

Abstract
Convincing concepts of redox control of gene transcription have been worked out for prokaryotes and lower eukaryotes, whereas the knowledge on complex mammalian systems still resembles a patchwork of poorly connected findings. The article, therefore, reviews principles of redox regulation with special emphasis on chemical feasibility, kinetic requirements, specificity, and physiological context, taking well investigated mammalian transcription factor systems, nuclear transcription factor of bone marrow-derived lymphocytes (NF-κB), and kelch-like ECH-associated protein-1 (Keap1)/Nrf2, as paradigms. Major conclusions are that (i) direct signaling by free radicals is restricted to O2•− and •NO and can be excluded for fast reacting radicals such as •OH, •OR, or Cl•; (ii) oxidant signals are H2O2, enzymatically generated lipid hydroperoxides, and peroxynitrite; (iii) free radical damage is sensed via generation of Michael acceptors; (iv) protein thiol oxidation/alkylation is the prominent mechanism to modulate function; (v) redox sensors must be thiol peroxidases by themselves or proteins with similarly reactive cysteine or selenocysteine (Sec) residues to kinetically compete with glutathione peroxidase (GPx)- and peroxiredoxin (Prx)-type peroxidases or glutathione-S-transferases, respectively, a postulate that still has to be verified for putative mammalian sensors. S-transferases and Prxs are considered for system complementation. The impact of NF-κB and Nrf2 on hormesis, management of inflammatory diseases, and cancer prevention is critically discussed. Antioxid. Redox Signal. 15, 2335–2381.
“It was clear as mud but it covered the ground and the confusion made the brain go 'round.”
—Harry Belafonte (27)
I. Introduction
Activation of gene transcription has for long been considered to be primarily, if not exclusively, regulated by cascades of protein phosphorylation and de-phosphorylation. Screening the reviews that were written in the nineties (18, 114, 116, 117, 492) or even during the present decade (115, 118, 431, 433) by pioneers in protein phosphatase (PP) or kinase research such as Edmund Fischer, Joseph Schlessinger, or Axel Ullrich, it is hard to detect any hint that gene activation could be driven by anything else but phosphorylation, de-phosphorylation of receptors, and/or downstream signaling molecules. During the nineties, though, a second area was recognized to be intimately related to transcriptional regulation, the ubiquitin/proteasome system (69, 71, 73, 458, 480), that had been shown to either degrade transcription factors such as c-Fos and C-Jun (220, 463), p53 (420), hypoxia-inducible factor 1α (HIF-1α) (230), Nrf2 (323), β-catenin (2), and nuclear hormone receptors (342) or inhibitory cytosolic complexes of transcription factors such as the complex of nuclear transcription factor of bone marrow-derived lymphocytes (NF-κB) with its inhibitor IκB (240, 524). Other proteolytic systems such as the calpains (220, 364) and caspases (359) followed, and it became clear that these proteolytic systems complemented the phosphorylation cascades (178, 240, 359). In parallel, the impact of redox processes on transcriptional gene activation became obvious, although the main focus of the oxygen clubs and free radical associations remained the concern about the potential hazards of the reactive oxygen species (ROS) (453).
In retrospect, the reluctance to accept oxidants as mediators or modulators of regulatory processes is hard to understand. Already in the early nineties vanadate plus H2O2 (177) or peroxovanadium compounds were reported to mimic insulin action, were recognized to act as phosphatase inhibitors (381), and became widely used to enhance protein phosphorylation in the analysis of kinase cascades in general. That this analytical trick could have a physiological correlate could also have been guessed from publications of the seventies claiming H2O2 to be a second messenger of insulin signaling (81, 319, 320). However, the concept that gene transcription might be controlled by redox reactions remained dormant until an important transcription factor in eukaryotes, the nuclear transcription factor in B-cells NF-κB (19, 444, 455, 464), was shown to be activated by compounds known to trigger production of superoxide/H2O2 (440, 444) or by oxidants themselves, inter alia by H2O2, and inhibited by antioxidants (15, 440). Conceptually, an oxidative inactivation of phosphatases leading to enhanced signal transduction emerged as a likely mechanism (128).
Oxidative inactivation of phosphatases in signaling cascades, however, did not for long remain the only possible mechanism how oxidants could affect transcription. Microbiologists demonstrated that a direct oxidation of the transcription factor OxyR may orchestrate the transcription of defensive genes (11, 68). Other concepts followed, for example, activation of protein kinases (PKs), redox-dependent noncovalent binding of thioredoxin (Trx), thiol modification of proteins that form cytosolic complexes with transcription factors, or heterodimer formation of glutathione peroxidase (GPx)- and peroxiredoxin (Prx)-type peroxidases with transcription factors [reviewed in refs. (123, 134), see section II.D.1].
The multiple ways of redox regulations that became obvious over the last two decades lead us to presume that most, if not all, of the classical routes to transcriptional activation are modulated by redox processes or even critically depend on oxidant signals (Table 1). In this article we will briefly summarize pertinent mechanistic principles. In this context, insights from microbiology, which as usual is leading the field, will be discussed in respect to their possible relevance to the more complex mammalian systems. We then will focus on the redox-sensitive mammalian pathways of gene activation, choosing the two best investigated ones, the Nrf2 and NF-κB systems, as paradigms of redox-controlled transcriptional activation and basis for hormetic responses in higher organisms.
Table 1.
Mammalian Transcription Factors Regulated by Redox Events
| Transcription factor systems | Main regulatory redox events | Physiological consequences | Refs. |
|---|---|---|---|
| AhR | Hypoxia, competition of HIF and AhR for ARNT is discussed | Inhibition of DNA binding | (119) |
| AP-1 | Enhanced phosphorylation of c-Jun and c-Fos by activation of upstream kinases (e.g., RTKs, MAPKs, PKCs and/or inactivation of PPAs) | Enhanced association of c-Jun and c-Fos with CBP/p300 and transcriptional activation (inhibition of DNA binding of c-Jun when phosphorylated by GSK3β) | (475) |
| Activation of ASK-1 due to oxidation of associated Trx (or Grx) | De-inhibition of ASK-1 and activation of JNK | (415) | |
| Oxidant-induced dissociation of GSTπ or μ from ASK-1 and JNK | Activation of JNK | (488) | |
| Oxidation of Cys154 in c- Fos and Cys272 in c-Jun | Inhibition of DNA binding | (1) | |
| Reversal of Cys oxidation in c-Fos and c-Jun by Trx and APE1/Ref-1 | Prerequisite for DNA binding | (475, 520) | |
| β-catenin | Thiol oxidation in nucleoredoxin | Release of nucleoredoxin from dishevelled which activates β-catenin | (139) |
| Egr-1 | Oxidation of cysteines in the 3 Zn-fingers reversible by APE1/Ref-1 | Inhibition of DNA binding | (193, 467) |
| FOXO | Cysteine oxidation of PTP1b | Activation of Akt, phosphorylation and nuclear export of FOXO | (83) |
| Intramolecular disulfide formation between Cys297 and Cys311 in Akt | Affinity increase of Akt for PP2A, de-phosphorylation and inactivation of Akt. FOXO remains in the nucleus | (83) | |
| Oxidative activation of JNK | Phosphorylation of FOXO, in this case prevention of nuclear export thus counteracting Akt. | (83) | |
| GR | Zn-finger oxidation, reversible by Trx | Inhibition of DNA binding | (159) |
| HIF-1α | Oxidation of Fe2+ to Fe3+ in HPH | Inactivation of HPH, prevention of hydroxylation and degradation of HIF, activation of HIF | (155, 475) |
| Reduction of Cys800 in HIF by APE1/Ref-1 and Trx/TrxR | Increase in transactivation capacity of HIF | (248, 293) | |
| P53 | Low oxidative activation of MAPKs | Low phosphorylation/activation of p53, induction of antioxidant and defense systems, cell growth | (412, 467, 475) |
| Enhanced oxidative activation of MAPKs | Enhanced phosphorylation and activation of p53, elimination of cells with mutations by apoptosis | (412) | |
| Oxidation of conserved cysteines in the DNA-binding domain and Zn-fingers | Inhibition of DNA binding | (391) | |
| Reduction of Zn-coordinating cysteines in p53 by APE1/Ref-1 and Trx/TrxR | Maintenance of DNA-binding capacity | (54, 221) | |
| Pax-5, −8 | Oxidation of Cys45 and 57 in Pax-8, reversible by APE1/Ref-1 | Inhibition of DNA binding | (55, 426, 475) |
| Intramolecular disulfide formation in Pax-5, reversible by APE1/Ref-1 | Inhibition of DNA binding | (476) | |
| NF-κB | Formation of an intermolecular disulfide bond in LC8, reversible by TRP14 | Enabling phosphorylation of IκBα by IKKβ followed by IκB degradation. Release and activation of NF-κB | (227) |
| Reduction of Cys62 in p50 by APE1/Ref-1 and Trx/TrxR in the nucleus | Enhancement of DNA binding | (159, 228) | |
| Others | See section IV | ||
| Nrf2 | Modulation of Cys272 and Cys288 in Keap1 | Conformational change of Keap1, prevention of Nrf2 ubiquitination, activation of Nrf2 | (97) |
| Modulation of Cys151 | Prevention of ubiquitination and degradation of Nrf2, activation of Nrf2 | (389) | |
| Others | See section III | ||
| Sp1 | Oxidation of cysteines in 3 Zn-fingers | Inhibition of DNA binding | (508, 519) |
| TTF | Oxidation of cysteines in the Zn-finger domain, reversible by APE1/Ref-1 | Inhibition of DNA binding | (467, 475) |
| USF | Cys229 and Cys248 cross-linking by oxidants | Inhibition of DNA binding | (377) |
| Interaction of HMG-1 (high mobility group protein-1) with reduced USF | Enhancement of DNA binding | (310) |
ARNT, aryl hydrocarbon receptor nuclear translocator; ASK-1, apoptosis signal-regulating kinase-1; CBP, CREB-binding protein; Egr, early growth response; FOXO, Forkhead box O; GR=glucocorticoid receptor; Grx, glutaredoxin; GSK3β, glycogen synthase kinase-3β; GST, glutathione-S-transferase; HIF-1, hypoxia-inducible factor 1; HPH, HIF prolyl hydroxylase; IKK, IκB kinase; JNK, c-jun N-terminal kinase; Keap1, kelch-like ECH-associated protein-1; LC8, dynein light chain 8; MAPK, mitogen-activated protein kinase; NF-κB, nuclear transcription factor of bone marrow-derived lymphocytes; PKC, protein kinase C; PTP, protein tyrosine phosphatase; RTK, receptor tyrosine kinase; TRP14, thioredoxin-related protein-14; Trx/TrxR, thioredoxin/thioredoxin reductase; TTF, thyroid transcription factor; USF, upstream stimulatory factor.
II. Mechanistic Principles in Redox Regulation
A. Indispensable components of regulatory circuits
As in technology in general, a biological regulatory circuit needs a minimum set of elements to adapt the metabolic system to special requirements: a signal and a sensor to switch-on the adaptive process, a transducer, a modulator of sensitivity, an effector, and a switch-off device. As is common for biological processes, also the biochemistry of regulatory circuits is more complicated. At best in prokaryotes, simple versions resembling technical regulatory circuits appear to be realized (Fig. 1). With increasing complexity of the organism, the regulatory systems have to cross-talk with different compartments of the cell, with the entire organism, and its environment. They have no chance to operate in splendid isolation; moreover, the resting position is not an equilibrium but a snap shot of steady states of competing reactions within the metastable and open system that defines life. The complexity, thus created, has two important implications: (i) each regulatory step under consideration has to be kinetically competitive with a realm of competing reactions and (ii) the signal has to be specifically transduced to the effector despite possible side reactions and cross-talks between signaling cascades. As will become evident, these two aspects are particularly relevant in redox regulation when the technical terms of a circuit are to be translated into defined biochemical entities.
FIG. 1.

Scheme of transcriptional regulation and its implementation in bacterial redox control. (A) A minimalistic scheme of a regulatory circuit with the required elements: signaling molecule (black with white letters); sensor (white box); transducer(s) (light gray); and effector (typically the target gene). Termination reactions, which are subject to modulation, are indicated by reversed arrows. The labeling code is maintained in all following figures, if appropriate. (B) Demonstrates how this scheme is used in the OxyR regulation of prokaryotes. The signaling molecule is H2O2 targeting reduced OxyR as sensor. OxyR is first oxidized at Cys199 (with the oxidized sensor marked by a star) and stepwised further oxidized by H2O2. OxyR turns into a transducer (OxyRS8) and binds to its effector, the responsive element in the DNA, and allows the RNA polymerase (RNAP) to effect gene expression that removes the signal via enhanced alkyl hydroperoxide reductase (AhpC/AhpF) synthesis, thus terminating or modulating the sensing process. Prevention or termination of transduction is also achieved by reducing oxidized OxyR by glutaredoxin A (GrxA). The Grx system is modulated by glutathione (GSH) regeneration. In all eukaryotic systems additional transducers that are distinct from modified sensors are involved (see Fig. 2 and others).
Whereas, for example, the signal/sensor interaction in cytokine signaling, that is, binding of a peptide to its receptor, is unproblematic in respect to specificity, it is enigmatic how signaling by promiscuously reacting ROS or radicals complies with the specificity requirement of a meaningful redox regulation. The other problem is raised by the abundance of superoxide dismutases (SOD), heme-based peroxidases, GPxs, and Prxs, which eliminate most of the ROS at rates that are hard to beat. SODs dismutate O2•− with rate constants around 109 M−1 s−1 (251, 321); for peroxidases, the bimolecular rate constants k+1, which defines the oxidation of the reduced enzymes by ROOH in analogy to Eq. 1 or 4, are around 107 M−1 s−1 in case of heme enzymes (59); they range from 104 to 107 M−1 s−1 for Prxs (489) and GPx-type peroxidases (487) working with sulfur catalysis; they reach 108 M−1 s−1 for GPxs working with selenium catalysis (487), and rate constants beyond 107 M−1 s−1 were also reported for peroxynitrite reduction by Prxs (101, 489). Whatever the signaling molecule is, it should hit the sensor selectively despite competition with a realm of extremely fast enzymes.
B. Radicals or hydroperoxides as oxidant signals in redox signaling?
At least one radical, •NO, is an accepted signaling molecule. It reversibly binds to heme prosthetic groups in guanylate cyclases, thereby triggering a broad spectrum of physiological events (136, 196, 302, 331), but signaling by •NO is not commonly subsumed under the term redox signaling, as long as •NO is not previously transformed to peroxynitrite, which is an oxidant signaling molecule indeed but no longer a radical (112, 430). Like •NO, the superoxide radical anion O2•− has the potential to bind reversibly to heme (322), but evidence on regulatory consequence of this ability is scarce. Its affinity to iron complexes is, however, widely used for redox sensing by iron sulfur [Fe-S] clusters in bacteria. The transcription factor SoxR responds to O2•− with transcription of MnSOD and other protective proteins (351, 518) and its binuclear (247) cluster proved to be functionally essential for superoxide sensing (182), the mechanism being a univalent oxidation of the complexed iron by O2•− (82).
A recent thoughtful analysis by Forman et al. concluded that there is neither evidence nor likelihood that redox regulation is directly mediated by any of the fast reacting oxygen-centered or other radicals that may arise from chain reactions initiated by the primary physiological radicals (134). Their persuasive, if not convincing, argument is that aggressive radicals such as •OH, RO•, or halogen radicals derived from the myeloperoxidase reaction, which react with almost all kind of biomolecules at nearly diffusion-limited rate constants (∼2×1010 M−1 s−1), simply lack the ability to modify regulatory proteins with the mandatory selectivity. Instead, they consider H2O2 as the key oxidant signal in redox signaling, because it can selectively oxidize particularly sensitive SH groups.
This seemingly provocative article may benefit from minor amendments, but convinces in so far as very few of the protein modifications that have up to now been identified in ROS signaling disclose sequelae of direct reactions between radicals and target proteins. An exception seems to be the H2O2-dependent de-repression of the repressor PerR (50, 332). In these proteins iron-co-ordinated histidines are oxidized to oxo-histidines, which reminds of a Fenton chemistry-mediated hydroxylation. Accordingly, the •OH radical is considered the ultimately reacting ROS (82). It is, however, not a free •OH that is sensed by the repressor protein, but rather a crypto-OH (421, 527), that is, an •OH that is likely formed locally from an iron peroxo complex and targeted to its intimate environment, which is here the imidazole moiety of a histidine (274). More commonly, the modifications detected in oxidized sensors are easily explained by two-electron withdrawal from thiolates.
The most common one of these reactions is the oxidation of cysteine residues by H2O2 to sulfenic acids (Eq. 1) followed by disulfide formation with glutathione (GSH) or protein thiols (Eqs. 2 and 3). The same sequence of reactions is achieved by the primary products of lipoxygenases (LOX) or cyclo-oxygenases (COX), which are alkylhydroperoxides (ROOH) (Eq. 4), and also by peroxynitrite (ONOO−), which is spontaneously formed from •NO and O2•− at nearly diffusion-limited rates (Eqs. 5 and 6). The efficiencies of these compounds in oxidizing thiols differ, peroxynitrite being the most and H2O2 the least reactive one, whereas lipid hydroperoxides occupy interim positions. Also, the residue R in lipid hydroperoxides may prevent or facilitate the interaction with a particular protein thiol, thus lending some selectivity to these signaling molecules. The qualitative equivalence of H2O2, ROOH, and ONOO− in oxidizing protein thiols to sulfenic acids has, however, been amply demonstrated in the initial step of the catalytic cycles of Prx- and GPx-type peroxidases, which is the oxidation of the peroxidatic cysteine CP, as shown in Eqs. 1, 4, and 5 (105, 301, 379, 380, 487, 489). Analogous reactions of exposed and dissociated thiols in other proteins cannot reasonably be doubted, whereas a radical-initiated sulfhydryl oxidation would likely yield unspecific protein damage, if not terminated by the extremely unlikely event of meeting another thiyl radical to form a disulfide bond. In short, glutathionylation or intra- and intermolecular disulfide bond formation in regulatory proteins does not result from any free radical attack, but most likely from an electron pair transition from nonradical ROS such as H2O2, ROOH, or ONOO−. Hydroperoxides including ONOO− and not radicals themselves may, thus, be considered oxidant signals in ROS signaling, the sensing process being the oxidation of susceptible cysteine residues to cysteine sulfenic acids (123, 133, 134). The intimate downstream transduction step, the reaction of the sulfenic acid with another thiol according to Eqs. 2 and 3, respectively, is analogous to the reaction of the oxidized CP in thiol peroxidases with their resolving cysteine CR. This coreacting CR is typically not dissociated and not reactive enough to sense an ROOH by itself. However, it readily reacts with the sulfenic acid if this is sterically possible. Therefore, the analogous transduction of an oxidant signal, sensed as a sulfenic acid residue (Eq. 3), appears to be exclusively determined by sterical fit, which lends further specificity to the overall signaling process.
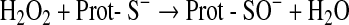 |
(1) |
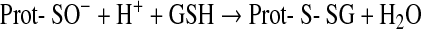 |
(2) |
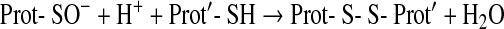 |
(3) |
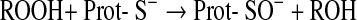 |
(4) |
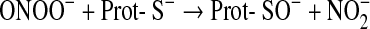 |
(5) |
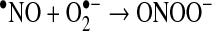 |
(6) |
Protein thiol modification as sensing mechanism may also be achieved by different ways (250). Theoretically, glutathionylation may result from thiol/disulfide exchange between oxidized glutathione (glutathione disulfide [GSSG]) and protein thiol (47, 48).
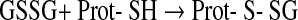 |
(7) |
However, the comparatively low cellular GSSG concentrations render this reaction less likely than the direct protein SH oxidation followed by reaction with GSH (Eqs. 1 and 2). Further, disulfides can be formed by nitroso glutathione (GSNO) and protein SH groups or, inversely, from S-nitrosylated proteins and GSH (Eqs. 8 and 9) (513).
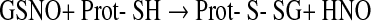 |
(8) |
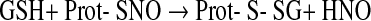 |
(9) |
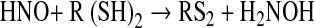 |
(10) |
Thereby, products derived from •NO and oxygen with the potential of S-nitrosation, the reactive nitrogen species (RNS) (112), have to be considered as possible signaling molecules, the most likely candidates being the nitroxyl cation NO+ and N2O3, whereas the nitroxyl HNO formed according to Eq. 8 might again contribute to disulfide formation (Eq. 10) (328). As in the case of ROS signaling, not the •NO radical itself is used as thiol modifier in RNS signaling but nonradical derivatives thereof.
Apart from O2•−, the only oxygen-centered radical that is directly used as signaling molecule appears to be molecular dioxygen itself, a bi-radical that, though, is hardly ever addressed as ROS (see section II.D.6).
C. Signals of free radical damage
The statement that free radicals are not ideal signaling molecules seemingly conflicts with the widely accepted view that the organism reacts to free radical formation and radical-driven processes with adaptive responses. As is evident from the previous section, the often used term “free radical signaling” can claim its justification from the fact that most of the oxidant signals so far identified are indeed derived from the two important natural radicals •NO and O2•−. Remains the question how the organism responds if these radicals are not channeled into smooth physiological pathways for, for example, hormone or growth factor signaling, but cause tissue damage due to unbalanced production in conditions that deserve the name oxidative stress or nitrosative stress. In pathological conditions such as septicemia or reperfusion injury or any other kind of fulminant inflammation, free radical-dependent tissue damage is obvious and it seems straight forward that the organism should somehow sense the abundance of radicals. Apparently, however, the radicals themselves are not sensed either under these extreme conditions. Instead, the outcome of the radical attack is sensed by a nonradical sensing mechanism.
Protein thiols identified as likely sensors in redox signaling offer chemical options beyond thiol oxidation and thiol/disulfide exchange. If dissociated, they are powerful nucleophiles and, thus, may react with a large variety of compounds with reactive double bonds such a α, β-unsaturated aldehydes or ketones and other Michael acceptors. This huge list of compounds inter alia comprises well-documented products of enzymatic or free-radical-driven lipid peroxidation, the most prominent examples being 4-hydroxy-nonenal (HNE) and 15-deoxy-Δ12,14-prostaglandin J2 (15d-PGJ2). Such compounds have been amply documented to alkylate particular protein thiols under oxidative or nitrosative stress and therefore may be implicated as stress signaling molecules that are sensed via S-alkylation. The best known example of a regulatory protein modified this way is kelch-like ECH-associated protein-1 (Keap1), which plays a pivotal role in responding to oxidative challenge with an adaptive response via activation of the transcription factor Nrf2 (6, 94–98, 254, 479, 481) (see section III), but analogous stress sensing has also been implicated in the NF-κB pathway (408) and in apoptosis (12).
D. Sensing and transducing proteins
As outlined, the main problem of redox signaling is seen in rendering specificity to oxidant signals. Since thiol oxidation and alkylation appear to be the prevailing sensing mechanisms in redox regulation, proteins with highly reactive thiols must be sensors of choice. Such thiols have to fulfill three requirements: they have to be surface exposed, dissociated, and kinetically competent to compete with peroxidases and, if S-alkylation is involved, also with glutathione-S-transferases (GSTs). Beyond, the resulting thiol modification must lead to a structural change of the sensor to allow specific signal transduction.
1. Thiol peroxidases as sensors
The rate constants for the oxidation of freely accessible SH groups in low–molecular-weight compounds by H2O2, even if extrapolated to full SH dissociation, do hardly exceed 50 M−1 s−1 (512) and, thus, fall short by orders of magnitude when compared to those of GPx- or Prx-type peroxidases (see section II.A). Protein-bound cysteines are by no means more reactive (31, 123, 134), unless they are embedded in a micro-architecture that facilitates cleavage of the hydroperoxy bond by polarization and proton shuttling as in the thiol peroxidases (378, 487). Evolution has designed these proteins for highly efficient hydroperoxide reduction. Accordingly, they do not only deserve interest as hydroperoxide-detoxifying enzymes, but also as ideal sensors for ROOH.
In recent years, a sensor/transducer function of peroxidases has indeed been elucidated in transcriptional regulation of yeasts (135). In Saccharomyces cerevisiae a GPx-type peroxidase Orp1 senses H2O2 in being oxidized to its sulfenic acid form, as in Eq. 1. The cysteine sulfenic acid residue of Orp1 then forms a disulfide bridge with a particular thiol of the transcription factor activating protein-1 (AP-1)-like transcription factor from yeast (Yap1), thereby directly transducing the oxidant signal (86) (Fig. 2). The physiological meaning of the H2O2 sensing by the peroxidase is seen in transducing the oxidant signal to a defined target protein with the specificity typical for protein/protein interactions (86). In another S. cerevisiae strain, the 2-Cys Prx Tsa1 appears to activate Yap1 in an analogous way (357, 407), and in Schizosaccharomyces pombe Tsa1 is the major peroxidase that reacts with the transcription factor Pap1, which is an homolog of Yap1 (333). Also, in S. pombe Tsa1, upon having sensed H2O2, forms a disulfide bridge with the stress kinase Sty1, thereby transducing the oxidant signal to the transcription factor Atf1 (333, 497).
FIG. 2.

Sensing and transducing the H2O2 signal by the Orp/AP1-like transcription factor from yeast (Yap) system in yeast. In this system the glutathione peroxidase (GPx)-type peroxidase Orp1 act as sensor for H2O2 and becomes oxidized at Cys36 to a sulfenic acid. The sulfenic acid then forms a heterodimeric disulfide bridge with Cys598 in Yap1, which in turn is resolved by two more thiol/disulfide exchange reactions (not shown), ultimately resulting in oxidized Yap1 (Yap1S2) and reduced Orp1. Oxidized Yap1 can bind to the respective enhancer elements in the promoter of target genes, which protect against oxidative challenge. This way Yap1 acts as the transducer of the H2O2 signal. An alternative reaction of Orp1 sulfenic acid leads to an intramolecular disulfide bridge (Orp1S2) that is reduced by thioredoxin (Trx). In this role of a Trx peroxidase Orp1 acts as a modulator of this system. Trx also terminates signaling by reducing Yap1S2.
A case of H2O2 sensing by a Prx has also been demonstrated in Kinetoplastida. In these unicellular parasites a universal minicircle sequence binding protein (UMCSBP), which is a Zn-finger protein, binds to the peculiar kinetoplast DNA in a Zn- and redox-dependent manner, thereby regulating replication [reviewed in ref. (452)]. DNA binding of UMCSBP is abrogated by H2O2, which is sensed by the Prx-type tryparedoxin peroxidase with rate constants that, depending on species, range from 105 to 107 M−1 s−1 (489). The peroxidase directly transduces the oxidation equivalents to CxxC motifs of the Zn-finger domain. The reductive reactivation of UMCSBP depends on the trypanothione redox cascade comprising trypanothione reductase, trypanothione, and tryparedoxin, the latter being a thioredoxin-related protein (TRP) specialized for protein disulfide reduction (349).
Analogous ROOH sensing by peroxidases in mammals have not yet been reported. However, the idea that at least some of the GPx- or Prx-type peroxidases could similarly act as sensors for H2O2, alkyl hydroperoxides, and peroxynitrite in mammals is intriguing in several respects. It would comply with the observation that redox signaling also takes place at physiological H2O2 fluxes; it could explain why in some cases Prxs improve rather than inhibit signaling (75, 238), and it finally might help to explain oxidative modification of proteins with SH group reactivities that are simply not competitive enough to occur without enzymatic support (134). Mechanistically, transducing mechanisms analogous to those worked out for peroxidases of yeasts and Kinetoplastida would be straight forward for all 2-Cys Prxs. Upon oxidation of their peroxidatic cysteine CP to a sulfenic acid, they can either form an internal disulfide bridge with their resolving cysteine CR, which in the ROOH removing function is then reduced by a redoxin, or an intermolecular disulfide bridge with another protein, thus acting as thiol-modifying agent with regulatory consequences. Examples of Prxs found disulfide-linked to other proteins are abundant (93, 187, 333), but also GPx-type peroxidases devoid of a CR can modify protein thiols, as has been demonstrated for mammalian GPx4. In shortage of GSH, the oxidized active site selenium (UP) can selenylate proteins, a process that so far has not been implicated in transcriptional regulation, but is pivotal to the differentiation of spermatids into spermatozoa (121, 318, 428, 493) (Fig. 3).
FIG. 3.

Possible routes of hydroperoxide sensing by thiols or selenols. (A) General reaction scheme of GPx-catalyzed reduction of hydroperoxides. The selenol is oxidized by ROOH to a selenenic acid, which is step-wise reduced back to selenol by two molecules GSH. A role of selenols in ROOH sensing has so far not been reported. (B) The selenenic acid in GPx4 has been shown to react not only with GSH but also with thiol groups of other proteins. The enzyme, thus, transduces a signal to another protein target and acts as a thiol modifying agent. This mechanism is the molecular basis for the unique transformation of GPx4 into a structural protein during sperm maturation (493). Theoretically, the reaction sequence could be used in redox signaling with GPx4 as sensor and Se-S linked heterodimers as transducers. The sensor-reduced GPx4 could be regenerated by GSH (as in A) or by SH groups of the same or another protein (lower line). (C) 2-Cys peroxiredoxins (Prxs) react with a hydroperoxide at the peroxidatic cysteine (SP) to the sulfenic acid. Oxidized SP can then form an intra- or intermolecular S-S bridge with the resolving cysteine (SR). This bridge is typically reduced by a redoxin (Trx, TXN, or Grx). The oxidized SP can, however, also react with another protein, thus acting as a thiol modifying agent, as demonstrated in the redox control of yeasts (333). (D) Certain cysteines in protein phosphatases (PPs) react with a hydroperoxide to form a sulfenic acid. This either reacts with a second cysteine to form an intramolecular disulfide bridge in analogy to (C). Alternatively (E), the sulfenic acid reacts with a nitrogen in the peptide bond to a sulfenyl amide. Both forms can be reduced by GSH and Grx. The oxidized enzymes are typically inhibited. As discussed in section II.D.3, the known reaction constants for the reaction of ROOH with thiols in phosphatases are low. Therefore, the possibility that the oxidative signal is first sensed by a GPx- or Prx-type peroxidase and then transduced to a PP, as shown in (B) or (C), respectively, merits consideration.
Prxs can, however, also sense ROOH and transduce the signal in a more indirect way, just by means of their conventional role in removing hydroperoxides. Most of the mammalian Prxs, as well as the nonmammalian 2-Cys Prxs and many nonmammalian GPxs, use Trx or related redoxins as reductants. Under high peroxide flux, they thereby reduce the steady-state of reduced redoxins that are often used as terminators of transcriptional gene activation or interrupters of signaling cascades. Inactivating reduction of OxyR (100) or Yap1 (86, 333) by glutaredoxin (Grx) or Trx, respectively, and reduction of GSH mixed disulfides by Grx (250, 292) may suffice as examples. Further, oxidation of a redoxin may promote signaling due to reversal of inhibition, as has first been shown for the apoptosis signal-regulating kinase-1 (ASK-1), which is blocked by noncovalently bound reduced Trx, but not by the oxidized one (415). Similar functions are ascribed to nucleoredoxin (Nrx) in the wingless and Int 1 (Wnt) pathway (139), and to the dynein light chain 8 (LC8)/Trx-like 14 protein couple in the NF-κB system (227) (see section IV and Fig. 4). Although metabolic regulation by Trx and redoxins, in general, has for long been a widely accepted concept (131), it appears to be still persistently overlooked that oxidation of redoxins in vivo does not likely occur spontaneously, but is catalyzed by Prxs, which came to stage much later (399). Although the pKa of the exposed cysteine in their CxxC motif is quite low, their rate constants for oxidation by ROOH is usually two orders of magnitude smaller than those of the most abundant Prxs that efficiently oxidize redoxins (489, 490). It therefore appears not too risky to consider most, if not all, regulatory events that are attributed to Trx oxidation to be downstream events of hydroperoxide sensing by a thiol peroxidase, a Prx being the more likely candidate in mammals (123, 134).
FIG. 4.

Redoxins in sensing mechanisms. (A) Reduced Trx has been demonstrated to inhibit apoptosis signaling by binding (and inhibiting) apoptosis signal-regulating kinase-1 (ASK-1). Oxidation of Trx relieves the block (415). Since redoxins are excellent substrates of Prxs, a hypothetical Prx is inserted as real sensor of the system as in (B–D). (B) Nucleoredoxin (Nrx) in the reduced state binds to dishevelled (Dvl), a phosphoprotein that transduces wingless and Int 1 (Wnt) signals from the Frizzled receptor. Upon oxidation, Nrx is released and Dvl can associate with the β-catenin degradadion complex, where it inhibits glycogen synthase kinase-3β (GSK3β) and phosphorylation, ubiquitination, and degradation of β-catenin. β-Catenin moves into the nucleus and activates genes required for proliferation (138). (C) Reduced Nrx associates with flightless-I (Fli-I) and myeloid differentiation factor 88 (MyD88), thereby preventing recruitment of MyD88 to the TLR4 after lipopolysaccharide stimulation (171). This way unnecessary stimulation is avoided. After removal of Nrx, MyD88 can be recruited, and the TLR mediate its signals. Although in this system oxidative processes have not yet been studied, the requirement for oxidants in TLR stimulation may be based in the need to oxidatively remove Nrx from the Fli-I/MyD88 complex, as in (B). (D) Reduced dynein light chain (LC8) binds to IκB and prevents its degradation. Upon oxidation to an intermolecular disulfide LC8 dissociates and gives the way free for IκB degradation and activation of nuclear transcription factor of bone marrow-derived lymphocytes (NF-κB). The oxidation is reversed by the Trx homolog thioredoxin-related protein-14 (TRP14), which is reduced by cytosolic thioredoxin reductase (TrxR1). In this system either LC9 itself is the sensor or is oxidized by an upstream sensing thiol peroxidase (224).
Finally, a role of Prxs in sensing ROOH may be seen in their ability to switch from a peroxidase to a chaperone, as was first demonstrated in yeast (217, 333) but has also been implicated for mammalian Prx1 (403). The chaperone function of Prxs itself appears to be independent from their peroxidase activity (403), as it prevails in the peroxidatically inactive high-molecular-weight forms having their CP oxidized to a sulfinic acid. The functional switch to the chaperone, however, is initiated via fast ROOH sensing by CP. Once transformed into a chaperone, the Prx may affect many regulatory pathways, enhanced cytokine expression through Toll-like receptor-4 (TLR4)-mediated NF-κB activation being a revealing example (403).
2. Transcription factors as sensors for hydroperoxides
Certainly, also proteins that are not annotated as peroxidases may sense ROOH through thiol oxidation, if their cysteine residues are activated by neighboring basic residues or other mechanisms. A well-documented example is the bacterial transcription factor OxyR (100). Its Cys199 may be considered kind of CP, as it is directly attacked by H2O2 with a bimolecular rate constant of ∼105 M−1 s−1 (11), which comes close to those of peroxidases. The initial oxidation of Cys199, like in the thiol peroxidases, is followed by a disulfide formation that facilitates oxidation of the remaining cysteines to the fully oxidized active transcription factor, whereas the inactivation is achieved by Grx-catalyzed reduction by GSH (Fig. 1).
Similarly, direct sensing of organic hydroperoxides has been implicated for organic hydroperoxide resistance repressor (OhrR), a repressor of the Ohr gene found in many bacteria (100). Although OhrR is phylogenetically unrelated to any of the peroxidase families, it shares the mechanism with atypical 2-cysteine Prxs (281): it is oxidized by ROOH (not by H2O2) at its CP, which is the arginine-coordinated Cys60 (Pseudomonas aeroginosa sequence). The oxidation of Cys60 to a sulfenic acid, which suffices for de-repression, leads to formation of an intramolecular disulfide bond with Cys124, which can be reduced by dithiothreitol, the natural reductant being unknown.
Any direct interaction of a hydroperoxide or any other oxidant signal with a mammalian transcription factor has so far not been convincingly demonstrated.
3. PPs and PKs as potential sensors
Phosphory-lation and de-phosphorylation are key events in the regulation of enzyme activity in cellular signaling and certainly also dominate mammalian transcription factor activation. In most of the cases, the phosphorylated kinases and phosphatases represent the active modifications, whereas de-phosphorylation leads to inactivation of these enzymes. Exceptions to the rule are, for instance, glycogen synthase (GS) and glycogen synthase kinase-3β (GSK3β), which are active in the de-phosphorylated state. Protein kinases and, more importantly, PPs are widely assumed to sense H2O2, other oxidants, or S-alkylants, and it may be guessed that most, if not all, phosphorylation/de-phosphorylation balances are redox-controlled via cysteine modification (Fig. 3D, E).
In receptor protein kinases, a highly conserved MxxCW motif is considered as the general switch that, upon cysteine oxidation, starts the tyrosine phosphorylation required for catalytic activity (339). Oxidative activation is also observed in the various forms of protein kinase C (PKC) that are characterized by Zn-fingers in their regulatory domain, in which two Zn++ ions are tetrahedrically coordinated with six conserved cysteine and two histidine residues (77, 194, 258). The Zn-finger is assumed to work as kind of hinge wherein the Zn++ functions as a linchpin. Oxidation of the Zn-coordinated cysteines was shown to cause Zn++ release (259) and is assumed to favor the active kinase conformation, phosphorylation, binding to Ca++, and phosphatidylserine and, thus, membrane recruitment of these enzymes (258, 309). Similarly, Zn++ release associated with activation of kinase activity due to oxidation of Zn-coordinated cysteines was reported for c-RAF (190). Since PKCs inter alia mediate oxidative responses such as the oxidative burst of phagocytes (see section IV.D.1), the inhibitory function of Zn++ in these enzymes may well explain the still mysterious antioxidant function of the ion, which by itself is redox-inert under physiological condition (49, 309, 382).
In contrast, PPs are more or less readily inhibited by cysteine oxidation/modification. PPs are specific for protein tyrosine phosphates, serine (Ser)/threonine (Thr) phosphates, or for both, and, accordingly, are classified into tyrosine PPs (protein tyrosine phosphatase [PTPs]), Ser/Thr PPs (PSPs), and dual specificity PPs (DSPs). Their mechanism of action differs and so does their sensitivity to oxidants.
PTPs are generally susceptible to oxidative inactivation. They are characterized by an HCx5R motif that comprises the PTP loop that binds the phosphate groups of phosphotyrosines [(472); reviewed in ref. (67)]. The cysteine in this motif, due to its low pKa value, is nucleophilic, which is a prerequisite for both substrate de-phosphorylation and oxidative inactivation. More than 100 PTPs of the human genome (486) also include the DSPs that share with the typical PTPs the characteristic HCx5R motif and may, therefore, be suspected to be equally prone to oxidative inactivation. The DSP family not only de-phosphorylates tyrosine, Ser, and Thr residues, but also further comprises phosphatases acting on nonprotein substrates, such as phosphoinositol phospholipids (PIPs). Examples for the latter specificity are the Src homology-2 (SH2)-domain-containing inositide phosphatases (SHIPs) (149), which de-phosphorylate the membrane-bound PI3K-generated key signaling lipid PI(3,4,5)P3 at position 5 to PI(3,4)P2, and the phosphatase and tensin homologue (PTEN), which dephosphorylates the 3 position of both PI(3,4,5)P3 and PI(3,4)P2 (304). Sharing the active site motif with the PTP/DSP family, PTEN is a typical example of a redox-sensitive phosphatase (280).
A particularly reactive cysteine being unidentified in PSPs, this type of phosphatase can not a priori be rated as redox sensitive. However, also Ser/Thr phosphatases such as PP2A have been shown to undergo reversible oxidation. PP2A contains 10 cysteine residues in the catalytic subunit including a vicinal pair at positions 266–269, which reminds of a redoxin motif. This CxxC motif proved to be sensitive to oxidation in vitro, and its oxidation resulted in a decreased activity (130). In addition, PSPs can have metal ions in their active center, which are essential for their enzymatic function (449). Thus, apart from redox modification of cysteine residues, also an oxidation of the metal clusters appears conceivable.
Mechanistically, the oxidative inactivation of phosphatases via cysteine oxidation involves reactions known from GPx or Prx mechanisms (127). A pivotal cysteine appears to be oxidized to a sulfenic acid (Eq. 1). This unstable oxidation form typically forms a disulfide with another cysteine residue (Eq. 3), which reminds of the analogous reaction of CP and CR in Prx or GPx catalysis (Fig. 3). In oxidized PTEN a disulfide bridge between the catalytic Cys124 and the neighboring Cys71 was detected (275, 418). Similarly, in the DSP Cdc25 phosphatase B, a second cysteine resides in the active site, which in the oxidized form is disulfide-linked to the nucleophilic cysteine of the signature motif (51). In contrast, in PTP1B, the cysteine in the active site motif [I/V]HCxxGxxR[S/T] forms a cyclic sulfenyl-amide (419, 495) in analogy to the redox cycle of the GPx mimic ebselen (423) (Fig. 3E). As in the catalytic cycles of Prxs (378) and GPxs (487), the physiological meaning of conserving the labile oxidation state of the sulfenic acid in a more stable form is seen in the prevention of sulfur oxidation to the sulfinic or sulfonic forms that would be hard to reverse. As in the enzymatic cycles, the oxidized phosphatases are readily reactivated by physiological thiol compounds such as GSH, Trx, Grx, or other redoxins.
However, the almost generally agreed assumption that protein kinases and phosphatases may sense oxidants via cysteine oxidation is overshadowed by lack of any confirming kinetic data. None has been reported for any kinase and the few available rate constants for a direct oxidation of the critical thiols in PPs by H2O2 do not exceed 50 M−1 s−1 (31, 87, 459) and, thus, are by far too small to corroborate any physiological relevance of the process. For three redox-sensitive PTPs (PTP1, leukocyte-common antigen-related (phosphatase) [LAR], and Vaccinia H1-related (phosphatase) [VHR]) reaction rates for oxidative inactivation were insufficient (10–20 M−1 s−1); for three Ser/Thr phosphatases (PP2Cα, calcineurin, and lambda phosphatase) they were evidently too slow to be quantified (87). For the PTP-type Cdc25 phosphatases the rate was a bit more promising, being 15-fold higher than for PTP1B (459). In the latter case, the authors stress that the reaction of H2O2 with Cdc25 is 400 times faster than with GSH, which though is a poor argument, since the spontaneous reaction of GSH with H2O2 is physiologically irrelevant. For the relevant GPx reaction the rate constant is around 108 M−1 s−1.
According to Forman et al. (134), the available kinetic data predict that even the nonenzymatic reaction of H2O2 with GSH would completely compete out the oxidation of the active site cysteine of PPs. However, since oxidation of PTP1B and other PPs, as evidenced by glutathionylation (Eqs. 1 and 2), evidently occurs under in vivo conditions (250, 405), oxidant sensing by PPs must be more complex. It could be envisaged that any of the numerous thiol peroxidases first senses ROOH and then, in analogy to the yeast GPx- and Prx-type peroxidases, forms a disulfide bridge with the target phosphatase that is cleaved by GSH, leaving the glutathionyl residue at the target protein. Grx has been implicated in protein glutathionylation but appears to rather de-glutathionylate (229). Alternatively, the rate constants, which were obtained in vitro with isolated enzymes, could be grossly misleading, as they might in vivo be substantially improved by protein/protein interaction within regulatory complexes. Human Prx6 may be taken as an example for a dramatic change in thiol reactivity due to association with another protein: it displays a significant GPx activity only when associated with a GSH-loaded GSTπ (439). Before the discrepancies between in vitro data and in vivo observations have not been convincingly explained, we should cautiously address redox-sensitive PPs and kinases as putative ROOH sensors.
4. Redox sensing by cytosolic inhibitory complexes of transcription factors
In eukaryotes, transcription factors are commonly sequestered in the cytosol in form of multicomponent complexes from which they have to be released for gene activation in the nucleus. The activation of the transcription factor requires modification of one or more components of the complex to enable release of the transcription factor and its nuclear import. Almost regularly, a redox modification of a component is followed by ubiquitination and proteasomal degradation of the same or another component (183). A typical example of this reaction scheme is the stress-signaling Nrf2/Keap1 system, wherein Nrf2 is the transcription factor and Keap1 the complex partner that prevents Nrf2 activation (see section III for details). In this particular case, the inhibitory protein Keap1 not only prevents transcription factor activity by keeping it in the cytosol, but also by targeting it for the proteasomal degradation pathway (254, 531).
Keap1, which again is a Zn-finger protein, is considered to be the redox sensor of the system. Its reactive cysteines may be oxidized to sulfenic acids, form disulfides, or be alkylated by electrophiles (Michael acceptors) such as HNE (201), other α, β-unsaturated aldehydes, or ketones (282, 410), a realm of phytochemicals (469) or other electrophilic xenobiotics (104). The consequence of cysteine modification in Keap1 is a dramatic conformational change resulting in a destabilization of the complex and prevention of Nrf2 degradation. As generally observed in Zn-finger proteins (see section II.D.3), oxidation of Zn-coordinated cysteines leads to release of Zinc and consecutive unfolding of the finger structure. The structural change of Keap1 then prevents ubiquitination and proteasomal degradation (see section III.C.2).
The ubiquitination process proceeds via the conventional scheme: a coordinated cascade of 3 enzymes, the ubiquitin-activating enzymes E1, the ubiquitin-conjugating enzymes E2, and the ubiquitin ligases E3 (Fig. 5). Of the known E3 ligases, the really interesting new gene (RING) family is the largest. The RING motif in these enzymes interacts with E2 and facilitates the transfer of ubiquitin from E2 to a lysine in the target protein (370). One subtype of the RING family is the Cullin family. Cullin-RING E3 ligases utilize 1–7 Cullin scaffolds to assemble several substrate-specific adaptors that recognize and position the target, here Nrf2, in the cullin-E3 complex for proper ubiquitination. The unmodified Keap1 serves as an adapter for the RING box protein (Rbx1)-bound Cullin-3 (Cul3)-based E3 ligase, which targets the Nrf2 within the Keap1/Nrf2 complex (80, 140, 141, 255, 372, 532). Cysteine modification in Keap1 disrupts the presentation of Nrf2 for ubiquitination.
FIG. 5.

