

Abstract
We isolated mutations in Drosophila E2F and DP that affect chorion gene amplification and ORC2 localization in the follicle cells. In the follicle cells of the ovary, the ORC2 protein is localized throughout the follicle cell nuclei when they are undergoing polyploid genomic replication, and its levels appear constant in both S and G phases. In contrast, when genomic replication ceases and specific regions amplify, ORC2 is present solely at the amplifying loci. Mutations in the DNA-binding domains of dE2F or dDP reduce amplification, and in these mutants specific localization of ORC2 to amplification loci is lost. Interestingly, a dE2F mutant predicted to lack the carboxy-terminal transcriptional activation and RB-binding domain does not abolish ORC2 localization and shows premature chorion amplification. The effect of the mutations in the heterodimer subunits suggests that E2F controls not only the onset of S phase but also origin activity within S phase.
Keywords: DNA replication, ORC, E2F, DP, Drosophila, oogenesis
The E2F transcription factor plays a pivotal role in the control of S-phase entry. This transcription factor is a heterodimer of E2F and DP proteins, and there are several forms of both proteins in mammalian cells (Weinberg 1995; Dyson 1998). Ectopic expression of the best-characterized E2F protein, E2F-1, is sufficient to drive serum-starved, quiescent fibroblasts through S phase, suggesting a positive role for E2F in S-phase entry (Johnson et al. 1993; Qin et al. 1994). E2F activates the transcription of genes whose products regulate the initiation of replication (Ohtani et al. 1996). Its activity is also needed for a normal rate of replication, because E2F activates the transcription of components of the DNA replication machinery such as DNA polymerase α (Nevins 1992). E2F has not been demonstrated to have a role in controlling replication once S phase has initiated. It has been shown, however, that inactivation of E2F by cyclin A is needed for the completion of DNA replication and S phase (Krek et al. 1995).
E2F can repress transcription of genes needed for S phase when complexed with a pocket protein such as the retinoblastoma protein (RB) (Weintraub et al. 1992). The E2F/DP heterodimer tethers RB to E2F-dependent promoters through a small RB-binding region embedded in the transactivation domain of the E2F protein (Helin et al. 1993). In addition to occluding the E2F activation domain, RB represses transcription actively at these promoters and is thought to affect the state of chromatin accessibility through an interaction with a histone deacetylase (Weintraub et al. 1995; Brehm et al. 1998; Luo et al. 1998; Magnaghi-Jaulin et al. 1998). Late in G1, phosphorylation of RB by cyclin D/CDK4,6 and cyclin E/CDK2 frees E2F/DP and triggers the expression of E2F-dependent genes including cyclin E (Ewen et al. 1993; Kato et al. 1993; Ohtani et al. 1995; Botz et al. 1996; Geng et al. 1996). The cyclin E/CDK2 complex then creates a positive feedback loop that is thought to result in an irrevocable commitment to S phase (Sherr 1996).
Drosophila homologs to E2F, DP, and an RB-like protein (RBF) have been identified (Dynlacht et al. 1994; Ohtani and Nevins 1994; Hao et al. 1995; Du et al. 1996a). We will refer to the individual subunits of E2F as dE2F and dDP and will use the term E2F to designate the heterodimer transcription factor. As in mammalian cells, ectopic dE2F expression is sufficient to drive quiescent cells into S phase (Asano et al. 1996; Du et al. 1996b). We have shown that null mutations in dE2F and dDP cause lethality late in development with some tissues being underdeveloped or absent (Royzman et al. 1997). A positive role for the E2F heterodimer in cell cycle progression was inferred from these defects. This role has been confirmed by analysis of E2F function in developing wing discs (Neufeld et al. 1998). In addition, G1–S transcription of cyclin E and genes encoding replication functions such as proliferating cell nuclear antigen (PCNA) and ribonucleotide reductase 2 (RNR2), observed in late embryogenesis normally, is missing in the dE2F and dDP mutants (Duronio et al. 1995, 1998; Royzman et al. 1997). Despite the pronounced effect on E2F-dependent G1–S transcription, replication in the mutants was only slowed and a block to replication was not observed.
Drosophila oogenesis makes it possible to examine aspects of DNA replication that are not readily apparent during embryogenesis (for reviews, see Spradling 1993; Royzman and Orr-Weaver 1998). Ovarian follicle cells undergo a set of mitotic divisions before switching to an endo cycle (a cycle consisting of only S phase and a gap phase) and becoming polyploid. Genomic replication ceases after four endo cycles, but two genomic regions that contain clusters of chorion genes continue to replicate so that the chorion genes are amplified as much as 80-fold relative to genomic DNA. The chorion genes encode the eggshell proteins. Amplification of the chorion genes is needed to produce sufficient chorion protein for a normal eggshell, and amplification occurs by repeated rounds of initiation of DNA replication and fork movement to produce a gradient of amplified DNA extending ∼100 kb (for reviews, see Orr-Weaver 1991; Calvi and Spradling 1999). Mutants with reduced amplification have a phenotype of thin eggshells and female sterility.
Chorion gene amplification appears to use components that are required normally for initiating DNA replication. Origin recognition complex (ORC) is a complex of six subunits and is required for initiation of replication (for review, see Dutta and Bell 1997). Mutations in the Drosophila orc2 gene disrupt amplification (Landis et al. 1997). Overexpression and inhibition studies indicate that cyclin E is needed for amplification also (Calvi et al. 1998). Because the levels of Cyclin E protein oscillate with genomic replication but remain constant in follicle cells undergoing amplification, Calvi et al. (1998) postulated that the high Cyclin E activity blocks genomic replication and that some mechanism permits the amplicons to escape this block to rereplication.
Here we report the identification and analysis of new mutations in Drosophila dE2F that cause cell-cycle defects in oogenesis. These mutations, in addition to a female-sterile allele of dDP isolated previously (Royzman et al. 1997), allowed us to analyze the role of E2F in DNA replication in follicle cells. We find that E2F influences chorion amplification and affects localization of ORC within the nucleus.
Results
Female-sterile mutations in the dE2F gene
We recovered two viable mutations in the gene for the dE2F subunit of the E2F transcription factor, dE2Fi1 and dE2Fi2. These mutants were identified in a screen of mutagenized third chromosome lines (Moore et al. 1998) that affected PCNA expression in embryos. When placed in trans to a deficiency or a null dE2F allele, the dE2Fi1 mutation caused reduced viability, and the recovered adults were female sterile and had eye and bristle defects. The mutant females were able to lay eggs, but the eggs failed to develop. These phenotypes were fully recessive and not seen in dE2Fi1/TM3 control siblings (TM3 is a balancer chromosome that carries a wild-type copy of the dE2F gene). These phenotypes resembled closely those observed for a weak allele of dDP, dDPa1 (Royzman et al. 1997). The dE2Fi2 mutation was fully viable in trans to a deficiency or null dE2F allele; these adults had slightly rough eyes and the females had reduced fertility. These phenotypes were fully recessive. Unexpectedly, the two alleles were viable and fertile in trans to each other.
We sequenced the dE2F coding regions from the mutants; both mutants had changes in the dE2F open reading frame (ORF) (Fig. 1). The dE2Fi1 mutation is a G → A nucleotide transition that converts amino acid Asp-296 of the dE2F DNA-binding domain to Asn. This residue lies in a region that is highly similar between the E2F and DP families of proteins, and this Asp is conserved in all known Drosophila and vertebrate E2F and DP proteins (Fig. 1B) (Hao et al. 1995; Bandara et al. 1997). The finding that the dE2Fi1 mutation is analogous to the missense change in the DNA-binding region in the dDPa1 allele may explain the similarity of the phenotypes exhibited by the two mutants (see Fig. 1B). The mutation in dE2Fi2 is a C → T change that converts Gln-527 to a stop codon. The resultant E2F protein is predicted to lack the carboxy-terminal RB-binding and transcriptional activation region shown to be both necessary and sufficient for transcriptional activation in Drosophila SL2 cells (Ohtani and Nevins 1994).
Figure 1.

Position and nature of new dE2F alleles. (A) A schematic of the dE2F protein with the conserved regions indicated: the DNA-binding domain, leucine zipper, transcriptional activation domain, and the embedded RB-binding region (black). The positions and the amino acid changes in dE2Fi1 and dE2Fi2 are shown relative to dE2F91, the previously characterized null allele (Duronio et al. 1995; Royzman et al. 1997). (B) The dE2F DNA-binding domain includes a region of considerable similarity between the DP and E2F families of proteins. This block of amino acids is called the DEF box in the DP gene, and it is important for E2F/DP dimerization and the recognition of E2F binding sites in the promoters of E2F regulated genes (Bandara et al. 1993; Hao et al. 1995). The DEF consensus for Drosophila DP and E2F proteins is shown (modified from Bandara et al. 1997). A broader consensus to include mammalian DPs (1–3) and E2Fs (1–5), as well as the recently identified E2F-like protein, E2F6/EMA (Morkel et al. 1997; Trimarchi et al. 1998), is indicated in bold. The positions of mutations dDPa1, dE2Fi1, and dDPa2 are shown within the DEF consensus. dDPa1 is an amino acid substitution, whereas dDPa2 is a stop codon. Note that the affected residues are perfectly conserved in all known Drosophila and vertebrate E2F and DP families of proteins.
Phenotypic analysis of Drosophila E2F91, a null allele that lacks all the recognized functional domains of the E2F family of proteins, established that dE2F is essential for viability and normal growth (Duronio et al. 1995; Royzman et al. 1997). Viability and growth are not affected in the dE2Fi2 mutant, and thus the dE2Fi2 mutant appears to retain significant physiological function. We also tested whether reducing by half the dosage of the cyclin E gene, a critical target of E2F in Drosophila (Duronio et al. 1996), would effect the viability of dE2Fi2/dE2F91 flies and found that it had no effect. In contrast, the same reduction of cyclin E enhanced the lethality of the dE2Fi1/dE2F91 mutant organisms, as we were not able to recover dE2Fi1/dE2F91 adults that were heterozygous for a strong but completely recessive cyclin E mutation.
