

Abstract
Faithful inheritance of genetic information requires that DNA be copied only once each cell cycle. Initiation of DNA replication involves the establishment of a prereplication complex (pre-RC) and subsequent activation by CDK/cyclins, converting the pre-RC to a post-RC. The origin recognition complex (ORC), Cdc6p, and the MCM proteins are required for establishing the pre-RC. We show that all six ORC subunits remain bound to chromatin throughout the cell cycle, whereas the MCM proteins cycle on and off, corresponding precisely to transitions of the RC. A newly isolated cdc6 mutant displays promiscuous initiation of DNA replication, increased nuclear DNA content, and constant MCM protein association with chromatin throughout the cell cycle. This gain-of-function cdc6 mutant ignores the negative controls imposed normally on initiation by the CDK/cyclins, suggesting that Cdc6p is a key mediator of once-per-cell-cycle control of DNA replication.
Keywords: DNA replication, ORC, Cdc6, MCM, cell cycle, S. cerevisiae
All eukaryotic cells duplicate the entire genome during the S phase of every cell cycle by initiating DNA synthesis from a large number of origins of DNA replication in each chromosome. Initiation is controlled by cis-acting replicators that are recognized by initiator proteins. To insure equal inheritance of all genetic material to each progeny cell, initiation at each origin must be tightly regulated so that it occurs just once during each S-phase.
Eukaryotic cell DNA replication has been studied most extensively in the budding yeast Saccharomyces cerevisiae, in which replicators have been defined genetically, origins of DNA replication have been physically mapped, and many initiation proteins and regulators have been identified (for review, see Diffley 1996; Stillman 1996). Homologs of some of the yeast initiation proteins have been identified recently in higher eukaryotes, and shown to be required for DNA replication (for review, see Chong et al. 1996; Jallepalli and Kelly 1997). In many species, however, the cis-acting elements (replicators) that may interact with these initiation proteins have not been defined.
The yeast initiator protein, origin recognition complex (ORC), binds directly to autonomously replicating sequences (ARS), the yeast replicators (Bell and Stillman 1992), and determines the location and frequency of firing of replication origins in chromosomes (Fox et al. 1995; Liang et al. 1995). Genomic footprints over ARS A and B1 elements from isolated nuclei (Diffley and Cocker 1992; Diffley et al. 1994) resemble the nuclease digestion patterns of purified ORC bound to naked ARS DNA (Bell and Stillman 1992). This pattern of nuclease protection suggests, but does not demonstrate, that ORC remains bound to replicators throughout the cell cycle. The implied protein complex has been referred to as the post-replication complex (post-RC) and exists during S, G2, and early M phases of the cell cycle. At the M → G1 transition, however, a characteristic deoxyribonuclease I (DNase I) hypersensitive site in B1 is lost and an extended footprint appears (Diffley et al. 1994). This is believed to reflect the formation of a prereplication complex (pre-RC) containing additional proteins bound to the ARS or to ORC during late M and G1 phases.
Cdc6p is required for formation and maintenance of the pre-RC, as defined by the absence of the hypersensitive site at ARSs (Diffley et al. 1994; Cocker et al. 1996; Detweiler and Li 1997). The six sequence-related, minichromosome maintenance (MCM) proteins may also be part of the pre-RC, as they are required for DNA replication (Hennessy et al. 1991; Yan et al. 1993; Dalton and Whitbread 1995; C. Liang and B. Stillman, unpubl.). The MCM family members have been shown to bind to chromatin in yeast (Donovan et al. 1997), Xenopus egg extracts (Chong et al. 1995; Kubota et al. 1995; Madine et al. 1995; Coleman et al. 1996), and mammalian cells (Kimura et al. 1995; Todorov et al. 1995). Current models suggest that formation of a pre-RC at each origin determines that the replicator is competent for initiation of DNA replication at that locus. Once formed, the pre-RC needs to be activated by protein kinases Cdc28p/Clb5p(/Clb6p) and Cdc7p/Dbf4p, among other proteins, to trigger the onset of S-phase. Upon initiation, part of the pre-RC is thought to be destroyed, reverting to a post-RC. Rebuilding of the pre-RC is prohibited by the high kinase activities of Cdc28/Clb1-6 proteins in the S, G2, and early M phases of the cell cycle, therefore rereplication is prevented until the next cell cycle (for review, see Nasmyth 1996; Stillman 1996; Jallepalli and Kelly 1997).
In this report, we show that ORC binds chromatin across the cell cycle, supporting the notion that ORC is a landing pad for other initiation proteins. In contrast, the chromatin binding of MCM proteins is tightly cell cycle regulated. MCM chromatin association and dissociation coincide with transitions in the state of the replication complex at origins. A newly isolated cdc6 mutant displays promiscuous initiation of DNA replication, increases in nuclear DNA content, and constant MCM protein association with chromatin throughout the cell cycle, despite the presence of high mitotic CDK activities, suggesting that the cdc6 mutant cells ignore the negative controls normally imposed on initiation of DNA replication by mitotic CDK cyclins.
Results
An assay for protein chromatin binding
A chromatin-binding assay (Fig. 1A) was developed to examine chromatin association of initiation proteins. Cells were spheroplasted and lyzed in a buffer containing Triton X-100. Chromatin, together with nuclear matrix and cell debris was spun down at low speed. The crude chromatin pellet at this stage is equivalent to that in the chromatin-binding assay with Xenopus egg extracts (Kubota et al. 1995; Coleman et al. 1996), and to a recently described assay in yeast (Donovan et al. 1997). To make sure that proteins in the crude pellet were actually bound to chromatin, we released ∼2- to 15-kb-long polynucleosomes from the pellet by limited micrococal nuclease (MNase) digestion, and spun down the remaining material at low speed. Solubilized polynucleosomes were then pelleted by high speed ultracentrifugation. If a protein is found in the high speed pellet fraction, it should be chromatin bound.
Figure 1.

A protein chromatin-binding assay. (A) Diagram of the chromatin-binding assay. Fractions are as follows. (1. WCE) whole cell extract from the total lysate; (2. Sup) first low speed supernatant containing nonchromatin-binding proteins; (3. Pel) crude chromatin pellet; (4. Sup) polynucleosome-containing supernatant after limited MNase digestion of the crude chromatin pellet; (5. Pel) remaining solid after MNase digestion; (6. Sup) supernatant from ultracentrifugation; (7. Pel) high-speed pellet containing polynucleosomes. (B) All seven fractions from the chromatin-binding assay (except that the first low-speed pellet was not washed) using asynchronous W303-1A cells were Western blotted for Orc3 and Orc5 subunits. The same cell equivalent was loaded in all seven fractions. (X) cross-reacting band by the Orc3p antibody. (C) The same blot used in B was probed for Mcm2p. The arrow points to the upper band of endogenous Mcm2p, which comigrates with purified, baculovirus-expressed Mcm2p from sf9 cells (data not shown, but see Fig. 2F). The lower band is likely degradation product of Mcm2p.
Proteins in all seven fractions were resolved by gel electrophoresis and probed for ORC with a mixture of monoclonal antibodies (Fig. 1B; only Orc3p and Orc5p were assayed in this experiment). ORC was found in the whole cell extract (WCE; lane 1) and low-speed pellet (lane 3), but almost none in the low-speed supernatant (lane 2). Therefore, most of the cellular ORC was associated with chromatin or nuclear matrix, or both. A protein of ∼70 kD that cross-reacted with the Orc3p antibody (labeled X) was partitioned to the low speed supernatant, thus serving as a nonchromatin-binding protein control. About half of the ORC in the low-speed pellet was released into solution by MNase digestion (lane 4) and the rest remained in the pellet (lane 5). Limited, instead of complete, MNase digestion was used so that the released polynucleosomes were sufficiently large to be pelleted later by ultracentrifugation (lane 7). ORC was detected in the high-speed pellet (lane 7), but not in the supernatant (lane 6). Therefore, ORC is a chromatin-bound protein.
Mcm2p also bound chromatin (Fig. 1C). It was detected by a monoclonal antibody in the WCE (lane 1), low-speed supernatant (lane 2), and low-speed pellet (lane 3). Therefore, not all of the cellular Mcm2p bound chromatin at any given time. The MNase-released Mcm2p (lane 4) could be pelleted by ultracentrifugation (lane 7). Similar data (not shown) were obtained for Mcm3p.
To examine chromatin association of initiation proteins during the cell cycle, we assayed the crude chromatin fractions from the first low-speed centrifugation. Because all of the ORC and MCM released by MNase from the crude chromatin pellets can be spun down by ultracentrifugation (Fig. 1B,C), it is reasonable to assume that most, if not all, of ORC and MCM proteins found in the crude chromatin pellets are chromatin bound.
