

Abstract
Aging, or senescence, is the progressive deterioration of every bodily function over time. A fundamental question that applies to all life forms, including growth-arrested bacteria, is why growing older by necessity causes organisms to grow more fragile. In this work, we demonstrate that the levels of oxidized proteins is correlated to the age of a stationary-phase Escherichia coli culture; both disulfide bridge formation of a cytoplasmic leader-less alkaline phosphatase and protein carbonyl levels increase during stasis. The stasis-induced increase in protein oxidation is enhanced in cells lacking the global regulators OxyR and ςs. Some proteins were found to be specifically susceptible to stasis-induced oxidation; notably several TCA cycle enzymes, glutamine synthetase, glutamate synthase, pyruvate kinase, DnaK, and H-NS. Evidence that oxidation of target proteins during stasis serves as the signal for stationary-phase, developmental, induction of the heat shock regulon is presented by demonstrating that this induction is mitigated by overproducing the superoxide dismutase SodA. In addition, cells lacking cytoplasmic superoxide dismutase activity exhibit superinduction of heat shock proteins. The possibility that oxidative sensitivity of TCA cycle enzymes serves as a feedback mechanism down-regulating toxic respiration is discussed.
Keywords: Stationary phase, protein carbonylation, disulfide bonds, OxyR, RpoS, DnaK
Reactive oxygen species (ROS) such as hydrogen peroxide, superoxide anion, and hydroxyl radicals have been implicated in determining the life span of several organisms, and cancer, chronic inflammation, and cardiovascular, neurodegenerative, and pulmonary diseases have been associated with age-related oxidative stress (e.g., Stadtman 1992). External environmental factors including ionizing radiation and a variety of chemicals can generate ROS (Cooper and Zika 1983; Petasne and Zika 1987). Importantly, they are also produced during normal metabolic electron transport by mitochondria, chloroplasts, and respiring bacteria (Fridovich 1978) and cells have developed antioxidant defense systems aimed at protecting against these ROS (e.g., Halliwell 1974; Demple and Halbrook 1983; Christman et al. 1985; Greenberg et al. 1990; Tsaneva and Weiss 1990). However, the defense systems are not leak proof and ROS can escape and attack subcellular components, including nucleic acids, fatty acids, and proteins (Halliwell and Gutteride 1984; Davies et al. 1987). Proteins are one of the major targets for this attack and oxidation of amino acids can generate non-native cysteine disulfide bonds (Davies et al. 1987), methionine sulfoxide (Levine et al. 1996), and introduce carbonyl groups at lysine, arginine, proline, and threonine residues (Farber and Levine 1986; Amici et al. 1989). These modifications may inhibit or alter the proteins’ activities and increase their susceptibility to proteolytic attack (e.g., Rivett and Levine 1990; Bertlett and Stadtman 1997).
It has been demonstrated that growth arrest of Escherichia coli caused by starvation for different nutrients elicits an increased production of proteins that are normally made during oxidative stress (e.g., H2O2 exposure) (Jenkins et al. 1988). As a consequence, starved, aging E. coli cells become markedly resistant to H2O2 treatments (Jenkins et al. 1988). The development of this resistance is, at least partly, dependent on the transcription factor sigma-S (σs) encoded by rpoS (Lange and Hengge-Aronis 1991; McCann et al. 1991; Von Ossowski et al. 1991) and several rpoS-dependent genes have been shown to encode proteins with specific roles in oxidative-stress resistance; they include catalase HPII (katE) (Von Ossowski et al. 1991), exonuclease III (xthA) (Sak et al. 1989), and the DNA-binding protein Dps (dps) (Almiron et al. 1992; Martinez and Kolter 1997). What is the physiological significance of this oxidative-stress resistance elicited by growth arrest? It has been suggested that induction of the rpoS-dependent network during entry into the stationary phase may increase the chance of survival in future situations that do not allow protein production (Hengge-Aronis 1993; Kolter et al. 1993). Thus, the role of oxidative-stress protein production at the onset of stasis could be to protect the cell against oxidant exposure that it may later encounter during the time of growth arrest (Hengge-Aronis 1993). Alternatively, oxidative-stress proteins may be produced during growth arrest because oxidative attack of macromolecules is a problem caused by continued metabolic activities in the growth-arrested cell. Superoxide radicals arise as byproducts of all aerobic respiration but inactive ROS-damaged macromolecules are rapidly diluted as long as the environment promotes growth and continued translation. Gonzalez-Flecha and Demple (1995) measured the rates of hydrogen peroxide and superoxide anion production during aerobic growth of E. coli and demonstrated that a considerable production of these ROS continued in stationary phase (Gonzalez-Flecha and Demple 1995). Thus, it is possible that during growth arrest of metabolically active cells, such as E. coli, the levels of oxidized proteins increase simply because they cannot be diluted by de novo protein synthesis.
Several heat shock proteins are produced increasingly during growth arrest (Jenkins et al. 1988, 1991; Nyström 1994), indicating that this regulon, like the hydrogen peroxide defense stimulon, may respond to some starvation developmental signal as well as a true stress-inducible signal elicited by heat (or ethanol). Using the gene–protein database of E. coli (VanBogelen et al. 1990), we have identified eight stasis proteins as being the heat shock proteins GrpE, GroEL, DnaK, GroES, HtpH, HtpO, ClpB, and Lon (Nyström 1994). Matin and coworkers (Jenkins et al. 1991) demonstrated that the heat shock σ factor σ32 is required also for developmental induction of the heat shock regulon but the regulatory mechanisms involved in this induction have not been elucidated. In addition, the physiological significance of this regulon during stasis is not clear but may be argued to either prepare the cell for future cataclysmic changes in the environment or to actually have a role in stasis survival. However, cells lacking DnaK die rapidly during carbon starvation-induced stasis (Spence et al. 1990); fail to develop starvation-induced resistance to heat, oxidation, and osmotic stress; and exhibit reduced levels of σs in stationary phase (Rockarbrand et al. 1998).
The objective of this study was to test whether stasis, per se, causes increased oxidation of cytoplasmic proteins and whether oxidative-defense regulons known to be induced during stasis have a role in minimizing such oxidation in growth-arrested, aging bacteria. Protein oxidation was analyzed with respect to protein disulfide bridge formation in the cytoplasm and protein carbonylation. It is demonstrated that (1) the activity of a leader-less alkaline phosphatase (Δ2-22AP) increases, aerobically but not anaerobically, during stasis in wild-type E. coli in parallel to an increase in protein carbonylation; (2) the increase in Δ2-22AP activity observed during stasis is enhanced in cells lacking glutathione reductase or σs, whereas increased carbonylation is enhanced in both rpoS and oxyR mutants; (3) stasis-induced oxidation targets specific proteins, including tricarboxylic acid (TCA) cycle enzymes, glutamine synthetase, glutamate synthase, pyruvate kinase, DnaK, and H-NS; and (4) reducing the levels of superoxide in stationary-phase cells by overexpressing superoxide dismutase A (SodA) decreases protein carbonylation and mitigates developmental induction of the heat shock regulon. The results explain, in part, how and why the stationary-phase bacterial cell becomes resistant to both oxidants and heat and have implications for our understanding of the forces causing the deterioration of growth-arrested cells.