The role of dE2F and dDP in chorion gene amplification
In follicle cells, chorion gene clusters escape restrictions on rereplication, and amplification of these regions continues after the cessation of general genomic replication (Calvi et al. 1998). The amplification of chorion genes in follicle cells is required for the rapid synthesis of the eggshell (chorion), thus mutations that disrupt this process cause female sterility and thin eggshells. Consistent with a defect in chorion amplification, eggs laid by dDPa1/Df mutant mothers had abnormally thin eggshells (data not shown). To establish that this eggshell defect was caused by reduced DP function, we created a fly strain predicted to have reduced dDP levels. This strain contained a null dDP mutation (dDPa2) that was partially rescued from lethality by basal expression from an hsp70–dDP transgene. The recovered dDPa2/Df; hs–dDP mutant adults had a thin eggshell phenotype identical to the dDPa1/Df mutant (data not shown), confirming that the eggshell defect in dDPa1 is caused by reduced DP activity. The eggshells produced by the dE2Fi1/dE2F91mutant (affecting the DNA-binding region) were also very thin (data not shown), but the eggshell defect was less pronounced than in the dDP mutants. The dE2Fi2 mutant had the opposite effect on eggshell morphology; the eggshell was much thicker and rougher than normal (data not shown). Although the eggshell defects contribute to the female sterility observed in these mutants, they are not the sole cause of sterility, as there are nurse-cell defects that affect fertility (I. Royzman and T. Orr-Weaver, in prep.).
The eggshell phenotypes observed for the dDP and dE2F mutants predict a block or reduction in chorion gene amplification in the dE2Fi1/dE2F91, dDPa1/Df, and dDPa2/Df; hs–dDP mutants, and indicate a possible increase in amplification in the dE2Fi2 mutant. To test the role of E2F in chorion gene amplification directly, we analyzed BrdU incorporation in whole-mount ovaries (Figs. 2 and 3; Table 1).
Figure 2.
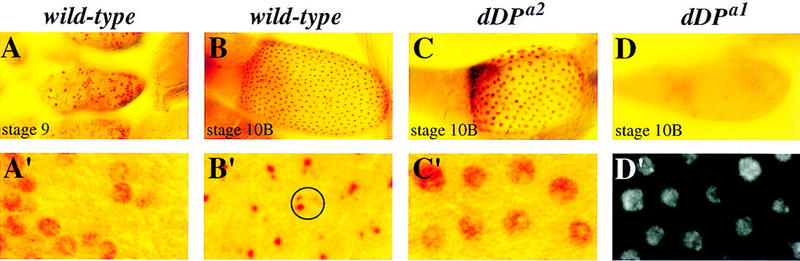
dDP mutants affect amplification and the shutoff of genomic replication. (A–D) DNA replication was assayed by BrdU labeling observed by antibody staining detected by the HRP reaction. BrdU incorporation is indicated by the red staining. (A–D) The same magnification, and shown below (A′–D′) at a higher magnification. (A,A′) In stage 9 control dDPa1/+ egg chambers genomic replication is asynchronous among follicle cell nuclei. The egg chamber contains follicle cells that are labeled (S phase) with BrdU and those that are not labeled (G phase). The same pattern of nuclear BrdU incorporation is observed in the female-sterile dE2F and dDP mutants. (B,B′) In stage-10B control egg chambers, polyploidization of the follicle cell genome is no longer observed. Instead, synchronous subnuclear BrdU incorporation is seen. The subnuclear dots correspond to amplifying chorion clusters. The nucleus of a single follicle cell is indicated by a circle. The different intensity and size of foci (one large, one medium, and two small) reflect the extent of gene amplification (Calvi et al. 1998). (C,C′) A stage-10B dDPa2/Df; hs–dDP/+ mutant egg chamber is shown. Genomic replication is occurring synchronously in the follicle cells of this mutant egg chamber. The same phenotypes were observed for the dDPa1/Df mutant. Note that the nuclei are much larger than at stage 9, indicating higher ploidy due to inappropriate genomic replication. (D) A stage-10B dDPa1/Df egg chamber is shown. Genomic replication has been shutoff, but amplification is not observed. The same replication defect was observed for dDPa2/Df; hs–dDP/+. (D′) A magnified DAPI image of D showing that the follicle cell nuclei are present and polyploid.
Figure 3.
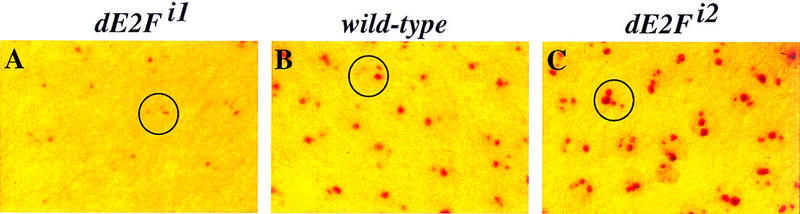
dE2F mutants affect the level of BrdU incorporation at amplicons. DNA replication in stage-10B egg chambers was assayed by incorporation of BrdU (red staining). (A–C) The same magnification. In dE2Fi1/Df mutant egg chambers (A) amplifying chorion loci are reduced in size and intensity as compared to wild-type (B). In the wild-type egg chamber most follicle cell nuclei (circle) have four spots of different intensities. It is sometimes difficult to see them in the same focal plane. In dE2Fi2/Df mutant egg chambers (C) the size and intensity of the BrdU foci are increased greatly. The mutant phenotype makes it easier to see the four subnuclear dots within each follicle cell nucleus (circle).
Table 1.
Replication phenotypes of dE2F and dDP mutants in follicle cells
| Genotype
|
Inappropriate genomic replication (%)a (Fig. 2C)
|
Chorion amplification foci (%)b
|
Total no. of 10B–11 egg chambers
|
|||
|---|---|---|---|---|---|---|
| absent (fig. 2D)
|
reduced (Fig. 3A)
|
normal (Fig. 2B)
|
increased (Fig. 3C)
|
|||
| dDPa1/+ | 0 | 16 | 8 | 76 | 0 | 38 |
| dDPa1/Df | 18c | 82 | 0 | 0 | 0 | 51 |
| dE2Fi1/dE2F91or dE2F7172 d | 5e | 43 | 30 | 21 | 0 | 61 |
| dE2Fi2/dE2F91or Dff | 0 | 10 | 11 | 19 | 60 | 42 |
Genomic follicle cell replication is inappropriate at stages 10B–11.
The percent of egg chambers with given phenotype.
Genomic replication occurred in synchrony in all of the follicle cells.
The data are combined for genotypes dE2Fi1/dE2F91and dE2Fi1/dE2F7172.
Genomic replication persisted in a few follicle cells.
The data are combined for genotypes dE2Fi2/dE2F91and dE2Fi2/Df.
In wild-type ovaries the shift from general genomic replication to amplification of chorion genes is readily observed. Consistent with the findings of Calvi et al. (1998), we observed BrdU incorporation throughout the entire follicle cell nucleus during genomic replication (shown for stage 9) (Fig. 2A,A′). Groups of follicle cells in the same egg chamber undergo BrdU incorporation at different times to give an asynchronous pattern (Fig. 2A,A′). During amplification (stages 10B–13), we observed continuous BrdU incorporation in all the follicle cells but only at four subnuclear foci (Fig. 2B,B′, see circle in B′). Two of these BrdU-labeled foci correspond to the amplified chorion gene clusters of the X and third chromosome, and the other two amplifying regions have not been identified yet (Calvi et al. 1998). In the dDP and dE2F mutant ovaries, replication prior to stage 10B is in the wild-type pattern; both labeled (S phase) and unlabeled (G phase) follicle cell nuclei were observed (data not shown). However, the mutants deviated from wild type and each other during amplification (stage 10B and later) (Table 1).
Two different patterns of BrdU labeling were observed in both the dDPa1/Df and the dDPa2/Df; hs–dDP/+ mutants (Fig. 2; Table 1). In the majority of stage-10B egg chambers for both dDP alleles, the follicle cell nuclei failed to initiate amplification (shown for dDPa1/Df but dDPa2/Df; hs–dDP/+ is the same, Fig. 2D,D′). In some stage-10B egg chambers, both alleles showed genomic replication rather than amplification (Fig. 2C,C′; shown for dDPa2/Df; hs–dDP/+ but dDPa1/Df shows the same pattern). dDPa1/CyO or dDPa2/CyO control siblings were examined, and the replication phenotypes of these mutants are recessive (CyO is a balancer chromosome that carries a wild-type copy of the dDP gene).
There are multiple observations indicating that the dDP mutant stage-10B egg chambers undergo inappropriate genomic replication and are not delayed with respect to development or cell-cycle timing. First, it is striking that the mutant stage 10B genomic replication is synchronous in all the follicle cells, a property associated with amplication but not genomic replication (Fig. 2, cf. C′ to A′ and B′). Thus the genomic replication is not likely to be the consequence of follicle-cell genomic replication being delayed relative to egg chamber development and persisting into stage 10B. Second, it is unlikely that this genomic replication results from a slower S phase, with replicating follicle cells accumulating until replicating cells are continuous across the cell layer. We saw either stage-10B egg chambers with no replicating follicle cells or all of the follicle cells replicating, no evidence of a gradual increase in follicle cells in S phase. Third, these egg chambers are at stage 10B by morphological criteria, the oocyte and nurse cell size, and centripetal migration of the follicle cells. Fourth, we see the stage-appropriate change in Cyclin E protein distribution in the mutant stage-10B nurse cells (see below and Fig. 6, below). We postulate that the difference between the phenotypes shown in Figure 2, C and D is from variation in the levels of active dDP among egg chambers. Taken together, the phenotypes suggest that dDP plays a dual role in the regulation of replication in follicle cells. It is needed to activate chorion gene amplification, but it also may be required to inhibit follicle-cell genomic replication. Importantly, the results of the BrdU analysis correlate directly with the severe eggshell defects observed for the dDP mutants.
Figure 6.