ORC binds chromatin throughout the cell cycle
Cells were synchronized in G1 by α factor, then released into the cell cycle at 25 °C and at various times, were processed for the chromatin-binding assay (Fig. 2). For ORC, only the chromatin fractions are shown (Fig. 2A), because very little ORC was in the supernatant (data not shown, but see Fig. 1B). Orc1, Orc3, Orc5, and Orc6 subunits were detected in the chromatin pellets from all of the time points (Fig. 2A). Orc2 and Orc4 subunits were also found on chromatin across the cell cycle with darker exposures (data not shown). We conclude that ORC is associated with chromatin throughout the cell cycle.
Figure 2.

ORC binds chromatin throughout the cell cycle, whereas MCM chromatin association is cell cycle regulated. (A–E) W303-1A cells were synchronized in G1 with α factor (0 min) and then released into fresh YPD medium at 25°C. Samples were taken at various times (10–120 min) after the release and processed to the first low-speed centrifugation of the chromatin-binding assay. S phase was identified by FACS (B) as the period when DNA increases from 1C to 2C. Western blots are shown for ORC in crude chromatin pellet (fraction 3. Pel; A) and for Mcm3p in fractions 1. WCE (C), 2. Sup (D) and 3. Pel (E). About the same amount of proteins was loaded for each time point in each gel, and the same cell equivalent was loaded between WCE, Sup, and Pel (i.e., the signals from WCE should equal to those from Sup and Pel combined). bORC (A), bMCM3 (C–E), and bMCM2 (F) are purified baculovirus-expressed ORC, Mcm3p, and Mcm2p, respectively, from sf9 cells. Each ORC subunit, including the shifting of the Orc6p band, has been confirmed with individual antibodies (data not shown). Baculovirus-expressed Orc6p is phosphorylated, therefore, comigrates with Orc5p. (65K) degradation product of Orc1p. Endogenous Orc2p and Orc4p bands are visible in all lanes in darker exposures (not shown). The arrows in C–E point to the upper band of the endogenous Mcm3p as well as bMcm3p. The lower band is a likely degradation product of Mcm3p. (F) dbf2-2 homozygous (strain J2) cells were arrested in mitosis at 37°C (0 min), and then released into the cell cycle by shifting to 25°C. Samples were taken at various times (10 min–210 min) after the shift and fractionated into low-speed supernatant and pellet. Only the pellet fractions are shown. S-phases were identified by FACS (not shown). The post- and pre-RC are marked according to Diffley et al. (1994).
The Orc6p band shifted up (almost merges with the Orc5p band in this blot) as cells enter S-phase [30 min; see fluorescence-activated cell sorter (FACS) in Fig. 2B] and shifted down at 90 min. The upper band of Orc6p is most likely attributable to phosphorylation, because Orc6p is phosphorylated in vivo and mutating all potential Cdc28p phosphorylation sites in ORC6 resulted in only the lower band (M. Weinreich, C. Liang, and B. Stillman, unpubl.).
Association of MCM with chromatin is cell cycle regulated and MCM is part of the pre-RC
A Mcm3p monoclonal antibody detected the protein at roughly constant levels across the cell cycle in the WCE (Fig. 2C) and supernatant (Fig. 2D). Most of the cellular Mcm3p was not chromatin bound, as the levels in the supernatant were only slightly less than those in the WCE. Only small amounts were in the pellet (Fig. 2E). In contrast to ORC, the chromatin association of Mcm3p was cell cycle dependent (Fig. 2E). Mcm3p bound chromatin in G1 (α factor or 0-, 10-, and 20-min time points), left chromatin as cells entered and progressed through S-phase (30–40 min), and bound chromatin again at 90 min, the same time when Orc6p became dephosphorylated (Fig. 2A). Similar data (not shown) were obtained for Mcm2p.
The timing of MCM proteins starting to leave chromatin (Fig. 2E) as well as Orc6p phosphorylation (Fig. 2A) corresponded to the G1 → S transition, 30–40 min after cells were released from the α factor block. This is known to be the transition point from the proposed pre-RC to post-RC at the G1/S boundary (Diffley et al. 1994).
MCM proteins reappearing on chromatin and Orc6p dephosphorylation at 90 min after the release from the α factor correlated with reestablishment of the pre-RC at the end of mitosis. To test this possibility more vigorously, we performed chromatin-binding experiments with a homozygous dbf2-2 diploid strain that can be arrested in mitosis at the restrictive temperature of 37°C and then released synchronously into the next cell cycle at the permissive temperature of 25°C. This same strain was used previously for genomic footprinting experiments to define the timing of the transition from post-RC to pre-RC and then from pre-RC to post-RC (Diffley et al. 1994). At the restrictive temperature, dbf2-2 cells arrested in mitosis and had a post-RC footprint. The pre-RC was established ∼20 min after the cells were shifted to the permissive temperature. The transition occurred at the end of mitosis (anaphase), right before mitotic spindles broke down. At 40 min after the release, as cells entered S-phase, the pre-RC then converted back to post-RC (Diffley et al. 1994).
We used the same strain and conditions to examine MCM protein chromatin binding (Fig. 2F). At 37°C (0 min) or 10 min after the release, when the post-RC is known to be present, Mcm2p was not associated with chromatin. Mcm2p was on chromatin 20 min after release, the same time when the pre-RC was established. At the onset of S-phase, 40 min after the release, Mcm2p began to disappear from the chromatin as the pre-RC converted to the post-RC state. Similar data (not shown) were obtained for Mcm3p. These precise temporal correlations between MCM protein binding to chromatin and loss of the hypersensitive site in the genomic footprint (and vice versa) suggest that MCM proteins are part of the pre-RC. Reprobing for Orc6p on the same blot used for Figure 2F showed that Orc6p dephosphorylation and phosphorylation also coincided with transition from the post-RC to pre-RC states and from the pre- to post-RC states, respectively (data not shown). Therefore, it is likely that the post-RC contains ORC with phosphorylated Orc6p and the absence of MCM proteins, whereas MCM and ORC with dephosphorylated Orc6p are present in the pre-RC.
New temperature-sensitive alleles of cdc6
Several recent studies indicate that Cdc6p is a key factor in DNA replication. Cdc6p is required for formation and maintenance of the pre-RC and loading of MCM onto chromatin, is required for initiation of replication, and cooperates with ORC on individual replicators to determine the frequency of initiation in the genome (Diffley et al. 1994; Liang et al. 1995; Cocker et al. 1996; Coleman et al. 1996; Piatti et al. 1996; Detweiler and Li 1997; Donovan et al. 1997). The levels of CDC6 mRNA and Cdc6 protein (at least the epitope tag version of Cdc6p) fluctuate in the cell cycle; both are high in late M and early G1, and very low in S, G2, and early M phases (Zhou and Jong 1990; Zwerschke et al. 1994; Piatti et al. 1995; Detweiler and Li 1997). CDC6 has also been implicated in controlling mitosis entry (Bueno and Russell 1992). To investigate further the roles of Cdc6p in DNA replication, we carried out a PCR-based random mutagenesis of the gene followed by plasmid shuffling to uncover the properties of potential cdc6 mutants. Several new alleles of cdc6 temperature-sensitive mutants were thus isolated. Two of them (cdc6-2 and cdc6-3) show promiscuous replication phenotypes and were further characterized.
Cells containing a cdc6-2 plasmid as the sole source of cdc6p are very sick even at 25°C (Fig. 3A) and can accumulate >2C DNA (data not shown), and we have not been able to integrate the mutant gene into the chromosome. cdc6-3 has been integrated into the chromosome, and the mutant has been characterized in more detail. cdc6-3 cells are viable at 25°C and lose viability at 37°C (Fig. 3A). The temperature-sensitive lethal phenotype of cdc6-3 was recessive; however, the mutant had a second phenotype of promiscuous replication even at the permissive temperature, which was dominant in the heterozygous diploid mutant cells.
Figure 3.