Simplified scheme of ubiquitination of substrates in mammalian cells. In a first step ubiquitin is activated at the C-terminal glycine by the ATP-dependent formation of a thioester at a cysteine of the E1 enzyme. E1 transfers ubiquitin to a cysteine of the ubiquitin-conjugating enzyme E2 and is released from the complex. The ubiquitin-loaded E2 then forms a complex with the E3 enzyme to which the respective substrate (S) is bound. The really interesting new gene (RING) box protein-1 (Rbx1) targets the substrate for ubiquitination. Ubiquitin can then be passed either directly to a lysine of the RING-bearing E3-bound substrate (Rbx1) or, in case of the homologous to E6AP C terminus (HECT)-domain bearing E3, to another cysteine on E3 and from there to the substrate lysine (186).
The ubiquitination machinery, here briefly introduced as a downstream event of an oxidative Keap1 modification, has also been rated as redox sensitive. Both the ubiquitin-activating enzyme E1 and the ubiqutin-conjugating E2 were shown to be reversibly inactivated by glutathionylation due to treatment with H2O2 or diamide (212, 355). The ubiquitin ligases, of which about 600 different ones were identified in man, are mostly characterized by a RING finger domain, in which two Zn++ ions are coordinated with seven cysteine and one histidine residues. Clearly, this structural element, like the Zn-finger of PKCs and Keap1, is suggestive of a site for redox regulation, an option that, however, has so far been left unexplored (88). Similarly, the redox regulation of SUMOylation, the analogous protein modification by small ubiquitin-like modifiers (SUMO), appears not to have been investigated to any significant extend (511).
Ubiquitination followed by degradation is also used for de-inhibition of transcription factors in other redox-sensitive cytosolic complexes, but the sensing step and the ubiquitinated entities as well as the regulatory principles of the systems differ substantially (Fig. 6). In the hypoxia response system it is also the transcription factor itself that is permanently ubiquitinated and degraded under normal conditions, that is, under normoxia. The redox sensor, however, is not an inhibitory protein such as Keap1 (Fig. 6A), but an enzyme: the proline hydroxylase that hydroxylates the transcription factor HIF-1α and targets it for ubiquitination (Fig. 6B). Similarly, the transcription factor β-catenin in the Wnt pathway is steadily ubiquitinated and degraded under resting conditions. In this context Nrx is assumed to be the redox sensor; in the reduced state it blocks the Wnt pathway by binding to the upstream adapter protein disheveled (Dvl), while allowing nuclear import of β-catenin upon oxidation of its CxxC motif (138, 139) (Fig. 6C). In the canonical NF-κB system, the inhibitory NF-κB-binding protein IκB has to be ubiquitinated and degraded to allow nuclear import of p50/p65. IκB is targeted for ubiquitinating by phosphorylation through IκB kinase (IKK) (Fig. 6D). The redox sensor of the system might be the LC8, which in its reduced states, is bound to IκB and protects IκB from attack by IKK, but allows IκB phosphorylation and degradation upon oxidative dimerization via intersubunit disulfide formation (Fig. 4D and section IV.D.3) (227). For lipopolysaccharide (LPS)-induced NF-κB activation via TLR 4, again Nrx has been implicated as negatively regulating sensor by binding to Fli-1 (flightless), thereby preventing recruitment of the essential adaptor myeloid differentiation factor 88 (MyD88) to the receptor (171).
FIG. 6.

Distinct roles of redox-dependent ubiquitination in the regulation of transcription factor activity. (A) In the Nrf2/kelch-like ECH-associated protein-1 (Keap1) system, oxidant sensing by Keap1 terminates the ubiquintination and degradation of Nrf2 and allows its nuclear translocation and transcriptional activation of target genes. (B) In the hypoxia-inducible factor 1 (HIF) system, under normoxic conditions HIF-1α is hydroxylated by HIF prolyl hydroxylase (HPH), ubiquitinated, and degraded. In hypoxia HPH is inactive, ubiquitination is prevented, and HIF-1α can translocate into the nucleus and activate gene expression. (C) In the Wnt/β-catenin pathway, GSK3β phosphorylates β-catenin, thereby facilitating its ubiquitination and degradation and preventing β-catenin-mediated gene expression. Dvl is captured by Nrx. Upon oxidation of Nrx, Dvl is released to inhibit GSK3β activity. β-Catenin is no longer phosphorylated and is translocated into the nucleus. (D) In the NF-κB system, LC8 is associated to IκB, thereby preventing its degradation and release of NF-κB. Nuclear translocation of NF-κB can take place after oxidative modification and release of LC8 and degradation of the inhibitor IκB.
It is needless to state that kinetic data for component interactions in these complex systems do not exist at all and will hard to be obtained. The mentioned redox-sensitive proteins, which, in principle, can directly be oxidized by any ROOH, might therefore not be the real sensors, but, instead, be oxidized by an upstream thiol peroxidase sensing the oxidant.
5. Sensing by selenocysteine-containing proteins?
A sensor's competence to sense ROOH or an alkylant selectively would certainly be enhanced if its sensing sulfur were replaced by the super-sulfur selenium (120). Many selenoproteins display signatures of redox proteins (146) and most of those with an established enzymatic function are indeed oxidoreductases with selenocysteine (Sec) as pivotal catalytic entity (122). As a rule, these seleno-enzymes are substantially faster than their homologs working with sulfur catalysis (33). Simply by chemical reasoning, therefore, selenoproteins would have an optimum chance to beat thiol-based redox sensors in terms of speed and specificity.
Circumstantial evidence has for long suggested that selenoproteins are relevant to transcriptional activation. In selenium-deficient animals a huge number of nonselenoproteins, now overwhelmingly known as Nrf2 targets, were found elevated since the late seventies (78, 79, 270, 395–397). The unexpected phenomenon could not be convincingly explained by GPx1 deficiency alone (396). However, at least one of the selenoproteins had to be involved in Nrf2 activation, since a complete loss of all selenoproteins by an organ-specific removal of the gene encoding selenocysteine tRNA (Trsp) (56) resulted in the induction of GST isoforms in liver, of GSTP1 and NADPH quinone oxidoreductase 1 (NQO1) in liver and macrophages (471) and of heme oxygenase-1 (HO-1) in liver (446). Also a moderate selenium deficiency in which only selenoprotein W, GPx1, and selenoprotein H and M were markedly decreased (249) led to an increase in NQO1, GSTs, sulfotransferases, and UDP-glucuronyl transferases (UGT) as well as HO-1, Prx1, sulfiredoxin-1 (Srx1), and γ-glutamyl-cysteine synthase (335). A firm link between selenium deficiency and Nrf2 activation was finally established by Burk et al. by demonstrating a strong increase in electrophile-responsive element (EpRE)-driven reporter gene activity in livers of selenium-deficient wild-type but not in Nrf2−/− mice (52).
Certainly, these findings do not strongly corroborate a role of selenoproteins as sensors, as they may be equally explained as adaptive response to impaired hydroperoxide metabolism. The hypothesis of a direct sensing function has not yet been established for any of the selenoproteins, but remains attractive from a chemical point of view. H2O2-dependent selenylation of protein thiols by GPx4 (see section II.D.1) is mechanistically equivalent to H2O2 sensing by thiol peroxidases and reveals that selenoproteins can in principle act as combined redox sensors and transducers (121, 123). After all, the mammalian Trx reductases, which all are selenoproteins, may be indirectly involved in ROOH sensing by regenerating Trxs as key redox regulators and reducing substrates of Prxs. These aspects certainly deserve more interest when searching for the biological roles for the more than 25 distinct mammalian selenoproteins (167).
6. Oxygen sensing
[Fe-S] clusters in proteins have for long been known to be susceptible to oxidation (314) and may therefore be considered bona fide sensors for oxidants. In bacteria [Fe-S]-cluster proteins have not only been reported to sense O2•− (see section II.B), but also molecular oxygen. The transcription factor FNR (for fumarate nitrate reduction), which is responsible for the adaptation to oxygen restriction in bacteria, directly reacts with O2, resulting in loss of transcriptional activity. Its [4Fe-4S]2+ cluster is thereby transformed into a [2Fe-2S]2+ cluster (247). Mammalian [Fe-S] proteins also respond to oxidative stress, the moonlighting of aconitase to the transcription factor iron response element-binding protein 1 (IRP1) being a prominent example (74, 334).
Beyond, any of the numerous oxidoreductases using O2 as acceptor (all EC numbers 1.1.3–1.10.3 having three in the third position as well as the oxygenase group EC 1.13) may be considered to be oxygen sensors, if their respective products have regulatory functions. Of particular importance in our context are the NADPH oxidases (NOX) that produce O2•− (see section IV.D.1), and the prolylhydroxylases that hydroxylate proline residues in the HIF (383, 443). In the hypoxia response, O2 serves as substrate of prolylhydroxylases that hydroxylate specific proline residues in the α-subunits of HIF depending on O2 pressure, thereby priming HIF for ubiquitination and proteasomal degradation (442).
E. Adjusting sensitivity by competing systems
As mentioned above, redox signaling triggered or modulated by hydroperoxides has to compete with peroxidases that reduce ROOH. In fact, it appears enigmatic how thiol oxidation in regulatory proteins can at all occur in the presence of peroxidases. The most commonly heard explanation is that all redox regulations must be events restricted to cellular microenvironments, which are hard to describe in kinetic or thermodynamic terms, an argument that is also used to explain regulatory phenomena in general (432). However, genetic tools have meanwhile revealed that redox regulation is also linked to, and controlled by, the overall cellular hydroperoxide metabolism. As a rule, suppression of Prx expression or Prx inhibition facilitates transduction through phosphorylation cascades, whereas overexpression of Prx dampens it, as compiled by Sue Goo Rhee and others (161, 198, 218, 236, 400). Similarly, overexpression of GPx1 in tissue culture inhibits tumor necrosis factor α (TNFα)-induced NF-κB activation by affecting the phosphorylation state of IκB (261). Also, overexpression of GPx4 abrogated lipid hydroperoxide- and interleukin-1 (IL-1)-induced NF-κB activation (45, 46). Most remarkably, a mouse systemically overexpressing GPx1 became insulin-resistant (277). Although these observations are open to different interpretations (218), they collectively suggest that peroxidases efficiently compete for ROOH that is needed in signaling cascades, thereby determining their sensitivity or setting the threshold at which they respond. An extreme version of this view is the flood gate theory, which assumes that hydroperoxide scavenging by Prxs has to be overcome by reversible oxidative inactivation of the latter to save ROOH for signaling (516). However, the flood gate can be opened not only by over-oxidation of Prxs but also by phosphorylation of the latter: Prx1 activity is inhibited by phosphorylation at Thr90 by cyclin-dependent kinases (CDKs), saving H2O2 to signal cell cycle progression (60) or at Tyr194 by a Src kinase within a growth factor receptor complex, thus enhancing platelet-derived growth factor (PDGF)- or epidermal growth factor (EGF)-triggered signaling due to local increase of H2O2 (514).
When being alkylated, the thiols of regulatory proteins also have to face competition. In mammals, seven classes of GSTs have been identified that efficiently compete with thiol groups in regulatory proteins for endogenous stress signals such as HNE or structurally related xenobiotics (175). Although the alkylation-susceptible SH groups in some regulatory proteins have been identified (256), the kinetics of interaction with alkylants is poorly investigated. The rate constants for the reaction of GSH with various Michael acceptors range between 1 and 350 M−1 s−1 (410) and may be higher by a factor up to 100 for a fully dissociated thiol. Nevertheless, the GSTs, which in toto can make up 5% the cellular protein, represent a substantial hurdle, which has to be overcome before S-alkylation of regulatory proteins can come into play. Accordingly, damage signaling via S-alkylation is dampened by GSTs and facilitated by localized or general GSH depletion (175), as it may happen in severe oxidative stress. The importance of GSTs in counteracting stress signaling through S-alkylation has been corroborated by inverse genetics. As compiled by Hayes et al. (175), knockout of several of the GSTs results in exaggerated response to xenobiotics and oxidative stress. Inversely, a conditioned knockout of the selenocysteyl-t-RNA gene, which disrupts the entire selenium-dependent antioxidant defense, leads to a compensatory increase of GST alpha, mu, and theta, which is likely mediated by activation of the Nrf2/Keap1 system due to increased H2O2, ROOH, and/or alkylating damage signals (see section II.D.5).
F. The problems beyond signals and sensors
Trying to present generally applicable concepts as to how proteins with regulatory potential may work in a complex eukaryotic system is an almost hopeless adventure. There is not any rule emerging without exceptions. As already evident from previous sections, peroxidases may act as sensors, transducers, or competitive modulators. Reduced Trx may act as inhibitor of signaling cascades by noncovalent binding as in the ASK-1 example (415), more commonly as terminator by reversing transcription factor binding to its target DNA as in the Orp1/Yap1 system (86) or as essential enhancer of transcription factor activity as in the NF-κB system. Grx is believed to play a key role in dampening phosphorylation by de-glutathionylation of counteracting phosphatases, but depending on the glutathionylated protein and the context may adapt an opposite role. Ubiquitination and proteasomal degradation facilitate import of transcription factors, if they attack their cytosolic inhibitors, but inhibit translocation, if the factor itself is degraded. We therefore refer to the following sections, which elaborate on two prototypic mammalian systems of transcription with focus on putative or established roles of redox-responsive elements.
III. Orchestrating the Adaptive Response by the Keap1/Nrf2 System
A. Discovery
Nrf2, the nuclear factor erythroid 2 (NF-E2)-related transcription factor, is a member of the Cap ‘n’ Collar (CNC) family of basic leucine zipper (bZIP) proteins and was first described in 1994 by Moi et al. as stimulator of β-globin gene expression (329). It was recognized to transduce gene activation of phase II enzymes by binding to the α, β-naphtoflavone-responsive element (210), previously described by Rushmore and Picket and called antioxidant-responsive element (ARE), because it was also activated by phenolic antioxidants such as t-butyl hydroquinone (411). A similar set of antioxidants were shown to also induce γ-glutamyl-cysteine synthetase (40, 294) and the pertinent responsive element was termed EpRE for electrophile-responsive element (330), which is identical with ARE. After it had become clear that the name-giving antioxidants in vivo acted as oxidants due to redox cycling or, as their oxidized counterparts, as electrophiles, the misnomer ARE should better be replaced by EpRE, but still persists in the literature.
B. The physiological context of Nrf2-dependent gene activation
Nrf2, via activation of EpRE (ARE), regulates the expression of proteins that collectively favor cell survival. They comprise enzymes that directly or indirectly exert antioxidant functions (5, 21, 111, 202, 264, 373, 491), molecular chaperons (265), and proteins that enhance GSH synthesis and regeneration (164, 174, 210, 276, 330, 336), phase 2 detoxication, drug metabolism (169, 210, 326, 392, 424, 479), recognition, repair, and removal of damaged proteins (191, 264, 265, 393), and nucleotide excision repair (8), further proteins that regulate the expression of other transcription factors, growth factors, and receptors (113, 163), and inhibit cytokine-mediated inflammation (6, 209, 245, 385, 503) and autophagy (394). With its broad range of target genes, Nrf2 is certainly one of the most important transcription factors that protect the organism against exogenous stressors, be they poisonous food ingredients, physical damage, or infection. In line with the appreciation of Nrf2 as dominant inducer of the adaptive response, the realm of its inducers comprises endogenous signaling molecules associated with oxidative stress such as H2O2, ROOH, ONOO−, oxo-aldehydes, and ketones, as well as exogenous ones such as isothiocyanates, thiocarbamates, trivalent arsenicals, quinones, dithiolethiones, vicinal dimercaptanes, certain statins, and heavy metals (96, 97, 160, 460, 474). They are obviously recognized by the organism to be potentially hazardous and to be eliminated via enforced Nrf2-mediated gene activation. The only common denominator of these chemically heterogeneous compounds is their ability to modify cysteine residues. With these characteristics, the Nrf2 system builds the major molecular basis for an enigmatic toxicological phenomenon known as hormesis: it describes that subtoxic dosages of a poison may protect against toxic ones or even against other challenges (180, 305, 317). Thus, with a delay of some centuries the therapeutic wisdom of the ancient Philippus Theophrastus Bombastus von Hohenheim (1493–1541), who claimed that solum dosis facit venenum (the dose only makes the poison), has been corroborated by a scientific basis.
The protective role of Nrf2 is also demonstrated by inverse genetics. Nrf2−/− mice are vital and do not display any signs of an increased basal oxidation state (58). Only when challenged they have an impaired ability to respond (454). The Nrf2 system, thus, is an emergency device that comes into play upon severe challenge only. For instance, mice lacking Nrf2, when treated with dextran sulfate sodium, developed a more severe intestinal inflammation with increased aberrant crypts than controls (246, 360), which suggests a function of Nrf2 in prevention of inflammation and inflammation-mediated carcinogenesis. Activation of Nrf2 is therefore considered to contribute to phenomena such as development of tolerance against all kind of inflammatory stimuli and, accordingly, potent activators such as sulforaphane, by acting as kind of vaccine, hold great promise in chemoprevention.
However, also this coin has two sites, since Nrf2 activation may not be beneficial under all circumstances. Upregulation of enzymes metabolizing xenobiotics such as GSTs will not always improve detoxification, since these enzymes may also increase xenobiotics' toxicity, as reviewed by Hayes et al. (175). Also, if the xenobiotic is a drug, Nrf2 activation may negatively affect its bioavailability. Moreover, not only normal but also tumor cells may benefit from the protective function of Nrf2. In fact, tumor cells usually show a constitutively high Nrf2 activity; hence, given the physiological result thereof, that is, inhibition of autophagy and apoptosis and increase of proteasomal degradation of damaged proteins, tumors have acquired a superior survival chance. Accordingly, activation of Nrf2 by chemo-preventive dietary compounds can reasonably be expected to inhibit tumor initiation but is likely detrimental, once the tumor has been established (10, 268). In support of this view, a selenoprotein that is an Nrf2 target, GPx2 (21), impairs inflammation-driven intestinal carcinogenesis, as convincingly demonstrated in knockout mice (109), but, as xenografts, wild-type tumor cells grew up substantially faster with than those without GPx2 (22). Finally, a persistent overactivation of the Nrf2 system, as is observed in autophagy-deficient (Atg7 knockout) mice, causes, or contributes to, severe liver damage, as evidenced by abrogation of the liver pathology in Atg7/Nrf2 double knockout mice (257).
C. Mechanistic aspects of Nrf2 signaling
1. Basics of Nrf2 activation
As already mentioned under section II.D.4, Nrf2 belongs to the type of transcription factors that are kept in cytosolic complexes under resting conditions and is there permanently subjected to proteasomal degradation (344). Within the cytosolic complex Keap1 serves as bona fide sensor for oxidants or electrophiles. Modification and conformational change of Keap1 allows translocation of Nrf2 to the nucleus for transduction. The molecular details of Nrf2 and the pertinent signaling pathway have been summarized in many reviews (42, 104, 287, 348, 469) and will here be briefly recalled and amended as far as appropriate.
2. Keap1 as primary redox sensor of Nrf2 signaling
Keap1, sometimes also called INrf2 for inhibitor of Nrf2 (348), contains three characteristic domains, the broad complex/tramtrac/bric-a-brac (BTB) domain, the intervening region (IVR), and the kelch domain, also known as double glycine repeat (DGR) domain (3). The BTB domain binds Cul3 (see section II.D.4) (141) and is required for homodimerization (538), whereas the IVR contains cysteine residues that are in part Zn-coordinated (98) and are considered relevant to regulation (see below) and a nuclear export signal (NES) (498). The kelch/DGR domain is required for the interaction of Keap1 with the actin cytoskeleton (234, 235) and/or myosin VIIa (499), which immobilizes Keap1 in the cytoplasm. The kelch/DGR domain is also essential for binding of Nrf2, which interacts with Keap1 with its amino-terminal Nrf2-ECH homology-2 (Neh2) domain (208).
The structure of the kelch/DGR domain of Keap1 has recently been resolved (297, 362), which sheds new light on the Keap1/Nrf2 interaction (484) (Fig. 7). According to the hinge and latch model there are two distinct binding motifs in the Neh2 domain of Nrf2, each binding one molecule of a Keap1 homodimer (two-sites binding) (325, 484). The high affinity binding ETGE motif functions as a hinge to pin down Nrf2 to Keap1. The low affinity DLG motif functions as a latch. The link between the hinge and the latch motif is the lysine-rich central α-helix of Nrf2. In this helix six of seven lysine residues point to the same site (484). The Neh2 domain is locked in a position suitable for ubiquitination of the Lys residues (325, 485) which, hence, act as ubiquitin acceptors (532). The hinge and latch model (484), thus, complies with a substrate-presentation mechanism: presentation of Nrf2 for ubiquitination and subsequent proteasomal degradation.
FIG. 7.

Model for redox-regulated Nrf2 activation based on conformational change of Keap1. In the resting state (A), Keap1 forms a homodimer via the broad complex/tramtrac/bric-a-brac (BTB) domains and associates to Nrf2 with its kelch domains. The Nrf2-ECH homology-2 (Neh2) domain of Nrf2 contains two Keap1 binding motifs with different affinity to the kelch domains, with ETGE representing the amino acid sequence with the higher affinity and DLG, the sequence with a much lower affinity. This two-site binding locks the central α-helix of the Neh2 domain of Nrf2 with is seven lysine residues (7 Lys) into a position suitable for ubiquitination. The intervening regions (IVR) contain the most reactive cysteine residues (Cys273 and 288) and build the adapter region for the Cullin-3 (Cul3)-based E3 ligase system. Nrf2 is polyubiquitinated and degraded by the proteasome. Under stressed conditions (B), oxidative/electrophilic modification of Cys272 and 288 induces a conformational change of Keap1 resulting in the dissociation from the weaker binding DLG motif in Nrf2, whereas binding to the ETGE motif remains [hinge and latch model as proposed by (484)]. P21 interacts with the DLG motif and supports dissociation of Nrf2 from this site. In contrast, modification of Cys151 in the BTB domain does not change the conformation of Keap1 but rather disrupts BTB Cul3 interaction. By both modifications ubiquitin ligase activity is lost and newly synthesized and released Nrf2 can move into the nucleus.
The hinge and latch model can also explain how thiol-modification disturbs the presentation of Nrf2 for ubiquitination. Murine and rat Keap1 contain 25 cysteine residues, whereas human Keap1 has 27 (97, 98). Nine of these are predicted to be reactive due to their location adjacent to basic amino acids. The cysteines 257, 273, 288, and 297 lie in the IVR and, by MS analysis of tryptic peptides, were demonstrated to be most sensitive to alkylation by dexamethason mesylate in vitro (97). Further, optical monitoring of the reaction of Keap1 with dipyridyl disulfide revealed a sequential reaction of the cysteine residues, likely starting with an S-thiylation of the most reactive one and followed by thiol/disulfide exchange to finally form internal disulfides within Keap1. Competition experiments with a representative set of Nrf2 activators demonstrated that Keap1 modification capacity of these heterogeneous compounds roughly correlated with their in vivo activity (97). By these and many other thiol modification studies (256, 287) the hypothesis of Keap1 being the redox sensor of the Nrf2 system was corroborated. In vitro and in vivo mutation studies finally revealed that Cys273 and 288, which are the Zn-coordinated ones, are crucial for the response of Nrf2 to activators (282, 504, 531), whereas mutation of Cys151 mediates stressor-induced dissociation of Keap1 from Cul3 (389, 521) (Fig. 7).
However, the original hypothesis that the primary sensing event, that is, modification of critical cysteine residues, leads to instant release of Nrf2 has been revised: the hinge and latch model predicts that thiol modification of Keap1 changes its conformation in a way that only binding of Nrf2 through its DLG motif, the latch, is reversed, whereas Nrf2 remains attached to Keap1 by its hinge, the ETGE motif. Thereby, the presentation of Nrf2 for ubiquitination is impaired. Accordingly, the unmodified critical cysteines 273 and 288 are crucial for ubiquitination of Nrf2 (531) and the degradation of Nrf2 in the unstressed situation (254). Upon challenge, the still hinge-bound Nrf2 is stabilized by preventing ubiquitination and may finally be released by downstream events (see section III.C.3). Moreover, continuously newly synthesized Nrf2 can bypass the still Nrf2-loaded Keap1 for transduction (254).
How thiol modification leads to the conformational change is likely explained by the zinc finger nature of Keap1 (95). Zinc is bound to Keap1 with an impressive Kass of 1011 M−1. Zn binding to Keap1 evidently involves the critical cysteines, as thiylation of the latter by dipyridyl disulfide is accompanied by a stoichiometric release of Zn; by a Cys to Ala mutation, the Kass is dramatically decreased, and profound conformational changes are associated with Zn release, as evidenced by shifts in tryptophan fluorescence or depression of fluorescence of a hydrophobicity probe. The Zn-stabilized structure of Keap1, thus, is disturbed by derivatization of Zn-binding cysteines and equally by Zn chelation or Zn shortage, as has been similarly discussed for other redox-sensitive zinc proteins (82, 258, 309). However, it can also be envisaged that cysteine-coordinated Zn in Keap1 contributes to the pronounced reactivity of the cysteines, thereby facilitating Nrf2 activation. The pivotal role of zinc in Keap1 function might provide another lead to explain its antioxidant effect (49) (see also section II.D.3).
Finally, the realm of Nrf2 activators raises the question how their chemical heterogeneity complies with the expectation that a sensor should sense specifically. Emerging evidence indeed reveals that groups of activators act on different sets of cysteines in Keap1, which may be translated into a specific biological effect. From these observations the term cysteine code was created and breaking this code for each Nrf2 activator will help to understand the various effects resulting from Nrf2 activation (256, 521).
3. Downstream signaling events
The essence of the events following sensing by Keap1 is that free cytosolic Nrf2 has to reach its target EpRE in the nucleus. The nuclear transport of Nrf2 is mediated by the conventional set of nuclear importin and exportin proteins (156). Typical nuclear localization signals (NLS) or NES of cargo proteins to be recognized by the import and export machinery are found in the sequence of Nrf2: two NES, one located in the leucine zipper dimerization (Zip) domain and a second one in the transactivation domain (TAD) (215, 289), and three NLS (478).
The basic requirements for nuclear traffic being evident, we first have to elaborate on the questions how free Nrf2 is generated and primed for nuclear import (Fig. 8). As mentioned in the previous section, cysteine modification of Keap1 by itself is not considered sufficient to release Nrf2 from the cytosolic complex. The view favored at present therefore is that the Nrf2 that is synthesized de novo bypasses cytosolic Keap1 when the latter has been modified and hence loaded with stabilized Nrf2, whereas Nrf2 meeting Keap1 under resting conditions has a poor chance to escape from degradation (287). However, since permanent wasting of Nrf2 by the unchallenged organism appears uneconomic, mechanisms to release Nrf2 from the cytosolic complex merit consideration and do indeed exist. (i) PKC phosphorylates Nrf2 at Ser40 (192), the isoforms later being identified as PKCδ (283) and PKCι (350). Ser40 is located between the DLG and the ETGE motifs, and phosphorylation inhibits binding to Keap1 and simultaneously facilitates nuclear import (347, 348). It appears, however, to be unsettled if this phosphorylation is also achieved within the Keap1/Nrf2 complex (35, 192, 350). (ii) The CDK inhibitor p21 binds to stabilized Nrf2 at Keap1 and possibly facilitates Nrf2 release (63). (iii) Prothymosin α (239, 361), and (iv) the sequestosome 1 (SQSTM1; p62) (269) interact with Keap1 in a way that displacement of Nrf2 appears unavoidable. SQSTM1 was further shown to lower Keap1 levels by increasing its rate of degradation (76). Accordingly, overexpression of SQSTM1 results in transcriptional activation of Nrf2 target genes (76, 257). Collectively, these findings suggest that also the Keap1-bound Nrf2 that has been saved from ubiquitination can be made available for nuclear import (482). It has further also been proposed that Nrf2-loaded Keap1 enters the nucleus (498).
FIG. 8.

Nuclear translocation of Nrf2. Activation of Nrf2 does not necessarily require activation of a specific surface receptor. Stimuli can be exogenous or endogenously produced. In response to oxidative/electrophilic stress, the interaction of Keap1 and Nrf2 is disturbed and Nrf2 is no longer degraded. Nrf2, either released from Keap1 or newly synthesized, enters the nucleus. For translocation, Nrf2 has to be phosphorylated at Ser40. The phosphorylation is achieved by PKCδ or ι and/or other kinases (see text). Phosphorylated Nrf2 enters the nucleus, forms a complex with the basic leucine zipper (bZIP) factors CREB-binding protein (CBP)/p300, and activates gene expression by binding to the electrophile responsive element (EpRE). Phosphorylation at Tyr568 by the tyrosine kinase Fyn leads to nuclear export of Nrf2 and terminates the signal. It is not known whether Nrf2 has first to be de-phosphorylated at Ser40 or whether the bis-phosphate is exported. Exported Nrf2 can re-associate with Keap1.
Thus, nuclear import of Nrf2 is initiated by the key oxidative modification of Keap1 and further facilitated, inter alia, by PKC-dependent phosphorylation, which is also considered to be favored by thiol oxidation (Fig. 8; section II.D.3). Moreover, Nrf2 itself and, hence, its nuclear localization is redox sensitive (Fig. 9). Its activity can also be enhanced by inhibiting the nuclear export of Nrf2. The NES of Nrf2 are recognized by chromosome region maintenance (Crm1)/exportin, but in the nucleus are largely masked, probably by heterodimerization of Nrf2 with musculo-aponeurotic fibrosarcoma (Maf) proteins that retain Nrf2 in the nucleus. In consequence, the import/export balance is in favor of import even under basal conditions, which accounts for the constitutive Nrf2 activity that guarantees the basal expression of stress response genes. Whereas the NES in the bZIP domain is redox insensitive (289), oxidation of Cys183 in the NES in the TAD of human Nrf2 is discussed to inhibit the access and binding of Crm1 (288). Under oxidative stress conditions, therefore, the function of the redox-sensitive NES in the TAD is impaired and the overall weight of export signals is further decreased (287). Similarly, an oxidative modification within an NES of nuclear Keap1 can retain Nrf2 in the nucleus (498). Irrespective of the mechanism, the net effect is a nuclear accumulation of Nrf2. Cysteine modification, depending on its chemistry, may be reversible, for example, by GSH or a redoxin, and reversal of nuclear localization of Nrf2 by Trx has indeed been reported (162). Cys506 is critical for binding of Nrf2 to its responsive element and the interaction with the cofactor CREB-binding protein (CBP)/p300 (34). Apart from Cys506 also Cys119 and 235 (numbering in the mouse Nrf2) have been found to have similar functions (176). Hence, re-establishing reductive conditions in the nucleus deserves attention not only as required for DNA binding but also as shut-off mechanism of the system (see section III.C.5).
FIG. 9.