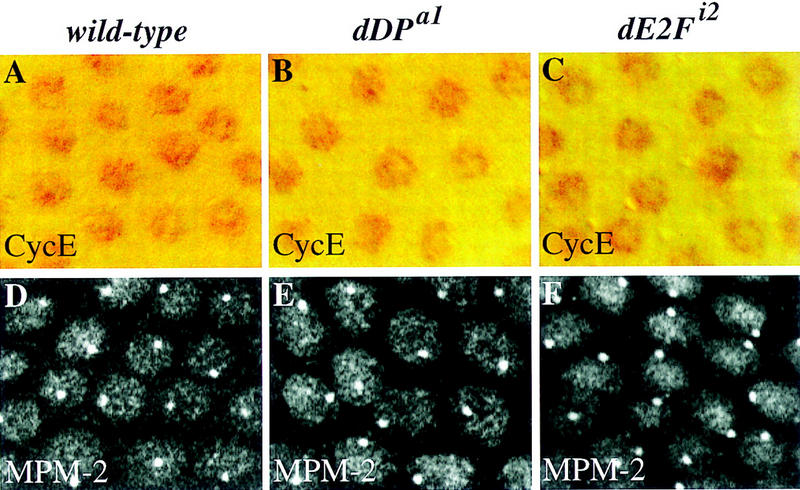
Cyclin E levels and activity are not affected in the dDP and dE2F mutants. A field of follicle cells from a stage-10B egg chamber is shown in each panel. (A–C) Cyclin E protein levels in wild-type and mutant follicle cells following staining with a monoclonal antibody (Richardson et al. 1995). The antibody staining is detected by the HRP reaction. (A) In wild-type follicle cells cyclin E is present at uniformly high levels in all the nuclei. (B,C) The levels of Cyclin E protein appear normal in mutant dDPa1/Df and dE2Fi2/Df follicle cells. (D–F) MPM2 staining reflects cyclin E activity in follicle cells (Calvi et al. 1998). MPM2 staining is visualized by confocal immunofluorescence. (D) In wild-type follicle cells a bright nuclear sphere is seen in all the follicle cell nuclei. (E,F) An identical MPM2 sphere is observed in the mutant dDPa1/Df and dE2Fi2/Df follicle cells.
BrdU labeling of dE2Fi1/Df mutant egg chambers showed that dE2F also is required for amplification; subnuclear BrdU incorporation was either absent or reduced greatly in these mutant egg chambers (Fig. 3, cf. A and B). The dE2Fi2 mutation, however, had a different effect on amplification. BrdU labeling of dE2Fi2 mutant ovaries appeared to have significantly larger labeled foci (stage 10B and in the later stages) than those observed in wild type (Fig. 3, cf. C and B). In addition to the over-amplifying follicle cells, some dE2Fi2 egg chambers had small patches that failed to incorporate BrdU (data not shown). The increased amount of BrdU incorporation in the dE2Fi2 mutant could explain the thicker eggshell, if additional copies of the chorion genes were amplified. The dE2Fi2 mutant phenotype also indicates that the carboxy-terminal transcriptional activation and RB-binding domains of dE2F are not required for formation of amplification foci. Both of the dE2F mutations are recessive for these amplification effects.
We quantitated the timing and level of amplification in the dDP and dE2F mutants. The dDP and dE2F mutations were isolated in stocks that had been isogenized for a single second or third chromosome, respectively. Consequently we determined the developmental pattern of amplification in these strain backgrounds by using dDPa1/CyO and dE2Fi1/TM3 control females, as these alleles are fully recessive. Because the dDPa1 and dE2Fi1 mutant flies have reduced viability, we developed a PCR assay that requires fewer egg chambers than Southern blots (see Materials and Methods) to compare the levels of amplification of the chorion clusters to the unamplified actin 5C gene (Fig. 4) or the rosy gene (data not shown). An essential 320-bp control element from the maximally amplified region of the third chromosome, ACE3, was assayed (Orr-Weaver et al. 1989). A 252-bp control fragment from within the ACE1 element of the X chromosome chorion cluster was also tested to confirm that the PCR assay was accurate (Spradling et al. 1987). To test the linearity and accuracy of the PCR assay, the amplification levels of dDPa1/CyO control egg chambers were quantified both by the PCR assay (Fig. 4) and by a quantitative Southern blot (data not shown). The developmental timing of amplification was identical in the two experiments and the levels of amplification were comparable.
Figure 4.

Quantitation of chorion gene amplification. Each row shows gels of the PCR products from egg chambers of stage 1–8, 10A, 10B, and 13. Within each egg chamber sample, a series of fourfold serial dilutions of egg chamber DNA were used for independent PCR reactions, and these PCR products are shown from left to right. The sizes of the ACE3, actin, and ACE1 products are shown on the right. The levels of amplification can be visualized by the intensity of the ACE3 or ACE1 products relative to the unamplified actin control. The genotype for each row is shown at left. In the dDPa1/CyO and dE2Fi1/TM3 controls amplification is observed readily in stage 10B, and ACE3 amplifies to higher levels than ACE1. In the dDPa1/Df mutant egg chambers, amplification occurs with correct developmental timing, but the levels are reduced. In contrast, in the dE2Fi2/Df mutant ACE3 is already amplified fourfold in stage 10A, and levels are elevated in later stages.
In agreement with previous results of Calvi et al. (1998), we found that in our strain backgrounds ACE1 did not amplify until stage 10B (Fig. 4). ACE1 was amplified fourfold in stage-10B egg chambers and sevenfold in stage-13 egg chambers. In contrast, we detected low levels, ∼1.5-fold, of amplification of ACE3 in stage 10A (Fig. 4). Calvi et al. (1998) previously demonstrated amplification of the third chromosome cluster in stage 10A, prior to amplification of the χ cluster. We found that ACE3 was amplified 8-fold in stage-10B egg chambers and 16-fold in stage-13 egg chambers (Fig. 4). Using the PCR assay, significant amplification of ACE3 and ACE1 occurs when BrdU incorporation is first evident at the amplifying loci, stage 10B.
Quantitative analysis of the dDPa1/Df mutant egg chambers showed that amplification occurred with correct developmental timing, but the levels of ACE3 amplification were reduced to twofold in stage-10B egg chambers and fourfold in stage-13 egg chambers (Fig. 4, dDPa1/Df row). This is consistent with the thin eggshells present in this mutant, and undetectable BrdU foci in the follicle cells. Although we did not detect BrdU incorporation in these follicle cells cytologically, the PCR assay is more sensitive and shows that amplification is not completely blocked.
In contrast to the reduced amplification in the dDPa1/Df mutant, in the dE2Fi2/Df mutant amplification initiated earlier and was amplified fourfold at stage 10A (Fig. 4, dE2Fi2/Df row). ACE3 was amplified 16-fold in stage-10B egg chambers from this mutant, whereas 32-fold amplification occurred at stage 13. The increased copy number of chorion genes in earlier egg chamber stages in the mutant can explain the thickened eggshells if these extra copies lead to increased levels of chorion proteins.
Expression of dE2F in the ovary
Because E2F is required for oogenesis, we examined the expression of dE2F and dDP in wild-type ovaries. The dDP transcript was constitutively high at all stages of oogenesis (data not shown). To examine dE2F transcription, we used a dE2F enhancer trap line, dE2Frm729/+ (Duronio et al. 1995; Brook et al. 1996). β-Galactosidase staining of ovaries from dE2Frm729/+ females revealed that there are high levels of the reporter protein in follicle and nurse-cell nuclei starting at stage 10A (Fig. 5A). To analyze the expression of dE2F protein in wild-type and mutant ovaries, we used antibodies against dE2F (Asano et al. 1996). These antibodies detect the dE2F protein specifically, because we did not see staining above background in the ovaries of dE2F91/Df females that were rescued to adulthood by transient expression of hs–dE2F (data not shown). In wild-type ovaries an increase in the levels of dE2F protein in follicle and nurse cell nuclei was observed at stage 10B, reflecting the induction of dE2F transcript (Fig. 5B,C). dE2F protein was also detected in ovaries from the dDPa1, dE2Fi1, and dE2Fi2 mutants, so neither the mutations in dE2F itself or in dDP affect dE2F protein stability significantly (Fig. 5D; data not shown). This is true even for the dE2Fi2 mutation that predicts a truncated protein. We conclude that the replication defects in the mutant follicle cells are not caused by loss of induction of expression or instability of the dE2F protein.
Figure 5.

dE2F expression correlates with E2F activity. A is at a lower magnification than panels B–D. (A) β-Galactosidase staining of egg chambers from dE2Frm729/+, a dE2F enhancer trap line in which the expression of β-galactosidase profiles the level of dE2F transcripts (Duronio et al. 1995; Brook et al. 1996). A stage-9 egg chamber is embedded between two stage-10 egg chambers. The β-galactosidase reporter is induced at stage 10 in all of the follicle cells of the egg chamber. This induction correlates with the transition to amplification in the follicle cells. The level of β-galactosidase is highest in the follicle cells covering the anterior region of the oocyte, correlating with a gradient of BrdU incorporation from anterior to posterior in the follicle cells of the egg chamber (data not shown). The small blue cells are the follicle cells (short arrow); the larger cells are nurse cells (large arrow). (B–D) dE2F protein levels in wild-type and mutant follicle cells following staining with a polyclonal antibody (Asano et al. 1996). The antibody staining was detected by a HRP reaction, and a field of follicle cells is shown in each panel at the same magnification. (B) In a stage-9 (and earlier stages) wild-type egg chambers some follicle cells are positive for dE2F staining and others are not. Note that even in the follicle cells that show dE2F expression, the levels of dE2F protein are low. (C) In wild-type stage-10 follicle cells there is an increase in the levels of dE2F protein in all the follicle cells. (D) dE2F protein is also detected in dE2Fi1 mutant follicle cells. The same staining pattern was observed for the dE2Fi2 and dDPa1 mutant.
The dDP and dE2F mutations do not affect cyclin E levels or activity
Given that cyclin E is an important target of the E2F transcription factor (Duronio and O’Farrell 1995), we sought to determine whether the effects of the dDP and dE2F mutants on chorion gene amplification result from a change in the levels or activity of Cyclin E. We did not observe a change in the levels of cyclin E transcripts in stage 10A or 10B follicle cells from either wild-type or the dDP and dE2F mutant ovaries (data not shown). We tested further for an effect of E2F on Cyclin E in follicle cells by monitoring Cyclin E protein in wild-type and mutant ovaries with a monoclonal antibody against Cyclin E (Richardson et al. 1995). Surprisingly, in dDPa1, dDPa2/Df; hs–dDP, dE2Fi1, and dE2Fi2 mutants the levels of Cyclin E protein were normal in follicle cells at all stages, including stage 10B (Fig. 6A–C; data not shown). It is difficult to measure the pool of active Cyclin E, but Calvi et al. (1998) have shown that mAb MPM2 staining responds to Cyclin E and is a marker for Cyclin E activity in follicle cells. Overproduction or inhibition of Cyclin E results in a corresponding change in MPM2 staining. In the dDP and dE2F mutants there was no effect on MPM2 staining in follicle cells at any stage (shown for stage 10B; Fig. 6D–F). Therefore, the dDP and dE2F mutant effects on chorion gene amplification appear not to occur via Cyclin E. We also note that two other expected transcriptional targets of E2F, PCNA and RNR2, were not induced in the follicle cells of either wild-type or the dDP and dE2F mutants (data not shown). This suggests that a G1–S transcriptional program is not driving amplification in the follicle cells.