Results
The rate of synthesis of oxidative-defense proteins increase during growth arrest
E. coli cells were pulse-labeled with [35S]methionine during exponential growth and at times after growth ceased because of glucose, nitrogen, or phosphate depletion. Analysis by two-dimensional gel electrophoresis revealed that the rates of synthesis of at least 10 proteins normally induced by H2O2 exposure were induced also during the starvation conditions studied. A significant overlap between starvation stimulons and the H2O2 stimulon of E. coli has been observed previously by Jenkins et al. (1988). Unfortunately, a correlation between this study and the study of glucose-starvation proteins by Matin and coworkers (Reeve et al. 1984; Jenkins et al. 1988, 1991) was impossible because of differences in the two-dimensional protocols. Six of the 10 oxidative-stress proteins identified here as produced increasingly during carbon starvation were found to be members of the oxyR regulon, that is, an oxyR mutant failed to increase the synthesis of these proteins during H2O2 exposure and these proteins were synthesized constitutively at high rates in an oxyR2 mutant that is constitutive for expression of the oxidative-stress regulon (not shown). The members of the OxyR regulon were induced also by phosphate and nitrogen starvation (not shown). The location of these proteins on the E. coli reference two-dimensional gel and the extent of induction during carbon starvation is depicted in Figure 1A and B, respectively. Two of the OxyR-dependent proteins were identified as glutathione reductase (GorA) and glutaredoxin 1 (GrxA), both involved in disulfide bond reduction. We found that both OxyR and σs were required for the increased production of GorA and GrxA during stasis (Fig. 1C). In addition, the kinetics of oxidative-stress protein induction during starvation was different from the typical stationary-phase protein induction. Specifically, most stress proteins (e.g., UspA; Fig. 1D) are induced as soon as growth is inhibited but no induction of oxidative-defense proteins (e.g., GrxA; Fig. 1D) could be observed prior to 15 min of starvation, indicating that the encoding genes do not respond to starvation per se. Also, the rate of GorA and GrxA production was not an effect of slow growth because the production rates were found to be indistinguishable at growth rates between k = 0.3 and 1.9 (Fig. 1E). For a comparison, the rate of SdhA production, known to be inversely dependent on growth rate (Nyström 1994), is depicted in Figure 1E.
Figure 1.
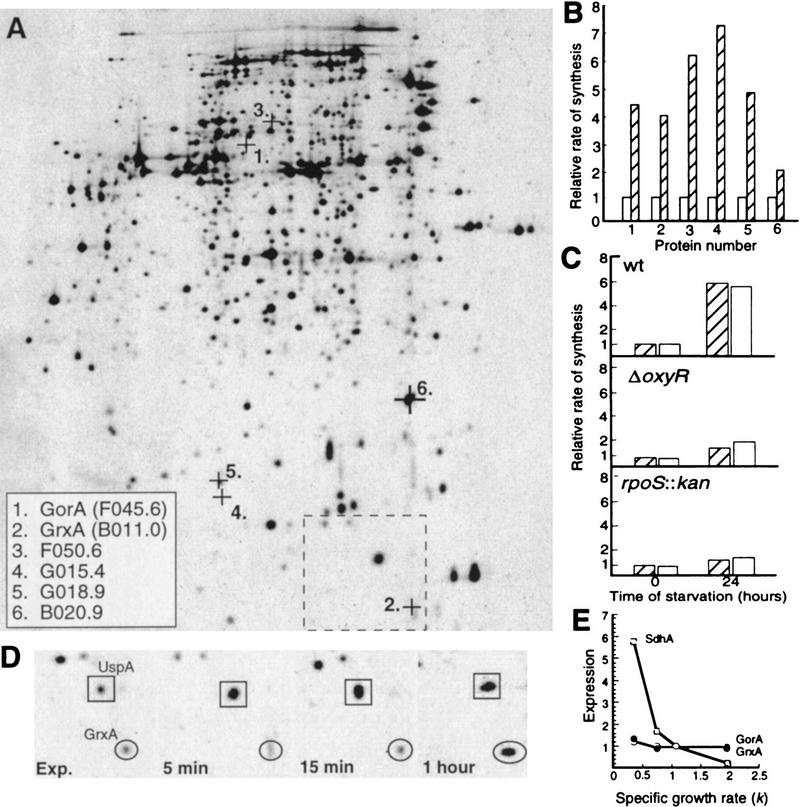
Synthesis of oxidative-defense proteins during starvation. (A) Location on the two-dimensional reference gel of the OxyR-dependent oxidative-stress proteins identified as inducible by carbon starvation. The alpha-numeric designations and protein names of identified proteins are shown in the inset box. The box with broken lines denotes the area shown in D. (B) Relative rate of synthesis of oxidative-stress proteins during growth (OD = 0.4 ± 0.05) (open bars) and after 16 hr carbon starvation (hatched bars). The rate of synthesis during exponential growth was assigned a value of 1.0. (C) Relative rate of GorA (hatched bars) and GrxA (open bars) synthesis during growth and starvation in wild-type, ΔoxyR, and rpoS::kan backgrounds. The rate of synthesis of GorA and GrxA during exponential growth in the wild-type background was assigned a value of 1.0. (D) Kinetics of GrxA synthesis in comparison to UspA during carbon starvation. (E) Relative rates of synthesis of GorA (•) and GrxA (○) in comparison to SdhA (□) at varying growth rates. The cultures were grown exponentially in minimal MOPS media supplemented with acetate, glycerol, or glucose as carbon sources and glucose plus amino acids, nucleotides, and vitamins for rich medium. The rates of protein synthesis are plotted relative to the rate of synthesis in glucose minimal media which was assigned a value of 1.0. Growth rates in the different media are expressed as k, the first-order growth-rate constant. The analysis was repeated three times to confirm reproducibility. Representative results are presented in the figure and the standard deviation was always <10% in the measurements of rates of individual protein synthesis.
The results, together with the fact that growth-arrested cells accumulate glutathione (Nyström 1995), indicate that the cell’s capacity to reduce cysteine disulfide bridges increases during stasis (Prinz et al. 1997). The question of whether this is caused by an increased need for these systems during growth arrest or if the described response prepares the cell for possible future oxidative conditions was approached by determining the occurrence of disulfide bridge formation in the cytoplasm as described below.