Organization of Nrf2. The Nrf2 protein comprises six domains (Neh1–6) with different functionalities. Neh2: Keap1 binding site; Neh4 and 5: transactivation (TA) site harbors the nuclear export signal (NES) with the redox-sensitive C183, and is required for binding of Nrf2 to its responsive element and interaction with the coactivator CBP/p300; Neh6: linker, site responsible for Keap1-independent nuclear degradation under oxidative stress (324); Neh1: contains the Cap ‘n’ Collar region (CNC), the redox-insensitive NES, the nuclear localization signal (NLS) with the redox-sensitive C506, and the bZIP motif; Neh3: the C-terminal domain contains Y568 and is required for transactivation (345). S40 is the phosphorylation site for PKCδ and ι; Y568, the phosphorylation site for Fyn. Arrows mark putative sites for casein kinases. Two novel NLSs spanning amino acids 42–53 and 587–593 identified in the mouse sequences are not included for sake of clarity (478). For numbering of different species see ref. (176).
Further, nuclear export of Nrf2 is regulated by phosphorylates (Fig. 7). The Fyn kinase phosphorylates Nrf2 at Tyr568 in the nucleus which leads to the strengthening of the interaction with Crm1 and to an enhanced nuclear export of Nrf2 (213, 348). A translocation of Fyn into the nucleus is observed several hours after Nrf2 activation and requires the phosphorylation of Fyn at a so far unknown Thr residue by GSK3β. For this activity GSK3β has to be phosphorylated at Tyr216, which in contrast to the phosphorylation at Ser9 by Akt, which inactivates GSK3β (327), keeps GSK3β in the active state. The Tyr216-specific kinase is still unknown, but it is activated by H2O2 (214). This way GSK3β may contribute to the termination of the Nrf2 signal.
4. Nrf2-mediated transduction events
In the nucleus Nrf2 dimerizes with small Maf proteins (207) or other bZIP transcriptions factor such as c-Jun or activation transcription factor-4 (ATF4). The heterodimer binds to the Nrf2-responsive element EpRE. The Nrf2/bZIP complex then recruits the CBP and p300 (536) and initiates transcription of target genes (Fig. 8). Formation of a heterodimer with c-Fos or c-Fos-related antigen-1 (Fra-1) inhibits Nrf2 activity due to binding to the AP-1 site within the EpRE. Also, overexpression of small Maf proteins leading to Maf homodimers and possibly unproductive Maf-Nrf2 heterodimers inhibited Nrf2 activity (90). The Nrf2 gene itself carries an EpRE and is transcriptionally stimulated by Nrf2 activators (263).
Binding of Nrf2 to EpRE is competed out by the BTB and CNC homology-1 (Bach1) transcription factor (91). Oxidative conditions inactivate Bach1 and allow Nrf2 binding to EpRE (203). Inactivation of Bach1 is mediated by a redox-sensitive phosphorylation at Tyr486 by an unknown kinase, which leads to rapid export of Bach1 (243).
5. Shut-off mechanisms
Apart from nuclear export of Nrf2 (see above, section III.C.3), termination of Nrf2-mediated transcription may be achieved by various mechanisms. (i) Also the nucleus contains Keap1, whose function is not fully clarified, but it may be envisaged that Keap1 also targets the nuclear Nrf2 for degradation by the nuclear proteasome (348). (ii) Polymerization of the actin skeleton is considered to be essential for retaining Nrf2 in the cytosol, thus preventing Nrf2 activity. PI3K inter alia regulates the response to oxidants by de-polymerization of actin, which is considered to allow Nrf2 to escape from Keap1 and to enter the nucleus together with actin (234). Re-polymerization of actin allows Nrf2 to exit the nucleus. In this context it might be revealing that glutathionylation of actin leads to depolymerization and is reversed by Grx (267, 506). (iii) Nrf2 activates transcription of its own cytosolic inhibitors such as Cul3, Rbx1, and Keap1 (242). (iv) The most efficient shut-off is certainly achieved by means of the myriad of enzymes simply eliminating the system's signals or preventing their formation.
Notably, Nrf2 targets such as Trx and Trx reductase, Srx and γ-glutamyl-cysteine synthetase synergize in improving the capacity of the entire Prx- and GPx-mediated hydroperoxide metabolism and thus eliminate oxidant signals and prevent the formation of damage signals arising from oxidative stress (125, 200). The scenario is complemented by induction of particular peroxidases such as Prx1 (200) and GPx2 (21). Similarly, the induction of many GSTs prevents activation of the system by eliminating the nonoxidant alkylants (175). Apart from eliminating Nrf2-activating signals, however, the expression of antioxidant enzymes may also interfere directly with ongoing Nrf2 transduction: murine liver txnrd1−/− cells that lack the selenoprotein Trx reductase-1 do not display any gross changes in the cellular redox homoeostasis, likely because GPxs can easily substitute for the Trx-dependent Prxs under most conditions. However, the txnrd1−/− cells displayed a pronounced persistence of Nrf2 in the nucleus. This, for the first time, points to the requirement of reduced Trx for switching off Nrf2 transduction (470) and provides a mosaic stone possibly explaining the role of selenium in Nrf2 signaling (see section II.D.5).
6. Modulation of Nrf2 function by cross-talk with phosphorylation cascades
In the previous paragraphs several phosphorylation/de-phosphorylation steps have already been mentioned that appear not to belong to the mainstream activation of the Nrf2 system. The functional relevance of phosphorylation events to the Nrf2 system has for long been established by demonstrating enhanced activation by broad-spectrum phosphatase inhibitors such as ocadaic acid (344) or genistein (213). Cross-talk of phosphorylation cascades has to be considered in particular when the Nrf2 system responds to oxidative signals produced endogenously upon activation of Toll-like or TNF receptors (TNFRs) as in infections, physical injury, or acute inflammation. Receptor activation by pathogen-derived molecules and pro-inflammatory cytokines is essentially signaled via phosphorylation and/or protease cascades but consistently accompanied by massive production of oxidants via activation of NOX and LOX or mitochondrial O2•− generation (see section IV.D.1). Under these conditions, therefore, the oxidant and alkylant signals, which are provided in concert with enhanced phosphorylating activities, activate Nrf2, whereas the outcome of Nrf2 activation feeds back to dampen the oxidative responses to inflammatory stimuli. In this situation the limited specificity of protein kinases and phosphatases provides ample opportunities for cross-talk between seemingly unrelated signaling pathways (298). Beyond, the oxidant conditions generally favor protein phosphorylation (see section II.D.3).
The following examples of enhanced Nrf2 activation due to phosphorylation are compiled below (Fig. 8):
(i) From the mitogen-activated protein kinase (MAPK) pathway, extracellular signal-regulated kinase (ERK) and c-Jun N-terminal kinase (JNK) stimulate Nrf2 activity. However, site-directed mutagenesis in putative MAPK phosphorylation sites in Nrf2 did not influence Nrf2 activity (448) and Ser/Thr residues that have been identified as MAPKs' targets in vivo did not significantly contribute to Nrf2 activation (468). Thus, indirect mechanisms have been suggested. The assumption is corroborated by earlier findings showing that ERK-mediated phosphorylation of CBP, which binds to the Nrf2 TA in the nucleus, stimulated transactivation activity of Nrf2 (448) and that electrophiles activate JNK by the inhibition of a JNK-specific phosphatase (64).
(ii) Casein kinase2 (CK2) phosphorylates Nrf2 at several sites, which are located in the TADs. Phosphorylation at the site resulting in an Nrf2 form with a molecular weight of 98 facilitates nuclear import of Nrf2, whereas the phosphorylated form with the molecular weight of 118 appears to be degraded more readily (9, 371).
(iii) The protein kinase R (PKR)-like endoplasmic reticulum kinase (PERK) phosphorylates Nrf2, triggers nuclear import, and inhibits re-association to Keap1 (80). However, the target amino acid has not been identified yet.
(iv) As mentioned (Fig. 8), phosphorylation of Nrf2 at Ser40 by PKCδ and PKCι facilitates nuclear import of Nrf2 (283, 347, 350).
(v) Phosphorylation of the initiation factor elF2α is implicated in the enhanced translation of Nrf2 mRNA in response to the Nrf2 activators H2O2 and sulforaphane (387).
(vi) Several Nrf2 activators have been shown to stimulate phosphorylation of Akt, the key player of the PI3K pathway. Activation of the PI3K pathway inactivates GSK3β, in turn preventing activation of Fyn with consequences discussed in sections III.C.3 and III.C.5 (213, 214, 234, 311, 417).
Phosphorylation reactions can also lead to an inhibition of Nrf2-mediated transduction (i) by phosphorylation of Nrf2 through p38 mitogen-activated kinases in an as yet unclear way (528); (ii) by GSK3β affecting nuclear export via Fyn-mediated Nrf2 phosphorylation at Tyr568 (see section III.C.3) (214, 417); and (iii) by stabilizing Keap1 via phosphorylation at Tyr141 (216).
D. Synopsis of the Nrf2 system
In view of the complexity of the Keap1/Nrf2 system and its ramifications a wood-carved summary of the essentials might be helpful. The system responds to oxidative stress with expression of a broad range of cytoprotective enzymes. It responds to H2O2, organic hydroperoxides, peroxynitrite, and electrophiles that are generated by oxidative tissue damage, the common denominator of these compounds being their ability to oxidize or alkylate thiols. The bona fide sensor is the Nrf2 inhibitor Keap1, which sequesters Nrf2 in the cytosol in a way that it is permanently degraded by the ubiquitin/proteasome system. The sensing mechanism consists of oxidation or alkylation of critical cysteine residues in Keap1. Downstream signaling is achieved by conformational changes of Keap1, whereby the transcription factor Nrf2 is made available for nuclear import and transduction by activation of EpRE (ARE). Termination of Nrf2 signaling is achieved by nuclear export, autoregulatory feedback loops, and re-establishing the redox homoeostasis by de novo synthesis of enzymes that eliminate the activating signals.
The Keap1/Nrf2 system, thus, is the only one that uses oxidants or electrophilic products of oxidative processes as primary signals. Beyond, several steps of the system, in particular the nuclear trafficking, critically depend on reversible phosphorylation and accordingly may be modulated by redox biochemistry.
Signaling through Keap1 has to overcome the competition by enzymes such as GPxs, Prxs, and GSTs, which eliminate the signaling molecules ROOH and electrophiles, respectively, and thus determine the threshold for the response. The scope of Nrf2 target genes allows an adjustment of the system's threshold and guarantees improved defense against more severe oxidative challenges. These characteristics place the system into the context of physiological adaptation to oxidative stress, as triggered in infectious diseases, acute inflammation, or injury (see section IV).
The endogenous signaling molecules of the system are mimicked by a wide range of chemicals and xenobiotics that activate the system either by generating H2O2 via redox cycling or simply by their ability to S-alkylate thiols. Such Nrf2 activators, in particular the redox-inert natural ones, are currently discussed for safe adaptation to oxidative challenges and the prevention of related diseases.
IV. NF-κB, a Key Regulator of the Immune Response
A. Discovery and definitions
NF-κB was discovered in 1986 by Sen and Baltimore (444) as a transcription factor of B lymphocytes (19). Over the years the term NF-κB has been applied to different protein complexes made up from homo- or heterodimers of five distinct proteins: p65 (RelA), RelB, c-Rel, p50, and its precursor p105, and p52 and its precursor p100 [reviewed in ref. (172)]. These proteins have an N-terminal Rel homology domain (RHD) that is responsible for binding to DNA and other proteins and harbor an NLS. As Nrf2 also NF-κB belongs to the type of transcription factors that is kept in the cytosol by complexation with inhibitors which have to be removed for activation. The family of NF-κB inhibitors (IκB) comprises IκBα, IκBβ, IκBγ, IκBɛ, and BCL-3. They contain 6–7 ankyrin repeats (240) that mediate binding to the RHD and interfere with its NLS function. The prevalent composition of the cytoplasmic NF-κB appears to be the p50/p65/IκBα complex.
NF-κB was the first mammalian transcription factor shown to be redox regulated (461) and suggested to be directly activated by a variety of ROS (438). Meanwhile, however, the NF-κB system has been recognized to be primarily activated by cytokines and nonoxidant foreign stressors via TNF, Toll-like, and other receptors.
B. The biological context of NF-κB
The pivotal role of the NF-κB system consists of the activation of an innate immune response upon challenge by micro-organisms or mimicking ligands. The main receptors of the system are TLRs, of which so far 13 distinct ones have been identified in mammals, 4 residing in endosomal vesicles, the other ones at the cell surface (262, 450). Most of the TLRs (TLR1, 2, 4–6 and 11) respond to microbial structures known as pathogen-associated molecular pattern (PAMP), the most prominent examples being the LPS, which are components of the outer membrane of Gram-negative bacteria (4, 29). The TLRs 3, 7, and 9 are specialized for recognition of double-stranded or single-stranded RNA or CpG-containing DNA, respectively, and thus can sense viral infection (4), and TLR 2 and 4 are implicated in the response to cellular damage by recognizing damage-associated molecular patterns (DAMPs) (144). NF-κB is also activated by inflammatory mediators such as IL-1β and TNFα via their respective receptors as well as by certain growth factor receptor tyrosine kinases (RTKs), by G-protein-coupled receptors (GPCRs) and antigen receptors (38, 147, 172, 173) (Fig. 10).
FIG. 10.

Different ways to activate NF-κB. Receptor tyrosine kinases (RTK): upon growth factor binding, RTKs associate forming dimers, are autophosphorylated at multiple tyrosines, and recruit additional signaling proteins. Among recruited proteins is phosphatidyl-inositol-3-kinase (PI3K). In the atypical pathway, PI3K phosphorylates spleen tyrosine kinase (SYK), which in turn phosphorylates IκB at Tyr42, the signal for IκB degradation. Alternatively, PI3K-activated Akt can transduce signals through the typical pathway by directly phosphorylating IκB kinase β (IKKβ). Interleukin-1 receptor-1 (IL-1R1): after binding of IL-1 to the receptor, MyD88 is rapidly recruited and associates to the receptor via interaction of Toll/IL-1 receptor (TIR) domains present in both IL-1R and MyD88. IL-1R-associated kinase 1 (IRAK1) and IRAK4 are recruited to MyD88, where IRAK4 phosphorylates IRAK1, resulting in hyperphosphorylated IRAK1 by autophosphorylation. The kinases dissociate from MyD88. An intermediate cytosolic complex composed of the E3 ligase pellino-1, IRAK1, IRAK2, and IRAK4 is formed, whereas TNF receptor (TNFR)-associated factor 6 (TRAF6) becomes ubiquitinated, which serves as a platform for TGFβ-activated kinase (TAK1). The same complex is formed in TLR signaling (not shown) [reviewed in ref. (157)]. TAK1 then activates IKKβ. Toll-like receptors (TLR): with the exception of TLR3 all TLRs similarly to IL-1R activate TAK1 through MyD88, activation of IRAK1 by IRAK4, IRAK1 autophosphorylation, and TRAF6 recruitment. In addition, Toll-receptor-associated activator of interferon (TRIF)-related adaptor molecule (TRAM), TRIF, and TIR-domain-containing adaptor protein (TIRAP) interact with the receptor. If the typical pathway is taken, TRAF6 activates TAK1. If the signal enters the alternative pathway, NIK1 becomes activated. NIK1 directly phosphorylates IKKα, which initiates proteolytic cleavage of p100, the precursor of p52, by phosphorylation at Ser866 and 870. Finally, the RelB/p52 heterodimer can enter the nucleus. TNFR: TNF binding to TNFR1 triggers the association of the adapter protein TNFR1-associated death domain protein (TRADD) to the receptor. Then, TRAF2, TRAF5, and the kinase receptor interacting protein-1 (RIP1) are recruited. RIP1 can activate TAK1 in the typical and NIK in the alternative pathway.
The involvement of such diverse receptor and signaling systems implies that the response to different NF-κB activators can hardly be uniform and must vary with the cell type and stimulus. However, the overall outcome of the NF-κB activation is an inflammatory response characterized by enhanced expression of pro-inflammatory cytokines such as TNFα, IL-1, and IL-6, chemokines such as monocyte chemotactic protein-1 (MCP-1), IL-8, and macrophage inflammatory protein-1α (MIP-1α), adhesion molecules such as intercellular adhesion molecule-1 (ICAM-1), vascular cell adhesion molecule (VCAM)-1, and endothelium leukocyte adhesion molecule (ELAM), growth factors, and enzymes that produce secondary inflammatory mediators such as COX-2 and induced NO synthase (iNOS). The proteins induced by NF-κB activation, thus, comprise several activators of NF-κB itself, which implies an amplification of the initial response. However, the NF-κB-dependent expression pattern, apart from the potentially self-destructive cocktail of the innate immune response, also includes self-protective proteins and inhibitors of autophagy and apoptosis (13, 38, 99, 172, 173). Collectively, therefore, the system serves to guarantee survival in a hostile environment although sometimes by using risky strategies (see section V).
The risky side of NF-κB has best been characterized in inflammation and cancer. A causal relationship between inflammation and cancer has been suspected already in the 19th century by Virchow [reviewed in ref. (17)]. Virchow's hypothesis that carcinogenesis may result from chronic inflammation has been amply confirmed for various forms of cancer. Among the inflammatory mediators, NF-κB ranks high in the list of potential culprits. NF-κB could indeed be shown to be crucial for malignant transformation (158, 300, 374). NF-κB links inflammation to carcinogenesis by at least three distinct roles: (i) By enforcing and sustaining an inflammatory response, the mutagenic potential of superoxide, H2O2 and lipid hydroperoxides comes into play, which can certainly not be ignored in the initiation process. (ii) In established tumors, the Janus-faced tumor-associated inflammation has to be considered. It may kill tumor cells via massive oxidative attack induced by TNFα (151); after all killing tumors was the name-giving activity of the TNF. On the other hand, NF-κB-mediated expression of inflammatory mediators such as TNFα, IL-1, IL-6, or prostaglandin E2 (PGE2), depending on the tumor type, may promote tumor growth. (iii) NF-κB activates gene expression for antiapoptotic factors such as cellular inhibitor of apoptosis proteins (cIAPs), X-linked inhibitor of apoptosis protein (XIAP), A20, also known as TNFα-induced protein 3, Bcl-2, survivin proteins, and MnSOD (41, 188), which collectively support tumor development. The consequences of an NF-κB activation, therefore, depend on type and stage of tumor.
C. Basics of NF-κB activation
The basics of NF-κB activation consist of liberation of the free transcription factor from its cytosolic complex with the inhibitor of NF-κB (IκB) to allow nuclear import and gene activation (Figs. 10 and 11). In the absence of stimuli, NF-κB dimers are tightly associated to IκB. IκB masks the NLS in NF-κB and, thus, keeps it in the cytosol. The central event in NF-κB activation, the release of IκB from the cytosolic NF-κB complex, is typically the consequence of an IκB phosphorylation by IKK (but see below). IκB is then ubiquitinated and subsequently degraded by proteasome 26S. The IKKs form a complex consisting of IKKα, IKKβ, and IKKγ. Therein two molecules of IKKγ linked through disulfide bonds between Cys54 and Cys347 (181) form the NF-κB essential modulator (NEMO) to which IKKα and IKKβ bind in the resting state (409, 522). Conformational changes and/or ubiquitination of NEMO due to binding to a receptor interacting protein (RIP) kinase leads to the phosphorylation/activation of IKK by an IKK kinase such as TGFβ-activated kinase (TAK1) (103, 173, 205, 425, 451, 517). With IκB phosphorylation by activated IKKs, nuclear import of NF-κB is initiated. In the nucleus NF-κB, typically the p50/p65 complex, binds to its promoter sequence together with the transcriptional coactivators CBP and p300. CBP and p300 have histone acetylase (HAT) activity. Further, NF-κB displaces histone deacetylase (HDAC) from the promoter. Thereby p65, p50 themselves, and, more importantly, histones become acetylated, the latter event leading to uncoiling of chromatin, which allows access of the transcription factor to its canonical and other promoter regions (147). Inversely, deacetylation of histones by HDACs leads to silencing of gene transcription and, further, deacetylation of NF-κB promotes its association with IκB and export from the nucleus (61).
FIG. 11.

Activation of NF-κB via the canonical pathway and putative sites for redox-regulation. TAK1, activated by IL-1R, TNFR, or TLR signaling, phosphorylates IKKβ in the IKK complex (see Fig. 10). IKKβ phosphorylates IκB at serines (Ser) 32 and 36 leading to the ubiquitination and degradation of IκB. Deliberated p65/p50 becomes phosphorylated by protein kinase A, catalytic subunit (PKAc) at Ser276 in p65, and is translocated into the nucleus. There it binds together with the coactivators CBP and p300 to the NF-κB responsive element in the promoters of target genes. For binding to DNA, cysteine 62 in p50, which can be oxidized to a sulfenic acid forming a mixed disulfide with GSH in the cytosol, has to be reduced in the nucleus by the Trx/TrxR or Grx system or by the base excision repair enzyme apurinic/apyrimidinic endonuclease/redox factor-1 (APE1/Ref-1).
The routes toward this largely common downstream event vary considerably with the cell type, the upstream signal and in particular with the receptor involved. The multiple channels by which NF-κB activation can be initiated are reflected in multiple activation pathways that can roughly be classified into three: the typical, the atypical, and the alternative one (147) (Fig. 10).
(i) The typical pathway (also called classical or canonical) relies on the phosphorylation of IκBα on Ser32 and 36 by TAK1-activated IKKβ (92), which creates a binding site for the β-transducing repeat containing protein (β-TrCP), the receptor subunit of an SCFβ−Trcp E3 ubiquitin ligase. E3 catalyzes ubiquitination of IκB for degradation through the 26S proteasome (65, 172, 204, 296, 406). This pathway is triggered, for example, by TNF, IL-1, LPS, and other PAMPs (89) and initiates the innate immune response (38). The receptors involved are TLRs, IL-1 receptor (IL-1R), and TNFRs. They have in common that they are not receptor kinases by themselves, but work through associated kinases, as, for example, described for recruitment of the IL-1R-associated kinase (IRAK) in IL-1 signaling (312). In some cell types the genuine system receptors may be by-passed by H2O2 (358).
(ii) In the atypical pathway IκBα is phosphorylated at Tyr42 or on Ser residues in the PEST domain (197, 437). The phosphorylation at Tyr42 is IKK independent and, instead, catalyzed by the spleen tyrosine kinase (SYK) (473), which is activated by kinases of the PI3K pathway. Phosphorylation of IκBα at Tyr42 is followed by its dissociation and degradation. Dissociation can also be obtained by direct interaction of activated PI3K with the Tyr-phosphorylated IκB, thereby removing it from NF-κB (28). The atypical pathway is triggered by growth factors like EGF, ciliary neurotrophic factor (CNTF), or nerve growth factor (NGF) binding to RTKs, but receptor occupation can be mimicked by H2O2 and PP inhibitors (147). Activation of the atypical pathway is mainly T cell-specific and depends on the presence of the phosphatase SHIP-1 (149). Inhibition of SHIP-1 by either H2O2 or PP inhibitors maintains phosphorylation of IκBα at Tyr42 and allows activation of NF-κB. In cells lacking SHIP-1 such as Jurkat cells, H2O2 mainly leads to IKK-dependent Ser-phosphorylation and degradation of IκB via the proteasome with only minor or absent phosphorylation at Tyr42 (149).
(iii) In the alternative pathway (also called noncanonical pathway) IKKα is activated by the NF-κB inducing kinase-1 (NIK1). Activated IKKα phosphorylates p100, resulting in its ubiquitination and processing to p52 (241, 337, 445). The pathway is activated by certain members of the TNF-receptor superfamily, such as CD40L (166), and thought to be necessary for the adaptive immune response (38). Interestingly, the alternative pathway also leads to the assembly of different NF-κB elements in the nucleus (368).
For a more detailed description of these pathways, the reader is referred to recent reviews in which also ubiquitination events are considered in more detail (147, 173, 368).
D. Redox regulation of NF-κB activation
In the years after the discovery of NF-κB, a series of studies revealed that (i) most, if not all, agents activating NF-κB trigger the formation of O2•−/H2O2 via NOX or are oxidants by themselves such as superoxide, H2O2 or LOX products; (ii) in some cell lines, NF-κB activation can be triggered by H2O2 or organic hydroperoxides in the absence of any physiological stimulus; (iii) NF-κB activation is inhibited by a broad range of chemically unrelated antioxidants. The findings were compiled in a hypothesis paper of 1997 (128) with the above title and summarized as follows: “This complex cascade of phosphorylation and dephosphorylation is modulated by redox reactions of unknown nature in the sense that the oxidant status increases the phosphorylation and degradation of IκB. NF-κB action, however, requires a Trx-dependent reduced status in the nucleus. Upstream kinase(s) and or phosphatase(s) prone to thiolation or oxidation of vicinal SH groups are at present considered the best candidates mediating the redox regulation of NF-κB.” In essence, the hypothesis has remained attractive, could be corroborated by detailed insights, and can be amended by novel system components.
Nevertheless, the seemingly convincing picture of a strict dependence of NF-κB activation on oxidants of the early nineties has since been controversially discussed. Main arguments were, for instance, that compounds labeled as antioxidants exerted effects that were unrelated with their antioxidant potential. For instance, the presumed antioxidant pyrrolidine dithiocarbamate (PDTC) inhibited IL-1-mediated NF-κB activation not due to an antioxidant function but rather by the modification of thiols in the IL-1R/IRAK1 complex (477). Similarly, an attempt to correlate the antioxidant function of N-acetyl cysteine (NAC) and PDTC with their potential to inhibit TNFα-induced NF-κB activation failed. Instead, NAC decreased the affinity of the receptor for TNFα and PDTC inhibited IκB ubiquitin ligase (168). Conflicting observations were also made with H2O2 itself. A critical review analyzing 40 articles on NF-κB activation by H2O2 alone or in conjunction with suitable cytokines revealed that H2O2 did not consistently lead to NF-κB activation: an activation of NF-κB was observed just in one third of the investigations, whereas in one-third H2O2 inhibited NF-κB activation and had no effect whatsoever in another third (358). This puzzling scenario is hard to explain. Revealingly, bolus administrations of 1 mM and beyond tended to inhibit NF-κB activation, and the authors recommended more physiological approaches in analyzing the problem (84, 358). The most decisive aspect of the role of H2O2, however, appeared to be the cellular system and the prevailing NF-κB-activating pathway (358). A regulatory function of H2O2 or similar oxidants in NF-κB activation has, nevertheless, to be inferred from experiments with genetically modified organisms and cells overproducing or being deficient in peroxidases. In mice deficient in GPx1, inflammatory responses, which likely are mediated by NF-κB, are consistently enhanced irrespective of being triggered by a PAMP-like LPS (211) or by viral infections (23, 24). Similarly, mice deficient in both GPx1 and GPx2 spontaneously developed an ileocolitis, when the gut was only colonized with apathogenic bacteria (108). Inversely, cells overexpressing catalase (434), GPx1 (261), GPx4 (45, 46), or Prx2 (237) displayed a dampened NF-κB activation, whereas modulation of SOD activity remained ambiguous (306, 434). Collectively, the meanwhile accumulated knowledge does no longer corroborate the concept that H2O2 acts as a direct activator or obligatory mediator of NF-κB activation, but rather supports the activation by other stimuli (148).
1. The source of the oxidants
In most of the schemes illustrating redox-regulated NF-κB activation, activating ROS, like a deus ex machina, show up in the center of an exploding star, leaving open the problems of the chemical nature and biological source of the magic activator. Intriguingly, however, a list of compounds known to induce O2•− formation in phagocytes already in 1985 (126) almost coincides with the one of NF-κB enhancers published by Schreck et al. in 1991 (438). It comprises a variety of PAMPs, antibodies, calcium ionophores, phorbolesters, substrates/products of LOX, and lipid mediators of inflammation. The two lists become practically congruent just by adding the pro-inflammatory cytokines IL-1 and TNFα to the older one. It incidentally was this pronounced similarity of activators of NOX and NF-κB enhancers that led to the concept of NF-κB activation by oxidants (128). It now corroborates that PAMPs or pro-inflammatory cytokines, when activating their respective receptors, simultaneously activate NOX and/or LOX enzymes.
The ability of calcium and PKC activators such as phorbol esters to trigger an oxidative burst in phagocytes had for long revealed a role of PKC-type enzymes in linking receptor occupation to the activation of NOX. Members of the PKC family are known to regulate many cellular processes (258) (see sections II.D.3 and III.C.3) and have meanwhile been shown to activate NOX- and LOX-type enzymes (see section IV.D.1), and to also interfere with the NF-κB system at other sites (see section IV.D.2). As mentioned in section II.D.3, they are characterized by redox-sensitive zinc finger domains. While they are activated by oxidation of the cysteine residues that are coordinated to zinc (152, 153), they may be inactivated by oxidation or alkylation of their catalytic cysteines (154). It, thus, appears conceivable that they amplify an NOX- or LOX-mediated oxidative response but contribute to its termination under stronger oxidative conditions.
Involvement of NOX has been convincingly shown in p47phox−/− mice. p47phox is an indispensable component of the phagocytic NOX complex that further contains p22phox, p40phox, the GTP-binding protein Rac-1, and the flavo-cytochrome gp91phox as the O2•− producing NOX2 (14). Accordingly, in the lungs of mice deficient in this essential activator the activation of NF-κB elicited by pathogens or LPS was impaired (253, 414). Similarly, in human aortic endothelial cells TLR4-mediated superoxide production and NF-κB activation were shown to depend on the homologous NOX4. A knockdown of NOX4 inhibited IκB degradation and binding of p65 to DNA (365). Mechanistically, an interaction of the C-terminal region of NOX4 with the Toll/IL-1 receptor (TIR) domain of TLR4 was discussed which now is known to be linked by MyD88 (290). Also, the source of IL-1-induced O2•−/H2O2 production in MCF7 cells was shown to be an NOX. The H2O2 thereby produced was reported to activate NIK-mediated phosphorylation on IκB probably by the inhibition of a phosphatase, since ocadaic acid mimicked the IL-1 effect (284).
The precise link between ligand occupation of TLRs and the activation of NOX systems is not yet entirely clear. In the possibly analogous growth factor signaling the activation of NOX1 is achieved by sequential activation of phosphatidyl-inositol-3-kinase (PI3K), formation of GTP-loaded Rac-1, and recruitment of phagocytic oxidase (phox) subunits to the membrane to assemble the active NOX complex (366). The PI3K pathway appears also to be generally activated through TLRs (313), whereby MyD88 links PI3K to the receptor (266) and PI3K produces the phosphatidyl inositol phosphates to form the platform where phox subunits can bind (106, 232) (Fig. 12). A similar sequence may, therefore, be envisaged for Rac-1 activation for NOX2 activation in phagocytes. The pivotal TLR-activated kinase for the phosphorylation of p47phox (30) and p67phox (533) in human monocytes is PKCδ, which in turn is activated by SYK and Src kinases associated with the zymosan-recognizing receptor Dectin-1, which in turn cooperates with TLR2 in the induction of inflammatory responses (142).
FIG. 12.

Upstream kinases and redox-sensitive phosphatases in NF-κB activation. Upon binding of stimuli to their receptors, which can be RTKs, TLRs or TNFRs, PI3-kinase is recruited to the membrane and binds to RTKs via its Src homolog-2 (SH2) domain to autophosphorylated tyrosines (72), to TLR via direct interaction with the receptor and MyD88 [reviewed in ref. (290)], and to TNFR via riboflavin kinase (525). Once localized at the membrane, the p110 catalytic subunit of PI3K phosphorylates phosphatidylinositol-4,5-bisphosphate (PIP2) at the 3′ position of its inositol residue forming PIP3. PIP3 recruits Akt and phosphoinositide-dependent protein kinase-1 (PDK1), the latter phosphorylating Akt at T308 and S493 in the regulatory domain. Akt thus activated phosphorylates multiple downstream targets one of them being IKKβ. Other IKKβ phosphorylating enzymes are TAK1, activated in the canonical pathway, and mitogen-activated protein kinase (MAPK) kinase kinase-3 (MEKK3), which can be activated by G-protein-coupled receptors (GPCR). PPs counteracting the involved kinases are the dual substrate phosphatases PP2A and PP2C. PP2A has been shown to colocalize with Akt and this way inhibits PDK1 action. It also reverses MEKK3 activation. PP2Cβ reverses Akt-catalyzed IKK phosphorylation and PP2Cη-2 de-phosphorylates TAK1-phosphorylated IKK. The lipid phosphatases PTEN and Src homology-2 (SH2)-domain-containing inositide phosphatase (SHIP-1) reverse the PI3K signal by removing the phosphate groups at position 3′ (phosphatase and tensin homologue [PTEN]) or 5′ (SHIP-1). All phosphatases in the scheme can be inactivated by oxidative modification of cysteine residues. The oxidizing signal comes from NADPH oxidase (NOX) enzymes that are also activated by receptor-bound PI3K there producing the inositol phospholipid products to which phagocytic oxidase (phox) subunits can bind (107, 232).
Novel mechanisms linking NF-κB activating receptors to NOX have recently been proposed. The TNFR proved to be coupled to NOX1 via riboflavin kinase (RFK), formerly known as flavokinase. RFK bridges TNFR1 and NOX1 via binding of the TNFR1-death-domain to the NOX subunit p22phox. In cells deficient in RFK, TNF-induced O2•− production was inhibited (525). Oakley et al. (353) highlighted the involvement of lipid rafts in the synchronized activation of NF-κB and NOX2 by IL-1β. Both IL-1R1 and NOX2 were found colocalized in lipid rafts. Upon IL-1 stimulation MyD88 was recruited to IL-1R1 and endocytosed into endosomes together with Rac-1 and NOX2 in a caveolin-dependent manner. H2O2 produced in this complex facilitated TNFR-associated factor (TRAF6) association with the receptor complex, thereby building a redox-active signaling platform that was called redoxosome (354) (Fig. 13). Recruitment of receptors together with NOX enzymes into specific membrane domains could be the missing link between receptor activation and the mysterious coactivation of O2•−/H2O2-producing enzymes. Compartmentalized NOX activation and redox signaling has recently been reviewed by Ushio-Fukai (494).
FIG. 13.

Redoxosome formation in IL-1 signaling. Redoxosome (redox active endosomes which produce O2•−/H2O2) formation has been demonstrated for the activation of NOX2 by IL-1 in MCF7 cells (354). After docking of IL-1 to IL-1R1 MyD88 is recruited as effector, and initiates endosome formation. Then, Rac-1 in its GTP-bound conformation together with superoxide dismutase 1 (SOD1) is transferred to the membrane, resulting in the recruitment of the second effector, NOX2 with all its subunits. O2•− produced by NOX leaves the endosome via anionic channels (AC) and is dismutated to H2O2 by SOD1. This creates an oxidative environment, which promotes docking of IRAK and TRAF6 to the receptor complex finally leading to NF-κB activation. An analogous pathway works in TNF signaling where TRADD and RIP1 are recruited instead of MyD88 and TRAF2 instead of TRAF6.
The NOX family is, however, not the only one of interest in the context of redox regulation of NF-κB. A steadily increasing number of observations document that a coactivation of LOX may be equally important. Overexpression of GPx4, which preferentially reduces lipid hydroperoxides, almost abrogated IL-1β-induced NF-κB activation in endothelial cells, whereas changes in GPx1 had a minor effect (45). Similarly, lipid peroxidation and not H2O2 was shown to enhance the activation of NF-κB by TNF in the human endothelial cell line ECV304, whereas it appeared not to be involved in IL-1-induced NF-κB activation (43). The 5-LOX inhibitor AA861 inhibited NF-κB activation in parallel with leukotriene B4 production in A549 cells (70). By similar approaches Bonizzi et al. demonstrated that NF-κB activation by IL-1β depends on 5-LOX in lymphoid cells, but not in epithelial cells, whereas it depended on NOX in monocytes (39). More recently, activation of TLR8 was demonstrated to induce the phosphorylation of the cytosolic phospholipase A2α (cPLA2α) and to promote 5-LOX translocation, thus stimulating the synthesis of inflammatory leukotrienes (165). The pivotal kinase involved in activating 5-LOX is likely PKCα, which has for long been known to phosphorylate cPLA2 (285).
The precise mechanisms how the activation of TNFRs or TLRs coactivates NOXs and/or LOXs likely vary with the receptor type, the stimulus, and the tissue. In any case, however, the NF-κB activation consistently occurs under conditions in which the production of O2•−/H2O2 and lipid hydroperoxides is drastically enhanced, which warrants a serious consideration of the interference of these oxidants with the NF-κB-activating cascades.
2. Modulation of NF-κB activation via redox-sensitive phosphorylation
As outlined NF-κB activation appears to benefit from oxidative events in the cytosol that result in increased phosphorylation of many of its components, thus facilitating nuclear import and ultimately transactivation. A general PTK/PTP imbalance causing an increased NF-κB activity is indeed observed in aged and LPS-treated rats (226). In principle, such enhanced phosphorylation state may be achieved by oxidative activation of protein kinases or oxidative inactivation of phosphatases (see section II.D.3). In particular, the p65 subunit of NF-κB requires phosphorylation at multiple sites for translocation, transactivation, and other activities (Fig. 14). These phosphorylations involve several kinases, as has been amply reviewed in refs. (147, 172, 173, 363, 501). Phosphorylation of p65 and release from IκB were, in fact, the first regulatory steps of the cascade observed (341, 343) and found to be required for DNA binding of NF-κB and the recruitment of coactivators. The possible regulation of NF-κB via oxidative kinase activation has lately been reviewed under thorough consideration of possible methodological artefacts and tissue specificities (363). A somewhat bold summary of this review would be that oxidative modification of kinases has so far not been convincingly shown to be involved in in vivo NF-κB activation, whereas oxidative inactivation is commonly observed.
FIG. 14.