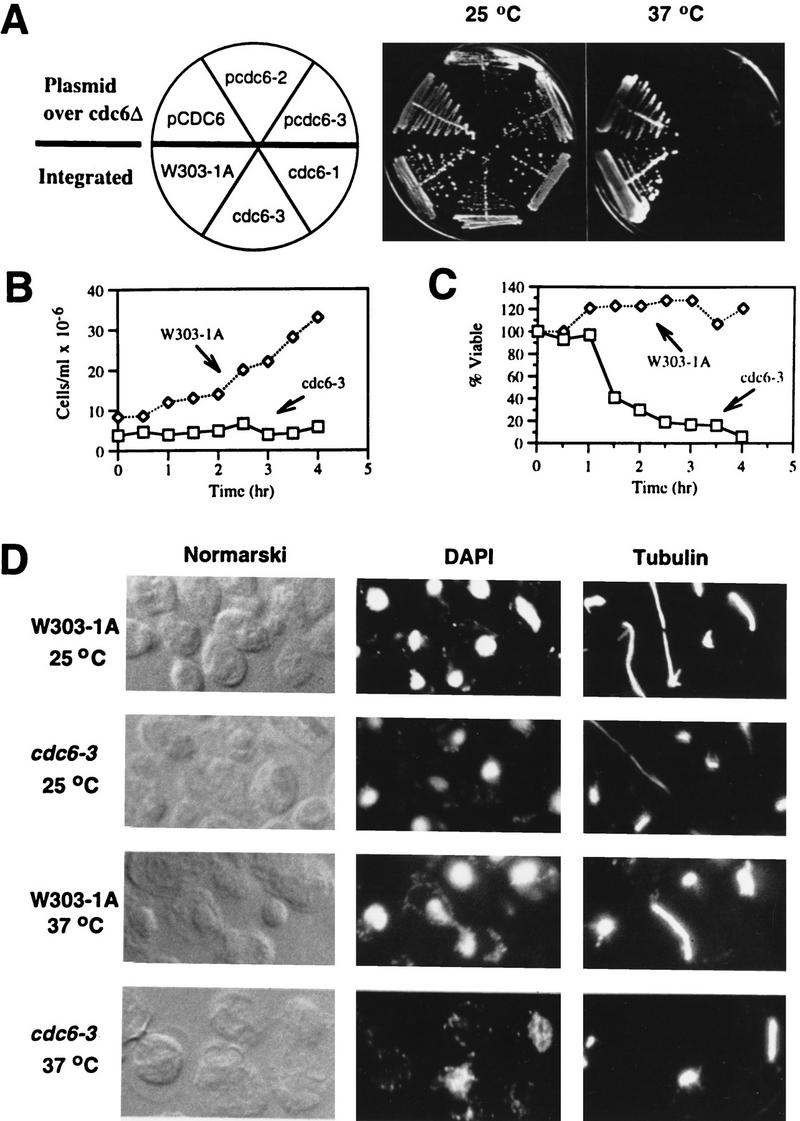
New alleles of cdc6 temperature-sensitive mutants. (A) top, cdc6Δ strains harboring either pCDC6, pcdc6-2, or pcdc6-3 plasmid, and bottom, integrated strains (W303-1A as the isogenic wild-type cdc6-3, and for comparison, cdc6-1) were streaked on YPD plates and incubated for 3 days at 25 or 37°C. (B) W303-1A and cdc6-3 cells were grown asynchronously at 25°C and then shifted to 37°C. Aliquots of cells were harvested at various times and cell numbers counted. (C) Aliquots of cells were retrieved from 37°C at various times as in B, single cells were plated and incubated at 25°C. Percent viable is the fraction of cells able to form colonies at 25°C, normalized to the number of viable cells at time zero (i.e., 25°C). (D) W303-1A and cdc6-3 cells were grown asynchronously at 25°C and then shifted to 37°C for 5 hr (data from 3.5 hr at 37°C were similar; not shown). Cells were fixed, prepared, stained with DAPI (for nuclei) and antitubulin antibody (for mitotic spindles), and visualized by Normarski optics and fluorescence microscopy.
When asynchronous cultures were shifted from 25°C to 37°C, the wild-type cells (strain w303-1A) grew, whereas cdc6-3 cells did not (Fig. 3B), indicating a first cell-cycle arrest of the mutant cells. This was confirmed by incubating single cells on plates at 37°C; the mutant cells did not divide as seen under a microscope. Moreover, the mutant cells lost viability at 37°C after an initial 1-hr delay (Fig. 3C).
After incubation at 37°C, the wild-type cells divided normally, whereas cdc6-3 cells were arrested with large buds (dumb bell), undivided single nuclei either in one of the two buds or between the two buds, and short mitotic spindles (Fig. 3D; 37°C). Therefore, the mutant cells failed to undergo mitosis at 37°C.
Flow cytometry (FACS) was used to measure DNA content of the wild-type and cdc6-3 cells (Fig. 4). To avoid possible interference from mitochondrial DNA replication, we used corresponding ρ° strains lacking mitochondrial DNA for FACS analysis. When released from the G1 α factor block into fresh medium at 37°C, wild-type cells progressed through the cell cycle and eventually became asynchronous (Fig. 4A; shaded). The mutant cells also entered S-phase, although with a delay (cf. time points at 60 and 90 min to wild type), and in later time points most cells accumulated with >2C DNA (Fig. 4A; unshaded; 160 min and 240 min). Similar data were obtained with ρ+ strains of w303-1A and cdc6-3, although both the wild-type and mutant cells entered S-phase slightly faster than the respective ρ° strains (data not shown).
Figure 4.
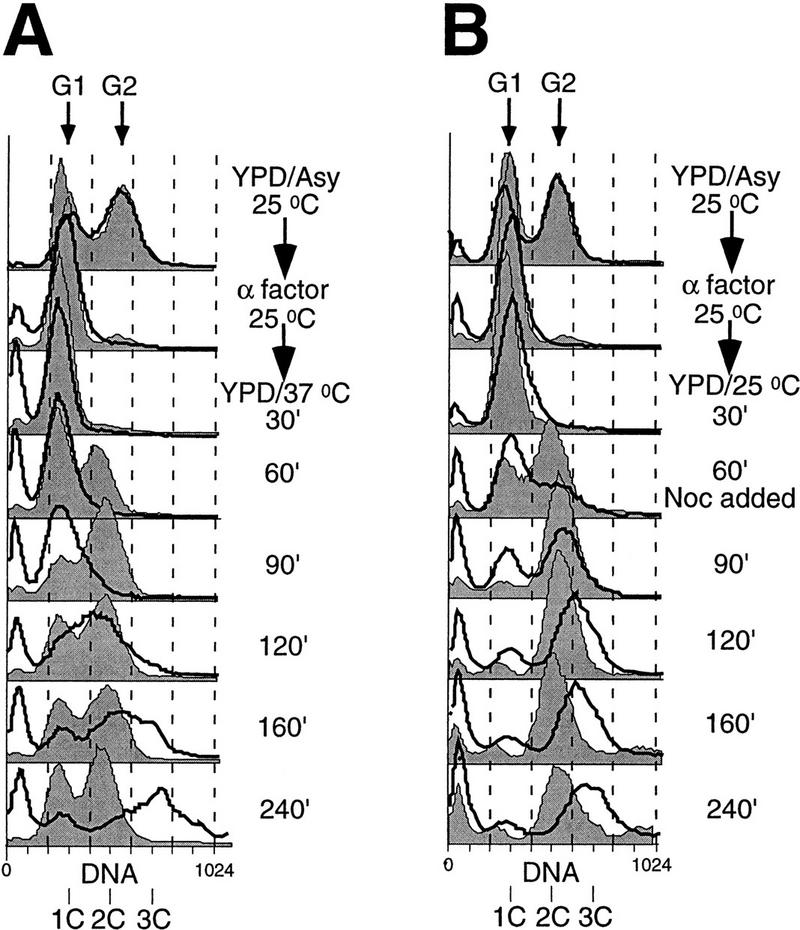
cdc6-3 mutant cells accumulate more than 2C DNA. ρ° strains of W303-1A (thin lines and shaded area) and cdc6-3 (thick lines and unshaded area) were grown asynchronously in YPD at 25°C (YPD/Asy/25°C), blocked in G1 with α factor (α factor/25°C), and then released into fresh YPD at 37°C (A), or at 25°C and then nocodazole (Noc) was added 60 min postrelease (B). Aliquots of cells were taken at various times after the release and analyzed by FACS. DNA contents of 1C and 2C, representing G1 and G2 cells, respectively, are marked. The peaks very close to the vertical axis (no DNA) are attributable to broken (from sonication) and dead cells, mainly of the mutant.
