinetics of signaling processes within small networks of individually interacting proteins and lipids. Many of these approaches utilize Monte Carlo2 methods (27; 28) to incorporate the random thermal fluctuations governing molecular Brownian motion and reactive encounters. With few exceptions (29; 30), the molecules are treated as spatially structureless entities and only their approximate radius is (sometimes) taken into account. Studies that aim at elucidating the cooperative behavior of receptor complexes in different states of activation frequently introduce the simplification of assigning those complexes positions on the nodes of spatial grids where only neighboring grid points can influence each other, to make the simulations computationally tractable for large numbers of individual receptors (31). Such methods do establish a model that corresponds in some degree to data from experimental studies of molecular interactions based on distance-sensitive methods such as FRET (Foerster Resonance Energy Transfer), but the specific geometry of the grid affects the simulation outcome and the chosen geometry may not provide a close approximation of the environment in which the actual molecular interactions occur, limiting the correspondence between model and biological system.
These particle-based methods can be very detailed and realistic in terms of space (32), but they are simply too computationally expensive in applications that include many molecular species with high concentrations embedded into spatial domains that go beyond just the size of small bacteria. Reaction-diffusion simulations of large networks of molecules aimed at investigating more than just a few seconds of signaling dynamics have to give up the advantages of particle-based approaches in terms of dynamical detail and conceptual simplicity in favor of the computational efficiency of molecule number- or concentration-based modeling techniques – that is, treatment of the bulk or average behavior of a molecular species rather than of each individual element. Such techniques, if they include spatial aspects, have to divide the relevant cellular space in which the biochemical events are being simulated (cytoplasmic domains, intracellular compartments, or membrane regions) into subvolumes small enough to justify the assumption of local homogenous concentrations, because this assumption underlies all concentration- or particle number-based simulations. Individual molecular Brownian motion simulated in Monte-Carlo approaches is replaced by the phenomenological concept of diffusion, driven by concentration differences. Instead of strictly localized bimolecular interactions, mass action-based equations are used to simulate reactions that modify the chemical composition of the modeled system (33–35).
Spatially-resolved simulations without stochasticity (that is, without explicit treatment of the substantial fluctuations characteristic of systems with small numbers of components in which ‘average’ behavior is not an adequate description), use discretized partial differential equations (PDEs) describing the effects of diffusion and of molecular reactions to calculate how molecule concentrations change as a function of space and time. Without having to pay the cost associated with simulating stochastic effects, such simulations can afford to calculate extended concentration time courses for spatial domains comprising entire cells or even groups of cells embedded into representations of extracellular space (36–38), provided that the signaling networks are not too complicated. However, for networks that comprise too many interacting components and/or components that have many functional binding sites mediating interactions with other molecules, the resulting systems of equations describing the rates of change of the concentrations in the system may become too large to be simulated with spatial resolution. Examples of signaling networks with great complexity are the pathways downstream of immunoreceptor or EGF receptor ligation. In fact, these pathways may be so complex that differential equation-based approaches trying to simulate kinetics down to the activation of transcription factors face serious problems with regard to just formulating the reaction kinetics in ways that permit communicating the model’s assumptions in a human readable format. The problem is simple to understand – if each receptor has multiple sites for phosphorylation (ITAMs and so on), and each phosphorylated site can bind adaptor molecules that in turn recruit kinases or phosphatases subsequently interacting with downstream targets, and if the phosphorylation of each set of sites in the cascade is heterogeneous, then the state of the system is represented by a combinatorial expansion that rapidly grows to enormous numbers. The phenomenon of such combinatorial complexity arising in models with components that have many functional binding sites has been extensively discussed (39; 40). As an alternative to describing such systems in terms of hundreds or thousands of differential equations, one can in principle describe their properties in terms of functional molecular binding sites and their interactions, just as we did in the previous sentence, and leave the task of unfolding the resulting networks of reactions to the computer simulating their concentration kinetics (36; 41–43).
All the basic simulation techniques discussed so far – particle- and concentration-based, stochastic and deterministic, spatial and non-spatial – take as inputs formal, computer readable descriptions of the quantitative interactions between signaling components and of parameters such as initial concentrations and – for spatially-resolved models – their initial distribution in the simulated (sub-)cellular domains. This in turn places two categories of demands on the investigator. The first is to collect the necessary quantitative data to constrain the many parameters used to define the model. One needs to know how many molecules of each species exist in what volume and, preferably, in what state of biochemical modification (phosphorylated or not, ubiquitinylated or not …), their affinities of interaction, the rates of enzymatic transformation, and so on. One also needs a reasonably complete picture of the molecular interactions in the (sub) pathway being modeled. For this reason, a diverse set of high-throughput data acquisition methods is needed to collect the necessary quantitative and organizational data on the components to be included in the model, and much of the remainder of this review is devoted to a discussion of the relevant technologies, their uses, and limitations.
The second issue relates to the modeling and simulation itself. Many modeling efforts in immunology in particular and biology in general have relied on expert mathematicians and computer scientists to develop equations, computer scripts or code to handle these formal descriptions and conduct the calculations necessary for simulating a model of interest to the biologist studying the question at hand. While such collaborations between theorists and experimental biologists reflect the inherent multi-disciplinary nature of systems biology, they always carry with them the danger of losing in translation important biological aspects of a model. Moreover, complex realistic models of extensive systems, in particular those that cover multiple scales, are often difficult to implement even with expert computational skills. For both of these reasons, substantial effort has been devoted to developing computer software that eases the translation of biological thinking into the equations necessary for quantitative simulations. Some approaches allow the user to define reactions in tabular form or by connecting nodes of a graphically-created network of signaling complexes (44–48) and to simulate the dynamics of those networks. An even more accessible approach is provided by novel software that performs automated generation of reaction networks from the simple biologist-defined bi-molecular interactions that underlie all reaction systems, input via graphical user interfaces offering iconic representations of molecules and their interactions that aim at resembling classical signaling diagrams (36; 49) and with which the biologist is comfortable. When combined with user-friendly software that also permits developing spatially-resolved computational models of cellular behavior (36; 50), such techniques remove many of the impediments that in the past have prevented experimental biologists from exploring the validity of their hypotheses through quantitative computer simulations. Furthermore, the output in these tools can take the form of graphs and charts that resemble the output of bench experiments, allowing the biologist to easily understand the predictions made by the model and simulation and to relate this output to data that can be derived from new experiments corresponding to the conditions chosen for the computer simulation.
Molecular-scale Modeling of Immune Receptor Interactions and Proximal Signaling Events
These various modeling approaches for simulating molecular events have been applied in an increasing number of laboratories to explore the basis for immune cell signaling. The ability of lymphocytes to quickly and strongly react to the engagement of even very few immune receptors by pathogen-derived ligands while filtering out the overwhelmingly more frequent ambient stimuli originating from uninfected host tissue has challenged modelers for decades. For T cell activation, the two main unsolved questions are 1) what, from the point of view of the T cell receptor, distinguishes the binding of an agonist peptide-MHC from the binding of a non-agonist ligand and 2) how does the T cell translate this difference in the nature of the primary stimulus into a useful cellular response? The majority of immunologists today believes that agonist stimulation differs from stimulation through non-agonists only with regard to the association and dissociation rates of the interaction between the TCR and the peptide-MHC complex although other concepts have been proposed (51; 52). A higher association rate would mean that agonist peptide-MHCs can trigger TCRs with a higher frequency whereas a lower dissociation rate would give the intercellular TCR-peptide-MHC complex a longer lifetime. If the TCR (or its immediate downstream signaling partners) keep the memory of a previous ligation for a non-negligible amount of time, both types of kinetic advantage of agonists over non-agonists would mean that the TCR is activated over greater periods of time, even between binding events (53), giving it the opportunity to activate downstream signaling components more effectively. This memory must, however, not last too long since it would otherwise blur the discrimination between the kinetics of agonist and non-agonist binding.
Full phosphorylation of the ITAMs in the TCR-associated ζ-chain by Lck is the hallmark of successful TCR activation. For this to occur, the binding of the TCR through peptide-MHC has to (transiently) shift the balance between the activities of the kinases directly or indirectly inducing this phosphorylation and the phosphatases reverting it in favor of the kinases. Many modeling efforts have been directed towards identifying the mechanisms that can achieve this outcome with high sensitivity (in order to allow for responses to low densities of agonist peptide-MHCs [pMHCs] on APCs) while at the same time retaining the capacity seen in real cells of an exquisite ability to distinguish between agonists and non-agonists. To accommodate the latter requirement, the concept of kinetic proofreading through the TCR signaling network was introduced (54), according to which ligation of the TCR induces a sequence of state transitions that are reverted once the TCR:pMHC bond breaks. Stably bound (or, if the system has memory, rapidly rebinding) ligands (see (55) for a quantitative exploration) would complete the transition sequence to the end point of full TCR activation whereas weak ligands would only lead to progression through part of the sequence before dissociating and causing the signaling system to fall back to its basal state. However, the question of which signaling components and mechanisms can account for this behavior and whether the above-mentioned shift towards higher kinase activity is part of the proofreading has remained controversial. Based on the notion that close contact between T cell and APC membrane surface necessary for the formation of TCR:pMHC complexes would exclude the phosphatase CD45 due to its large bulky extracellular domain (thus limiting dephosphorylation of Lck and the ITAMs in the CD3 and ζ-chains), van der Merwe and colleagues suggested that kinetic segregation of kinases and phosphatases could be the mechanism creating (relatively) kinase-rich domains promoting initiation of the proofreading cascade (56). To support their model these authors performed particle-based Monte Carlo simulations where the particles, representing pMHC and TCR, were confined to movement on the nodes of a 2D spatial grid representing the membranes. The simulations demonstrated that the segregation model can achieve ligand discrimination but did not reproduce the experimentally observed ability of T cells to perform this discrimination within less than a minute as biochemical studies show is the case (18). In order to reproduce these reported time scales computational models may have to focus on the smallest biochemical scale, the scale of signaling microclusters (57; 58) or even individual TCRs interacting locally with a highly dynamic actin skeleton (59; 60). Such models would require experimental data that combine high-resolution microscopy techniques with careful quantitative analysis of molecular movement and interactions (61).
Whereas the models discussed so far focused on specific biochemical aspects of membrane-proximal TCR signaling, other modeling efforts have aimed at elucidating the more kinetic or biophysical aspects of the interactions between the receptors of conjugated T cells and antigen presenting cells. Goldstein and colleagues investigated the relationships between TCR-pMHC dissociation rates and the ability of single pMHC to engage many TCRs, demonstrating that a model with physiological rates for molecular interactions and diffusion could confirm earlier experimental estimates of substantial serial engagement (62–64). Focusing entirely on the physical aspects of the T-antigen presenting cell interface, Chakraborty and coworkers were able to show that the characteristic spatial pattern – the ‘immunological synapse’ (for review see, e.g. (65)) – seen for the distribution of immune- and adhesion receptors could, in theory, emerge just based on physical membrane properties and receptor size differences (66).
Downstream of the immediate receptor-proximal ligand discrimination, T cells engage a complex signaling network, enforcing its cell-wide consequences (activation or adaptation) through various feedback mechanisms. Among these, the competing activation of the phosphatase SHP-1 and the kinase Erk was shown to play an important role (67). Based on this experimental work, several computational models were developed. Since many of the molecules involved in these networks are cytosolic, most models assume that spatial aspects at this later stage of the cellular response are less important than during its first seconds, in spite of growing evidence that intracellular signaling cascades such as the one activating MAPK involve spatial coordination of molecular interactions with the help of scaffolds (68; 69). Chan et al. (70) used a simple differential equation model to investigate the role of phosphatase/kinase feedbacks and showed how such mechanisms would be able to take advantage of self- (or null-) peptides and could also account for the phenomenon of antagonism. A far more complex model with explicit simulation of many of the molecular interactions downstream of the TCR allowed Altan-Bonnet and Germain (71) to make detailed predictions about the dynamics of SHP-1 and Erk activation that could be tested experimentally. One of the key findings of this work was that the Erk response of single T cells was digital – on or off – in contrast to the more continuous distribution of Erk phosphorylation levels observed in populations of stimulated cells. Using a stochastic simulation based on Gillespie’s Monte Carlo algorithm (72), Chakraborty, Weiss, and colleagues subsequently showed that feedback activation by Ras-GTP of the Ras-specific nucleotide exchange factor SOS (73) can lead to such all-or-nothing cellular responses in the Erk pathway and a filter-like sharp distinction between weak and strong signals coming from the TCR (61). Although postulated originally for TCR signaling, the concept of kinetic proofreading was subsequently shown to be relevant for B cell receptor ligand discrimination as well and mathematical models were used to explore its limitations and point out the possibility of low affinity ligands escaping this ‘quality control’ (74).
While these initial efforts at molecular-scale modeling and simulation in immune systems have been productive, they fall short of combining all the elements that would make the work more robust. Careful proteomic studies that provide protein abundances and interaction rates, combined with extensions of ongoing elegant work involving single-cell imaging that reports molecular re-organization and signaling behavior in a spatio-temporal manner with high resolution, will allow more complete models to be built and simulated. These will more closely reflect biological reality and hence, have more power to predict system behavior and guide experimental advances. Recent work exploring apoptotic signaling in non-lymphoid cells has clearly demonstrated the possibilities and benefits of such efforts (75–77).
Modeling Cellular Behavior
Many of the issues discussed so far for the modeling of molecular behavior and interaction networks – stochastic versus deterministic treatment, discrete simulations versus differential equations, spatial versus non-spatial approaches – apply similarly to computational models at the level of cellular behavior and cellular interaction networks. Three main modeling categories on different spatial and biological scales have emerged that treat biological systems in which the cell is the smallest element of function. The first uses mainly differential equation-based, mathematical models to investigate organism-wide cell population dynamics, in particular in response to pathogenic challenges such as viral or bacterial infection. These are typically what can be called ‘compartment’ models, in which the differential equations report the change over time in the concentration (number) of particular elements (cells, pathogens) in defined organ (spatial) compartments such as lymph node, or liver, or lung, as well as in functional compartments such as ‘central memory’ vs ‘effector’ T cells. The second approach – typically focusing on cell population-wide consequences of single cell decisions – explores the balance between cell-autonomous programmatic behavior on one hand and cellular crosstalk on the other hand in the regulation of processes such as cell activation, differentiation, division, or death. The third category takes direct advantage of recent advances in high-resolution in vivo microscopy and of ever-increasing computer power to study how individual cells react to external mechanical and chemical stimuli with the help of image-guided computational models that incorporate aspects of dynamic single-cell morphology.
Operating on the highest, organism-wide scale, the first category has the longest history in mathematical immunology – not only because the types of experimental data for such studies, mainly derived from cell counts in blood samples or dissociated organs, have been available for many decades – but also because many of the models in this category are based on the ancient family of predator-prey models (78) describing how interacting species (such as foxes and rabbits, or here: immune cells and pathogens) influence each other’s population sizes. The basic idea behind these models is that proliferation and death rates of one species depend on the size of the populations of other species with which it interacts: when there are a lot of rabbits, foxes find enough food to proliferate quickly. When their appetite for rabbits starts causing a reduction of their food source this, in turn, causes a decrease in the proliferation of foxes. The smaller number of foxes then allows the rabbits to recover, starting a new cycle. The transfer to immunology is in principle straightforward – with the interesting twist that in HIV infection the roles of the foxes and the rabbits are not clearly assigned since both (immune cells and virus) are sometimes predator, sometimes prey. In spite of their simplicity, these approaches have contributed much to our understanding of host-pathogen dynamics because of the way they sharpened our intuition with regard to cell population dynamics. Modeling studies of the kinetics of the loss of T cells after SIV/HIV infection (79; 80) and the (only incomplete) recovery following HAART (highly active anti-retroviral therapy) have prompted controversial discussions and led to experiments that moved the field from the concept of increased T cell proliferation after infection as a homeostatic response following direct virus-induced depletion towards a focus on uncontrolled broad immune activation (81; 82) and gradual loss of central memory T cells (83) as the driving force behind the progression to AIDS.
Almost as a side effect of the HIV modeling efforts (84), theorists developed differential equation-based population models to understand the regulation of lymphocyte homeostasis. The experimental inputs for such models typically consist of population sizes (cell counts) of cells of specific types or differentiation states. Since the goal of these studies is to infer relationships between the proliferation, differentiation, and death rates of interdependent cell types from fitting the corresponding parameters of equations describing the change over time of the population sizes, the ideal data consist of longitudinal studies measuring the relevant cell counts at several consecutive time points. However, longitudinal studies are difficult to do with human subjects and expensive with non-human primates. Moreover, the combined effects of proliferation, differentiation, and death are difficult to extract from cell count time series alone because, for instance, a decrease in the size of one population could stem from an increase of death or differentiation (leaving one population and entering another) or reduced proliferation or influx from other populations. Many modeling efforts therefore take advantage of data that report cell proliferation histories in addition to cell counts. Cell labels such as BrdU and CFSE become diluted by a factor of 2 every time a cell divides. Following the fluorescence intensity of such markers in different cell populations thus allows the experimentalist and modeler to distinguish, for instance, between increased proliferation and reduced death (85–88). Careful analysis of the time evolution of CFSE intensity profiles in single dividing cell populations has moreover yielded valuable insights about the role played by cell intrinsic variability and its heritability between mother and daughter cells in lymphocyte dynamics (85; 89).
While kinetic studies of cell population sizes can provide insights into high-level regulatory dynamics and the relative contributions of cell intrinsic properties and external regulation, they cannot readily explore the cell biological foundations for those dynamics: knowing that cell population A will proliferate with a rate that is inversely proportional to the death rate of population B does not immediately help us understand on what factors a cell of a particular type will base its decision to die, proliferate, differentiate, or change its location (previous history and contemporaneous exposure cytokines, chemokines and direct cell-cell contacts among many others). Some degree of detail of cellular communication can be incorporated into differential equation models, for instance, by allowing differentiation rates to depend on extracellular concentrations of cytokines and modeling the time course of such concentrations alongside the cell population sizes (90). However, cytokine and chemokine concentrations may be strongly location dependent just as are cell densities and hence opportunities for direct cell-cell communication will frequently be distributed non-homogenously in vivo (91), a property of the system that is not captured with ordinary differential equations that are the basis for most such models.
Although differential equation-based approaches can be extended to include multiple spatial compartments with specific cellular and molecular composition (92), agent-based models (which simulate individual cells as discrete agents that interact with other agents and specify their specific locations and states) can capture such spatial aspects more easily. Since the seminal work of Celada and Seiden who developed their simulator ImmSim to study immune receptor signal-based cellular behavior (93; 94) with bitstring representation for receptor specificities, agent-based modeling in immunology has been used in particular to study phenomena that involve the formation of specific spatial cellular arrangements. Among the classical examples in this category are the formation of germinal centers and the concomitant evolution of B cell receptor affinities (95; 96), the dynamics of pathogen control through granuloma formation in the immune response to Mycobacterium tuberculosis infection (97) and the maturation of T cells during their migration through the spatially heterogeneous structures of the thymus (98). Whereas the majority of agent-based approaches neglect the influence of mechanical interactions between cells and constrain the positions of the simulated cells to the nodes of a spatial grid, recent modeling efforts, building on the pioneering work by Graner and Glazier (99), have begun to incorporate cellular morphological dynamics into simulations. Among many potential applications, the present focus of these new approaches has been to reproduce data from in vivo microscopic observation of the interactions between T-cells and APCs with high fidelity, to permit more accurate estimation of the duration of these interactions and how they are influenced by the stromal networks within lymph nodes (100; 101).
Network Models for Molecular and Cellular Interactions
In the previous two sections we discussed various modeling approaches for systems of interacting molecular species or cell types. In many cases, such systems have natural representations as networks. This is obvious for molecular signaling processes. The nodes of the models of such networks represent concentrations or activation states of the different molecular species, whereas the links (or edges) encode interactions and state transitions (think of phosphorylation of a molecular species, for instance). Simulating such models then simply means updating the concentrations (or activation states) according to the interactions (and rates) associated with the links. Some modelers introduce the simplification that the nodes can only be in two states – on or off. Such networks, which have been applied to immunological systems such as in the analysis of TCR activation (102), are called Boolean networks because the rules for update determine the on- or off state for the next iteration, based on operations that produce 0 or 1 values and take as input logical combinations of 0 and 1 values. For example, a node may switch to on if the neighbor nodes it is linked to are all on (logical AND operation). While such networks obviously have far fewer parameters than the number that must be provided for a simulation of continuous state networks, the lack of graded responses of single nodes seriously limits the possible dynamical modes of such networks and hence, how well they reflect biological reality.
Many of the above remarks on molecular networks apply to network models of interacting cells types. In cases in which describing the state of a cell type in a model involves several parameters (as opposed to just one for concentration or activation), cellular interaction networks describe rules for interactions and induced transformations between multi-state entities, sometimes called automata. The simulation tool Simmune (36) combines molecular interaction networks and cellular state transitions by allowing users to couple particular biochemical states (concentration thresholds) to whole-cell responses such as division, death, movement or secretion of new molecules.
In addition to the dynamic approaches we discussed above, networks are also used to represent more static (or at least slowly changing) relationships between biological entities. Typically, the goal of those models is to find the most likely network of interdependencies between, for instance, the transcriptional states of genes that is compatible with mRNA expression data obtained under various experimental conditions. A major mathematical/statistical approach used for such network construction is Bayesian inference. For a more thorough discussion, the reader is referred to the bioinformatics literature (103).
Large-scale Data Acquisition Technologies
To construct models at various biological scales (cells, tissues, the whole organism) using the approaches and tools discussed above, a variety of data sets are needed. Knowledge of the entire set of expressed proteins and other molecules (the proteome, glycome, lipidome etc.), as well as the quantitative rules governing their interactions and chemical transformations, of gene activity (the transcriptome) and epigenetic state, and of metabolic state are needed for generation of comprehensive models at any of these scales. However, as already indicated above, the construction of ‘complete’ models is for the distant future except perhaps in the realm of metabolomics. There is nevertheless still great value in generating models based on less complete data and focused on only a defined aspect of a cell, tissue, or organ that nevertheless seek to understand the properties of an ensemble of elements rather than each component on its own. In pursuit of this goal, an increasing number of large-scale and in some cases global data acquisition technologies have been developed that enable collection of the information necessary to undertake useful model building and simulation. Each technology has its benefits and limitations, and an understanding of these competing aspects of the methodologies is key to devising an effective and reliable approach to a systems analysis at any level of biological resolution. In this section, we summarize these technologies, essential aspects of their proper use, some examples in which they have been applied, and some of their limitations. An understanding of just what tools are presently available, what advances are needed to address deficiencies in the current methods, and when these techniques can be well-employed, is essential for an investigator to develop an effective systems approach to immunological questions of interest.
Analysis of Gene and miRNA Expression
Nucleic acid-based technologies provide the most comprehensive analysis of cell and tissue state. As such they are the most well-developed and available methods for generating the ‘parts list’ of a cell. Transcript profiling using microarray technology has been the most widely used ‘omic’ technology and has provided significant insights into immunological development, homeostasis, and transcriptional dynamics during the immune response to antigens and to pathogens. However, this method is an incomplete reporter of cell state - there can be a substantial difference between the transcriptome and proteome in a cell (104–106); and immunologists are well acquainted with the existence of stored mRNA that is only translated upon cellular stimulation (107). Therefore complementary methods are needed to develop an effective model of cellular behavior based on the actual set of expressed proteins, for example. At the same time, transcriptomic technology can provide an extraordinarily useful and extensive, though incomplete, picture of the differentiation state of a cell or tissue and it is a very effective means of monitoring changes in this state induced by exposure to a stimulus. Both fine grained models of cellular biochemistry and coarse-grained models involving tissue and organ function are often built on pathway maps emerging from microarray analysis of transcriptional states and small RNA expression, in some cases linked to genetic mapping of quantitative trait loci in a technique called eQTL mapping (103; 108). This oldest of the omic–scale nucleic acids methods in now being supplanted by the emergence of next generation sequencing, which promises to revolutionize not only mRNA transcript profiling but also the analysis of genome sequence variation, small RNA expression and function, epigenetic modification and protein-nucleic acid interactions.
Transcript Profiling
It is now 15 years since the first microarray techniques were described using cDNA spotted on filter paper (109; 110) and this technology has been refined at a remarkable rate with the development first of cDNA-based and later of oligonucleotide-based, imaging-friendly microarrays that permitted genome-wide profiling of mRNA expression levels under varying experimental conditions. Oligonucleotide arrays have progressed from features with a 3′ bias that were unable to discriminate splice variants to currently available exon-tiling arrays that cover the entire transcript of each gene and provide significant insight to the specific splice variants expressed (111; 112). Moreover, with the explosion of available whole genome sequences, it has become commonplace to generate arrays specific for multiple organisms, including pathogens (113), which permits the simultaneous tracking of expression changes in both host and pathogen during an infection. Microarray technology has made a huge contribution to our understanding of the dynamic transcriptome in normal and disease states, and in clinical applications such as cancer cell profiling and disease prognosis, it has shown remarkable predictive capacity. Its ability to identify ‘signatures’ in samples from patients with acute myeloid and lymphocytic leukemia that correlated with disease outcome was one of the first demonstrations of the power of non-biased large-scale data gathering approaches to provide insight that would have been almost impossible to obtain from more reductionist methods (114). These initial successes were followed with the clear demonstration that expression profiles could guide therapy, through correlation of signatures with clinical outcomes in many tumors, both solid and hematological (115; 116). The immune system has proven to be particularly accessible in this regard, through analysis of signatures from whole blood, PBMCs, or from specific hematopoietic cell populations. The obvious value of high quality transcript data repositories to biological research has led to numerous efforts to generate comprehensive databases of mRNA expression maps in most experimental model systems. Following characterization of transcriptional responses to pathogens in innate immune cells (117–120), analysis of the blood transcriptome of patients has produced comprehensive catalogs of gene changes characteristic of autoimmune and inflammatory disorders (121–126) and specific responses to viruses (127–130), bacteria (131–133), and parasites (118; 134). The ImmGen project is attempting to provide the field with a highly qualified catalog of gene expression in nearly all identified hematopoietic cell types in the ‘resting’ state and after defined stimulation with ligands such as those engaging TLRs or various cytokine receptors (14).