The activity of a cytoplasmic alkaline phosphatase increases during stasis
To determine the extent of disulfide bond formation in the cytoplasm during growth and stasis, the signal sequence-less Δ2-22 alkaline phosphatase (AP) was expressed and assayed for activity. Wild-type AP is a periplasmic homodimeric enzyme that contains two interchain disulfide bridges in each monomer that are required for enzymatic activity. The amino-terminal signal sequence targets AP to the periplasm, where the disulfide bonds are formed. The Δ2-22AP is deleted for its signal sequence and therefore remains in the cytoplasm (Prinz et al. 1997). Normally, the cytoplasm, being a reducing environment, does not support disulfide bond formation and Δ2-22AP can therefore be used to assess the potential for disulfide bond formation in this compartment during different environmental conditions and in different mutant backgrounds. Prinz et al. (1997) used this system to demonstrate that mutants missing components of the thioredoxin reductase and glutathione reductase systems allowed disulfide bridges to form in the cytoplasm of growing E. coli.
As depicted in Figure 2A, the Δ2-22AP activity of the wild-type WP551 strain increased significantly during a period of two days in stationary phase. To ensure that this activity was derived from the cytoplasmic compartment, the activity measurements were repeated after subjecting the cells to an osmotic shock. No significant reduction in Δ2-22AP activity was observed after this treatment (Fig. 2A). In contrast, the same treatment almost totally abolished wild-type AP activity, demonstrating that the periplasmic shock treatment worked properly (Fig. 2B). No increase in Δ2-22AP activity during growth arrest was observed when cells were grown and starved anaerobically or aerobically in the presence of the reductant DTT (4 mm) (Fig. 2A), demonstrating that an oxidative environment is required for the increased Δ2-22AP activity observed in stationary phase.
Figure 2.

(A) Δ2-22AP activity during growth (time zero) and in stationary phase (2 days). AP activity measured in aerobically grown cells was normalized to total cell mass (OD600) before (open bars) and after an osmotic shock treatment (hatched bars). Activity was measured also in cells grown and starved anaerobically (shaded bars) and aerobically in the presence of 4 mm DTT (black bars). (B) Wild-type AP activity before (open bars) and after an osmotic shock (hatched bars). (C) Determination of Δ2-22AP activity after separation of crude extract by gel filtration on Sephacryl HR 100. The relative levels of Δ2-22AP in the eluted fractions were determined by Western blotting using polyclonal antibodies against AP and quantification of Δ2-22AP bands was performed electronically using the ImageQuant (Molecular Dynamics) software. (D) Effect of DTT (4 mm) on the in vitro Δ2-22 AP activity. See text for details. The analysis was repeated three times to confirm reproducibility. Representative results are presented and the standard deviation was always <7% in the measurements of AP activity.
A crude extract of WP551 cells starved for two days was obtained using a French press and the proteins were subsequently separated using a Sephacryl S-100 HR (Pharmacia) gel-filtration column. As shown in Figure 2C, Δ2-22AP activity was detected in the fraction of Δ2-22AP containing the larger, presumably dimeric, and as expected, less abundant Δ2-22AP subpopulation. The smaller, presumably monomeric, Δ2-22AP subfraction possessed no AP activity (Fig. 2C). The Δ2-22AP activity of fraction 7 (Fig. 2C) was significantly reduced by the addition of 4 mm DTT (Fig. 2D), further demonstrating that the activity requires disulfide bonds.
gorA and rpoS but not oxyR mutants exhibit increased Δ2-22AP activity during stasis
Next, the Δ2-22AP activity was determined in mutant cells lacking functional glutathione reductase encoded by gorA. As depicted in Figure 3, the gorA mutant culture exhibited a slightly higher Δ2-22AP activity as compared to the wild-type parent in exponential growth but the difference was enhanced with the time in stationary phase (Fig. 3). Thus, the growth-phase-dependent increase in GorA synthesis appears to be physiologically relevant as cells lacking gorA are subjected to an increased oxidative disulfide bond formation during stasis. As described above, increased gorA expression (Becker-Hapak and Eisenstark 1995) and GorA synthesis (Fig. 1B) during stationary phase requires both functional σs and OxyR. Therefore, the effect of a oxyR and rpoS mutation on Δ2-22AP activity was determined during growth and stasis. As shown (Fig. 3), the rpoS but not oxyR mutant culture exhibited enhanced Δ2-22AP activity in stationary phase.
Figure 3.
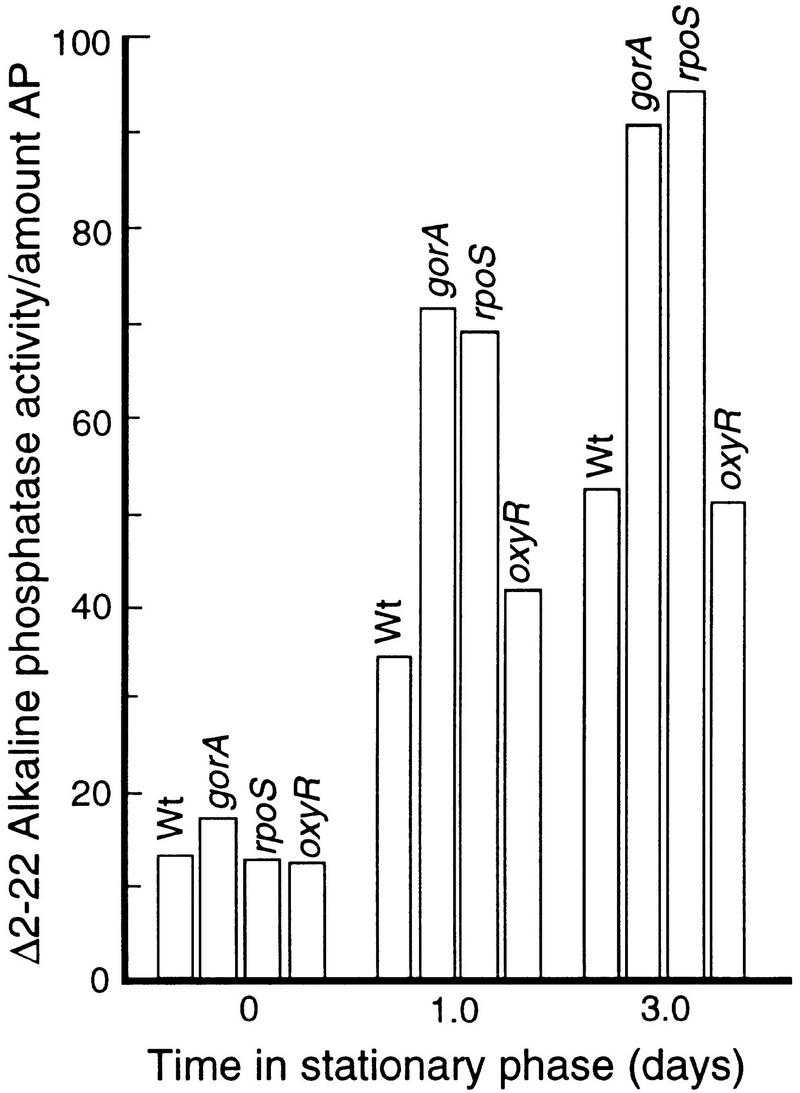
Δ2-22AP activity in wild-type, and gorA, oxyR, and rpoS mutants during growth (time zero) and stationary phase. All activity measurements were normalized to Δ2-22AP levels determined by Western blotting as described in Materials and Methods. The analysis was repeated three times to confirm reproducibility. The standard deviation was <8%.