Localization of ORC and MCMs in follicle cells
Given the replication defects in the dDP and dE2F mutants and because these mutations did not affect Cyclin E activity or PCNA and RNR2 expression in the follicle cells, we determined whether these mutations affected two other replication proteins, ORC and MCM. The ORC and MCM proteins are required to form the prereplication complex that is needed for the initiation of replication (for review, see Dutta and Bell 1997). Drosophila ORC2 was an especially good candidate, because in the orc2 female-sterile mutant the eggshells are thin and amplification of chorion gene clusters is defective (Landis et al. 1997; Calvi et al. 1998). The orc2 transcripts in follicle cells were not induced at stage 10 when the transition to amplification occurs (data not shown). Thus, the burst of E2F expression at this time does not trigger a detectable change in orc2 transcription. We then examined ORC2 protein levels in follicle cells using an antibody against Drosophila ORC2 that recognizes a single band on an immunoblot of Drosophila protein extracts (data not shown). During follicle cell mitotic divisions as well as subsequent follicle cell genomic polyploidization, the levels of ORC2 were uniformly nuclear and constant in all the follicle cells (Fig. 7A). This is in contrast to the pattern of BrdU incorporation at these stages (refer to Fig. 2A,A′). Thus, prior to stage 10, ORC2 expression is the same in follicle cells that are undergoing S phase and those that are in a gap phase of the mitotic or endo cell cycle. This is consistent with the observations in other organisms that ORC is bound to chromatin throughout the cell cycle (Dutta and Bell 1997).
Figure 7.
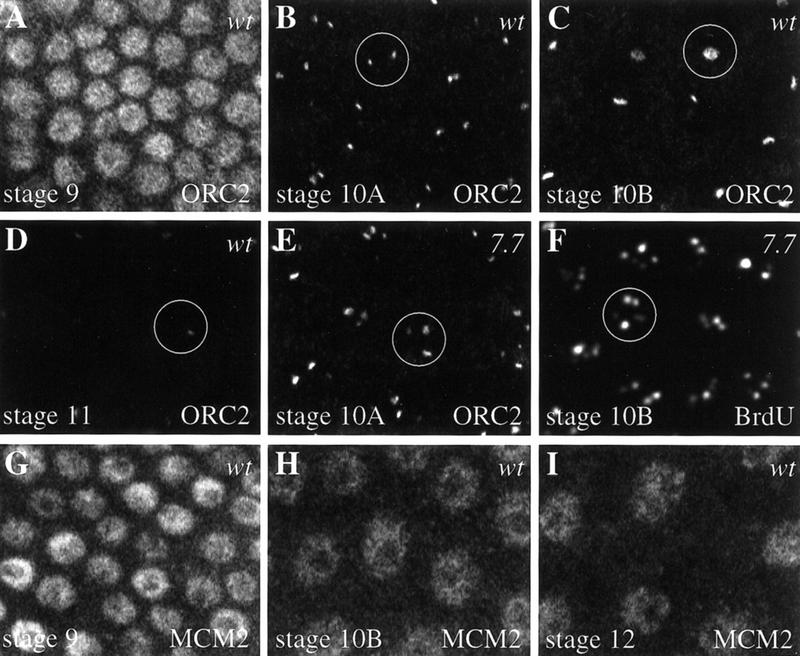
ORC and MCM localization during amplification in follicle cells. The ORC and MCM proteins were visualized by confocal immunofluorescence, and a field of follicle cells is shown in each panel. (A–D) The developmental sequence of changes in ORC2 localization in wild-type follicle cells. (A) ORC2 localization was uniform and constant in all the follicle cell nuclei of early egg chambers (shown for stage 9). Note that ORC localization does not reflect the pattern of BrdU incorporation at this stage (see Fig. 2A′). (B) At stage 10A, ORC2 protein (just prior to the onset of amplification as detected by BrdU) changes from uniform nuclear staining to specific localization at the amplifying loci. Two signals of approximately equal size and intensity were seen clearly and consistently in every follicle cell nucleus. A single follicle cell nucleus is designated by a circle. (C) At stage 10B only one ORC2 focus is observed. (D) By stage 11, ORC2 signal was greatly diminished, and it was no longer detectable at stage 12. (E) Three ORC2 signals were seen consistently at stage 10A in the fly strain with an additional 7.7-kb chorion fragment. Note that the third dot is smaller than the other two ORC2 foci. (F) Amplification is detected by BrdU incorporation in the fly strain carrying the amplifying 7.7-kb transposon. Five BrdU foci were observed consistently in all the follicle cell nuclei (one large dot, two intermediate, and two small). This is in contrast to BrdU detection in wild-type follicle cells in which only four dots were observed (one large, one intermediate, and two small) (see Fig. 2B′). (G,H) MCM2 localization in wild-type follicle cells is shown using a polyclonal antibody (Su et al. 1996; Su and O’Farrell 1998). (G) In stage-9 follicle cells MCM2 expression had a mosaic pattern with some follicle cell nuclei staining stronger than others. (H,I) MCM2 expression was lower in all the follicle cells at the onset of amplification (stage 10B). Similar staining was observed with MCM4 and MCM5.
At stage 10A we observed a striking change in the localization of ORC2. ORC2 was no longer present uniformly throughout the follicle cell nucleus. Instead we saw two foci of the same size in all follicle cells (Fig. 7, cf. B and A). In some follicle cells we were able to see three or four foci (data not shown). The ORC2 staining at stage 10A is in the pattern of BrdU incorporation observed later, at stage 10B. To determine the relationship between the ORC2 and BrdU foci, we performed double-labeling experiments (Fig. 8). The ORC foci were present prior to BrdU incorporation in stage 10A (Fig. 8A). ORC localization overlapped sites of BrdU incorporation in small, early stage-10B egg chambers (Fig. 8B). In larger (later) stage-10B egg chambers there was only a single focus of ORC labeling (Fig. 7C), and it colocalized with one of the sites of BrdU incorporation (Fig. 8C). By stage 11, the intensity of the single remaining ORC focus was reduced significantly (Fig. 7D), and ORC2 was no longer detectable by stage 12 (data not shown).
Figure 8.
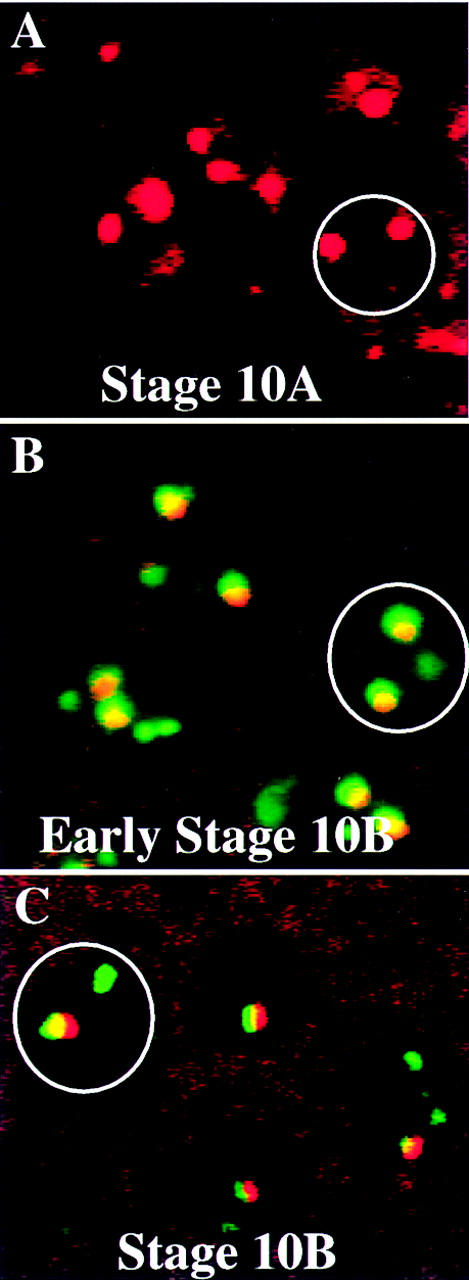
ORC2 and BrdU double labeling. Egg chambers were double labeled with anti-ORC2 and BrdU and staged according to Spradling (1993). ORC2 staining is shown in red and BrdU labeling in green. A field of follicle cells is shown in each panel. (A) In stage-10A egg chambers two large foci of ORC2 staining are present in each follicle cell nucleus (designated by a circle), but there is no BrdU incorporation. (B) Early stage-10B egg chambers are small, only slightly larger than stage 10A, and in these the two large ORC2 foci coincide with sites of BrdU incorporation. (C) In stage-10B egg chambers that are large and developmentally later, only a single focus of ORC2 is present and it corresponds to the largest dot of incorporated BrdU.
To demonstrate that ORC2 binds to amplifying regions specifically, we examined ORC2 localization in a Drosophila transformant strain with a 7.7-kb chorion fragment from the third chromosome cluster that is inserted onto the second chromosome and undergoes amplification (Orr-Weaver and Spradling 1986). In this strain we observed consistently an additional third ORC2 signal at stage 10A in all the follicle cells (Fig. 7, cf. E and B). The intensity of BrdU foci in this transformant line reflects the known levels of amplification with one large dot (presumably the endogenous third chromosome cluster), an intermediate dot (the amplified transposon), a smaller dot (the less amplified X cluster), and two faint dots (Fig. 7F). Thus both a new focus of ORC2 localization and BrdU labeling are produced by the transposon with the 7.7-kb chorion fragment, supporting the conclusion that ORC2 localizes specifically to the amplifying regions.
Because the MCMs associate with chromatin in an ORC-dependent manner, we also examined the localization of the MCMs in follicle cells. MCM2 was located throughout the nucleus and showed staining similar to that seen with MCM proteins in human and Xenopus replication systems (for review, see Chong et al. 1996; Dutta and Bell 1997) (Fig. 7). We observed similar staining with Drosophila MCMs 4 and 5 (data not shown). During the mitotic divisions and subsequent follicle-cell genomic polyploidization, MCM staining appeared bright in some follicle cells and faint in others (Fig. 7G). This staining pattern has been reported previously for embryonic and larval tissues, and the bright MCM signal may correlate with binding of MCM to chromatin prior to replication (Su and O’Farrell 1997, 1998). In contrast to ORC, MCM protein remained nuclear at stage 10 and discrete subnuclear foci were not observed (Fig. 7, cf. H and B). Note that MCM staining is faint and diffuse in all the follicle cell nuclei of stage-10 egg chambers and all subsequent stages.
dDP and dE2F affect ORC localization
We determined whether the levels or localization of MCMs and ORC2 were affected in the dDP and dE2F mutants. Immunostaining with anti-MCM2 and MCM5 showed that there was no effect in all of the mutants examined (dDPa1, dE2Fi1, and dE2Fi2) (Fig. 9B; data not shown). During the mitotic divisions and follicle cell genomic polyploidization (prior to stage 10A), immunostaining with anti-ORC2 revealed that the levels of ORC2 protein in all mutants were identical to wild type (data not shown).