Although microarray technology will remain an important contributor to integrative biology programs for the foreseeable future, it has some inherent drawbacks. Firstly it is dependent on prior knowledge of a given transcriptome, and is thus limited to organisms with fully sequenced genomes. Secondly, the dependence on nucleic acid hybridization contributes an unavoidable degree of noise in the transcript profile, and a limited dynamic range leading to poor performance in quantifying less abundant transcripts. Some of these drawbacks can be addressed using direct analysis of specific transcripts with quantitative PCR, although this is more limited in the number of genes that can be analyzed in a single experiment. These limitations can be improved by use of microfluidics to parallelize hundreds of reactions in a single assay (135), multiplexed bead-based assays that permit simultaneous detection of up to 100 transcripts (136), and the recent development of the ‘Nanostring’ technology that can directly capture up to 500 different non-amplified mRNA transcripts using a unique molecular barcoding approach (137). However, at a genome-wide level, all of these technologies will likely be superseded in the coming years by the recent advances in nucleic acid sequencing that will provide not only more comprehensive coverage but more quantitative data suited to using the information gathered not only for qualitative descriptions of cell or tissue state, but for quantitative modeling of changing gene activity correlated with alterations in chromatin state and transcription factor availability in the nucleus.
Next Generation Sequencing
Next generation sequencing (NGS) has already made a significant contribution to many fields of biology, accelerating the analysis of genomic sequence variation and disease linkage, epigenetics, transcriptomics, and small RNA function. NGS sequencers, initially developed by 454 and Illumina, and followed soon after by ABI and Helicos, all use massively parallel analysis of individually amplified DNA fragments (138; 139). While a run on the capillary-based sequencers used for the human genome project could provide approximately 100 reads of 800bp, NGS machines produce shorter reads of 35–400bp but the number of reads can reach 107, thus exceeding 1 gigabase of sequence per run. This has an enormous impact on the potential experimental applications of sequencing approaches. Genome-wide analyses can be undertaken that do not entail sequencing the entire genome, but instead that which is represented as expressed mRNA, modified DNA (e.g., methylated), nucleic acid bound to a specific protein (e.g., a transcription factor), or DNA sensitive to enzymatic degradation (2).
As a direct alternative to microarrays for mRNA profiling, cDNA reverse transcribed from poly A+ RNA can be deep sequenced directly (RNA-seq; (140)) after adapter ligation either from one end (single end sequencing) or both (paired end sequencing), the latter being particularly important in the analysis of splice variants (this should be further facilitated as the length of NGS sequence reads increase). Mapping each individual read back to a reference genome and counting hits at each site leads to generation of a quantitative distribution of expressed mRNA across the genome. This has a much greater dynamic range than array technology (over 5 orders of magnitude), permitting more effective identification of low abundance transcripts and it essentially eliminates the background noise associated with hybridization. It is also less prone to variation between research groups using different array platforms, although it is not without its drawbacks. Data storage requirements are significant, and considerable bioinformatic resources are needed to manage the data and deal with alignment challenges such as those associated with repetitive elements (141). There are also sequencing errors that must be dealt with; these are especially important in studies of the microbiome where alignment to reference sequences is not possible.
The most developed NGS application has been in the analysis of nucleic acid bound to a specific protein, though use of Chromatin Immunoprecipitation (ChIP). This global method is ideal for developing gene regulatory network maps for model building, including models that link intracellular biochemical changes induced by extracellular stimuli to changes in gene expression. In ChIP, an antibody recognizing a specific protein (such as RNA polymerase, a sequence-specific transcription factor, or a modified histone) is used to affinity-purify the protein chemically cross-linked to its associated nucleic acid, which is then separated from the conjugated protein and subject to deep sequencing (142). In the case of RNA pol II-based ChIP-seq, this provides a dynamic complement to RNA-seq as it identifies those genes being actively transcribed under specific experimental conditions, and this can be taken a step further by sequencing ribosome-protected mRNA to provide a quantitative measure of translation. Similarly, applications of ChIP-seq with transcriptional regulatory elements and chromatin-associated proteins are becoming commonplace and promise to revolutionize our understanding of how transcription factor binding and epigenetic modifications control gene expression on a system-wide level (143; 144).
In the context of disease characterization, genome-wide association studies are also being enriched by NGS. The analysis of genomes of patients for common genetic polymorphisms that correlate with specific traits, such as complex or multifactorial disease, has until now been done by profiling of several hundred thousand SNPs using array or bead-based technology (145). Ultimately, NGS will increase SNP coverage significantly beyond these most common markers, and efforts such the 1000 Genomes Project promise to expand significantly our knowledge of human genomic variation and disease linkage (146). However, most complex diseases cannot be explained by a single gene mutation, and are instead caused by multiple complex factors. Combined analysis of transcript profiles with genetic variation has shown that gene expression can be significantly influenced by polymorphisms in regulatory elements. Such quantitative traits based on statistically significant changes in transcript abundance have been termed expression QTLs (eQTLs; (103; 108)). Although accelerated data generation by NGS and even newer sequencing technologies will make this an area of rapid progress in the next few years, existing eQTL maps will need to move beyond the use of EBV- derived lymphoblastoid cell lines to more relevant tissue-specific sources (147). Also eQTL mapping can only take us so far, and will require integration of meta data, such as analysis of regulatory small RNAs and protein modification of regulatory factors, to permit a more comprehensive interpretation of the consequences of genetic sequence variation in disease susceptibility.
miRNA Profiling
Since their relatively recent discovery in plants and nematodes, microRNAs (miRNAs) have been shown to have key roles in the regulation of the genome/proteome in numerous cell types, including those of hematopoietic origin (148; 149). As such, they are key elements in attempts to build comprehensive models that link signaling to change of cell state, and because they often target transcription factors, to gene expression. The cellular processes engaged during siRNA-mediated gene knockdown are part of the endogenous pathway for processing of miRNAs, which are initially expressed as pol II-based primary transcripts of several hundred nucleotides. Processing by the ribonucleases Drosha and Dicer produces a mature miRNA, a dsRNA duplex of 20–23nt, which binds to cognate target mRNAs, primarily within their 3′UTR, through a 6–8nt ‘seed’ sequence towards the 5′end of the mature miRNA leading strand. This leads to a reduction in protein expression, usually through a combination of mRNA destabilization and translational repression (150). Several hundred miRNA genes have been identified, and many have been classified into families based on seed sequence homology. Although this would suggest overlap in target mRNAs and some degree of functional redundancy, experimental evidence suggests a significant amount of (still poorly understood) functional specificity among family members. Attempts to model the changing expression of molecules (in particular proteins) in a cell in response to extracellular stimuli will require integration of methods such as those detailed above for tracking protein-encoding transcripts with information on the post-transcriptional regulation of translation by miRNAs, along with proteomic methods for direct assessment of the translation products, as discussed in a subsequent section.
Due to their RNA-based structure and Watson-Crick base pairing dependent mechanism, the technology to profile and probe the function of miRNAs has been developed rapidly through modification of well-established nucleic acid-based methods. This has led to the rapid generation of miRNA expression profiles across cell types in normal and disease states (151). Similarly, analysis of the function of specific miRNAs has been facilitated by the development of antisense-based inhibitors (‘antagomirs’) and chemically synthesized miRNA mimics (152; 153). Since these reagents are small RNAs themselves, they can be transfected into cells using the same delivery protocols as for siRNA, and many screening groups now routinely add these reagents targeting all known miRNAs to their siRNA screening pipelines to provide a more direct means to identifying the miRNAs influencing the cellular process under study. The advantage with this approach is that it can identify miRNAs important for maintaining a cellular state that do not necessarily change their expression level between experimental conditions (and thus would not be picked up by profiling studies).
Experimental investigations of miRNA expression and function have been complemented by computational approaches for miRNA target prediction. Based primarily on the analysis of miRNA conservation across species and the conservation of seed sequences among orthologous 3′UTRs, target prediction algorithms based on seed matching have improved their predictive accuracy and have reduced their false prediction rate to less than 25% (150). However, these algorithms still have a significant false negative rate as the influence of sequence context beyond the seed match, both in the miRNA and the target mRNA, is poorly understood. Thus, experimental approaches to the identification of miRNA targets are in need of further development. Immunoprecipitation of the protein components of the RNA-induced silencing complex (RISC-IP), followed by array deep sequencing analysis to identify the miRNA-bound mRNA holds significant promise, but has yet to be applied to large-scale studies (154). The coupling of mRNA profiling (by microarray or RNA-seq) with quantitative proteomics under conditions of differential miRNA expression is another approach to target identification, and the feasibility of this method has been demonstrated in two recently published studies (155; 156). These approaches are vital to determine the scope of action of individual miRNAs and to infer the functional consequence of their altered expression on a broader scale.
Certain characteristics of miRNA function are emerging. A single site miRNA/mRNA interaction rarely leads to a >50% reduction in expression, and more commonly averages around 30%. Ninety percent of conserved interactions are single site, although most mRNAs have at least one further site for a different miRNA that can promote further repression. Since a 50% change in expression of a protein may not result in a marked phenotype, as evidenced by the rarity of haploinsufficient mutants, this might explain why very few phenotypes have been observed in systematic screens for miRNA mutants, although some important immunological exceptions to this view have recently emerged in cases where single miRNAs target multiple key signaling molecules (reviewed in (149)). The degree of influence of a miRNA on a given process may also be exaggerated in laboratory settings by the fact that the described technologies for modulating miRNA expression can lead to more dramatic alterations than occur physiologically. Laboratory conditions are likely more favorable to maintaining the stability of mutant phenotypes, but conditions that better simulate the environment that shaped evolution may be more suited to uncovering the importance of miRNAs in regulating the proteome. This brings us to the question of the mechanism by which miRNAs influence mammalian cell function. It has been proposed that rather than promoting large changes in the expression of a few proteins, they function on a broader scale to ‘tune’ the proteome. The high degree of conservation of miRNA target sites, and the selective avoidance of seed sequences in the 3′UTRs of mRNA that are co-expressed with a given miRNA suggests that precise control of protein concentration has significant implications for organismal ‘fitness’. Such subtle changes in protein concentrations may sensitize regulatory networks. These consideration re-emphasize the point made above that miRNAs and their activities are crucial to development of informative models relating stimulus to response, because network properties clearly involve not only the protein interactome we more typically map and whose component modifications we routinely consider, but also the influence of temporally changing miRNA expression on the balance of protein components within such networks.
Another notable pattern that has emerged from miRNA studies, particularly in immune cells, is the conservation of target sites for a single miRNA, or miRNA family, within proteins involved in a common process. For example, several key signaling components involved in the response of monocytic cells to bacterial endotoxin, such as IRAK1 and TRAF6, are targets for miR146 (157), while phosphatases shown to downregulate signaling through the T cell receptor (SHP2, PTPN22, DUSP5 and DUSP6) are targets for miR181 (158). Since these targets have a coherent influence on signal flow through the pathway (i.e., either activating or inhibiting the process), these findings suggest that these miRNAs, which have both been shown to change expression in response to cellular stimulation, act as negative or positive feedback regulators. This is consistent with their tuning function, where they can adjust thresholds for activation under various conditions. They may also have key roles in mediating crosstalk between inflammatory mediators, as evidenced by recent data suggesting the anti-inflammatory effects of IL-10 are promoted through direct inhibition of LPS-induced miR155 expression (159) These observations emphasize the importance of developing quantitative models to predict accurately the influence of miRNA expression changes on cellular function.
RNAi Screening
Although microarrays and NGS report on what genes (or miRNAs) are present in a cell or tissue, or the state of chromatin, or the binding behavior of transcription factors, and can inform us about how these elements change over time, these methods are descriptive and correlative, failing to provide a direct link between transcript, translated product, and biologic function. Establishing such connections has traditionally been the realm of genetic screens in mutant cell lines or organisms. Such studies have contributed significantly to the identification of specific elements that play a role in signaling or metabolic pathways, maintain cellular homeostasis, regulate cell structure, intracellular transport, and capacity for migration, modify gene expression, and in other ways contribute to normal physiology, or, when abnormal in form or amount, give rise to disease. Genetic assessments of this type have been especially valuable in model organisms such as Drosophila where common phenotypes were often found to be caused by genes acting in a single linked signaling pathway, associated with recognizable organelles or structural elements of the cell, or comprising a linked gene regulatory pathway (160). However, this approach tends to identify genes contributing to core functionality conserved across species rather than the components and mechanisms responsible for the subtleties of cell-type specificity and context-dependent cellular function. Thus, our understanding of pathways remains incomplete, and discovery of unknown pathway components has been hampered by canonical bias in experimental design and reagent availability (161). Non-biased approaches are therefore vital to fill in the gaps in networks to provide a more complete framework upon which we can base predictive models, while at the same time pruning the large parts lists generated by global methods of components unlinked to a direct test of functional relevance.
The discovery of RNA interference (RNAi) and the major advances in the understanding of small RNA biology in the past decade have provided researchers with an invaluable tool for wide-scale and rapid genetic screening that represents a less biased means of probing the role of various elements in cellular biology (162). As a research tool, RNAi takes advantage of endogenous RNA processing machinery, which permits the silencing of mRNA transcripts with small complementary dsRNA sequences. In C. elegans and Drosophila, this can be achieved through introduction of relatively long pieces of gene specific dsRNA (>100nt); however as this would induce an interferon response in mammalian cells, the gene-specific dsRNA must not exceed 21nt and is thus introduced as short interfering RNA (siRNA; (163)). siRNA transfection is the simplest and most direct application of RNAi in mammalian cells; however, in less tractable cell types and in cases where longer term silencing is desirable, the siRNA sequence can be expressed from plasmid or viral vectors as a short hairpin RNA (shRNA). This works especially well when the shRNA is designed to mimic an endogenous primary miRNA transcript, with the region that is processed to the mature miRNA replaced with gene-specific targeting sequence (164).
Soon after the technical application of RNAi was established for the study of individual genes, its discovery potential on a broader scale became evident, leading to the development of genome-scale libraries of reagents for several organisms, including human and mouse (162; 165). Although the potential of these broad screening tools has attracted many researchers, the practicalities of genome-wide screening are challenging and they require the acquisition of new techniques and capabilities for most biologists, as well as substantial monetary commitments. Furthermore, despite the discovery potential of RNAi, the technology has its limitations and certain practical criteria must be met for a gene to be detected in an RNAi screen:
The expression of the target gene must be reduced by the siRNA to a level that promotes an observable phenotype.
The protein must have a short enough half-life to permit depletion in the time course of the experiment.
The biological function must not be supported by multiple redundant factors.
The depletion of the target gene must not be toxic to the cells under study.
Because of the growing importance of this technique in systems biology research and because even more so than other methods, it has rather strict quality control requirements and interpretive limitations, we devote some space here to a detailed consideration of the different types of siRNA screens and the technical aspects of effective use of the methodology.
Screening formats and assay design
The most commonly used approach in large-scale RNA studies is the arrayed screen, where each well of a microtiter plate contains siRNA(s) or shRNA(s) against a single gene. This permits a straightforward correlation of phenotype to target, but it has the drawback that the time and expense required for a genome-wide screen is significant. An alternative approach that can be used with virally-expressed shRNAs is the pooled screen, where large populations of cells are transduced with complex mixtures of viruses expressing shRNAs against all genes. Clonally isolated cells showing the desired phenotype can then be sequenced to determine which shRNA they encode. If the output assay can be designed to select for a cell growth or survival phenotype, a more efficient means to identify hits is to use unique nucleic acid ‘barcodes’ within each shRNA vector that can be screened for enrichment or depletion by either microarray or sequencing technology. Such pooled screens are powerful, especially for applications in human primary cells of limited availability, but it remains difficult to ensure full genome coverage. These pooled shRNA methods have been productively applied to analysis of the signaling pathways involved in growth and viability of human B cell lymphomas, as one example (166–168).
The screening format is also in part dependent on the cell type chosen, as easy-to-transfect cells are more amenable to arrayed siRNA screens while less tractable cell lines and primary cells may require infection with shRNA-expressing viruses. Achieving satisfactory levels of gene knockdown remains a major technical hurdle in assay development and often forces researchers to choose a non- physiological cell type for the process they are studying, bringing up the concern that the physiological relevance of hits identified in a highly transfectable cell line may be questionable. In some cases, optimal transfection conditions can be identified in more physiological cell lines if an efficient and relatively high throughput knockdown assay [such as the targeting of a stably-expressed reporter gene (169)] can be developed that permits in investigator to assess rapidly and conveniently a wide range of transfection parameters and reagents.
The design and validation of a robust and specific assay is the most critical, and often the most time consuming, aspect of an RNAi screen. It can be extremely laborious, requiring attention to detail and continual assay repetition with minor alterations to optimize performance; however careful development of a robust assay pays off in the quality and discovery potential of the screening results. There are several considerations in choosing and designing a screening assay, such as whether the readout should be endpoint or time-resolved, single feature or multiplex, and whether data should be collected from populations or single cells. In each of these cases, the latter option would clearly produce a richer dataset, however the value of the type of data collected is often inversely proportional to the ease of assay design and implementation. Thus, a compromise has to be reached taking into consideration the overall logistics of running the screen, the cost, and the researcher’s expectations from the data. Most of the initially published genome- wide screens used relatively simple assays, such as cell lethality, and to a certain degree the goal was primarily to establish a proof of principle for the RNAi technology. Screens using more complex assays and readouts are now being published, but there remain relatively few examples in the literature of screens using live cells, multiplexed readouts, or single cell-based data collection.
Continued improvements in fluorescent reporter technology and high content imaging platforms provide the most versatile experimental platforms for data-rich screening assays. High content assays involve the collection of multiple functional parameters of cell state such as cell shape, location of structural markers and organelles, signaling response readouts and/or gene expression changes, often by use of multiple spectrally distinct fluorescent reporters. This opens up the possibility of tracking multiple readouts in a biological process in single cells over an extended time course. This approach, though clearly powerful, poses numerous technical challenges and published reports of screens with multiple assay readouts have, until recently, been limited to sub-genome scale efforts. Three recent reports have combined a mammalian genome-wide siRNA screen with multi-parametric image analysis. Zerial and colleagues studied the endocytosis and trafficking of either a recycling (transferrin) or late endosome/lysosome cargo (EGF receptor) in HeLa cells co-stained with nuclear and cytosolic markers (170). They identified hits enriched in specific pathways such as Wnt, TGFb and Notch, and a Bayesian network analysis led them to predict that numerous kinetic and spatial aspects of endocytosis are highly regulated. Ellenberg and colleagues used a live cell assay to profile cell division parameters in HeLa cells expressing GFP-labeled histone 2B (171). They used an siRNA microarray approach which involves plating cells in chambers with up to 384 siRNA, which has the advantage of standardizing cell culture conditions across the array (172). Although they collected data from only a single fluorescent channel, they were able to classify up to 16 morphological phenotypic characteristics based on the shape of the labeled nuclei, and their data analysis identified numerous genes involved in cell division and migration. Boutros and colleagues assessed the effect of siRNA transfection in HeLa cells co-stained for DNA and the cytoskeletal proteins actin and tubulin (173). Clustering of gene perturbations by phenotypic similarity allowed the identification of novel players in the DNA damage response. Use of multi-readout, high-content screening assays brings up additional challenges in the development of assay controls, which are vital to screening success. In addition to low-noise negative controls, it is desirable to identify multiple positive control siRNAs that provide a graded perturbation of the selected assay readout. If only strong positive controls are selected, evaluation of screen quality is biased and partial phenotypes of biological interest will likely be missed. Of course in a multiplex assay with data collected over time, the comparative strength of controls for different readouts will vary, which in itself can provide new biological insight prior to the screen. Ultimately, the more comprehensive the group of positive controls identified during development of the screening assay, the more valuable the control data will be during data analysis.
A popular statistical metric used to evaluate a screening assay is the Z′ factor. Originally developed for chemical screening, it is essentially a measure of how reliably the negative and positive controls can be distinguished in the dataset3. High Z′ factors of 0.5–1.0 are considered good, whereas low Z′ factors of <0 are indicative of a poor assay. While 0.5 is considered a minimum for a good chemical screening assay, recent data suggest that this is overly stringent for RNAi screens, which are inherently more noisy, and Z′ factors of >0 are considered acceptable (174). The comparatively higher noise in RNAi screen can be attributed to several factors; potential ‘off-target’ effects (discussed below), longer (48–72hr) siRNA incubation times required to achieve target knockdown, cell stress from the transfection procedure and the fact that, in contrast to a chemical library, every reagent in the RNAi library has a cellular target and thus a potential impact on cellular behavior.
Once an assay has been established with satisfactory statistical performance criteria, pilot screens with a few hundred genes are important to determine if the assay performance from the validation step is replicated in the screen. Since this is often not the case, this step provides a further opportunity to optimize the process prior to the significant investment of time and resources required for a genome-wide screen.
Whole genome considerations
One reason many of the published screens to date have used relatively simple assays is that the scale of genome-wide screening poses numerous additional unfamiliar challenges for most academic researchers. These screens benefit greatly from experimental automation and robotic workflows, which up until recently had been restricted primarily to groups in the pharmaceutical and biotechnology industries. A significant resource for RNAi screeners has been the recent establishment of small molecule screening infrastructure in the academic and government sectors (175), such that at many research sites, the expansion of these facilities into RNAi screening has been a natural progression. Prior high-throughput standard operating procedures for screening have thus already established the importance of detailed scheduling and tracking of day-to-day workflow, to permit correlation of data irregularities with procedural variations in screen execution. The importance of recording key screen parameters to allow comparison of RNAi screening datasets between different laboratories is also emphasized by an ongoing effort to establish community standards for the minimal information that should be recorded from an RNAi screening experiment (http://miare.sourceforge.net/HomePage).
While the use of laboratory automation in RNAi screening has drawn heavily from established expertise and technology in the small molecule screening arena, the process of confirmation of RNAi screening hits and data analysis has posed some unique challenges. As mentioned above, RNAi screens have a degree of inherent noise, as their nucleic acid base-pairing dependent mechanism will always suffer from a degree of non-specificity. So called ‘off-target’ effects can occur due to the hybridization of an siRNA to a non-intended target, especially within the ‘seed’ sequence, the first 8nt from the 5′ end of the siRNA leading strand (176). Although this has been addressed by improvements in sequence selection algorithms to increase siRNA specificity and chemical modifications of the siRNA duplex to limit interference by the siRNA passenger strand, it will remain a factor to consider (177). For practical reasons, primary genome-wide arrayed screens often use a pool of several siRNA reagents in the same well. To address the issue of whether hits in primary screen are due to an off-target effect from one of the siRNAs in the pool, it is common practice to re-screen the primary hits with the individual siRNAs from each scoring pool. Cases where only one of the siRNAs from the pool reproduces the phenotype are considered likely off-target hits and are dropped from further analysis. Another similar approach is to re-screen hits with alternative siRNAs from another source. Incremental improvements in the efficacy and specificity of genome-wide siRNA libraries should in time provide reagents that reliably diminish all genes in mammalian cells, with enough reagents available per gene to permit effective filtering of ‘off-target’ phenotypes during the analysis process.
Data analysis and hit identification
The data processing and statistical approaches currently favored for analyzing RNAi screening data have been recently reviewed (174), so we will not cover them in depth here. Briefly, the analysis process follows a series of keys steps: 1) Data triage and pre-processing, carried out during and/or immediately after completion of the screen to identify any unexpected patterns in the data that might indicate experimental or procedural issues; 2) Normalization, usually carried out within each plate to remove systematic errors caused by day-to-day experimental drift over the period of time required to complete the screen; 3) Calculation of quality metrics, such as Z or Z′ factor, to evaluate assay reproducibility and consistency of controls; 4) Hit identification. The most popular method of hit identification is mean +/− k standard deviation, which sets a threshold of deviation from the normalized mean beyond which a gene is considered a hit. This is satisfactory for normally distributed data, but it performs poorly in assays prone to outliers, where methods such as median +/− k median absolute deviation are more appropriate. Many of these data analysis steps can be carried out within freely available software packages appropriate for RNAi screens (178–180).
Importantly, the statistical methods used so far for hit identification in RNAi screening data have been primarily tested using single readout assays. The development of more complex assays, such as those used in high content screens, will require the parallel development of appropriate statistical methods to evaluate the data and weight hits according to their effects on multiple readouts.
A key question for many screeners from their primary screen data is what constitutes a biologically meaningful hit. The setting of a definitive threshold for classification of genes as hits or not can be inadvisable, as genes which score as a positive in an initial primary screen may drop below the threshold in a replicate screen. A better approach may be the relative weighting of all genes’ phenotypic contribution such as in a gene vs. phenotype relationship, which is less likely to miss weak but biologically meaningful hits (160). For practical purposes, a subset of genes may have to be chosen for follow up through secondary screens, but the importance of retention of all the data in a queryable database will be vital to future integrative analysis. This type of quantitative data can be lost in screens that assign qualitative scoring of genes as ‘yes/no’. An alternative approach to avoid overly stringent filtering of data is to assign hits into multiple categories with varying levels of confidence assigned (162).