The carbonyl content of proteins increase during stasis
To further determine if proteins were subjected to increased oxidative damage during stasis, an immunochemical assay for protein carbonyl groups was used. Carbonyl groups are introduced into protein side chains by site-specific oxidative modifications and protein carbonyl levels have been used to determine the oxidation status of proteins (e.g., Tamarit et al. 1998). Crude protein extracts were obtained during growth and at different times in stationary phase; the extracts reacted with the carbonyl reagent, DNPH; dot blotted onto a PVDF membrane; and oxidatively modified proteins were detected with anti-DNP antibodies. The carbonyl content of total proteins was measured densitometrically, which demonstrated a four- to fivefold increase in carbonyl content in wild-type cells during a period of 2 days in stationary phase (Fig. 4A,B). A gorA mutation did not affect the degree of carbonylation during either growth or stasis (Fig. 4A,B) indicating that GorA is specifically required for reduction of disulfide bonds rather than being a general oxidative-defense protein. However, both rpoS and oxyR mutants exhibited enhanced carbonyl content during stasis (Fig. 4A,B). No increase in carbonyl levels was detected when cells were grown and growth-arrested anaerobically (not shown).
Figure 4.
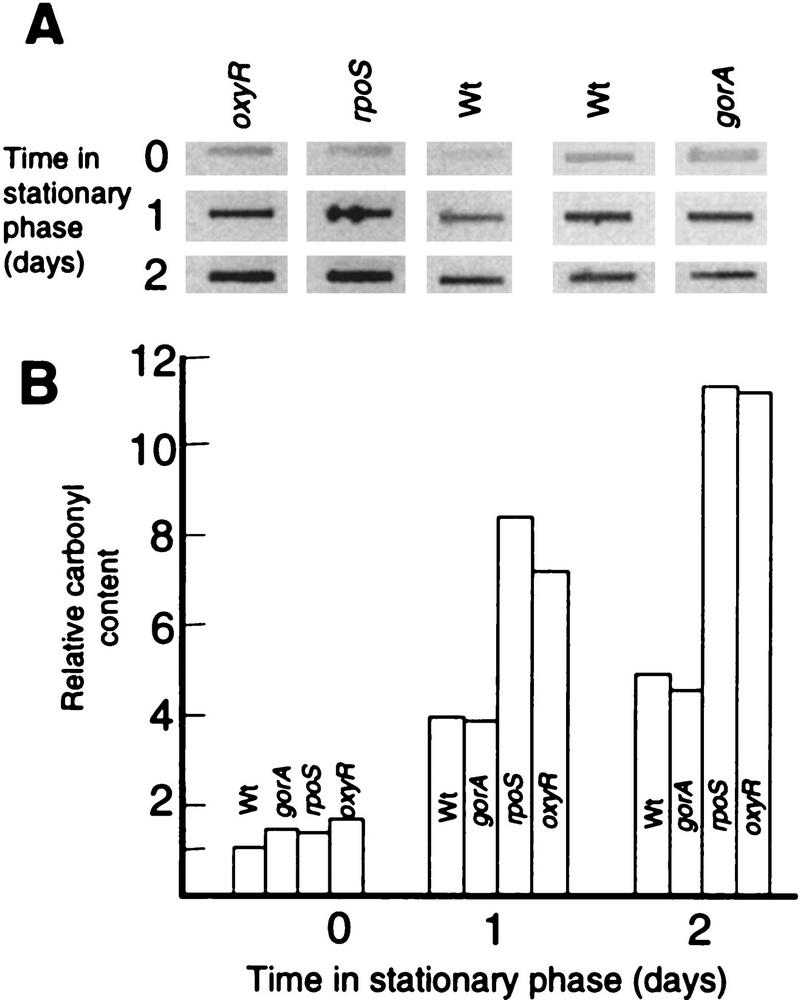
(A) Autoradiograph showing protein carbonyl levels in wild-type cells and different mutants, as indicated, during growth (time zero, OD = 0.5 ± 0.05) and stationary phase (1 and 2 days). Equal amounts of protein were loaded in each slot. (B) Quantification of carbonyl levels using the ImageQuant software (Molecular Dynamics). The analysis was repeated three times to confirm reproducibility. Representative results are presented.
Stasis-induced oxidative carbonylation targets specific proteins
Two-dimensional electrophoresis in combination with immunochemical assay for protein carbonyl groups demonstrated that some proteins are specifically susceptible to stasis-induced oxidation. Specifically, glutamate synthase, glutamine synthetase, pyruvate kinase, and the TCA cycle enzymes and subunits isocitrate dehydrogenase, dihydrolipoamide succinyltransferase, dihydrolipoamide acetyltransferase, succinyl CoA ligase, and malate dehydrogenase were found to be highly carbonylated (Fig. 5A). The identity of malate dehydrogenase as one of the carbonylated proteins was confirmed with amino-terminal sequencing yielding the sequence, MKVAVLGAAGI. As shown in Figure 5B, these proteins are not very abundant in the growth-arrested cell and we have previously demonstrated that TCA cycle enzymes are repressed markedly in stationary phase (Nyström 1994; Nyström et al. 1996). In addition, DnaK of the heat shock regulon was found to be markedly oxidized in response to stasis, whereas other members of the regulon, including GroEL, were not (it should be noted that the molar concentration of DnaK and GroEL are very similar under these conditions). Other proteins, including EF-Tu, FabB, PtsI, UspA, and H-NS were also found to be highly carbonylated. However, some of these proteins are very abundant in the growth-arrested cell (Fig. 5B) and it is therefore difficult to judge whether these proteins are specifically sensitive to oxidation.
Figure 5.

Specific protein carbonylation determined by two-dimensional Western blot immunoassay. (A) An autoradiograph obtained after carbonyl immunoassay of proteins from a wild-type E. coli culture starved for 1 day; (B) the same protein extract blotted to PVDF membrane and stained with Coomassie brilliant blue. (GltD) Glutamate synthase; (GlnA) glutamine synthetase; (Icd) isocitrate dehydrogenase; (SucB) dihydrolipoamide succinyltransferase; (Mdh) malate dehydrogenase; (AceF) dihydrolipoamide acetyltransferase; (SucC) succinyl CoA ligase; (PtsI) phosphoenolpyruvate–protein phosphotransferase; (Pyk) pyruvate kinase; (UspA) universal stress protein A; (FabB) β-ketoacyl-[acyl carrier protein] synthetase; (EF-Tu) elongation factor Tu. The analysis was repeated two times to confirm reproducibility. Representative results are presented.