Phosphorylation sites of p65 and kinases and phosphatases involved. Phosphorylation at Ser276 prevents interaction of the Rel homology domain (RHD) with the C-terminus of p65. Thereby, DNA binding and interaction with CBP and p300 is facilitated. Similarly, Ser311 phosphorylation enhances the interaction with CBP/p300. Phosphorylation at Thr435 promoter- and cell-type-specifically decreases histone deacetylase (HDAC) binding after TNF stimulation. Dephosphorylation by PP4 (526) appears to be cell type and promoter specific. Phosphorylation at Ser468 supports nuclear import. Phosphorylation at Thr505 inhibits transactivation activity due to increased association with HDAC1. The consequence of Ser529 phosphorylation is unclear; that of Ser536 affects transcriptional acitivity in a cell-specific way. For more detailed information see text (see section IV.D.2).
The Ser/Thr kinase Akt, which itself is activated by phosphorylation at Thr308 and Ser473 via the PI3K pathway, phosphorylates p65 in its DNA-binding domain, the precise site being unknown (62) (Fig. 14). Akt itself is redox sensitive, being inactivated by H2O2 due to disulfide formation between Cys297 and 311 and reactivated by Grx (338). The cAMP-dependent kinase PKAc phosphorylates p65 in the cytosol at Ser276 (535), as do the mitogen- and stress-activated protein kinases (MSK)-1/2 in the nucleus (500). These phosphorylations facilitate release from the cytosolic complex or enhance transcriptional activity, respectively (500, 534). The activity of protein kinase A (PKA), though, is inhibited by oxidants, whereas the redox-sensitivity of the MSKs (and ribosomal S6 kinase 1 [RSK-1], see below) appears not to be investigated (363). PKC-type kinases have been reported to be activated by H2O2 and other oxidants (153) due to oxidation of Zn-coordinated cysteines in their N-terminal regulatory domain (152), but inactivated by cysteine oxidation/alkylation of the catalytic domains, the prevailing event at a particular phase of the activation process remaining unclear (154). PKCζ, which interacts with the NF-κB activation cascade at multiple sites, also phosphorylates Ser311 of p65 (279), thereby promoting association with the coactivator CBP (102), but, like other PKCs, also PKCζ is inactivated, for example, by peroxynitrite (252). CK2 phosphorylates p65 at Ser529 with so far unknown consequences (32, 505) and at Thr435, which decreases HDAC binding (352). Apparently, the redox-sensitivity of this kinase also remains to be investigated. IKKβ and IKKɛ phosphorylate p65 at Ser468, which supports nuclear import (316), whereas phosphorylation by GSK3β rather inhibited the activity of p65 (53). More recently, Ser468-phosphorylated p65 has been shown to be preferentially ubiquitinated and degraded in the nucleus, leading to the termination of NF-κB-dependent gene expression (143, 307). Finally, p65 is phosphorylated at Ser536 by RSK-1 and, more typically, by IKKs. Phosphorylation at Ser536 is widely considered to be the decisive event that facilitates the release of p50/p65 from IκB in the cytosol (16, 303, 457), although in T-cells it appears to delay nuclear import (316). However, the phosphorylation at Ser536 also plays an important role in the nucleus, where it prevents recruitment of HDAC C3 to chromatin, allows its own acetylation at Lys310 by p300 (185) and impairs affinity to nuclear IκB and, thus, export from the nucleus (36). IKKα has indeed been reported to be recruited to the nucleus together with p65 and to there phosphorylate chromatin-bound p65 (185). IKKβ, the key player in NF-κB activation (16, 416), is redox sensitive in a sense that it cannot account for an activation of NF-κB by oxidants either. IKKβ can become directly oxidized by H2O2 at Cys179. This cysteine residue is located between the Ser residues 177 and 181, which have to be phosphorylated for activation. In consequence, oxidation of the residue prevents phosphorylation and results in inhibition of IKKβ (260, 363). By analogy, oxidative inactivation may also be assumed for IKKα and IKKɛ. A seemingly contradictory report claiming an oxidative activation of IKKα and β associated with enhanced phosphorylation at Ser180 or 181, respectively (231), is likely explained by H2O2-mediated inhibition of phosphatases (see below). In this context it may be revealing that Cys179 of IKKβ can also be alkylated by 15d-PGJ2. The modification of IKKβ with this bulky residue proved to be equally inhibitory, likely due to prevention of IKKβ phosphorylation. Since 15d-PGJ2 is a product of NF-κB-induced COX2, it is considered to contribute to the resolution of inflammation via IKKβ modification (408).
Thus, the enhanced phosphorylation state observed during, and obligatory for, NF-κB activation cannot likely be attributed to oxidative modification of any of the kinases with the possible exception of PKCs and oxidatively activated RTKs that initiate the atypical pathway (339) (see also section II.D.3). In contrast, oxidative inhibition of PPs at each level of the activation cascade may well account for the often seen enhanced NF-κB activation under oxidizing conditions. A systematic RNAi screen of phosphatases in mouse astrocytes identified a total of 19 phosphatases regulating NF-κB transcriptional activity. In particular, the PP2A-type enzymes were found associated with the IKK, NF-κB, and TRAF2 complexes (286). Depending on the cell type, the NF-κB activation mechanism and cross-talking signaling cascades, even more and other phosphatases may come into play (Fig. 12).
The central event in the typical pathway, IKKβ activation by phosphorylation via TAK1, could be reversed by PP2A (92) or PP2Cη-2 (179) and that of IKKα by PP2Cβ (384). Equally, IKKβ phosphorylation by the lysophosphatidic acid-induced MAP kinase kinase kinase (MEKK3) is counteracted by PP2A (465). TNFα-mediated IKKβ activation is reversed by PP2Cα and β (466). Similarly, the phosphorylation of p65 (Fig. 14) at Ser468 is reversed by PP1/PP2A (53), whereas the nuclear Ser/Thr-specific phosphatase PP4 was shown to act on Thr435 (526). Most recently, the wild-type p53-induced phosphatase-1 (WIP1), also known as PP2Cδ, has been identified as a key phosphatase for the most important p65 phosphorylation site Ser536 and, accordingly, is considered to act as a shut-off device of the NF-κB system (66). Interestingly, NF-κB upregulates WIP1 expression, thus contributing to the termination of its activity (299). Upstream events, in particular the PI3K/Akt pathway that substantially contributes to NF-κB activation by IL-1β, TNFα, and LPS (233), offer further possibilities for oxidative interference via phosphatase inhibition (Fig. 12): PI3K phosphorylates phosphatidyl inositol 3,4 bisphosphate [PIP(3,4)P2] to phosphatidyl inositol 3,4,5 trisphosphate [PIP(3,4,5)P3]. Interference with NF-κB activation by phosphatases is possible already at these early steps, since formation of PIP(3,4,5)P3 is reversed by the action of PTEN and SHIP-1. PIP(3,4,5)P3 in turn activates the phosphoinositide-dependent protein kinase-1 (PDK1). PDK1 phosphorylates Akt at Thr308 and Ser473, and its inactivating de-phosphorylation is again achieved by PP2A (291). Akt can by itself phosphorylate IKKα at Tyr23 and this phosphorylation is required for the phosphorylation of IκB (16).
Out of the numerous phosphatases that are possibly involved in counteracting NF-κB activation (298) a great deal can be inactivated by reversible or irreversible thiol oxidation, glutathionylation, or other mechanism involving oxidative processes, PTEN, SHIP-1, PP2A-, and PP2C-type enzymes being the most quoted suspects.
3. Regulation of NF-κB by redoxin systems
Trx, the prototype of the redoxins that is characterized by the WCGPC motif, has for long been recognized to interfere with NF-κB activation (128); transient cytosolic overexpression of Trx dampened NF-κB activation upon stimulation by phorbolester (429), whereas in the nucleus Trx was demonstrated to be of pivotal importance to keep the p50 subunit of NF-κB in the reduced state, which is required for DNA binding (170). These early findings for the first time revealed that the NF-κB system responds to redox events that are at best indirectly related to the then already widely accepted redox sensitivity of the phosphorylation/de-phosphorylation balance.
In contrast to the situation in apoptotic signaling (see section II.D.1) (415), a direct interaction of cytosolic Trx with any of the components of the NF-κB system could not be corroborated. In this context Trx appears to be replaced by other redoxins (see below). Inhibition of NF-κB activation by cytosolic Trx is now likely explained by its role as substrate of Prx I and II, which compete for hydroperoxides that facilitate NF-κB activation via other mechanisms (398) (see section II.E). In the nucleus, however, Trx directly interacts with p50. While in the redox circuits of lower organisms reduced Trx is often used to shut off redox signaling (see Figs. 1 and 2; section II), an opposite role is adopted in the mammalian NF-κB system: it reduces critical cysteines of p50, thereby enabling DNA binding of the p50/p65 complex and initiating target gene activation. Cys62 within the N-terminal region of p50 has been identified as the pivotal residue (483), which was a surprise, since this cysteine is most easily oxidized in the cytoplasm. However, when NF-κB has entered the nucleus, Cys62 is rapidly reduced by either Trx/TrxR (169, 184, 315) or apyrimidinic endonuclease 1/redox factor 1 (APE1/Ref-1) (7, 346). Similarly, NF-κB, which is translocated to mitochondria upon stimulation by apoptotic signals, including TNFα (529), is kept active via reduction of p65 by Trx2 (386). The response is complex: NF-κB negatively regulates the expression of proteins for oxidative phosphorylation (OXPHOS). Simultaneously, however, Trx2 interacts with the glucocorticoid receptor, which enhances OXPHOS expression. Also, TNFα-driven mitochondrial O2•− formation and apoptosis is inhibited by Trx2 (386). It is further discussed that activated mitochondrial NF-κB might be translocated back to the cytosol and the nucleus to initiate transcription of antiapoptotic genes such as MnSOD (386). The mechanism of p65/Trx2 interaction in mitochondria has not yet been clarified but is thought to be analogous to that of p50/Trx1 interaction, because only oxidized p65 was found to be bound to (reduced) Trx2, which suggests the conventional disulfide reductase activity to be involved (386).
Also, Grxs, characterized by the CPYC motif, interact with the NF-κB system at several levels. Apart from de-glutathionylating kinases and phosphatases (see sections II.D.3 and IV.D.2), Grx have been implicated in direct interaction with p50 and p65. Oxidation of the critical Cys62 to a sulfenic acid followed by S-glutathionylation has been elucidated as further mechanism for inhibition of DNA binding (376). Also, S-glutathionylation of the p65 subunit inhibited its binding to DNA (388). The reacting cysteine has not been identified; it might be Cys38 in the DNA-binding loop, since this cysteine is also a target for nitrosylation (147). Both glutathionylations are reversed by Grx (376, 388).
Another redoxin, the TRP 14 (TRP14), which is characterized by a WCPDC motif, was shown to inhibit TNFα-induced NF-κB activation by the typical pathway (223) (Fig. 4D). As its congeners, TRP14 acts as a disulfide reductase, its substrate being the LC8. In thereduced form LC8 binds to IκB and inhibits its phosphorylation by IKKs and subsequent degradation. By maintaining LC8 in the reduced state, TRP14 prevents cytosolic activation of NF-κB. Exposure to oxidants or treatment with TNFα, IL-1, or LPS known to signal via H2O2 production leads to the formation of LC8 dimers in which two molecules are linked via a disulfide bridge (515), resulting in its dissociation from IκB (224, 227). This mode of redox-dependent inhibition of protein activities by interaction with binding partners is analogous to the association of Trx to ASK-1, an upstream activator of the c-Jun N-terminal kinase (INK) and p-38 MAPK signaling pathways (415) (Fig. 4A). In contrast, in the NF-κB system it is not the redoxin itself that blocks signaling but the redoxin substrate LC8. This seemingly tiny difference opens up the still unresolved question how regulation by TRP14/LC8 interaction is integrated into the metabolic environment. Shutting-off NF-κB signaling thus activated appears to be clarified. Oxidized TRP14 is reduced by the cytosolic form of Trx reductase TrxR1, but not by the mitochondrial TrxR2 (222, 224). Remains the questions how the activating oxidation of LC8 is achieved. Certainly, LC8 could be considered the sensor for ROOH. However, oxidative linking of two remote cysteines in a dimeric protein (see Fig. 4D) is a priori not likely to happen spontaneously, nor has this possibility so far been supported experimentally. TRP14 having a redox potential (−257 mV) (222) between those of Trxs and Grxs might be able to catalyze thiol/disulfide exchange both ways and thus might be the ROOH acceptor that oxidizes LC8, but the reaction of TRP14 with H2O2 is reportedly just four times as fast as that of Trx, which has been rated as disappointingly slow (224). The possibility that TRP14, in analogy to other redoxins, might be oxidized by an ROOH-sensing Prx has evidently been ruled out experimentally (224). We are thus left with the options that LC8 itself has the capacity to sense ROOH or is oxidized by a still unknown upstream redox sensor.
Most recently, also Nrx has been demonstrated to be a specific negative regulator of LPS-induced TLR4-mediated signaling (171). Nrx, like tryparedoxin (see section II.D.1), belongs to the redoxin subfamily that is characterized by a WCPPC motif. In mammals, Nrx appears to be the only redoxin displaying this particular motif (137). It had so far been primarily discussed as a redox-sensitive regulator of the Wnt/β-catenin pathway (Fig. 4B). In this context reduced Nrx noncovalently binds to the adaptor protein Dvl, thereby silencing Wnt-responsive signaling. Oxidation of the WCPPC motif in Nrx leads to dissociation from Dvl and, in consequence, to activation of the transcription factor β-catenin. In TLR4-dependent signaling Nrx was found to be bound to the adaptor protein Fli-1 (for flightless in Drosophila), and control experiments with truncated mutants revealed that the WCPPC motif is essential for binding to Fli-1 (171). Moreover, LPS-triggered TLR4 stimulation and NF-κB activation was substantially enhanced in cells from Nrx−/− mice, which corroborates the functional relevance of Nrx/Fli-1 interaction (Fig. 4C). In respect to the mechanism of the inhibition of TLR4 signaling by Nrx, complex formation between Fli-1 and the adaptor protein MyD88 comes into play. MyD88 is essential for NF-κB activation mediated by TLRs and IL-1R where it recruits IRAKs to the receptor complexes (Fig. 10) (219, 244, 290, 427). Fli-1 inhibits TLR signaling through binding to MyD88 (507) and sequestering the adaptor in the cytosol. This sequestration of MyD88 has now been shown to require Nrx forming a ternary complex of Fli-1, MyD88, and Nrx (171).
Whether this novel adaptor function of Nrx is redox-controlled remains to be established. It is tempting to speculate that Nrx, like in the Wnt/β-catenin pathway, is released by oxidation, thus allowing TLR signaling to proceed. Interestingly, two more sequence-related proteins were shown to similarly bind to Fli-1: the rod-derived cone viability factor (RdCVF), expressed by photoreceptors and the chromosome 9 open reading frame 121 (C9orf121) protein (171). The former has the active site motif ACPQC, which can be rated as potentially active in disulfide reduction, in the latter the homologous sequence is RCAPS, which is incompatible with disulfide reductase activity but nevertheless could lead to the formation of mixed disulfides. However, also inactive Cys to Ser mutants of Nrx did bind to Fli-1, which implies that the active site of the redoxins is not likely involved in their interaction with Fli-1, but leaves open that redox-state-dependent structural changes affect protein/protein interaction. Alternative mechanisms, though, cannot be ruled out, since Nrx also interacts with other proteins such as, for example, HDAC6, Dvl1-3 (171), and the PP2A (137). The routes leading to oxidation and reduction of Nrx remain to be worked out. For the prototype tryparedoxin the oxidizing partner is a Prx, whereas the reduction is achieved by a typical flavin-dependent disulfide reductase mediated by the low-molecular-weight thiol trypanothione (199, 349). The search for an analogous metabolic context may be helpful in finally defining the precise role of this novel redox mediator in transcriptional activation.
E. Termination of NF-κB signaling
Out of the 39,679 publications with the keyword NF-κB (PubMed by July 29, 2010) only a vanishing proportion deals with the termination of signaling, which therefore is still poorly understood (173). This obvious lack of interest is quite surprising, since termination of the signaling cascade is pivotal to the problem why an exposure to PAMPs, DAMPs, or pro-inflammatory cytokines only exceptionally turns into a fulminant or chronic inflammation. In fact, the over 100 genes activated by NF-κB comprise also those encoding pro-inflammatory cytokines, whereby NF-κB would trigger perpetuation and even amplification of an inflammatory response, if not adequately balanced. Clearly, the organism requires tools that discriminate between an irrelevant exposure to bacterial structures or local tissue damage from systemic infections or polytrauma, respectively.
An essential way to terminate NF-κB signaling is the re-synthesis of IκBs, which is controlled by NF-κB itself (128, 401). Thereby, not only the resting state of the NF-κB system can be re-established by sequestering the transcription factor in the cytosol; also nuclear degradation of p65 is initiated. In particular IκBα binds to NF-κB dimers in the nucleus and transports them back to the cytosol as inactive complex. Nuclear export of p65 via IκBα binding is facilitated by nitration of Tyr66 and Tyr152 and nitrosation of Cys38 on p65 (147, 367). IκBα also dissociates p65 from DNA, facilitates its proteasomal degradation in the nucleus, and, thus, terminates transcription (340, 413). Simultaneously, NF-κB activity may be terminated by binding p50/p50 dimers, which have for long been recognized to inhibit NF-κB-mediated transcription (15). Oxidation or nitrosation of the critical Cys62 of p50 also contributes to nuclear export of NF-κB (147). Finally, binding of NF-κB to IκBα and nuclear export is facilitated by de-acetylation of p65 through the HDAC3. HDAC3 appears to be negatively affected by phosphorylation, nitration, or carbonylation (206, 523).
Surprisingly, IKKα, which is part of the central activation complex in the typical pathway and essential for activating the alternative pathway (see section IV.C), proved to adopt the role of a terminator in TLR4-mediated typical NF-κB signaling (271, 272). In macrophages stimulated with LPS, IKKα repressed NF-κB activity by enhancing the proteasomal degradation of p65. The biological relevance of this novel role of IKKα is evidenced by a sepsis-like response and enhanced mortality upon LPS stimulation as well as enhanced bacterial clearance in mice bearing an inactive IKKα variant (272).
The NF-κB system not only manages its own termination by de novo synthesis of system components that have been degraded during the activation process, but also via expression of other target genes. Particular interesting examples are iNOS, LOXs, and COX2. The iNOS product •NO, as long as it does not react to ONOO− by simultaneous O2•− formation, is clearly protective in the context of inflammation (196). The main products of 5-LOX, leukotriene B4 and D4, are clearly pro-inflammatory, but the lipoxins derived from 5-LOX or other LOX are currently considered to be key mediators of the resolution of inflammation (447). COX2, generally rated as the prototype of a pro-inflammatory enzyme, is, however, also Janus-faced, since some of its many products play distinct roles depending on the inflammatory phase: (i) PGE2, being responsible for the classical signs of inflammation such as pain, swelling, and redness, also inhibits superoxide formation in chemokine-stimulated neutrophils (375). PGE2 also interferes with NF-κB activation by selectively inhibiting nuclear import of p65, thus favoring a nuclear accumulation of inhibitory p50/p50 dimers in synovial fibroblasts (150). (ii) Prostacyclin (PGI2) known to strongly inhibit platelet aggregation and to cause vasodilatation (496), also inhibits leukocyte adherence to the endothelial layer (44). Further, metabolically stable PGI2 analogs (129) were shown to dampen PAMP-triggered O2•− formation in polymorphonuclear leukocytes (PMNs) (462) and protected rats against a lethal endotoxin dosage (435). The massive rise in PGI2 levels consistently observed in septic conditions may therefore be re-interpreted to beneficially interfere with the deadly scenario resulting from a massive TLR4-mediated NF-κB activation rather than to kill patients (124, 295, 530). (iii) An activation of Nrf2 and a simultaneous inhibition of NF-κB activity were observed in smooth muscle cells overexpressing GPx4 or 15-LOX (20). Subsequent upregulation of HO-1 inhibited IL-1-induced NF-κB activation and expression of VCAM-1. Similarly, disruption of Nrf2 enhanced NF-κB activity, production of pro-inflammatory cytokines, and ICAM expression in the brain of mice (225). (iv) More recently, COX2-derived cyclopentenone prostaglandins, in particular 15d-PGJ2, have been implicated in the resolution of inflammation (145). 15d-PGJ2 is a ligand of PPARγ (195) and thereby acts as a repressor of LPS-stimulated AP-1, STAT1, and NF-κB activation (402). Being a strong electrophile, 15d-PGJ2 can react with susceptible cysteines in a set of cytosolic and nuclear proteins (422), one of them being Keap1 (189). By targeting Keap1, 15d-PGJ2 activates Nrf2 and, thus, initiates gene transcription with an overall anti-inflammatory result (see sections III.B and III.D) (Fig. 15).
FIG. 15.