Secondary screening and hit validation
The validation of gene hits from a primary RNAi screen is vital to provide a measure of confidence in the data prior to attempting a predictive bioinformatic analysis of the dataset. Calculation of false positive and false negative rates is a popular validation method in high throughput screening, however this approach remains difficult in RNAi studies due to the lack of reliable data relating the quantitative effect of each siRNA on its target (and potential off-targets) to the observed phenotype. It has been common to use relatively subjective criteria to generate smaller lists of genes for secondary screens, although some recent studies have demonstrated that a well-designed primary screen with alternative selection criteria can generate a higher confidence list of primary hits (181). As discussed above, re-screening with deconvoluted siRNA pools and alternate sources of silencing reagents has been shown to be an excellent first step in the hit validation process. Another method that is impractical at a genome-wide scale but possible in secondary screening is to measure the extent of transcript depletion (or even better, protein expression loss) with each siRNA and then determine the correlation of knockdown with phenotype. Though expensive, transcript/protein profiling with multiple siRNAs per gene can also help validate hits or identify unexpected altered gene clusters that may contribute to off-target phenotypes. Rescue of a phenotype by ectopic expression of an RNAi resistant version of a target gene is considered the ultimate validation strategy, however this remains difficult to implement in a practical manner for a large number of genes (162).
Use of a secondary assay is a valuable means to not only validate hits, but to identify different degrees of dependence of the primary and secondary assay readouts on functionally diverse genes, which can lead to new biological insight as well as informing the subsequent classification of hits. Secondary assays with a small number of genes also provide the opportunity to introduce a more technically laborious but perhaps more data rich and potential insightful assay. Alternatively, a secondary assay might be designed specifically to remove non-specific hits from the gene set, such as genes involved in the general transcription/translation process identified in a screen using an expressed reporter as readout. The need for a secondary assay to validate primary screen hits also emphasizes the value of more complex multi-readout primary assays, as they essentially provide a secondary dataset across the entire genome. It should be obvious that there is a critical trade- off in establishing the cut-offs in primary and secondary screens for ‘hits’ – too lax a set of standards will led to many false positives in the resulting data set, leading to attempts to build networks that include spurious components, whereas too stringent a set of criteria will led to many false negatives, leaving gaps in the network construction as compared to biological reality.
Immunology applications
In the immunology and infectious disease arenas, the primary application of genome-wide RNAi to date has been in the screening for host factors involved in viral or bacterial infection. Many early studies of this type were carried out using Drosophila cells as a model for infection due to the technical simplicity of target gene knockdown in this organism. Since insect cells do not have an interferon response upon challenge with dsRNA, a long sequence of dsRNA complementary to the target gene can be introduced into cells and processing of this nucleic acid by the RNAi pathway produces a complex mixture of siRNAs that leads to robust target gene depletion. Drosophila siRNA screens have identified several of the signaling components involved in the fly systemic response to infection through the Toll, IMD and Jak/STAT pathways, and screens with live bacteria and viruses have identified specific host factors that, in some cases, are conserved in mammalian cells (reviewed in (182)). However there is a growing consensus that the integrated cellular responses to pathogens varies significantly between fly and human and that screens in mammalian cells are required to obtain data more relevant to human disease.
Genome-wide siRNA screens have recently been carried out in human cells to assess the response to several viruses and bacterial pathogens. Table 2 summarizes screens that have been published to date. The potential of these studies to identify candidate drug targets for disease treatment or vaccine therapies has been questioned on the grounds that multiple studies analyzing the same pathogen have produced largely non-overlapping gene lists. However, considering that there was no significant effort to control for experimental differences between laboratories in these studies, there are numerous reasons that might explain non-correlation:
TABLE 2.
Details of siRNA screens for host factors influencing pathogen infection in mammalian cells
| Pathogen screened | Strain | Cell line | siRNA reagent source | Genes targeted | Primary screen assay | Scoring method for hits | Total hits | Citation |
|---|---|---|---|---|---|---|---|---|
| Influenza virus | PR8 (H1N1); Udorn (H3N2) | HBEC | Dharmacon | 1,745 | Infection of 293T with HBEC viral supernatant | Z-score; 2-fold cutoff | 616 | (226) |
| Influenza virus | PR8 (H1N1) | U2OS | Dharmacon | 17,877 | HA detection on infected cell surface | 45% reduction in infected cells | 133 | (317) |
| Influenza virus | Recombinant WSN (H1N1) | A549 | Qiagen, Invitrogen, IDT | 19,628 | HA replaced with luciferase | Z-score; 2-fold cutoff by at least 2 independent siRNA/gene | 295 | (318) |
| Influenza virus | WSN (H1N1) | A549 | Qiagen | 22,843 | NP staining | Z-score; 2-fold cutoff | 287 | (319) |
| HIV | IIIB strain of HIV-1 | HeLa transgenic | Dharmacon | 21,121 | p24 staining | Z-score; 2-fold cutoff | 273 | (320) |
| HIV | Recombinant HIV-1 | HEK 293T | Not stated | 20,000 | Inhibition of luciferase expressed by recombinant virus | Statistically significant signal reduction by at least 2 independent siRNA/gene | 213 | (321) |
| HIV | HXB2 HIV-1 | HeLa transgenic | Rosetta/Merck | 19,709 | HIV LTR-driven b- gal reporter | 2 x SSMD | 390 | (322) |
| West Nile virus | WNV strain2471 | HeLa | Dharmacon | 21,121 | Viral envelope protein staining | 2-fold infection reduction | 305 | (323) |
| Mycobacterium tuberculosis | H37Rv | THP1 | Dharmacon | 18,174 | Altered bacterial CFU values 21 days post infection | Z-score; 2-fold cutoff | 275 | (324) |
| Mycobacterium tuberculosis | H37Rv | J774.1 | Qiagen | 1,032 | Altered bacterial CFU values 21 days post infection | Z-score; 2-fold cutoff | 41 | (325) |
| Hepatitis C virus | 1b N strain/NS3+NS5B subgenomic replicon | Huh-7 | Dharmacon | 4,000 | NS3-NS5B alkaline phosphatase reporter | >60% reduction in viral replication | 9 | (326) |
| Hepatitis C virus | 1b N strain/NS3+NS5B subgenomic replicon | Huh-7 | Dharmacon | 21,094 | NS3-NS5B luciferase reporter | Storey’s false discovery rate threshold of 0.10 | 96 | (327) |
Different gene sensitivities of cellular pathways to perturbation due to variable expression level of genes across the range of cell types used.
Variability in siRNA efficacy between different library sources.
Differences in the specific strains of virus used and additional variations in screening assay design and experimental protocols.
In an effort to address the disparities in gene hit lists from the multiple screens that have been carried out for HIV infection, a meta-analysis of the HIV siRNA datasets, incorporating HIV-host protein-protein interaction data and SNP contribution to HIV susceptibility, was recently carried out by Chanda and colleagues (183). This analysis initially compared all 1254 human genes identified in the genome-wide surveys to the 1434 genes in the NCBI HIV interaction database, and found that the overlap of 257 genes had a high degree of statistical significance. Network enrichment analysis of the functional categories over-represented in this gene set highlighted several gene ontology groups as being most important for HIV entry and replication, including nuclear pore/transport, GTP-binding proteins, protein complex assembly, intracellular transport, proteasome function, and RNA splicing. A closer analysis of the three siRNA screens found that of the 34 genes identified as HIV host factors in at least 2 screens, 29 were expressed in CD4+ cells and/or tissue, and thus would have the opportunity to influence the host response to virus in these key HIV target cells. Of the 8 genes in this set considered ‘druggable’, two proteins, CXCR4 and Akt1, are already targeted in established HIV treatment regimens, supporting the drug discovery potential of these screening approaches.
Similarly, a recent review of the data from multiple screens that have been carried out for influenza host factors revealed a greater overlap of hits when viewed in terms of gene ontology (GO) categories than was true when comparing individual genes (184). Of the 1449 genes identified in the published influenza screens, 128 were identified by at least two screens. Interestingly, the highest degree of pairwise overlap was observed between the Konig and Karlas screens, which used the same A549 epithelial cell line and variants of the WSN/H1N1 virus, supporting the likelihood that differences in experimental approach have a significant influence on hit correlation between independent studies. Combination of the screen hits with interaction data between flu and host proteins was used to generate a ‘host-virus network’ which showed enrichment for primary cellular processes required for the viral life cycle; endocytosis, nuclear transport, transcription/translation and viral assembly and budding. However, many of the genes highlighted by this type of analysis are likely to be poor drug targets as they are necessary for fundamental cellular processes. Selection of genes linked to cellular processes known to be important for the influenza viral life cycle may be less fruitful than taking a more non-biased approach to the screening data. For example, greater insight might be gained by combining the unfiltered siRNA datasets with genetic variation data to look for correlation between screen hits and polymorphisms linked to infection susceptibility.
This brings up an important issue with regards to the value of siRNA screening at a systems level. While individual screens in isolation may be limited in their ability to identify specific targets for drug or vaccine development, the genome-wide datasets may prove to have greater value in properly characterizing the broader cellular response and identifying the host and pathogen factors influencing survival vs. pathology in real infection scenarios, particularly when combined with metadata from the additional approaches we describe below. The value of entire screening datasets for use in subsequent integrative analyses emphasizes the need for published siRNA screens to adhere to community agreed standards on dataset content such as those described in the MIARE guidelines (http://miare.sourceforge.net/HomePage).
In addition to these studies focused on pathogenic host factors, siRNA screens have also contributed significantly to identification of key components involved in immune cell biology. RNAi screening led to the discovery of several molecules involved in the process of store-operated Ca2+ influx through plasma membrane CRAC channels and the subsequent activation of the transcription factor NFAT, which are fundamental to lymphocyte activation and function. Examples include the CRAC pore-forming protein Orai (185–187), the ER Ca2+ depletion sensor Stim (188; 189), and NFAT regulators such as NRON and the Dyrk family kinases (190; 191). RNAi has demonstrated a specific role for Akt1 in the regulation of intracellular bacterial growth (192), a screen for T cell inhibitory molecules in Hodgkin’s lymphoma identified the immunoregulatory glycan-binding protein Galectin-1 as a key factor in promoting immune privilege for neoplastic Reed Sternberg cells (193), and screens using virally-expressed shRNAs in less tractable immune cell types have identified an unexpected reliance of multiple myeloma cells on the transcription factor IRF4 (194), as well as providing significant insight to chronic antigen receptor signaling and the aberrant activation of NFkB in diffuse large B cell lymphoma (166–168). Further recent discoveries using RNAi include demonstration of key roles for the kinase RIP3 in programmed necrosis following viral inflammation and AIM2 in sensing of cytoplasmic DNA(195)(196). These examples highlight the potential of RNAi screening technology both in the discovery of individual proteins with essential functions in immune responses, and also the broader characterization of the cellular and organismal response to pathogenic challenge.
Interactomics
If microarray or NGS technologies gets us descriptive lists, and RNAi screening identifies gene products with functional relevance, we still need information on connectivity among the identified elements to develop a more complete picture of how these components work together to support biological function as we aim towards developing useful computational models of molecular pathways and networks.
Most of our insight into cellular processes is based on identifying and characterizing the interactions between cellular proteins and other biomolecules and discrete modeling of increasingly complex biological phenomena will require a detailed understanding not only of the potential binding partners of each molecule, but also the context-dependence of the interaction(s) as well as their kinetic characteristics. Although interactions among molecules of diverse chemical composition are important (for example, proteins with lipids or carbohydrates), at present, most efforts are devoted to initially producing a comprehensive view of the protein interactome. There are three principal methods in use for the cataloging of protein-protein interactions (PPIs). Yeast 2-hybrid (Y2H) remains the most practical high-throughput method for generation of interactome data, affinity purification of protein complexes followed by mass spectrometry (AP/MS) is the preferred approach for the characterization of multi-protein complexes, while protein complementation assays (PCA) are emerging as a means to measure interactions in a better preserved cellular context, with important applications for assessing real- time PPI dynamics (for reviews see (197–199)). While none of these methods stand alone as an optimal approach, together they have the potential to generate a comprehensive knowledge base in the emerging field of ‘interactomics’. In time, they will arguably generate a higher confidence (and certainly a more complete) dataset than literature curation, which has its own significant drawbacks, such as subjective interpretation of data quality, narrow focus on canonical pathway components, experimental variability between laboratories, and non-standard nomenclature across the published literature. Although most scientists tend to attribute higher intrinsic quality to the low-throughput experimental data that populate literature-curated databases, recent analyses suggest that the overlap between the most widely used databases is surprisingly low, and remarkably, >85% of the interactions are supported by only a single publication (200). A noteworthy advantage of literature curation is that biological function can often be inferred directly from the curated study; however, this ‘study bias’ can also hamper novel biological insight. Data-driven high throughput PPI approaches, although lacking a direct link to a biological question, usually avoid study bias, their systematic methodology can reduce inter experiment variability, and since negative data can also be collected, they permit an estimation of interactome coverage. Thus, although the total number of expressed genes between Drosophila, C. elegans and human is not significantly different, the respective interactomes have been estimated at 65K, 200K, and 650K, respectively (201), and may therefore provide a more meaningful barometer for organismal complexity.
Yeast 2-hybrid screens
Y2H methodology was first developed in 1989 (202). It involves the creation of yeast strains expressing ‘bait’ proteins fused with the DNA binding domain (BD) of a split transcription factor (such as Gal4), and the mating these yeast with strains expressing ‘prey’ proteins tagged with the transcription factor’s activation domain (AD). Interaction of a bait-prey ‘2-hybrid’ is then detected through expression of a reporter gene driven by a promoter responsive to the chosen reconstituted transcription factor. The original lacZ-based colorometric identification of interactions remains a popular reporter in Y2H assays, but for large-scale screening, protocols have evolved to incorporate some method of positive selection such that only cells hosting an interaction can survive in selective media (203). Examples of positive selection reporters include HIS3 and URA3, which permit growth in histidine- or uracil-lacking media, respectively. Use of these latter reporters can also be combined with competitive inhibitors and additional reagents that allow screeners to titrate for interaction strength or convert the reporter to negative selection to identify inhibitors of a given interaction.
The most sensitive screening approach is to mate each BD strain against a matrix of all available AD strains, to ensure that all pairwise combinations are tested. However, this becomes practically challenging in large screens where some form of AD strain pooling strategy is often used, followed by clone selection through reporter activation. However, sensitivity of a screen involving a large pooled library can be limited by sampling sensitivity, so a compromise approach has become popular in which small pools are screened in a matrix and positive pools are re-screened with individual BD/AD pairs (199).
Early versions of the assay were prone to identifying false positive interactions, often from auto-activating BD fusions. However, the technique has been improved considerably by careful control of fusion expression levels and the use of multiple reporters to increase the biological validity of the identified interactions. The development of recombination-based GATEWAY-type cloning technology also had a significant impact on the transition to genome-scale Y2H screening, as this permitted more efficient parallelization of ORF cloning to generate genome-scale BD and AD strain collections. Building upon these technical advances, genome-wide screens have been carried out for yeast (204; 205), Plasmodium falciparum (206), C. elegans (207), Drosophila (208), and more recently, approximately a 3rd of the human genome has been screened in this manner (209; 210).
Despite this enormous experimental effort, the limited degree of overlap observed in large-scale Y2H datasets involving proteins from the same organism remains a concern. Although some have argued that this points to a high false- positive rate, recent analysis suggests it is more likely due to poor sampling sensitivity (211). Improvement of Y2H screening efficiency is thus a continuing challenge, but may be attained with developments in accessory techniques, such as NGS for improved clone sequencing throughput. The filtering of false positives is another important aspect of Y2H data validation. Technical validation of whether an identified interaction can occur can be addressed with orthogonal technologies, such as affinity purification of the two proteins in a complex from cells. However, whether an interaction actually does occur with a function role in the organism is more difficult to address. Bioinformatic methods have been developed that use known attributes of the proteins to derive a statistical confidence score, which have been shown to correlate well with biological relevance in Drosophila studies (208).
In addition to these data validation challenges, several inherent limitations of the methodology require that Y2H datasets be complemented by additional approaches. Y2H interactions must take place in the yeast nucleus and thus often lack the appropriate spatial subcellular context, only binary interactions are detected (missing those that require accessory proteins in a larger complex), and the technique rarely detects the numerous interactions that depend on post- translational modification of one or more components.
Affinity purification/mass spectrometry (AP/MS)
AP/MS technology can address some of the deficiencies of Y2H as it involves the immunoprecipitation of a protein complex in a semi-native state followed by the MS-based identification of the complex constituents (see below). This is usually achieved by expressing the bait protein of interest with an affinity tag that can be recognized by a specific antibody to permit isolation of the bait in complex with associated proteins. Initial studies with simple affinity tags were prone to false negatives due to the stringent wash conditions required for effective bait purification. The development of tandem affinity purification (TAP) tags has addressed this by using a protease cleavable tag sequence with two affinity tags (such as protein A and a calmodulin-binding peptide). Two rounds of affinity purification with different tags significantly reduces non-specific binding and permits the use of less stringent purification conditions in each step, which has been shown to be more sensitive in identifying lower affinity interactions (197). One technical drawback is that the bait protein often must be heterologously expressed to introduce the affinity tag, and it is difficult to ensure that the expression level of the tagged protein is comparable to the endogenous untagged form. In yeast, this can be addressed by using homologous recombination at the native locus, and accordingly, genome-wide yeast strains have been generated expressing an affinity tagged version of every gene for AP/MS studies (212; 213). A similar resource for mammalian cells is some way off, but a technique has been developed using ‘BAC transgenomics’, which permits the tagging of mammalian genes at their native locus within a bacterial artificial chromosome construct, which is then stably transduced into a cell line allowing the tagged gene to be expressed from its native promoter (214). Generation of BAC libraries tagging all human and mouse genes in this manner would provide an invaluable resource for systematic interaction analysis and the potential of this approach is emphasized in a recent study of mitotic interaction complexes in human cells (215).
AP/MS promises to generate vital interactome data, especially for proteins that participate in numerous heterogeneous and often, multiplex complexes. Coupled with compartment-specific cell fractionation approaches, this could provide insights into how promiscuous proteins are ‘shared’ in the context of the whole cell PPI network. However, due to the technical challenges in affinity tagging large numbers of baits, AP/MS-based studies of mammalian interactomes have not reached the scale of Y2H studies. Some excellent focused studies have been carried out on specific modules known to have key roles in normal and disease states, such as the TGFβ (216) and TNFα/NFkB(217) subnetworks and these studies have highlighted the ability of affinity purification strategies to identify the dynamic interactions involved in signal relay through such subnetworks. A broader study of over 400 ‘disease relevant’ human proteins heterologously expressed from a CMV promoter in HEK293 cells identified over 6000 interactions, less than might be expected if the predicted 650K total interactome is accurate (218). However, the initial interaction dataset in this study was >24K, which may suggest overly stringent filtering of putative false positives during the informatic analysis. This emphasizes the importance of evaluating the process of data triage, although this will undoubtedly improve as the number of available high quality datasets increases.
Protein complementation assays (PCA)
PCAs provide another approach to high-throughput generation of PPI data. This is the most physiological of the described technologies as interactions can be detected in the cell that normally expresses the protein of interest, with post- translational modifications intact and without the problems associated with the affinity demands of the TAP method. However, like Y2H, the approach is best suited to the detection of directly interacting proteins, whereas TAP can identify molecular machines composed of multiple elements without the need for the tagged elements to directly interact with each of these other components. PCAs use a similar bait- prey premise as Y2H (hence the alternative ‘mammalian 2-hybrid’ nomenclature), but the tags expressed on the proteins represent two domains of an enzymatic or fluorescent reporter, which provides a signal only when a detected PPI brings the domains into sufficiently close proximity. Examples of reporters include DHFR, β-lactamase, and GFP (198). A recent analysis of interactions between >5K yeast ORFs by this method suggested that the confidence scores within the interaction data were improved over the other two technologies, with better enrichment of compartment-specific interactions that might be inaccessible to Y2H and AP/MS (219).
With ever improving availability of curated ORFs for human and (to a lesser extent) mouse genes, and the use of recombination technologies that permit the straightforward ‘shuttling’ of ORFs into expression platforms for all of the above approaches, there are ever fewer technical limitations to the use of PPI technologies to carry out genome scale analyses in mammalian cells.
Immunology applications
In addition to the characterization of cellular networks among host proteins, PPI techniques have key applications in the infectious disease field in determining the mechanisms of immune response evasion through the interaction of pathogen- derived proteins with the host proteome. Our knowledge of immune cell signaling, particularly in cells of the innate immune system, has been informed to a considerable extent by the mechanisms used by pathogens, especially those with type III secretion systems, to disrupt key signaling pathways and establish infection. Since pathogens often express relatively few proteins for direct interaction with the host proteome, global screens to determine host protein binding partners for pathogenic proteins have been and will continue to be highly informative in efforts to develop models of host-pathogen interactions. Since viruses are dependent on interactions within host cells for their survival and pathogenicity, elucidation of virus-host interactomes as well as intraviral interactions are of particular interest. Again, due to its amenability to high-throughput studies, Y2H has been employed much more than the other techniques described above. Intraviral Y2H screens have been carried out for numerous viruses, especially herpesviruses (VZV, KSHV, HSV-1, mCMV and EBV) due to their larger genomes providing more ORFs (70 to 170) for screening (220–223). Analysis of the intraviral network in this family has suggested a high degree of conservation of core interactions that support the viral life cycle, such as the involvement of the HSV-1 UL33, UL31 and UL34 orthologs across all herpesviruses (regardless of sequence conservation) in capsid envelopment and nuclear egress (224).
Large-scale viral-host screens against human proteins have thus far been limited to hepatitis C virus (HCV) (225), EBV (220), and influenza (226). The HCV screen used 27 baits screened against two human cDNA libraries and identified 314 interactions. This was expanded with literature curation to a network involving 11 HCV proteins with 420 human interactors. The NS3 and NS5A proteins of HCV were found to be most highly connected and the Jak/STAT and TGFβ networks were the most significantly enriched in the dataset. The EBV screen was more comprehensive, including 85 of the 89 known EBV proteins screened against 105–106 human AD strains covering most of the human genome. This identified 173 high confidence interactions between 40 EBV proteins and 112 human proteins. The influenza screen tested 10 viral proteins against approximately 12,000 human ORFs and identified 135 pairwise interaction involving 87 human proteins. In all of these virus-host screens, there was a significant enrichment for human proteins previously shown to be highly connected in the human interactome. Targeting of such ‘hub’ proteins by viruses could represent a conserved mechanism used by viruses to hijack the cellular communication machinery to promote viral replication. Recent theories of scale-free cellular network structure suggest that signal flow should be relatively robust to perturbation of single proteins, but selective targeting of multiple highly connected nodes by pathogen infection could have a significant impact.
A bioinformatic analysis of all published host-pathogen interactions showed that of >10K reported interactions, >98% involve viruses and >77% are from HIV studies (227). This emphasizes the lack of data addressing bacterial-host interactomes, which could be particularly informative if focused on secreted effectors that promote virulence. Interaction networks based on Y2H data were recently published for anthrax, Francisella and Yersinia, and interestingly came to a similar conclusion as the viral screens with regard to preferential interaction with hub proteins (228). However, depth of human genome coverage in the screened library was not reported in this study, so it is difficult to assess whether the hub preference arises from an unbiased screening pool.
These examples emphasize both the value of the data obtainable from host- pathogen interaction screens, but also the current lack of comprehensive datasets. Almost all the published large-scale studies use Y2H, and comparative analysis of these data has emphasized the low coverage obtained with this technique. Future efforts must not only focus on genome-wide analysis, but also integrate a combination of the approaches described to obtain a more comprehensive evaluation of host-pathogen interactomes and facilitate their use in building useful models of infection.
Proteomics
As emphasized in the preceding section on the ‘interactome’, system-wide analysis of the proteome has been progressing at a slower pace than that involving nucleic acid-related aspects of biology and fewer tools exist to study the proteome than the genome. At the roots of this discrepancy lie the natural properties of nucleic acids and proteins. The physicochemical properties of nucleic acids permit their recognition and replication by complementary base pairing, which provides the means of expanding the amount of material in hand from rare sources; DNA is also a relatively stable molecule and RNA can often be converted into DNA for experimental manipulation. In contrast, no widely useful method exists for amplifying proteins from cellular sources. Furthermore, although the folded structure of a protein is inherent in its primary sequence, protein function depends more on tertiary structure and especially on numerous post-translational modifications, such as phosphorylation, as compared with nucleic acid products of the cell (although we are beginning to appreciate how RNA folding and not just sequence contribute greatly to the performance of this class of biomolecules and DNA methylation is certainly an important post-synthetic modification). Because proteins cannot be amplified in a way resembling PCR for nucleic acids, methods for measurements of proteins and their properties rely either on very precise, sensitive physical measurements (mass spectrometry) or on signal amplification with antibodies (ELISA-based assays, protein microarrays, flow cytometry, confocal imaging). Proteomic and protein chemical studies are essential in developing quantitative models because they report on the actual molecular constituents of the cell, not just on the coding potential that is determined using transcriptomic methods, because they provide information on the biochemical state of the proteins and not just their existence, and because they can be used to quantify the number of protein molecules in a cellular compartment and hence, their effective concentrations. They can also ascertain the association/dissociation rates for molecular pairs and higher order complexes, and provide information on catalytic rates. These latter parameters are critical for fine-grained modeling of such systems as signaling networks. Some of the same methods used for proteomic studies are also the basis for other large-scale study of biomolecules in cells and fluids, especially mass spectrometry, which is a primary tool for metabolomic and lipidomic investigations.