When the previously characterized cdc6-1 mutant cells were shifted to the restrictive temperature of 37°C, they accumulated initially in G1 with 1C DNA, and then some cells went through reductional mitosis without replicating their DNA and ended up with <1C DNA (Piatti et al. 1995; C. Liang and B. Stillman, unpubl.). This is similar to cells deprived of Cdc6p. Therefore, cdc6-1 is a loss-of-function mutation. On the other hand, cdc6-3 (also plasmid-borne cdc6-2; data not shown) mutant cells failed to undergo mitosis at 37°C (Fig. 3D) and had >2C content of DNA (Fig. 4). Therefore, these are gain-of-function mutations. It is important to point out that other replication mutants such as orc2-1, orc5-1, cdc6-1, or mcm mutants did not accumulate >2C DNA as measured by FACS (Gibson et al. 1990; Hennessy et al. 1991; Bell et al. 1993; Loo et al. 1995; C. Liang and B. Stillman, unpubl.), indicating that the phenotype was unique to cdc6-3.
Promiscuous replication of cdc6-3 mutant at the permissive temperature
The increase in nuclear DNA content above the normal post-S-phase 2C content in haploid cells could also be observed when cdc6-3 cells were released from the α factor block into the cell cycle at the permissive temperature of 25°C. To prevent mitosis, nocodazole was added at 60 min postrelease. Wild-type cells entered the cell cycle and had a 2C DNA content by 90 min (Fig. 4B; shaded). cdc6-3 cells also entered the cell cycle, and most cells had accumulated ⩾2C DNA by 120 min. By 240 min, however, most of the mutant cells had ∼3C DNA (Fig. 4B; unshaded).
Cell size had little effect on the DNA profiles in the FACS analysis. Although cdc6-3 cells are larger than the wild type in asynchronous cultures at 25°C (by cell size measurement in FACS and microscopy observations; data not shown), the G1 and G2 DNA peaks were in the same position for the wild-type and mutant cells (Fig. 4A, B; YPD/Asy/25°C). Furthermore, when cells were released from the α-factor block into fresh medium and then blocked in G2/M by nocodazole, the wild-type cells reached the same size as the mutant cells (data not shown), yet only the mutant cells showed >2C peaks (Fig. 4B). Therefore, the rightward shift in the DNA peaks beyond 2C in the haploid mutant cells was attributable to extranuclear DNA rather than other biomass or fluorescence artifacts.
Two-dimensional replicon-mapping gels (2-D gels) were used to examine directly the initiation of DNA replication in the wild-type and cdc6-3 cells. Wild-type (W303-1A) or cdc6-3 mutant haploid cells either were grown asynchronously in the rich medium YPD at 25°C (Fig. 5A,B), or were incubated for 4 hr at 25°C in YPD containing the microtubule inhibitor nocodazole, which blocked cells at G2/M (Fig. 5C,D). Cells were harvested, total DNA was prepared, digested with NcoI, and enriched for replication intermediates. The enriched fractions from the same amount of total genomic DNA for each sample were resolved on 2-D gels. Blots of 2-D gels were probed for the 5-kb NcoI fragment containing ARS1, a normally fully active replication origin in wild-type cells (Liang et al. 1995 and references therein).
Figure 5.

cdc6-3 cells initiate replication even in G2/M and this phenotype is dominant. Haploid (W303-1A and cdc6-3; A–D) and diploid strains (W303-1, cdc6-3/W303-1B and cdc6-3/cdc6-3; E–J) were grown asynchronously in YPD at 25°C (A, B, E–G), or incubated in YPD containing nocodazole at 25°C for 4 hr (C,D, H–J). Small aliquots of cells were analyzed by FACS and the remaining cells were processed for two-dimensional gels, which were probed for the 5-kb NcoI fraction containing ARS1 on the chromosome.
When grown in YPD, the vast majority of wild-type cells initiated replication from ARS1 in almost every cell cycle, as the two-dimensional gel shows an expected clear bubble-to-fork arc transition and very little fork migration through the origin (Fig. 5A). cdc6-3 cells gave a bubble arc in addition to a complete fork arc (Fig. 5B), indicating a lower frequency of initiation at ARS1 and subsequent passive replication through this locus, which was similar to results previously observed with cdc6-1, orc2-1, or orc5-1 mutants (Fox et al. 1995; Liang et al. 1995). In nocodazole-containing medium, wild-type cells lacked a bubble arc as expected, as cells were arrested in G2/M (Fig. 5C; the fork arc probably represents a small number of cells not yet arrested at G2/M). In striking contrast, cdc6-3 cells still had a clearly visible bubble arc, although they had been arrested in a G2-like state with ⩾2C DNA (Fig. 5D).
These data suggest that, unlike the wild-type, cdc6-3 cells initiated DNA replication in G2/M, resulting in a DNA content more than the normal 2C amount. This promiscuous replication phenotype can explain why the mutant cells have difficulty entering into mitosis (∼11% of asynchronous mutant cells have long mitotic spindles and ∼80% budded vs. ∼23% for the wild type with ∼40% budded) and lose viability in a fraction of the population (20–25%), even at the permissive temperature.
To determine whether the promiscuous initiation phenotype was recessive or dominant, the initiation of DNA replication in homozygous wild type (W303-1), heterozygous (cdc6-3/W303-1B), and homozygous (cdc6-3/cdc6-3) strains was investigated using the same procedures used for haploid cells. These analyses showed that this phenotype was dominant (Fig. 5E–J). Thus, the cdc6-3 mutant is a complex one, with a recessive, temperature-sensitive phenotype for growth, yet at the same time is a weak gain-of-function allele. When the gain-of-function phenotype was too penetrant, as in cdc6-2, this resulted in more over-replication and decreased strain viability (data not shown; see Fig. 3A). Because haploid cells also displayed the promiscuous replication phenotypes, we conclude that cdc6-3 is a dominant gain-of-function allele.
MCM protein chromatin association is cell cycle independent in cdc6-3 cells
Because MCM proteins appear to be components of the pre-RC, the binding of these proteins to chromatin was investigated in cdc6-3 cells during the cell cycle. Only the chromatin-bound fractions are shown (Fig. 6). As in wild-type cells, ORC was associated with chromatin throughout the cell cycle in cdc6-3 cells at 25°C (Fig. 6A). The timing of Orc6p phosphorylation and dephosphorylation during the cell cycle was similar to the wild type (cf. Figs. 2A and 6A), indicating that cdc6-3 cells progressed through some aspects of the cell cycle with similar kinetics as wild-type (also see FACS; Figs. 2B and 6B). Unlike in wild-type cells, however, Mcm3p was bound to chromatin throughout the entire cell cycle in cdc6-3 cells (Fig. 6C). Similar data (not shown) were obtained for Mcm2p. This was not attributable to the low frequency of initiation in cdc6-3, because most, if not all, of cdc6-3 cells entered S phase and accumulated a fully replicated ⩾2C amount of DNA after release from the α factor block (Fig. 6B). Moreover, cdc6-1 and orc5-1 cells also have a lower than normal frequency of initiation at the permissive temperature (Liang et al. 1995) similar to that of cdc6-3, yet the chromatin association of MCM proteins in cdc6-1 and orc5-1 mutants was cell cycle regulated in a similar manner as found in wild-type cells; that is, Mcm2p (Fig. 7A) and Mcm3p (data not shown) were not chromatin bound in S and G2. Also notice that unlike cdc6-3, orc5-1 or cdc6-1 cells did not accumulate >2C DNA when they were released from the α factor block (Fig. 7B). These data demonstrate that the continuous binding of MCM proteins in cdc6-3 cells was not attributable to slow progression through S phase, but rather is specific for this particular form of mutant cdc6 protein.
Figure 6.

MCM protein chromatin association is cell cycle independent in cdc6-3 cells. Chromatin-binding experiments were carried out with cdc6-3 cells in the same way as W303-1A cells in Fig. 2. Only the crude chromatin pellet fractions were shown for ORC (A) and Mcm3p (C). The endogenous ORC (Orc2 and Orc4 bands visible only in darker exposures; not shown) and Mcm3p were detected in all lanes. Small aliquots of cells were analyzed by FACS (B).
Figure 7.

MCM protein chromatin binding is cell cycle regulated in orc5-1 and cdc6-1 mutants. Chromatin-binding experiments were carried out with W303-1a, orc5-1 or cdc6-1 cells across the cell cycle, and the chromatin bound Mcm2p was assayed (A). Small aliquots of cells were analyzed by FACS (B). (Asy) asynchronous cells; (0 min) α factor blocked cells.
Mcm2 and Mcm3 proteins were also found in the high-speed pellet (polynucleosomes) from cdc6-3 cells arrested at G2/M by nocodazole (data not shown), confirming that the MCM proteins found in the low-speed crude chromatin pellets from S and G2 cdc6-3 cells (Fig. 6C) were actually chromatin bound.
Together, these data indicate that the competent state of replicators in S and G2 in cdc6-3 cells includes chromatin-bound MCM proteins, probably together with the mutant cdc6-3 protein. We have not been able to detect clearly endogenous Cdc6p or cdc6-3p in crude yeast extracts or chromatin fractions with the available Cdc6p antibodies.