NF-κB and Nrf2, the Yin and Yang of the inflammatory response. Occupation of receptors by pathogen-associated molecular patterns, damage-associated molecular pattern (DAMPs), TNFα, or IL-1 leads to superoxide formation in the redoxosome by NOX. H2O2 formed by SOD favors activation of NF-κB at multiple sites though enhancing protein phosphorylation, but also oxidizes Keap1 in the Nrf2 system. While NF-κB tends to enhance and perpetuate the inflammatory response by triggering the expression of pro-inflammatory cytokines, Nrf2 activation through Keap1 oxidation dampens pro-inflammatory signaling by expression of peroxidases and other anti-inflammatory proteins. As E3-ligase, Keap1 also primes IKKβ to degradation via ubiquitination, thereby directly interfering with NF-κB activation. For sake of clarity, only NOX-derived H2O2 is shown as oxidant signal. Depending on the cellular system and the inflammatory stimulus, NOX-derived H2O2 may be supported or replaced by mitochondrial H2O2, lipoxygenase products, and S-alkylating electrophiles derived there from.
The latest concept on resolution of inflammation implicates NOX-derived H2O2 as the major mediator to terminate inflammatory processes (441). This proposal obviously conflicts with the wide-spread belief that the role of the coactivation of NOXs by cytokine, TNF, and TLR occupation consist in fortifying signaling to NF-κB. However, by means of a model of sterile lung inflammation triggered by intratracheal application of zymosan or LPS, the authors unambiguously demonstrated that inflammation was exaggerated and more progressive in mice deficient in phagocytic NOX2 as compared to wild-type mice. In parallel, whole lung NF-κB activation was dramatically increased and persisted for at least 6 days in the NOX2-deficient mice; in consequence, the release of NF-κB-dependent production of cytokines such as TNFα, granulocyte colony-stimulating factor (G-CSF), and IL-17 was similarly increased and prolonged in broncho-alveolar lavage macrophages from the deficient mice. The enhanced response was observed irrespective of p47phox or gp91phox being knocked out, and practically identical results were obtained with peripheral blood monocytes of chronic granulomatous disease patients with a defective NOX. Clearly, NOX-derived H2O2 in macrophages and monocytes rather inhibited than supported NF-κB activation. The mechanism remains to be established, but conceivably oxidative inhibition of NF-κB-activating kinases in these systems is more important than that of the counteracting phosphatases. Also, Keap1, the key player in Nrf2 activation, has recently been shown to downregulate NF-κB activation by priming IKKβ to degradation via ubiquitination (273). More importantly, the classical role of Keap1 as sensor in the Nrf2 system in part explains the unexpected role of H2O2 as terminator of NF-κB activation. Nrf2 activation in NOX-deficient macrophages stimulated with zymosan was completely abrogated, whereas it was clearly detectable in control cells already 1 h after zymosan exposure (441). Moreover, Nrf2−/− mice behaved similar to NOX-deficient mice, and the wild-type phenotype could almost be restored in NOX-deficient mice by administration of a compound (1-[2-cyano-3,12-dioxooleana-1,9(11)-dien-28-oyl]imidazole) that activates Nrf2 via direct, that is, H2O2-independent Keap1 modification (441).
In conclusion, activation of the Keap1/Nrf2 system be it by products of unspecific lipid peroxidation, cyclopentenone prostaglandins, or NOX-derived H2O2, appears to be the prominent mechanism to terminate the NF-κB-driven immune response.
F. Synopsis of the NF-κB system
NF-κB is a pleiotropic transcription factor that regulates expression of more than hundred genes that collectively dominate the innate and acquired immune response and associated inflammation. NF-κB is activated via different receptor families which signal to NF-κB by distinct phosphorylation cascades, the common denominator being release from an inhibitory cytosolic complex and nuclear import of the active transcription factor, which typically is the p50/p65 heterodimer. Receptor occupation that leads to NF-κB activation is regularly associated with activation of NOX-type enzymes, the nature of which depends on the receptor type. Concomitant activation of LOX has also been observed. Redox regulation of the activating phosphorylation cascades has been debated since the discovery of the system and remains a matter of debate 20 years thereafter. Likely this issue has to be more critically viewed under consideration of the heterogeneity of the pathways. By triggering the production of its own stimuli such as IL-1 and TNFα, the system tends to become self-amplifying, which raises the question of termination. Restoring the resting state involves nuclear export, de novo synthesis of cascade components that were degraded during the activation process and synthesis of enzymes that produce molecules cross-talking to antagonizing systems, in particular to the anti-inflammatory Keap1/Nrf2 system. In this context, Nrf2 activation via modification or oxidation of Keap1 by COX products such as 15d-PGJ2, H2O2 or other oxidants arising from coactivation of NOX- and LOX-type enzymes are most attractive candidates. The NF-κB system, thus, generates the signals required for Nrf2 activation and thereby complements the hormetic response to a broad scope of exogenous challenges.
V. Loose Ends and Perspectives
It was neither our intention nor a realistic option to in-depth review what has been published on redox-dependent transcriptional gene activation since this topic became a hot issue about two decades ago. We rather tried to clarify basic chemical principles that might reasonably work in redox-regulation of biological processes and to demonstrate the relevance of these principles to two seemingly well-documented examples of redox-regulated mammalian transcription systems, the Nrf2 and the NF-κB systems, which, acting in concert, appear to be the key players in the hormetic and inflammatory responses to exogenous stressors (456). Needless to state that we ended up with a considerable number of loose ends.
Plausible regulatory circuits to adapt life to environmental oxidant changes have been worked out for procaryotes and lower eukaryotes such as yeast (333) (see sections II.A and II.D.1) and trypanosomatids (452). In these cases, not more than two dozen of solid investigations were needed to generate a concise and satisfying picture on the signaling molecules, the sensors, the transducer(s), affected genes, and modulating mechanisms (see Figs. 1 and 2). When switching to the best investigated redox-responsive mammalian transcription systems we chose as paradigms, the Keap1/Nrf2 (see section III) and the NF-κB system (see section IV), we felt confronted with a horror scenario: thousands of related original publications and hundreds of reviews were unable to generate a comprehensive and generally accepted concept and the daily increment of about 10 publications, which often conflicted with seemingly established views or opened up new perspectives, permanently forced us to revise our article. We therefore have to apologize for just delivering a snap shot of current opinions, which, beyond, is biased by a chemical/enzymological point of view.
For sure, the difficulties in understanding redox regulation in mammalian organisms are easily explained by the complexity of the systems. However, these inherent difficulties are not the only reasons for the persistent confusion. Major deficiencies one stumbles across in the field are (i) lack of chemical precision, (ii) lack of (bio)chemical kinetics, and (iii) insufficient consideration of time and spatial organization of biological processes.
To (i): The persistent reluctance to name the chemical entity supposed to exert a biological effect does not solve problems; the current talking of signaling by poorly defined chemicals such as ROS, NRS, or radical species simply widens the range of chemical reactions to unpredictability. It is, however, neither impossible nor irrelevant to define the chemical entity that is supposed to signal. As outlined in section II, signaling by radicals is rather the exception than the rule and is likely restricted to •NO and O2•−; the most common oxidant signaling molecule is H2O2; it may be a LOX product, under special conditions also ONOO−, and under severe oxidative challenge an electrophilic break-down product of oxidized lipids. Knowing this, the possibly resulting chemistry is defined and its relevance under biological conditions amply documented by related enzymological studies. Making better use of the accumulated knowledge on the reaction mechanisms of, for example, thiol peroxidases and S-transferases will certainly be helpful to further define modifications of proteins relevant to regulatory processes. Another source of confusion is the uncritical use of the term “antioxidant” for a huge number of chemically unrelated compounds that have the only common denominator to react with most aggressive oxygen-centered radicals such as •OH in physiologically meaningless in vitro settings. In a biological context, these compounds may indeed act as antioxidants in the chemical definition, which means compounds interfering with an oxidant-initiated free-radical-mediated chain reaction. Yet, experimental evidence supporting this concept is very scarce. More likely the antioxidants are simply reductants, S-modifying agents, redox-cyclers, cofactors, or constituents of enzymes, or display their own pharmacodynamic profile that sometimes is unrelated to redox chemistry. Facing these uncertainties, one can hardly expect conclusive data from uncritically exposing tissue cultures or animals to massive dosages of antioxidants. A revealing example is the misleading historical name ARE for EpRE, the target element of Nrf2 (see section III.B). The term “antioxidant-responsive element” was coined because the element responded to test-tube antioxidants that in vivo produced O2•− or H2O2 due to autoxidation or redox cycling.
To (ii): The lack of kinetic data in redox regulation is a problem indeed. It has become fashionable to declare signaling components with reactive cysteines to be sensors for H2O2, ROOH, peroxynitrite, or alkylants (see section II.D). These assignments, however, are overwhelmingly based on swampy grounds. After having screened the pertinent literature, we are aware of a total of 3 investigations (11, 87, 459) that documented the reactivity of such cysteines in presumed sensors that are not peroxidases by appropriate kinetic measurements: the only satisfying case was OxyR. With a rate constant of 105 M−1 s−1 this sensor is kinetically competitive enough to sense H2O2 in the presence of competing peroxidases. In contrast, the known rate constants for oxidative inactivation of phosphatases had to be rated as noncompetitive (87, 459) (see section II.D.3). For all remaining thiol-based H2O2 sensors experimental data that would corroborate their kinetic competiveness are missing. The kinetoplast system (see section II.D.1) works with a tryparedoxin peroxidase as H2O2 sensor for which competitive rate constants have been established (349, 489, 490). The H2O2 sensors of the yeast systems (see section II.D.1) may by analogy be regarded as competitive, as they are related Prxs or equally efficient GPx-type peroxidases (487). A sensing function of the mammalian Keap1 is corroborated by circumstantial evidence. For the phosphatases, kinases, redoxins, and others, a direct sensing function is rather unlikely (134), which promises future surprises. Moreover, the interpretation of redox changes in regulatory proteins obtained in vitro is often impeded by the experimental settings in which they are generated. Tissue cultures, if not adequately supplemented, tend to be deficient in selenium and, thus, in GPxs (278) and Trx reductases (308). Being thereby deprived of both GSH- and Trx-dependent peroxidase systems, protein oxidations may occur upon H2O2 challenge that are physiologically irrelevant, but even well-supplemented culture cells, when suspended a medium up to 10 mM H2O2 (358), may be anticipated to have their reductive capacities (GSH and Trx pools) exhausted within minutes, allowing the excess H2O2 to attack protein thiols that physiologically would never be affected. In short, whereas the chemical principles of redox regulation by protein thiol modifications have quite convincingly been worked out, the physiological relevance of individual proposals may often be questioned, because of missing kinetic data and testing conditions that favor the generation of artefacts.
To (iii): The most challenging task ahead, however, is the resolution of system kinetics at a more extended time scale. The activation of transcription factors generally operates by hit and run mechanisms: receptor occupation followed by downstream processes. Receptor activation and downstream signaling requires milliseconds to minutes; target genes expression extends into hours and days; and the consequences thereof, if cell recruitment and differentiation is involved, may take weeks. Cross-talk between different transcription factor systems already occurs in the early phases, if signaling components are shared, but regularly dominates later stages. The need of a more serious consideration of these aspects is, again, revealed by the Yin and Yang interplay of NF-κB and Nrf2 (Fig. 15). Depending on kind, strength, and persistence of a PAMP or DAMP exposure, a hormetic response without any obvious inflammatory process, a self-healing or chronic inflammation, or a deadly septic crisis may be triggered. The adaptive response to a subcritical challenge likely reflects the early and delayed counteracting activation of Nrf2. The latter, in concert with various growth factor systems, also contributes to self-healing inflammation. The switch to chronic inflammation, if not explained by persistent challenge, is poorly understood. Septic shock, finally, the prototype of an oxidative stress disease, presents as an extreme dysbalance of several transcription factor systems and may become lethal at various stages and by different pathogenic mechanisms. The early phase is undoubtedly caused by a massive overactivation of NF-κB due to PAMP or DAMP exposure with oxidative burst in phagocytes and excessive expression of adhesion factors, which, in combination with vasoconstriction and intravascular coagulation, leads to endothelial dysfunction, collapse of the microcirculation, tissue hypoxia, and multiorgan failure. Later counteracting events such as activation of the fibrinolytic system and vasodilation due to exorbitant production of PGI2 and •NO may resolve the circulatory block, but inevitably result in a critical drop in blood pressure, but even if the early crises of septicemia are overcome, patients tend to die weeks later in a status of immune paralysis (37). Evidently, the final outcome of a septic shock has nothing in common with the initial event which in essence is the beneficial activation of the innate immune response to cope with intruded pathogens or tissue damage. The concommittant oxidative burst will not only activate Nrf2 but also enhance signaling through all pathways that positively respond to oxidants; NF-κB not only induces circulatory collaps but also promotes TNFα-mediated apoptosis; tissue ischemia must trigger the hypoxic response; resumption of circulation may result in reperfusion injury; the neuronal and hormonal alarm that accompanies the entire process alerts a lot more signaling cascades; and with progressing time primary target cells such as PMNs and macrophages undergo apoptosis and are replaced by populations of lymphocytes responding differently to different signals, anti-inflammatory cytokines becoming predominant. As net outcome, the exaggerated activation of the self-protective innate immune response turns over weeks into a life-threatening anergy of the immune system (37, 369).
A clinical exploitation of the emerging knowledge clearly demands a better understanding of the long-term consequences of a transcription factor activation, which result from multiple and poorly understood cross-talks with other systems. Nevertheless, modulation of the NF-κB or Nrf2 system has for long been, and still is, an attractive clinical perspective, in particular for the treatment of inflammatory diseases and the prevention of cancer. However, the complexity of an inflammatory response we could only briefly address, as well as the fragmentary knowledge, hampers a fast clinical implementation, and sepsis may again serve to demonstrate the inherent difficulties. Already in the eighties therapeutic success in sepsis, polytrauma, and reperfusion injury was expected from preventing tissue damage due to PAMP- or DAMP-induced oxidative burst and acute inflammatory responses (85, 404). Numerous animal experiments were performed with application of SOD alone (436) or together with catalase (510) to remove excess O2•−, H2O2, or ONOO−(25), thiol- or selenium-based antioxidants (509), iron chelators to prevent Fenton chemistry (25), TNFα (132) or LPS antibodies (390, 404) or IL-1 antagonists (356, 502), mostly with encouraging results in well defined experimental settings. But none of these intervention strategies has so far been crowned by clinically success. The major reason for the failure of these strategies has to be seen in their limited feasibility under clinical conditions: first of all, at the time of clinical diagnosis, the self-amplifying inflammatory cascades are already too advanced for a meaningful intervention. Second, animal sepsis models that are designed for, for example, a precisely defined LPS dosage and an exact time point of intervention after challenge can hardly be extrapolated to the clinical situation with usually unknown onset and overlapping phases of the inflammatory process. Finally, all experimental strategies mentioned above focus on the correction of early dysregulation in septicaemia and it remains to be explored if any intervention with the early oxidant inflammatory processes can improve the overall clinical outcome. If so, a prophylactic blockade of the early inflammatory response still deserves interest in the treatment of polytrauma, where the sepsis-like syndrome develops with a predictable delay of two to 3 days.
More convincingly, hopes for better therapy of inflammation are based on the hormetic character of the NF-κB - Nrf2 interplay. It has for long been known that a sub-critical dosage of LPS induces tolerance to a lethal one (26), whereby pronounced cross-tolerance between LPS, other PAMPs, hyperbaric oxygen and inflammatory cytokines (IL-1, TNF) is observed (57). The mechanism of tolerance development is not fully understood (110). Inhibition of NF-κB transcriptional activity by p50 homodimers appears to be involved (537). However, circumstantial evidence suggests that this kind of tolerance development is, in part at least, caused by oxidative activation of the Keap1/Nrf2 system (see section III). As the latter can also be activated by natural and presumably safe electrophiles that by themselves do not necessarily induce any oxidative damage, such compounds are being considered for prophylactic treatment of high risk groups. However, this option still awaits a systematic clinical exploration.
A logical extrapolation of this hormetic concept seems to be the application of Nrf2 activators for prevention of inflammation-associated carcinogenesis. Indeed, many natural compounds activating Nrf2 such as sulfuraphane, curcumin, resveratrol, flavonoles and catechins are currently being promoted for chemoprevention. However, since evolution has designed the Keap1/Nrf2 axis as a system that only works on demand and for a limited time, there are good reasons to be concerned about the long term effects of a permanent Nrf2 activation. Liver damage due to persistent Nrf2 activation, although so far only documented for autophagy-deficient mice (257), may be taken as a warning.
The few spot-light may suffice to demonstrate clinical spin-offs are showing up at the horizon. However, as outlined in this article, the regulatory systems are extremely complex and a lot of gray and black areas have to be filled with solid data, before reliable predictions on any interference strategy can safely be made. The steps from bench to bedside, therefore, will hardly be quick ones.
Abbreviations Used
- 15d-PGJ2
15-deoxy-Δ12,14-prostaglandin J2
- AhR
aryl hydrocarbon receptor
- AP-1
activating protein-1
- ARE
antioxidant responsive element
- ARNT
aryl hydrocarbon receptor nuclear translocator
- ASK-1
apoptosis signal-regulating kinase-1
- Atf1
activating transcription factor-1, cyclic AMP dependent
- ATF4
activation transcription factor-4
- Atg
autophagy-related
- β-TrCP
β-transducing repeat containing protein
- Bach1
BTB and CNC homology-1
- BTB
broad complex/tramtrac/bric-a-brac
- bZIP
basic leucine zipper
- CBP
CREB-binding protein
- Cdc25
cell division cycle 25, dual specificity phosphatase from yeast
- CDK
cyclin-dependent kinase
- CDNB
chloro-dinitrobenzene
- c-FOS
cellular protooncogene which forms with c-jun the AP-1 transcription factor complex
- cIAPs
cellular inhibitor of apoptosis proteins
- CK2
casein kinase-2
- CNC
Cap'n Collar
- CNTF
ciliary neurotrophic factor
- COX
cyclooxygenase
- CREB
cAMP response element binding protein
- Crm1
chromosome region maintenance
- Cul3
Cullin-3
- Cys
cysteine
- DAMP
damage-associated molecular pattern
- DGR
double glycine repeat
- DLG
Asp-Leu-Gly
- DSP
dual specificity phosphatase
- Dvl
dishevelled
- ECH
erythroid cell-derived protein with CNC homology
- EGF
epidermal growth factor
- Egr
early growth response
- ELAM
endothelium leukocyte adhesion molecule
- EpRE
electrophile responsive element
- ERK
extracellular signal-regulated kinase
- ETGE
Glu-Thr-Gly-Glu
- Fli-1
flight less (drosophila)
- FOXO
Forkhead box O
- Fra-1
c-Fos-related antigen 1
- Fyn
membrane associated nonreceptor protein tyrosine kinase, of the Src family
- G-CSF
granulocyte colony-stimulating factor
- GPCR
G-protein-coupled receptor
- GPx
glutathione peroxidase
- GR
glucocorticoid receptor
- Grx
glutaredoxin
- GSH
glutathione
- GSK3β
glycogen synthase kinase-3β
- GSSG
glutathione disulfide
- GST
glutathione‐S‐transferase
- HAT
histone acetylase
- HDAC
histone deacetylase
- HIF‐1
hypoxia‐inducible factor 1
- HNE
4‐hydroxy‐nonenal
- HO‐1
heme oxygenase‐1
- ICAM
intercellular adhesion molecule‐1
- IKK
IκB kinase
- IL‐1
interleukin‐1
- iNOS
induced NO synthase
- IRAK
IL‐1R‐associated kinase
- IRP1
iron response element‐binding protein 1
- IVR
intervening region
- JNK
c‐jun N‐terminal kinase
- Keap1
kelch‐like ECH‐associated protein‐1
- LAR
leukocyte‐common antigen‐related (phosphatase)
- LC8
dynein light chain 8
- LOX
lipoxygenase
- LPS
lipopolysaccharide
- Maf
musculo‐aponeurotic fibrosarcoma
- MAPK
mitogen‐activated protein kinase
- MCP‐1
monocyte chemotactic protein‐1
- MEKK
MAP kinase kinase kinase
- MIP‐1
macrophage inflammatory protein‐1
- MnSOD
manganese superoxide dismutase
- MSK
mitogen‐ and stress‐activated protein kinase
- MyD88
myeloid differentiation factor 88
- NAC
N‐acetyl cysteine
- Neh
Nrf2‐ECH homology
- NEMO
NF‐κB essential modulator
- NES
nuclear export signal
- NF‐κB
nuclear transcription factor of bone marrow‐derived lymphocytes
- NGF
nerve growth factor
- NLS
nuclear localization signal
- NOX
NADPH oxidase
- NQO1
NADPH quinone oxidoreductase
- Nrx
nucleoredoxin
- Ohr
organic hydroperoxide resistance
- OhrR
organic hydroperoxide resistance repressor
- Orp1
GPx3‐like protein in Saccharomyces cerevisiae
- OxyR
transcription factor which regulates genes induced by oxidative stress in Salmonella typhimurium and Escherichia coli
- p300
transcriptional coactivator with HAT activity
- PAMP
pathogen‐associated molecular pattern
- Pap1
transcription factor regulation hydrogen peroxide induced responses in S. pombe
- PDGF
platelet‐derived growth factor
- PDK1
phosphoinositide‐dependent protein kinase‐1
- PDTC
pyrrolidine dithiocarbamate
- PERK
PKR‐like endoplasmic reticulum kinase
- PerR
transcription factor regulating gene expression in response to H2O2
- PEST
proline glutamic acid serine and threonine‐rich peptide sequence
- PGE2
prostaglandin E2
- PGI2
prostacyclin
- Phox
phagocytic oxidase
- PI3K
phosphatidyl‐inositol‐3‐kinase
- PIP
phosphoinositol phospholipid
- PKAc
protein kinase A, catalytic domain
- PKC
protein kinase C
- PKR
protein kinase R, eukaryotic translation initiation factor‐2α kinase‐2
- PMN
polymorpho nuclear leukocytes
- PP
protein phosphatase
- Prx
peroxiredoxin
- PSP
protein serine/threonine phosphatase
- PTEN
phosphatase and tensin homologue
- PTP
protein tyrosine phosphatase
- Rac
small GTPase of the family of Rac proteins
- Rbx1
RING box protein 1
- RFK
riboflavin kinase
- RHD
Rel homology domain
- RING
really interesting new gene
- RIP
receptor interacting protein
- RNAP
RNA polymerase
- RNS
reactive nitrogen species
- ROS
reactive oxygen species
- RSK
ribosomal S6 kinase
- RTK
receptor tyrosine kinase
- Ser
serine
- SH2
Src homology‐2
- SHIP
SH2‐domain‐containing inositide phosphatase
- SOD
superoxide dismutase
- SoxR
transcriptional regulator in response to superoxide generating compounds
- SQSTM1
sequestosome 1
- Src
cellular and sarcoma, tyrosine kinase
- Srx1
sulfiredoxin‐1
- SUMO
small ubiquitin‐like modifier
- SYK
spleen tyrosine kinase
- TAD
transactivation domain
- TAK1
TGFβ‐activated kinase
- Thr
threonine
- TIR
Toll/IL‐1 receptor
- TIRAP
TIR‐domain‐containing adaptor protein
- TLR
Toll‐like receptor
- TNF
tumor necrosis factor
- TNFR
TNF receptor
- TRADD
TNF‐associated receptor death domain
- TRAF
TNFR‐associated factor
- TRAM
TRIF‐related adaptor molecule
- TRIF
Toll‐receptor‐associated activator of interferon
- TRP14
thioredoxin‐related protein‐14
- Trx
thioredoxin
- Tsa1
first characterized peroxiredoxin in S. cerevisiae
- TTF
thyroid transcription factor
- Tyr
tyrosine
- UGT
UDP‐glucuronyl transferase
- UMCSBP
universal minicircle sequence binding protein
- USF
upstream stimulatory factor
- VCAM
vascular cell adhesion molecule
- VHR
Vaccinia H1‐related (phosphatase)
- XIAP
X‐linked inhibitor of apoptosis protein
- WIP1
wild‐type p53‐induced phosphatase‐1
- Wnt
wingless and Int 1
- Yap1
AP‐1 like transcription factor from yeast
Acknowledgments
The work was supported by the German Research Council (DFG; Grant BR778/8-1) and the European Union (Grant CM0801).
References
- 1.Abate C. Patel L. Rauscher FJ., 3rd Curran T. Redox regulation of fos and jun DNA-binding activity in vitro. Science. 1990;249:1157–1161. doi: 10.1126/science.2118682. [DOI] [PubMed] [Google Scholar]
- 2.Aberle H. Bauer A. Stappert J. Kispert A. Kemler R. beta-Catenin is a target for the ubiquitin-proteasome pathway. EMBO J. 1997;16:3797–3804. doi: 10.1093/emboj/16.13.3797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Adams J. Kelso R. Cooley L. The kelch repeat superfamily of proteins: propellers of cell function. Trends Cell Biol. 2000;10:17–24. doi: 10.1016/s0962-8924(99)01673-6. [DOI] [PubMed] [Google Scholar]
- 4.Akira S. Takeda K. Toll-like receptor signalling. Nat Rev Immunol. 2004;4:499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- 5.Alam J. Stewart D. Touchard C. Boinapally S. Choi AM. Cook JL. Nrf2, a Cap'n'Collar transcription factor, regulates induction of the heme oxygenase-1 gene. J Biol Chem. 1999;274:26071–26078. doi: 10.1074/jbc.274.37.26071. [DOI] [PubMed] [Google Scholar]
- 6.Aleksunes LM. Manautou JE. Emerging role of Nrf2 in protecting against hepatic and gastrointestinal disease. Toxicol Pathol. 2007;35:459–473. doi: 10.1080/01926230701311344. [DOI] [PubMed] [Google Scholar]
- 7.Ando K. Hirao S. Kabe Y. Ogura Y. Sato I. Yamaguchi Y. Wada T. Handa H. A new APE1/Ref-1-dependent pathway leading to reduction of NF-kappaB and AP-1, and activation of their DNA-binding activity. Nucleic Acids Res. 2008;36:4327–4336. doi: 10.1093/nar/gkn416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aoki Y. Hashimoto AH. Amanuma K. Matsumoto M. Hiyoshi K. Takano H. Masumura K. Itoh K. Nohmi T. Yamamoto M. Enhanced spontaneous and benzo(a)pyrene-induced mutations in the lung of Nrf2-deficient gpt delta mice. Cancer Res. 2007;67:5643–5648. doi: 10.1158/0008-5472.CAN-06-3355. [DOI] [PubMed] [Google Scholar]
- 9.Apopa PL. He X. Ma Q. Phosphorylation of Nrf2 in the transcription activation domain by casein kinase 2 (CK2) is critical for the nuclear translocation and transcription activation function of Nrf2 in IMR-32 neuroblastoma cells. J Biochem Mol Toxicol. 2008;22:63–76. doi: 10.1002/jbt.20212. [DOI] [PubMed] [Google Scholar]
- 10.Arlt A. Bauer I. Schafmayer C. Tepel J. Muerkoster SS. Brosch M. Roder C. Kalthoff H. Hampe J. Moyer MP. Folsch UR. Schafer H. Increased proteasome subunit protein expression and proteasome activity in colon cancer relate to an enhanced activation of nuclear factor E2-related factor 2 (Nrf2) Oncogene. 2009;28:3983–3996. doi: 10.1038/onc.2009.264. [DOI] [PubMed] [Google Scholar]
- 11.Aslund F. Zheng M. Beckwith J. Storz G. Regulation of the OxyR transcription factor by hydrogen peroxide and the cellular thiol-disulfide status. Proc Natl Acad Sci U S A. 1999;96:6161–6165. doi: 10.1073/pnas.96.11.6161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Awasthi YC. Sharma R. Sharma A. Yadav S. Singhal SS. Chaudhary P. Awasthi S. Self-regulatory role of 4-hydroxynonenal in signaling for stress-induced programmed cell death. Free Radic Biol Med. 2008;45:111–118. doi: 10.1016/j.freeradbiomed.2008.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Azad MB. Chen Y. Gibson SB. Regulation of autophagy by reactive oxygen species (ROS): implications for cancer progression and treatment. Antioxid Redox Signal. 2009;11:777–790. doi: 10.1089/ars.2008.2270. [DOI] [PubMed] [Google Scholar]
- 14.Babior BM. The respiratory burst oxidase. Curr Opin Hematol. 1995;2:55–60. doi: 10.1097/00062752-199502010-00008. [DOI] [PubMed] [Google Scholar]
- 15.Baeuerle PA. Henkel T. Function and activation of NF-kappa B in the immune system. Annu Rev Immunol. 1994;12:141–179. doi: 10.1146/annurev.iy.12.040194.001041. [DOI] [PubMed] [Google Scholar]
- 16.Bai D. Ueno L. Vogt PK. Akt-mediated regulation of NFkappaB and the essentialness of NFkappaB for the oncogenicity of PI3K and Akt. Int J Cancer. 2009;125:2863–2870. doi: 10.1002/ijc.24748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Balkwill F. Mantovani A. Inflammation and cancer: back to Virchow? Lancet. 2001;357:539–545. doi: 10.1016/S0140-6736(00)04046-0. [DOI] [PubMed] [Google Scholar]
- 18.Ballotti R. Baron V. Gautier N. Hainaut P. Scimeca JC. Dolais-Kitabgi J. Lammers R. Schlessinger J. Ullrich A. Van Obberghen E. Activation and regulation of the insulin receptor kinase. Diabete Metab. 1992;18:98–103. [PubMed] [Google Scholar]
- 19.Baltimore D. Discovering NF-kB. Cold Spring Harb Perspect Biol 2009. 2010;1(a000026):1–3. doi: 10.1101/cshperspect.a000026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Banning A. Brigelius-Flohé R. NF-kappaB, Nrf2, and HO-1 interplay in redox-regulated VCAM-1 expression. Antioxid Redox Signal. 2005;7:889–899. doi: 10.1089/ars.2005.7.889. [DOI] [PubMed] [Google Scholar]
- 21.Banning A. Deubel S. Kluth D. Zhou Z. Brigelius-Flohé R. The GI-GPx gene is a target for Nrf2. Mol Cell Biol. 2005;25:4914–4923. doi: 10.1128/MCB.25.12.4914-4923.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Banning A. Kipp A. Schmitmeier S. Lowinger M. Florian S. Krehl S. Thalmann S. Thierbach R. Steinberg P. Brigelius-Flohé R. Glutathione peroxidase 2 inhibits cyclooxygenase-2-mediated migration and invasion of HT-29 adenocarcinoma cells but supports their growth as tumors in nude mice. Cancer Res. 2008;68:9746–9753. doi: 10.1158/0008-5472.CAN-08-1321. [DOI] [PubMed] [Google Scholar]
- 23.Beck MA. Selenium and viral infections. In: Hatfield DL, editor; Berry MJ, editor; Gladyshev VN, editor. Selenium Its Molecular Biology and Role in Human Health. New York: Springer; 2006. pp. 287–298. [Google Scholar]
- 24.Beck MA. Shi Q. Morris VC. Levander OA. Rapid genomic evolution of a non-virulent coxsackievirus B3 in selenium-deficient mice results in selection of identical virulent isolates. Nat Med. 1995;1:433–436. doi: 10.1038/nm0595-433. [DOI] [PubMed] [Google Scholar]
- 25.Beckman JS. Beckman TW. Chen J. Marshall PA. Freeman BA. Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide. Proc Natl Acad Sci U S A. 1990;87:1620–1624. doi: 10.1073/pnas.87.4.1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Beeson PB. Tolerance To Bacterial Pyrogens: I. Factors influencing its development. J Exp Med. 1947;86:29–38. doi: 10.1084/jem.86.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Belafonte H. Rollins JK. Man Piaba, Recording #1, p. Master Nr.:E4BV4186-4181. 1954.
- 28.Beraud C. Henzel WJ. Baeuerle PA. Involvement of regulatory and catalytic subunits of phosphoinositide 3-kinase in NF-kappaB activation. Proc Natl Acad Sci U S A. 1999;96:429–434. doi: 10.1073/pnas.96.2.429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Beutler B. Rietschel ET. Innate immune sensing and its roots: the story of endotoxin. Nat Rev Immunol. 2003;3:169–176. doi: 10.1038/nri1004. [DOI] [PubMed] [Google Scholar]
- 30.Bey EA. Xu B. Bhattacharjee A. Oldfield CM. Zhao X. Li Q. Subbulakshmi V. Feldman GM. Wientjes FB. Cathcart MK. Protein kinase C delta is required for p47phox phosphorylation and translocation in activated human monocytes. J Immunol. 2004;173:5730–5738. doi: 10.4049/jimmunol.173.9.5730. [DOI] [PubMed] [Google Scholar]
- 31.Bindoli A. Fukuto JM. Forman HJ. Thiol chemistry in peroxidase catalysis and redox signaling. Antioxid Redox Signal. 2008;10:1549–1564. doi: 10.1089/ars.2008.2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bird TA. Schooley K. Dower SK. Hagen H. Virca GD. Activation of nuclear transcription factor NF-kappaB by interleukin-1 is accompanied by casein kinase II-mediated phosphorylation of the p65 subunit. J Biol Chem. 1997;272:32606–32612. doi: 10.1074/jbc.272.51.32606. [DOI] [PubMed] [Google Scholar]
- 33.Birringer M. Pilawa S. Flohé L. Trends in selenium biochemistry. Nat Prod Rep. 2002;19:693–718. doi: 10.1039/b205802m. [DOI] [PubMed] [Google Scholar]
- 34.Bloom D. Dhakshinamoorthy S. Jaiswal AK. Site-directed mutagenesis of cysteine to serine in the DNA binding region of Nrf2 decreases its capacity to upregulate antioxidant response element-mediated expression and antioxidant induction of NAD(P)H:quinone oxidoreductase1 gene. Oncogene. 2002;21:2191–2200. doi: 10.1038/sj.onc.1205288. [DOI] [PubMed] [Google Scholar]
- 35.Bloom DA. Jaiswal AK. Phosphorylation of Nrf2 at Ser40 by protein kinase C in response to antioxidants leads to the release of Nrf2 from INrf2, but is not required for Nrf2 stabilization/accumulation in the nucleus and transcriptional activation of antioxidant response element-mediated NAD(P)H:quinone oxidoreductase-1 gene expression. J Biol Chem. 2003;278:44675–44682. doi: 10.1074/jbc.M307633200. [DOI] [PubMed] [Google Scholar]
- 36.Bohuslav J. Chen LF. Kwon H. Mu Y. Greene WC. p53 induces NF-kappaB activation by an IkappaB kinase-independent mechanism involving phosphorylation of p65 by ribosomal S6 kinase 1. J Biol Chem. 2004;279:26115–26125. doi: 10.1074/jbc.M313509200. [DOI] [PubMed] [Google Scholar]
- 37.Bone RC. Grodzin CJ. Balk RA. Sepsis: a new hypothesis for pathogenesis of the disease process. Chest. 1997;112:235–243. doi: 10.1378/chest.112.1.235. [DOI] [PubMed] [Google Scholar]
- 38.Bonizzi G. Karin M. The two NF-kappaB activation pathways and their role in innate and adaptive immunity. Trends Immunol. 2004;25:280–288. doi: 10.1016/j.it.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 39.Bonizzi G. Piette J. Schoonbroodt S. Greimers R. Havard L. Merville MP. Bours V. Reactive oxygen intermediate-dependent NF-kappaB activation by interleukin-1beta requires 5-lipoxygenase or NADPH oxidase activity. Mol Cell Biol. 1999;19:1950–1960. doi: 10.1128/mcb.19.3.1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Borroz KI. Buetler TM. Eaton DL. Modulation of gamma-glutamylcysteine synthetase large subunit mRNA expression by butylated hydroxyanisole. Toxicol Appl Pharmacol. 1994;126:150–155. doi: 10.1006/taap.1994.1101. [DOI] [PubMed] [Google Scholar]
- 41.Bours V. Bentires-Alj M. Hellin AC. Viatour P. Robe P. Delhalle S. Benoit V. Merville MP. Nuclear factor-kappa B, cancer, and apoptosis. Biochem Pharmacol. 2000;60:1085–1089. doi: 10.1016/s0006-2952(00)00391-9. [DOI] [PubMed] [Google Scholar]
- 42.Boutten A. Goven D. Boczkowski J. Bonay M. Oxidative stress targets in pulmonary emphysema: focus on the Nrf2 pathway. Expert Opin Ther Targets. 2010;14:329–346. doi: 10.1517/14728221003629750. [DOI] [PubMed] [Google Scholar]
- 43.Bowie AG. Moynagh PN. O'Neill LA. Lipid peroxidation is involved in the activation of NF-kappaB by tumor necrosis factor but not interleukin-1 in the human endothelial cell line ECV304. Lack of involvement of H2O2 in NF-kappaB activation by either cytokine in both primary and transformed endothelial cells. J Biol Chem. 1997;272:25941–25950. doi: 10.1074/jbc.272.41.25941. [DOI] [PubMed] [Google Scholar]
- 44.Boxer LA. Allen JM. Schmidt M. Yoder M. Baehner RL. Inhibition of polymorphonuclear leukocyte adherence by prostacyclin. J Lab Clin Med. 1980;95:672–678. [PubMed] [Google Scholar]
- 45.Brigelius-Flohé R. Friedrichs B. Maurer S. Schultz M. Streicher R. Interleukin-1-induced nuclear factor kappa B activation is inhibited by overexpression of phospholipid hydroperoxide glutathione peroxidase in a human endothelial cell line. Biochem J. 1997;328:199–203. doi: 10.1042/bj3280199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brigelius-Flohé R. Maurer S. Lotzer K. Bol G. Kallionpaa H. Lehtolainen P. Viita H. Yla-Herttuala S. Overexpression of PHGPx inhibits hydroperoxide-induced oxidation, NFkappaB activation and apoptosis and affects oxLDL-mediated proliferation of rabbit aortic smooth muscle cells. Atherosclerosis. 2000;152:307–316. doi: 10.1016/s0021-9150(99)00486-4. [DOI] [PubMed] [Google Scholar]
- 47.Brigelius R. Mixed disulfides: biological functions and increase in oxidative stress. In: Sies H, editor. Oxidative Stress. London, Orlando, San Diego, New York, Toronto, Montreal, Sydney, Tokyo: Academic Press; 1985. pp. 243–272. [Google Scholar]
- 48.Brigelius R. Muckel C. Akerboom TP. Sies H. Identification and quantitation of glutathione in hepatic protein mixed disulfides and its relationship to glutathione disulfide. Biochem Pharmacol. 1983;32:2529–2534. doi: 10.1016/0006-2952(83)90014-x. [DOI] [PubMed] [Google Scholar]
- 49.Bruno RS. Song Y. Leonard SW. Mustacich DJ. Taylor AW. Traber MG. Ho E. Dietary zinc restriction in rats alters antioxidant status and increases plasma F2 isoprostanes. J Nutr Biochem. 2007;18:509–518. doi: 10.1016/j.jnutbio.2006.09.001. [DOI] [PubMed] [Google Scholar]
- 50.Bsat N. Herbig A. Casillas-Martinez L. Setlow P. Helmann JD. Bacillus subtilis contains multiple Fur homologues: identification of the iron uptake (Fur) and peroxide regulon (PerR) repressors. Mol Microbiol. 1998;29:189–198. doi: 10.1046/j.1365-2958.1998.00921.x. [DOI] [PubMed] [Google Scholar]
- 51.Buhrman G. Parker B. Sohn J. Rudolph J. Mattos C. Structural mechanism of oxidative regulation of the phosphatase Cdc25B via an intramolecular disulfide bond. Biochemistry. 2005;44:5307–5316. doi: 10.1021/bi047449f. [DOI] [PubMed] [Google Scholar]
- 52.Burk RF. Hill KE. Nakayama A. Mostert V. Levander XA. Motley AK. Johnson DA. Johnson JA. Freeman ML. Austin LM. Selenium deficiency activates mouse liver Nrf2-ARE but vitamin E deficiency does not. Free Radic Biol Med. 2008;44:1617–1623. doi: 10.1016/j.freeradbiomed.2008.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Buss H. Dorrie A. Schmitz ML. Frank R. Livingstone M. Resch K. Kracht M. Phosphorylation of serine 468 by GSK-3beta negatively regulates basal p65 NF-kappaB activity. J Biol Chem. 2004;279:49571–49574. doi: 10.1074/jbc.C400442200. [DOI] [PubMed] [Google Scholar]
- 54.Buzek J. Latonen L. Kurki S. Peltonen K. Laiho M. Redox state of tumor suppressor p53 regulates its sequence-specific DNA binding in DNA-damaged cells by cysteine 277. Nucleic Acids Res. 2002;30:2340–2348. doi: 10.1093/nar/30.11.2340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cao X. Kambe F. Ohmori S. Seo H. Oxidoreductive modification of two cysteine residues in paired domain by Ref-1 regulates DNA-binding activity of Pax-8. Biochem Biophys Res Commun. 2002;297:288–293. doi: 10.1016/s0006-291x(02)02196-4. [DOI] [PubMed] [Google Scholar]
- 56.Carlson BA. Novoselov SV. Kumaraswamy E. Lee BJ. Anver MR. Gladyshev VN. Hatfield DL. Specific excision of the selenocysteine tRNA[Ser]Sec (Trsp) gene in mouse liver demonstrates an essential role of selenoproteins in liver function. J Biol Chem. 2004;279:8011–8017. doi: 10.1074/jbc.M310470200. [DOI] [PubMed] [Google Scholar]