Mass Spectrometry
Mass spectrometry has long been a method of choice for identification and characterization of single proteins, either from pure preparations or from SDS-PAGE bands or spots. Any soluble protein can in principle be analyzed and unambiguously characterized based on peptide mass and sequence determination. The basis of mass spectrometry is the measurement of mass-to-charge ratio (m/z) of ions in gas phase. Therefore, a mass spectrometer must contain an ion source for conversion of the sample into gas phase ions, a mass analyzer for the separation of ions based on the m/z, and a detector measuring the number of ions in the sample (usually with amplification, since the number of ions passing through the instrument is quite small). Initially, mass spectrometry was developed for small molecules, and the methods and instrumentation for small molecule analysis are still in wide use. More recently, however, protein analysis using mass spectrometry has been markedly enhanced by the development of soft ionization techniques, such as electrospray ionization (ESI) (229), used for ionization of liquid samples, and matrix-assisted laser desorption-ionization (MALDI) (230), used for ionization of solid samples bound to specific matrices.
These ion sources can be coupled with different mass analyzers. Mass analyzers sort the ions based on the m/z by applying electromagnetic fields, but the details of their construction and applied physics can vary, leading to differences in performance. The most commonly used mass analyzers for protein analysis are quadrupoles (Q), ion traps (quadrupole/three-dimensional, QIT, or linear, LTQ), time-of-flight (TOF), and Fourier-transform ion cyclotron resonance (FT-ICR) devices (an excellent summary of the properties of different types of mass spectrometers can be found in (231)). Protein identification is usually achieved by tandem mass spectrometry (MS/MS), in which the m/z ratios of peptides derived from enzymatic (usually tryptic) digestion of the proteins in the source sample are determined in the MS mode by the first stage spectrometer, and then peptides corresponding to particular m/z ratios are sequenced after energetic fragmentation. The spectrum determination of the fragmented peptides is performed by an additional mass analyzer (the second ‘MS’ in the name of this method). Therefore, many of the instruments used in proteomics are hybrids combining different types of mass analyzers, for example triple quadrupole, quadrupole/time-of-flight, or quadrupole/ion trap. The selection of the instrument for an experiment depends strongly on the application, which for proteomic studies falls primarily into three groups: determination of protein expression levels (globally and in specific cellular compartments), identification and quantification of post-translational modifications, and characterization of protein-protein interactions. Recently, a new type of mass analyzer, called the Orbitrap, has been developed (232) and implemented in a hybrid instrument with a linear ion trap and a C-trap (233). Its superior sensitivity, accuracy and resolution (234), combined with MS/MS capability and robustness, makes the LTQ-Orbitrap ideal for high sensitivity, high-throughput peptide analysis (235) and these properties have quickly made it an instrument of choice for many proteomic studies (236–238).
The fragmentation method used for sequencing depends on the application. The most popular fragmentation method, collision-induced dissociation (CID), is effective for protein identification. The peptide ions in the gas phase are allowed to collide with molecules of a neutral gas (for example, helium or argon). The collision causes bond breakage in the peptide and causes the fragmentation of the peptide ion into shorter fragment ions. By analyzing the m/z ratios of the entire set of fragments from a given peptide, it is possible to deduce the amino acid sequence due to the characteristic masses of individual amino acids, the loss of which during fragmentation at different positions leads to a ladder of fragment masses based on the identity and position of each amino acid in the peptide. Although some amino acids have the same mass, the derived sequence can be matched to a protein sequence data base and many such ambiguities are resolved by this informatics alignment process. This is not always the case, however, and hence, multiple peptides from a single protein are usually needed for unambiguous identification of the parent molecule. Post-translational modifications (PTMs) of the side chains of the amino acids also can complicate the sequence calling but the characteristic masses of such modifications can also be used to identify these sites within a protein. Labile PTMs, such as serine and threonine phosphorylation, require lower energy to dissociate them from the peptide than the energy needed to break the peptide bond. Therefore, when CID is used, the phosphate groups are removed leaving the peptide intact and not identified. The peptides with PTMs can be effectively analyzed with use of electron capture dissociation (ECD), using free electrons (239) and, especially, electron transfer dissociation (ETD), using radicals such as anthracene, to cleave the peptide backbone leaving the PTMs intact (240). C- trap dissociation (HCD), using much higher voltages to create fragment ions, has been developed for the LTQ-Orbitrap and resulted in the improved identification of small (100–200 Daltons) fragment ions. This fragmentation method is particularly effective combined with CID in achieving both good relative quantification with chemical tags (see below) and accurate peptide sequence identification (241).
Typically, the ion spectra generated by any of these instruments from the peptide input are scanned periodically (the time spent on one MS spectrum, or dwell time, is counted in milliseconds). Of the entire set of peptides injected into the instrument at any moment, only three to five of the most abundant ions detected in this short period of scan time are chosen for fragmentation and therefore identified. To enable more distinct peptides to be identified, ions of high intensity present in several consecutive spectra can be automatically excluded in subsequent scans and the next most abundant ions sequenced. However, this exclusion approach does not assure that all the peptides present as ion peaks in the MS spectra will be sequenced, and therefore many peptides present in the sample remain unidentified. This sampling issue is a major problem in mass spectrometry, especially of such samples as serum in which albumin and immunoglobulins are present at orders of magnitude greater concentrations than other elements of interest such as cytokines or shed surface proteins that might serve as biomarkers. Without depletion of the predominant species or extensive fractionation, nearly all the major peptides in each spectra will derive from the predominant species and be resequenced, preventing identification of the rarer entities. Even with efforts to enrich for rare species, it is often the case that the repeat analysis of the same sample will yield different results for overall content, simply due to incomplete sampling in each run. For protein phosphorylation measurements, a situation in which the species of interest can be rare indeed relative to the overall concentration of the parent molecule or other components in the sample, enrichment methods, such as precipitation with anti-phosphotyrosine antibodies or utilizing phosphopeptide affinity to metals and metal oxides (Immobilized Metal Affinity Chromatography, IMAC, and Metal Oxide Affinity Chromatography, MOAC) have been developed, permitting isolation of phosphopeptides from more abundant non-phosphorylated peptides in the sample. Enrichment methods of this type are also typically coupled with high performance liquid chromatography for further separation of the peptides directly prior to injection into the mass spectrometer. Two-dimensional gel electrophoresis (2D-PAGE), or chromatographic methods such as strong cation exchange (SCX) can be used for additional sample pre-fractionation. These additional fractionation steps further decrease the number of peptides entering the mass spectrometer at a given time and increases the probability that the less abundant peptides will be detected, sequenced, and identified.
In contrast to experiments in which ions for identification are selected in a largely random manner based on overall abundance, with the procedures outlined above added on to detect low abundance species, instruments with single reaction monitoring (SRM) and multiple reaction monitoring (MRM) capacity are used in quantitative experiments for reproducible detection of defined (pre-determined) sets of peptides. As already noted, only the most intense ions from the MS spectra are likely to be chosen for fragmentation in the MS/MS mode without user intervention. In SRM/MRM, the peptides from a list generated by the user are preferentially sequenced even if more intense contaminants are present in the sample. For successful SRM/MRM, the m/z of the peptides of interest as well as characteristic, non-complementary, stable fragment ions (“transitions”) have to be carefully chosen from an initial experiment conducted in ‘discovery’ mode. The dwell time of each MS peak increases with the list of transitions, so the experimental conditions have to be carefully designed, but they ensure reproducible datasets that can be compared between analyses (242).
The mere identification of species using mass spectrometry is not enough to support development of quantitative models for simulation; identification methods help establish the parts list for model building, but one still needs quantitative parameters to be used during simulation if dynamic modeling in undertaken. In the most commonly used relative comparative methods (Table 3), proteins from cells or animals representing the different experimental conditions of interest are labeled with distinct mass tags; once a peptide species from a given protein is identified, a comparison of the ion current peaks for the two isotopic versions from the two cell/animal states allows the relative quantification of this species in the two states. To achieve the necessary isotopic tagging, the proteins can be labeled metabolically, in vivo, for example, by SILAC (Stable-Isotope Labeling with Amino-Acids in Culture) (243) or post-extraction in vitro, for example, by iTRAQ (isobaric Tags for Relative and Absolute Quantification) (244; 245).
TABLE 3.
Examples of quantitative mass spectrometry methods.
| Method name | Description | Relative/absolute | Label/standard used? | Limitations | Citation |
|---|---|---|---|---|---|
| SILAC (Stable Labeling of Amino Acids in Cell Culture) | Metabolic isotope labeling; multiplexing up to three samples per analysis | Relative | Yes | Practically limited to cell culture | (243) |
| iTRAQ/TMT | Chemical post-processing labeling method; up to eight samples per analysis | Relative | Yes | Sample handling prior to labeling can introduce error | (244; 245) |
| AQUA | Synthetic heavy-labeled stanard peptides are used | Absolute | Yes | Expensive, some peptides are difficult to synthesize | (328) |
| QConCAT | Biologically expressed heavy-labeled standard peptides are used | Absolute | Yes | Expression in E.coli – PTMs are different | (251) |
| Direct comparison | Two or more separate analyses on the same platform are compared | Relative | No; Label-free | Error-prone | (329) |
| Spiking with standard | Samples are spiked with known quantities of standard peptides and compared | Absolute | No; Label-free | Needs careful computational approach | (330) |
| Targeted proteomics combined with labeled peptide standards | SRM approach with crude synthetic peptide libraries | Absolute | Yes | Limited to several dozens of peptides | (248) |
| Intensity combined with spectral counting and SRM | Data-mining computational approach | Absolute | No; label-free | Currently not applicable to PTMs; needs adaptation to platform used | (249) |
SILAC relies on incorporation of heavy elements into proteins through culture of cells in medium containing “heavy” or “light” amino acids (usually arginine or lysine labeled with stable heavy isotopes). The heavy and light labeled samples, after standard processing, are mixed and yield tryptic peptides, which display mass differences of several m/z and can be distinguished in the MS mode, where the mass to charge ratios of whole peptides are displayed. The comparison of peak intensities gives the ratio of labeled to unlabeled samples. The use of “medium” labels facilitates the relative quantification of three samples in one analysis. The caveats of SILAC include incomplete labeling (multiple cell divisions are required to replace amino acids with their labeled analogs) and the limitation of routine application to cultured cells (labeling of whole organisms, for example, mice, by feeding them labeled amino acid mix has been reported but it is not in wide use, probably due to high cost and unknown incorporation throughout the body (246)).
Chemical post-processing protein modification methods such as iTRAQ are widely used to label samples from virtually any source. The iTRAQ reagent in its original form consists of four reactive mass labels of varying isotopic composition, used to modify primary amines in peptides. The tag molecules consist of the same amine reactive group, and the 4 different isotopic labels are each balanced with a different linker so that the 4 tags are isobaric, adding the same mass to each peptide. Therefore, the peptides from the four samples labeled with different tags and then mixed before fractionation elute from the chromatographic gradient together and are observed as one peak in the MS spectra. The quantification is performed in the MS/MS mode, together with peptide identification, when the co-eluted tagged peptides are fragmented. The fragmentation breaks bonds within the peptide as well as between the peptide and mass tag in a way that separates the tag from the mass balancing component bound to the labeled amino acid. This yields peaks in the MS/MS spectra corresponding to the tags in a low molecular mass range (between 100 and 120 Daltons) and these can be compared to provide a measure of the relative quantity of the species in the various labeled samples. Recently developed iTRAQ 8-plex (245) allows parallel quantification of up to eight samples.
For accurate modeling of cellular processes, relative quantification of the type obtained by methods such as SILAC and iTRAQ in their standard use mode is not sufficient and absolute quantification of the total number of protein molecules or modified proteins in a sample is required. Accurate absolute quantification methodologies in mass spectrometry-based proteomics are very recent and are still being tested (247–250). These methods usually combine spectral counting, calibration with internal standards and advanced analytical software to avoid errors coming from the differential ionization of peptides and comparisons across experiments (Table 3). Spectral counting (a frequency-based analysis approach using the number of observed spectra) alone is not reliable and is referred to rather as “semi-quantitative”. Typically, absolute quantification methods rely on the addition of heavy-isotope labeled peptides to the biological sample to serve as internal standards of known concentration.
In the AQUA (Absolute QUAntification) method (250), isotope-labeled peptides corresponding to defined tryptic fragments of protein species of interest are chemically synthesized. Peptide selection for synthesis is based upon the careful examination of the protein sequence and often done empirically based on results obtained in non-quantitative MS/MS experiments that determine the set of detectable peptides that are generated from the protein of interest. The synthetic peptide has exactly the same sequence as the enzymatically-generated peptide, but differs in molecular mass because of stable isotope incorporation. The retention time and fragmentation of the synthetic peptide are determined by LC-MS/MS and the fragment ions (transitions) for SRM are chosen. Then, the known quantities of AQUA peptides are added to the sample mixture and the SRM experiments designed to detect the paired sample analyte-AQUA peptide are performed. This way, the concentrations of analyte peptides can be determined based on a standard curve derived from AQUA peptide additions and whole protein concentrations can be calculated from the peptide values.
Because chemical synthesis of AQUA peptides is expensive and time consuming, and not all peptide sequences can be successfully synthesized and resolubilized, the method is not perfect. An alternative approach is the production of isotopically labeled reference peptides in Escherichia coli (251). In this method, termed QconCAT, the bacteria express the concatenated peptides of interest from a synthetic gene and are grown in a medium that allows isotopic labeling of the synthesized material. The artificial protein is purified and digested to yield peptides that can be used to spike the sample as internal standards and employed in SRM as with AQUA peptides (252). The caveats of QconCAT are expression and solubilization problems, and getting the equimolar amounts of all the peptides from the concatenate during enzymatic digestion. Recently, this issue has been addressed through an equalizer peptide (EtEP) strategy, which allows equalization of internal standard peptides with a single peptide, whose concentration has been exactly determined by amino acid analysis (253). Amino acid analysis, a gold standard for the accurate absolute quantification of peptides, cannot be routinely done for all standard peptides used for quantification in mass spectrometry; for this reason one equalizer peptide is typically used as reference. This method combines the benefits of AQUA (chemical synthesis of peptides) and ConCAT (equalized peptides are kept in solution to avoid problematic resolubilization).
Mass spectrometry in systems biology is also a tool for metabolomic analysis of small-molecule metabolites from biological samples, especially for biomarker discovery. The technology differs in details from the ones used for proteomics, because of the size difference in the analytes. The most often-used instrument combination is gas chromatography coupled with mass spectrometry (GC-MS), although in some cases the molecules can be directly infused into the mass spectrometer. An excellent review of developments in mass spectrometry-based metabolomics has been published recently (254).
Mass spectrometry as a systems biology tool has several weaknesses. It is low-throughput – analyses of complex samples require laborious pre-processing and/or long HPLC gradients and time-consuming data analysis. The common “bottom-up” approach to protein and post-translational modification identification and quantification, based on proteolytic digests, requires abundant starting material (for T cells, which are small and do not have a lot of cytoplasm, often a minimum of 107 cells per analysis). In addition, even with near complete digestion efficiency, the peptides in the digest may not be optimally ionized or could be too large to be detected (the typical detection range for MS is from m/z ~400 to ~2000), so only a fraction of the sample is eventually analyzed. Finally, the results are measurements of population averages and there is no current possibility of single-cell measurements. The high number of cells needed for each analysis have made experiments on primary cells a challenge and most often, cultured cells have been analyzed; only recently have quantitative analyses of material from primary cells begun to appear (238; 255).
Nevertheless, the advantages of mass spectrometry are hard to overestimate: comparisons of thousands of proteins or phosphorylation sites in one analysis and multiplexing of up to 8 samples, lack of limitations connected with antibody availability (as for the affinity methods described below) plus measurement accuracy are alls reason it is one of the most successful proteomic methods.
Antibody-based Technologies
Population-based array methods
Antibodies are a staple in a large set of affinity-based protein analysis methods. The high specificity of properly selected and validated antibodies makes them an ideal tool for detection and quantification of proteins solubilized from virtually any kind of sample, including solid tissue, but the downside is that all antibody-based methods are limited by the availability of selective and properly validated antibodies for the molecular species in question.
Immunoblot (Western blot) and ELISA (enzyme-linked immunosorbent assay) are two examples of widely used, low-throughput, assays, although recently several groups have begun to develop ways of conducting these assays in a high- throughput mode suitable for data gathering for quantitative modeling, in combination with other methods, such as flow cytometry and live-cell assays (75). Immunoblot relies on separation of proteins in the gel by mass and/or charge, transfer to membrane and detection with antibodies. ELISA usually utilizes multi- well plates with immobilized antibodies to capture proteins, which are then detected directly (if labeled) or indirectly with another set of antibodies. Detection in both methods often involves signal enhancement; chemiluminescence or fluorescence are commonly used. For both these assays, the required quantities of sample per assay point are typically small and can be derived from a modest number of cells (104 –105 cells), but detection of low-abundance analytes is hard and multiplexing is limited. The measurements are also average abundances in cell populations, not single cell results.
Several techniques have been developed to increase the multiplexing ability of affinity-based assays. Many of them are based on bead technology. In bead-based flow assays (such as Luminex), the target proteins are captured from solution with immobilized antibodies on microparticle beads that are labeled, usually with different amounts of a single fluorochrome to allow identification. The captured proteins are detected by a second antibody set and quantified in a modified flow cytometer. The labeling of beads displaying one specific antibody with unique dye signature allows for parallel quantification of up to 100 different proteins in a single experiment. High selectivity (two antibodies per antigen) is combined here with fairly high throughput.
More sophisticated antibody-based assays have much in common with these two prototypic methods and are the result of efforts to improve throughput and accuracy and to lower detection limits. Nanofluidic Proteomic Immunoassay (NIA) separates proteins by isoelectric focusing (IEF) in a short length of a nanocapillary tube (256). Resolved proteins are captured by photochemically-activated molecules attached to the capillary wall and then detected with specific antibodies. Immobilized protein-antibody complexes are detected with chemiluminescence reagents flowing through the tube. NIA can be automated in principle, it is highly sensitive (proteins from as few as 25 cells, or fewer than 500 molecules of HRP gave good signal to noise ratio) and only a small sample volume is needed, but the technology has so far been low-throughput (up to 12 samples per day, using the prototype setup), expensive, and the analyzed proteins must be soluble. Probably the most popular emerging technology of the affinity type is protein microarray technology. There are many variations of microarrays and considerable efforts are continuously made to augment the existing protocols, because of the promise of extremely high throughput, automatization, and multiplexing. Protein microarrays are similar to DNA microarrays. Proteins are automatically spotted and immobilized on a solid surface of various compositions. The immobilized fraction can consist of capture molecules, such as purified recombinant ligands, peptides, antibodies, antibody fragments, antibody mimicking aptamers, or, in the case of reverse phase protein microarrays, a complex solution of sample cell lysates. When capture molecules are immobilized, the sample solution is applied over the spots. When the lysate is immobilized, it is probed with soluble capture molecules. The latter provides an advantage in miniaturization: a single spot can contain as little protein as from a single cell. On the other hand, the stability of microarrays has to be considered: spotted antibodies are usually more stable and less affected by prolonged storage than immobilized whole lysates.
Variability of applied material captured in individual spots is a challenge in printing all microarrays and the engineering efforts to circumvent this problem are visible in the automation area: non-contact robots delivering precise, nano-to-picoliter droplets are superior to contact printers, where carry-over and pin alignment are an issue (257; 258). The stability and, to some extent, the variability issues are prevented in a recently developed protein microarray method called nucleic acid programmable protein array (NAPPA), where functional proteins are translated in situ from DNA printed on the spots with the use of in vitro transcription-translation system (IVTT) (259). The coding region of the cDNA corresponding to a protein to be expressed is engineered into an RNA expression plasmid and fused to a co-expressed GST-tag. Plasmid DNA is cross-linked to a psoralen-biotin conjugate with the use of ultraviolet light, and arrayed onto glass slides together with avidin and polyclonal GST antibody. At this stage, the array is dried and can be stored at room temperature. To activate and use the array, the IVTT (such as reticulocyte lysate with T7 polymerase to generate synthetic RNA) is added to the array. The transcribed RNA is translated by the IVTT and the expressed proteins are immediately immobilized in their spots by the capture of the GST-tag by anti-GST antibody. Independently, similar alternative strategies have been developed: MIST (multiple spotting technique) spots the IVTT from E.coli on a printed PCR template; in DAPA (DNA array to protein array) proteins translated on the cDNA array diffuse through a membrane infused with cell-free extract to a surface with capture molecules; and high density peptide-and protein chips can be produced by immediate immobilization of proteins synthesized on stalled ribosomes with puromycin-grafted oligonucleotides (260). Such approaches ensure reproducible, fresh, high-yield protein microarrays for each experiment. Also, the expression clones can be stored in large collection in cassettes, permitting facile examination of the proteins encoded by interesting clones after their expression in live cells using a mammalian expression vector containing the same insert. Protein microarrays can be used to quantify protein abundance in the samples as well as to explore protein function, for example kinase activity measured by substrate phosphorylation levels.
Immunoassays, like mass spectrometry, measure averages in the population of molecules obtained from cells. Because even synchronized cells of the same origin, not to mention tissue samples, are most often heterogeneous and their response to stimuli can be diverse, measurements at the single cell level provide complementary and crucial information.
Single-cell flow cytometric methods
Flow cytometry permits multiparameter analysis, single-cell resolution, and close to real-time measurements under limited conditions. Initially used for detection of cell surface markers on live cell populations, nowadays this technology is widely used to measure intracellular protein and DNA concentrations and cellular processes in fixed cells. The multiplexing ability of flow methods, which depend on fluorescently labeled antibodies, has been limited by the factors connected with antibody use in addition to sample acquisition throughput and much effort concentrates on overcoming this limitation.
Phosphoflow, a flow cytometric approach to determine phosphoepitope levels, is an essential tool in single-cell proteomics for mapping cell signaling networks (261). This method involves cell fixation and permeabilization prior to staining with anti-phosphoprotein antibodies. Carefully developed procedure with specific reagents compatible with flow cytometry and use of combinations of phospho-specific antibodies labeled with different fluorophores provides a rapid and efficient means to measure the levels of a variety of intracellular phosphoepitopes (262). The number of distinct phosphoepitopes depends only on the current capabilities of flow cytometry (as of now, 17 markers can be measured simultaneously (3)). To increase sample throughput, a cell-based multiplexing approach, called fluorescent cell barcoding (FCB), was developed especially for phosphoflow (263) and is based on a similar principle to that used for multiplexing in bead-based cytokine assays. In FCB, samples are labeled with fluorophore to variable intensity, which is achieved by treatment with different concentrations of the reactive form of the fluorophore. In this way, each sample gets a unique signature of fluorescence intensity. With one fluorophore, 4–6 different samples can be analyzed together. Because multiple fluorophores can be used to label the cells, the number of different fluorescent signatures increases geometrically (16–36 samples can be monitored with the combination of two markers). The method has been reported to increase the resolution when used with 96 samples simultaneously, at the same time reducing antibody consumption and acquisition time.
Still, the overlapping fluorophore spectra require compensation and the experiments become more difficult to perform with each additional color. The alternatives for fluorophores in multiparameter detection include Surface Enhanced Raman Scattering (SERS), which has been implemented in flow cytometry and microscopy in several variants (264–266), and a newly reported technology called mass cytometry (CyTOF) (267). CyTOF replaces fluorescent labels with specially designed multiatom elemental (lanthanide) antibody tags. These labels are detected by an inductively coupled plasma time-of-flight mass spectrometer (ICP-TOF-MS) that has recently become commercially available. The ICP source produces transient ion clouds from cells or beads, and the ions from the generated supersonic plasma jet are detected by a fast TOF ion detector. The signal is amplified and digitized, so even low intensity signals can be detected. The availability of many stable isotopes for the antibody tags facilitates simultaneous detection of many target molecules in the sample – the authors demonstrate the method as applied to 20 antigens in model leukemia cell lines and acute myeloid leukemia patient samples, but the method can be broadened to analyze many more antigens on individual cells in a single experiment. The initial device and methods do not permit cell sorting, however, because particles are completely disintegrated during the analysis, but techniques are being developed to allow elution of the labels and timed collection of the eluted material to maintain correspondence to the cell of origin in the flow stream, so this limitation may change in the near future. Other limitations include the analysis of only a subset (presently 30% but soon to be >70%) of the applied cell sample), slower cell throughput than conventional flow cytometers, and reduced sensitivity on the very low end of expression. Nevertheless, the ability to analyze dozens of target antigens with no concern of spectral overlap or the need for complex compensation methods makes this an extremely promising new method that may help generate highly quantitative data on a single cell level in multiplexed form for use in modeling studies. On the other hand, the greatly increased number of parameters being examined makes data analysis much more complex; even downloading the files from the instrument pushes the limits of current computers and new configurations of processors in specialized devices are being developed to handle the data throughput, while new computational algorithms for automatic clustering of the many parameters being measured will enable rapid and unbiased assessment of the multidimensional data (263).
Beyond identification and quantification of specific molecular moieties in individual cells, flow-based methods are also useful for assessing molecular interactions using methods such as FRET or protein complementation, described above in the section on interactome determination.