Figure 9.
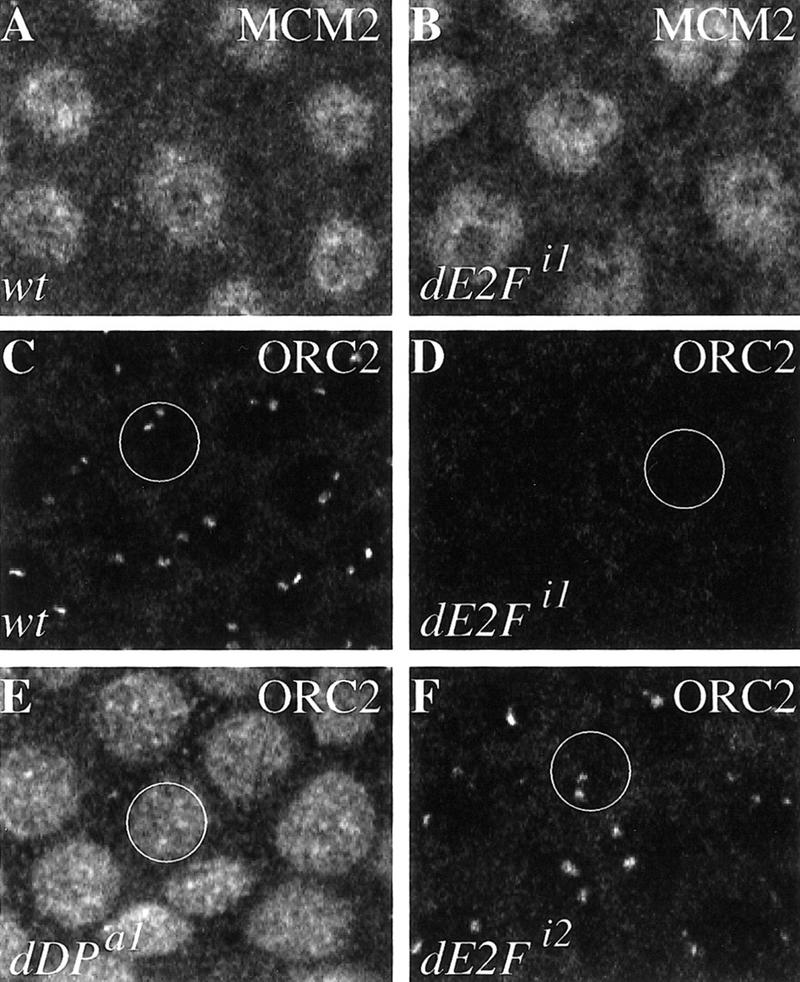
The dDP and dE2F mutants affect the localization of ORC but not MCMs. The ORC and MCM proteins were visualized by confocal immunofluorescence, and a field of follicle cells is shown in each panel. (A,B) The localization of MCM-2 is identical in wild-type (A) and dE2Fi1 mutant stage-10 (B) follicle cells. Similar staining was seen with dE2Fi2 and dDPa1. (C) ORC2 localization in wild-type stage-10A follicle cells. A single follicle cell nucleus is designated by a circle. (D) In the dE2Fi1 mutant ORC2 localization was not detected in follicle cells at stage 10A or subsequent stages. (E) In the dDPa1 mutant ORC2 localization persists inappropriately throughout the follicle cell nucleus (cf. Fig. 7A). (F) The dE2Fi2 mutant does not appear to affect ORC2 localization.
Interestingly, we detected significant differences in the localization of ORC in the dDP and dE2F mutants during amplification (stages 10 and later). In the dE2Fi1 mutant, ORC2 localization was not detectable in follicle cells at stage 10A and all subsequent stages (Fig. 9, cf. D and C). Thus, the greatly diminished or failed localization of ORC2 can explain the significant reduction in BrdU incorporation at the amplifying foci in the dE2Fi1 mutant. Although we cannot detect ORC localization, it is possible that an amount of ORC insufficient to observe by immunofluorescence is localized, and this accounts for the weak BrdU incorporation seen in the mutant. In the dDPa1 mutant nuclear ORC2 staining persisted inappropriately in stage 10 and later stages (Fig. 9E). It is possible that ORC2 is localized to the amplifying loci in these mutant egg chambers but that this localization cannot be distinguished above the background of total genomic staining. The lack of specific ORC2 localization could explain the reduced amplification and inappropriate genomic replication phenotypes observed for this mutant. Our inability to detect specific localization of ORC2 to the amplicons in the dDPa1 and dE2Fi1 mutants is not because of the inability to visualize ORC2 immunofluorescence on DNA amplified to lower levels. By PCR analysis of our wild-type strains, ACE1 is not amplified in stage-10A egg chambers and ACE3 is amplified only 1.5-fold (Fig. 4), but ORC2 localization is readily visible. Thus, both the dDP and the dE2F DNA-binding mutations have a pronounced effect on ORC2 localization.
We analyzed the effect on ORC2 localization in the dE2Fi2 mutant that is predicted to have a truncated dE2F protein. In this mutant amplification was increased at stages 10A and B (Figs. 3C and 4), and the eggshell was thicker. Consistent with the continued amplification observed in this mutant, no effect on the localization of ORC2 protein was observed and the levels of staining were not dramatically different from wild type (Fig. 9F). Therefore, ORC2 immunostaining in all three dDP and dE2F mutants shows a correlation between the BrdU foci, amplification levels, and ORC2 localization.
Discussion
In the follicle cells ORC2 is present throughout the nucleus when the endo cycle is occurring, whereas it is restricted to sites of amplification once genomic replication ceases. Mutations in dDP or dE2F affect levels of amplification and ORC2 localization in corresponding ways. These results indicate that E2F influences ORC localization either via an E2F transcriptional target(s) or possibly via a more direct mechanism. The replication phenotypes of the mutants and their effects on ORC localization suggest that E2F is not only important for the onset of S phase but the control of origin activity within S phase.
ORC localization and amplification
At developmental stages when the follicle cells are in the S–G endo cycle and genomic replication is occurring, prior to stage 10A, ORC2 is present throughout the nucleus. ORC2 is detected in all of the follicle cells of each egg chamber, yet the endo cycle occurs across the follicle cell layer asynchronously, with only some groups of follicle cells incorporating BrdU in any given egg chamber. Thus it appears that in the endo cycle, as in the archetypal cycle, ORC is present in the nuclei whether they are in S or G phase. In Drosophila the six ORC proteins form a stable complex (Gossen et al. 1995); thus, ORC2 localization is likely to reflect the presence of the ORC complex.
After the endo cycle and genomic replication cease in the follicle cells, ORC2 is present at specific nuclear foci in all of the follicle cells (stage 10A). We used an additional transposon copy of chorion DNA to show that at least two of the sites of ORC2 localization correspond to chorion amplicons. In stage 10B the ORC2 foci overlap sites of BrdU incorporation. In Drosophila mitotic cells and salivary gland polytene chromosomes ORC2 has been observed localized preferentially to the centric heterochromatin, but we did not observe this in the follicle cells (Pak et al. 1997).
The developmental pattern of ORC2 localization provides new insights into the mechanism of chorion gene amplification. First, the specific localization of ORC2 precedes detectable BrdU labeling or amplification of ACE1 by quantitative assays. It also precedes most of ACE3 amplification. This suggests that binding of ORC to the amplification origins is a prerequisite for repeated initiation and amplification. Second, the disappearance of both genomic ORC2 staining and amplicon localization are very striking. Between stages 9 and 10A ORC2 changes abruptly from uniform distribution in the nucleus to specific localization to amplifying foci. By late stage 10B a single site of ORC2 localization is detectable; this overlaps the largest focus of BrdU incorporation, the site of the third chromosome chorion gene cluster (Calvi et al. 1998). By stage 11 this focus of ORC2 has nearly disappearred. In contrast, the two large and two small foci of BrdU incorporation persist until at least cycle 13. Because a 1-hr pulse of BrdU labeling was done, and development from stage 10B to stage 13 takes 7.5 hr (Spradling 1993), the BrdU seen in stage 12 and 13 egg chambers is not because of development of stage 10B egg chambers labeled during the pulse. Thus nucleotide incorporation persists after ORC2 dissociation, and perhaps reflects an elongation rather than an initiation event.
In summary, these observations raise the possibility that delocalization of ORC from the genome is linked to the cessation of genomic replication. Our results indicate that ORC localization to the amplicons in stage 10A permits the repeated initiation of amplification. BrdU incorporation begins in stage 10B, and in early 10B egg chambers ORC2 localization is coincident with sites of BrdU incorporation. Later in stage 10B, ORC2 appears to be present solely at the third chromosome chorion cluster, the one that amplifies to highest levels. By quantitative analysis we find that at stage 10B ACE3, a fragment known to be near the preferred amplification origin (Delidakis and Kafatos 1989; Heck and Spradling 1990), has already undergone several rounds of amplification. After stage 11, ORC2 is no longer detectable. Thus we propose that ORC localizes to amplicons in stage 10A, permits repeated initiation, and that by stage 10B nearly all of the initiation events have occurred. This is marked by the dissociation of ORC. Because the third chromosome chorion cluster amplifies to higher levels it undergoes a larger number of initiation events, and ORC persists the longest at this site. Our model proposes that during stages 11–13 the BrdU incorporation observed results from elongation of previously initiated replication forks, producing the 100-kb gradient of amplified DNA found in stage-13 egg chambers (Spradling 1981).
E2F and ORC localization
We found that mutations in dDP or dE2F affect both amplification and ORC2 localization (Table 2). In contrast to the striking effects on ORC2, the dE2F and dDP mutations do not affect either the MCM proteins or Cyclin E. Cyclin E appears to be necessary for amplification (Calvi et al. 1998). The fact that the levels and activity of Cyclin E are not affected in dDP or dE2F mutant follicle cells suggests that the role of Cyclin E in amplification is either parallel to or upstream of E2F.
Table 2.