Promiscuous initiation occurs despite high Cdc28/Clb kinase activities
The available evidence suggests that Cdc28/Clb5(/Clb6) kinases normally activate replication at the G1/S boundary and then the mitotic CDK cyclins inhibit rereplication in S and G2 (Schwob et al. 1994; Dahmann et al. 1995; Piatti et al. 1996). Moreover, Cdc28p interacts with Cdc6p (Elsasser et al. 1996; Piatti et al. 1996). Similar interactions occur with the equivalent proteins in Schizosaccaromyces pombe (Jallepalli and Kelly 1997). Therefore, it was conceivable that in wild-type cells, once the pre-RC converted to a post-RC state after initiation, the high CDK activities in S and G2 prevented reassociation of MCM proteins with chromatin by blocking Cdc6p function. Because we found that the MCM proteins remained chromatin bound during the cell cycle in cdc6-3 cells, we asked whether there was an absence of high CDK activities in cdc6-3 cells, or promiscuous initiation occurs despite the high CDK activities.
Either wild-type or cdc6-3 cells were released into the cell cycle at 25°C from the G1 α factor block, and samples were taken at various times after the release. Native proteins were extracted, Cdc28p was precipitated with p13/Suc1–agarose beads, and assayed for histone H1 kinase activity (Fig. 8).
Figure 8.

The CDK activities in S and G2 in cdc6-3 are as high as in the wild-type cells. W303-1A and cdc6-3 cells were grown asynchronously in YPD at 25°C (Asy), synchronized in G1 with α factor (0 min) and then released into fresh YPD at 25°C. Samples were taken at various times (20–120 min) after the release. The last lanes (Noc) were from cells incubated in YPD containing nocodazole at 25°C for 4 hr. Native proteins were extracted and used for p13 bead precipitation. The precipitates were assayed for histone H1 kinase activities.
Control glutathion–agarose beads did not pull down the kinase (Fig. 8; Ctrl bead). The histone H1 kinase activity across the cell cycle in the wild-type cells was found to be as expected (Schwob et al. 1994 and references therein). It was low in α factor-blocked cells, increased at 20 min after the release, and rose to higher levels after 40 min. The last lane shows the expected high kinase activity when cells were arrested at G2/M with nocodazole for 4 hr at 25°C. The kinase activity during the cell cycle for the cdc6-3 cells was very similar to those for wild-type cells. Notice that the kinase activity from cdc6-3 cells that were in nocodazole-containing medium was as high as wild type, yet cdc6-3, but not wild-type cells, were able to initiate DNA replication under these conditions (see Fig. 5C,D). Therefore, MCM protein chromatin association and initiation of DNA replication in cells arrested at G2/M in cdc6-3 cells in the presence of the normal level of Cdc28 kinase activities. These results suggest that the negative control by mitotic cyclins on establishing the pre-RC is mediated through Cdc6p. cdc6-3 mutant cells ignore this negative control and promiscuous initiation of DNA replication occurs.
Discussion
ORC is a sequence-specific DNA-binding protein that binds to replicators that determine the location of origins of DNA replication in S. cerevisiae (Bell and Stillman 1992). Genomic footprinting experiments have suggested that ORC binds the replicator throughout most, if not all, of the cell cycle (Diffley and Cocker 1992; Diffley et al. 1994). Our chromatin-binding assay provides direct physical evidence that all ORC subunits remain bound to chromatin at all times, and thus ORC has the potential to serve as the DNA-bound landing pad for other initiation proteins. It is most likely that ORC is bound to replicator sequences in chromatin, a result demonstrated recently by Tanaka et al. (1997). On the other hand, MCM proteins bind chromatin in a cell cycle-dependent fashion, consistent with them being part of the mechanism that licenses replication in yeast, as has been demonstrated in Xenopus egg extracts (for review, see Chong et al. 1996). A recent chromatin-binding study using cell cycle inhibitors and cdc mutants shows that MCM is on chromatin in G1 (α factor and cdc7), much less so in S (hydroxyurea), and not at all in G2/M (nocodazole and cdc15) (Donovan et al. 1997).
The idea of two states (pre- and post-RC) of proteins bound to the replicator has come from genomic footprinting studies, principally from the absence and presence, respectively, of a DNase I hypersensitive site within the ARS B1 element (Diffley et al. 1994). We have shown that MCM proteins bind chromatin when the hypersensitive site is absent (pre-RC) in late M and G1, and are not on chromatin when the hypersensitive site is present (post-RC) in S, G2, and early M phases. Coupled with the observation that immunoprecipitation of tagged Mcm7p coprecipitated replication origin containing DNA (Tanaka et al. 1997), these data suggest strongly that chromatin-bound MCM proteins are part of the pre-RC and contribute to protection at the B1 site from DNase I.
The number of ORC molecules (∼600) in the cell roughly equals the number of replication origins, but the MCM proteins are at least 20 times more abundant (Lei et al. 1996; Donovan et al. 1997). The amount of chromatin-bound Cdc6p in G1 also approximates that of the number of replication origins (Donovan et al. 1997). Paradoxically, Mcm2p appears to be limiting for replication (Lei et al. 1996). Only a small fraction, however, of the cellular MCM proteins bind chromatin. Therefore, it is likely that the fraction of chromatin-bound MCM proteins that is detected by our procedure and by others (Donovan et al. 1997; Tanaka et al. 1997), as well as ORC and Cdc6 proteins, acts at individual replicators, rather than globally on the chromosomes. Consistent with this notion, mutants in five of the MCM family of genes have reduced frequencies of initiation (Yan et al. 1993; C. Liang and B. Stillman, unpubl.).
Our analysis of the cdc6-3 gain-of-function mutants revealed three interesting and related phenotypes. First, the mutant cells accumulate abnormal amounts of nuclear DNA without passing through mitosis. Second, two-dimensional gel analyses suggest that initiation of DNA replication occurs late in the cell cycle when it is normally inhibited in wild-type cells. Third, the MCM proteins remain bound to the chromatin at times when they are not bound in other cells. Combined, these phenotypes suggest that the cell cycle control of initiation of DNA replication is awry in the cdc6-3 mutant cells, resulting in over-replication of the genome, leading to genetic instability and inviability.
Cdc6p interacts with ORC, and is required for loading of MCM proteins onto chromatin, formation of the pre-RC, and initiation of DNA replication (Liang et al. 1995; Cocker et al. 1996; Coleman et al. 1996; Piatti et al. 1996; Detweiler and Li 1997; Donovan et al. 1997). Cdc6p can execute its function between the end of anaphase and late G1, before activation of replication by Cdc28p/Clb5p and Clb6p kinases (Piatti et al. 1996). Formation of the pre-RC in the rest of the cell cycle is prevented by high CDK activities of Cdc28p/Clb cyclins (Dahmann et al. 1995), and high levels of Cdk2/cyclin E inhibit Mcm3p chromatin binding and replication in Xenopus egg extracts (Hua et al. 1997). Because a cdc6 mutant can bypass the CDK inhibition of rereplication, we propose that this negative regulation is mediated through Cdc6p.
cdc6-3 mutant cells have a lower frequency of initiation of DNA replication at any given origin, similar to cdc6-1 and several orc (Fox et al. 1995; Liang et al. 1995) and mcm mutants (Yan et al. 1993; C. Liang and B. Stillman, unpubl.), probably attributable to lower concentrations (Bell et al. 1993; Loo et al. 1995) or lower activity of these initiation proteins. When shifted to the restrictive temperature, the frequency of initiation is further reduced so that these mutant cells are arrested in the cell cycle and die. However, unlike cdc6-1 or orc mutants, cdc6-3 cells continue to initiate DNA replication even after 3 hr at the restrictive temperature in the presence of nocodazole, albeit at a lower frequency than wild type, leading to accumulation of ⩾3C DNA and eventually cell inviability. The loss of cell viability, as detected by the inability to form colonies on plates at the permissive temperature, is probably attributable to the increased genetic instability in the cdc6-3 mutants as a result of the promiscuous initiation of replication.
The promiscuous initiation of DNA replication in the cdc6-3 mutant was also observed even at the permissive temperature in a single cell cycle. This phenotype was dominant, but probably not fully penetrant because such a mutant would cause too much genome instability and cell death. The cdc6-2 mutant over-replicated its DNA more than cdc6-3 (data not shown), and these cells were less viable than cdc6-3 cells.