- 57.Cavaillon JM. Adrie C. Fitting C. Adib-Conquy M. Endotoxin tolerance: is there a clinical relevance? J Endotoxin Res. 2003;9:101–107. doi: 10.1179/096805103125001487. [DOI] [PubMed] [Google Scholar]
- 58.Chan K. Lu R. Chang JC. Kan YW. NRF2, a member of the NFE2 family of transcription factors, is not essential for murine erythropoiesis, growth, and development. Proc Natl Acad Sci U S A. 1996;93:13943–13948. doi: 10.1073/pnas.93.24.13943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chance B. Sies H. Boveris A. Hydroperoxide metabolism in mammalian organs. Physiol Rev. 1979;59:527–605. doi: 10.1152/physrev.1979.59.3.527. [DOI] [PubMed] [Google Scholar]
- 60.Chang TS. Jeong W. Choi SY. Yu S. Kang SW. Rhee SG. Regulation of peroxiredoxin I activity by Cdc2-mediated phosphorylation. J Biol Chem. 2002;277:25370–25376. doi: 10.1074/jbc.M110432200. [DOI] [PubMed] [Google Scholar]
- 61.Chen LF. Greene WC. Regulation of distinct biological activities of the NF-kappaB transcription factor complex by acetylation. J Mol Med. 2003;81:549–557. doi: 10.1007/s00109-003-0469-0. [DOI] [PubMed] [Google Scholar]
- 62.Chen LF. Greene WC. Shaping the nuclear action of NF-kappaB. Nat Rev Mol Cell Biol. 2004;5:392–401. doi: 10.1038/nrm1368. [DOI] [PubMed] [Google Scholar]
- 63.Chen W. Sun Z. Wang XJ. Jiang T. Huang Z. Fang D. Zhang DD. Direct interaction between Nrf2 and p21(Cip1/WAF1) upregulates the Nrf2-mediated antioxidant response. Mol Cell. 2009;34:663–673. doi: 10.1016/j.molcel.2009.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chen YR. Han J. Kori R. Kong AN. Tan TH. Phenylethyl isothiocyanate induces apoptotic signaling via suppressing phosphatase activity against c-Jun N-terminal kinase. J Biol Chem. 2002;277:39334–39342. doi: 10.1074/jbc.M202070200. [DOI] [PubMed] [Google Scholar]
- 65.Chen ZJ. Parent L. Maniatis T. Site-specific phosphorylation of IkappaBalpha by a novel ubiquitination-dependent protein kinase activity. Cell. 1996;84:853–862. doi: 10.1016/s0092-8674(00)81064-8. [DOI] [PubMed] [Google Scholar]
- 66.Chew J. Biswas S. Shreeram S. Humaidi M. Wong ET. Dhillion MK. Teo H. Hazra A. Fang CC. Lopez-Collazo E. Bulavin DV. Tergaonkar V. WIP1 phosphatase is a negative regulator of NF-kappaB signalling. Nat Cell Biol. 2009;11:659–666. doi: 10.1038/ncb1873. [DOI] [PubMed] [Google Scholar]
- 67.Chiarugi P. PTPs versus PTKs: the redox side of the coin. Free Radic Res. 2005;39:353–364. doi: 10.1080/10715760400027987. [DOI] [PubMed] [Google Scholar]
- 68.Christman MF. Storz G. Ames BN. OxyR, a positive regulator of hydrogen peroxide-inducible genes in Escherichia coli and Salmonella typhimurium, is homologous to a family of bacterial regulatory proteins. Proc Natl Acad Sci U S A. 1989;86:3484–3488. doi: 10.1073/pnas.86.10.3484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chun KT. Mathias N. Goebl MG. Ubiquitin-dependent proteolysis and cell cycle control in yeast. Prog Cell Cycle Res. 1996;2:115–127. doi: 10.1007/978-1-4615-5873-6_12. [DOI] [PubMed] [Google Scholar]
- 70.Chung SW. Toriba A. Chung HY. Yu BP. Kameda T. Tang N. Kizu R. Hayakawa K. Activation of 5-lipoxygenase and NF-kappa B in the action of acenaphthenequinone by modulation of oxidative stress. Toxicol Sci. 2008;101:152–158. doi: 10.1093/toxsci/kfm252. [DOI] [PubMed] [Google Scholar]
- 71.Ciechanover A. The ubiquitin-proteasome pathway: on protein death and cell life. EMBO J. 1998;17:7151–7160. doi: 10.1093/emboj/17.24.7151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Clerkin JS. Naughton R. Quiney C. Cotter TG. Mechanisms of ROS modulated cell survival during carcinogenesis. Cancer Lett. 2008;266:30–36. doi: 10.1016/j.canlet.2008.02.029. [DOI] [PubMed] [Google Scholar]
- 73.Conaway RC. Brower CS. Conaway JW. Emerging roles of ubiquitin in transcription regulation. Science. 2002;296:1254–1258. doi: 10.1126/science.1067466. [DOI] [PubMed] [Google Scholar]
- 74.Condo I. Malisan F. Guccini I. Serio D. Rufini A. Testi R. Molecular control of the cytosolic aconitase/IRP1 switch by extramitochondrial frataxin. Hum Mol Genet. 2010;19:1221–1229. doi: 10.1093/hmg/ddp592. [DOI] [PubMed] [Google Scholar]
- 75.Conway JP. Kinter M. Dual role of peroxiredoxin I in macrophage-derived foam cells. J Biol Chem. 2006;281:27991–28001. doi: 10.1074/jbc.M605026200. [DOI] [PubMed] [Google Scholar]
- 76.Copple IM. Lister A. Obeng AD. Kitteringham NR. Jenkins RE. Layfield R. Foster BJ. Goldring CE. Park BK. Physical and functional interaction of sequestosome 1 with Keap1 regulates the Keap1-Nrf2 cell defense pathway. J Biol Chem. 2010;285:16782–16788. doi: 10.1074/jbc.M109.096545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Corbalan-Garcia S. Gomez-Fernandez JC. Protein kinase C regulatory domains: the art of decoding many different signals in membranes. Biochim Biophys Acta. 2006;1761:633–654. doi: 10.1016/j.bbalip.2006.04.015. [DOI] [PubMed] [Google Scholar]
- 78.Correia MA. Burk RF. Hepatic heme metabolism in selenium-deficient rats: effect of phenobarbital. Arch Biochem Biophys. 1976;177:642–644. doi: 10.1016/0003-9861(76)90476-8. [DOI] [PubMed] [Google Scholar]
- 79.Correia MA. Burk RF. Defective utilization of haem in selenium-deficient rat liver. Biochem J. 1983;214:53–58. doi: 10.1042/bj2140053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Cullinan SB. Zhang D. Hannink M. Arvisais E. Kaufman RJ. Diehl JA. Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol Cell Biol. 2003;23:7198–7209. doi: 10.1128/MCB.23.20.7198-7209.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Czech MP. Lawrence JC., Jr. Lynn WS. Evidence for the involvement of sulfhydryl oxidation in the regulation of fat cell hexose transport by insulin. Proc Natl Acad Sci U S A. 1974;71:4173–4177. doi: 10.1073/pnas.71.10.4173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.D'Autreaux B. Toledano MB. ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis. Nat Rev Mol Cell Biol. 2007;8:813–824. doi: 10.1038/nrm2256. [DOI] [PubMed] [Google Scholar]
- 83.de Keizer PLJ. Burgering BMT. Dansen TB. Forkhead Box O as a sensor, mediator, and regulator of redox signaling. Antioxid Redox Signal. 2011;14:1093–1106. doi: 10.1089/ars.2010.3403. [DOI] [PubMed] [Google Scholar]
- 84.de Oliveira-Marques V. Cyrne L. Marinho HS. Antunes F. A quantitative study of NF-kappaB activation by H2O2: relevance in inflammation and synergy with TNF-alpha. J Immunol. 2007;178:3893–3902. doi: 10.4049/jimmunol.178.6.3893. [DOI] [PubMed] [Google Scholar]
- 85.Decker K. Basic mechanisms of the inflammatory response. In: Sies H, editor; Flohé L, editor; Zimmer G, editor. Molecular Aspects of Inflammation. Heidelberg: Springer-Verlag; 1991. pp. 1–23. [Google Scholar]
- 86.Delaunay A. Pflieger D. Barrault MB. Vinh J. Toledano MB. A thiol peroxidase is an H2O2 receptor and redox-transducer in gene activation. Cell. 2002;111:471–481. doi: 10.1016/s0092-8674(02)01048-6. [DOI] [PubMed] [Google Scholar]
- 87.Denu JM. Tanner KG. Specific and reversible inactivation of protein tyrosine phosphatases by hydrogen peroxide: evidence for a sulfenic acid intermediate and implications for redox regulation. Biochemistry. 1998;37:5633–5642. doi: 10.1021/bi973035t. [DOI] [PubMed] [Google Scholar]
- 88.Deshaies RJ. Joazeiro CA. RING domain E3 ubiquitin ligases. Annu Rev Biochem. 2009;78:399–434. doi: 10.1146/annurev.biochem.78.101807.093809. [DOI] [PubMed] [Google Scholar]
- 89.Devin A. Cook A. Lin Y. Rodriguez Y. Kelliher M. Liu Z. The distinct roles of TRAF2 and RIP in IKK activation by TNF-R1: TRAF2 recruits IKK to TNF-R1 while RIP mediates IKK activation. Immunity. 2000;12:419–429. doi: 10.1016/s1074-7613(00)80194-6. [DOI] [PubMed] [Google Scholar]
- 90.Dhakshinamoorthy S. Jaiswal AK. Small maf (MafG and MafK) proteins negatively regulate antioxidant response element-mediated expression and antioxidant induction of the NAD(P)H:Quinone oxidoreductase1 gene. J Biol Chem. 2000;275:40134–40141. doi: 10.1074/jbc.M003531200. [DOI] [PubMed] [Google Scholar]
- 91.Dhakshinamoorthy S. Jain AK. Bloom DA. Jaiswal AK. Bach1 competes with Nrf2 leading to negative regulation of the antioxidant response element (ARE)-mediated NAD(P)H:quinone oxidoreductase 1 gene expression and induction in response to antioxidants. J Biol Chem. 2005;280:16891–16900. doi: 10.1074/jbc.M500166200. [DOI] [PubMed] [Google Scholar]
- 92.DiDonato JA. Hayakawa M. Rothwarf DM. Zandi E. Karin M. A cytokine-responsive IkappaB kinase that activates the transcription factor NF-kappaB. Nature. 1997;388:548–554. doi: 10.1038/41493. [DOI] [PubMed] [Google Scholar]
- 93.Dietz KJ. The dual function of plant peroxiredoxins in antioxidant defence and redox signaling. Subcell Biochem. 2007;44:267–294. doi: 10.1007/978-1-4020-6051-9_13. [DOI] [PubMed] [Google Scholar]
- 94.Dinkova-Kostova AT. Talalay P. Relation of structure of curcumin analogs to their potencies as inducers of phase 2 detoxification enzymes. Carcinogenesis. 1999;20:911–914. doi: 10.1093/carcin/20.5.911. [DOI] [PubMed] [Google Scholar]
- 95.Dinkova-Kostova AT. Holtzclaw WD. Wakabayashi N. Keap1, the sensor for electrophiles and oxidants that regulates the phase 2 response, is a zinc metalloprotein. Biochemistry. 2005;44:6889–6899. doi: 10.1021/bi047434h. [DOI] [PubMed] [Google Scholar]
- 96.Dinkova-Kostova AT. Massiah MA. Bozak RE. Hicks RJ. Talalay P. Potency of Michael reaction acceptors as inducers of enzymes that protect against carcinogenesis depends on their reactivity with sulfhydryl groups. Proc Natl Acad Sci U S A. 2001;98:3404–3409. doi: 10.1073/pnas.051632198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Dinkova-Kostova AT. Holtzclaw WD. Cole RN. Itoh K. Wakabayashi N. Katoh Y. Yamamoto M. Talalay P. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc Natl Acad Sci U S A. 2002;99:11908–11913. doi: 10.1073/pnas.172398899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Dinkova-Kostova AT. Liby KT. Stephenson KK. Holtzclaw WD. Gao X. Suh N. Williams C. Risingsong R. Honda T. Gribble GW. Sporn MB. Talalay P. Extremely potent triterpenoid inducers of the phase 2 response: correlations of protection against oxidant and inflammatory stress. Proc Natl Acad Sci U S A. 2005;102:4584–4589. doi: 10.1073/pnas.0500815102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Djavaheri-Mergny M. Amelotti M. Mathieu J. Besancon F. Bauvy C. Souquere S. Pierron G. Codogno P. NF-kappaB activation represses tumor necrosis factor-alpha-induced autophagy. J Biol Chem. 2006;281:30373–30382. doi: 10.1074/jbc.M602097200. [DOI] [PubMed] [Google Scholar]
- 100.Dubbs JM. Mongkolsuk S. Peroxiredoxins in bacterial antioxidant defense. Subcell Biochem. 2007;44:143–193. doi: 10.1007/978-1-4020-6051-9_7. [DOI] [PubMed] [Google Scholar]
- 101.Dubuisson M. Vander Stricht D. Clippe A. Etienne F. Nauser T. Kissner R. Koppenol WH. Rees JF. Knoops B. Human peroxiredoxin 5 is a peroxynitrite reductase. FEBS Lett. 2004;571:161–165. doi: 10.1016/j.febslet.2004.06.080. [DOI] [PubMed] [Google Scholar]
- 102.Duran A. Diaz-Meco MT. Moscat J. Essential role of RelA Ser311 phosphorylation by zetaPKC in NF-kappaB transcriptional activation. EMBO J. 2003;22:3910–3918. doi: 10.1093/emboj/cdg370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ea CK. Deng L. Xia ZP. Pineda G. Chen ZJ. Activation of IKK by TNFalpha requires site-specific ubiquitination of RIP1 and polyubiquitin binding by NEMO. Mol Cell. 2006;22:245–257. doi: 10.1016/j.molcel.2006.03.026. [DOI] [PubMed] [Google Scholar]
- 104.Eggler AL. Gay KA. Mesecar AD. Molecular mechanisms of natural products in chemoprevention: induction of cytoprotective enzymes by Nrf2. Mol Nutr Food Res. 2008;52(Suppl 1):S84–S94. doi: 10.1002/mnfr.200700249. [DOI] [PubMed] [Google Scholar]
- 105.Ellis HR. Poole LB. Novel application of 7-chloro-4-nitrobenzo-2-oxa-1,3-diazole to identify cysteine sulfenic acid in the AhpC component of alkyl hydroperoxide reductase. Biochemistry. 1997;36:15013–15018. doi: 10.1021/bi972191x. [DOI] [PubMed] [Google Scholar]
- 106.Ellson C. Davidson K. Anderson K. Stephens LR. Hawkins PT. PtdIns3P binding to the PX domain of p40phox is a physiological signal in NADPH oxidase activation. EMBO J. 2006;25:4468–4478. doi: 10.1038/sj.emboj.7601346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ellson CD. Gobert-Gosse S. Anderson KE. Davidson K. Erdjument-Bromage H. Tempst P. Thuring JW. Cooper MA. Lim ZY. Holmes AB. Gaffney PR. Coadwell J. Chilvers ER. Hawkins PT. Stephens LR. PtdIns(3)P regulates the neutrophil oxidase complex by binding to the PX domain of p40(phox) Nat Cell Biol. 2001;3:679–682. doi: 10.1038/35083076. [DOI] [PubMed] [Google Scholar]
- 108.Esworthy RS. Binder SW. Doroshow JH. Chu FF. Microflora trigger colitis in mice deficient in selenium-dependent glutathione peroxidase and induce Gpx2 gene expression. Biol Chem. 2003;384:597–607. doi: 10.1515/BC.2003.067. [DOI] [PubMed] [Google Scholar]
- 109.Esworthy RS. Yang L. Frankel PH. Chu FF. Epithelium-specific glutathione peroxidase, Gpx2, is involved in the prevention of intestinal inflammation in selenium-deficient mice. J Nutr. 2005;135:740–745. doi: 10.1093/jn/135.4.740. [DOI] [PubMed] [Google Scholar]
- 110.Fan H. Cook JA. Molecular mechanisms of endotoxin tolerance. J Endotoxin Res. 2004;10:71–84. doi: 10.1179/096805104225003997. [DOI] [PubMed] [Google Scholar]
- 111.Favreau LV. Pickett CB. The rat quinone reductase antioxidant response element. Identification of the nucleotide sequence required for basal and inducible activity and detection of antioxidant response element-binding proteins in hepatoma and non-hepatoma cell lines. J Biol Chem. 1995;270:24468–24474. doi: 10.1074/jbc.270.41.24468. [DOI] [PubMed] [Google Scholar]
- 112.Ferrer-Sueta G. Radi R. Chemical biology of peroxynitrite: kinetics, diffusion, and radicals. ACS Chem Biol. 2009;4:161–177. doi: 10.1021/cb800279q. [DOI] [PubMed] [Google Scholar]
- 113.Finkel T. Holbrook NJ. Oxidants, oxidative stress and the biology of ageing. Nature. 2000;408:239–247. doi: 10.1038/35041687. [DOI] [PubMed] [Google Scholar]
- 114.Fischer EH. Cellular regulation by protein phosphorylation: a historical overview. Biofactors. 1997;6:367–374. doi: 10.1002/biof.5520060307. [DOI] [PubMed] [Google Scholar]
- 115.Fischer EH. Phosphorylase and the origin of reversible protein phosphorylation. Biol Chem. 2010;391:131–137. doi: 10.1515/bc.2010.011. [DOI] [PubMed] [Google Scholar]
- 116.Fischer EH. Charbonneau H. Tonks NK. Protein tyrosine phosphatases: a diverse family of intracellular and transmembrane enzymes. Science. 1991;253:401–406. doi: 10.1126/science.1650499. [DOI] [PubMed] [Google Scholar]
- 117.Fischer EH. Charbonneau H. Cool DE. Tonks NK. Tyrosine phosphatases and their possible interplay with tyrosine kinases. Ciba Found Symp. 1992;164:132–140. doi: 10.1002/9780470514207.ch9. discussion 140–144, [DOI] [PubMed] [Google Scholar]
- 118.Fischer OM. Hart S. Gschwind A. Ullrich A. EGFR signal transactivation in cancer cells. Biochem Soc Trans. 2003;31:1203–1208. doi: 10.1042/bst0311203. [DOI] [PubMed] [Google Scholar]
- 119.Fleming CR. Billiard SM. Di Giulio RT. Hypoxia inhibits induction of aryl hydrocarbon receptor activity in topminnow hepatocarcinoma cells in an ARNT-dependent manner. Comp Biochem Physiol C Toxicol Pharmacol. 2009;150:383–389. doi: 10.1016/j.cbpc.2009.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Flohé L. Sulfur and selenium catalysis as paradigms for redox regulations. In: Forman J, editor; Torres M, editor; Fukuto JM, editor. Signal Transduction by Reactive Oxygen and Nitrogen Species. Dordrecht: Kluwer Academic Publishers; 2003. pp. 15–32. [Google Scholar]
- 121.Flohé L. Selenium in mammalian spermiogenesis. Biol Chem. 2007;388:987–995. doi: 10.1515/BC.2007.112. [DOI] [PubMed] [Google Scholar]
- 122.Flohé L. The labour pains of biochemical selenology: the history of selenoprotein biosynthesis. Biochim Biophys Acta. 2009;1790:1389–1403. doi: 10.1016/j.bbagen.2009.03.031. [DOI] [PubMed] [Google Scholar]
- 123.Flohé L. Changing paradigms in thiology: from antioxidant defence towards redox regulation. Methods Enzymol. 2010;473:1–39. doi: 10.1016/S0076-6879(10)73001-9. [DOI] [PubMed] [Google Scholar]
- 124.Flohé L. Giertz H. Endotoxins, arachidonic acid, and superoxide formation. Rev Infect Dis. 1987;9(Suppl 5):S553–S561. doi: 10.1093/clinids/9.supplement_5.s553. [DOI] [PubMed] [Google Scholar]
- 125.Flohé L. Brigelius-Flohé R. Selenoproteins of the glutathione system. In: Hatfield DL, editor. Selenium Its Molecular Biology and Role in Human Health. Boston, Dordrecht, London: Kluwer Academic Publishers; 2001. pp. 157–178. [Google Scholar]
- 126.Flohé L. Beckmann R. Giertz H. Loschen G. Oxygen-centered free radicals as mediators of inflammation. In: Sies H, editor. Oxidative Stress. London, Orlando, San Diego, New York, Toronto, Montreal, Sydney, Tokyo: Academic Press; 1985. pp. 403–435. [Google Scholar]
- 127.Flohé L. Toppo S. Cozza G. Ursini F. A comparison of thiol peroxidase mechanisms. Antioxid Redox Signal. 2011;15:763–780. doi: 10.1089/ars.2010.3397. [DOI] [PubMed] [Google Scholar]
- 128.Flohé L. Brigelius-Flohé R. Saliou C. Traber MG. Packer L. Redox regulation of NF-kappa B activation. Free Radic Biol Med. 1997;22:1115–1126. doi: 10.1016/s0891-5849(96)00501-1. [DOI] [PubMed] [Google Scholar]
- 129.Flohé L. Böhlke H. Frankus E. Kim SM. Lintz W. Loschen G. Michel G. Müller B. Schneider J. Seipp U. Vollenberg W. Wilsmann K. Designing prostacyclin analogues. Arzneimittelforschung. 1983;33:1240–1248. [PubMed] [Google Scholar]
- 130.Foley TD. Petro LA. Stredny CM. Coppa TM. Oxidative inhibition of protein phosphatase 2A activity: role of catalytic subunit disulfides. Neurochem Res. 2007;32:1957–1964. doi: 10.1007/s11064-007-9394-x. [DOI] [PubMed] [Google Scholar]
- 131.Follmann H. Häberlein I. Thioredoxins: universal, yet specific thiol-disulfide redox cofactors. Biofactors. 1995;5:147–156. [PubMed] [Google Scholar]
- 132.Fong Y. Tracey KJ. Moldawer LL. Hesse DG. Manogue KB. Kenney JS. Lee AT. Kuo GC. Allison AC. Lowry SF, et al. Antibodies to cachectin/tumor necrosis factor reduce interleukin 1 beta and interleukin 6 appearance during lethal bacteremia. J Exp Med. 1989;170:1627–1633. doi: 10.1084/jem.170.5.1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Forman HJ. Fukuto JM. Torres M. Redox signaling: thiol chemistry defines which reactive oxygen and nitrogen species can act as second messengers. Am J Physiol Cell Physiol. 2004;287:C246–C256. doi: 10.1152/ajpcell.00516.2003. [DOI] [PubMed] [Google Scholar]
- 134.Forman HJ. Maiorino M. Ursini F. Signaling functions of reactive oxygen species. Biochemistry. 2010;49:835–842. doi: 10.1021/bi9020378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Fourquet S. Huang ME. D'Autreaux B. Toledano MB. The dual functions of thiol-based peroxidases in H2O2 scavenging and signaling. Antioxid Redox Signal. 2008;10:1565–1576. doi: 10.1089/ars.2008.2049. [DOI] [PubMed] [Google Scholar]
- 136.Friebe A. Koesling D. The function of NO-sensitive guanylyl cyclase: what we can learn from genetic mouse models. Nitric Oxide. 2009;21:149–156. doi: 10.1016/j.niox.2009.07.004. [DOI] [PubMed] [Google Scholar]
- 137.Funato Y. Miki H. Nucleoredoxin, a novel thioredoxin family member involved in cell growth and differentiation. Antioxid Redox Signal. 2007;9:1035–1057. doi: 10.1089/ars.2007.1550. [DOI] [PubMed] [Google Scholar]
- 138.Funato Y. Miki H. Redox regulation of Wnt signalling via nucleoredoxin. Free Radic Res. 2010;44:379–388. doi: 10.3109/10715761003610745. [DOI] [PubMed] [Google Scholar]
- 139.Funato Y. Michiue T. Asashima M. Miki H. The thioredoxin-related redox-regulating protein nucleoredoxin inhibits Wnt-beta-catenin signalling through dishevelled. Nat Cell Biol. 2006;8:501–508. doi: 10.1038/ncb1405. [DOI] [PubMed] [Google Scholar]
- 140.Furukawa M. Xiong Y. BTB protein Keap1 targets antioxidant transcription factor Nrf2 for ubiquitination by the Cullin 3-Roc1 ligase. Mol Cell Biol. 2005;25:162–171. doi: 10.1128/MCB.25.1.162-171.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Furukawa M. He YJ. Borchers C. Xiong Y. Targeting of protein ubiquitination by BTB-Cullin 3-Roc1 ubiquitin ligases. Nat Cell Biol. 2003;5:1001–1007. doi: 10.1038/ncb1056. [DOI] [PubMed] [Google Scholar]
- 142.Gantner BN. Simmons RM. Canavera SJ. Akira S. Underhill DM. Collaborative induction of inflammatory responses by dectin-1 and Toll-like receptor 2. J Exp Med. 2003;197:1107–1117. doi: 10.1084/jem.20021787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Geng H. Wittwer T. Dittrich-Breiholz O. Kracht M. Schmitz ML. Phosphorylation of NF-kappaB p65 at Ser468 controls its COMMD1-dependent ubiquitination and target gene-specific proteasomal elimination. EMBO Rep. 2009;10:381–386. doi: 10.1038/embor.2009.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Gill R. Tsung A. Billiar T. Linking oxidative stress to inflammation: toll-like receptors. Free Radic Biol Med. 2010;48:1121–1132. doi: 10.1016/j.freeradbiomed.2010.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Gilroy DW. Colville-Nash PR. Willis D. Chivers J. Paul-Clark MJ. Willoughby DA. Inducible cyclooxygenase may have anti-inflammatory properties. Nat Med. 1999;5:698–701. doi: 10.1038/9550. [DOI] [PubMed] [Google Scholar]
- 146.Gladyshev VN. Kryukov GV. Evolution of selenocysteine-containing proteins: significance of identification and functional characterization of selenoproteins. Biofactors. 2001;14:87–92. doi: 10.1002/biof.5520140112. [DOI] [PubMed] [Google Scholar]
- 147.Gloire G. Piette J. Redox regulation of nuclear post-translational modifications during NF-kappaB activation. Antioxid Redox Signal. 2009;11:2209–2222. doi: 10.1089/ars.2009.2463. [DOI] [PubMed] [Google Scholar]
- 148.Gloire G. Legrand-Poels S. Piette J. NF-kappaB activation by reactive oxygen species: fifteen years later. Biochem Pharmacol. 2006;72:1493–1505. doi: 10.1016/j.bcp.2006.04.011. [DOI] [PubMed] [Google Scholar]
- 149.Gloire G. Charlier E. Rahmouni S. Volanti C. Chariot A. Erneux C. Piette J. Restoration of SHIP-1 activity in human leukemic cells modifies NF-kappaB activation pathway and cellular survival upon oxidative stress. Oncogene. 2006;25:5485–5494. doi: 10.1038/sj.onc.1209542. [DOI] [PubMed] [Google Scholar]
- 150.Gomez PF. Pillinger MH. Attur M. Marjanovic N. Dave M. Park J. Bingham CO., 3rd Al-Mussawir H. Abramson SB. Resolution of inflammation: prostaglandin E2 dissociates nuclear trafficking of individual NF-kappaB subunits (p65, p50) in stimulated rheumatoid synovial fibroblasts. J Immunol. 2005;175:6924–6930. doi: 10.4049/jimmunol.175.10.6924. [DOI] [PubMed] [Google Scholar]
- 151.Goossens V. Grooten J. De Vos K. Fiers W. Direct evidence for tumor necrosis factor-induced mitochondrial reactive oxygen intermediates and their involvement in cytotoxicity. Proc Natl Acad Sci U S A. 1995;92:8115–8119. doi: 10.1073/pnas.92.18.8115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Gopalakrishna R. Anderson WB. Ca2+- and phospholipid-independent activation of protein kinase C by selective oxidative modification of the regulatory domain. Proc Natl Acad Sci U S A. 1989;86:6758–6762. doi: 10.1073/pnas.86.17.6758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Gopalakrishna R. Jaken S. Protein kinase C signaling and oxidative stress. Free Radic Biol Med. 2000;28:1349–1361. doi: 10.1016/s0891-5849(00)00221-5. [DOI] [PubMed] [Google Scholar]
- 154.Gopalakrishna R. Gundimeda U. Antioxidant regulation of protein kinase C in cancer prevention. J Nutr. 2002;132:3819S–3823S. doi: 10.1093/jn/132.12.3819S. [DOI] [PubMed] [Google Scholar]
- 155.Gorlach A. Kietzmann T. Superoxide and derived reactive oxygen species in the regulation of hypoxia-inducible factors. Methods Enzymol. 2007;435:421–446. doi: 10.1016/S0076-6879(07)35022-2. [DOI] [PubMed] [Google Scholar]
- 156.Görlich D. Kutay U. Transport between the cell nucleus and the cytoplasm. Annu Rev Cell Dev Biol. 1999;15:607–660. doi: 10.1146/annurev.cellbio.15.1.607. [DOI] [PubMed] [Google Scholar]
- 157.Gottipati S. Rao NL. Fung-Leung WP. IRAK1: a critical signaling mediator of innate immunity. Cell Signal. 2008;20:269–276. doi: 10.1016/j.cellsig.2007.08.009. [DOI] [PubMed] [Google Scholar]
- 158.Greten FR. Eckmann L. Greten TF. Park JM. Li ZW. Egan LJ. Kagnoff MF. Karin M. IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell. 2004;118:285–296. doi: 10.1016/j.cell.2004.07.013. [DOI] [PubMed] [Google Scholar]
- 159.Grippo JF. Holmgren A. Pratt WB. Proof that the endogenous, heat-stable glucocorticoid receptor-activating factor is thioredoxin. J Biol Chem. 1985;260:93–97. [PubMed] [Google Scholar]
- 160.Habeos IG. Ziros PG. Chartoumpekis D. Psyrogiannis A. Kyriazopoulou V. Papavassiliou AG. Simvastatin activates Keap1/Nrf2 signaling in rat liver. J Mol Med. 2008;86:1279–1285. doi: 10.1007/s00109-008-0393-4. [DOI] [PubMed] [Google Scholar]
- 161.Hall A. Karplus PA. Poole LB. Typical 2-Cys peroxiredoxins—structures, mechanisms and functions. FEBS J. 2009;276:2469–2477. doi: 10.1111/j.1742-4658.2009.06985.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Hansen JM. Watson WH. Jones DP. Compartmentation of Nrf-2 redox control: regulation of cytoplasmic activation by glutathione and DNA binding by thioredoxin-1. Toxicol Sci. 2004;82:308–317. doi: 10.1093/toxsci/kfh231. [DOI] [PubMed] [Google Scholar]
- 163.Harding HP. Zhang Y. Zeng H. Novoa I. Lu PD. Calfon M. Sadri N. Yun C. Popko B. Paules R. Stojdl DF. Bell JC. Hettmann T. Leiden JM. Ron D. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell. 2003;11:619–633. doi: 10.1016/s1097-2765(03)00105-9. [DOI] [PubMed] [Google Scholar]
- 164.Harvey CJ. Thimmulappa RK. Singh A. Blake DJ. Ling G. Wakabayashi N. Fujii J. Myers A. Biswal S. Nrf2-regulated glutathione recycling independent of biosynthesis is critical for cell survival during oxidative stress. Free Radic Biol Med. 2009;46:443–453. doi: 10.1016/j.freeradbiomed.2008.10.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Hattermann K. Picard S. Borgeat M. Leclerc P. Pouliot M. Borgeat P. The Toll-like receptor 7/8-ligand resiquimod (R-848) primes human neutrophils for leukotriene B4, prostaglandin E2 and platelet-activating factor biosynthesis. FASEB J. 2007;21:1575–1585. doi: 10.1096/fj.06-7457com. [DOI] [PubMed] [Google Scholar]
- 166.Hauer J. Puschner S. Ramakrishnan P. Simon U. Bongers M. Federle C. Engelmann H. TNF receptor (TNFR)-associated factor (TRAF) 3 serves as an inhibitor of TRAF2/5-mediated activation of the noncanonical NF-kappaB pathway by TRAF-binding TNFRs. Proc Natl Acad Sci U S A. 2005;102:2874–2879. doi: 10.1073/pnas.0500187102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Hawkes WC. Alkan Z. Regulation of redox signaling by selenoproteins. Biol Trace Elem Res. 2010;134:235–251. doi: 10.1007/s12011-010-8656-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Hayakawa M. Miyashita H. Sakamoto I. Kitagawa M. Tanaka H. Yasuda H. Karin M. Kikugawa K. Evidence that reactive oxygen species do not mediate NF-kappaB activation. EMBO J. 2003;22:3356–3366. doi: 10.1093/emboj/cdg332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Hayashi A. Suzuki H. Itoh K. Yamamoto M. Sugiyama Y. Transcription factor Nrf2 is required for the constitutive and inducible expression of multidrug resistance-associated protein 1 in mouse embryo fibroblasts. Biochem Biophys Res Commun. 2003;310:824–829. doi: 10.1016/j.bbrc.2003.09.086. [DOI] [PubMed] [Google Scholar]
- 170.Hayashi T. Ueno Y. Okamoto T. Oxidoreductive regulation of nuclear factor kappa B. Involvement of a cellular reducing catalyst thioredoxin. J Biol Chem. 1993;268:11380–11388. [PubMed] [Google Scholar]
- 171.Hayashi T. Funato Y. Terabayashi T. Morinaka A. Sakamoto R. Ichise H. Fukuda H. Yoshida N. Miki H. Nucleoredoxin negatively regulates Toll-like receptor 4 signaling via recruitment of flightless-I to myeloid differentiation primary response gene (88) J Biol Chem. 2010;285:18586–18593. doi: 10.1074/jbc.M110.106468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Hayden MS. Ghosh S. Signaling to NF-kappaB. Genes Dev. 2004;18:2195–2224. doi: 10.1101/gad.1228704. [DOI] [PubMed] [Google Scholar]
- 173.Hayden MS. Ghosh S. Shared principles in NF-kappaB signaling. Cell. 2008;132:344–362. doi: 10.1016/j.cell.2008.01.020. [DOI] [PubMed] [Google Scholar]
- 174.Hayes JD. McLellan LI. Glutathione and glutathione-dependent enzymes represent a co-ordinately regulated defence against oxidative stress. Free Radic Res. 1999;31:273–300. doi: 10.1080/10715769900300851. [DOI] [PubMed] [Google Scholar]
- 175.Hayes JD. Flanagan JU. Jowsey IR. Glutathione transferases. Annu Rev Pharmacol Toxicol. 2005;45:51–88. doi: 10.1146/annurev.pharmtox.45.120403.095857. [DOI] [PubMed] [Google Scholar]
- 176.He X. Ma Q. NRF2 cysteine residues are critical for oxidant/electrophile-sensing, Kelch-like ECH-associated protein-1-dependent ubiquitination-proteasomal degradation, and transcription activation. Mol Pharmacol. 2009;76:1265–1278. doi: 10.1124/mol.109.058453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Heffetz D. Bushkin I. Dror R. Zick Y. The insulinomimetic agents H2O2 and vanadate stimulate protein tyrosine phosphorylation in intact cells. J Biol Chem. 1990;265:2896–2902. [PubMed] [Google Scholar]
- 178.Henchoz S. Chi Y. Catarin B. Herskowitz I. Deshaies RJ. Peter M. Phosphorylation- and ubiquitin-dependent degradation of the cyclin-dependent kinase inhibitor Far1p in budding yeast. Genes Dev. 1997;11:3046–3060. doi: 10.1101/gad.11.22.3046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Henmi T. Amano K. Nagaura Y. Matsumoto K. Echigo S. Tamura S. Kobayashi T. A mechanism for the suppression of interleukin-1-induced nuclear factor kappaB activation by protein phosphatase 2Ceta-2. Biochem J. 2009;423:71–78. doi: 10.1042/BJ20090208. [DOI] [PubMed] [Google Scholar]
- 180.Henschler D. The origin of hormesis: historical background and driving forces. Hum Exp Toxicol. 2006;25:347–351. doi: 10.1191/0960327106ht642oa. [DOI] [PubMed] [Google Scholar]
- 181.Herscovitch M. Comb W. Ennis T. Coleman K. Yong S. Armstead B. Kalaitzidis D. Chandani S. Gilmore TD. Intermolecular disulfide bond formation in the NEMO dimer requires Cys54 and Cys347. Biochem Biophys Res Commun. 2008;367:103–108. doi: 10.1016/j.bbrc.2007.12.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Hidalgo E. Demple B. An iron-sulfur center essential for transcriptional activation by the redox-sensing SoxR protein. EMBO J. 1994;13:138–146. doi: 10.1002/j.1460-2075.1994.tb06243.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Hilt W. Wolf DH. Proteasomes: destruction as a programme. Trends Biochem Sci. 1996;21:96–102. [PubMed] [Google Scholar]
- 184.Hirota K. Murata M. Sachi Y. Nakamura H. Takeuchi J. Mori K. Yodoi J. Distinct roles of thioredoxin in the cytoplasm and in the nucleus. A two-step mechanism of redox regulation of transcription factor NF-kappaB. J Biol Chem. 1999;274:27891–27897. doi: 10.1074/jbc.274.39.27891. [DOI] [PubMed] [Google Scholar]
- 185.Hoberg JE. Popko AE. Ramsey CS. Mayo MW. IkappaB kinase alpha-mediated derepression of SMRT potentiates acetylation of RelA/p65 by p300. Mol Cell Biol. 2006;26:457–471. doi: 10.1128/MCB.26.2.457-471.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Hochstrasser M. Lingering mysteries of ubiquitin-chain assembly. Cell. 2006;124:27–34. doi: 10.1016/j.cell.2005.12.025. [DOI] [PubMed] [Google Scholar]
- 187.Hofmann B. Hecht HJ. Flohé L. Peroxiredoxins. Biol Chem. 2002;383:347–364. doi: 10.1515/BC.2002.040. [DOI] [PubMed] [Google Scholar]
- 188.Hormi-Carver K. Zhang X. Zhang HY. Whitehead RH. Terada LS. Spechler SJ. Souza RF. Unlike esophageal squamous cells, Barrett's epithelial cells resist apoptosis by activating the nuclear factor-kappaB pathway. Cancer Res. 2009;69:672–677. doi: 10.1158/0008-5472.CAN-08-3703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Hosoya T. Maruyama A. Kang MI. Kawatani Y. Shibata T. Uchida K. Warabi E. Noguchi N. Itoh K. Yamamoto M. Differential responses of the Nrf2-Keap1 system to laminar and oscillatory shear stresses in endothelial cells. J Biol Chem. 2005;280:27244–27250. doi: 10.1074/jbc.M502551200. [DOI] [PubMed] [Google Scholar]
- 190.Hoyos B. Imam A. Korichneva I. Levi E. Chua R. Hämmerling U. Activation of c-Raf kinase by ultraviolet light. Regulation by retinoids. J Biol Chem. 2002;277:23949–23957. doi: 10.1074/jbc.M110750200. [DOI] [PubMed] [Google Scholar]
- 191.Hu R. Xu C. Shen G. Jain MR. Khor TO. Gopalkrishnan A. Lin W. Reddy B. Chan JY. Kong AN. Gene expression profiles induced by cancer chemopreventive isothiocyanate sulforaphane in the liver of C57BL/6J mice and C57BL/6J/Nrf2 (-/-) mice. Cancer Lett. 2006;243:170–192. doi: 10.1016/j.canlet.2005.11.050. [DOI] [PubMed] [Google Scholar]
- 192.Huang HC. Nguyen T. Pickett CB. Phosphorylation of Nrf2 at Ser-40 by protein kinase C regulates antioxidant response element-mediated transcription. J Biol Chem. 2002;277:42769–42774. doi: 10.1074/jbc.M206911200. [DOI] [PubMed] [Google Scholar]
- 193.Huang RP. Adamson ED. Characterization of the DNA-binding properties of the early growth response-1 (Egr-1) transcription factor: evidence for modulation by a redox mechanism. DNA Cell Biol. 1993;12:265–273. doi: 10.1089/dna.1993.12.265. [DOI] [PubMed] [Google Scholar]
- 194.Hubbard SR. Bishop WR. Kirschmeier P. George SJ. Cramer SP. Hendrickson WA. Identification and characterization of zinc binding sites in protein kinase C. Science. 1991;254:1776–1779. doi: 10.1126/science.1763327. [DOI] [PubMed] [Google Scholar]
- 195.Ide T. Egan K. Bell-Parikh LC. FitzGerald GA. Activation of nuclear receptors by prostaglandins. Thromb Res. 2003;110:311–315. doi: 10.1016/s0049-3848(03)00418-3. [DOI] [PubMed] [Google Scholar]
- 196.Ignarro LJ. Activation and regulation of the nitric oxide-cyclic GMP signal transduction pathway by oxidative stress. In: Forman HJ, editor; Cadenas E, editor. Oxidative Stress and Signal Transduction. New York: Chapman & Hill; 1997. pp. 3–31. [Google Scholar]
- 197.Imbert V. Rupec RA. Livolsi A. Pahl HL. Traenckner EB. Mueller-Dieckmann C. Farahifar D. Rossi B. Auberger P. Baeuerle PA. Peyron JF. Tyrosine phosphorylation of I kappa B-alpha activates NF-kappa B without proteolytic degradation of I kappa B-alpha. Cell. 1996;86:787–798. doi: 10.1016/s0092-8674(00)80153-1. [DOI] [PubMed] [Google Scholar]
- 198.Immenschuh S. Baumgart-Vogt E. Peroxiredoxins, oxidative stress, and cell proliferation. Antioxid Redox Signal. 2005;7:768–777. doi: 10.1089/ars.2005.7.768. [DOI] [PubMed] [Google Scholar]
- 199.Irigoin F. Cibils L. Comini MA. Wilkinson SR. Flohé L. Radi R. Insights into the redox biology of Trypanosoma cruzi: Trypanothione metabolism and oxidant detoxification. Free Radic Biol Med. 2008;45:733–742. doi: 10.1016/j.freeradbiomed.2008.05.028. [DOI] [PubMed] [Google Scholar]
- 200.Ishii T. Yanagawa T. Stress-induced peroxiredoxins. Subcell Biochem. 2007;44:375–384. doi: 10.1007/978-1-4020-6051-9_18. [DOI] [PubMed] [Google Scholar]
- 201.Ishii T. Itoh K. Ruiz E. Leake DS. Unoki H. Yamamoto M. Mann GE. Role of Nrf2 in the regulation of CD36 and stress protein expression in murine macrophages: activation by oxidatively modified LDL and 4-hydroxynonenal. Circ Res. 2004;94:609–616. doi: 10.1161/01.RES.0000119171.44657.45. [DOI] [PubMed] [Google Scholar]
- 202.Ishii T. Itoh K. Takahashi S. Sato H. Yanagawa T. Katoh Y. Bannai S. Yamamoto M. Transcription factor Nrf2 coordinately regulates a group of oxidative stress-inducible genes in macrophages. J Biol Chem. 2000;275:16023–16029. doi: 10.1074/jbc.275.21.16023. [DOI] [PubMed] [Google Scholar]
- 203.Ishikawa M. Numazawa S. Yoshida T. Redox regulation of the transcriptional repressor Bach1. Free Radic Biol Med. 2005;38:1344–1352. doi: 10.1016/j.freeradbiomed.2005.01.021. [DOI] [PubMed] [Google Scholar]
- 204.Israel A. A role for phosphorylation and degradation in the control of NF-kappa B activity. Trends Genet. 1995;11:203–205. doi: 10.1016/s0168-9525(00)89045-9. [DOI] [PubMed] [Google Scholar]
- 205.Israel A. The IKK complex, a central regulator of NF-kappaB activation. Cold Spring Harb Perspect Biol. 2010;2:a000158. doi: 10.1101/cshperspect.a000158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Ito K. Lim S. Caramori G. Chung KF. Barnes PJ. Adcock IM. Cigarette smoking reduces histone deacetylase 2 expression, enhances cytokine expression, and inhibits glucocorticoid actions in alveolar macrophages. FASEB J. 2001;15:1110–1112. [PubMed] [Google Scholar]
- 207.Itoh K. Igarashi K. Hayashi N. Nishizawa M. Yamamoto M. Cloning and characterization of a novel erythroid cell-derived CNC family transcription factor heterodimerizing with the small Maf family proteins. Mol Cell Biol. 1995;15:4184–4193. doi: 10.1128/mcb.15.8.4184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Itoh K. Wakabayashi N. Katoh Y. Ishii T. Igarashi K. Engel JD. Yamamoto M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999;13:76–86. doi: 10.1101/gad.13.1.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Itoh K. Mochizuki M. Ishii Y. Ishii T. Shibata T. Kawamoto Y. Kelly V. Sekizawa K. Uchida K. Yamamoto M. Transcription factor Nrf2 regulates inflammation by mediating the effect of 15-deoxy-Delta(12,14)-prostaglandin j(2) Mol Cell Biol. 2004;24:36–45. doi: 10.1128/MCB.24.1.36-45.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Itoh K. Chiba T. Takahashi S. Ishii T. Igarashi K. Katoh Y. Oyake T. Hayashi N. Satoh K. Hatayama I. Yamamoto M. Nabeshima Y. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun. 1997;236:313–322. doi: 10.1006/bbrc.1997.6943. [DOI] [PubMed] [Google Scholar]
- 211.Jaeschke H. Ho YS. Fisher MA. Lawson JA. Farhood A. Glutathione peroxidase-deficient mice are more susceptible to neutrophil-mediated hepatic parenchymal cell injury during endotoxemia: importance of an intracellular oxidant stress. Hepatology. 1999;29:443–450. doi: 10.1002/hep.510290222. [DOI] [PubMed] [Google Scholar]
- 212.Jahngen-Hodge J. Obin MS. Gong X. Shang F. Nowell TR., Jr. Gong J. Abasi H. Blumberg J. Taylor A. Regulation of ubiquitin-conjugating enzymes by glutathione following oxidative stress. J Biol Chem. 1997;272:28218–28226. doi: 10.1074/jbc.272.45.28218. [DOI] [PubMed] [Google Scholar]
- 213.Jain AK. Jaiswal AK. Phosphorylation of tyrosine 568 controls nuclear export of Nrf2. J Biol Chem. 2006;281:12132–12142. doi: 10.1074/jbc.M511198200. [DOI] [PubMed] [Google Scholar]
- 214.Jain AK. Jaiswal AK. GSK-3beta acts upstream of Fyn kinase in regulation of nuclear export and degradation of NF-E2 related factor 2. J Biol Chem. 2007;282:16502–16510. doi: 10.1074/jbc.M611336200. [DOI] [PubMed] [Google Scholar]
- 215.Jain AK. Bloom DA. Jaiswal AK. Nuclear import and export signals in control of Nrf2. J Biol Chem. 2005;280:29158–29168. doi: 10.1074/jbc.M502083200. [DOI] [PubMed] [Google Scholar]
- 216.Jain AK. Mahajan S. Jaiswal AK. Phosphorylation and dephosphorylation of tyrosine 141 regulate stability and degradation of INrf2: a novel mechanism in Nrf2 activation. J Biol Chem. 2008;283:17712–17720. doi: 10.1074/jbc.M709854200. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 217.Jang HH. Lee KO. Chi YH. Jung BG. Park SK. Park JH. Lee JR. Lee SS. Moon JC. Yun JW. Choi YO. Kim WY. Kang JS. Cheong GW. Yun DJ. Rhee SG. Cho MJ. Lee SY. Two enzymes in one; two yeast peroxiredoxins display oxidative stress-dependent switching from a peroxidase to a molecular chaperone function. Cell. 2004;117:625–635. doi: 10.1016/j.cell.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 218.Janssen-Heininger YM. Mossman BT. Heintz NH. Forman HJ. Kalyanaraman B. Finkel T. Stamler JS. Rhee SG. van der Vliet A. Redox-based regulation of signal transduction: principles, pitfalls, and promises. Free Radic Biol Med. 2008;45:1–17. doi: 10.1016/j.freeradbiomed.2008.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Janssens S. Beyaert R. A universal role for MyD88 in TLR/IL-1R-mediated signaling. Trends Biochem Sci. 2002;27:474–482. doi: 10.1016/s0968-0004(02)02145-x. [DOI] [PubMed] [Google Scholar]
- 220.Jariel-Encontre I. Salvat C. Steff AM. Pariat M. Acquaviva C. Furstoss O. Piechaczyk M. Complex mechanisms for c-fos and c-jun degradation. Mol Biol Rep. 1997;24:51–56. doi: 10.1023/a:1006804723722. [DOI] [PubMed] [Google Scholar]
- 221.Jayaraman L. Murthy KG. Zhu C. Curran T. Xanthoudakis S. Prives C. Identification of redox/repair protein Ref-1 as a potent activator of p53. Genes Dev. 1997;11:558–570. doi: 10.1101/gad.11.5.558. [DOI] [PubMed] [Google Scholar]
- 222.Jeong W. Yoon HW. Lee SR. Rhee SG. Identification and characterization of TRP14, a thioredoxin-related protein of 14 kDa. New insights into the specificity of thioredoxin function. J Biol Chem. 2004;279:3142–3150. doi: 10.1074/jbc.M307932200. [DOI] [PubMed] [Google Scholar]
- 223.Jeong W. Chang TS. Boja ES. Fales HM. Rhee SG. Roles of TRP14, a thioredoxin-related protein in tumor necrosis factor-alpha signaling pathways. J Biol Chem. 2004;279:3151–3159. doi: 10.1074/jbc.M307959200. [DOI] [PubMed] [Google Scholar]