Microscopic Methods
While flow cytometry can reveal much about the identities and amounts of components of a cell, and even something about local molecular interactions, it does not provide means of resolving topographically where in the cell the labeled molecules reside, it cannot trace changes in the location or number of molecules in a single cell over time (it only provides a single time point for data collection from each cell), it cannot relate signaling events to changes in cell shape, dynamics, or motility, and it cannot examine cells in complex environments. For all these aspects of biology that one might wish to incorporate in models and simulations, microscopic imaging is the method of choice, with implementations ranging from ultrahigh resolution electron microscopic tomography to define structural details for models of such processes as neurotransmitter release in a synapse (268; 269), to super-resolution light imaging to better understand the location of molecules in a single cell (270), to TIRF-imaging to investigate components involved in membrane- related events with counterligands displayed in planar membrane surfaces (271), to live cell confocal and 2-photon imaging in 2 and 3-D environments (272; 273) and intravital imaging using 2-photon instruments to assess cell behavior and signaling in complex tissues and organs (274). Especially through the use of methods of fluorescence microscopy that allow specific molecules to be tagged with light- emitting proteins (275), it is possible to collect quantitative spatiotemporal information about protein expression, molecular movements, and cytoskeletal changes, among other parameters, in living cells over prolonged time frames. Coupled with the use of emerging microfluidic methods that parallelize the treatment of individual members of cell population with the same stimulus and allow for simultaneous imaging of this cell population, one can develop extensive datasets on multiple target molecules through color multiplexing and with additional information about intercellular heterogeneity within the cell population in terms of both molecular expression and cell behavior in response to the stimulus (276; 277).
Fluorescence microscopy is by far the most commonly employed technique in systems studies. The broad spectrum of fluorophores emitting light of different colors allows simultaneous detection of several molecules at the same time. Basic epi-fluorescence microscopy is affordable and requires simple instruments, but the blurring of the image due to the light emission from different depths of the sample is a constant challenge, especially for correct quantitative measurements. This problem is overcome by confocal microscopy, where only the light from the focal plane is collected. The resulting sharp images allow the reconstruction of the three-dimensional image by stacking a series of optical x-y section in the z-plane (z-stacks). Confocal microscopy can be used for quantitative measurements of cell motility and dynamic intracellular events in time-lapse experiments (278), although there are limitations. High-energy excitation photons from the lasers used on confocal microscopes can lead to phototoxicity and photobleaching, and scattering limits the thickness of the sample to 80 μm (278). The alternative technology is multiphoton laser scanning fluorescence microscopy (279), which used lower energy light in short pulses, compensating the energy by simultaneous absorption of multiple photons for excitation of the sample. Two-photon microscopy, where two photons of infrared light are enough to cause light emission from the fluorophore, yields superb results in thicker (up to 1 mm) samples of live, explanted immune tissues with limited photodamage. Although often considered ‘confocal’ inherently, 2-photon imaging does have a lower axial (z-dimension) resolution as compared to confocal designs and for isolated cells, the latter is often preferable if bleaching and phototoxicity are not limitations in the experiment.
Resolution in fluorescence microscopy has until recently been constrained by the diffraction limit, which is dependent on the numerical aperture of the lens and the wavelength of light being used. More recently, so-called super-resolution methods have been introduce that allow computational techniques to discriminate point sources below this diffraction limit. Two such high-resolution techniques are photoactivated localization microscopy (PALM) (280) and stochastic optical reconstruction microscopy (STORM) (281). PALM resolves molecules separated only by a few nanometers by serial photoactivation and subsequent bleaching of many sparse subsets of photoactivatable fluorescent protein (PA-FP) molecules. Each source spot at each photoconversion cycle is treated as a Gaussian intensity distribution and the center of the Gaussian peak is calculated, improving the resolution from around 200 to ~20–40 nM. The method works well for separating the images of molecules densely packed within cell samples and is constantly being improved with introduction of new photochromes and methods for performing the excitation, collection, and bleach cycles at faster and faster speeds, thus allowing the method to image molecules in motion (282). In the closely related and similar method of STORM, the fluorescent image is constructed from high-accuracy localization of individual fluorescent molecules that are switched on and off using different color light.
More examples and a detailed review of modern microscopic technologies used for imaging the immune system can be found in (283). Here we highlight some examples of the applications of the instruments and techniques mentioned above. The recruitment of molecules to the immunological synapse has been elucidated in three-dimensional detail using digital fluorescence microscopy (284; 285), which led to subsequent modeling of the molecular patterns during immunological synapse formation (66) and speculation about the role of the cSMAC as a site of TCR signaling inactivation rather than activation, as originally proposed (286; 287). Intravital two-photon microscopy has yielded data on cell dynamics in vivo that have served as the basis for several computational exercises examining issues such as the search pattern of T cells within lymph nodes and the role of stromal elements in this behavior (288), the relationship between signal intensity and the transition between the motile and static states seen during the initiation of T cell adaptive responses (289–291), the possible roles of guided vs. random walk patterns in ensuring effective activation of T cells during a single passage through a lymph node (292), and the validity of older models of migration between the dark and light zones of the germinal center in affinity-based selection (293).
FRET has not only been used in flow studies, but also exploited using microscopic imaging to assess protein-protein interactions within the live cell environment. For example, differences in TCR recruitment to the immunological synapse and CD8-TCR interaction after T-cell induction with different APCs (294), characterization of the TCRα chain connecting peptide motif allowing binding to CD8 and efficient signal initiation (295), and the formation of a complex leading to B cell antigen receptor signaling upon binding of multivalent antigens (296) have been studied using this method.
Surface Plasmon Resonance Methods
The methods described above allow generation of a catalog of expressed molecules, especially proteins, their quantification, and the description of their chemical modification, the detection of molecular interactions, and the generation of time- and space-resolved datasets involving molecular interactions, modification, and intracellular relocation. All of these types of information are critical for the generation of fine-grained models of cellular behavior, especially signaling pathways leading to gene activation that can be assessed by the nucleic acid technologies also described here, but using this information for model building and simulation is still impeded by the absence of many of the quantitative parameters that can constrain the substantial degrees of freedom in such models, particularly those involving the dynamics of molecular interactions and of enzymatic processes. Only a few of the required numbers exist in a validated form in the literature, in part because until recently it was unclear to investigators why time and resources should be spent determining affinity constants and catalytic rates. With the advent of an increased emphasis on modeling in immunology, such numbers have become much more important, though still sparse. A number of tools exist for acquiring the necessary data, though few have been developed as robust, multiplexed methods suitable for the scale of typical systems biology model building.
The binding constants necessary for detailed molecular interaction models are possible to obtain with the use of surface plasmon resonance (SPR) technology, implemented in instruments such as Biacore (GE) or ProteOn (BioRad). The phenomenon of surface plasmon resonance occurs when the plasmons (electron charge density waves) on the planar surface of the metal in a special chip (such as gold) are excited by a change in the boundary of this surface with a medium of different refractive index (such as buffer). This occurs when the mass of material at the buffer-chip interface changes, as will be the case when an applied substance binds to molecules immobilized on the chip surface. Since the resultant resonance change is not affected by sample color or opacity, SPR can used to determine the interactions between many different types of ligands and their binding partners, to search for novel protein-protein, DNA–protein and DNA-RNA interactions and to determine drug targets (297). At the present, a major limitation of this method is the substantial amount of effort required to produce very pure protein in a form suitable for either linkage to the chip surface or for adding as the analyte to a derivatized chip. The generation of such purified reagents is typically a slow process involving one molecule at a time and the production of a reliable dataset can take months. This is clearly inadequate for use in parameterizing complex models with many components. However, improvements in chip design, especially the multiplexing capacity of microfluidic versions of these instruments, allow many assays to be performed in parallel. There is the prospect of combining some of the technology of NAPPA for in vitro synthesis of proteins from expression clones with such microfluidic multiplexed SPR devices to enable analysis of dozens of molecular interactions in a short (several week) period without the need for individual molecule purification, albeit with less precision than would be the case for carefully purified individual components. However, the nature of molecular models is such that small errors in such measurements typically have little effect on model performance, but they do need to be in the right overall numerical range; sensitivity testing of the model can determine which parameters are most critical to simulated outcomes and these can point to specific interactions that might require re- measurement by the more precise methods involving highly purified material. This combination of multiplexed SPR with NAPPA-style synthesis of relevant molecules can open the door to the collection of interaction parameters needed for fine-grained modeling at a pace and to an extent that will allow a substantial reduction in the degrees of freedom in molecular models.
Applications in Immunology
Progress in proteomic instrumentation and technology has made possible many novel studies of primary immune system cells. One of the areas that has yielded especially intriguing results is the study of the dendritic cell proteome. Dendritic cells (DCs), a heterogeneous population of professional antigen presenting cells, consist of a number of distinct subtypes, distinguishable by multiple surface markers. These DC subtypes differ substantially in origin and function. Luber et al. (238) looked at the differences in protein abundance in some of the commonly described DC subsets using a label-free quantitative mass spectrometry approach with an in-house quantification algorithm. They profiled protein abundance differences among conventional DC subsets, which they defined as conventional CD8+, CD4+, and double negative DCs separated by flow cytometry, to a depth of more than 5000 proteins and requiring only 1.5–2.0 × 106 purified cells. The results were validated with surface markers of known abundance in DC subtypes and comparison to the results of previous microarray studies. Based on the latter comparisons, the authors conclude that this approach is a reliable method to study closely related cell types in vivo. The analysis revealed mutually exclusive expression of some pattern recognition pathways (differential expression of members of the NLR, TLR, and RLH signaling pathways in response to flu virus and Sendai virus), which might explain differences in response to different pathogens. These new data provided new insight into the regulation of PRRs in infected cells. A more focused approach was used by Segura et al. (298), who isolated the plasma membranes of mouse spleen CD8+ and CD8− DCs by immunoaffinity methods and analyzed their protein composition using mass spectrometry. The results were only semi-quantitative, because only the spectral counting method was used. Nevertheless, the comparative analysis suggested that many known or potential pathogen-recognition receptors and immunomodulatory molecules are differentially expressed, and these results were subsequently confirmed by flow cytometry and Western blot analyses. At this point in time, however, these profiling experiments are mainly cataloguing exercises that yield parts lists for future studies of a more integrated and functional nature.
Phosphorylation in immune cell signaling has not been analyzed frequently by mass spectrometry using primary cells because of the difficulty of obtaining these cells in sufficient number. Most examinations of phophorylation sites involved in T-cell signaling have thus utilized the leukemic Jurkat T cell line (299). However, recent increases in instrument sensitivity and improved enrichment methods now allow analysis of primary cells and reports of phosphoproteomics in non- transformed T-cells have begun to appear. Iwai et al. examined the tyrosine phosphorylation network in T-cell receptor signaling in cells from diabetes-prone and normal mice, identifying and quantifying over 100 tyrosine phosphorylation sites using iTRAQ labeling (255). More frequently, phospho-specific flow cytometry has been utilized to analyze phosphorylation-based signaling in the immune system cells. Hale et al. (300) applied phosphoflow to examine the changes in cytokine signal transduction through the STAT family of transcription factors during clinical progression of systemic lupus erythematosus (SLE) in human patients. The measurements of signaling responses at the single-cell level in five immune cell types to a panel of 10 cytokines thought to play essential roles in SLE led to a highly multiplexed view of cytokine signal processing in adaptive and innate immune cells. Robust changes, including an increase in the T-cell response to IL-10 and cessation of STAT molecule responses to multiple cytokines, were found during progression of SLE, providing evidence for negative feedback regulation and existence of a cytokine-driven oscillator, which may regulate periodic changes in SLE activity. In early rheumatoid arthritis (ERA), 15 signaling effectors were examined by phospho- flow in peripheral blood mononuclear cells, identifying patterns of effector activation by phosphorylation characteristic for rheumatoid arthritis (distinct from the patterns observed in osteoarthritis), but the same in ERA as in the late RA (301). For example the ratio of p-AKT to p-p38 was significantly elevated in patients with RA, and may be used for diagnostic purposes. To date, these data have not been used to derive models suitable for simulation and testing of intracellular signaling, but rather serve primarily as tools for biomarker development.
The use of various types of protein arrays has facilitated many recent systems-type immunological studies. Merbl et al. (302) combined antigen- microarray device and a genetic algorithm analysis to investigate large-scale patterns in the antibody repertoire reflecting the state of metastatic or non- metastatic tumors in C57BL/6 mice. The analysis of IgG and IgM autoantibodies binding to over 300 self-antigens revealed informative antibody patterns responding to the growth of the tumor cells and distinguishing between animals bearing metastatic and non-metastatic tumors. Nucleic acid-programmable protein microarrays (NAPPA) were successfully used (303) to identify Pseudomonas aeruginosa proteins that trigger an adaptive immune response in cystic fibrosis. Out of 262 P. aeruginosa outer membrane and exported proteins examined, antibodies to 12 were detected in statistically significant number of patients and confirmed by ELISA as potential candidates for future vaccine development. Ceroni et al. (304) used the same technology to investigate the adaptive immune response to infection with Varicella zoster virus. All 69 virus proteins were studied in sera from 68 infected individuals and the IgG antibodies against many viral antigens were detected and validated by Western blot. These antibodies were only present in the subset of VZV-positive patients, demonstrating the complexity of the humoral immune response to the viral infection and validating the NAPPA approach for the development of diagnostic tests. Protein arrays were used also for systemic, large-scale profiling of the specificity of antibody responses against autoantigens in a panel of autoimmune diseases (305) and allergies (306; 307).
Concluding Remarks
In this review, we have attempted to present one major aspect in the still emerging field of systems biology, namely the use of dynamic computational models for simulating biological behavior. In particular, we have emphasized the importance of integrating high-throughput data collection methods and highly quantitative experimental techniques with mathematical approaches to develop an enhanced understanding of how the immune system operates at various scales.
It is apparent from the text that much more has been accomplished to date at the level of ‘omic scale’ data collection than in the development and use of models and simulation. This is not to say that important contributions have not come from the modeling efforts we and many others have undertaken, but rather, that the number and power of the models created to date are rather limited and that this approach is not yet the primary way high content data are used to develop new insights into immune system function. In part, this limitation comes from the recent emergence of the experimental capacity to collect the necessary high-quality, extensive datasets required for computational analysis. It also arises from a gap in the skills sets of experimental biologists and the only very recent develop of software platforms that empower these investigators to undertake complex model building. Finally, it reflects a ‘cultural’ bias that places primary emphasis on the discovery nature of biological research and accords modeling less value.
Indeed, a primary goal of this review has been to lay out the rationale for the critical importance going forward of incorporating more computational methods into biological studies. The overall complexity of all biological systems, especially the immune system with its hundreds of cell types and regulatory and effector molecules, necessitates such an approach if we are to place these multitude of components into a scheme that helps us understand and eventually predict how they interact to yield biological behavior. We have a rather limited ability to visualize the very non-linear ways in which concatenated feedback circuits so characteristic of the immune system affect its output. This demands the use of formal mathematical treatments to associate modest differences in component concentrations, binding affinities, enzymatic activity, cell replication and death rates, and the like with the effects of gene polymorphism and mutation or drug treatments on system performance. The recent evidence for digital behavior in the central Erk signaling pathway (308–310), for the role of very modest differences in transcription factor concentration in controlling developmental cell fate of lymphocytes (311–313), or for small differences in the ratio of regulatory vs. effector cells in determining the presence or absence of autoimmune disease (314) all speak to the need to develop more quantitative methods for integrating our fine-grained knowledge of immune components into larger schemes that better reflect system output.
A variety of activities and events will impact the rate at which modeling and simulation are integrated into the immunological mainstream. First, nothing succeeds like success and as various groups produce models and simulations that make useful predictions about biology and these predictions are validated by experiment, others will seek to adopt such approaches in their own work. Second, the dissemination of improved tools for constructing complex models, conducting simulations, and sharing the results, together with greater access to ever cheaper mass computing power, will empower the community to work together in building and utilizing better and bigger models. Finally, the changes already in progress in the way students are educated, with the inclusion of more statistics, mathematics, and computer programming at various stages of secondary, college, and post-graduate education, will make it easier for new investigators to incorporate these methods into their research activities, just as the familiarity of newly minted MDs and PhDs with ‘omic’ technologies has led over the past decade to the facile inclusion of such methods into their projects. We therefore close on an optimistic note, suggesting that within a generation, modeling and simulation will have become a mainstream component of biological research, comfortable for many if not most investigators. This will result in the movement from cartoon representations of biological systems to instantiated quantitative models shared by the field and capable of high quality predictive analysis of how small and large scale aspects of the immune system will behave when perturbed experimentally, by genetic variation, or by medical intervention.
Figure 1.
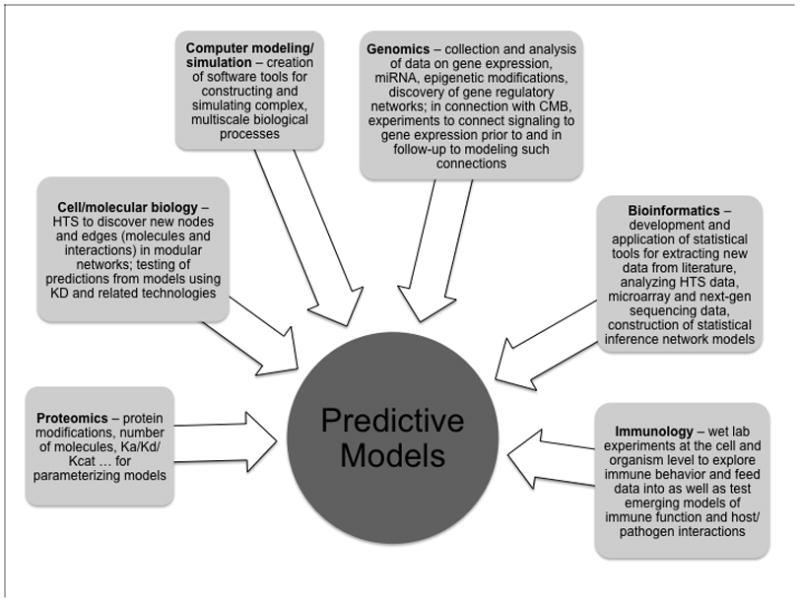
Team approach to modeling.
The figure illustrates the various technical, data gathering, and biological components of an integrated research approach to computational systems biology with a focus on fine-grained, dynamic modeling and simulation of processes such as cell signaling.
Acknowledgments
The authors wish to thank many colleagues, especially Gregoire Altan- Bonnet, or discussions and research interactions that were critical to our developing the perspective espoused in this review. We apologize to those in the field whose papers and contributions were not explicitly cited due to limitations of length. This work was supported by the Intramural Research Program of NIAID, NIH.
Footnotes
The term ‘ridiculome’ comes from a talk by Andrea Califano of Columbia University.
The name was chosen with reference to the Monte Carlo gambling casino, due to the element of chance used in the approach to calculate the probability of particular events – such as molecular movements or interactions – at any time point.
The Z′ factor is calculated from the following equation: Where SD+ = positive control standard deviation, SD− = negative control standard deviation, μ+ = positive control mean, μ− = negative control mean.