Stasis-induced expression of heat shock genes is mitigated by overproducing the superoxide dismutase SodA
Aberrant proteins have been suggested to be the signals that induce the heat shock response (e.g., Bukau 1993) and the best inducers of this response, namely heat and ethanol, are thought to elicit the response by generating denatured, partially or completely unfolded proteins (e.g., Bukau 1993; Gross 1996). The fact that cytoplasmic proteins are increasingly oxidized during stasis made us hypothesize that this oxidation of target polypeptides may cause misfolding of proteins which, by titrating out the heat shock modulators, DnaK, DnaJ, and GrpE, elicit the stationary-phase induction of the regulon. To test this hypothesis, we measured the levels of DnaK and the induction of a groEL–lacZ fusion during stationary-phase conditions in the presence and absence of SodA overproduction, arguing that reducing superoxide levels may, at least partly, suppress induction of the regulon. Cells harboring a chromosomal groEL–lacZ fusion were transformed with plasmid pDT1-19 which is pBR322 carrying lacIq and an operon fusion between the tac promoter and the sodA gene. Cells were grown in presence of 0.1 mm MnCl2 (SodA requires Mn2+ for activity) and with or without 1 mm IPTG and samples were taken for measurements of β-galactosidase and DnaK levels. As expected groEL–lacZ was induced in stationary phase. Moreover, as shown in Figure 6, cells overexpressing sodA exhibited a markedly decreased induction of groEL (Fig. 6A) and levels of DnaK (Fig. 6B) in response to growth arrest. In addition, by measuring protein carbonyl levels it was demonstrated that overproduction of SodA mitigated the stationary-phase-induced increase in oxidative carbonylation of proteins (Fig. 6C). Finally, cells lacking cytoplasmic superoxide dismutase activity (sodA sodB double mutant) exhibit superinduction of groEL–lacZ (Fig. 6D) and DnaK (Fig. 6E). The decreased β-galactosidase activity in the sodA sodB mutant after 1 day in stationary phase may be caused by the poor ability of this double mutant to survive stasis. It has been shown that the die-off rate of sodA sodB mutants in early stationary phase (up to 3 days) is much higher than for the wild-type (Benov and Fridovich 1995).
Figure 6.
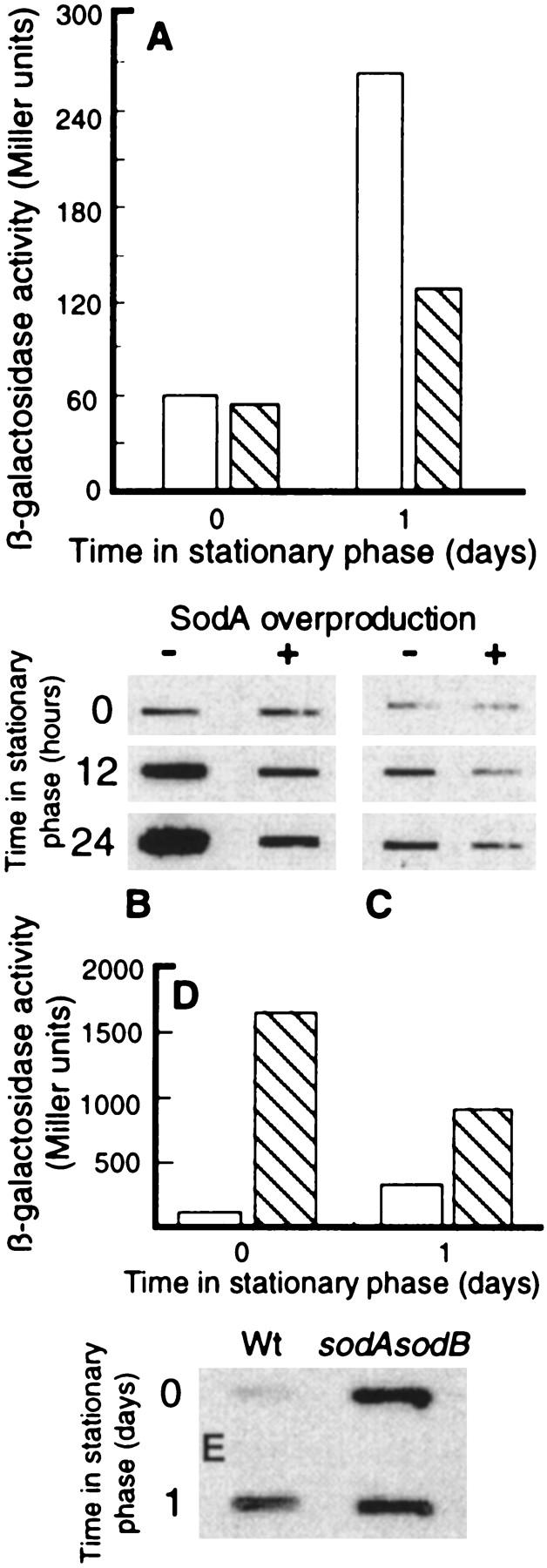
Effects of overproducing SodA on stasis-induced heat shock gene expression. (A) groEL promoter activity in the absence (open bars) and presence (hatched bars) of elevated SodA levels during growth (time zero, OD = 0.3 ± 0.05) and in stationary phase (day 1). Expression of GroEL was determined by measuring β-galactosidase activity in cells harboring a chromosomal groEL–lacZ fusion. (B) Levels of DnaK during growth (time zero) and at times in stationary phase in the absence or presence of elevated levels of SodA. The levels of DnaK was determined by Western blot analysis using monoclonal mouse anti-DnaK antibodies. (C) Protein carbonyl levels during growth (time zero) and at times in stationary phase in the absence or presence of elevated levels of SodA. Equal amounts of protein were loaded in each slot. (D) groEL promoter activity in the wild-type strain (open bars) and a sodA sodB double mutant (hatched bars) during growth (time zero, OD = 0.3 ± 0.05) and stationary phase (1 day). (E) Levels of DnaK during growth (time zero) and after 1 day of stationary phase in wild-type and sodA sodB double mutant strains. The levels of DnaK was determined by Western blot analysis using monoclonal mouse anti-DnaK antibodies. The analysis was repeated three times (Miller units) or two times (Western blot) to confirm reproducibility. Representative results are presented in the figure and the standard deviation was always <10%.
Discussion
Long-term growth-arrested cells, which have little ability to produce new polypeptides, must strive to maintain a pool of functional proteins. The increased levels of the heat shock proteins (Jenkins et al. 1988; Spence et al. 1990), l-isoaspartyl protein methyltransferase (Pcm) (Li and Clarke 1992), peptide methionine sulfoxide reductase (MsrA) (Moskovitz et al. 1995), glutathione (GSH), glutathione reductase (Nyström 1995), catalase HPII (Von Ossowski et al. 1991), and superoxide dismutase A (T. Nyström, unpubl.) during stasis indicate that the cell increases its ability to deal with oxidative denaturation and spontaneous aging of proteins. In addition, the ArcA-dependent reduced production of respiratory substrates and components of the aerobic respiratory apparatus during stasis may be another integral component of a defense system aimed at minimizing potential damaging effects of oxygen during stasis (Nyström et al. 1996). However, it is not clear whether these responses are devoted to avoiding and repairing damage by ROS during stasis or if the increased levels of the oxidative-defense proteins are rather aimed at increasing the ability of the cells to survive possible stresses that may be encountered in the future. However, the fact that protein oxidation, measured as disulfide bond formation of a cytoplasmic alkaline phosphatase and protein carbonyl levels, increase during stasis and that mutants of the rpoS and oxyR regulons exhibit enhanced carbonylation levels demonstrates that both these regulons are indeed involved in protecting the cell against endogenously derived oxidation of target macromolecules during stasis.