Persistent initiation of DNA replication and MCM protein chromatin association in cdc6-3 cells occur although the Cdc28/Clb kinase activities in S and G2 in the mutant cells were as high as in wild-type cells. It is possible that the function of Cdc6p to load MCM proteins is regulated by the CDK cyclins, perhaps through the ATP-binding site in Cdc6p, and that the mutant cdc6 protein is locked into a constitutively active conformation, rendering it immune to the inhibitory effects of the mitotic cyclins. As a result, either the MCM proteins remain bound on replicated chromatin and initiation of DNA replication can occur even in G2/M, or that initiation complexes containing MCM proteins can be reformed continually, even in the presence of high mitotic CDK activity. This was specific for cdc6-3 cells, and was not observed in other mutants that have a lower frequency of initiation than wild-type cells. Thus, the promiscuous initiation of replication and constant chromatin binding of MCM protein in cdc6-3 cells were not attributable to slow S-phase progression.
Rereplication phenotypes in S. cerevisiae have been reported in the ubiquitin hydrolase (Singer et al. 1996) and cdc16 and cdc27 mutants (Heichman and Roberts 1996). Currently, the relationship between these and CDC6 genes is unknown. Overexpression of the Cdc6-related protein called cdc18+ in S. pombe, either directly or indirectly, causes multiple rounds of replication of the genome without mitosis (Nishitani and Nurse 1995; Muzi-Falconi et al. 1996). In all of these cases, over-replication could be attributable to the high levels of proteins that are normally degraded by ubiquitin-mediated proteolysis indirectly affecting CDK/cyclin activity late in the cell cycle. This could result in a block to mitosis but allow multiple rounds of S phases in these cells.
The phenotype of the cdc6-3 mutant is remarkably similar to the promiscuous initiation of DNA replication found in the Escherichia coli DnaAcos mutant (Katayama 1994; Katayama and Kornberg 1994). The DnaA initiator protein binds to OriC origin in its active ATP-bound form, slowly hydrolyzes the ATP to form an inactive ADP-bound form that cannot initiate replication (for review, see Skarstad and Boye 1994). DnaAcos is a revertant of the DnaA46 temperature-sensitive mutant and like cdc6-3, contains multiple mutations. The DnaAcos protein causes promiscuous initiation of DNA replication in vivo and in vitro by overcoming an unknown negative regulation of initiation. Interestingly, the mutant protein cannot bind ATP. We suggest that the normal function of Cdc6p may also be sensitive to ATP, just like the DnaA initiator and its eukaryotic sequence relative in the ORC complex Orc1p (Bell et al. 1995; Klemm et al. 1997). Therefore, it is possible that cdc6-3p may have unregulated ATP binding, causing an altered conformation and resistance to negative control by the CDK cyclins. Finally, we consider it probable that Cdc6p may control the switch to amplification cycles during development in certain cell types, such as the ORC-dependent amplification of the chorion genes in Drosophila (Landis et al. 1997).
Materials and methods
Yeast strains and plasmids
All yeast strains used were, or derivatives of, W303-1A (Mata), W303-1B (Matα) (ho, ade2, trp1, can1, leu2, his3, GAL, psi+), or W303-1 (W303-1A/W303-1B), except for strain J2 (dbf2-2/dbf2-2) (Diffley et al. 1994) used in Figure 2F. All cdc6Δ strains were derived from strain GS1 (W303-1; cdc6Δ::hisG-URA3–hisG/CDC6), kindly provided by Gavin Sherlock and Bruce Futcher of Cold Spring Harbor Laboratory, in which all but the first and last 50 bp of one copy of the CDC6-coding sequence has been deleted on the basis of the PCR-directed gene disruption method (Schneider et al. 1996). The URA3 gene used to disrupt CDC6 was later inactivated by use of 5-fluoro-orotic acid. The following strains are cdc6Δ in W303-1A containing pRS426–CDC6 (YB0209), pRS415–2.1CDC6 (YB0321), pRS415–cdc6-2 (YB0275), or pRS415–cdc6-3 (YB0276). YB0297 is cdc6-3 integrated in W303-1A by transplacement, and YB0044 is Mata, cdc6-1 (backcrossed four times to W303; Liang et al. 1995). ρ° strains (YB0433 from W303-1A and YB0434 from cdc6-3) were generated by growing the ρ+ strains in SCM medium containing 20 μg/ml of EtBr overnight at 25°C and then selecting for inability to grow on glycerol plates. Diploid strains were YB0325 (cdc6-3/W303-1B) and YB0323 (cdc6-3/cdc6-3).
pRS426–CDC6 is the 3.1-kb MluI–EcoRI CDC6 fragment cloned in pRS426 (2μ ARS, URA3) by exchanging the vectors with pRS415–CDC6 (Liang et al. 1995). pKS-2.1CDC6 was constructed by ligating the 2.1-kb HindIIINarI CDC6 fragment from pRS415-CDC6 (HindIII site is in the vector just upstream of the MluI site of the CDC6 promotor) to pBluescript/KS+ (Strategene) cut with HindIII and ClaI. pRS415–2.1CDC6 (shorthanded as pCDC6) was constructed by ligating the 2.1-kb HindIII–XhoI (both sites on vector) fragment containing CDC6 from pKS–2.1CDC6 to pRS415 (CEN6, H4 ARS, LEU2) cut with the same enzymes. pRS415–cdc6-2 (as pcdc6-2) and pRS415–cdc6-3 (as pcdc6-3) were derived from pRS415–2.1CDC6 by random mutagenesis.
Cell synchronization and release
Cells were grown in YPD at 25°C to early log phase (∼5 × 106 cells/ml), α factor was then added to 5 μg/ml, and the culture was incubated with shaking for 1.5 hr at 25°C. Another 5 μg/ml of α factor was added, and the culture was incubated for another 1.5 hr at 25°C. To release, cells were spun down, and resuspended in fresh YPD. Rapid shifting to 37°C was achieved by swirling culture flasks in ∼50°C water. Nocodazole (20 μg/ml) was used to arrest cells in G2/M. YPD with 4% dextrose was used for the ρ° strains.
Protein chromatin-binding assay
About 1 × 109 cells at ∼2 × 107 cells/ml were harvested and sodium azide was added to 0.1%. Cells were spheroplasted according to Conradt et al. (1992) with modifications. Cells were incubated at room temperature for 10 min in 3 ml of prespheroplasting buffer [100 mm PIPES (pH 9.4), 10 mm DTT], followed by incubation in 2 ml of spheroplasting buffer [50 mm KH2PO4/K2HPO4 (pH 7.5), 0.6 m Sorbitol, 10 mm DTT] containing 80 μl of 1 mg/ml of Oxalyticase (Enzogenetics) at 30°C for 10–15 min with occasional mixing, until the OD600 of a 1:100 dilution of the cell suspension (in water) dropped to <10% of the value before digestion. Sphereoplasts, without incubation for recovery and regrowth, were washed with 1 ml of ice-chilled wash buffer [100 mm KCl, 50 mm HEPES-KOH (pH 7.5), 2.5 mm MgCl2, and 0.4 m Sorbitol], pelleted at 4000 rpm for 1 min in a microcentrifuge at 4°C, and resuspended in an equal pellet volume (∼80 μl) of extraction buffer [EB; 100 mm KCl, 50 mm HEPES-KOH (pH 7.5), 2.5 mm MgCl2, 50 mm NaF, 5 mm Na4P2O7, 0.1 mm NaVO3], and protease inhibitors (1 mm PMSF, 20 μg/ml of leupeptin, 2 μg/ml of pepstatin, 2 mm benzamidine HCl, and 0.2 mg/ml of bacitracin). The suspension was split into three tubes, each with 25% (for fraction 1. WCE), 25% (for fractions 2. Sup and 3. Pel), and 50% (for fractions 4–7) of the total. Spheroplasts were lyzed by adding Triton X-100 to 0.25% and incubating on ice for 5 min with gentle mixing. Lysate was underlayered with 50% volume of 30% sucrose (volume refers to the volume of the spheroplast suspension in EB; the same below), and spun at 12,000 rpm for 10 min at 4°C. Pellet was washed with 25% volume of EB containing 0.25% Triton X-100 (EBX), and spun again at 10,000 rpm for 5 min at 4°C. The crude chromatin pellet for fractions 4–7 was incubated in 50% volume of EBX at 37°C for 2 min. CaCl2 (to 1 mm) and 600 μm units of MNase were added, and incubation continued for 1 min with mixing. Digestion was stopped by adding EGTA to 1 mm, and spun at 10,000 rpm for 2 min at 4°C. Pellet was digested once more with 200 μm units of MNase in 25% volume of EBX and spun as above. Supernatants were combined, and then split equally into two: one for fraction 4 (MNase Sup) and the other for high speed fractions 6 and 7. The latter was centrifuged at 100,000 g (50,000 rpm in Beckman TLA-100.3 rotor) for 1 hr at 4°C. All pellet fractions were resuspended in EBX, and the volumes of all fractions were adjusted with EBX (∼60 μl final volume) to reflect the same cell equivalent. Equal volumes of 2× Laemmli’s buffer were added to each fraction. Samples were boiled for 3 min, and spun at 10,000 rpm for 1 min before loading to 12.5% SDS-PAGE gels.