- 224.Jeong W. Jung Y. Kim H. Park SJ. Rhee SG. Thioredoxin-related protein 14, a new member of the thioredoxin family with disulfide reductase activity: implication in the redox regulation of TNF-alpha signaling. Free Radic Biol Med. 2009;47:1294–1303. doi: 10.1016/j.freeradbiomed.2009.07.021. [DOI] [PubMed] [Google Scholar]
- 225.Jin W. Wang H. Yan W. Xu L. Wang X. Zhao X. Yang X. Chen G. Ji Y. Disruption of Nrf2 enhances upregulation of nuclear factor-kappaB activity, proinflammatory cytokines, and intercellular adhesion molecule-1 in the brain after traumatic brain injury. Mediators Inflamm. 2008;2008:725174. doi: 10.1155/2008/725174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Jung KJ. Lee EK. Yu BP. Chung HY. Significance of protein tyrosine kinase/protein tyrosine phosphatase balance in the regulation of NF-kappaB signaling in the inflammatory process and aging. Free Radic Biol Med. 2009;47:983–991. doi: 10.1016/j.freeradbiomed.2009.07.009. [DOI] [PubMed] [Google Scholar]
- 227.Jung Y. Kim H. Min SH. Rhee SG. Jeong W. Dynein light chain LC8 negatively regulates NF-kappaB through the redox-dependent interaction with IkappaBalpha. J Biol Chem. 2008;283:23863–23871. doi: 10.1074/jbc.M803072200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Kabe Y. Ando K. Hirao S. Yoshida M. Handa H. Redox regulation of NF-kappaB activation: distinct redox regulation between the cytoplasm and the nucleus. Antioxid Redox Signal. 2005;7:395–403. doi: 10.1089/ars.2005.7.395. [DOI] [PubMed] [Google Scholar]
- 229.Kalinina EV. Chernov NN. Saprin AN. Involvement of thio-, peroxi-, and glutaredoxins in cellular redox-dependent processes. Biochemistry (Mosc) 2008;73:1493–1510. doi: 10.1134/s0006297908130099. [DOI] [PubMed] [Google Scholar]
- 230.Kallio PJ. Wilson WJ. O'Brien S. Makino Y. Poellinger L. Regulation of the hypoxia-inducible transcription factor 1alpha by the ubiquitin-proteasome pathway. J Biol Chem. 1999;274:6519–6525. doi: 10.1074/jbc.274.10.6519. [DOI] [PubMed] [Google Scholar]
- 231.Kamata H. Manabe T. Oka S. Kamata K. Hirata H. Hydrogen peroxide activates IkappaB kinases through phosphorylation of serine residues in the activation loops. FEBS Lett. 2002;519:231–237. doi: 10.1016/s0014-5793(02)02712-6. [DOI] [PubMed] [Google Scholar]
- 232.Kanai F. Liu H. Field SJ. Akbary H. Matsuo T. Brown GE. Cantley LC. Yaffe MB. The PX domains of p47phox and p40phox bind to lipid products of PI(3)K. Nat Cell Biol. 2001;3:675–678. doi: 10.1038/35083070. [DOI] [PubMed] [Google Scholar]
- 233.Kane LP. Shapiro VS. Stokoe D. Weiss A. Induction of NF-kappaB by the Akt/PKB kinase. Curr Biol. 1999;9:601–604. doi: 10.1016/s0960-9822(99)80265-6. [DOI] [PubMed] [Google Scholar]
- 234.Kang KW. Lee SJ. Park JW. Kim SG. Phosphatidylinositol 3-kinase regulates nuclear translocation of NF-E2-related factor 2 through actin rearrangement in response to oxidative stress. Mol Pharmacol. 2002;62:1001–1010. doi: 10.1124/mol.62.5.1001. [DOI] [PubMed] [Google Scholar]
- 235.Kang MI. Kobayashi A. Wakabayashi N. Kim SG. Yamamoto M. Scaffolding of Keap1 to the actin cytoskeleton controls the function of Nrf2 as key regulator of cytoprotective phase 2 genes. Proc Natl Acad Sci U S A. 2004;101:2046–2051. doi: 10.1073/pnas.0308347100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Kang SW. Rhee SG. Chang TS. Jeong W. Choi MH. 2-Cys peroxiredoxin function in intracellular signal transduction: therapeutic implications. Trends Mol Med. 2005;11:571–578. doi: 10.1016/j.molmed.2005.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Kang SW. Chae HZ. Seo MS. Kim K. Baines IC. Rhee SG. Mammalian peroxiredoxin isoforms can reduce hydrogen peroxide generated in response to growth factors and tumor necrosis factor-alpha. J Biol Chem. 1998;273:6297–6302. doi: 10.1074/jbc.273.11.6297. [DOI] [PubMed] [Google Scholar]
- 238.Kang SW. Chang TS. Lee TH. Kim ES. Yu DY. Rhee SG. Cytosolic peroxiredoxin attenuates the activation of Jnk and p38 but potentiates that of Erk in Hela cells stimulated with tumor necrosis factor-alpha. J Biol Chem. 2004;279:2535–2543. doi: 10.1074/jbc.M307698200. [DOI] [PubMed] [Google Scholar]
- 239.Karapetian RN. Evstafieva AG. Abaeva IS. Chichkova NV. Filonov GS. Rubtsov YP. Sukhacheva EA. Melnikov SV. Schneider U. Wanker EE. Vartapetian AB. Nuclear oncoprotein prothymosin alpha is a partner of Keap1: implications for expression of oxidative stress-protecting genes. Mol Cell Biol. 2005;25:1089–1099. doi: 10.1128/MCB.25.3.1089-1099.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Karin M. Ben-Neriah Y. Phosphorylation meets ubiquitination: the control of NF-[kappa]B activity. Annu Rev Immunol. 2000;18:621–663. doi: 10.1146/annurev.immunol.18.1.621. [DOI] [PubMed] [Google Scholar]
- 241.Karin M. Lin A. NF-kappaB at the crossroads of life and death. Nat Immunol. 2002;3:221–227. doi: 10.1038/ni0302-221. [DOI] [PubMed] [Google Scholar]
- 242.Kaspar JW. Jaiswal AK. An autoregulatory loop between Nrf2 and Cul3-Rbx1 controls their cellular abundance. J Biol Chem. 2010;285:21349–21358. doi: 10.1074/jbc.M110.121863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Kaspar JW. Jaiswal AK. Antioxidant-induced phosphorylation of tyrosine 486 leads to rapid nuclear export of Bach1 that allows Nrf2 to bind to the antioxidant response element and activate defensive gene expression. J Biol Chem. 2010;285:153–162. doi: 10.1074/jbc.M109.040022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Kawai T. Akira S. TLR signaling. Cell Death Differ. 2006;13:816–825. doi: 10.1038/sj.cdd.4401850. [DOI] [PubMed] [Google Scholar]
- 245.Kensler TW. Wakabayashi N. Biswal S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu Rev Pharmacol Toxicol. 2007;47:89–116. doi: 10.1146/annurev.pharmtox.46.120604.141046. [DOI] [PubMed] [Google Scholar]
- 246.Khor TO. Huang MT. Kwon KH. Chan JY. Reddy BS. Kong AN. Nrf2-deficient mice have an increased susceptibility to dextran sulfate sodium-induced colitis. Cancer Res. 2006;66:11580–11584. doi: 10.1158/0008-5472.CAN-06-3562. [DOI] [PubMed] [Google Scholar]
- 247.Khoroshilova N. Popescu C. Munck E. Beinert H. Kiley PJ. Iron-sulfur cluster disassembly in the FNR protein of Escherichia coli by O2: [4Fe-4S] to [2Fe-2S] conversion with loss of biological activity. Proc Natl Acad Sci U S A. 1997;94:6087–6092. doi: 10.1073/pnas.94.12.6087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Kietzmann T. Gorlach A. Reactive oxygen species in the control of hypoxia-inducible factor-mediated gene expression. Semin Cell Dev Biol. 2005;16:474–486. doi: 10.1016/j.semcdb.2005.03.010. [DOI] [PubMed] [Google Scholar]
- 249.Kipp A. Banning A. van Schothorst EM. Meplan C. Schomburg L. Evelo C. Coort S. Gaj S. Keijer J. Hesketh J. Brigelius-Flohe R. Four selenoproteins, protein biosynthesis, and Wnt signalling are particularly sensitive to limited selenium intake in mouse colon. Mol Nutr Food Res. 2009;53:1561–1572. doi: 10.1002/mnfr.200900105. [DOI] [PubMed] [Google Scholar]
- 250.Klatt P. Lamas S. Regulation of protein function by S-glutathiolation in response to oxidative and nitrosative stress. Eur J Biochem. 2000;267:4928–4944. doi: 10.1046/j.1432-1327.2000.01601.x. [DOI] [PubMed] [Google Scholar]
- 251.Klug D. Rabani J. Fridovich I. A direct demonstration of the catalytic action of superoxide dismutase through the use of pulse radiolysis. J Biol Chem. 1972;247:4839–4842. [PubMed] [Google Scholar]
- 252.Knapp LT. Kanterewicz BI. Hayes EL. Klann E. Peroxynitrite-induced tyrosine nitration and inhibition of protein kinase C. Biochem Biophys Res Commun. 2001;286:764–770. doi: 10.1006/bbrc.2001.5448. [DOI] [PubMed] [Google Scholar]
- 253.Koay MA. Christman JW. Segal BH. Venkatakrishnan A. Blackwell TR. Holland SM. Blackwell TS. Impaired pulmonary NF-kappaB activation in response to lipopolysaccharide in NADPH oxidase-deficient mice. Infect Immun. 2001;69:5991–5996. doi: 10.1128/IAI.69.10.5991-5996.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Kobayashi A. Kang MI. Watai Y. Tong KI. Shibata T. Uchida K. Yamamoto M. Oxidative and electrophilic stresses activate Nrf2 through inhibition of ubiquitination activity of Keap1. Mol Cell Biol. 2006;26:221–229. doi: 10.1128/MCB.26.1.221-229.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Kobayashi A. Kang MI. Okawa H. Ohtsuji M. Zenke Y. Chiba T. Igarashi K. Yamamoto M. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol Cell Biol. 2004;24:7130–7139. doi: 10.1128/MCB.24.16.7130-7139.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Kobayashi M. Li L. Iwamoto N. Nakajima-Takagi Y. Kaneko H. Nakayama Y. Eguchi M. Wada Y. Kumagai Y. Yamamoto M. The antioxidant defense system Keap1-Nrf2 comprises a multiple sensing mechanism for responding to a wide range of chemical compounds. Mol Cell Biol. 2009;29:493–502. doi: 10.1128/MCB.01080-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Komatsu M. Kurokawa H. Waguri S. Taguchi K. Kobayashi A. Ichimura Y. Sou YS. Ueno I. Sakamoto A. Tong KI. Kim M. Nishito Y. Iemura S. Natsume T. Ueno T. Kominami E. Motohashi H. Tanaka K. Yamamoto M. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol. 2010;12:213–223. doi: 10.1038/ncb2021. [DOI] [PubMed] [Google Scholar]
- 258.Korichneva I. Zinc dynamics in the myocardial redox signaling network. Antioxid Redox Signal. 2006;8:1707–1721. doi: 10.1089/ars.2006.8.1707. [DOI] [PubMed] [Google Scholar]
- 259.Korichneva I. Hoyos B. Chua R. Levi E. Hämmerling U. Zinc release from protein kinase C as the common event during activation by lipid second messenger or reactive oxygen. J Biol Chem. 2002;277:44327–44331. doi: 10.1074/jbc.M205634200. [DOI] [PubMed] [Google Scholar]
- 260.Korn SH. Wouters EF. Vos N. Janssen-Heininger YM. Cytokine-induced activation of nuclear factor-kappa B is inhibited by hydrogen peroxide through oxidative inactivation of IkappaB kinase. J Biol Chem. 2001;276:35693–35700. doi: 10.1074/jbc.M104321200. [DOI] [PubMed] [Google Scholar]
- 261.Kretz-Remy C. Mehlen P. Mirault ME. Arrigo AP. Inhibition of I kappa B-alpha phosphorylation and degradation and subsequent NF-kappa B activation by glutathione peroxidase overexpression. J Cell Biol. 1996;133:1083–1093. doi: 10.1083/jcb.133.5.1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Kumar H. Kawai T. Akira S. Toll-like receptors and innate immunity. Biochem Biophys Res Commun. 2009;388:621–625. doi: 10.1016/j.bbrc.2009.08.062. [DOI] [PubMed] [Google Scholar]
- 263.Kwak MK. Itoh K. Yamamoto M. Kensler TW. Enhanced expression of the transcription factor Nrf2 by cancer chemopreventive agents: role of antioxidant response element-like sequences in the nrf2 promoter. Mol Cell Biol. 2002;22:2883–2892. doi: 10.1128/MCB.22.9.2883-2892.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Kwak MK. Wakabayashi N. Greenlaw JL. Yamamoto M. Kensler TW. Antioxidants enhance mammalian proteasome expression through the Keap1-Nrf2 signaling pathway. Mol Cell Biol. 2003;23:8786–8794. doi: 10.1128/MCB.23.23.8786-8794.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Kwak MK. Wakabayashi N. Itoh K. Motohashi H. Yamamoto M. Kensler TW. Modulation of gene expression by cancer chemopreventive dithiolethiones through the Keap1-Nrf2 pathway. Identification of novel gene clusters for cell survival. J Biol Chem. 2003;278:8135–8145. doi: 10.1074/jbc.M211898200. [DOI] [PubMed] [Google Scholar]
- 266.Laird MH. Rhee SH. Perkins DJ. Medvedev AE. Piao W. Fenton MJ. Vogel SN. TLR4/MyD88/PI3K interactions regulate TLR4 signaling. J Leukoc Biol. 2009;85:966–977. doi: 10.1189/jlb.1208763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Landino LM. Moynihan KL. Todd JV. Kennett KL. Modulation of the redox state of tubulin by the glutathione/glutaredoxin reductase system. Biochem Biophys Res Commun. 2004;314:555–560. doi: 10.1016/j.bbrc.2003.12.126. [DOI] [PubMed] [Google Scholar]
- 268.Lau A. Villeneuve NF. Sun Z. Wong PK. Zhang DD. Dual roles of Nrf2 in cancer. Pharmacol Res. 2008;58:262–270. doi: 10.1016/j.phrs.2008.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Lau A. Wang XJ. Zhao F. Villeneuve NF. Wu T. Jiang T. Sun Z. White E. Zhang DD. A non-canonical mechanism of Nrf2 activation by autophagy deficiency: a direct interaction between Keap1 and p62. Mol Cell Biol. 2010;30:3275–3285. doi: 10.1128/MCB.00248-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Lawrence RA. Parkhill LK. Burk RF. Hepatic cytosolic non selenium-dependent glutathione peroxidase activity: its nature and the effect of selenium deficiency. J Nutr. 1978;108:981–987. doi: 10.1093/jn/108.6.981. [DOI] [PubMed] [Google Scholar]
- 271.Lawrence T. Fong C. The resolution of inflammation: anti-inflammatory roles for NF-kappaB. Int J Biochem Cell Biol. 2010;42:519–523. doi: 10.1016/j.biocel.2009.12.016. [DOI] [PubMed] [Google Scholar]
- 272.Lawrence T. Bebien M. Liu GY. Nizet V. Karin M. IKKalpha limits macrophage NF-kappaB activation and contributes to the resolution of inflammation. Nature. 2005;434:1138–1143. doi: 10.1038/nature03491. [DOI] [PubMed] [Google Scholar]
- 273.Lee DF. Kuo HP. Liu M. Chou CK. Xia W. Du Y. Shen J. Chen CT. Huo L. Hsu MC. Li CW. Ding Q. Liao TL. Lai CC. Lin AC. Chang YH. Tsai SF. Li LY. Hung MC. KEAP1 E3 ligase-mediated downregulation of NF-kappaB signaling by targeting IKKbeta. Mol Cell. 2009;36:131–140. doi: 10.1016/j.molcel.2009.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Lee JW. Helmann JD. The PerR transcription factor senses H2O2 by metal-catalysed histidine oxidation. Nature. 2006;440:363–367. doi: 10.1038/nature04537. [DOI] [PubMed] [Google Scholar]
- 275.Lee SR. Yang KS. Kwon J. Lee C. Jeong W. Rhee SG. Reversible inactivation of the tumor suppressor PTEN by H2O2. J Biol Chem. 2002;277:20336–20342. doi: 10.1074/jbc.M111899200. [DOI] [PubMed] [Google Scholar]
- 276.Lee TD. Yang H. Whang J. Lu SC. Cloning and characterization of the human glutathione synthetase 5′-flanking region. Biochem J. 2005;390:521–528. doi: 10.1042/BJ20050439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Lei XG. Cheng W-H. New roles of glutathione peroxidase-1 in oxidative stress and diabetes. In: Hatfield DL, editor; Berry MJ, editor; Gladyshev VN, editor. Selenium Its Molecular Biology and Role in Human Health. New York: Springer; 2006. pp. 173–182. [Google Scholar]
- 278.Leist M. Raab B. Maurer S. Rosick U. Brigelius-Flohé R. Conventional cell culture media do not adequately supply cells with antioxidants and thus facilitate peroxide-induced genotoxicity. Free Radic Biol Med. 1996;21:297–306. doi: 10.1016/0891-5849(96)00045-7. [DOI] [PubMed] [Google Scholar]
- 279.Leitges M. Sanz L. Martin P. Duran A. Braun U. Garcia JF. Camacho F. Diaz-Meco MT. Rennert PD. Moscat J. Targeted disruption of the zetaPKC gene results in the impairment of the NF-kappaB pathway. Mol Cell. 2001;8:771–780. doi: 10.1016/s1097-2765(01)00361-6. [DOI] [PubMed] [Google Scholar]
- 280.Leslie NR. Bennett D. Lindsay YE. Stewart H. Gray A. Downes CP. Redox regulation of PI 3-kinase signalling via inactivation of PTEN. EMBO J. 2003;22:5501–5510. doi: 10.1093/emboj/cdg513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Lesniak J. Barton WA. Nikolov DB. Structural and functional characterization of the Pseudomonas hydroperoxide resistance protein Ohr. EMBO J. 2002;21:6649–6659. doi: 10.1093/emboj/cdf670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Levonen AL. Landar A. Ramachandran A. Ceaser EK. Dickinson DA. Zanoni G. Morrow JD. Darley-Usmar VM. Cellular mechanisms of redox cell signalling: role of cysteine modification in controlling antioxidant defences in response to electrophilic lipid oxidation products. Biochem J. 2004;378:373–382. doi: 10.1042/BJ20031049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Li B. Wang X. Rasheed N. Hu Y. Boast S. Ishii T. Nakayama K. Nakayama KI. Goff SP. Distinct roles of c-Abl and Atm in oxidative stress response are mediated by protein kinase C delta. Genes Dev. 2004;18:1824–1837. doi: 10.1101/gad.1223504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Li Q. Engelhardt JF. Interleukin-1beta induction of NFkappaB is partially regulated by H2O2-mediated activation of NFkappaB-inducing kinase. J Biol Chem. 2006;281:1495–1505. doi: 10.1074/jbc.M511153200. [DOI] [PubMed] [Google Scholar]
- 285.Li Q. Subbulakshmi V. Oldfield CM. Aamir R. Weyman CM. Wolfman A. Cathcart MK. PKCalpha regulates phosphorylation and enzymatic activity of cPLA2 in vitro and in activated human monocytes. Cell Signal. 2007;19:359–366. doi: 10.1016/j.cellsig.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 286.Li S. Wang L. Berman MA. Zhang Y. Dorf ME. RNAi screen in mouse astrocytes identifies phosphatases that regulate NF-kappaB signaling. Mol Cell. 2006;24:497–509. doi: 10.1016/j.molcel.2006.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Li W. Kong AN. Molecular mechanisms of Nrf2-mediated antioxidant response. Mol Carcinog. 2009;48:91–104. doi: 10.1002/mc.20465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Li W. Yu SW. Kong AN. Nrf2 possesses a redox-sensitive nuclear exporting signal in the Neh5 transactivation domain. J Biol Chem. 2006;281:27251–27263. doi: 10.1074/jbc.M602746200. [DOI] [PubMed] [Google Scholar]
- 289.Li W. Jain MR. Chen C. Yue X. Hebbar V. Zhou R. Kong AN. Nrf2 Possesses a redox-insensitive nuclear export signal overlapping with the leucine zipper motif. J Biol Chem. 2005;280:28430–28438. doi: 10.1074/jbc.M410601200. [DOI] [PubMed] [Google Scholar]
- 290.Li X. Jiang S. Tapping RI. Toll-like receptor signaling in cell proliferation and survival. Cytokine. 2010;49:1–9. doi: 10.1016/j.cyto.2009.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Liao Y. Hung MC. A new role of protein phosphatase 2a in adenoviral E1A protein-mediated sensitization to anticancer drug-induced apoptosis in human breast cancer cells. Cancer Res. 2004;64:5938–5942. doi: 10.1158/0008-5472.CAN-04-1533. [DOI] [PubMed] [Google Scholar]
- 292.Lillig CH. Berndt C. Holmgren A. Glutaredoxin systems. Biochim Biophys Acta. 2008;1780:1304–1317. doi: 10.1016/j.bbagen.2008.06.003. [DOI] [PubMed] [Google Scholar]
- 293.Liu Q. Berchner-Pfannschmidt U. Moller U. Brecht M. Wotzlaw C. Acker H. Jungermann K. Kietzmann T. A Fenton reaction at the endoplasmic reticulum is involved in the redox control of hypoxia-inducible gene expression. Proc Natl Acad Sci U S A. 2004;101:4302–4307. doi: 10.1073/pnas.0400265101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Liu RM. Hu H. Robison TW. Forman HJ. Increased gamma-glutamylcysteine synthetase and gamma-glutamyl transpeptidase activities enhance resistance of rat lung epithelial L2 cells to quinone toxicity. Am J Respir Cell Mol Biol. 1996;14:192–197. doi: 10.1165/ajrcmb.14.2.8630270. [DOI] [PubMed] [Google Scholar]
- 295.Liu SF. Malik AB. NF-kappa B activation as a pathological mechanism of septic shock and inflammation. Am J Physiol Lung Cell Mol Physiol. 2006;290:L622–L645. doi: 10.1152/ajplung.00477.2005. [DOI] [PubMed] [Google Scholar]
- 296.Liu YC. Ubiquitin ligases and the immune response. Annu Rev Immunol. 2004;22:81–127. doi: 10.1146/annurev.immunol.22.012703.104813. [DOI] [PubMed] [Google Scholar]
- 297.Lo SC. Li X. Henzl MT. Beamer LJ. Hannink M. Structure of the Keap1:Nrf2 interface provides mechanistic insight into Nrf2 signaling. EMBO J. 2006;25:3605–3617. doi: 10.1038/sj.emboj.7601243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Lopez-Otin C. Hunter T. The regulatory crosstalk between kinases and proteases in cancer. Nat Rev Cancer. 2010;10:278–292. doi: 10.1038/nrc2823. [DOI] [PubMed] [Google Scholar]
- 299.Lowe JM. Cha H. Yang Q. Fornace AJ., Jr Nuclear factor-kappaB (NF-kappaB) is a novel positive transcriptional regulator of the oncogenic Wip1 phosphatase. J Biol Chem. 2010;285:5249–5257. doi: 10.1074/jbc.M109.034579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Luo JL. Maeda S. Hsu LC. Yagita H. Karin M. Inhibition of NF-kappaB in cancer cells converts inflammation-induced tumor growth mediated by TNFalpha to TRAIL-mediated tumor regression. Cancer Cell. 2004;6:297–305. doi: 10.1016/j.ccr.2004.08.012. [DOI] [PubMed] [Google Scholar]
- 301.Ma LH. Takanishi CL. Wood MJ. Molecular mechanism of oxidative stress perception by the Orp1 protein. J Biol Chem. 2007;282:31429–31436. doi: 10.1074/jbc.M705953200. [DOI] [PubMed] [Google Scholar]
- 302.Ma W. Smigel A. Verma R. Berkowitz GA. Cyclic nucleotide gated channels and related signaling components in plant innate immunity. Plant Signal Behav. 2009;4:277–282. doi: 10.4161/psb.4.4.8103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Madrid LV. Mayo MW. Reuther JY. Baldwin AS., Jr Akt stimulates the transactivation potential of the RelA/p65 Subunit of NF-kappa B through utilization of the Ikappa B kinase and activation of the mitogen-activated protein kinase p38. J Biol Chem. 2001;276:18934–18940. doi: 10.1074/jbc.M101103200. [DOI] [PubMed] [Google Scholar]
- 304.Maehama T. Dixon JE. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem. 1998;273:13375–13378. doi: 10.1074/jbc.273.22.13375. [DOI] [PubMed] [Google Scholar]
- 305.Maher J. Yamamoto M. The rise of antioxidant signaling—the evolution and hormetic actions of Nrf2. Toxicol Appl Pharmacol. 2010;244:4–15. doi: 10.1016/j.taap.2010.01.011. [DOI] [PubMed] [Google Scholar]
- 306.Manna SK. Zhang HJ. Yan T. Oberley LW. Aggarwal BB. Overexpression of manganese superoxide dismutase suppresses tumor necrosis factor-induced apoptosis and activation of nuclear transcription factor-kappaB and activated protein-1. J Biol Chem. 1998;273:13245–13254. doi: 10.1074/jbc.273.21.13245. [DOI] [PubMed] [Google Scholar]
- 307.Mao X. Gluck N. Li D. Maine GN. Li H. Zaidi IW. Repaka A. Mayo MW. Burstein E. GCN5 is a required cofactor for a ubiquitin ligase that targets NF-kappaB/RelA. Genes Dev. 2009;23:849–861. doi: 10.1101/gad.1748409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Marcocci L. Flohe L. Packer L. Evidence for a functional relevance of the selenocysteine residue in mammalian thioredoxin reductase. Biofactors. 1997;6:351–358. doi: 10.1002/biof.5520060305. [DOI] [PubMed] [Google Scholar]
- 309.Maret W. Zinc coordination environments in proteins as redox sensors and signal transducers. Antioxid Redox Signal. 2006;8:1419–1441. doi: 10.1089/ars.2006.8.1419. [DOI] [PubMed] [Google Scholar]
- 310.Marmillot P. Scovell W. Enhancement of transcription factor, USF, binding to the adenovirus major late promoter: effect of dithiothreitol and high mobility group protein-1. Biochim Biophys Acta. 1998;1395:228–236. doi: 10.1016/s0167-4781(97)00153-x. [DOI] [PubMed] [Google Scholar]
- 311.Martin D. Rojo AI. Salinas M. Diaz R. Gallardo G. Alam J. De Galarreta CM. Cuadrado A. Regulation of heme oxygenase-1 expression through the phosphatidylinositol 3-kinase/Akt pathway and the Nrf2 transcription factor in response to the antioxidant phytochemical carnosol. J Biol Chem. 2004;279:8919–8929. doi: 10.1074/jbc.M309660200. [DOI] [PubMed] [Google Scholar]
- 312.Martin M. Böl GF. Eriksson A. Resch K. Brigelius-Flohé R. Interleukin-1-induced activation of a protein kinase co-precipitating with the type I interleukin-1 receptor in T cells. Eur J Immunol. 1994;24:1566–1571. doi: 10.1002/eji.1830240717. [DOI] [PubMed] [Google Scholar]
- 313.Martin M. Schifferle RE. Cuesta N. Vogel SN. Katz J. Michalek SM. Role of the phosphatidylinositol 3 kinase-Akt pathway in the regulation of IL-10 and IL-12 by Porphyromonas gingivalis lipopolysaccharide. J Immunol. 2003;171:717–725. doi: 10.4049/jimmunol.171.2.717. [DOI] [PubMed] [Google Scholar]
- 314.Maskiewicz R. Bruice TC. Bartsch RG. The acid-base properties and kinetics of dissolution of the Fe4S4 cores of Chromatin ferredoxin and high potential iron protein. Biochem Biophys Res Commun. 1975;65:407–412. doi: 10.1016/s0006-291x(75)80108-2. [DOI] [PubMed] [Google Scholar]
- 315.Matthews JR. Wakasugi N. Virelizier JL. Yodoi J. Hay RT. Thioredoxin regulates the DNA binding activity of NF-kappa B by reduction of a disulphide bond involving cysteine 62. Nucleic Acids Res. 1992;20:3821–3830. doi: 10.1093/nar/20.15.3821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Mattioli I. Sebald A. Bucher C. Charles RP. Nakano H. Doi T. Kracht M. Schmitz ML. Transient and selective NF-kappa B p65 serine 536 phosphorylation induced by T cell costimulation is mediated by I kappa B kinase beta and controls the kinetics of p65 nuclear import. J Immunol. 2004;172:6336–6344. doi: 10.4049/jimmunol.172.10.6336. [DOI] [PubMed] [Google Scholar]
- 317.Mattson MP. Hormesis defined. Ageing Res Rev. 2008;7:1–7. doi: 10.1016/j.arr.2007.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Mauri P. Benazzi L. Flohé L. Maiorino M. Pietta PG. Pilawa S. Roveri A. Ursini F. Versatility of selenium catalysis in PHGPx unraveled by LC/ESI-MS/MS. Biol Chem Hoppe Seyler. 2003;384:575–588. doi: 10.1515/BC.2003.065. [DOI] [PubMed] [Google Scholar]
- 319.May JM. de Haen C. Insulin-stimulated intracellular hydrogen peroxide production in rat epididymal fat cells. J Biol Chem. 1979;254:2214–2220. [PubMed] [Google Scholar]
- 320.May JM. de Haen C. The insulin-like effect of hydrogen peroxide on pathways of lipid synthesis in rat adipocytes. J Biol Chem. 1979;254:9017–9021. [PubMed] [Google Scholar]
- 321.McAdam ME. Levelle F. Fox RA. Fielden EM. A pulse-radiolysis study of the manganese-containing superoxide dismutase from Bacillus stearothermophilus. Biochem J. 1977;165:81–87. doi: 10.1042/bj1650081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.McCord JM. Fridovich I. Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein) J Biol Chem. 1969;244:6049–6055. [PubMed] [Google Scholar]
- 323.McMahon M. Itoh K. Yamamoto M. Hayes JD. Keap1-dependent proteasomal degradation of transcription factor Nrf2 contributes to the negative regulation of antioxidant response element-driven gene expression. J Biol Chem. 2003;278:21592–21600. doi: 10.1074/jbc.M300931200. [DOI] [PubMed] [Google Scholar]
- 324.McMahon M. Thomas N. Itoh K. Yamamoto M. Hayes JD. Redox-regulated turnover of Nrf2 is determined by at least two separate protein domains, the redox-sensitive Neh2 degron and the redox-insensitive Neh6 degron. J Biol Chem. 2004;279:31556–31567. doi: 10.1074/jbc.M403061200. [DOI] [PubMed] [Google Scholar]
- 325.McMahon M. Thomas N. Itoh K. Yamamoto M. Hayes JD. Dimerization of substrate adaptors can facilitate cullin-mediated ubiquitylation of proteins by a “tethering” mechanism: a two-site interaction model for the Nrf2-Keap1 complex. J Biol Chem. 2006;281:24756–24768. doi: 10.1074/jbc.M601119200. [DOI] [PubMed] [Google Scholar]
- 326.McMahon M. Itoh K. Yamamoto M. Chanas SA. Henderson CJ. McLellan LI. Wolf CR. Cavin C. Hayes JD. The Cap‘n’Collar basic leucine zipper transcription factor Nrf2 (NF-E2 p45-related factor 2) controls both constitutive and inducible expression of intestinal detoxification and glutathione biosynthetic enzymes. Cancer Res. 2001;61:3299–3307. [PubMed] [Google Scholar]
- 327.McManus EJ. Sakamoto K. Armit LJ. Ronaldson L. Shpiro N. Marquez R. Alessi DR. Role that phosphorylation of GSK3 plays in insulin and Wnt signalling defined by knockin analysis. EMBO J. 2005;24:1571–1583. doi: 10.1038/sj.emboj.7600633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328.Miller TW. Cherney MM. Lee AJ. Francoleon NE. Farmer PJ. King SB. Hobbs AJ. Miranda KM. Burstyn JN. Fukuto JM. The effects of nitroxyl (HNO) on soluble guanylate cyclase activity: interactions at ferrous heme and cysteine thiols. J Biol Chem. 2009;284:21788–21796. doi: 10.1074/jbc.M109.014282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329.Moi P. Chan K. Asunis I. Cao A. Kan YW. Isolation of NF-E2-related factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the beta-globin locus control region. Proc Natl Acad Sci U S A. 1994;91:9926–9930. doi: 10.1073/pnas.91.21.9926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.Moinova HR. Mulcahy RT. An electrophile responsive element (EpRE) regulates beta-naphthoflavone induction of the human gamma-glutamylcysteine synthetase regulatory subunit gene. Constitutive expression is mediated by an adjacent AP-1 site. J Biol Chem. 1998;273:14683–14689. doi: 10.1074/jbc.273.24.14683. [DOI] [PubMed] [Google Scholar]
- 331.Moncada S. Palmer RM. Higgs EA. The discovery of nitric oxide as the endogenous nitrovasodilator. Hypertension. 1988;12:365–372. doi: 10.1161/01.hyp.12.4.365. [DOI] [PubMed] [Google Scholar]
- 332.Mongkolsuk S. Helmann JD. Regulation of inducible peroxide stress responses. Mol Microbiol. 2002;45:9–15. doi: 10.1046/j.1365-2958.2002.03015.x. [DOI] [PubMed] [Google Scholar]
- 333.Morgan BA. Veal EA. Functions of typical 2-Cys peroxiredoxins in yeast. Subcell Biochem. 2007;44:253–265. doi: 10.1007/978-1-4020-6051-9_12. [DOI] [PubMed] [Google Scholar]
- 334.Muckenthaler MU. Galy B. Hentze MW. Systemic iron homeostasis and the iron-responsive element/iron-regulatory protein (IRE/IRP) regulatory network. Annu Rev Nutr. 2008;28:197–213. doi: 10.1146/annurev.nutr.28.061807.155521. [DOI] [PubMed] [Google Scholar]
- 335.Mueller M. Banning A. Brigelius-Flohé R. Kipp A. Nrf2 target genes are induced under marginal selenium-deficiency. Genes Nutr. 2010;5:297–307. doi: 10.1007/s12263-010-0168-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336.Mulcahy RT. Gipp JJ. Identification of a putative antioxidant response element in the 5′-flanking region of the human gamma-glutamylcysteine synthetase heavy subunit gene. Biochem Biophys Res Commun. 1995;209:227–233. doi: 10.1006/bbrc.1995.1493. [DOI] [PubMed] [Google Scholar]
- 337.Muller JR. Siebenlist U. Lymphotoxin beta receptor induces sequential activation of distinct NF-kappa B factors via separate signaling pathways. J Biol Chem. 2003;278:12006–12012. doi: 10.1074/jbc.M210768200. [DOI] [PubMed] [Google Scholar]
- 338.Murata H. Ihara Y. Nakamura H. Yodoi J. Sumikawa K. Kondo T. Glutaredoxin exerts an antiapoptotic effect by regulating the redox state of Akt. J Biol Chem. 2003;278:50226–50233. doi: 10.1074/jbc.M310171200. [DOI] [PubMed] [Google Scholar]
- 339.Nakashima I. Takeda K. Kawamoto Y. Okuno Y. Kato M. Suzuki H. Redox control of catalytic activities of membrane-associated protein tyrosine kinases. Arch Biochem Biophys. 2005;434:3–10. doi: 10.1016/j.abb.2004.06.016. [DOI] [PubMed] [Google Scholar]
- 340.Natoli G. Chiocca S. Nuclear ubiquitin ligases, NF-kappaB degradation, and the control of inflammation. Sci Signal. 2008;1:pe1. doi: 10.1126/stke.11pe1. [DOI] [PubMed] [Google Scholar]
- 341.Naumann M. Scheidereit C. Activation of NF-kappa B in vivo is regulated by multiple phosphorylations. EMBO J. 1994;13:4597–4607. doi: 10.1002/j.1460-2075.1994.tb06781.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342.Nawaz Z. Lonard DM. Dennis AP. Smith CL. O'Malley BW. Proteasome-dependent degradation of the human estrogen receptor. Proc Natl Acad Sci U S A. 1999;96:1858–1862. doi: 10.1073/pnas.96.5.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343.Neumann M. Grieshammer T. Chuvpilo S. Kneitz B. Lohoff M. Schimpl A. Franza BR., Jr. Serfling E. RelA/p65 is a molecular target for the immunosuppressive action of protein kinase A. EMBO J. 1995;14:1991–2004. doi: 10.1002/j.1460-2075.1995.tb07191.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344.Nguyen T. Sherratt PJ. Huang HC. Yang CS. Pickett CB. Increased protein stability as a mechanism that enhances Nrf2-mediated transcriptional activation of the antioxidant response element. Degradation of Nrf2 by the 26 S proteasome. J Biol Chem. 2003;278:4536–4541. doi: 10.1074/jbc.M207293200. [DOI] [PubMed] [Google Scholar]
- 345.Nioi P. Nguyen T. Sherratt PJ. Pickett CB. The carboxy-terminal Neh3 domain of Nrf2 is required for transcriptional activation. Mol Cell Biol. 2005;25:10895–10906. doi: 10.1128/MCB.25.24.10895-10906.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346.Nishi T. Shimizu N. Hiramoto M. Sato I. Yamaguchi Y. Hasegawa M. Aizawa S. Tanaka H. Kataoka K. Watanabe H. Handa H. Spatial redox regulation of a critical cysteine residue of NF-kappa B in vivo. J Biol Chem. 2002;277:44548–44556. doi: 10.1074/jbc.M202970200. [DOI] [PubMed] [Google Scholar]
- 347.Niture SK. Jain AK. Jaiswal AK. Antioxidant-induced modification of INrf2 cysteine 151 and PKC-delta-mediated phosphorylation of Nrf2 serine 40 are both required for stabilization and nuclear translocation of Nrf2 and increased drug resistance. J Cell Sci. 2009;122:4452–4464. doi: 10.1242/jcs.058537. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 348.Niture SK. Kaspar JW. Shen J. Jaiswal AK. Nrf2 signaling and cell survival. Toxicol Appl Pharmacol. 2010;244:37–42. doi: 10.1016/j.taap.2009.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349.Nogoceke E. Gommel DU. Kiess M. Kalisz HM. Flohe L. A unique cascade of oxidoreductases catalyses trypanothione-mediated peroxide metabolism in Crithidia fasciculata. Biol Chem. 1997;378:827–836. doi: 10.1515/bchm.1997.378.8.827. [DOI] [PubMed] [Google Scholar]
- 350.Numazawa S. Ishikawa M. Yoshida A. Tanaka S. Yoshida T. Atypical protein kinase C mediates activation of NF-E2-related factor 2 in response to oxidative stress. Am J Physiol Cell Physiol. 2003;285:C334–C342. doi: 10.1152/ajpcell.00043.2003. [DOI] [PubMed] [Google Scholar]
- 351.Nunoshiba T. Hidalgo E. Amabile Cuevas CF. Demple B. Two-stage control of an oxidative stress regulon: the Escherichia coli SoxR protein triggers redox-inducible expression of the soxS regulatory gene. J Bacteriol. 1992;174:6054–6060. doi: 10.1128/jb.174.19.6054-6060.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352.O'Shea JM. Perkins ND. Thr435 phosphorylation regulates RelA (p65) NF-kappaB subunit transactivation. Biochem J. 2010;426:345–354. doi: 10.1042/BJ20091630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 353.Oakley FD. Smith RL. Engelhardt JF. Lipid rafts and caveolin-1 coordinate interleukin-1beta (IL-1beta)-dependent activation of NFkappaB by controlling endocytosis of Nox2 and IL-1beta receptor 1 from the plasma membrane. J Biol Chem. 2009;284:33255–33264. doi: 10.1074/jbc.M109.042127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354.Oakley FD. Abbott D. Li Q. Engelhardt JF. Signaling components of redox active endosomes: the redoxosomes. Antioxid Redox Signal. 2009;11:1313–1333. doi: 10.1089/ars.2008.2363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355.Obin M. Shang F. Gong X. Handelman G. Blumberg J. Taylor A. Redox regulation of ubiquitin-conjugating enzymes: mechanistic insights using the thiol-specific oxidant diamide. FASEB J. 1998;12:561–569. doi: 10.1096/fasebj.12.7.561. [DOI] [PubMed] [Google Scholar]
- 356.Ohlsson K. Bjork P. Bergenfeldt M. Hageman R. Thompson RC. Interleukin-1 receptor antagonist reduces mortality from endotoxin shock. Nature. 1990;348:550–552. doi: 10.1038/348550a0. [DOI] [PubMed] [Google Scholar]
- 357.Okazaki S. Naganuma A. Kuge S. Peroxiredoxin-mediated redox regulation of the nuclear localization of Yap1, a transcription factor in budding yeast. Antioxid Redox Signal. 2005;7:327–334. doi: 10.1089/ars.2005.7.327. [DOI] [PubMed] [Google Scholar]
- 358.Oliveira-Marques V. Marinho HS. Cyrne L. Antunes F. Role of hydrogen peroxide in NF-kappaB activation: from inducer to modulator. Antioxid Redox Signal. 2009;11:2223–2243. doi: 10.1089/ars.2009.2601. [DOI] [PubMed] [Google Scholar]
- 359.Orrenius S. Zhivotovsky B. Nicotera P. Regulation of cell death: the calcium-apoptosis link. Nat Rev Mol Cell Biol. 2003;4:552–565. doi: 10.1038/nrm1150. [DOI] [PubMed] [Google Scholar]
- 360.Osburn WO. Karim B. Dolan PM. Liu G. Yamamoto M. Huso DL. Kensler TW. Increased colonic inflammatory injury and formation of aberrant crypt foci in Nrf2-deficient mice upon dextran sulfate treatment. Int J Cancer. 2007;121:1883–1891. doi: 10.1002/ijc.22943. [DOI] [PubMed] [Google Scholar]
- 361.Padmanabhan B. Nakamura Y. Yokoyama S. Structural analysis of the complex of Keap1 with a prothymosin alpha peptide. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2008;64:233–238. doi: 10.1107/S1744309108004995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 362.Padmanabhan B. Tong KI. Ohta T. Nakamura Y. Scharlock M. Ohtsuji M. Kang MI. Kobayashi A. Yokoyama S. Yamamoto M. Structural basis for defects of Keap1 activity provoked by its point mutations in lung cancer. Mol Cell. 2006;21:689–700. doi: 10.1016/j.molcel.2006.01.013. [DOI] [PubMed] [Google Scholar]
- 363.Pantano C. Reynaert NL. van der Vliet A. Janssen-Heininger YM. Redox-sensitive kinases of the nuclear factor-kappaB signaling pathway. Antioxid Redox Signal. 2006;8:1791–1806. doi: 10.1089/ars.2006.8.1791. [DOI] [PubMed] [Google Scholar]
- 364.Pariat M. Salvat C. Bebien M. Brockly F. Altieri E. Carillo S. Jariel-Encontre I. Piechaczyk M. The sensitivity of c-Jun and c-Fos proteins to calpains depends on conformational determinants of the monomers and not on formation of dimers. Biochem J. 2000;345(Pt 1):129–138. [PMC free article] [PubMed] [Google Scholar]
- 365.Park HS. Chun JN. Jung HY. Choi C. Bae YS. Role of NADPH oxidase 4 in lipopolysaccharide-induced proinflammatory responses by human aortic endothelial cells. Cardiovasc Res. 2006;72:447–455. doi: 10.1016/j.cardiores.2006.09.012. [DOI] [PubMed] [Google Scholar]
- 366.Park HS. Jung HY. Park EY. Kim J. Lee WJ. Bae YS. Cutting edge: direct interaction of TLR4 with NAD(P)H oxidase 4 isozyme is essential for lipopolysaccharide-induced production of reactive oxygen species and activation of NF-kappa B. J Immunol. 2004;173:3589–3593. doi: 10.4049/jimmunol.173.6.3589. [DOI] [PubMed] [Google Scholar]
- 367.Park SW. Huq MD. Hu X. Wei LN. Tyrosine nitration on p65: a novel mechanism to rapidly inactivate nuclear factor-kappaB. Mol Cell Proteomics. 2005;4:300–309. doi: 10.1074/mcp.M400195-MCP200. [DOI] [PubMed] [Google Scholar]
- 368.Perkins ND. Integrating cell-signalling pathways with NF-kappaB and IKK function. Nat Rev Mol Cell Biol. 2007;8:49–62. doi: 10.1038/nrm2083. [DOI] [PubMed] [Google Scholar]
- 369.Perl M. Chung CS. Garber M. Huang X. Ayala A. Contribution of anti-inflammatory/immune suppressive processes to the pathology of sepsis. Front Biosci. 2006;11:272–299. doi: 10.2741/1797. [DOI] [PubMed] [Google Scholar]
- 370.Petroski MD. Deshaies RJ. Function and regulation of cullin-RING ubiquitin ligases. Nat Rev Mol Cell Biol. 2005;6:9–20. doi: 10.1038/nrm1547. [DOI] [PubMed] [Google Scholar]
- 371.Pi J. Bai Y. Reece JM. Williams J. Liu D. Freeman ML. Fahl WE. Shugar D. Liu J. Qu W. Collins S. Waalkes MP. Molecular mechanism of human Nrf2 activation and degradation: role of sequential phosphorylation by protein kinase CK2. Free Radic Biol Med. 2007;42:1797–1806. doi: 10.1016/j.freeradbiomed.2007.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 372.Pickart CM. Eddins MJ. Ubiquitin: structures, functions, mechanisms. Biochim Biophys Acta. 2004;1695:55–72. doi: 10.1016/j.bbamcr.2004.09.019. [DOI] [PubMed] [Google Scholar]
- 373.Pietsch EC. Chan JY. Torti FM. Torti SV. Nrf2 mediates the induction of ferritin H in response to xenobiotics and cancer chemopreventive dithiolethiones. J Biol Chem. 2003;278:2361–2369. doi: 10.1074/jbc.M210664200. [DOI] [PubMed] [Google Scholar]
- 374.Pikarsky E. Porat RM. Stein I. Abramovitch R. Amit S. Kasem S. Gutkovich-Pyest E. Urieli-Shoval S. Galun E. Ben-Neriah Y. NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature. 2004;431:461–466. doi: 10.1038/nature02924. [DOI] [PubMed] [Google Scholar]
- 375.Pillinger MH. Philips MR. Feoktistov A. Weissmann G. Crosstalk in signal transduction via EP receptors: prostaglandin E1 inhibits chemoattractant-induced mitogen-activated protein kinase activity in human neutrophils. Adv Prostaglandin Thromboxane Leukot Res. 1995;23:311–316. [PubMed] [Google Scholar]
- 376.Pineda-Molina E. Klatt P. Vazquez J. Marina A. Garcia de Lacoba M. Perez-Sala D. Lamas S. Glutathionylation of the p50 subunit of NF-kappaB: a mechanism for redox-induced inhibition of DNA binding. Biochemistry. 2001;40:14134–14142. doi: 10.1021/bi011459o. [DOI] [PubMed] [Google Scholar]
- 377.Pognonec P. Kato H. Roeder RG. The helix-loop-helix/leucine repeat transcription factor USF can be functionally regulated in a redox-dependent manner. J Biol Chem. 1992;267:24563–24567. [PubMed] [Google Scholar]
- 378.Poole LB. The catalytic mechanism of peroxiredoxins. Subcell Biochem. 2007;44:61–81. doi: 10.1007/978-1-4020-6051-9_4. [DOI] [PubMed] [Google Scholar]
- 379.Poole LB. Claiborne A. The non-flavin redox center of the streptococcal NADH peroxidase. II. Evidence for a stabilized cysteine-sulfenic acid. J Biol Chem. 1989;264:12330–12338. [PubMed] [Google Scholar]
- 380.Poole LB. Ellis HR. Identification of cysteine sulfenic acid in AhpC of alkyl hydroperoxide reductase. Methods Enzymol. 2002;348:122–136. doi: 10.1016/s0076-6879(02)48632-6. [DOI] [PubMed] [Google Scholar]