Disulfide bond formation and GorA synthesis in rpoS versus oxyR mutants deserves some comments. As demonstrated by two-dimensional gel analysis, GorA synthesis failed to increase during stasis in both an rpoS and oxyR mutant (this work). This is in accord with previous results demonstrating that stasis-induced gorA promoter activity requires both σs and OxyR (Becker-Hapak and Eisenstark 1995). However, GorA activity increases during stasis in an oxyR mutant but not an rpoS mutant (Becker-Hapak and Eisenstark 1995). This should be compared with the data demonstrating that disulfide bond formation during stasis was enhanced in gorA and rpoS but not oxyR mutants (Fig. 3). Taken together, the results suggest that GorA activity can increase during stasis in an σs-dependent fashion in the absence of increased gorA transcription (Becker-Hapak and Eisenstark 1995) and translation (this paper). In addition, Becker-Hapak and Eisenstark (1995) have shown that GorA does not confer general resistance to different oxidants. This is consistent with the fact that gorA mutants did not exhibit enhanced carbonylation indicating that this gene may be involved specifically with preventing disulfide bond formation in the cytoplasm, whereas other members of the oxyR and rpoS regulons are devoted to a general protection against protein oxidation. However, it should be recognized that the role of GorA by itself is difficult to interpret because the lack of GorA can be compensated for, at least partly, with an increased activity of the thioredoxin reductase (trxB) pathway (Prinz et al. 1997). For example, in a gorA trxB double mutant, Δ2-22AP activity increased rapidly during stasis to extremely high levels that were well above the sum of the activity levels in single mutants (not shown, but see Prinz et al. 1997). Also, these double mutants died rapidly in stationary-phase (not shown). Again, these results are difficult to interpret with respect to stationary-phase phenotypes since the trxB gorA double mutant also grows extremely poorly unless DTT is added to the medium (Prinz et al. 1997).
Whereas it appears clear that the oxyR and rpoS regulons are involved in a defense against oxidation of target macromolecules during stasis, it is equally clear that these two regulatory systems cannot fully prevent oxidation of proteins during growth arrest of cells under the conditions studied. Wild-type cells were found to exhibit gradually increased levels of active Δ2-22AP and protein carbonyl groups during stationary phase. This gradual increase in protein oxidation may help explain both the oxyR and rpoH dependent gene expression during stasis. It has been demonstrated that OxyR becomes active through the oxidative formation of a disulfide bond between two of its cysteine residues (Zheng et al. 1998). It is feasible that cysteine residues on OxyR, like Δ2-22AP, may be subjected to gradual oxidation during stasis, which eventually may give rise to a high enough titer of oxidized OxyR to activate gene expression. This would mechanistically explain the OxyR-dependent increase in gorA expression (Becker-Hapak and Eisenstark 1995), and GorA and GrxA synthesis (this work) during stasis.
Likewise, we suggest that the stationary phase, developmental, induction of heat shock genes (Jenkins et al. 1988; Nyström 1994) is connected to oxidative attack on target proteins. The best inducers of the heat shock response, namely heat and ethanol, are thought to elicit the response by generating aberrant, partially or completely unfolded proteins (e.g., Bukau 1993; Gross 1996). Thus, aberrant proteins have been suggested to be the signals that induce the heat shock response, a hypothesis that is strongly supported by the demonstration that the production of heterologous proteins, mutant proteins, or protein fragments induce a heat shock response (Goff and Goldberg 1985; Ito et al. 1986; Parsell and Sauer 1989). The demonstration that the levels of oxidized proteins increase during stasis may form the basis for heat shock induction during stationary phase. Presumably, this oxidation would result in increasing levels of denatured or misfolded proteins that may titrate out the heat shock modulators DnaJ, DnaK, and GrpE and allow an increased stability of σ32, whose levels have been demonstrated to increase during starvation-induced growth arrest (Jenkins et al. 1991). In addition, the observed oxidation of DnaK itself may well damage the protein and reduce its well-known ability to participate in the pathway leading to σ32 degradation (e.g., Bukau 1993; Gross 1996). In support of this oxidative-signaling model, we demonstrate here that stationary-phase induction of heat shock genes (and protein carbonylation) can be mitigated markedly by overproducing SodA. Thus, the sensor-signal mechanisms involved in induction of the heat shock regulon during heat shock and stationary-phase conditions may well be identical but the signal (aberrant proteins) is generated by different pathways. A schematic description of the model is depicted in Figure 7. The fact that cells lacking cytoplasmic superoxide dismutase activity exhibit superinduction of heat shock genes further substantiates the notion that oxidative attack of proteins may serve as a signal mechanism eliciting increased expression of the heat shock regulon.
Figure 7.

Schematic representation of the model for stasis-induced, developmental, induction of the heat shock regulon. The model is very much the same as the proposed signal-response transduction pathway for heat shock induction presented by Bukau (1993) with the addition of DnaJ, DnaK, and GrpE, as negative modulators of the response (for review, see Bukau 1993; Gross 1996). Central to this model is that the heat shock proteins DnaJ, DnaK, and GrpE, have dual functions; one is to aid refolding of denatured or misfolded proteins and the other is to promote proteolytic degradation of the heat shock regulator σ32. An increase in the cellular level of denatured proteins as a consequence of heat or ethanol treatment will sequester the DnaJ, DnaK, and GrpE components (provided at least one of them is limiting) through binding to their denatured substrate. This will allow stabilization of σ32, which will trigger increased transcription of the heat shock genes (e.g., Bukau 1993). During stasis, we propose that oxidative damage of target proteins will similarly sequester these heat shock modulators allowing induction of the regulon. This notion is supported by the fact that heat shock induction is markedly suppressed by overproducing SodA during stasis. In addition, the fact that DnaK appears to be specifically sensitive to oxidative carbonylation opens up the possibility that oxidative damage of DnaK itself may be involved in stasis-induced heat shock gene expression. Presumably, oxidation of DnaK will destroy or reduce its ability to participate in the degradation of σ32 which, in turn, will activate heat shock promoters.
It is interesting that some TCA cycle enzymes appear to be specifically susceptible to oxidative attack and it is tempting to speculate that this is physiologically significant. It may be argued that specific oxidation sensitivity of key enzymes of the TCA cycle will feedback control the rate of respiration so that when the rate of electron transport and ROS production becomes too high the oxidation, and presumably loss of activity, of TCA cycle enzymes will reduce respiration by limiting its substrate, NADH. [It should be noted that carbonylation of glutamine synthetase in vitro correlates well with a loss of enzyme activity (Levine 1983).] However, at this point it is difficult, if not impossible, to interpret whether specific sensitivity to oxidation is the result of design rather than necessity or chance. For example, it is possible that metal-catalyzed oxidation will be an intrinsic problem for all proteins containing, or being associated with, metals such as iron (see Fucci et al. 1983; Tamarit et al. 1998). In addition, the apparent sensitivity of some proteins may be caused by the fact that these proteins happen to be in close proximity to ROS-generating sites.