Random mutagenesis of CDC6 and isolation of new cdc6 temperature-sensitive mutants
The CDC6 gene was amplified from pKS–CDC6 by PCR using universal sequencing (T7) and reverse sequence primers from the vector. PCR was performed using five units of AmpliTag polymerase (Perkin-Elmer) in 100 μl of standard buffer from the manufacturer plus 0.1 ng of template, 50 pmoles each of primers and 20 mm each of dNTP for 25 cycles of (94°C, 1 min; 54°C, 1 min; 72°C, 3 min) after an initial 2 min at 94°C. PCR products were cut with XhoI and HindIII, and the 2.1-kb fragment was gel purified and cloned into pRS415 cut with the same enzymes. Plasmids were transformed into E. coli and recovered from ∼60,000 pooled primary transformants.
Plasmids (5 μg) from the mutagenized library were transformed into YB0209. After incubation at 25°C for 4 days, ∼14,000 transformants that were in the range of 200–300 colonies/plate were replica-plated onto two sets of FOA–Leu plates, and incubated at 25 or 37°C. Plasmids from temperature-sensitive colonies were retested by the same plasmid shuffle. Two mutants were sequenced with ABI377 sequencer (Perkin Elmer): cdc6-2 has A320S (GCA → tCA), L338L (TTA → cTA) and L450S (TTA → TcA), and cdc6-3 has H144T (CAT → tAT) and L258S (TTA → TcA).
Fluorescence microscopy, two-dimensional gels, and FACS
Fluorescence microscopy (Diffley et al. 1994) and two-dimensional gels (Liang et al. 1995) were performed essentially as described. For FACS, ∼5 × 106 cells for each sample were fixed in 70% ethanol, resuspended in 50 mm sodium citrate (pH 7.2), briefly sonicated, digested with 0.25 mg/ml RNase in the same buffer followed by 1 mg/ml of proteinase K, each for 1 hr at 50°C. DNA was stained with 10 μg/ml of propidium iodide, and 50,000 cells from each sample were scanned with Elite (Coulter) or FACSCalibur (Becton-Dickinson) FACS machine.
Histone H1 kinase assay and Western blotting
Suc1/p13 bead precipitation and histone H1 kinase assay were according to Elsasser et al. (1996). Extract containing 250 μg of proteins from each sample was used for the kinase assay. For Western blotting, proteins were transferred to nitrocellulose, and blocked with 5% dry milk in TBST (0.1% Tween 20). Monoclonal antibodies (mAb) from ascites against Orc1–Orc6 subunits were SB35, 46, 3, 12, 5, and 49 in dilutions of 1:1000, 1:500, 1:50,000, 1:2000, 1:2000, and 1:2000, respectively, plus SB16 against Orc1p in 1:2000. Mcm2 and Mcm3 mAb were 28 and 18, respectively, both in 1:10,000. Horseradish peroxidase-coupled anti-mouse secondary antibody was used in 1:10,000 in TBST containing 5% dry milk. Reactive bands were visualized with the SuperSignal or SuperSignal ULTRA chemiluminescent kit (Pierce).
Acknowledgments
We thank Michael Weinreich for providing bORC and orc6 phosphorylation mutant, and for discussions and reading of the manuscript, Masahiro Akiyama for MCM antibodies and bMCM, Stephen Bell for ORC antibodies, and Gavin Sherlock and Bruce Futcher for the cdc6Δ strain, mostly before publication. We also thank Corine Driessens for technical assistance, Pat Burfiend and Maria Coronesi for FACS analysis, Gavin Sherlock for help with fluorescence microscopy, Alain Verreault for discussion on MNase digestion, James Duffy for art work, and Philip Renna for photography. This work was supported by a grant from National Institutes of Health (GM45436 to B.S.) and a postdoctoral fellowship from the Cancer Research Fund of the Damon Runyon-Walter Winchell Foundation (DRG-1308 to C.L.).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL stillman@cshl.org; FAX (516) 367-8879.
References
- Bell SP, Stillman B. ATP-dependent recognition of eukaryotic origins of DNA replication by a multiprotein complex. Nature. 1992;357:128–134. doi: 10.1038/357128a0. [DOI] [PubMed] [Google Scholar]
- Bell SP, Kobayashi R, Stillman B. Yeast origin recognition complex functions in transcription silencing and DNA replication. Science. 1993;262:1844–1870. doi: 10.1126/science.8266072. [DOI] [PubMed] [Google Scholar]
- Bell SP, Mitchell J, Leber J, Kobayashi R, Stillman B. The multidomain structure of ORC1p reveals similarity to regulators of DNA replication and transcriptional silencing. Cell. 1995;83:563–568. doi: 10.1016/0092-8674(95)90096-9. [DOI] [PubMed] [Google Scholar]
- Bueno A, Russell P. Dual functions of CDC6: A yeast protein required for DNA replication also inhibits nuclear division. EMBO J. 1992;11:2167–2176. doi: 10.1002/j.1460-2075.1992.tb05276.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong JPJ, Mahbubani HM, Khoo C-Y, Blow JJ. Purification of an MCM-containing complex as a component of the DNA replication licensing system. Nature. 1995;375:418–421. doi: 10.1038/375418a0. [DOI] [PubMed] [Google Scholar]
- Chong JPJ, Thommes P, Blow JJ. The role of MCM/P1 proteins in the licensing of DNA replication. Trends Biochem Sci. 1996;21:102–106. [PubMed] [Google Scholar]
- Cocker JH, Piatti S, Santocanale C, Nasmyth K, Diffley JFX. An essential role for the Cdc6 protein in forming the pre-replicative complexes of budding yeast. Nature. 1996;379:180–182. doi: 10.1038/379180a0. [DOI] [PubMed] [Google Scholar]
- Coleman TR, Carpenter PB, Dunphy WG. The Xenopus Cdc6 protein is essential for the initiation of a single round of DNA replication in cell-free extracts. Cell. 1996;87:53–63. doi: 10.1016/s0092-8674(00)81322-7. [DOI] [PubMed] [Google Scholar]
- Conradt B, Shaw J, Vida T, Emr S, Wickner W. In vitro reactions of vacuole inheritance in Saccharomyces cerevisiae. J Cell Biol. 1992;119:1469–1479. doi: 10.1083/jcb.119.6.1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahmann C, Diffley JFX, Nasmyth KA. S-phase-promoting cyclin-dependent kinases prevent re-replication by inhibiting the transition of replication origins to a pre-replicative state. Curr Biol. 1995;5:1257–1269. doi: 10.1016/s0960-9822(95)00252-1. [DOI] [PubMed] [Google Scholar]
- Dalton S, Whitbread L. Cell cycle-regulated nuclear import and export of Cdc47, a protein essential for initiation of DNA replication in budding yeast. Proc Natl Acad Sci. 1995;92:2514–2518. doi: 10.1073/pnas.92.7.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Detweiler CS, Li JJ. Cdc6p establishes and maintains a state of replication competence during G1 phase. J Cell Sci. 1997;110:753–763. doi: 10.1242/jcs.110.6.753. [DOI] [PubMed] [Google Scholar]
- Diffley JFX. Once and only once upon a time: Specifying and regulating origins of DNA replication in eukaryotic cells. Genes & Dev. 1996;10:2819–2830. doi: 10.1101/gad.10.22.2819. [DOI] [PubMed] [Google Scholar]
- Diffley JFX, Cocker JH. Protein–DNA interactions at a yeast replication origin. Nature. 1992;357:169–172. doi: 10.1038/357169a0. [DOI] [PubMed] [Google Scholar]
- Diffley JFX, Cocker JH, Dowell SJ, Rowley A. Two steps in the assembly of complexes at yeast replication origins in vivo. Cell. 1994;78:303–316. doi: 10.1016/0092-8674(94)90299-2. [DOI] [PubMed] [Google Scholar]
- Donovan S, Harwood J, Drury LS, Diffley JFX. Cdc6p-dependent loading of Mcm proteins onto pre-replicative chromatin in budding yeast. Proc Natl Acad Sci. 1997;94:5611–5616. doi: 10.1073/pnas.94.11.5611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elsasser S, Lou F, Wang B, Campbell JL, Jong A. Interaction between yeast Cdc6 protein and B-type cyclin/Cdc28 kinases. Mol Biol Cell. 1996;7:1723–1735. doi: 10.1091/mbc.7.11.1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox CA, Loo S, Dillin A, Rine J. The origin recognition complex has essential functions in transcriptional silencing and chromosomal replication. Genes & Dev. 1995;9:911–924. doi: 10.1101/gad.9.8.911. [DOI] [PubMed] [Google Scholar]