Literature Cited
- 1.Hoheisel JD. Microarray technology: beyond transcript profiling and genotype analysis. Nat Rev Genet. 2006;7:200–10. doi: 10.1038/nrg1809. [DOI] [PubMed] [Google Scholar]
- 2.Wold B, Myers RM. Sequence census methods for functional genomics. Nat Methods. 2008;5:19–21. doi: 10.1038/nmeth1157. [DOI] [PubMed] [Google Scholar]
- 3.Perfetto SP, Chattopadhyay PK, Roederer M. Seventeen-colour flow cytometry: unravelling the immune system. Nat Rev Immunol. 2004;4:648–55. doi: 10.1038/nri1416. [DOI] [PubMed] [Google Scholar]
- 4.Aebersold R, Mann M. Mass spectrometry-based proteomics. Nature. 2003;422:198–207. doi: 10.1038/nature01511. [DOI] [PubMed] [Google Scholar]
- 5.Benoist C, Germain RN, Mathis D. A plaidoyer for ‘systems immunology’. Immunol Rev. 2006;210:229–34. doi: 10.1111/j.0105-2896.2006.00374.x. [DOI] [PubMed] [Google Scholar]
- 6.Noble D. Modeling the heart--from genes to cells to the whole organ. Science. 2002;295:1678–82. doi: 10.1126/science.1069881. [DOI] [PubMed] [Google Scholar]
- 7.Alon U. Network motifs: theory and experimental approaches. Nat Rev Genet. 2007;8:450–61. doi: 10.1038/nrg2102. [DOI] [PubMed] [Google Scholar]
- 8.Herzenberg LA, Black SJ. Regulatory circuits and antibody responses. Eur J Immunol. 1980;10:1–11. doi: 10.1002/eji.1830100102. [DOI] [PubMed] [Google Scholar]
- 9.Germain RN. The art of the probable: system control in the adaptive immune system. Science. 2001;293:240–5. doi: 10.1126/science.1062946. [DOI] [PubMed] [Google Scholar]
- 10.Callard R, Hodgkin P. Modeling T- and B-cell growth and differentiation. Immunol Rev. 2007;216:119–29. doi: 10.1111/j.1600-065X.2006.00498.x. [DOI] [PubMed] [Google Scholar]
- 11.Meier-Schellersheim M, Fraser ID, Klauschen F. Multiscale modeling for biologists. Wiley Interdiscip Rev Syst Biol Med. 2009;1:4–14. doi: 10.1002/wsbm.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Davidson EH. Network design principles from the sea urchin embryo. Curr Opin Genet Dev. 2009;19:535–40. doi: 10.1016/j.gde.2009.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Alon U. An Introduction to Systems Biology: Design Principles of Biological Circuits. Chapman & Hall/CRC; 2007. [Google Scholar]
- 14.Heng TS, Painter MW. The Immunological Genome Project: networks of gene expression in immune cells. Nat Immunol. 2008;9:1091–4. doi: 10.1038/ni1008-1091. [DOI] [PubMed] [Google Scholar]
- 15.Shaw DE, Deneroff MM, Dror RO, Kuskin JS, Larson RH, et al. Anton, a special-purpose machine for molecular dynamics simulation. Commun Acm. 2008;51:91–7. [Google Scholar]
- 16.Klepeis JL, Lindorff-Larsen K, Dror RO, Shaw DE. Long-timescale molecular dynamics simulations of protein structure and function. Curr Opin Struct Biol. 2009;19:120–7. doi: 10.1016/j.sbi.2009.03.004. [DOI] [PubMed] [Google Scholar]
- 17.Varma R, Campi G, Yokosuka T, Saito T, Dustin ML. T cell receptor-proximal signals are sustained in peripheral microclusters and terminated in the central supramolecular activation cluster. Immunity. 2006;25:117–27. doi: 10.1016/j.immuni.2006.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huse M, Klein LO, Girvin AT, Faraj JM, Li QJ, et al. Spatial and temporal dynamics of T cell receptor signaling with a photoactivatable agonist. Immunity. 2007;27:76–88. doi: 10.1016/j.immuni.2007.05.017. [DOI] [PubMed] [Google Scholar]
- 19.Batista FD, Arana E, Barral P, Carrasco YR, Depoil D, et al. The role of integrins and coreceptors in refining thresholds for B-cell responses. Immunol Rev. 2007;218:197–213. doi: 10.1111/j.1600-065X.2007.00540.x. [DOI] [PubMed] [Google Scholar]
- 20.Delon J, Kaibuchi K, Germain RN. Exclusion of CD43 from the immunological synapse is mediated by phosphorylation-regulated relocation of the cytoskeletal adaptor moesin. Immunity. 2001;15:691–701. doi: 10.1016/s1074-7613(01)00231-x. [DOI] [PubMed] [Google Scholar]
- 21.Faure S, Salazar-Fontana LI, Semichon M, Tybulewicz VL, Bismuth G, et al. ERM proteins regulate cytoskeleton relaxation promoting T cell-APC conjugation. Nat Immunol. 2004;5:272–9. doi: 10.1038/ni1039. [DOI] [PubMed] [Google Scholar]
- 22.Gomez TS, Billadeau DD. T cell activation and the cytoskeleton: you can’t have one without the other. Adv Immunol. 2008;97:1–64. doi: 10.1016/S0065-2776(08)00001-1. [DOI] [PubMed] [Google Scholar]
- 23.Badour K, Zhang J, Shi F, Leng Y, Collins M, Siminovitch KA. Fyn and PTP-PEST-mediated regulation of Wiskott-Aldrich syndrome protein (WASp) tyrosine phosphorylation is required for coupling T cell antigen receptor engagement to WASp effector function and T cell activation. J Exp Med. 2004;199:99–112. doi: 10.1084/jem.20030976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sohn HW, Tolar P, Pierce SK. Membrane heterogeneities in the formation of B cell receptor-Lyn kinase microclusters and the immune synapse. J Cell Biol. 2008;182:367–79. doi: 10.1083/jcb.200802007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Treanor B, Depoil D, Gonzalez-Granja A, Barral P, Weber M, et al. The membrane skeleton controls diffusion dynamics and signaling through the B cell receptor. Immunity. 2010;32:187–99. doi: 10.1016/j.immuni.2009.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Andrews SS, Bray D. Stochastic simulation of chemical reactions with spatial resolution and single molecule detail. Phys Biol. 2004;1:137–51. doi: 10.1088/1478-3967/1/3/001. [DOI] [PubMed] [Google Scholar]
- 27.Saxton MJ. Modeling 2D and 3D diffusion. Methods Mol Biol. 2007;400:295–321. doi: 10.1007/978-1-59745-519-0_20. [DOI] [PubMed] [Google Scholar]
- 28.Kerr RA, Bartol TM, Kaminsky B, Dittrich M, Chang JC, et al. Fast Monte Carlo Simulation Methods for Biological Reaction-Diffusion Systems in Solution and on Surfaces. SIAM J Sci Comput. 2008;30:3126. doi: 10.1137/070692017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tolle DP, Le Novere N. Meredys, a multi-compartment reaction-diffusion simulator using multistate realistic molecular complexes. BMC Syst Biol. 2010;4:24. doi: 10.1186/1752-0509-4-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gruenert G, Ibrahim B, Lenser T, Lohel M, Hinze T, Dittrich P. Rule-based spatial modeling with diffusing, geometrically constrained molecules. BMC Bioinformatics. 2010;11:307. doi: 10.1186/1471-2105-11-307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chan C, George AJ, Stark J. Cooperative enhancement of specificity in a lattice of T cell receptors. Proc Natl Acad Sci U S A. 2001;98:5758–63. doi: 10.1073/pnas.101113698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sosinsky GE, Deerinck TJ, Greco R, Buitenhuys CH, Bartol TM, Ellisman MH. Development of a model for microphysiological simulations: small nodes of ranvier from peripheral nerves of mice reconstructed by electron tomography. Neuroinformatics. 2005;3:133–62. doi: 10.1385/NI:3:2:133. [DOI] [PubMed] [Google Scholar]
- 33.Ander M, Beltrao P, Di Ventura B, Ferkinghoff-Borg J, Foglierini M, et al. SmartCell, a framework to simulate cellular processes that combines stochastic approximation with diffusion and localisation: analysis of simple networks. Syst Biol (Stevenage) 2004;1:129–38. doi: 10.1049/sb:20045017. [DOI] [PubMed] [Google Scholar]
- 34.Schaff J, Fink CC, Slepchenko B, Carson JH, Loew LM. A general computational framework for modeling cellular structure and function. Biophys J. 1997;73:1135–46. doi: 10.1016/S0006-3495(97)78146-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hattne J, Fange D, Elf J. Stochastic reaction-diffusion simulation with MesoRD. Bioinformatics. 2005;21:2923–4. doi: 10.1093/bioinformatics/bti431. [DOI] [PubMed] [Google Scholar]
- 36.Meier-Schellersheim M, Xu X, Angermann B, Kunkel EJ, Jin T, Germain RN. Key role of local regulation in chemosensing revealed by a new molecular interaction-based modeling method. PLoS Comput Biol. 2006;2:e82. doi: 10.1371/journal.pcbi.0020082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schaff JC, Slepchenko BM, Loew LM. Physiological modeling with virtual cell framework. Methods Enzymol. 2000;321:1–23. doi: 10.1016/s0076-6879(00)21184-1. [DOI] [PubMed] [Google Scholar]
- 38.Schneider IC, Haugh JM. Quantitative elucidation of a distinct spatial gradient-sensing mechanism in fibroblasts. J Cell Biol. 2005;171:883–92. doi: 10.1083/jcb.200509028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hlavacek WS, Faeder JR, Blinov ML, Perelson AS, Goldstein B. The complexity of complexes in signal transduction. Biotechnol Bioeng. 2003;84:783–94. doi: 10.1002/bit.10842. [DOI] [PubMed] [Google Scholar]
- 40.Hlavacek WS, Faeder JR. The complexity of cell signaling and the need for a new mechanics. Sci Signal. 2009;2:pe46. doi: 10.1126/scisignal.281pe46. [DOI] [PubMed] [Google Scholar]
- 41.Lok L, Brent R. Automatic generation of cellular reaction networks with Moleculizer 1.0. Nat Biotechnol. 2005;23:131–6. doi: 10.1038/nbt1054. [DOI] [PubMed] [Google Scholar]
- 42.Hlavacek WS, Faeder JR, Blinov ML, Posner RG, Hucka M, Fontana W. Rules for modeling signal-transduction systems. Sci STKE. 2006;2006:re6. doi: 10.1126/stke.3442006re6. [DOI] [PubMed] [Google Scholar]
- 43.Feret J, Danos V, Krivine J, Harmer R, Fontana W. Internal coarse-graining of molecular systems. Proc Natl Acad Sci U S A. 2009;106:6453–8. doi: 10.1073/pnas.0809908106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Takahashi K, Ishikawa N, Sadamoto Y, Sasamoto H, Ohta S, et al. E-Cell 2: multi-platform E-Cell simulation system. Bioinformatics. 2003;19:1727–9. doi: 10.1093/bioinformatics/btg221. [DOI] [PubMed] [Google Scholar]
- 45.Dhar P, Meng TC, Somani S, Ye L, Sairam A, et al. Cellware--a multi-algorithmic software for computational systems biology. Bioinformatics. 2004;20:1319–21. doi: 10.1093/bioinformatics/bth067. [DOI] [PubMed] [Google Scholar]
- 46.Hoops S, Sahle S, Gauges R, Lee C, Pahle J, et al. COPASI--a COmplex PAthway SImulator. Bioinformatics. 2006;22:3067–74. doi: 10.1093/bioinformatics/btl485. [DOI] [PubMed] [Google Scholar]
- 47.Hucka M, Finney A, Sauro HM, Bolouri H, Doyle J, Kitano H. The ERATO Systems Biology Workbench: enabling interaction and exchange between software tools for computational biology. Pacific Symposium on Biocomputing. 2002:450–61. doi: 10.1142/9789812799623_0042. [DOI] [PubMed] [Google Scholar]
- 48.Shapiro BE, Levchenko A, Meyerowitz EM, Wold BJ, Mjolsness ED. Cellerator: extending a computer algebra system to include biochemical arrows for signal transduction simulations. Bioinformatics. 2003;19:677–8. doi: 10.1093/bioinformatics/btg042. [DOI] [PubMed] [Google Scholar]
- 49.Hu B, Matthew Fricke G, Faeder JR, Posner RG, Hlavacek WS. GetBonNie for building, analyzing and sharing rule-based models. Bioinformatics. 2009;25:1457–60. doi: 10.1093/bioinformatics/btp173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Moraru, Schaff JC, Slepchenko BM, Blinov ML, Morgan F, et al. Virtual Cell modelling and simulation software environment. IET Syst Biol. 2008;2:352–62. doi: 10.1049/iet-syb:20080102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Minguet S, Schamel WW. A permissive geometry model for TCR-CD3 activation. Trends Biochem Sci. 2008;33:51–7. doi: 10.1016/j.tibs.2007.10.008. [DOI] [PubMed] [Google Scholar]
- 52.Ma Z, Janmey PA, Finkel TH. The receptor deformation model of TCR triggering. FASEB J. 2008;22:1002–8. doi: 10.1096/fj.07-9331hyp. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Germain RN. T-cell signaling: the importance of receptor clustering. Curr Biol. 1997;7:R640–4. doi: 10.1016/s0960-9822(06)00323-x. [DOI] [PubMed] [Google Scholar]
- 54.McKeithan TW. Kinetic proofreading in T-cell receptor signal transduction. Proc Natl Acad Sci U S A. 1995;92:5042–6. doi: 10.1073/pnas.92.11.5042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dushek O, Das R, Coombs D. A role for rebinding in rapid and reliable T cell responses to antigen. PLoS Comput Biol. 2009;5:e1000578. doi: 10.1371/journal.pcbi.1000578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Burroughs NJ, Lazic Z, van der Merwe PA. Ligand detection and discrimination by spatial relocalization: A kinase-phosphatase segregation model of TCR activation. Biophys J. 2006;91:1619–29. doi: 10.1529/biophysj.105.080044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bunnell SC, Hong DI, Kardon JR, Yamazaki T, McGlade CJ, et al. T cell receptor ligation induces the formation of dynamically regulated signaling assemblies. J Cell Biol. 2002;158:1263–75. doi: 10.1083/jcb.200203043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Campi G, Varma R, Dustin ML. Actin and agonist MHC-peptide complex- dependent T cell receptor microclusters as scaffolds for signaling. J Exp Med. 2005;202:1031–6. doi: 10.1084/jem.20051182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dushek O, Mueller S, Soubies S, Depoil D, Caramalho I, et al. Effects of intracellular calcium and actin cytoskeleton on TCR mobility measured by fluorescence recovery. PLoS One. 2008;3:e3913. doi: 10.1371/journal.pone.0003913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cairo CW, Das R, Albohy A, Baca QJ, Pradhan D, et al. Dynamic regulation of CD45 lateral mobility by the spectrin-ankyrin cytoskeleton of T cells. J Biol Chem. 285:11392–401. doi: 10.1074/jbc.M109.075648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Das J, Ho M, Zikherman J, Govern C, Yang M, et al. Digital signaling and hysteresis characterize ras activation in lymphoid cells. Cell. 2009;136:337–51. doi: 10.1016/j.cell.2008.11.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wofsy C, Coombs D, Goldstein B. Calculations show substantial serial engagement of T cell receptors. Biophys J. 2001;80:606–12. doi: 10.1016/S0006-3495(01)76041-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Coombs D, Kalergis AM, Nathenson SG, Wofsy C, Goldstein B. Activated TCRs remain marked for internalization after dissociation from pMHC. Nat Immunol. 2002;3:926–31. doi: 10.1038/ni838. [DOI] [PubMed] [Google Scholar]
- 64.Valitutti S, Muller S, Cella M, Padovan E, Lanzavecchia A. Serial triggering of many T-cell receptors by a few peptide-MHC complexes. Nature. 1995;375:148–51. doi: 10.1038/375148a0. [DOI] [PubMed] [Google Scholar]
- 65.Padhan K, Varma R. Immunological synapse: a multi-protein signalling cellular apparatus for controlling gene expression. Immunology. 129:322–8. doi: 10.1111/j.1365-2567.2009.03241.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Qi SY, Groves JT, Chakraborty AK. Synaptic pattern formation during cellular recognition. Proc Natl Acad Sci U S A. 2001;98:6548–53. doi: 10.1073/pnas.111536798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Stefanova I, Hemmer B, Vergelli M, Martin R, Biddison WE, Germain RN. TCR ligand discrimination is enforced by competing ERK positive and SHP-1 negative feedback pathways. Nat Immunol. 2003;4:248–54. doi: 10.1038/ni895. [DOI] [PubMed] [Google Scholar]
- 68.Kolch W. Coordinating ERK/MAPK signalling through scaffolds and inhibitors. Nat Rev Mol Cell Biol. 2005;6:827–37. doi: 10.1038/nrm1743. [DOI] [PubMed] [Google Scholar]
- 69.Levchenko A, Bruck J, Sternberg PW. Scaffold proteins may biphasically affect the levels of mitogen-activated protein kinase signaling and reduce its threshold properties. Proc Natl Acad Sci U S A. 2000;97:5818–23. doi: 10.1073/pnas.97.11.5818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chan C, Stark J, George AJ. Feedback control of T-cell receptor activation. Proc Biol Sci. 2004;271:931–9. doi: 10.1098/rspb.2003.2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Altan-Bonnet G, Germain RN. Modeling T cell antigen discrimination based on feedback control of digital ERK responses. PLoS Biol. 2005;3:e356. doi: 10.1371/journal.pbio.0030356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gillespie DT. A general method for numerically simulating the stochastic time evolution of coupled chemical reactions. J Comp Phys. 1976;22:403–34. [Google Scholar]
- 73.Margarit SM, Sondermann H, Hall BE, Nagar B, Hoelz A, et al. Structural evidence for feedback activation by Ras.GTP of the Ras-specific nucleotide exchange factor SOS. Cell. 2003;112:685–95. doi: 10.1016/s0092-8674(03)00149-1. [DOI] [PubMed] [Google Scholar]
- 74.Hlavacek WS, Redondo A, Metzger H, Wofsy C, Goldstein B. Kinetic proofreading models for cell signaling predict ways to escape kinetic proofreading. Proc Natl Acad Sci U S A. 2001;98:7295–300. doi: 10.1073/pnas.121172298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Albeck JG, Burke JM, Aldridge BB, Zhang M, Lauffenburger DA, Sorger PK. Quantitative analysis of pathways controlling extrinsic apoptosis in single cells. Mol Cell. 2008;30:11–25. doi: 10.1016/j.molcel.2008.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Albeck JG, Burke JM, Spencer SL, Lauffenburger DA, Sorger PK. Modeling a snap-action, variable-delay switch controlling extrinsic cell death. PLoS Biol. 2008;6:2831–52. doi: 10.1371/journal.pbio.0060299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Albeck JG, MacBeath G, White FM, Sorger PK, Lauffenburger DA, Gaudet S. Collecting and organizing systematic sets of protein data. Nat Rev Mol Cell Biol. 2006;7:803–12. doi: 10.1038/nrm2042. [DOI] [PubMed] [Google Scholar]
- 78.Volterra V. Variazioni e fluttuazioni del humero d’individui in specie animali conviventi [Variations and fluctuations of the number of individuals in animal species living together] Mem Acad Lincei. 1926;2:31–113. [Google Scholar]
- 79.Mohri H, Bonhoeffer S, Monard S, Perelson AS, Ho DD. Rapid turnover of T lymphocytes in SIV-infected rhesus macaques. Science. 1998;279:1223–7. doi: 10.1126/science.279.5354.1223. [DOI] [PubMed] [Google Scholar]
- 80.De Boer RJ, Mohri H, Ho DD, Perelson AS. Turnover rates of B cells, T cells, and NK cells in simian immunodeficiency virus-infected and uninfected rhesus macaques. J Immunol. 2003;170:2479–87. doi: 10.4049/jimmunol.170.5.2479. [DOI] [PubMed] [Google Scholar]
- 81.Grossman Z, Meier-Schellersheim M, Sousa AE, Victorino RM, Paul WE. CD4+ T-cell depletion in HIV infection: are we closer to understanding the cause? Nat Med. 2002;8:319–23. doi: 10.1038/nm0402-319. [DOI] [PubMed] [Google Scholar]
- 82.Brenchley JM, Douek DC. HIV infection and the gastrointestinal immune system. Mucosal Immunol. 2008;1:23–30. doi: 10.1038/mi.2007.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Okoye A, Meier-Schellersheim M, Brenchley JM, Hagen SI, Walker JM, et al. Progressive CD4+ central memory T cell decline results in CD4+ effector memory insufficiency and overt disease in chronic SIV infection. J Exp Med. 2007;204:2171–85. doi: 10.1084/jem.20070567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Grossman Z, Paul WE. The impact of HIV on naive T-cell homeostasis. Nat Med. 2000;6:976–7. doi: 10.1038/79667. [DOI] [PubMed] [Google Scholar]
- 85.Hawkins ED, Hommel M, Turner ML, Battye FL, Markham JF, Hodgkin PD. Measuring lymphocyte proliferation, survival and differentiation using CFSE time-series data. Nat Protoc. 2007;2:2057–67. doi: 10.1038/nprot.2007.297. [DOI] [PubMed] [Google Scholar]
- 86.De Boer RJ, Ganusov VV, Milutinovic D, Hodgkin PD, Perelson AS. Estimating lymphocyte division and death rates from CFSE data. Bull Math Biol. 2006;68:1011–31. doi: 10.1007/s11538-006-9094-8. [DOI] [PubMed] [Google Scholar]
- 87.Yates A, Chan C, Strid J, Moon S, Callard R, et al. Reconstruction of cell population dynamics using CFSE. BMC Bioinformatics. 2007;8:196. doi: 10.1186/1471-2105-8-196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Luzyanina T, Roose D, Schenkel T, Sester M, Ehl S, et al. Numerical modelling of label-structured cell population growth using CFSE distribution data. Theor Biol Med Model. 2007;4:26. doi: 10.1186/1742-4682-4-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Dowling MR, Hodgkin PD. Modelling naive T-cell homeostasis: consequences of heritable cellular lifespan during ageing. Immunol Cell Biol. 2009;87:445–56. doi: 10.1038/icb.2009.11. [DOI] [PubMed] [Google Scholar]
- 90.Yates A, Callard R, Stark J. Combining cytokine signalling with T-bet and GATA-3 regulation in Th1 and Th2 differentiation: a model for cellular decision-making. J Theor Biol. 2004;231:181–96. doi: 10.1016/j.jtbi.2004.06.013. [DOI] [PubMed] [Google Scholar]
- 91.Busse D, de la Rosa M, Hobiger K, Thurley K, Flossdorf M, et al. Competing feedback loops shape IL-2 signaling between helper and regulatory T lymphocytes in cellular microenvironments. Proc Natl Acad Sci U S A. 2010;107:3058–63. doi: 10.1073/pnas.0812851107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Regoes RR, Barber DL, Ahmed R, Antia R. Estimation of the rate of killing by cytotoxic T lymphocytes in vivo. Proc Natl Acad Sci U S A. 2007;104:1599–603. doi: 10.1073/pnas.0508830104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Celada F, Seiden PE. A computer model of cellular interactions in the immune system. Immunol Today. 1992;13:56–62. doi: 10.1016/0167-5699(92)90135-T. [DOI] [PubMed] [Google Scholar]
- 94.Celada F, Seiden PE. Affinity maturation and hypermutation in a simulation of the humoral immune response. Eur J Immunol. 1996;26:1350–8. doi: 10.1002/eji.1830260626. [DOI] [PubMed] [Google Scholar]
- 95.Kesmir C, De Boer RJ. A spatial model of germinal center reactions: cellular adhesion based sorting of B cells results in efficient affinity maturation. J Theor Biol. 2003;222:9–22. doi: 10.1016/s0022-5193(03)00010-9. [DOI] [PubMed] [Google Scholar]
- 96.Figge MT, Garin A, Gunzer M, Kosco-Vilbois M, Toellner KM, Meyer-Hermann M. Deriving a germinal center lymphocyte migration model from two- photon data. J Exp Med. 2008;205:3019–29. doi: 10.1084/jem.20081160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ray JC, Flynn JL, Kirschner DE. Synergy between individual TNF-dependent functions determines granuloma performance for controlling Mycobacterium tuberculosis infection. J Immunol. 2009;182:3706–17. doi: 10.4049/jimmunol.0802297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Efroni S, Harel D, Cohen IR. Emergent dynamics of thymocyte development and lineage determination. PLoS Comput Biol. 2007;3:e13. doi: 10.1371/journal.pcbi.0030013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Graner F, Glazier JA. Simulation of biological cell sorting using a two-dimensional extended Potts model. Phys Rev Lett. 1992;69:2013–6. doi: 10.1103/PhysRevLett.69.2013. [DOI] [PubMed] [Google Scholar]
- 100.Beltman JB, Maree AF, de Boer RJ. Spatial modelling of brief and long interactions between T cells and dendritic cells. Immunol Cell Biol. 2007;85:306–14. doi: 10.1038/sj.icb.7100054. [DOI] [PubMed] [Google Scholar]
- 101.Beltman JB, Henrickson SE, von Andrian UH, de Boer RJ, Maree AF. Towards estimating the true duration of dendritic cell interactions with T cells. J Immunol Methods. 2009;347:54–69. doi: 10.1016/j.jim.2009.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Saez-Rodriguez J, Simeoni L, Lindquist JA, Hemenway R, Bommhardt U, et al. A logical model provides insights into T cell receptor signaling. PLoS Comput Biol. 2007;3:e163. doi: 10.1371/journal.pcbi.0030163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Schadt EE, Monks SA, Drake TA, Lusis AJ, Che N, et al. Genetics of gene expression surveyed in maize, mouse and man. Nature. 2003;422:297–302. doi: 10.1038/nature01434. [DOI] [PubMed] [Google Scholar]
- 104.Unwin RD, Whetton AD. Systematic proteome and transcriptome analysis of stem cell populations. Cell Cycle. 2006;5:1587–91. doi: 10.4161/cc.5.15.3101. [DOI] [PubMed] [Google Scholar]
- 105.Gerling IC, Singh S, Lenchik NI, Marshall DR, Wu J. New data analysis and mining approaches identify unique proteome and transcriptome markers of susceptibility to autoimmune diabetes. Mol Cell Proteomics. 2006;5:293–305. doi: 10.1074/mcp.M500197-MCP200. [DOI] [PubMed] [Google Scholar]
- 106.de Sousa Abreu R, Penalva LO, Marcotte EM, Vogel C. Global signatures of protein and mRNA expression levels. Mol Biosyst. 2009;5:1512–26. doi: 10.1039/b908315d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Stamou P, Kontoyiannis DL. Posttranscriptional regulation of TNF mRNA: a paradigm of signal-dependent mRNA utilization and its relevance to pathology. Curr Dir Autoimmun. 11:61–79. doi: 10.1159/000289197. [DOI] [PubMed] [Google Scholar]
- 108.Brem RB, Yvert G, Clinton R, Kruglyak L. Genetic dissection of transcriptional regulation in budding yeast. Science. 2002;296:752–5. doi: 10.1126/science.1069516. [DOI] [PubMed] [Google Scholar]
- 109.DeRisi J, Penland L, Brown PO, Bittner ML, Meltzer PS, et al. Use of a cDNA microarray to analyse gene expression patterns in human cancer. Nat Genet. 1996;14:457–60. doi: 10.1038/ng1296-457. [DOI] [PubMed] [Google Scholar]
- 110.Lockhart DJ, Dong H, Byrne MC, Follettie MT, Gallo MV, et al. Expression monitoring by hybridization to high-density oligonucleotide arrays. Nat Biotechnol. 1996;14:1675–80. doi: 10.1038/nbt1296-1675. [DOI] [PubMed] [Google Scholar]
- 111.Gardina PJ, Clark TA, Shimada B, Staples MK, Yang Q, et al. Alternative splicing and differential gene expression in colon cancer detected by a whole genome exon array. BMC Genomics. 2006;7:325. doi: 10.1186/1471-2164-7-325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Srinivasan K, Shiue L, Hayes JD, Centers R, Fitzwater S, et al. Detection and measurement of alternative splicing using splicing-sensitive microarrays. Methods. 2005;37:345–59. doi: 10.1016/j.ymeth.2005.09.007. [DOI] [PubMed] [Google Scholar]
- 113.Uttamchandani M, Neo JL, Ong BN, Moochhala S. Applications of microarrays in pathogen detection and biodefence. Trends Biotechnol. 2009;27:53–61. doi: 10.1016/j.tibtech.2008.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Golub TR, Slonim DK, Tamayo P, Huard C, Gaasenbeek M, et al. Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science. 1999;286:531–7. doi: 10.1126/science.286.5439.531. [DOI] [PubMed] [Google Scholar]
- 115.Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature. 2000;403:503–11. doi: 10.1038/35000501. [DOI] [PubMed] [Google Scholar]
- 116.Winnepenninckx V, Lazar V, Michiels S, Dessen P, Stas M, et al. Gene expression profiling of primary cutaneous melanoma and clinical outcome. J Natl Cancer Inst. 2006;98:472–82. doi: 10.1093/jnci/djj103. [DOI] [PubMed] [Google Scholar]
- 117.Boldrick JC, Alizadeh AA, Diehn M, Dudoit S, Liu CL, et al. Stereotyped and specific gene expression programs in human innate immune responses to bacteria. Proc Natl Acad Sci U S A. 2002;99:972–7. doi: 10.1073/pnas.231625398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Chaussabel D, Semnani RT, McDowell MA, Sacks D, Sher A, Nutman TB. Unique gene expression profiles of human macrophages and dendritic cells to phylogenetically distinct parasites. Blood. 2003;102:672–81. doi: 10.1182/blood-2002-10-3232. [DOI] [PubMed] [Google Scholar]
- 119.Huang Q, Liu D, Majewski P, Schulte LC, Korn JM, et al. The plasticity of dendritic cell responses to pathogens and their components. Science. 2001;294:870–5. doi: 10.1126/science.294.5543.870. [DOI] [PubMed] [Google Scholar]
- 120.Nau GJ, Richmond JF, Schlesinger A, Jennings EG, Lander ES, Young RA. Human macrophage activation programs induced by bacterial pathogens. Proc Natl Acad Sci U S A. 2002;99:1503–8. doi: 10.1073/pnas.022649799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Batliwalla FM, Baechler EC, Xiao X, Li W, Balasubramanian S, et al. Peripheral blood gene expression profiling in rheumatoid arthritis. Genes Immun. 2005;6:388–97. doi: 10.1038/sj.gene.6364209. [DOI] [PubMed] [Google Scholar]
- 122.Batliwalla FM, Li W, Ritchlin CT, Xiao X, Brenner M, et al. Microarray analyses of peripheral blood cells identifies unique gene expression signature in psoriatic arthritis. Mol Med. 2005;11:21–9. doi: 10.2119/2006-00003.Gulko. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Bennett L, Palucka AK, Arce E, Cantrell V, Borvak J, et al. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J Exp Med. 2003;197:711–23. doi: 10.1084/jem.20021553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Bomprezzi R, Ringner M, Kim S, Bittner ML, Khan J, et al. Gene expression profile in multiple sclerosis patients and healthy controls: identifying pathways relevant to disease. Hum Mol Genet. 2003;12:2191–9. doi: 10.1093/hmg/ddg221. [DOI] [PubMed] [Google Scholar]
- 125.Burczynski ME, Peterson RL, Twine NC, Zuberek KA, Brodeur BJ, et al. Molecular classification of Crohn’s disease and ulcerative colitis patients using transcriptional profiles in peripheral blood mononuclear cells. J Mol Diagn. 2006;8:51–61. doi: 10.2353/jmoldx.2006.050079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Pascual V, Allantaz F, Arce E, Punaro M, Banchereau J. Role of interleukin-1 (IL-1) in the pathogenesis of systemic onset juvenile idiopathic arthritis and clinical response to IL-1 blockade. J Exp Med. 2005;201:1479–86. doi: 10.1084/jem.20050473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Lempicki RA, Polis MA, Yang J, McLaughlin M, Koratich C, et al. Gene expression profiles in hepatitis C virus (HCV) and HIV coinfection: class prediction analyses before treatment predict the outcome of anti-HCV therapy among HIV-coinfected persons. J Infect Dis. 2006;193:1172–7. doi: 10.1086/501365. [DOI] [PubMed] [Google Scholar]
- 128.Reghunathan R, Jayapal M, Hsu LY, Chng HH, Tai D, et al. Expression profile of immune response genes in patients with Severe Acute Respiratory Syndrome. BMC Immunol. 2005;6:2. doi: 10.1186/1471-2172-6-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Simmons CP, Popper S, Dolocek C, Chau TN, Griffiths M, et al. Patterns of host genome-wide gene transcript abundance in the peripheral blood of patients with acute dengue hemorrhagic fever. J Infect Dis. 2007;195:1097–107. doi: 10.1086/512162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Zaas AK, Chen M, Varkey J, Veldman T, Hero AO, 3rd, et al. Gene expression signatures diagnose influenza and other symptomatic respiratory viral infections in humans. Cell Host Microbe. 2009;6:207–17. doi: 10.1016/j.chom.2009.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Ardura MI, Banchereau R, Mejias A, Di Pucchio T, Glaser C, et al. Enhanced monocyte response and decreased central memory T cells in children with invasive Staphylococcus aureus infections. PLoS One. 2009;4:e5446. doi: 10.1371/journal.pone.0005446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Jacobsen M, Repsilber D, Gutschmidt A, Neher A, Feldmann K, et al. Candidate biomarkers for discrimination between infection and disease caused by Mycobacterium tuberculosis. J Mol Med. 2007;85:613–21. doi: 10.1007/s00109-007-0157-6. [DOI] [PubMed] [Google Scholar]
- 133.Thompson LJ, Dunstan SJ, Dolecek C, Perkins T, House D, et al. Transcriptional response in the peripheral blood of patients infected with Salmonella enterica serovar Typhi. Proc Natl Acad Sci U S A. 2009;106:22433–8. doi: 10.1073/pnas.0912386106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Griffiths MJ, Shafi MJ, Popper SJ, Hemingway CA, Kortok MM, et al. Genomewide analysis of the host response to malaria in Kenyan children. J Infect Dis. 2005;191:1599–611. doi: 10.1086/429297. [DOI] [PubMed] [Google Scholar]
- 135.Wang J, Ramakrishnan R, Tang Z, Fan W, Kluge A, et al. Quantifying EGFR alterations in the lung cancer genome with nanofluidic digital PCR arrays. Clin Chem. 56:623–32. doi: 10.1373/clinchem.2009.134973. [DOI] [PubMed] [Google Scholar]
- 136.Flagella M, Bui S, Zheng Z, Nguyen CT, Zhang A, et al. A multiplex branched DNA assay for parallel quantitative gene expression profiling. Anal Biochem. 2006;352:50–60. doi: 10.1016/j.ab.2006.02.013. [DOI] [PubMed] [Google Scholar]
- 137.Geiss GK, Bumgarner RE, Birditt B, Dahl T, Dowidar N, et al. Direct multiplexed measurement of gene expression with color-coded probe pairs. Nat Biotechnol. 2008;26:317–25. doi: 10.1038/nbt1385. [DOI] [PubMed] [Google Scholar]
- 138.Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437:376–80. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Shendure J, Porreca GJ, Reppas NB, Lin X, McCutcheon JP, et al. Accurate multiplex polony sequencing of an evolved bacterial genome. Science. 2005;309:1728–32. doi: 10.1126/science.1117389. [DOI] [PubMed] [Google Scholar]
- 140.Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Hawkins RD, Hon GC, Ren B. Next-generation genomics: an integrative approach. Nat Rev Genet. 11:476–86. doi: 10.1038/nrg2795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Park PJ. ChIP-seq: advantages and challenges of a maturing technology. Nat Rev Genet. 2009;10:669–80. doi: 10.1038/nrg2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Farnham PJ. Insights from genomic profiling of transcription factors. Nat Rev Genet. 2009;10:605–16. doi: 10.1038/nrg2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Wang Z, Schones DE, Zhao K. Characterization of human epigenomes. Curr Opin Genet Dev. 2009;19:127–34. doi: 10.1016/j.gde.2009.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Hinds DA, Stuve LL, Nilsen GB, Halperin E, Eskin E, et al. Whole- genome patterns of common DNA variation in three human populations. Science. 2005;307:1072–9. doi: 10.1126/science.1105436. [DOI] [PubMed] [Google Scholar]
- 146.2008. The 1000 Genomes Project.