Finally, whereas this work demonstrates that the levels of oxidized proteins increase during stasis, it is not clear whether the life span of nongrowing wild-type E. coli populations is limited by oxidation-inflicted damage of target macromolecules such as proteins, lipids, or DNA. Reduced oxygen species may be one of the causal factors underlying the aging process in multicellular organisms and it has been demonstrated that overproduction of Sod alone, or together with a catalase, can significantly increase the maximal life span of some multicellular eukaryotic species (e.g., Tyler et al. 1993; Warner 1994). Similarly, it has been suggested that oxidative damage derived from endogenous metabolism may be a causal factor in bacterial self-destruction (Dodd et al. 1997). Also, it was demonstrated recently that oxidative damage during aging of the housefly targets the mitochondrial aconitase and that experimental manipulations that decrease aconitase activity also decreased the life span of the organism (Yan et al. 1997). To our knowledge, this is the first report demonstrating that bacterial stasis per se results in an increased and differential oxidation of target proteins. Furthermore, we believe that the identification of specific targets sensitive to oxidation in stationary phase reported here will greatly benefit our search for cellular functions associated with the senescence and deterioration of growth-arrested bacteria.
Materials and methods
Chemicals and reagents
Detection of carbonylated proteins was performed using the chemical and immunological reagents of the OxyBlot Oxidized Protein Detection Kit (Oncor). Antibodies (rabbit) against E. coli alkaline phosphatase were kindly provided by Lars Hederstedt (Lund University). Anti-DnaK monoclonal mouse antibodies were purchased from StressGen Biotechnologies Corp. Anti-mouse IgG peroxidase conjugates were from Sigma Immuno Chemicals. The chemiluminescence blotting substrate (POD) was obtained from Boehringer Mannheim and used according to instructions provided by the manufacturer. Immobilon-P polyvinylidene difluoride (PVDF) membrane was obtained from Millipore Corp. Protein assay reagents were from Pierce. All chemicals used for radiolabeling of proteins were from Amersham Corp. X-Omat AR-5 film was purchased from Eastman Kodak Co. The ampholines (Resolyte 4–8) used for two-dimensional electrophoresis were from BDH.
Bacterial strains and media
The E. coli K-12 strains used in this study are listed in Table 1. Cultures were grown aerobically or anearobically in liquid Luria–Bertani (LB) or glucose M9 (Miller 1972) medium in Erlenmeyer flasks in a rotary shaker. When appropriate, the medium was supplemented with kanamycin (50 μg/ml), carbenicillin (200 μg/ml), and/or tetracycline (20 μg/ml).
Table 1.
Strains and plasmids used in this work
| Bacterium or plasmid
|
Genotype or relevant characteristic(s)
|
Source
|
|---|---|---|
| E. coli strains | ||
| W3110 | F− IN[rrnD–rrnE]1 | F.C. Neidhardt (University of Michigan, Ann Arbor) |
| DHB4 | F′ lac–pro lacIQ/[ara–leu]7697 araD139 lacX74 galE galK rpsL phoR (phoA) PvuII malF3 thi | W. Prinz (Harvard University, Boston, MA) |
| WP551 | DHB4 + pAID135 | W. Prinz |
| WP552 | WP551 trxB::Km | W. Prinz |
| WP841 | WP551 gor522 mini-Tn10Tc | W. Prinz |
| TN5001 | WP551 rpoS::Km | this work |
| TN5002 | WP551 oxyR::Km | this work |
| SL396 | lacZ-350 galK2 galT22 rpsL179 λpF13 (PgroE::lacZ)F′ | C. Gross (UCSF Medical Center) |
| SD18 | SL396 Φ(sodA′–′lacZ)49 Φ(sodB–kan)Δ2 | this work |
| SD20 | SL396 katE::Tn10 katG::Tn10 | this work |
| SD22 | SD20 Φ(sodA′–′lacZ)49 Φ(sodB–kan)Δ2 | this work |
| SD16 | SL396 + pDT1-19 | this work |
| MG1655 | F− | D. Touati (University of Paris, France) |
| QC2413 | MG1655 oxyR::Km | D. Touati |
| QC2410 | MG1655 rpoS::Tn10 | D. Touati |
| Plasmids | ||
| pAID135 | Δ2-22AP under Ptac control | W. Prinz |
| pDT1–19 | pBR322 carrying the lacI gene and the SodA gene under Ptac control | D. Touati |
General methods
Plasmid DNA was purified using Qiagen columns according to the protocol provided by the manufacturer. P1 transductions and plasmid transformations were performed as described by Miller (1972) and Sambrook et al. (1989). Crude cell extracts were obtained using an SLM Aminco French Pressure Cell (SLM Instrument). Culture samples were processed to produce extracts for resolution on two-dimensional polyacrylamide gels by the methods of O’Farrell (1975) with modifications (VanBogelen and Neidhardt 1990). The two-dimensional gel apparatuses used for the O’Farrell methodology were that of Hoeffer Corp. and Millipore Corp. Separation of monomeric and dimeric Δ2-22AP by gel filtration was performed using a Sephacryl HR 100 (Pharmacia) column (2.6 × 95 cm). Proteins were eluted with 100 mm Tris (pH 8.0) containing 10 mm iodoacetamide at a flow rate of 0.66 ml/min. Fractions were collected at 5-min intervals. The presence of Δ2-22AP in the fractions were determined by Western blot analysis using polyclonal antibodies against AP. AP activity was measured as described below.
The β-galactosidase levels were measured as described by Miller (1972) with modifications (Albertson and Nyström 1994). Samples were measured spectrophotometrically at 420 nm (β-galactosidase). The β-galactosidase activity is expressed as follows: (OD420)/(OD600 culture) × (reaction time).
Amino-terminal sequencing of proteins recovered from two-dimensional gels was performed as described previously (Nyström et al. 1996).
Alkaline phosphatase assays
The strains were grown at 37°C in LB medium containing 200 μg/ml carbenicillin, and 5 mm isopropyl-1-thio-d-galactopyranoside (IPTG) to induce expression of alkaline phosphatase Δ2-22AP. Cells were washed twice in M9 salts (20) containing 10 mm iodoacetamide and subjected subsequently to an osmotic release of periplasmic proteins as described (Nossal and Heppel 1966) to eliminate measurement of possible activity from Δ2-22AP that may have leaked to the periplasm. After osmotic shock, the cells were centrifuged, resuspended, and assayed for AP activity as described (Prinz et al. 1997). Alkaline phosphatase activity was normalized to the actual levels of Δ2-22AP in the cells, which was determined using Western blot analysis with antibodies (rabbit) against AP and secondary goat anti-rabbit immunoglobin G-horseradish peroxidase conjugate antibodies. For quantification, autoradiograms were scanned using a UMAX S-12. Quantification was performed electronically using the ImageQuant (Molecular Dynamics) software. ΔAP activity is expressed using the formula: OD420/reaction time (min) × relative quantity ΔAP × volume (ml). The relative quantity of Δ2-22AP in exponentially growing wild-type W551 cells was assigned a value of 1.0.