- Gibson SI, Surosky RT, Tye B-K. The phenotype of the minichromosome maintenance mutant mcm3 is characteristic of mutants defective in DNA replication. Mol Cell Biol. 1990;10:5707–5720. doi: 10.1128/mcb.10.11.5707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heichman KA, Roberts JM. The yeast CDC16 and CDC27 genes restrict DNA replication to once per cell cycle. Cell. 1996;85:39–48. doi: 10.1016/s0092-8674(00)81080-6. [DOI] [PubMed] [Google Scholar]
- Hennessy KM, Lee A, Chen E, Botstein D. A group of interacting yeast DNA replication genes. Genes & Dev. 1991;5:958–969. doi: 10.1101/gad.5.6.958. [DOI] [PubMed] [Google Scholar]
- Hua XH, Yan H, Newport J. A role for Cdk2 kinase in negatively regulating DNA replication during S phase of the cell cycle. J Cell Biol. 1997;137:183–192. doi: 10.1083/jcb.137.1.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jallepalli PV, Kelly T. Cyclin-dependent kinase and initiation at eukaryotic origins: A replication switch? Curr Opin Cell Biol. 1997;9:358–363. doi: 10.1016/s0955-0674(97)80008-7. [DOI] [PubMed] [Google Scholar]
- Katayama T. The mutant DnaAcos protein which overinitiates replication of the Escherichia coli chromosome is inert to negative regulation for initiation. J Biol Chem. 1994;269:22072–22079. [PubMed] [Google Scholar]
- Katayama T, Kornberg A. Hyperactive initiation of chromosomal replication in vivo and in vitro by a mutant initiator protein, DnaAcos, of Escherichia coli. J Biol Chem. 1994;269:12698–12703. [PubMed] [Google Scholar]
- Kimura H, Takizawa N, Nozaki N, Sugimoto K. Molecular cloning of cDNA encoding mouse Cdc21 and CDC46 homologs and characterization of the products: Physical interaction between P1(MCM3) and CDC46 proteins. Nucleic Acids Res. 1995;23:2097–2104. doi: 10.1093/nar/23.12.2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klemm RD, Austin RJ, Bell SP. Coordinate binding of ATP and origin DNA regulates the ATPase activity of the origin recognition complex. Cell. 1997;88:493–502. doi: 10.1016/s0092-8674(00)81889-9. [DOI] [PubMed] [Google Scholar]
- Kubota Y, Mimura S, Nishimoto S-I, Takisawa H, Nojima H. Identification of the yeast MCM3-related protein as a component of Xenopus DNA replication licensing factor. Cell. 1995;81:601–609. doi: 10.1016/0092-8674(95)90081-0. [DOI] [PubMed] [Google Scholar]
- Landis G, Kelley R, Spradling AC, Tower J. The k43 gene, required for chorion gene amplification and diploid cell chromosome replication, encodes the Drosophila homolog of yeast origin recognition complex subunit 2. Proc Natl Acad Sci. 1997;94:3888–3892. doi: 10.1073/pnas.94.8.3888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei M, Kawasaki Y, Tye B-K. Physical interactions among Mcm proteins and effects of Mcm dosage on DNA replication in Saccharomyces cerevisiae. Mol Cell Biol. 1996;16:5081–5090. doi: 10.1128/mcb.16.9.5081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang C, Weinreich M, Stillman B. ORC and Cdc6p interact and determine the frequency of initiation of DNA replication in the genome. Cell. 1995;81:667–676. doi: 10.1016/0092-8674(95)90528-6. [DOI] [PubMed] [Google Scholar]
- Loo S, Fox CA, Rine J, Kobayashi R, Stillman B, Bell S. The origin recognition complex in silencing, cell-cycle progression and DNA replication. Mol Biol Cell. 1995;6:741–756. doi: 10.1091/mbc.6.6.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madine MA, Khoo C-Y, Mills AD, Laskey RA. MCM3 complex required for cell cycle regulation of DNA replication in vertebrate cells. Nature. 1995;375:421–424. doi: 10.1038/375421a0. [DOI] [PubMed] [Google Scholar]
- Muzi-Falconi M, Brown GTW, Kelly TJ. cdc18+ regulates initiation of DNA replication in Schizosaccharomyces pombe. Proc Natl Acad Sci. 1996;93:1566–1570. doi: 10.1073/pnas.93.4.1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasmyth K. Putting the cell cycle in order. Science. 1996;274:1643–1645. doi: 10.1126/science.274.5293.1643. [DOI] [PubMed] [Google Scholar]
- Nishitani H, Nurse P. p65cdc18 plans a major role controlling the initiation of DNA replication in fission yeast. Cell. 1995;83:397–405. doi: 10.1016/0092-8674(95)90117-5. [DOI] [PubMed] [Google Scholar]
- Piatti S, Lengauer C, Nasmyth K. Cdc6 is an unstable protein whose de novo synthesis in G1 is important for the onset of S phase and for preventing a “reductional” anaphase in the budding yeast Saccharomyces cerevisiae. EMBO J. 1995;1141:3788–3799. doi: 10.1002/j.1460-2075.1995.tb00048.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piatti S, Bohm T, Cocker JH, Diffley JFX, Nasmyth K. Activation of S-phase-promoting CDKs in late G1 defines a “point of no return” after which Cdc6 synthesis cannot promote DNA replication in yeast. Genes & Dev. 1996;10:1516–1531. doi: 10.1101/gad.10.12.1516. [DOI] [PubMed] [Google Scholar]
- Schneider BL, Stein B, Seufert W, Futcher AB. pMPY-ZAP: A reusable polymerase chain reaction-directed gene disruption cassette for Saccharomyces cerevisiae. Yeast. 1996;12:129–134. doi: 10.1002/(sici)1097-0061(199602)12:2<129::aid-yea891>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- Schwob E, Bohm T, Mendenhall MD, Nasmyth K. The B-type cyclin kinase inhibitor p40SIC1 controls the G1 to S transition in S. cerevisiae. Cell. 1994;79:233–244. doi: 10.1016/0092-8674(94)90193-7. [DOI] [PubMed] [Google Scholar]
- Singer JD, Manning BM, Formosa T. Coordinating DNA replication to produce one copy of the genome requires genes that act in ubiquitin metabolism. Mol Cell Biol. 1996;16:1356–1366. doi: 10.1128/mcb.16.4.1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skarstad K, Boye E. The initiator protein DnaA: Evolution, properties and function. Bioch Bioph Acta. 1994;1217:111–130. doi: 10.1016/0167-4781(94)90025-6. [DOI] [PubMed] [Google Scholar]
- Stillman B. Cell cycle control of DNA replication. Science. 1996;274:1659–1664. doi: 10.1126/science.274.5293.1659. [DOI] [PubMed] [Google Scholar]
- Tanaka T, Knapp D, Nasmyth K. Loading of an Mcm protein onto DNA replication origins is regulated by Cdc6p and CDKs. Cell. 1997;90:649–660. doi: 10.1016/s0092-8674(00)80526-7. [DOI] [PubMed] [Google Scholar]
- Todorov IT, Attaran A, Kearsey SE. BM28, a human member of the MCM2-3-5 family, is displaced from chromatin during DNA replication. J Cell Biol. 1995;129:1433–1445. doi: 10.1083/jcb.129.6.1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan H, Merchant AM, Tye B-K. Cell cycle-regulated nuclear localization of MCM2 and MCM3, which are required for the initiation of DNA synthesis at chromosomal replication origins in yeast. Genes & Dev. 1993;7:2149–2160. doi: 10.1101/gad.7.11.2149. [DOI] [PubMed] [Google Scholar]
- Zhou C, Jong A. CDC6 mRNA fluctuates periodically in the yeast cell cycle. J Biol Chem. 1990;265:19904–19909. [PubMed] [Google Scholar]
- Zwerschke W, Rottjakob H-W, Kuntzel H. The Saccharomyces cerevisiae CDC6 gene is transcribed at late mitosis and encodes a ATP/GTPase controlling S phase initiation. J Biol Chem. 1994;269:23352–23356. [PubMed] [Google Scholar]