- 147.Cookson W, Liang L, Abecasis G, Moffatt M, Lathrop M. Mapping complex disease traits with global gene expression. Nat Rev Genet. 2009;10:184–94. doi: 10.1038/nrg2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Chen CZ, Li L, Lodish HF, Bartel DP. MicroRNAs modulate hematopoietic lineage differentiation. Science. 2004;303:83–6. doi: 10.1126/science.1091903. [DOI] [PubMed] [Google Scholar]
- 149.Baltimore D, Boldin MP, O’Connell RM, Rao DS, Taganov KD. MicroRNAs: new regulators of immune cell development and function. Nat Immunol. 2008;9:839–45. doi: 10.1038/ni.f.209. [DOI] [PubMed] [Google Scholar]
- 150.Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–33. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Landgraf P, Rusu M, Sheridan R, Sewer A, Iovino N, et al. A mammalian microRNA expression atlas based on small RNA library sequencing. Cell. 2007;129:1401–14. doi: 10.1016/j.cell.2007.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Meister G, Landthaler M, Dorsett Y, Tuschl T. Sequence-specific inhibition of microRNA- and siRNA-induced RNA silencing. RNA. 2004;10:544–50. doi: 10.1261/rna.5235104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Krutzfeldt J, Rajewsky N, Braich R, Rajeev KG, Tuschl T, et al. Silencing of microRNAs in vivo with ‘antagomirs’. Nature. 2005;438:685–9. doi: 10.1038/nature04303. [DOI] [PubMed] [Google Scholar]
- 154.Karginov FV, Conaco C, Xuan Z, Schmidt BH, Parker JS, et al. A biochemical approach to identifying microRNA targets. Proc Natl Acad Sci U S A. 2007;104:19291–6. doi: 10.1073/pnas.0709971104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Baek D, Villen J, Shin C, Camargo FD, Gygi SP, Bartel DP. The impact of microRNAs on protein output. Nature. 2008;455:64–71. doi: 10.1038/nature07242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Selbach M, Schwanhausser B, Thierfelder N, Fang Z, Khanin R, Rajewsky N. Widespread changes in protein synthesis induced by microRNAs. Nature. 2008;455:58–63. doi: 10.1038/nature07228. [DOI] [PubMed] [Google Scholar]
- 157.Taganov KD, Boldin MP, Chang KJ, Baltimore D. NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc Natl Acad Sci U S A. 2006;103:12481–6. doi: 10.1073/pnas.0605298103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Li QJ, Chau J, Ebert PJ, Sylvester G, Min H, et al. miR-181a is an intrinsic modulator of T cell sensitivity and selection. Cell. 2007;129:147–61. doi: 10.1016/j.cell.2007.03.008. [DOI] [PubMed] [Google Scholar]
- 159.McCoy CE, Sheedy FJ, Qualls JE, Doyle SL, Quinn SR, et al. IL-10 inhibits miR-155 induction by toll-like receptors. J Biol Chem. 285:20492–8. doi: 10.1074/jbc.M110.102111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Friedman A, Perrimon N. Genetic screening for signal transduction in the era of network biology. Cell. 2007;128:225–31. doi: 10.1016/j.cell.2007.01.007. [DOI] [PubMed] [Google Scholar]
- 161.Fraser ID, Germain RN. Navigating the network: signaling cross-talk in hematopoietic cells. Nat Immunol. 2009;10:327–31. doi: 10.1038/ni.1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Boutros M, Ahringer J. The art and design of genetic screens: RNA interference. Nat Rev Genet. 2008;9:554–66. doi: 10.1038/nrg2364. [DOI] [PubMed] [Google Scholar]
- 163.Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 2001;411:494–8. doi: 10.1038/35078107. [DOI] [PubMed] [Google Scholar]
- 164.Zeng Y, Cullen BR. Sequence requirements for micro RNA processing and function in human cells. RNA. 2003;9:112–23. doi: 10.1261/rna.2780503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Moffat J, Sabatini DM. Building mammalian signalling pathways with RNAi screens. Nat Rev Mol Cell Biol. 2006;7:177–87. doi: 10.1038/nrm1860. [DOI] [PubMed] [Google Scholar]
- 166.Bidere N, Ngo VN, Lee J, Collins C, Zheng L, et al. Casein kinase 1alpha governs antigen-receptor-induced NF-kappaB activation and human lymphoma cell survival. Nature. 2009;458:92–6. doi: 10.1038/nature07613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Davis RE, Ngo VN, Lenz G, Tolar P, Young RM, et al. Chronic active B-cell-receptor signalling in diffuse large B-cell lymphoma. Nature. 463:88–92. doi: 10.1038/nature08638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Lam LT, Davis RE, Ngo VN, Lenz G, Wright G, et al. Compensatory IKKalpha activation of classical NF-kappaB signaling during IKKbeta inhibition identified by an RNA interference sensitization screen. Proc Natl Acad Sci U S A. 2008;105:20798–803. doi: 10.1073/pnas.0806491106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Carralot JP, Kim TK, Lenseigne B, Boese AS, Sommer P, et al. Automated high-throughput siRNA transfection in raw 264.7 macrophages: a case study for optimization procedure. J Biomol Screen. 2009;14:151–60. doi: 10.1177/1087057108328762. [DOI] [PubMed] [Google Scholar]
- 170.Collinet C, Stoter M, Bradshaw CR, Samusik N, Rink JC, et al. Systems survey of endocytosis by multiparametric image analysis. Nature. 464:243–9. doi: 10.1038/nature08779. [DOI] [PubMed] [Google Scholar]
- 171.Neumann B, Walter T, Heriche JK, Bulkescher J, Erfle H, et al. Phenotypic profiling of the human genome by time-lapse microscopy reveals cell division genes. Nature. 464:721–7. doi: 10.1038/nature08869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Neumann B, Held M, Liebel U, Erfle H, Rogers P, et al. High-throughput RNAi screening by time-lapse imaging of live human cells. Nat Methods. 2006;3:385–90. doi: 10.1038/nmeth876. [DOI] [PubMed] [Google Scholar]
- 173.Fuchs F, Pau G, Kranz D, Sklyar O, Budjan C, et al. Clustering phenotype populations by genome-wide RNAi and multiparametric imaging. Mol Syst Biol. 6:370. doi: 10.1038/msb.2010.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Birmingham A, Selfors LM, Forster T, Wrobel D, Kennedy CJ, et al. Statistical methods for analysis of high-throughput RNA interference screens. Nat Methods. 2009;6:569–75. doi: 10.1038/nmeth.1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Austin CP, Brady LS, Insel TR, Collins FS. NIH Molecular Libraries Initiative. Science. 2004;306:1138–9. doi: 10.1126/science.1105511. [DOI] [PubMed] [Google Scholar]
- 176.Birmingham A, Anderson EM, Reynolds A, Ilsley-Tyree D, Leake D, et al. 3′ UTR seed matches, but not overall identity, are associated with RNAi off-targets. Nat Methods. 2006;3:199–204. doi: 10.1038/nmeth854. [DOI] [PubMed] [Google Scholar]
- 177.Jackson AL, Burchard J, Leake D, Reynolds A, Schelter J, et al. Position- specific chemical modification of siRNAs reduces “off-target” transcript silencing. RNA. 2006;12:1197–205. doi: 10.1261/rna.30706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Pelz O, Gilsdorf M, Boutros M. web cellHTS2: a web-application for the analysis of high-throughput screening data. BMC Bioinformatics. 11:185. doi: 10.1186/1471-2105-11-185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Rieber N, Knapp B, Eils R, Kaderali L. RNAither, an automated pipeline for the statistical analysis of high-throughput RNAi screens. Bioinformatics. 2009;25:678–9. doi: 10.1093/bioinformatics/btp014. [DOI] [PubMed] [Google Scholar]
- 181.Major MB, Roberts BS, Berndt JD, Marine S, Anastas J, et al. New regulators of Wnt/beta-catenin signaling revealed by integrative molecular screening. Sci Signal. 2008;1:ra12. doi: 10.1126/scisignal.2000037. [DOI] [PubMed] [Google Scholar]
- 182.Cherry S. Genomic RNAi screening in Drosophila S2 cells: what have we learned about host-pathogen interactions? Curr Opin Microbiol. 2008;11:262–70. doi: 10.1016/j.mib.2008.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Bushman FD, Malani N, Fernandes J, D’Orso I, Cagney G, et al. Host cell factors in HIV replication: meta-analysis of genome-wide studies. PLoS Pathog. 2009;5:e1000437. doi: 10.1371/journal.ppat.1000437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Watanabe T, Watanabe S, Kawaoka Y. Cellular networks involved in the influenza virus life cycle. Cell Host Microbe. 7:427–39. doi: 10.1016/j.chom.2010.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Gwack Y, Srikanth S, Feske S, Cruz-Guilloty F, Oh-hora M, et al. Biochemical and functional characterization of Orai proteins. J Biol Chem. 2007;282:16232–43. doi: 10.1074/jbc.M609630200. [DOI] [PubMed] [Google Scholar]
- 186.Zhang SL, Yeromin AV, Zhang XH, Yu Y, Safrina O, et al. Genome-wide RNAi screen of Ca(2+) influx identifies genes that regulate Ca(2+) release- activated Ca(2+) channel activity. Proc Natl Acad Sci U S A. 2006;103:9357–62. doi: 10.1073/pnas.0603161103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Vig M, Peinelt C, Beck A, Koomoa DL, Rabah D, et al. CRACM1 is a plasma membrane protein essential for store-operated Ca2+ entry. Science. 2006;312:1220–3. doi: 10.1126/science.1127883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Liou J, Kim ML, Heo WD, Jones JT, Myers JW, et al. STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr Biol. 2005;15:1235–41. doi: 10.1016/j.cub.2005.05.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Roos J, DiGregorio PJ, Yeromin AV, Ohlsen K, Lioudyno M, et al. STIM1, an essential and conserved component of store-operated Ca2+ channel function. J Cell Biol. 2005;169:435–45. doi: 10.1083/jcb.200502019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Willingham AT, Orth AP, Batalov S, Peters EC, Wen BG, et al. A strategy for probing the function of noncoding RNAs finds a repressor of NFAT. Science. 2005;309:1570–3. doi: 10.1126/science.1115901. [DOI] [PubMed] [Google Scholar]
- 191.Gwack Y, Sharma S, Nardone J, Tanasa B, Iuga A, et al. A genome-wide Drosophila RNAi screen identifies DYRK-family kinases as regulators of NFAT. Nature. 2006;441:646–50. doi: 10.1038/nature04631. [DOI] [PubMed] [Google Scholar]
- 192.Kuijl C, Savage ND, Marsman M, Tuin AW, Janssen L, et al. Intracellular bacterial growth is controlled by a kinase network around PKB/AKT1. Nature. 2007;450:725–30. doi: 10.1038/nature06345. [DOI] [PubMed] [Google Scholar]
- 193.Juszczynski P, Ouyang J, Monti S, Rodig SJ, Takeyama K, et al. The AP1- dependent secretion of galectin-1 by Reed Sternberg cells fosters immune privilege in classical Hodgkin lymphoma. Proc Natl Acad Sci U S A. 2007;104:13134– 9. doi: 10.1073/pnas.0706017104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Shaffer AL, Emre NC, Lamy L, Ngo VN, Wright G, et al. IRF4 addiction in multiple myeloma. Nature. 2008;454:226–31. doi: 10.1038/nature07064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Cho YS, Challa S, Moquin D, Genga R, Ray TD, et al. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell. 2009;137:1112–23. doi: 10.1016/j.cell.2009.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Burckstummer T, Baumann C, Bluml S, Dixit E, Durnberger G, et al. An orthogonal proteomic-genomic screen identifies AIM2 as a cytoplasmic DNA sensor for the inflammasome. Nat Immunol. 2009;10:266–72. doi: 10.1038/ni.1702. [DOI] [PubMed] [Google Scholar]
- 197.Bauer A, Kuster B. Affinity purification-mass spectrometry. Powerful tools for the characterization of protein complexes. Eur J Biochem. 2003;270:570–8. doi: 10.1046/j.1432-1033.2003.03428.x. [DOI] [PubMed] [Google Scholar]
- 198.Michnick SW, Ear PH, Manderson EN, Remy I, Stefan E. Universal strategies in research and drug discovery based on protein-fragment complementation assays. Nat Rev Drug Discov. 2007;6:569–82. doi: 10.1038/nrd2311. [DOI] [PubMed] [Google Scholar]
- 199.Parrish JR, Gulyas KD, Finley RL., Jr Yeast two-hybrid contributions to interactome mapping. Curr Opin Biotechnol. 2006;17:387–93. doi: 10.1016/j.copbio.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 200.Cusick ME, Yu H, Smolyar A, Venkatesan K, Carvunis AR, et al. Literature-curated protein interaction datasets. Nat Methods. 2009;6:39–46. doi: 10.1038/nmeth.1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Stumpf MP, Thorne T, de Silva E, Stewart R, An HJ, et al. Estimating the size of the human interactome. Proc Natl Acad Sci U S A. 2008;105:6959–64. doi: 10.1073/pnas.0708078105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Fields S, Song O. A novel genetic system to detect protein-protein interactions. Nature. 1989;340:245–6. doi: 10.1038/340245a0. [DOI] [PubMed] [Google Scholar]
- 203.Walhout AJ, Vidal M. High-throughput yeast two-hybrid assays for large-scale protein interaction mapping. Methods. 2001;24:297–306. doi: 10.1006/meth.2001.1190. [DOI] [PubMed] [Google Scholar]
- 204.Ito T, Chiba T, Ozawa R, Yoshida M, Hattori M, Sakaki Y. A comprehensive two-hybrid analysis to explore the yeast protein interactome. Proc Natl Acad Sci U S A. 2001;98:4569–74. doi: 10.1073/pnas.061034498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Uetz P, Giot L, Cagney G, Mansfield TA, Judson RS, et al. A comprehensive analysis of protein-protein interactions in Saccharomyces cerevisiae. Nature. 2000;403:623–7. doi: 10.1038/35001009. [DOI] [PubMed] [Google Scholar]
- 206.LaCount DJ, Vignali M, Chettier R, Phansalkar A, Bell R, et al. A protein interaction network of the malaria parasite Plasmodium falciparum. Nature. 2005;438:103–7. doi: 10.1038/nature04104. [DOI] [PubMed] [Google Scholar]
- 207.Li S, Armstrong CM, Bertin N, Ge H, Milstein S, et al. A map of the interactome network of the metazoan C. elegans. Science. 2004;303:540–3. doi: 10.1126/science.1091403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Giot L, Bader JS, Brouwer C, Chaudhuri A, Kuang B, et al. A protein interaction map of Drosophila melanogaster. Science. 2003;302:1727–36. doi: 10.1126/science.1090289. [DOI] [PubMed] [Google Scholar]
- 209.Rual JF, Venkatesan K, Hao T, Hirozane-Kishikawa T, Dricot A, et al. Towards a proteome-scale map of the human protein-protein interaction network. Nature. 2005;437:1173–8. doi: 10.1038/nature04209. [DOI] [PubMed] [Google Scholar]
- 210.Stelzl U, Worm U, Lalowski M, Haenig C, Brembeck FH, et al. A human protein-protein interaction network: a resource for annotating the proteome. Cell. 2005;122:957–68. doi: 10.1016/j.cell.2005.08.029. [DOI] [PubMed] [Google Scholar]
- 211.Venkatesan K, Rual JF, Vazquez A, Stelzl U, Lemmens I, et al. An empirical framework for binary interactome mapping. Nat Methods. 2009;6:83–90. doi: 10.1038/nmeth.1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Gavin AC, Aloy P, Grandi P, Krause R, Boesche M, et al. Proteome survey reveals modularity of the yeast cell machinery. Nature. 2006;440:631–6. doi: 10.1038/nature04532. [DOI] [PubMed] [Google Scholar]
- 213.Krogan NJ, Cagney G, Yu H, Zhong G, Guo X, et al. Global landscape of protein complexes in the yeast Saccharomyces cerevisiae. Nature. 2006;440:637–43. doi: 10.1038/nature04670. [DOI] [PubMed] [Google Scholar]
- 214.Poser I, Sarov M, Hutchins JR, Heriche JK, Toyoda Y, et al. BAC TransgeneOmics: a high-throughput method for exploration of protein function in mammals. Nat Methods. 2008;5:409–15. doi: 10.1038/nmeth.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Hutchins JR, Toyoda Y, Hegemann B, Poser I, Heriche JK, et al. Systematic analysis of human protein complexes identifies chromosome segregation proteins. Science. 328:593–9. doi: 10.1126/science.1181348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Barrios-Rodiles M, Brown KR, Ozdamar B, Bose R, Liu Z, et al. High-throughput mapping of a dynamic signaling network in mammalian cells. Science. 2005;307:1621–5. doi: 10.1126/science.1105776. [DOI] [PubMed] [Google Scholar]
- 217.Bouwmeester T, Bauch A, Ruffner H, Angrand PO, Bergamini G, et al. A physical and functional map of the human TNF-alpha/NF-kappa B signal transduction pathway. Nat Cell Biol. 2004;6:97–105. doi: 10.1038/ncb1086. [DOI] [PubMed] [Google Scholar]
- 218.Ewing RM, Chu P, Elisma F, Li H, Taylor P, et al. Large-scale mapping of human protein-protein interactions by mass spectrometry. Mol Syst Biol. 2007;3:89. doi: 10.1038/msb4100134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Tarassov K, Messier V, Landry CR, Radinovic S, Serna Molina MM, et al. An in vivo map of the yeast protein interactome. Science. 2008;320:1465–70. doi: 10.1126/science.1153878. [DOI] [PubMed] [Google Scholar]
- 220.Calderwood MA, Venkatesan K, Xing L, Chase MR, Vazquez A, et al. Epstein-Barr virus and virus human protein interaction maps. Proc Natl Acad Sci U S A. 2007;104:7606–11. doi: 10.1073/pnas.0702332104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Lee JH, Vittone V, Diefenbach E, Cunningham AL, Diefenbach RJ. Identification of structural protein-protein interactions of herpes simplex virus type 1. Virology. 2008;378:347–54. doi: 10.1016/j.virol.2008.05.035. [DOI] [PubMed] [Google Scholar]
- 222.Rozen R, Sathish N, Li Y, Yuan Y. Virion-wide protein interactions of Kaposi’s sarcoma-associated herpesvirus. J Virol. 2008;82:4742–50. doi: 10.1128/JVI.02745-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Uetz P, Dong YA, Zeretzke C, Atzler C, Baiker A, et al. Herpesviral protein networks and their interaction with the human proteome. Science. 2006;311:239–42. doi: 10.1126/science.1116804. [DOI] [PubMed] [Google Scholar]
- 224.Fossum E, Friedel CC, Rajagopala SV, Titz B, Baiker A, et al. Evolutionarily conserved herpesviral protein interaction networks. PLoS Pathog. 2009;5:e1000570. doi: 10.1371/journal.ppat.1000570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.de Chassey B, Navratil V, Tafforeau L, Hiet MS, Aublin-Gex A, et al. Hepatitis C virus infection protein network. Mol Syst Biol. 2008;4:230. doi: 10.1038/msb.2008.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Shapira SD, Gat-Viks I, Shum BO, Dricot A, de Grace MM, et al. A physical and regulatory map of host-influenza interactions reveals pathways in H1N1 infection. Cell. 2009;139:1255–67. doi: 10.1016/j.cell.2009.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Dyer MD, Murali TM, Sobral BW. The landscape of human proteins interacting with viruses and other pathogens. PLoS Pathog. 2008;4:e32. doi: 10.1371/journal.ppat.0040032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Dyer MD, Neff C, Dufford M, Rivera CG, Shattuck D, et al. The human-bacterial pathogen protein interaction networks of Bacillus anthracis, Francisella tularensis, and Yersinia pestis. PLoS One. 5:e12089. doi: 10.1371/journal.pone.0012089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Fenn JB, Mann M, Meng CK, Wong SF, Whitehouse CM. Electrospray ionization for mass spectrometry of large biomolecules. Science. 1989;246:64–71. doi: 10.1126/science.2675315. [DOI] [PubMed] [Google Scholar]
- 230.Karas M, Hillenkamp F. Laser desorption ionization of proteins with molecular masses exceeding 10,000 daltons. Anal Chem. 1988;60:2299–301. doi: 10.1021/ac00171a028. [DOI] [PubMed] [Google Scholar]
- 231.Han X, Aslanian A, Yates JR., 3rd Mass spectrometry for proteomics. Curr Opin Chem Biol. 2008;12:483–90. doi: 10.1016/j.cbpa.2008.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Makarov A. Electrostatic axially harmonic orbital trapping: a high-performance technique of mass analysis. Anal Chem. 2000;72:1156–62. doi: 10.1021/ac991131p. [DOI] [PubMed] [Google Scholar]
- 233.Makarov A, Denisov E, Kholomeev A, Balschun W, Lange O, et al. Performance evaluation of a hybrid linear ion trap/orbitrap mass spectrometer. Anal Chem. 2006;78:2113–20. doi: 10.1021/ac0518811. [DOI] [PubMed] [Google Scholar]
- 234.Makarov A, Denisov E, Lange O, Horning S. Dynamic range of mass accuracy in LTQ Orbitrap hybrid mass spectrometer. J Am Soc Mass Spectrom. 2006;17:977–82. doi: 10.1016/j.jasms.2006.03.006. [DOI] [PubMed] [Google Scholar]
- 235.Yates JR, Cociorva D, Liao L, Zabrouskov V. Performance of a linear ion trap-Orbitrap hybrid for peptide analysis. Anal Chem. 2006;78:493–500. doi: 10.1021/ac0514624. [DOI] [PubMed] [Google Scholar]
- 236.Macek B, Waanders LF, Olsen JV, Mann M. Top-down protein sequencing and MS3 on a hybrid linear quadrupole ion trap-orbitrap mass spectrometer. Mol Cell Proteomics. 2006;5:949–58. doi: 10.1074/mcp.T500042-MCP200. [DOI] [PubMed] [Google Scholar]
- 237.Venable JD, Wohlschlegel J, McClatchy DB, Park SK, Yates JR., 3rd Relative quantification of stable isotope labeled peptides using a linear ion trap-Orbitrap hybrid mass spectrometer. Anal Chem. 2007;79:3056–64. doi: 10.1021/ac062054i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Luber CA, Cox J, Lauterbach H, Fancke B, Selbach M, et al. Quantitative proteomics reveals subset-specific viral recognition in dendritic cells. Immunity. 32:279–89. doi: 10.1016/j.immuni.2010.01.013. [DOI] [PubMed] [Google Scholar]
- 239.Zubarev RA, Horn DM, Fridriksson EK, Kelleher NL, Kruger NA, et al. Electron capture dissociation for structural characterization of multiply charged protein cations. Anal Chem. 2000;72:563–73. doi: 10.1021/ac990811p. [DOI] [PubMed] [Google Scholar]
- 240.Syka JE, Coon JJ, Schroeder MJ, Shabanowitz J, Hunt DF. Peptide and protein sequence analysis by electron transfer dissociation mass spectrometry. Proc Natl Acad Sci U S A. 2004;101:9528–33. doi: 10.1073/pnas.0402700101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Olsen JV, Macek B, Lange O, Makarov A, Horning S, Mann M. Higher-energy C-trap dissociation for peptide modification analysis. Nat Methods. 2007;4:709–12. doi: 10.1038/nmeth1060. [DOI] [PubMed] [Google Scholar]
- 242.Wolf-Yadlin A, Hautaniemi S, Lauffenburger DA, White FM. Multiple reaction monitoring for robust quantitative proteomic analysis of cellular signaling networks. Proc Natl Acad Sci U S A. 2007;104:5860–5. doi: 10.1073/pnas.0608638104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Ong SE, Blagoev B, Kratchmarova I, Kristensen DB, Steen H, et al. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol Cell Proteomics. 2002;1:376–86. doi: 10.1074/mcp.m200025-mcp200. [DOI] [PubMed] [Google Scholar]