Summary of dE2F−and dDP−defects
| Genotype
|
Mutant protein
|
Viability
|
Viability with reduced cyclin E dose
|
Eggshell morphology
|
Inappropriate genomic replication
|
Amplification (BrdU foci)
|
ORC2 foci
|
|---|---|---|---|---|---|---|---|
| dE2F91 |  |
− | −a | N.D. | N.D. | N.D. | N.D. |
| DNALeucineActivation & | |||||||
| bindingzipperRb binding | |||||||
| dE2Fi1 |  |
+ | − | thin | no | −/+b | − |
| DNALeucine | |||||||
| bindingzipper | |||||||
| dE2Fi2 |  |
+ | + | thick | no | ++c | + |
| DNA | |||||||
| binding | |||||||
| dDPa1 |  |
+ | N.D. | very thin | yes | − | −d |
These animals die earlier in development with a twofold reduction of the zygotic cyclin E contribution.
(−/+) Reduced, but not absent, levels of amplification.
(++) Increased levels of amplification relative to wild type.
Uniform nuclear ORC2 localization persists inappropriately.
In evaluating the mechanism by which dDP and dE2F affect ORC localization and DNA replication it is useful to consider each of the three alleles and the distinct effects separately. There are two aspects of ORC localization: clearing of ORC uniformly present in the follicle cell nuclei and subsequent specific localization of ORC to the amplicons. The dDPa1 mutation has the most severe effect in reducing BrdU incorporation and produces eggs with the thinnest shells. In addition, in some egg chambers continued follicle cell polyploidization occurs in place of amplification. The fact that in all the dDP mutant egg chambers nuclear localization of ORC2 persists, and ORC2 is not detectable specifically at amplifying foci could indicate that amplification requires that ORC be cleared from genomic chromatin and assembled at amplification origins. There are two outcomes from persistence of genomic ORC localization. It either blocks amplification or in a minority of egg chambers permits continued genomic replication. The clearing of ORC from genomic origins may be linked to a global change that permits rereplication and amplification of those loci that retain ORC binding.
The dE2Fi1 mutants have less severe phenotypic effects. ORC is cleared from genomic origins but is not localized to amplification origins. The outcome of this appears to be that genomic polyploidization appropriately stops, but amplification is reduced. These effects also support the idea that ORC concentration at amplifying foci is needed for rereplication. We propose that the dE2Fi1 defect is less severe than that of dDPa1 because a second dE2F gene exists (identified by D. Huen and N. Dyson, pers. comm.; Sawado et al. 1998) that is able to compensate partially for the dE2F mutant protein.
The absence of an effect of the dE2Fi2 mutation on ORC localization is consistent with the fact that in this mutant genomic replication ceases and amplification occurs. It is striking that amplification occurs earlier and has increased levels in mutant flies with a predicted truncated form of dE2F lacking the RB-binding domain. Thus restriction of the onset and extent of origin amplification may be regulated by E2F complexed with RB. It has been demonstrated that RB, when complexed to E2F, is capable of recruiting histone deacetylase and thereby converting chromatin to a compacted state (Brehm et al. 1998; Luo et al. 1998; Magnaghi-Jaulin et al. 1998). This state is correlated with inaccessibility to transcription factors, and it is reasonable to propose that it would also hinder binding of replication factors. Thus in this model, E2F in complex with RB would cause histone deacetylation in the vicinity of replication origins, leading to inhibition of amplification until stage 10B. The inability of dE2Fi2 protein to bind RB would prevent inhibition and result in premature amplification.
The differences between the three mutations in the E2F subunits provides insights into the mechanism by which E2F may influence ORC localization. This effect could be direct or indirect. Both the dDPa1 and dE2Fi1 mutations are predicted to weaken E2F DNA binding. Thus the known E2F activities should be present but at reduced levels. For example, these two mutant proteins should retain transactivation activity and the ability to bind RB, repress transcription, and alter chromatin structure. Despite these activities, ORC foci are not detected, implying that the ability of E2F to bind DNA is crucial for its ability to influence ORC localization. This conclusion is supported by the fact that ORC is localized properly in the dE2Fi2 mutant in which the protein has a normal DNA-binding motif and is predicted to lack the transactivation and RB-binding domains.
The suggestion that the critical activity of E2F in controlling ORC localization is DNA binding makes it possible that E2F has a direct interaction with ORC to localize it to amplification origins. There are candidate E2F-binding sites within the amplification control region for the third chromosome cluster. Using polyclonal antibodies against dE2F (Asano et al. 1996), we were unable to detect dE2F at discrete nuclear foci when amplification is occurring (I. Royzman and T. Orr-Weaver, unpubl.); however, dE2F may be more difficult to visualize in situ than ORC.
Another alternative is that E2F influences ORC by one of its transcriptional targets. There may be an E2F transcriptional target whose gene product affects ORC localization. Alternatively, the key target might be another subunit of ORC. In human cells ORC1, but not ORC2, is transcriptionally regulated by E2F (Ohtani et al. 1996). The observation that the truncated form of dE2F (dE2Fi2) is sufficient for ORC2 localization would then suggest that dE2F normally activates the transcription of the critical target gene by recruiting another positive regulator to the promoter or by displacing a negative regulator. Asano and Wharton found recently that the pattern of ORC1 localization in the follicle cells parallels that seen with ORC2 (Asano and Wharton 1999). Furthermore, overexpression of ORC1 in late-stage follicle cells results in its localization throughout the nucleus and a restoration of genomic replication. Because ORC1 is transcriptionally regulated by E2F, they propose that E2F influences ORC localization and origin activity via effects on the level of ORC1.
The mutations in dDP and dE2F reveal a previously unrecognized role for E2F in controlling replication origin activity within S phase by affecting ORC localization. These results both define a new cell cycle function for E2F and suggest that it affects replication complex assembly directly or via one of its targets. Defining this mechanism will greatly enhance our understanding of the regulation and developmental control of replication initiation.
Materials and methods
Fly strains
dDPa1 and dDPa2 alleles were described previously (Royzman et al. 1997). The deficiency uncovering dDP, Df(2R)vg56, was provided by R. Duronio and colleagues (1998). dE2F7172, dE2F91, dE2Frm729 alleles (Duronio et al. 1995), and the transgenic lines P[w+, hsp70–dE2F], P[w+, hsp70–dDP] were provided by N. Dyson (Duronio et al. 1996). The deficiency uncovering dE2F, Df(3R)e-BS2 was obtained from the Bloomington Stock Center. cyclin EPZ5 was provided by J. Roote (University of Cambridge, UK). The R7.7-6 chorion transformant has been described previously (Orr-Weaver and Spradling 1986).
New mutations in dE2F
dE2Fi1 and dE2Fi2 were isolated from third chromosome mutant lines established in the laboratory of R. Lehmann (Skirball Institute, New York University Medical Center, NY). The EMS mutagenesis and crosses to establish balanced stocks have been described (Moore et al. 1998). We screened these lines for mutations that disrupt G1–S transcription by the strategy previously described (Royzman et al. 1997).
To demonstrate that dE2Fi1 and dE2Fi2 are mutations in the dE2F gene, both mutant lines were sequenced. Genomic DNA was isolated from adults transheterozygous for the mutagenized chromosome and Df(3R)e-BS2. The dE2F ORF was amplified from the mutant genomic DNA, and the PCR products were sequenced directly by Research Genetics.
PCR amplification assay
Ovaries were dissected in EBR (Ephrussi and Beadle 1936) to isolate staged egg chambers. Approximately 15–30 egg chambers were used. Excess EBR was removed and was replaced with 500 μl of lysis buffer [50 mm HEPES (K+) at pH 7.5, 140 mm NaCl, 1 mm EDTA, 1% Triton X-100, 0.1% deoxycholate]. The egg chambers were disrupted by brief sonication, and the insoluble material was removed by centrifugation. The supernatant was digested with proteinase K, extracted with phenol/chloroform, and ethanol precipitated. The resulting pellet was dissolved in TE buffer at a volume of 2 μl per egg chamber. Fourfold serial dilutions were made of the dissolved pellet, and each dilution was analyzed by PCR. PCR reactions (50 μl), containing 5 μl of egg chamber DNA, Vent buffer (New England Biolabs), 200 μm dNTPs, 1 μm each primer, and Taq DNA polymerase, were subjected to 28 rounds of thermocycling. Gel electrophoresis, photography, densitometry, and quantitation, were as described previously (Aparicio et al. 1997). Levels of amplification were quantified for stage 10A, 10B, or 13 egg chambers by taking the ratio of the ACE3 or ACE1 product to that of the actin product and dividing by the chorion/actin ratio from control, preamplification stage 1–8 egg chambers. The dilutions used for quantitation were those from the dilution series that were in the linear range of the film and densitometer.
Primers used for PCR were CACTACCGTTTGAGTTCTTGTGCTG and GGAAGGGGAAAGCTACTTACATTTGG for actin; GGTACCCTGAGCCTGGCCAACATC and CCGCATAGTTTCGATCAGTATTGC for ACE3; TCTTCACTGGCTATCGCAGGAATGTTATC and CACCAAAAGCCATCGAGATCTCCGCCAC for rosy; GGAGCAACTATAATTTTACGGCCTC and CTCTAGTTGCAAAGAGATTTGAAGATG for ACE1.
Preparation of the ORC2 antibodies
The Drosophila ORC2 coding sequence was PCR amplified from a 4- to 8-hr embryonic cDNA library (Brown and Kafatos 1988). The PCR product was subcloned into pCYB2 (New England Biolabs) using NdeI and SmaI sites engineered into the termini of the PCR product. DmORC2 protein was expressed in bacteria and was purified using the Impact system (New England Biolabs) following the manufacturer’s instructions. The purified protein was used to raise antibodies in rabbits.
Cytological analysis and microscopy
In situ hybridizations (Tautz and Pfeifle 1989) were carried out with digoxigenin-labeled RNA probes exactly as described in Royzman et al. (1997). For whole-mount in situ hybridization to ovaries, ovaries were prepared as described by Ephrussi et al. (1991) and treated for 35 min in proteinase K. Subsequent steps were carried out as for embryos. The β-galactosidase expression pattern in dE2Frm729 ovaries was determined according to a standard protocol (Montell et al. 1992).
Ovaries were labeled with BrdU as described previously (Lilly and Spradling 1996). BrdU labeling was visualized using a goat anti-mouse antibody conjugated to horseradish peroxidase (HRP) (Bio-Rad) at a dilution of 1:200, or a DTAF goat anti-mouse secondary antibody (Jackson) at a dilution of 1:150. Nuclei were counterstained with DAPI as above.