- 381.Posner BI. Faure R. Burgess JW. Bevan AP. Lachance D. Zhang-Sun G. Fantus IG. Ng JB. Hall DA. Lum BS, et al. Peroxovanadium compounds. A new class of potent phosphotyrosine phosphatase inhibitors which are insulin mimetics. J Biol Chem. 1994;269:4596–4604. [PubMed] [Google Scholar]
- 382.Powell SR. The antioxidant properties of zinc. J Nutr. 2000;130:1447S–1454S. doi: 10.1093/jn/130.5.1447S. [DOI] [PubMed] [Google Scholar]
- 383.Prabhakar NR. Redox Pioneer: Professor Gregg L. Semenza. Antioxid Redox Signal. 2010;13:559–564. doi: 10.1089/ars.2010.3155. [DOI] [PubMed] [Google Scholar]
- 384.Prajapati S. Verma U. Yamamoto Y. Kwak YT. Gaynor RB. Protein phosphatase 2Cbeta association with the IkappaB kinase complex is involved in regulating NF-kappaB activity. J Biol Chem. 2004;279:1739–1746. doi: 10.1074/jbc.M306273200. [DOI] [PubMed] [Google Scholar]
- 385.Primiano T. Li Y. Kensler TW. Trush MA. Sutter TR. Identification of dithiolethione-inducible gene-1 as a leukotriene B4 12-hydroxydehydrogenase: implications for chemoprevention. Carcinogenesis. 1998;19:999–1005. doi: 10.1093/carcin/19.6.999. [DOI] [PubMed] [Google Scholar]
- 386.Psarra AM. Hermann S. Panayotou G. Spyrou G. Interaction of mitochondrial thioredoxin with glucocorticoid receptor and NF-kappaB modulates glucocorticoid receptor and NF-kappaB signalling in HEK-293 cells. Biochem J. 2009;422:521–531. doi: 10.1042/BJ20090107. [DOI] [PubMed] [Google Scholar]
- 387.Purdom-Dickinson SE. Sheveleva EV. Sun H. Chen QM. Translational control of nrf2 protein in activation of antioxidant response by oxidants. Mol Pharmacol. 2007;72:1074–1081. doi: 10.1124/mol.107.035360. [DOI] [PubMed] [Google Scholar]
- 388.Qanungo S. Starke DW. Pai HV. Mieyal JJ. Nieminen AL. Glutathione supplementation potentiates hypoxic apoptosis by S-glutathionylation of p65-NFkappaB. J Biol Chem. 2007;282:18427–18436. doi: 10.1074/jbc.M610934200. [DOI] [PubMed] [Google Scholar]
- 389.Rachakonda G. Xiong Y. Sekhar KR. Stamer SL. Liebler DC. Freeman ML. Covalent modification at Cys151 dissociates the electrophile sensor Keap1 from the ubiquitin ligase CUL3. Chem Res Toxicol. 2008;21:705–710. doi: 10.1021/tx700302s. [DOI] [PubMed] [Google Scholar]
- 390.Rachoin JS. Schorr CA. Dellinger RP. Targeting endotoxin in the treatment of sepsis. Subcell Biochem. 2010;53:323–338. doi: 10.1007/978-90-481-9078-2_15. [DOI] [PubMed] [Google Scholar]
- 391.Rainwater R. Parks D. Anderson ME. Tegtmeyer P. Mann K. Role of cysteine residues in regulation of p53 function. Mol Cell Biol. 1995;15:3892–3903. doi: 10.1128/mcb.15.7.3892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 392.Ramos-Gomez M. Kwak MK. Dolan PM. Itoh K. Yamamoto M. Talalay P. Kensler TW. Sensitivity to carcinogenesis is increased and chemoprotective efficacy of enzyme inducers is lost in nrf2 transcription factor-deficient mice. Proc Natl Acad Sci U S A. 2001;98:3410–3415. doi: 10.1073/pnas.051618798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 393.Rangasamy T. Cho CY. Thimmulappa RK. Zhen L. Srisuma SS. Kensler TW. Yamamoto M. Petrache I. Tuder RM. Biswal S. Genetic ablation of Nrf2 enhances susceptibility to cigarette smoke-induced emphysema in mice. J Clin Invest. 2004;114:1248–1259. doi: 10.1172/JCI21146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 394.Rao VA. Klein SR. Bonar SJ. Zielonka J. Mizuno N. Dickey JS. Keller PW. Joseph J. Kalyanaraman B. Shacter E. The antioxidant transcription factor Nrf2 negatively regulates autophagy and growth arrest induced by the anticancer redox agent mitoquinone. J Biol Chem. 2010;285:34447–34459. doi: 10.1074/jbc.M110.133579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 395.Reiter R. Wendel A. Selenium and drug metabolism—I. Multiple modulations of mouse liver enzymes. Biochem Pharmacol. 1983;32:3063–3067. doi: 10.1016/0006-2952(83)90250-2. [DOI] [PubMed] [Google Scholar]
- 396.Reiter R. Wendel A. Selenium and drug metabolism—II. Independence of glutathione peroxidase and reversibility of hepatic enzyme modulations in deficient mice. Biochem Pharmacol. 1984;33:1923–1928. doi: 10.1016/0006-2952(84)90548-3. [DOI] [PubMed] [Google Scholar]
- 397.Reiter R. Wendel A. Selenium and drug metabolism—III. Relation of glutathione-peroxidase and other hepatic enzyme modulations to dietary supplements. Biochem Pharmacol. 1985;34:2287–2290. doi: 10.1016/0006-2952(85)90783-x. [DOI] [PubMed] [Google Scholar]
- 398.Rhee SG. Woo HA. Multiple functions of peroxiredoxins: peroxidases, sensors and regulators of the intracellular messenger H2O2, and protein chaperones. Antioxid Redox Signal. 2011;15:781–794. doi: 10.1089/ars.2010.3393. [DOI] [PubMed] [Google Scholar]
- 399.Rhee SG. Chae HZ. Kim K. Peroxiredoxins: a historical overview and speculative preview of novel mechanisms and emerging concepts in cell signaling. Free Radic Biol Med. 2005;38:1543–1552. doi: 10.1016/j.freeradbiomed.2005.02.026. [DOI] [PubMed] [Google Scholar]
- 400.Rhee SG. Kang SW. Jeong W. Chang TS. Yang KS. Woo HA. Intracellular messenger function of hydrogen peroxide and its regulation by peroxiredoxins. Curr Opin Cell Biol. 2005;17:183–189. doi: 10.1016/j.ceb.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 401.Rice NR. Ernst MK. In vivo control of NF-kappa B activation by I kappa B alpha. EMBO J. 1993;12:4685–4695. doi: 10.1002/j.1460-2075.1993.tb06157.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 402.Ricote M. Li AC. Willson TM. Kelly CJ. Glass CK. The peroxisome proliferator-activated receptor-gamma is a negative regulator of macrophage activation. Nature. 1998;391:79–82. doi: 10.1038/34178. [DOI] [PubMed] [Google Scholar]
- 403.Riddell JR. Wang XY. Minderman H. Gollnick SO. Peroxiredoxin 1 stimulates secretion of proinflammatory cytokines by binding to TLR4. J Immunol. 2010;184:1022–1030. doi: 10.4049/jimmunol.0901945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 404.Rietschel ET. Kirikae T. Feist W. Loppnow H. Zabel P. Brade L. Ulmer AJ. Brade H. Seydel U. Zähringer U. Schlaak M. Flad HD. Schade U. Molecular aspects of the chemistryand biology of endotoxin. In: Sie H, editor; Flohé L, editor; Zimmer G, editor. Molecular Aspects of Inflammation. Heidelberg: Springer-Verlag; 1991. pp. 207–231. [Google Scholar]
- 405.Rinna A. Torres M. Forman HJ. Stimulation of the alveolar macrophage respiratory burst by ADP causes selective glutathionylation of protein tyrosine phosphatase 1B. Free Radic Biol Med. 2006;41:86–91. doi: 10.1016/j.freeradbiomed.2006.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 406.Roff M. Thompson J. Rodriguez MS. Jacque JM. Baleux F. Arenzana-Seisdedos F. Hay RT. Role of IkappaBalpha ubiquitination in signal-induced activation of NFkappaB in vivo. J Biol Chem. 1996;271:7844–7850. doi: 10.1074/jbc.271.13.7844. [DOI] [PubMed] [Google Scholar]
- 407.Ross SJ. Findlay VJ. Malakasi P. Morgan BA. Thioredoxin peroxidase is required for the transcriptional response to oxidative stress in budding yeast. Mol Biol Cell. 2000;11:2631–2642. doi: 10.1091/mbc.11.8.2631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 408.Rossi A. Kapahi P. Natoli G. Takahashi T. Chen Y. Karin M. Santoro MG. Anti-inflammatory cyclopentenone prostaglandins are direct inhibitors of IkappaB kinase. Nature. 2000;403:103–108. doi: 10.1038/47520. [DOI] [PubMed] [Google Scholar]
- 409.Rudolph D. Yeh WC. Wakeham A. Rudolph B. Nallainathan D. Potter J. Elia AJ. Mak TW. Severe liver degeneration and lack of NF-kappaB activation in NEMO/IKKgamma-deficient mice. Genes Dev. 2000;14:854–862. [PMC free article] [PubMed] [Google Scholar]
- 410.Rudolph TK. Freeman BA. Transduction of redox signaling by electrophile-protein reactions. Sci Signal. 2009;2:re1–re7. doi: 10.1126/scisignal.290re7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 411.Rushmore TH. Pickett CB. Transcriptional regulation of the rat glutathione S-transferase Ya subunit gene. Characterization of a xenobiotic-responsive element controlling inducible expression by phenolic antioxidants. J Biol Chem. 1990;265:14648–14653. [PubMed] [Google Scholar]
- 412.Sablina AA. Budanov AV. Ilyinskaya GV. Agapova LS. Kravchenko JE. Chumakov PM. The antioxidant function of the p53 tumor suppressor. Nat Med. 2005;11:1306–1313. doi: 10.1038/nm1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 413.Saccani S. Marazzi I. Beg AA. Natoli G. Degradation of promoter-bound p65/RelA is essential for the prompt termination of the nuclear factor kappaB response. J Exp Med. 2004;200:107–113. doi: 10.1084/jem.20040196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 414.Sadikot RT. Zeng H. Yull FE. Li B. Cheng DS. Kernodle DS. Jansen ED. Contag CH. Segal BH. Holland SM. Blackwell TS. Christman JW. p47phox deficiency impairs NF-kappa B activation and host defense in Pseudomonas pneumonia. J Immunol. 2004;172:1801–1808. doi: 10.4049/jimmunol.172.3.1801. [DOI] [PubMed] [Google Scholar]
- 415.Saitoh M. Nishitoh H. Fujii M. Takeda K. Tobiume K. Sawada Y. Kawabata M. Miyazono K. Ichijo H. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J. 1998;17:2596–2606. doi: 10.1093/emboj/17.9.2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 416.Sakurai H. Chiba H. Miyoshi H. Sugita T. Toriumi W. IkappaB kinases phosphorylate NF-kappaB p65 subunit on serine 536 in the transactivation domain. J Biol Chem. 1999;274:30353–30356. doi: 10.1074/jbc.274.43.30353. [DOI] [PubMed] [Google Scholar]
- 417.Salazar M. Rojo AI. Velasco D. de Sagarra RM. Cuadrado A. Glycogen synthase kinase-3beta inhibits the xenobiotic and antioxidant cell response by direct phosphorylation and nuclear exclusion of the transcription factor Nrf2. J Biol Chem. 2006;281:14841–14851. doi: 10.1074/jbc.M513737200. [DOI] [PubMed] [Google Scholar]
- 418.Salmeen A. Barford D. Functions and mechanisms of redox regulation of cysteine-based phosphatases. Antioxid Redox Signal. 2005;7:560–577. doi: 10.1089/ars.2005.7.560. [DOI] [PubMed] [Google Scholar]
- 419.Salmeen A. Andersen JN. Myers MP. Meng TC. Hinks JA. Tonks NK. Barford D. Redox regulation of protein tyrosine phosphatase 1B involves a sulphenyl-amide intermediate. Nature. 2003;423:769–773. doi: 10.1038/nature01680. [DOI] [PubMed] [Google Scholar]
- 420.Salvat C. Aquaviva C. Jariel-Encontre I. Ferrara P. Pariat M. Steff AM. Carillo S. Piechaczyk M. Are there multiple proteolytic pathways contributing to c-Fos, c-Jun and p53 protein degradation in vivo? Mol Biol Rep. 1999;26:45–51. doi: 10.1023/a:1006960021281. [DOI] [PubMed] [Google Scholar]
- 421.Samuni A. Aronovitch J. Chevion M. Capski G. Metal-mediated hdroxyl radical damage. A site-specific mechanism. In: Rotilio G, editor; Bannister JV, editor. Oxidative Damage and Related Enzymes. Chur, London, Paris, New York: Harwood Academic Publishers; 1983. pp. 39–47. [Google Scholar]
- 422.Sanchez-Gomez FJ. Cernuda-Morollon E. Stamatakis K. Perez-Sala D. Protein thiol modification by 15-deoxy-Delta12,14-prostaglandin J2 addition in mesangial cells: role in the inhibition of pro-inflammatory genes. Mol Pharmacol. 2004;66:1349–1358. doi: 10.1124/mol.104.002824. [DOI] [PubMed] [Google Scholar]
- 423.Sarma BK. Mugesh G. Antioxidant activity of the anti-inflammatory compound ebselen: a reversible cyclization pathway via selenenic and seleninic acid intermediates. Chemistry. 2008;14:10603–10614. doi: 10.1002/chem.200801258. [DOI] [PubMed] [Google Scholar]
- 424.Sasaki H. Sato H. Kuriyama-Matsumura K. Sato K. Maebara K. Wang H. Tamba M. Itoh K. Yamamoto M. Bannai S. Electrophile response element-mediated induction of the cystine/glutamate exchange transporter gene expression. J Biol Chem. 2002;277:44765–44771. doi: 10.1074/jbc.M208704200. [DOI] [PubMed] [Google Scholar]
- 425.Sato S. Sanjo H. Takeda K. Ninomiya-Tsuji J. Yamamoto M. Kawai T. Matsumoto K. Takeuchi O. Akira S. Essential function for the kinase TAK1 in innate and adaptive immune responses. Nat Immunol. 2005;6:1087–1095. doi: 10.1038/ni1255. [DOI] [PubMed] [Google Scholar]
- 426.Scaloni A. Tell G. Mass spectrometry approaches for the redox characterization of protein cysteine residues the case of the transcription factor Pax-8. Methods Enzymol. 2010;473:227–250. doi: 10.1016/S0076-6879(10)73012-3. [DOI] [PubMed] [Google Scholar]
- 427.Schauvliege R. Janssens S. Beyaert R. Pellino proteins: novel players in TLR and IL-1R signalling. J Cell Mol Med. 2007;11:453–461. doi: 10.1111/j.1582-4934.2007.00040.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 428.Scheerer P. Borchert A. Krauss N. Wessner H. Gerth C. Hohne W. Kuhn H. Structural basis for catalytic activity and enzyme polymerization of phospholipid hydroperoxide glutathione peroxidase-4 (GPx4) Biochemistry. 2007;46:9041–9049. doi: 10.1021/bi700840d. [DOI] [PubMed] [Google Scholar]
- 429.Schenk H. Klein M. Erdbrugger W. Dröge W. Schulze-Osthoff K. Distinct effects of thioredoxin and antioxidants on the activation of transcription factors NF-kappa B and AP-1. Proc Natl Acad Sci U S A. 1994;91:1672–1676. doi: 10.1073/pnas.91.5.1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 430.Schildknecht S. Ullrich V. Peroxynitrite as regulator of vascular prostanoid synthesis. Arch Biochem Biophys. 2009;484:183–189. doi: 10.1016/j.abb.2008.10.023. [DOI] [PubMed] [Google Scholar]
- 431.Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2000;103:211–225. doi: 10.1016/s0092-8674(00)00114-8. [DOI] [PubMed] [Google Scholar]
- 432.Schlessinger J. All signaling is local? Mol Cell. 2002;10:218–219. doi: 10.1016/s1097-2765(02)00607-x. [DOI] [PubMed] [Google Scholar]
- 433.Schlessinger J. Lemmon MA. Nuclear signaling by receptor tyrosine kinases: the first robin of spring. Cell. 2006;127:45–48. doi: 10.1016/j.cell.2006.09.013. [DOI] [PubMed] [Google Scholar]
- 434.Schmidt KN. Amstad P. Cerutti P. Baeuerle PA. The roles of hydrogen peroxide and superoxide as messengers in the activation of transcription factor NF-kappa B. Chem Biol. 1995;2:13–22. doi: 10.1016/1074-5521(95)90076-4. [DOI] [PubMed] [Google Scholar]
- 435.Schneider J. Salutary effects of the prostacyclin analogue CG 4203 in lethal endotoxemia in rats. Prostaglandins Leukot Essent Fatty Acids. 1988;31:139–146. doi: 10.1016/0952-3278(88)90110-x. [DOI] [PubMed] [Google Scholar]
- 436.Schneider J. Friderichs E. Heintze K. Flohé L. Effects of recombinant human superoxide dismutase on increased lung vascular permeability and respiratory disorder in endotoxemic rats. Circ Shock. 1990;30:97–106. [PubMed] [Google Scholar]
- 437.Schoonbroodt S. Ferreira V. Best-Belpomme M. Boelaert JR. Legrand-Poels S. Korner M. Piette J. Crucial role of the amino-terminal tyrosine residue 42 and the carboxyl-terminal PEST domain of I kappa B alpha in NF-kappa B activation by an oxidative stress. J Immunol. 2000;164:4292–4300. doi: 10.4049/jimmunol.164.8.4292. [DOI] [PubMed] [Google Scholar]
- 438.Schreck R. Rieber P. Baeuerle PA. Reactive oxygen intermediates as apparently widely used messengers in the activation of the NF-kappa B transcription factor and HIV-1. EMBO J. 1991;10:2247–2258. doi: 10.1002/j.1460-2075.1991.tb07761.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 439.Schremmer B. Manevich Y. Feinstein SI. Fisher AB. Peroxiredoxins in the lung with emphasis on peroxiredoxin VI. Subcell Biochem. 2007;44:317–344. doi: 10.1007/978-1-4020-6051-9_15. [DOI] [PubMed] [Google Scholar]
- 440.Schulze-Osthoff K. Beyaert R. Vandevoorde V. Haegeman G. Fiers W. Depletion of the mitochondrial electron transport abrogates the cytotoxic and gene-inductive effects of TNF. EMBO J. 1993;12:3095–3104. doi: 10.1002/j.1460-2075.1993.tb05978.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 441.Segal BH. Han W. Bushey JJ. Joo M. Bhatti Z. Feminella J. Dennis CG. Vethanayagam RR. Yull FE. Capitano M. Wallace PK. Minderman H. Christman JW. Sporn MB. Chan J. Vinh DC. Holland SM. Romani LR. Gaffen SL. Freeman ML. Blackwell TS. NADPH oxidase limits innate immune responses in the lungs in mice. PLoS One. 2010;5:e9631. doi: 10.1371/journal.pone.0009631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 442.Semenza GL. Life with oxygen. Science. 2007;318:62–64. doi: 10.1126/science.1147949. [DOI] [PubMed] [Google Scholar]
- 443.Semenza GL. Wang GL. A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol Cell Biol. 1992;12:5447–5454. doi: 10.1128/mcb.12.12.5447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 444.Sen R. Baltimore D. Inducibility of kappa immunoglobulin enhancer-binding protein Nf-kappa B by a posttranslational mechanism. Cell. 1986;47:921–928. doi: 10.1016/0092-8674(86)90807-x. [DOI] [PubMed] [Google Scholar]
- 445.Senftleben U. Cao Y. Xiao G. Greten FR. Krahn G. Bonizzi G. Chen Y. Hu Y. Fong A. Sun SC. Karin M. Activation by IKKalpha of a second, evolutionary conserved, NF-kappa B signaling pathway. Science. 2001;293:1495–1499. doi: 10.1126/science.1062677. [DOI] [PubMed] [Google Scholar]
- 446.Sengupta A. Carlson BA. Weaver JA. Novoselov SV. Fomenko DE. Gladyshev VN. Hatfield DL. A functional link between housekeeping selenoproteins and phase II enzymes. Biochem J. 2008;413:151–161. doi: 10.1042/BJ20080277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 447.Serhan CN. Yacoubian S. Yang R. Anti-inflammatory and proresolving lipid mediators. Annu Rev Pathol. 2008;3:279–312. doi: 10.1146/annurev.pathmechdis.3.121806.151409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 448.Shen G. Hebbar V. Nair S. Xu C. Li W. Lin W. Keum YS. Han J. Gallo MA. Kong AN. Regulation of Nrf2 transactivation domain activity. The differential effects of mitogen-activated protein kinase cascades and synergistic stimulatory effect of Raf and CREB-binding protein. J Biol Chem. 2004;279:23052–23060. doi: 10.1074/jbc.M401368200. [DOI] [PubMed] [Google Scholar]
- 449.Shi Y. Serine/threonine phosphatases: mechanism through structure. Cell. 2009;139:468–484. doi: 10.1016/j.cell.2009.10.006. [DOI] [PubMed] [Google Scholar]
- 450.Shi Z. Cai Z. Wen S. Chen C. Gendron C. Sanchez A. Patterson K. Fu S. Yang J. Wildman D. Finnell RH. Zhang D. Transcriptional regulation of the novel Toll-like receptor Tlr13. J Biol Chem. 2009;284:20540–20547. doi: 10.1074/jbc.M109.022541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 451.Shim JH. Xiao C. Paschal AE. Bailey ST. Rao P. Hayden MS. Lee KY. Bussey C. Steckel M. Tanaka N. Yamada G. Akira S. Matsumoto K. Ghosh S. TAK1, but not TAB1 or TAB2, plays an essential role in multiple signaling pathways in vivo. Genes Dev. 2005;19:2668–2681. doi: 10.1101/gad.1360605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 452.Shlomai J. Redox control of protein-dna interactions: from molecular mechanisms to significance in signal transduction, gene expression, and DNA replication. Antioxid Redox Signal. 2010;13:1429–1476. doi: 10.1089/ars.2009.3029. [DOI] [PubMed] [Google Scholar]
- 453.Sies H, editor. Oxidative Stress. London: Academic Press; 1985. [Google Scholar]
- 454.Singh A. Ling G. Suhasini AN. Zhang P. Yamamoto M. Navas-Acien A. Cosgrove G. Tuder RM. Kensler TW. Watson WH. Biswal S. Nrf2-dependent sulfiredoxin-1 expression protects against cigarette smoke-induced oxidative stress in lungs. Free Radic Biol Med. 2009;46:376–386. doi: 10.1016/j.freeradbiomed.2008.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 455.Singh H. Sen R. Baltimore D. Sharp PA. A nuclear factor that binds to a conserved sequence motif in transcriptional control elements of immunoglobulin genes. Nature. 1986;319:154–158. doi: 10.1038/319154a0. [DOI] [PubMed] [Google Scholar]
- 456.Singh S. Vrishni S. Singh BK. Rahman I. Kakkar P. Nrf2-ARE stress response mechanism: a control point in oxidative stress-mediated dysfunctions and chronic inflammatory diseases. Free Radic Res. 2010;44:1267–1288. doi: 10.3109/10715762.2010.507670. [DOI] [PubMed] [Google Scholar]
- 457.Sizemore N. Leung S. Stark GR. Activation of phosphatidylinositol 3-kinase in response to interleukin-1 leads to phosphorylation and activation of the NF-kappaB p65/RelA subunit. Mol Cell Biol. 1999;19:4798–4805. doi: 10.1128/mcb.19.7.4798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 458.Smith SE. Koegl M. Jentsch S. Role of the ubiquitin/proteasome system in regulated protein degradation in Saccharomyces cerevisiae. Biol Chem. 1996;377:437–446. [PubMed] [Google Scholar]
- 459.Sohn J. Rudolph J. Catalytic and chemical competence of regulation of cdc25 phosphatase by oxidation/reduction. Biochemistry. 2003;42:10060–10070. doi: 10.1021/bi0345081. [DOI] [PubMed] [Google Scholar]
- 460.Spencer SR. Xue LA. Klenz EM. Talalay P. The potency of inducers of NAD(P)H:(quinone-acceptor) oxidoreductase parallels their efficiency as substrates for glutathione transferases. Structural and electronic correlations. Biochem J. 1991;273:711–717. doi: 10.1042/bj2730711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 461.Staal FJ. Roederer M. Herzenberg LA. Intracellular thiols regulate activation of nuclear factor kappa B and transcription of human immunodeficiency virus. Proc Natl Acad Sci U S A. 1990;87:9943–9947. doi: 10.1073/pnas.87.24.9943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 462.Stahlberg HJ. Loschen G. Flohé L. Effects of prostacyclin analogs on cyclic adenosine monophosphate and superoxide formation in human polymorphonuclear leukocytes stimulated by formyl-methionyl-leucyl-phenylalanine. Biol Chem Hoppe Seyler. 1988;369:329–336. doi: 10.1515/bchm3.1988.369.1.329. [DOI] [PubMed] [Google Scholar]
- 463.Stancovski I. Gonen H. Orian A. Schwartz AL. Ciechanover A. Degradation of the proto-oncogene product c-Fos by the ubiquitin proteolytic system in vivo and in vitro: identification and characterization of the conjugating enzymes. Mol Cell Biol. 1995;15:7106–7116. doi: 10.1128/mcb.15.12.7106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 464.Staudt LM. Singh H. Sen R. Wirth T. Sharp PA. Baltimore D. A lymphoid-specific protein binding to the octamer motif of immunoglobulin genes. Nature. 1986;323:640–643. doi: 10.1038/323640a0. [DOI] [PubMed] [Google Scholar]
- 465.Sun W. Wang H. Zhao X. Yu Y. Fan Y. Wang X. Lu X. Zhang G. Fu S. Yang J. Protein phosphatase 2A acts as a mitogen-activated protein kinase kinase kinase 3 (MEKK3) phosphatase to inhibit lysophosphatidic acid-induced IkappaB kinase beta/nuclear factor-kappaB activation. J Biol Chem. 2010;285:21341–21348. doi: 10.1074/jbc.M110.104224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 466.Sun W. Yu Y. Dotti G. Shen T. Tan X. Savoldo B. Pass AK. Chu M. Zhang D. Lu X. Fu S. Lin X. Yang J. PPM1A and PPM1B act as IKKbeta phosphatases to terminate TNFalpha-induced IKKbeta-NF-kappaB activation. Cell Signal. 2009;21:95–102. doi: 10.1016/j.cellsig.2008.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 467.Sun Y. Oberley LW. Redox regulation of transcriptional activators. Free Radic Biol Med. 1996;21:335–348. doi: 10.1016/0891-5849(96)00109-8. [DOI] [PubMed] [Google Scholar]
- 468.Sun Z. Huang Z. Zhang DD. Phosphorylation of Nrf2 at multiple sites by MAP kinases has a limited contribution in modulating the Nrf2-dependent antioxidant response. PLoS One. 2009;4:e6588. doi: 10.1371/journal.pone.0006588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 469.Surh YJ. Kundu JK. Na HK. Nrf2 as a master redox switch in turning on the cellular signaling involved in the induction of cytoprotective genes by some chemopreventive phytochemicals. Planta Med. 2008;74:1526–1539. doi: 10.1055/s-0028-1088302. [DOI] [PubMed] [Google Scholar]
- 470.Suvorova ES. Lucas O. Weisend CM. Rollins MF. Merrill GF. Capecchi MR. Schmidt EE. Cytoprotective Nrf2 pathway is induced in chronically txnrd 1-deficient hepatocytes. PLoS One. 2009;4:e6158. doi: 10.1371/journal.pone.0006158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 471.Suzuki T. Kelly VP. Motohashi H. Nakajima O. Takahashi S. Nishimura S. Yamamoto M. Deletion of the selenocysteine tRNA gene in macrophages and liver results in compensatory gene induction of cytoprotective enzymes by Nrf2. J Biol Chem. 2008;283:2021–2030. doi: 10.1074/jbc.M708352200. [DOI] [PubMed] [Google Scholar]
- 472.Tabernero L. Aricescu AR. Jones EY. Szedlacsek SE. Protein tyrosine phosphatases: structure-function relationships. FEBS J. 2008;275:867–882. doi: 10.1111/j.1742-4658.2008.06251.x. [DOI] [PubMed] [Google Scholar]
- 473.Takada Y. Mukhopadhyay A. Kundu GC. Mahabeleshwar GH. Singh S. Aggarwal BB. Hydrogen peroxide activates NF-kappa B through tyrosine phosphorylation of I kappa B alpha and serine phosphorylation of p65: evidence for the involvement of I kappa B alpha kinase and Syk protein-tyrosine kinase. J Biol Chem. 2003;278:24233–24241. doi: 10.1074/jbc.M212389200. [DOI] [PubMed] [Google Scholar]
- 474.Talalay P. De Long MJ. Prochaska HJ. Identification of a common chemical signal regulating the induction of enzymes that protect against chemical carcinogenesis. Proc Natl Acad Sci U S A. 1988;85:8261–8265. doi: 10.1073/pnas.85.21.8261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 475.Tell G. Quadrifoglio F. Tiribelli C. Kelley MR. The many functions of APE1/Ref-1: not only a DNA repair enzyme. Antioxid Redox Signal. 2009;11:601–620. doi: 10.1089/ars.2008.2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 476.Tell G. Zecca A. Pellizzari L. Spessotto P. Colombatti A. Kelley MR. Damante G. Pucillo C. An “environment to nucleus” signaling system operates in B lymphocytes: redox status modulates BSAP/Pax-5 activation through Ref-1 nuclear translocation. Nucleic Acids Res. 2000;28:1099–1105. doi: 10.1093/nar/28.5.1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 477.Tewes F. Böl GF. Brigelius-Flohé R. Thiol modulation inhibits the interleukin (IL)-1-mediated activation of an IL-1 receptor-associated protein kinase and NF-kappa B. Eur J Immunol. 1997;27:3015–3021. doi: 10.1002/eji.1830271139. [DOI] [PubMed] [Google Scholar]
- 478.Theodore M. Kawai Y. Yang J. Kleshchenko Y. Reddy SP. Villalta F. Arinze IJ. Multiple nuclear localization signals function in the nuclear import of the transcription factor Nrf2. J Biol Chem. 2008;283:8984–8994. doi: 10.1074/jbc.M709040200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 479.Thimmulappa RK. Mai KH. Srisuma S. Kensler TW. Yamamoto M. Biswal S. Identification of Nrf2-regulated genes induced by the chemopreventive agent sulforaphane by oligonucleotide microarray. Cancer Res. 2002;62:5196–5203. [PubMed] [Google Scholar]
- 480.Thomas D. Tyers M. Transcriptional regulation: kamikaze activators. Curr Biol. 2000;10:R341–R343. doi: 10.1016/s0960-9822(00)00462-0. [DOI] [PubMed] [Google Scholar]
- 481.Tjalkens RB. Luckey SW. Kroll DJ. Petersen DR. Alpha, beta-unsaturated aldehydes mediate inducible expression of glutathione S-transferase in hepatoma cells through activation of the antioxidant response element (ARE) Adv Exp Med Biol. 1999;463:123–131. doi: 10.1007/978-1-4615-4735-8_15. [DOI] [PubMed] [Google Scholar]
- 482.Toledano MB. The guardian recruits cops: the p53-p21 axis delegates prosurvival duties to the Keap1-Nrf2 stress pathway. Mol Cell. 2009;34:637–639. doi: 10.1016/j.molcel.2009.06.005. [DOI] [PubMed] [Google Scholar]
- 483.Toledano MB. Ghosh D. Trinh F. Leonard WJ. N-terminal DNA-binding domains contribute to differential DNA-binding specificities of NF-kappa B p50 and p65. Mol Cell Biol. 1993;13:852–860. doi: 10.1128/mcb.13.2.852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 484.Tong KI. Kobayashi A. Katsuoka F. Yamamoto M. Two-site substrate recognition model for the Keap1-Nrf2 system: a hinge and latch mechanism. Biol Chem. 2006;387:1311–1320. doi: 10.1515/BC.2006.164. [DOI] [PubMed] [Google Scholar]
- 485.Tong KI. Katoh Y. Kusunoki H. Itoh K. Tanaka T. Yamamoto M. Keap1 recruits Neh2 through binding to ETGE and DLG motifs: characterization of the two-site molecular recognition model. Mol Cell Biol. 2006;26:2887–2900. doi: 10.1128/MCB.26.8.2887-2900.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 486.Tonks NK. Protein tyrosine phosphatases: from genes, to function, to disease. Nat Rev Mol Cell Biol. 2006;7:833–846. doi: 10.1038/nrm2039. [DOI] [PubMed] [Google Scholar]
- 487.Toppo S. Flohé L. Ursini F. Vanin S. Maiorino M. Catalytic mechanisms and specificities of glutathione peroxidases: variations of a basic scheme. Biochim Biophys Acta. 2009;1790:1486–1500. doi: 10.1016/j.bbagen.2009.04.007. [DOI] [PubMed] [Google Scholar]
- 488.Townsend DM. Tew KD. The role of glutathione-S-transferase in anti-cancer drug resistance. Oncogene. 2003;22:7369–7375. doi: 10.1038/sj.onc.1206940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 489.Trujillo M. Ferrer-Sueta G. Thomson L. Flohé L. Radi R. Kinetics of peroxiredoxins and their role in the decomposition of peroxynitrite. Subcell Biochem. 2007;44:83–113. doi: 10.1007/978-1-4020-6051-9_5. [DOI] [PubMed] [Google Scholar]
- 490.Trujillo M. Budde H. Pineyro MD. Stehr M. Robello C. Flohé L. Radi R. Trypanosoma brucei and Trypanosoma cruzi tryparedoxin peroxidases catalytically detoxify peroxynitrite via oxidation of fast reacting thiols. J Biol Chem. 2004;279:34175–34182. doi: 10.1074/jbc.M404317200. [DOI] [PubMed] [Google Scholar]
- 491.Tsuji Y. Ayaki H. Whitman SP. Morrow CS. Torti SV. Torti FM. Coordinate transcriptional and translational regulation of ferritin in response to oxidative stress. Mol Cell Biol. 2000;20:5818–5827. doi: 10.1128/mcb.20.16.5818-5827.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 492.Ullrich A. Schlessinger J. Signal transduction by receptors with tyrosine kinase activity. Cell. 1990;61:203–212. doi: 10.1016/0092-8674(90)90801-k. [DOI] [PubMed] [Google Scholar]
- 493.Ursini F. Heim S. Kiess M. Maiorino M. Roveri A. Wissing J. Flohé L. Dual function of the selenoprotein PHGPx during sperm maturation. Science. 1999;285:1393–1396. doi: 10.1126/science.285.5432.1393. [DOI] [PubMed] [Google Scholar]
- 494.Ushio-Fukai M. Compartmentalization of redox signaling through NADPH oxidase-derived ROS. Antioxid Redox Signal. 2009;11:1289–1299. doi: 10.1089/ars.2008.2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 495.van Montfort RL. Congreve M. Tisi D. Carr R. Jhoti H. Oxidation state of the active-site cysteine in protein tyrosine phosphatase 1B. Nature. 2003;423:773–777. doi: 10.1038/nature01681. [DOI] [PubMed] [Google Scholar]
- 496.Vane JR. Clinical potential of prostacyclin. Adv Prostaglandin Thromboxane Leukot Res. 1983;11:449–456. [PubMed] [Google Scholar]
- 497.Veal EA. Findlay VJ. Day AM. Bozonet SM. Evans JM. Quinn J. Morgan BA. A 2-Cys peroxiredoxin regulates peroxide-induced oxidation and activation of a stress-activated MAP kinase. Mol Cell. 2004;15:129–139. doi: 10.1016/j.molcel.2004.06.021. [DOI] [PubMed] [Google Scholar]
- 498.Velichkova M. Hasson T. Keap1 regulates the oxidation-sensitive shuttling of Nrf2 into and out of the nucleus via a Crm1-dependent nuclear export mechanism. Mol Cell Biol. 2005;25:4501–4513. doi: 10.1128/MCB.25.11.4501-4513.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 499.Velichkova M. Guttman J. Warren C. Eng L. Kline K. Vogl AW. Hasson T. A human homologue of Drosophila kelch associates with myosin-VIIa in specialized adhesion junctions. Cell Motil Cytoskeleton. 2002;51:147–164. doi: 10.1002/cm.10025. [DOI] [PubMed] [Google Scholar]
- 500.Vermeulen L. De Wilde G. Van Damme P. Vanden Berghe W. Haegeman G. Transcriptional activation of the NF-kappaB p65 subunit by mitogen- and stress-activated protein kinase-1 (MSK1) EMBO J. 2003;22:1313–1324. doi: 10.1093/emboj/cdg139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 501.Viatour P. Merville MP. Bours V. Chariot A. Phosphorylation of NF-kappaB and IkappaB proteins: implications in cancer and inflammation. Trends Biochem Sci. 2005;30:43–52. doi: 10.1016/j.tibs.2004.11.009. [DOI] [PubMed] [Google Scholar]
- 502.Wakabayashi G. Gelfand JA. Burke JF. Thompson RC. Dinarello CA. A specific receptor antagonist for interleukin 1 prevents Escherichia coli-induced shock in rabbits. FASEB J. 1991;5:338–343. doi: 10.1096/fasebj.5.3.1825816. [DOI] [PubMed] [Google Scholar]
- 503.Wakabayashi N. Slocum SL. Skoko JJ. Shin S. Kensler TW. When NRF2 Talks, Who's Listening? Antioxid Redox Signal. 2010;13:1649–1663. doi: 10.1089/ars.2010.3216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 504.Wakabayashi N. Dinkova-Kostova AT. Holtzclaw WD. Kang MI. Kobayashi A. Yamamoto M. Kensler TW. Talalay P. Protection against electrophile and oxidant stress by induction of the phase 2 response: fate of cysteines of the Keap1 sensor modified by inducers. Proc Natl Acad Sci U S A. 2004;101:2040–2045. doi: 10.1073/pnas.0307301101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 505.Wang D. Baldwin AS., Jr Activation of nuclear factor-kappaB-dependent transcription by tumor necrosis factor-alpha is mediated through phosphorylation of RelA/p65 on serine 529. J Biol Chem. 1998;273:29411–29416. doi: 10.1074/jbc.273.45.29411. [DOI] [PubMed] [Google Scholar]
- 506.Wang J. Boja ES. Tan W. Tekle E. Fales HM. English S. Mieyal JJ. Chock PB. Reversible glutathionylation regulates actin polymerization in A431 cells. J Biol Chem. 2001;276:47763–47766. doi: 10.1074/jbc.C100415200. [DOI] [PubMed] [Google Scholar]
- 507.Wang T. Chuang TH. Ronni T. Gu S. Du YC. Cai H. Sun HQ. Yin HL. Chen X. Flightless I homolog negatively modulates the TLR pathway. J Immunol. 2006;176:1355–1362. doi: 10.4049/jimmunol.176.3.1355. [DOI] [PubMed] [Google Scholar]
- 508.Webster KA. Prentice H. Bishopric NH. Oxidation of zinc finger transcription factors: physiological consequences. Antioxid Redox Signal. 2001;3:535–548. doi: 10.1089/15230860152542916. [DOI] [PubMed] [Google Scholar]
- 509.Wendel A. Tiegs G. A novel biologically active seleno-organic compound—VI. Protection by ebselen (PZ 51) against galactosamine/endotoxin-induced hepatitis in mice. Biochem Pharmacol. 1986;35:2115–2118. doi: 10.1016/0006-2952(86)90578-2. [DOI] [PubMed] [Google Scholar]
- 510.Wendel A. Tiegs G. Werner C. Evidence for the involvement of a reperfusion injury in galactosamine/endotoxin-induced hepatitis in mice. Biochem Pharmacol. 1987;36:2637–2639. doi: 10.1016/0006-2952(87)90544-2. [DOI] [PubMed] [Google Scholar]
- 511.Wilkinson KA. Henley JM. Mechanisms, regulation and consequences of protein SUMOylation. Biochem J. 2010;428:133–145. doi: 10.1042/BJ20100158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 512.Winterbourn CC. Metodiewa D. Reactivity of biologically important thiol compounds with superoxide and hydrogen peroxide. Free Radic Biol Med. 1999;27:322–328. doi: 10.1016/s0891-5849(99)00051-9. [DOI] [PubMed] [Google Scholar]
- 513.Wong PS. Hyun J. Fukuto JM. Shirota FN. DeMaster EG. Shoeman DW. Nagasawa HT. Reaction between S-nitrosothiols and thiols: generation of nitroxyl (HNO) and subsequent chemistry. Biochemistry. 1998;37:5362–5371. doi: 10.1021/bi973153g. [DOI] [PubMed] [Google Scholar]
- 514.Woo HA. Yim SH. Shin DH. Kang D. Yu DY. Rhee SG. Inactivation of peroxiredoxin I by phosphorylation allows localized H(2)O(2) accumulation for cell signaling. Cell. 2010;140:517–528. doi: 10.1016/j.cell.2010.01.009. [DOI] [PubMed] [Google Scholar]
- 515.Woo JR. Kim SJ. Jeong W. Cho YH. Lee SC. Chung YJ. Rhee SG. Ryu SE. Structural basis of cellular redox regulation by human TRP14. J Biol Chem. 2004;279:48120–48125. doi: 10.1074/jbc.M407079200. [DOI] [PubMed] [Google Scholar]
- 516.Wood ZA. Poole LB. Karplus PA. Peroxiredoxin evolution and the regulation of hydrogen peroxide signaling. Science. 2003;300:650–653. doi: 10.1126/science.1080405. [DOI] [PubMed] [Google Scholar]
- 517.Wu CJ. Conze DB. Li T. Srinivasula SM. Ashwell JD. Sensing of Lys 63-linked polyubiquitination by NEMO is a key event in NF-kappaB activation [corrected] Nat Cell Biol. 2006;8:398–406. doi: 10.1038/ncb1384. [DOI] [PubMed] [Google Scholar]
- 518.Wu J. Weiss B. Two-stage induction of the soxRS (superoxide response) regulon of Escherichia coli. J Bacteriol. 1992;174:3915–3920. doi: 10.1128/jb.174.12.3915-3920.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 519.Wu X. Bishopric NH. Discher DJ. Murphy BJ. Webster KA. Physical and functional sensitivity of zinc finger transcription factors to redox change. Mol Cell Biol. 1996;16:1035–1046. doi: 10.1128/mcb.16.3.1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 520.Xanthoudakis S. Curran T. Identification and characterization of Ref-1, a nuclear protein that facilitates AP-1 DNA-binding activity. EMBO J. 1992;11:653–665. doi: 10.1002/j.1460-2075.1992.tb05097.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 521.Yamamoto T. Suzuki T. Kobayashi A. Wakabayashi J. Maher J. Motohashi H. Yamamoto M. Physiological significance of reactive cysteine residues of Keap1 in determining Nrf2 activity. Mol Cell Biol. 2008;28:2758–2770. doi: 10.1128/MCB.01704-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 522.Yamaoka S. Courtois G. Bessia C. Whiteside ST. Weil R. Agou F. Kirk HE. Kay RJ. Israel A. Complementation cloning of NEMO, a component of the IkappaB kinase complex essential for NF-kappaB activation. Cell. 1998;93:1231–1240. doi: 10.1016/s0092-8674(00)81466-x. [DOI] [PubMed] [Google Scholar]
- 523.Yang SR. Chida AS. Bauter MR. Shafiq N. Seweryniak K. Maggirwar SB. Kilty I. Rahman I. Cigarette smoke induces proinflammatory cytokine release by activation of NF-kappaB and posttranslational modifications of histone deacetylase in macrophages. Am J Physiol Lung Cell Mol Physiol. 2006;291:L46–L57. doi: 10.1152/ajplung.00241.2005. [DOI] [PubMed] [Google Scholar]
- 524.Yaron A. Gonen H. Alkalay I. Hatzubai A. Jung S. Beyth S. Mercurio F. Manning AM. Ciechanover A. Ben-Neriah Y. Inhibition of NF-kappa-B cellular function via specific targeting of the I-kappa-B-ubiquitin ligase. EMBO J. 1997;16:6486–6494. doi: 10.1093/emboj/16.21.6486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 525.Yazdanpanah B. Wiegmann K. Tchikov V. Krut O. Pongratz C. Schramm M. Kleinridders A. Wunderlich T. Kashkar H. Utermohlen O. Bruning JC. Schutze S. Kronke M. Riboflavin kinase couples TNF receptor 1 to NADPH oxidase. Nature. 2009;460:1159–1163. doi: 10.1038/nature08206. [DOI] [PubMed] [Google Scholar]
- 526.Yeh PY. Yeh KH. Chuang SE. Song YC. Cheng AL. Suppression of MEK/ERK signaling pathway enhances cisplatin-induced NF-kappaB activation by protein phosphatase 4-mediated NF-kappaB p65 Thr dephosphorylation. J Biol Chem. 2004;279:26143–26148. doi: 10.1074/jbc.M402362200. [DOI] [PubMed] [Google Scholar]
- 527.Youngman RJ. Elstner EF. Oxygen species in paraquat toxicity: the crypto-OH radical. FEBS Lett. 1981;129:265–268. doi: 10.1016/0014-5793(81)80180-9. [DOI] [PubMed] [Google Scholar]
- 528.Yu R. Chen C. Mo YY. Hebbar V. Owuor ED. Tan TH. Kong AN. Activation of mitogen-activated protein kinase pathways induces antioxidant response element-mediated gene expression via a Nrf2-dependent mechanism. J Biol Chem. 2000;275:39907–39913. doi: 10.1074/jbc.M004037200. [DOI] [PubMed] [Google Scholar]
- 529.Zamora M. Merono C. Vinas O. Mampel T. Recruitment of NF-kappaB into mitochondria is involved in adenine nucleotide translocase 1 (ANT1)-induced apoptosis. J Biol Chem. 2004;279:38415–38423. doi: 10.1074/jbc.M404928200. [DOI] [PubMed] [Google Scholar]
- 530.Zardi EM. Zardi DM. Dobrina A. Afeltra A. Prostacyclin in sepsis: a systematic review. Prostaglandins Other Lipid Mediat. 2007;83:1–24. doi: 10.1016/j.prostaglandins.2006.12.004. [DOI] [PubMed] [Google Scholar]
- 531.Zhang DD. Hannink M. Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol Cell Biol. 2003;23:8137–8151. doi: 10.1128/MCB.23.22.8137-8151.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 532.Zhang DD. Lo SC. Cross JV. Templeton DJ. Hannink M. Keap1 is a redox-regulated substrate adaptor protein for a Cul3-dependent ubiquitin ligase complex. Mol Cell Biol. 2004;24:10941–10953. doi: 10.1128/MCB.24.24.10941-10953.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 533.Zhao X. Xu B. Bhattacharjee A. Oldfield CM. Wientjes FB. Feldman GM. Cathcart MK. Protein kinase Cdelta regulates p67phox phosphorylation in human monocytes. J Leukoc Biol. 2005;77:414–420. doi: 10.1189/jlb.0504284. [DOI] [PubMed] [Google Scholar]
- 534.Zhong H. May MJ. Jimi E. Ghosh S. The phosphorylation status of nuclear NF-kappa B determines its association with CBP/p300 or HDAC-1. Mol Cell. 2002;9:625–636. doi: 10.1016/s1097-2765(02)00477-x. [DOI] [PubMed] [Google Scholar]
- 535.Zhong H. SuYang H. Erdjument-Bromage H. Tempst P. Ghosh S. The transcriptional activity of NF-kappaB is regulated by the IkappaB-associated PKAc subunit through a cyclic AMP-independent mechanism. Cell. 1997;89:413–424. doi: 10.1016/s0092-8674(00)80222-6. [DOI] [PubMed] [Google Scholar]
- 536.Zhu M. Fahl WE. Functional characterization of transcription regulators that interact with the electrophile response element. Biochem Biophys Res Commun. 2001;289:212–219. doi: 10.1006/bbrc.2001.5944. [DOI] [PubMed] [Google Scholar]
- 537.Ziegler-Heitbrock L. The p50-homodimer mechanism in tolerance to LPS. J Endotoxin Res. 2001;7:219–222. [PubMed] [Google Scholar]
- 538.Zipper LM. Mulcahy RT. The Keap1 BTB/POZ dimerization function is required to sequester Nrf2 in cytoplasm. J Biol Chem. 2002;277:36544–36552. doi: 10.1074/jbc.M206530200. [DOI] [PubMed] [Google Scholar]