- 244.Ross PL, Huang YN, Marchese JN, Williamson B, Parker K, et al. Multiplexed protein quantitation in Saccharomyces cerevisiae using amine- reactive isobaric tagging reagents. Mol Cell Proteomics. 2004;3:1154–69. doi: 10.1074/mcp.M400129-MCP200. [DOI] [PubMed] [Google Scholar]
- 245.Choe L, D’Ascenzo M, Relkin NR, Pappin D, Ross P, et al. 8-plex quantitation of changes in cerebrospinal fluid protein expression in subjects undergoing intravenous immunoglobulin treatment for Alzheimer’s disease. Proteomics. 2007;7:3651–60. doi: 10.1002/pmic.200700316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Kruger M, Moser M, Ussar S, Thievessen I, Luber CA, et al. SILAC mouse for quantitative proteomics uncovers kindlin-3 as an essential factor for red blood cell function. Cell. 2008;134:353–64. doi: 10.1016/j.cell.2008.05.033. [DOI] [PubMed] [Google Scholar]
- 247.Picotti P, Bodenmiller B, Mueller LN, Domon B, Aebersold R. Full dynamic range proteome analysis of S. cerevisiae by targeted proteomics. Cell. 2009;138:795–806. doi: 10.1016/j.cell.2009.05.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Picotti P, Rinner O, Stallmach R, Dautel F, Farrah T, et al. High-throughput generation of selected reaction-monitoring assays for proteins and proteomes. Nat Methods. 7:43–6. doi: 10.1038/nmeth.1408. [DOI] [PubMed] [Google Scholar]
- 249.Vogel C, Marcotte EM. Absolute abundance for the masses. Nat Biotechnol. 2009;27:825–6. doi: 10.1038/nbt0909-825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Kirkpatrick DS, Gerber SA, Gygi SP. The absolute quantification strategy: a general procedure for the quantification of proteins and post-translational modifications. Methods. 2005;35:265–73. doi: 10.1016/j.ymeth.2004.08.018. [DOI] [PubMed] [Google Scholar]
- 251.Pratt JM, Simpson DM, Doherty MK, Rivers J, Gaskell SJ, Beynon RJ. Multiplexed absolute quantification for proteomics using concatenated signature peptides encoded by QconCAT genes. Nat Protoc. 2006;1:1029–43. doi: 10.1038/nprot.2006.129. [DOI] [PubMed] [Google Scholar]
- 252.Mirzaei H, McBee JK, Watts J, Aebersold R. Comparative evaluation of current peptide production platforms used in absolute quantification in proteomics. Mol Cell Proteomics. 2008;7:813–23. doi: 10.1074/mcp.M700495-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Holzmann J, Pichler P, Madalinski M, Kurzbauer R, Mechtler K. Stoichiometry determination of the MP1-p14 complex using a novel and cost-efficient method to produce an equimolar mixture of standard peptides. Anal Chem. 2009;81:10254–61. doi: 10.1021/ac902286m. [DOI] [PubMed] [Google Scholar]
- 254.Koal T, Deigner HP. Challenges in mass spectrometry based targeted metabolomics. Curr Mol Med. 10:216–26. doi: 10.2174/156652410790963312. [DOI] [PubMed] [Google Scholar]
- 255.Iwai LK, Benoist C, Mathis D, White FM. Quantitative Phosphoproteomic Analysis of T Cell Receptor Signaling in Diabetes Prone and Resistant Mice. J Proteome Res. doi: 10.1021/pr100035b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.O’Neill RA, Bhamidipati A, Bi X, Deb-Basu D, Cahill L, et al. Isoelectric focusing technology quantifies protein signaling in 25 cells. Proc Natl Acad Sci U S A. 2006;103:16153–8. doi: 10.1073/pnas.0607973103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Borrebaeck CA, Ekstrom S, Hager AC, Nilsson J, Laurell T, Marko-Varga G. Protein chips based on recombinant antibody fragments: a highly sensitive approach as detected by mass spectrometry. Biotechniques. 2001;30:1126–30. 32. doi: 10.2144/01305dd05. [DOI] [PubMed] [Google Scholar]
- 258.Zhu H, Snyder M. Protein chip technology. Curr Opin Chem Biol. 2003;7:55–63. doi: 10.1016/s1367-5931(02)00005-4. [DOI] [PubMed] [Google Scholar]
- 259.Ramachandran N, Hainsworth E, Bhullar B, Eisenstein S, Rosen B, et al. Self-assembling protein microarrays. Science. 2004;305:86–90. doi: 10.1126/science.1097639. [DOI] [PubMed] [Google Scholar]
- 260.Tao SC, Zhu H. Protein chip fabrication by capture of nascent polypeptides. Nat Biotechnol. 2006;24:1253–4. doi: 10.1038/nbt1249. [DOI] [PubMed] [Google Scholar]
- 261.Irish JM, Kotecha N, Nolan GP. Mapping normal and cancer cell signalling networks: towards single-cell proteomics. Nat Rev Cancer. 2006;6:146–55. doi: 10.1038/nrc1804. [DOI] [PubMed] [Google Scholar]
- 262.Krutzik PO, Nolan GP. Intracellular phospho-protein staining techniques for flow cytometry: monitoring single cell signaling events. Cytometry A. 2003;55:61–70. doi: 10.1002/cyto.a.10072. [DOI] [PubMed] [Google Scholar]
- 263.Krutzik PO, Nolan GP. Fluorescent cell barcoding in flow cytometry allows high-throughput drug screening and signaling profiling. Nat Methods. 2006;3:361–8. doi: 10.1038/nmeth872. [DOI] [PubMed] [Google Scholar]
- 264.Goddard G, Martin JC, Naivar M, Goodwin PM, Graves SW, et al. Single particle high resolution spectral analysis flow cytometry. Cytometry A. 2006;69:842–51. doi: 10.1002/cyto.a.20320. [DOI] [PubMed] [Google Scholar]
- 265.Shachaf CM, Elchuri SV, Koh AL, Zhu J, Nguyen LN, et al. A novel method for detection of phosphorylation in single cells by surface enhanced Raman scattering (SERS) using composite organic-inorganic nanoparticles (COINs) PLoS One. 2009;4:e5206. doi: 10.1371/journal.pone.0005206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Watson DA, Brown LO, Gaskill DF, Naivar M, Graves SW, et al. A flow cytometer for the measurement of Raman spectra. Cytometry A. 2008;73:119–28. doi: 10.1002/cyto.a.20520. [DOI] [PubMed] [Google Scholar]
- 267.Bandura DR, Baranov VI, Ornatsky OI, Antonov A, Kinach R, et al. Mass cytometry: technique for real time single cell multitarget immunoassay based on inductively coupled plasma time-of-flight mass spectrometry. Anal Chem. 2009;81:6813–22. doi: 10.1021/ac901049w. [DOI] [PubMed] [Google Scholar]
- 268.Coggan JS, Bartol TM, Esquenazi E, Stiles JR, Lamont S, et al. Evidence for ectopic neurotransmission at a neuronal synapse. Science. 2005;309:446–51. doi: 10.1126/science.1108239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Shoop RD, Esquenazi E, Yamada N, Ellisman MH, Berg DK. Ultrastructure of a somatic spine mat for nicotinic signaling in neurons. J Neurosci. 2002;22:748–56. doi: 10.1523/JNEUROSCI.22-03-00748.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Smith CS, Joseph N, Rieger B, Lidke KA. Fast, single-molecule localization that achieves theoretically minimum uncertainty. Nat Methods. 7:373–5. doi: 10.1038/nmeth.1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Sohn HW, Tolar P, Brzostowski J, Pierce SK. A method for analyzing protein-protein interactions in the plasma membrane of live B cells by fluorescence resonance energy transfer imaging as acquired by total internal reflection fluorescence microscopy. Methods Mol Biol. 2010;591:159–83. doi: 10.1007/978-1-60761-404-3_10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Ding JB, Takasaki KT, Sabatini BL. Supraresolution imaging in brain slices using stimulated-emission depletion two-photon laser scanning microscopy. Neuron. 2009;63:429–37. doi: 10.1016/j.neuron.2009.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Park C, Hwang IY, Kehrl JH. Intravital two-photon imaging of adoptively transferred B lymphocytes in inguinal lymph nodes. Methods Mol Biol. 2009;571:199–207. doi: 10.1007/978-1-60761-198-1_13. [DOI] [PubMed] [Google Scholar]
- 274.Bajenoff M, Germain RN. Seeing is believing: a focus on the contribution of microscopic imaging to our understanding of immune system function. Eur J Immunol. 2007;37(Suppl 1):S18–33. doi: 10.1002/eji.200737663. [DOI] [PubMed] [Google Scholar]
- 275.Cabantous S, Terwilliger TC, Waldo GS. Protein tagging and detection with engineered self-assembling fragments of green fluorescent protein. Nat Biotechnol. 2005;23:102–7. doi: 10.1038/nbt1044. [DOI] [PubMed] [Google Scholar]
- 276.Gerber D, Maerkl SJ, Quake SR. An in vitro microfluidic approach to generating protein-interaction networks. Nat Methods. 2009;6:71–4. doi: 10.1038/nmeth.1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Tay S, Hughey JJ, Lee TK, Lipniacki T, Quake SR, Covert MW. Single-cell NF-kappaB dynamics reveal digital activation and analogue information processing. Nature. 466:267–71. doi: 10.1038/nature09145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Kim HD, Guo TW, Wu AP, Wells A, Gertler FB, Lauffenburger DA. Epidermal growth factor-induced enhancement of glioblastoma cell migration in 3D arises from an intrinsic increase in speed but an extrinsic matrix- and proteolysis-dependent increase in persistence. Mol Biol Cell. 2008;19:4249–59. doi: 10.1091/mbc.E08-05-0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Denk W, Strickler JH, Webb WW. Two-photon laser scanning fluorescence microscopy. Science. 1990;248:73–6. doi: 10.1126/science.2321027. [DOI] [PubMed] [Google Scholar]
- 280.Betzig E, Patterson GH, Sougrat R, Lindwasser OW, Olenych S, et al. Imaging intracellular fluorescent proteins at nanometer resolution. Science. 2006;313:1642–5. doi: 10.1126/science.1127344. [DOI] [PubMed] [Google Scholar]
- 281.Rust MJ, Bates M, Zhuang X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM) Nat Methods. 2006;3:793–5. doi: 10.1038/nmeth929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Fuchs J, Bohme S, Oswald F, Hedde PN, Krause M, et al. A photoactivatable marker protein for pulse-chase imaging with superresolution. Nat Methods. doi: 10.1038/nmeth.1477. [DOI] [PubMed] [Google Scholar]
- 283.Sarris M, Betz AG. Shine a light: imaging the immune system. Eur J Immunol. 2009;39:1188–202. doi: 10.1002/eji.200839026. [DOI] [PubMed] [Google Scholar]
- 284.Monks CR, Freiberg BA, Kupfer H, Sciaky N, Kupfer A. Three- dimensional segregation of supramolecular activation clusters in T cells. Nature. 1998;395:82–6. doi: 10.1038/25764. [DOI] [PubMed] [Google Scholar]
- 285.Lee KH, Holdorf AD, Dustin ML, Chan AC, Allen PM, Shaw AS. T cell receptor signaling precedes immunological synapse formation. Science. 2002;295:1539–42. doi: 10.1126/science.1067710. [DOI] [PubMed] [Google Scholar]
- 286.Cemerski S, Das J, Giurisato E, Markiewicz MA, Allen PM, et al. The balance between T cell receptor signaling and degradation at the center of the immunological synapse is determined by antigen quality. Immunity. 2008;29:414–22. doi: 10.1016/j.immuni.2008.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Cemerski S, Das J, Locasale J, Arnold P, Giurisato E, et al. The stimulatory potency of T cell antigens is influenced by the formation of the immunological synapse. Immunity. 2007;26:345–55. doi: 10.1016/j.immuni.2007.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Beltman JB, Maree AF, Lynch JN, Miller MJ, de Boer RJ. Lymph node topology dictates T cell migration behavior. J Exp Med. 2007;204:771–80. doi: 10.1084/jem.20061278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Henrickson SE, Mempel TR, Mazo IB, Liu B, Artyomov MN, et al. In vivo imaging of T cell priming. Sci Signal. 2008;1(pt 2) doi: 10.1126/stke.112pt2. [DOI] [PubMed] [Google Scholar]
- 290.Henrickson SE, Mempel TR, Mazo IB, Liu B, Artyomov MN, et al. T cell sensing of antigen dose governs interactive behavior with dendritic cells and sets a threshold for T cell activation. Nat Immunol. 2008;9:282–91. doi: 10.1038/ni1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Zheng H, Jin B, Henrickson SE, Perelson AS, von Andrian UH, Chakraborty AK. How antigen quantity and quality determine T-cell decisions in lymphoid tissue. Mol Cell Biol. 2008;28:4040–51. doi: 10.1128/MCB.00136-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Cahalan MD, Parker I. Choreography of cell motility and interaction dynamics imaged by two-photon microscopy in lymphoid organs. Annu Rev Immunol. 2008;26:585–626. doi: 10.1146/annurev.immunol.24.021605.090620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Hauser AE, Junt T, Mempel TR, Sneddon MW, Kleinstein SH, et al. Definition of germinal-center B cell migration in vivo reveals predominant intrazonal circulation patterns. Immunity. 2007;26:655–67. doi: 10.1016/j.immuni.2007.04.008. [DOI] [PubMed] [Google Scholar]
- 294.Yachi PP, Ampudia J, Zal T, Gascoigne NR. Altered peptide ligands induce delayed CD8-T cell receptor interaction--a role for CD8 in distinguishing antigen quality. Immunity. 2006;25:203–11. doi: 10.1016/j.immuni.2006.05.015. [DOI] [PubMed] [Google Scholar]
- 295.Mallaun M, Naeher D, Daniels MA, Yachi PP, Hausmann B, et al. The T cell receptor’s alpha-chain connecting peptide motif promotes close approximation of the CD8 coreceptor allowing efficient signal initiation. J Immunol. 2008;180:8211–21. doi: 10.4049/jimmunol.180.12.8211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Tolar P, Sohn HW, Pierce SK. The initiation of antigen-induced B cell antigen receptor signaling viewed in living cells by fluorescence resonance energy transfer. Nat Immunol. 2005;6:1168–76. doi: 10.1038/ni1262. [DOI] [PubMed] [Google Scholar]
- 297.Aslan K, Lakowicz JR, Geddes CD. Plasmon light scattering in biology and medicine: new sensing approaches, visions and perspectives. Curr Opin Chem Biol. 2005;9:538–44. doi: 10.1016/j.cbpa.2005.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Segura E, Kapp E, Gupta N, Wong J, Lim J, et al. Differential expression of pathogen-recognition molecules between dendritic cell subsets revealed by plasma membrane proteomic analysis. Mol Immunol. 47:1765–73. doi: 10.1016/j.molimm.2010.02.028. [DOI] [PubMed] [Google Scholar]
- 299.Kim JE, White FM. Quantitative analysis of phosphotyrosine signaling networks triggered by CD3 and CD28 costimulation in Jurkat cells. J Immunol. 2006;176:2833–43. doi: 10.4049/jimmunol.176.5.2833. [DOI] [PubMed] [Google Scholar]
- 300.Hale MB, Krutzik PO, Samra SS, Crane JM, Nolan GP. Stage dependent aberrant regulation of cytokine-STAT signaling in murine systemic lupus erythematosus. PLoS One. 2009;4:e6756. doi: 10.1371/journal.pone.0006756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Galligan CL, Siebert JC, Siminovitch KA, Keystone EC, Bykerk V, et al. Multiparameter phospho-flow analysis of lymphocytes in early rheumatoid arthritis: implications for diagnosis and monitoring drug therapy. PLoS One. 2009;4:e6703. doi: 10.1371/journal.pone.0006703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Merbl Y, Itzchak R, Vider-Shalit T, Louzoun Y, Quintana FJ, et al. A systems immunology approach to the host-tumor interaction: large-scale patterns of natural autoantibodies distinguish healthy and tumor-bearing mice. PLoS One. 2009;4:e6053. doi: 10.1371/journal.pone.0006053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Montor WR, Huang J, Hu Y, Hainsworth E, Lynch S, et al. Genome-wide study of Pseudomonas aeruginosa outer membrane protein immunogenicity using self-assembling protein microarrays. Infect Immun. 2009;77:4877–86. doi: 10.1128/IAI.00698-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Ceroni A, Sibani S, Baiker A, Pothineni VR, Bailer SM, et al. Systematic analysis of the IgG antibody immune response against varicella zoster virus (VZV) using a self-assembled protein microarray. Mol Biosyst. doi: 10.1039/c003798b. [DOI] [PubMed] [Google Scholar]
- 305.Robinson WH, DiGennaro C, Hueber W, Haab BB, Kamachi M, et al. Autoantigen microarrays for multiplex characterization of autoantibody responses. Nat Med. 2002;8:295–301. doi: 10.1038/nm0302-295. [DOI] [PubMed] [Google Scholar]
- 306.Harwanegg C, Laffer S, Hiller R, Mueller MW, Kraft D, et al. Microarrayed recombinant allergens for diagnosis of allergy. Clin Exp Allergy. 2003;33:7–13. doi: 10.1046/j.1365-2222.2003.01550.x. [DOI] [PubMed] [Google Scholar]
- 307.Hiller R, Laffer S, Harwanegg C, Huber M, Schmidt WM, et al. Microarrayed allergen molecules: diagnostic gatekeepers for allergy treatment. FASEB J. 2002;16:414–6. doi: 10.1096/fj.01-0711fje. [DOI] [PubMed] [Google Scholar]
- 308.Miletic AV, Sakata-Sogawa K, Hiroshima M, Hamann MJ, Gomez TS, et al. Vav1 acidic region tyrosine 174 is required for the formation of T cell receptor-induced microclusters and is essential in T cell development and activation. J Biol Chem. 2006;281:38257–65. doi: 10.1074/jbc.M608913200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Nolz JC, Gomez TS, Billadeau DD. The Ezh2 methyltransferase complex: actin up in the cytosol. Trends Cell Biol. 2005;15:514–7. doi: 10.1016/j.tcb.2005.08.003. [DOI] [PubMed] [Google Scholar]
- 310.Nolz JC, Gomez TS, Zhu P, Li S, Medeiros RB, et al. The WAVE2 complex regulates actin cytoskeletal reorganization and CRAC-mediated calcium entry during T cell activation. Curr Biol. 2006;16:24–34. doi: 10.1016/j.cub.2005.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Spooner CJ, Cheng JX, Pujadas E, Laslo P, Singh H. A recurrent network involving the transcription factors PU.1 and Gfi1 orchestrates innate and adaptive immune cell fates. Immunity. 2009;31:576–86. doi: 10.1016/j.immuni.2009.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Laslo P, Spooner CJ, Warmflash A, Lancki DW, Lee HJ, et al. Multilineage transcriptional priming and determination of alternate hematopoietic cell fates. Cell. 2006;126:755–66. doi: 10.1016/j.cell.2006.06.052. [DOI] [PubMed] [Google Scholar]
- 313.Georgescu C, Longabaugh WJ, Scripture-Adams DD, David-Fung ES, Yui MA, et al. A gene regulatory network armature for T lymphocyte specification. Proc Natl Acad Sci U S A. 2008;105:20100–5. doi: 10.1073/pnas.0806501105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Monteiro JP, Farache J, Mercadante AC, Mignaco JA, Bonamino M, Bonomo A. Pathogenic effector T cell enrichment overcomes regulatory T cell control and generates autoimmune gastritis. J Immunol. 2008;181:5895–903. doi: 10.4049/jimmunol.181.9.5895. [DOI] [PubMed] [Google Scholar]
- 315.Andrews SS, Addy NJ, Brent R, Arkin AP. Detailed simulations of cell biology with Smoldyn 2.1. PLoS Comput Biol. 6:e1000705. doi: 10.1371/journal.pcbi.1000705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Plimpton SJ, Slepoy A. Microbial cell modeling via reacting diffusing particles. Journal of Physics: Conference Series. 2005;16:305–9. [Google Scholar]
- 317.Brass AL, Huang IC, Benita Y, John SP, Krishnan MN, et al. The IFITM proteins mediate cellular resistance to influenza A H1N1 virus, West Nile virus, and dengue virus. Cell. 2009;139:1243–54. doi: 10.1016/j.cell.2009.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Konig R, Stertz S, Zhou Y, Inoue A, Hoffmann HH, et al. Human host factors required for influenza virus replication. Nature. 463:813–7. doi: 10.1038/nature08699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Karlas A, Machuy N, Shin Y, Pleissner KP, Artarini A, et al. Genome-wide RNAi screen identifies human host factors crucial for influenza virus replication. Nature. 463:818–22. doi: 10.1038/nature08760. [DOI] [PubMed] [Google Scholar]
- 320.Brass AL, Dykxhoorn DM, Benita Y, Yan N, Engelman A, et al. Identification of host proteins required for HIV infection through a functional genomic screen. Science. 2008;319:921–6. doi: 10.1126/science.1152725. [DOI] [PubMed] [Google Scholar]
- 321.Konig R, Zhou Y, Elleder D, Diamond TL, Bonamy GM, et al. Global analysis of host-pathogen interactions that regulate early-stage HIV-1 replication. Cell. 2008;135:49–60. doi: 10.1016/j.cell.2008.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Zhou H, Xu M, Huang Q, Gates AT, Zhang XD, et al. Genome-scale RNAi screen for host factors required for HIV replication. Cell Host Microbe. 2008;4:495–504. doi: 10.1016/j.chom.2008.10.004. [DOI] [PubMed] [Google Scholar]
- 323.Krishnan MN, Ng A, Sukumaran B, Gilfoy FD, Uchil PD, et al. RNA interference screen for human genes associated with West Nile virus infection. Nature. 2008;455:242–5. doi: 10.1038/nature07207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Kumar D, Nath L, Kamal MA, Varshney A, Jain A, et al. Genome-wide analysis of the host intracellular network that regulates survival of Mycobacterium tuberculosis. Cell. 140:731–43. doi: 10.1016/j.cell.2010.02.012. [DOI] [PubMed] [Google Scholar]
- 325.Jayaswal S, Kamal MA, Dua R, Gupta S, Majumdar T, et al. Identification of host-dependent survival factors for intracellular Mycobacterium tuberculosis through an siRNA screen. PLoS Pathog. 6:e1000839. doi: 10.1371/journal.ppat.1000839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.Ng TI, Mo H, Pilot-Matias T, He Y, Koev G, et al. Identification of host genes involved in hepatitis C virus replication by small interfering RNA technology. Hepatology. 2007;45:1413–21. doi: 10.1002/hep.21608. [DOI] [PubMed] [Google Scholar]
- 327.Tai AW, Benita Y, Peng LF, Kim SS, Sakamoto N, et al. A functional genomic screen identifies cellular cofactors of hepatitis C virus replication. Cell Host Microbe. 2009;5:298–307. doi: 10.1016/j.chom.2009.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328.Gerber SA, Rush J, Stemman O, Kirschner MW, Gygi SP. Absolute quantification of proteins and phosphoproteins from cell lysates by tandem MS. Proc Natl Acad Sci U S A. 2003;100:6940–5. doi: 10.1073/pnas.0832254100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329.Foster LJ, de Hoog CL, Zhang Y, Xie X, Mootha VK, Mann M. A mammalian organelle map by protein correlation profiling. Cell. 2006;125:187–99. doi: 10.1016/j.cell.2006.03.022. [DOI] [PubMed] [Google Scholar]
- 330.Mallick P, Schirle M, Chen SS, Flory MR, Lee H, et al. Computational prediction of proteotypic peptides for quantitative proteomics. Nat Biotechnol. 2007;25:125–31. doi: 10.1038/nbt1275. [DOI] [PubMed] [Google Scholar]