For antibody stainings, ovaries were fixed in 8% EM-grade formaldehyde (Ted Pella, Inc., Irvine, CA) for 5 min, and extracted for 2 hr in 1% Triton X-100 in PBS, and then blocked for 1 hr in 1× PBS, 1% BSA, 0.3% Triton X-100, and 2% normal goat serum. For detection of Cyclin E, mouse mAb 8B10 was provided by H. Richardson and used at a dilution of 1:5 (Richardson et al. 1995; Lilly and Spradling 1996). The HRP goat anti-mouse secondary antibody (Bio-Rad) was used at 1:200. MPM-2 antibody (Davis et al. 1983) was purchased from DAKO Corporation. MPM-2 was diluted 1:100 (Calvi et al. 1998) and detected with a Cy3-conjugated donkey anti-mouse secondary antibody (Jackson Immunoresearch Laboratories) at a dilution of 1:150. For E2F detection, a rabbit polyclonal antibody was provided by R. Wharton (Asano et al. 1996) and used at a dilution of 1:100. We showed that this antibody specifically recognizes dE2F in the ovary, because dE2F91/Df females rescued to the adult stage by hsE2F did not show protein in the ovary. An HRP-conjugated goat anti-rabbit secondary antibody (Jackson) was used at a dilution of 1:100. The MCM antibodies (affinity-purified MCM2 and MCM5 and MCM4 sera) were provided by T. T. Su and P. O’Farrell (Su et al. 1996). MCM2 and MCM5 were used at 1:50 and MCM4 was used at 1:500. The ORC2 sera were used at 1:2500. ORC2 and the MCM proteins were detected using a Cy3-conjugated donkey anti-rabbit secondary antibody (Jackson) at a dilution of 1:150.
For double labeling with BrdU and anti-ORC2 antibodies, the ovaries were labeled with BrdU, fixed, bound to anti-ORC2 antibodies and secondary antibodies as described above. Fixation was performed a second time for 20 min with 4:1:1 of 16% formaldehyde/buffer B/H2O. Buffer B is (100 mm KH2PO4/K2HPO4 at pH 6.8, 450 mM KCl, 150 mm NaCl, 20 mm MgCl2). Our standard protocol was then followed for detection of BrdU.
A Zeiss Axiophot microscope equipped with Nomarski optics and fluorescence was used to examine and photograph the ovaries. Plan-Neofluar 10×, 20×, and 40× objectives were used. The images shown in Figures 6, D–F, and 7–9 were captured with a MRC 600 Bio-Rad confocal laser scanning head equipped with a krypton/argon laser, mounted on a Zeiss Axioskop microscope. A 40× Plan Neofluar objective was used.
Acknowledgments
We are grateful to M. Asano and R. Wharton for the antibodies against dE2F and for sharing information prior to publication, N. Dyson for providing stocks and sharing unpublished information, H. Richardson for the cyclin E antibodies, T. Su and P. O’Farrell for the MCM antibodies, and B. Calvi and A. Spradling for communicating results prior to publication. We are grateful to the laboratory of R. Lehmann for sharing mutagenized lines. D. Fenger assisted with the confocal microscopy. J. Lees and D. Lee provided critical comments on the manuscript, and A. Amon, N. Dyson, M. Meyerson, I. Rebay, and D. Fenger gave helpful comments on earlier versions. We also thank J. Lees and A. Whittaker for many helpful discussions. I.R. was supported by a National Institutes of Health (NIH) predoctoral training grant. R.J.A. was supported by a postdoctoral fellowship from the NIH (GM18170).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked ‘advertisement’ in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL weaver@wi.mit.edu; FAX (617) 258-9872.
References
- Aparicio OM, Weinstein DM, Bell SP. Components and dynamics of DNA replication complexes in S. cerevisiae: Redistribution of MCM proteins and Cdc45p during S phase. Cell. 1997;91:59–69. doi: 10.1016/s0092-8674(01)80009-x. [DOI] [PubMed] [Google Scholar]
- Asano, M. and R.P. Wharton. 1999. E2F mediates developmental and cell cycle regulation of ORC1 in Drosophila. EMBO J. (in press). [DOI] [PMC free article] [PubMed]
- Asano M, Nevins JR, Wharton RP. Ectopic E2F expression induces S phase and apoptosis in Drosophila imaginal discs. Genes & Dev. 1996;10:1422–1432. doi: 10.1101/gad.10.11.1422. [DOI] [PubMed] [Google Scholar]
- Bandara LR, Buck VM, Zamanian M, Johnston LH, La Thangue NB. Functional synergy between DP-1 and E2F-1 in the cell cycle-regulating transcription factor DRTF1/E2F. EMBO J. 1993;12:4317–4324. doi: 10.1002/j.1460-2075.1993.tb06116.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandara LR, Girling R, LaThangue NB. Apoptosis induced in mammalian cells by small peptides that functionally antagonize the Rb-regulated E2F transcription factor. Nat Biotechnol. 1997;15:896–901. doi: 10.1038/nbt0997-896. [DOI] [PubMed] [Google Scholar]
- Botz J, Zerfass-Thome K, Spitkovsky D, Delius H, Vogt B, Eilers M, Hatzigeorgiou A, Jansen-Durr P. Cell cycle regulation of the murine cyclin E gene depends on an E2F binding site in the promoter. Mol Cell Biol. 1996;16:3401–3409. doi: 10.1128/mcb.16.7.3401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brehm A, Miska EA, McCance DJ, Reid JL, Bannister AJ, Kouzarides T. Retinoblastoma protein recruits histone deacetylase to repress transcription. Nature. 1998;391:597–601. doi: 10.1038/35404. [DOI] [PubMed] [Google Scholar]
- Brook A, Xie J-E, Du W, Dyson N. Requirements for dE2F function in proliferating cells and in post-mitotic differentiating cells. EMBO J. 1996;15:3676–3683. [PMC free article] [PubMed] [Google Scholar]
- Brown NH, Kafatos F. Functional cDNA libraries from Drosophila embryos. J Mol Biol. 1988;203:425–437. doi: 10.1016/0022-2836(88)90010-1. [DOI] [PubMed] [Google Scholar]
- Calvi BR, Spradling AC. Chorion gene amplification in Drosophila: A model for origins of DNA replication and S phase control. In: Fisher P, editor. Genetic approaches to eukaryotic replication and repair. New York, NY: Academic Press; 1999. (In press.) [DOI] [PubMed] [Google Scholar]
- Calvi BR, Lilly MA, Spradling AC. Cell cycle control of chorion gene amplification. Genes & Dev. 1998;12:734–744. doi: 10.1101/gad.12.5.734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong JP, Thommes JP, Blow JJ. The role of MCM/P1 proteins in the licensing of DNA replication. Trends Biochem Sci. 1996;21:102–106. [PubMed] [Google Scholar]
- Davis FM, Tsao TY, Fowler SK, Rao PN. Monoclonal antibodies to mitotic cells. Proc Natl Acad Sci. 1983;80:2926–2930. doi: 10.1073/pnas.80.10.2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delidakis C, Kafatos FC. Amplification enhancers and replication origins in the autosomal chorion gene cluster of Drosophila. EMBO J. 1989;8:891–901. doi: 10.1002/j.1460-2075.1989.tb03450.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du W, Vidal M, Xie J-E, Dyson N. RBF, a novel RB-related gene that regulates E2F activity and interacts with cyclin E in Drosophila. Genes & Dev. 1996a;10:1206–1218. doi: 10.1101/gad.10.10.1206. [DOI] [PubMed] [Google Scholar]
- Du W, Xie J-E, Dyson N. Ectopic expression of dE2F and dDP induces cell proliferation and death in the Drosophila eye. EMBO J. 1996b;15:3684–3692. [PMC free article] [PubMed] [Google Scholar]
- Duronio RJ, O’Farrell PH. Developmental control of the G1 to S transition in Drosophila: Cyclin E is a limiting downstream target of E2F. Genes & Dev. 1995;9:1456–1468. doi: 10.1101/gad.9.12.1456. [DOI] [PubMed] [Google Scholar]
- Duronio RJ, O’Farrell PH, Xie J-E, Brook A, Dyson N. The transcription factor E2F is required for S phase during Drosophila embryogenesis. Genes & Dev. 1995;9:1445–1455. doi: 10.1101/gad.9.12.1445. [DOI] [PubMed] [Google Scholar]
- Duronio RJ, Brook A, Dyson N, O’Farrell PH. E2F-induced S phase requires cyclin E. Genes & Dev. 1996;10:2505–2513. doi: 10.1101/gad.10.19.2505. [DOI] [PubMed] [Google Scholar]
- Duronio RJ, Bonnette PC, O’Farrell PH. Mutations of the Drosophila dDP, dE2F, and cyclin E genes reveal distinct roles for the E2F-DP transcription factor and cyclin E during the G1–S transition. Mol Cell Biol. 1998;18:141–151. doi: 10.1128/mcb.18.1.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta A, Bell SP. Initiation of DNA replication in eukaryotic cells. Annu Rev Cell Dev Biol. 1997;13:293–332. doi: 10.1146/annurev.cellbio.13.1.293. [DOI] [PubMed] [Google Scholar]
- Dynlacht BD, Brook A, Dembski M, Yenush L, Dyson N. DNA-binding and trans-activation properties of Drosophila E2F and DP proteins. Proc Natl Acad Sci. 1994;91:6359–6363. doi: 10.1073/pnas.91.14.6359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyson N. The regulation of E2F by pRB-family proteins. Genes & Dev. 1998;12:2245–2262. doi: 10.1101/gad.12.15.2245. [DOI] [PubMed] [Google Scholar]
- Ephrussi A, Dickinson LK, Lehmann R. oskar organizes the germ plasm and directs localization of the posterior determinant nanos. Cell. 1991;66:37–50. doi: 10.1016/0092-8674(91)90137-n. [DOI] [PubMed] [Google Scholar]
- Ephrussi B, Beadle G. A technique of transplantation for Drosophila. Am Nat. 1936;70:218–225. [Google Scholar]
- Ewen ME, Sluss HK, Sherr CJ, Matsushime H, Kato J, Livingston DM. Functional interactions of the retinoblastoma protein with mammalian D-type cyclins. Cell. 1993;73:487–497. doi: 10.1016/0092-8674(93)90136-e. [DOI] [PubMed] [Google Scholar]
- Geng Y, Eaton EN, Picon M, Roberts JM, Lundberg AS, Gifford A, Sardet C, Weinberg RA. Regulation of cyclin E transcription by E2Fs and retinoblastoma protein. Oncogene. 1996;12:1173–1180. [PubMed] [Google Scholar]
- Gossen M, Pak DTS, Hansen SK, Acharya JK, Botchan MR. A Drosophila homolog of the yeast origin recognition complex. Science. 1995;270:1674–1677. doi: 10.1126/science.270.5242.1674. [DOI] [PubMed] [Google Scholar]
- Hao XF, Alphey L, Bandara LR, Lam E, Glover D, LaThangue NB. Functional conservation of the cell cycle-regulating transcription factor DRTF1/E2F and its pathway of control in Drosophila melanogaster. J Cell Sci. 1995;108:2945–2954. doi: 10.1242/jcs.108.9.2945. [DOI] [PubMed] [Google Scholar]
- Heck M, Spradling A. Multiple replication origins are used during Drosophila chorion gene amplification. J Cell Biol. 1990;110:903–914. doi: 10.1083/jcb.110.4.903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helin K, Wu C-L, Fattaey AR, Lees JA, Dynlacht BD, Ngwu C, Harlow E. Heterodimerization of the transcription factors E2F-1 and DP-1 leads to cooperative trans-activation. Genes & Dev. 1993;7:1850–1861. doi: 10.1101/gad.7.10.1850. [DOI] [PubMed] [Google Scholar]
- Johnson DG, Schwarz JK, Cress WD, Nevins JR. Expression of transcription factor E2F1 induces quiescent cells to enter S phase. Nature. 1993;365:349–352. doi: 10.1038/365349a0. [DOI] [PubMed] [Google Scholar]
- Kato J, Matsushime H, Hiebert H, Ewen ME, Sherr CJ. Direct binding of cyclin D to the retinoblastoma gene product (pRb) and pRb phosphorylation by the cyclin D-dependent kinase CDK4. Genes & Dev. 1993;7:331–342. doi: 10.1101/gad.7.3.331. [DOI] [PubMed] [Google Scholar]
- Krek W, Xu G, Livingston DM. Cyclin A-kinase regulation of E2F-1 DNA binding function underlies suppression of an S phase checkpoint. Cell. 1995;83:1149–1158. doi: 10.1016/0092-8674(95)90141-8. [DOI] [PubMed] [Google Scholar]
- Landis G, Kelley R, Spradling AC, Tower J. The k43 gene, required for chorion gene amplification and diploid cell chromosome replication, encodes the Drosophila homolog of yeast origin recognition complex subunit 2. Proc Natl Acad Sci. 1997;94:3888–3892. doi: 10.1073/pnas.94.8.3888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lilly MA, Spradling AC. The Drosophila endocycle is controlled by Cyclin E and lacks a checkpoint ensuring S-phase completion. Genes & Dev. 1996;10:2514–2526. doi: 10.1101/gad.10.19.2514. [DOI] [PubMed] [Google Scholar]
- Luo RX, Postigo AA, Dean DC. Rb interacts with histone deacetylase to repress transcription. Cell. 1998;92:463–473. doi: 10.1016/s0092-8674(00)80940-x. [DOI] [PubMed] [Google Scholar]
- Magnaghi-Jaulin L, Groisman R, Naguibneva I, Robin P, Lorain S, Le Villain JP, Troalen F, Trouche D, Harel-Bellan A. Retinoblastoma protein represses transcription by recruiting a histone deacetylase. Nature. 1998;391:601–605. doi: 10.1038/35410. [DOI] [PubMed] [Google Scholar]
- Montell DJ, Rorth P, Spradling AC. slow border cells, a locus required for a developmentally regulated cell migration during oogenesis, encodes a Drosophila C/EBP. Cell. 1992;71:51–62. doi: 10.1016/0092-8674(92)90265-e. [DOI] [PubMed] [Google Scholar]
- Moore LA, Broihier HT, Van Doren M, Lunsford LB, Lehmann R. Identification of genes controlling germ cell migration and embryonic gonad formation in Drosophila. Development. 1998;125:667–678. doi: 10.1242/dev.125.4.667. [DOI] [PubMed] [Google Scholar]
- Morkel M, Wenkel J, Bannister AJ, Kouzarides T, Hagemeier C. An E2F-like repressor of transcription. Nature. 1997;390:567–568. doi: 10.1038/37507. [DOI] [PubMed] [Google Scholar]
- Neufeld TP, de la Cruz AF, Johnston LA, Edgar BA. Coordination of growth and cell division in the Drosophila wing. Cell. 1998;93:1183–1193. doi: 10.1016/s0092-8674(00)81462-2. [DOI] [PubMed] [Google Scholar]
- Nevins JR. E2F: A link between the Rb tumor suppressor protein and viral oncoproteins. Science. 1992;258:424–429. doi: 10.1126/science.1411535. [DOI] [PubMed] [Google Scholar]
- Ohtani K, Nevins JR. Functional properties of a Drosophila homolog of the E2F1 gene. Mol Cell Biol. 1994;14:1603–1612. doi: 10.1128/mcb.14.3.1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtani K, DeGregori J, Nevins JR. Regulation of the cyclin E gene by transcription factor E2F1. Proc Natl Acad Sci. 1995;92:12146–12150. doi: 10.1073/pnas.92.26.12146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtani K, DeGregori J, Leone G, Herendeen DR, Kelley TJ, Nevins JR. Expression of the HsOrc1 gene, a human ORC1 homolog, is regulated by cell proliferation via the E2F transcription factor. Mol Cell Biol. 1996;16:6977–6984. doi: 10.1128/mcb.16.12.6977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr-Weaver TL. Drosophila chorion genes: Cracking the eggshell’s secrets. BioEssays. 1991;13:97–105. doi: 10.1002/bies.950130302. [DOI] [PubMed] [Google Scholar]
- Orr-Weaver T, Spradling A. Drosophila chorion gene amplification requires an upstream region regulating s18 transcription. Mol Cell Biol. 1986;6:4624–4633. doi: 10.1128/mcb.6.12.4624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr-Weaver TL, Johnston CG, Spradling AC. The role of ACE3 in Drosophila chorion gene amplification. EMBO J. 1989;8:4153–4162. doi: 10.1002/j.1460-2075.1989.tb08600.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pak DTS, Pflumm M, Chesnokov I, Huang DW, Kellum R, Marr J, Romanowski P, Botchan MR. Association of the origin recognition complex with heterochromatin and HP1 in higher eukaryotes. Cell. 1997;91:311–323. doi: 10.1016/s0092-8674(00)80415-8. [DOI] [PubMed] [Google Scholar]
- Qin X-Q, Livingston DM, Kaelin WG, Adams PD. Deregulated E2F1 expression leads to S-phase entry and p53-mediated apoptosis. Proc Natl Acad Sci. 1994;91:10918–10922. doi: 10.1073/pnas.91.23.10918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson H, O’Keefe LV, Marty T, Saint R. Ectopic cyclin E expression induces premature entry into S phase and disrupts pattern formation in the Drosophila eye imaginal disc. Development. 1995;121:3371–3379. doi: 10.1242/dev.121.10.3371. [DOI] [PubMed] [Google Scholar]
- Royzman I, Orr-Weaver TL. S phase and differential DNA replication during Drosophila oogenesis. Genes Cells. 1998;3:767–776. doi: 10.1046/j.1365-2443.1998.00232.x. [DOI] [PubMed] [Google Scholar]
- Royzman I, Whittaker AJ, Orr-Weaver TL. Mutations in Drosophila DP and E2F distinguish G1–S progression from an associated transcriptional program. Genes & Dev. 1997;11:1999–2011. doi: 10.1101/gad.11.15.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawado T, Yamaguchi M, Nishimoto Y, Ohno K, Sakaguchi K, Matsukage A. dE2F2, a novel E2F-family transcription factor in Drosophila melanogaster. Biochem Biophys Res Commun. 1998;251:409–415. doi: 10.1006/bbrc.1998.9407. [DOI] [PubMed] [Google Scholar]
- Sherr CJ. Cancer cell cycles. Science. 1996;274:1672–1677. doi: 10.1126/science.274.5293.1672. [DOI] [PubMed] [Google Scholar]
- Spradling A. The organization and amplification of two clusters of Drosophila chorion genes. Cell. 1981;27:193–202. doi: 10.1016/0092-8674(81)90373-1. [DOI] [PubMed] [Google Scholar]
- Spradling AC. Developmental genetics of oogenesis. In: Bate M, Martinez-Arias A, editors. The development of Drosophila melanogaster. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1993. pp. 1–70. [Google Scholar]
- Spradling AC, de Cicco DV, Wakimoto BT, Levine JF, Kalfayan LJ, Cooley L. Amplification of the X-linked Drosophila chorion gene cluster requires a region upstream from the s38 chorion gene. EMBO J. 1987;6:1045–1053. doi: 10.1002/j.1460-2075.1987.tb04857.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su TT, O’Farrell PH. Chromosome association of minichromosome maintenance proteins in Drosophila mitotic cycles. J Cell Biol. 1997;139:13–22. doi: 10.1083/jcb.139.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ————— Chromosome association of minichromosome maintenance proteins in Drosophila endoreplication cycles. J Cell Biol. 1998;140:451–460. doi: 10.1083/jcb.140.3.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su TT, Feger G, O’Farrell PH. Drosophila MCM protein complexes. Mol Biol Cell. 1996;7:319–329. doi: 10.1091/mbc.7.2.319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tautz D, Pfeifle C. A non-radioactive in situ hybridization method for the localization of specific RNAs in Drosophila embryos reveals translational control of the segmentation gene hunchback. Chromosoma. 1989;98:81–85. doi: 10.1007/BF00291041. [DOI] [PubMed] [Google Scholar]
- Trimarchi JM, Fairchild B, Verona R, Moberg K, Andon N, Lees JA. E2F-6, a member of the E2F family that can behave as a transcriptional repressor. Proc Natl Acad Sci. 1998;95:2850–2855. doi: 10.1073/pnas.95.6.2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg RA. The retinoblastoma protein and cell cycle control. Cell. 1995;81:323–330. doi: 10.1016/0092-8674(95)90385-2. [DOI] [PubMed] [Google Scholar]
- Weintraub SJ, Prater CA, Dean DC. Retinoblastoma protein switches the E2F site from positive to negative element. Nature. 1992;358:259–261. doi: 10.1038/358259a0. [DOI] [PubMed] [Google Scholar]
- Weintraub SJ, Chow KNB, Luo RX, Zhang SH, He S, Dean DC. Mechanism of active transcriptional repression by the retinoblastoma protein. Nature. 1995;375:812–815. doi: 10.1038/375812a0. [DOI] [PubMed] [Google Scholar]