Carbonylation assays
The carbonyl groups in the protein side chains were derivatized, using the Oncor OxyBlot kit, to 2,4-dinitrophenylhydrazone (DNP-hydrazone) by reaction with 2,4-dinitrophenylhydrazine. The DNP-derivatized crude protein extracts were dot blotted onto PVDF membrane filters. The filters were incubated with primary antibody, specific to the DNP moiety of the proteins, and subsequently incubated with secondary (goat anti-rabbit) horseradish peroxidase-antibody conjugate directed against the primary antibody. Filters were subsequently treated with the chemiluminescence blotting substrate (POD) for detection. To detect specific carbonylated proteins, DNP-derivatized proteins were separated by two-dimensional SDS–polyacrylamide gel electrophoresis and transferred subsequently to PVDF membranes by using a semidry blotting system. For immunodetection, the same protocol was used as described above for dot-blotted protein extracts.
Measurement of rates of synthesis of individual proteins
A portion (1 ml) of a culture was removed and placed in a flask containing [3H]leucine (5 mCi/mmole, 100 μCi/ml). Incorporation was allowed to proceed for different lengths of time after which nonradioactive leucine (2.4 mm) was added for a chase. To this sample was added a portion of a culture of the same strain grown in [35S]methionine labeling medium as described (Nyström and Neidhardt 1992). The mixed sample was then prepared as described previously (VanBogelen and Neidhardt 1990) to produce extracts for resolution of the cellular proteins on two-dimensional gels. An autoradiogram was prepared to permit visualization of labeled proteins. Protein spots chosen for quantitative assay were sampled from the dried gel with a syringe needle and treated as described (Pedersen et al. 1976) to permit measurement of their 3H and 35S content by scintillation counting. The differential rate of synthesis of a sampled protein was defined as the 3H/35S ratio of the sampled spot divided by the same isotope ratio of unfractionated mixed extracts. The identifications of GorA and GrxA were performed by comigration with the purified proteins.
Acknowledgments
We thank Daniele Touati and Anne Farewell for strains, plasmids, and discussions throughout this work. Will Prinz, Fredrik Åslund, and Carol Gross are gratefully acknowledged for providing strains, plasmids, and purified GrxA. We thank Lars Hederstedt for providing AP antibodies and for valuable suggestions on this work and Mimmi Throne-Holst and Jenny Bengtsson for technical assistance. This work was supported by the Swedish Natural Science Research Council (NFR) to T.N. and by Suez-Lyonnaise des Eaux and a Federation of European Microbiological Societies fellowship to S.D.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked ‘advertisement’ in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL Thomas.Nystrom@mikrbiol.lu.se; FAX 46 046 157839.
References
- Albertson NH, Nyström T. Effects of starvation for exogenous carbon on functional mRNA stability and rate of peptide chain elongation in Escherichia coli. FEMS Microbiol Lett. 1994;117:181–187. doi: 10.1111/j.1574-6968.1994.tb06762.x. [DOI] [PubMed] [Google Scholar]
- Almiron M, Link AJ, Furlong D, Kolter R. A novel DNA-binding protein with regulatory and protective roles in starved Escherichia coli. Genes & Dev. 1992;6:2646–2654. doi: 10.1101/gad.6.12b.2646. [DOI] [PubMed] [Google Scholar]
- Amici A, Levine RL, Tsai L, Stadtman ER. Conversion of amino acid residues in proteins and amino acid homopolymers to carbonyl derivatives by metal-catalyzed oxidation reactions. J Biol Chem. 1989;264:3341–3346. [PubMed] [Google Scholar]
- Becker-Hapak M, Eisenstark A. Role of rpoS in the regulation of glutathione oxidoreductase (gor) in Escherichia coli. FEMS Microbiol Lett. 1995;134:39–44. doi: 10.1111/j.1574-6968.1995.tb07911.x. [DOI] [PubMed] [Google Scholar]
- Benov L, Fridovich I. A superoxide dismutase mimic protects sodA sodB Escherichia coli against aerobic heating and stationary-phase death. Arch Biochem Biophys. 1995;322:291–294. doi: 10.1006/abbi.1995.1465. [DOI] [PubMed] [Google Scholar]
- Berlett BS, Stadtman ER. Protein oxidation in aging, disease, and oxidative stress. J Biol Chem. 1997;272:20313–20316. doi: 10.1074/jbc.272.33.20313. [DOI] [PubMed] [Google Scholar]
- Bukau B. Regulation of the Escherichia coli heat-shock response. Mol Microbiol. 1993;9:671–680. doi: 10.1111/j.1365-2958.1993.tb01727.x. [DOI] [PubMed] [Google Scholar]
- Christman MF, Morgan RW, Jacobson FS, Ames BN. Positive control of a regulon for defense against oxidative stress and some heat shock proteins in Salmonella typhimurium. Cell. 1985;41:753–762. doi: 10.1016/s0092-8674(85)80056-8. [DOI] [PubMed] [Google Scholar]
- Cooper WJ, Zika RG. Photochemical formation of hydrogen peroxide in surface and ground waters exposed to sunlight. Science. 1983;220:711–712. doi: 10.1126/science.220.4598.711. [DOI] [PubMed] [Google Scholar]
- Davies KJA. Protein damage and degradation by oxygen radicals. J Biol Chem. 1987;262:9895–9901. [PubMed] [Google Scholar]
- Demple B, Halbrook J. Inducible repair of oxidative damage in Escherichia coli. Nature. 1983;304:466–468. doi: 10.1038/304466a0. [DOI] [PubMed] [Google Scholar]
- Dodd CER, Sharman RL, Bloomfield SF, Booth IR, Stewart GSAB. Inimical processes: Bacterial self-destruction and sub-lethal injury. Trends Food Sci Technol. 1997;8:238–241. [Google Scholar]
- Farber JM, Levine RL. Sequence of a peptide susceptible to mixed-function oxidation. Probable cation binding site in glutamine synthetase. J Biol Chem. 1986;261:4574–4578. [PubMed] [Google Scholar]
- Fridovich I. The biology of oxygen radicals. The superoxide radical is an agent of oxygen toxicity: Superoxide dismutases provide an important defense. Science. 1978;201:875–880. doi: 10.1126/science.210504. [DOI] [PubMed] [Google Scholar]
- Fucci L, Oliver CN, Coon MJ, Stadtman ER. Inactivation of key metabolic enzymes by mixed-function oxidation reactions: Possible implication in protein turnover and aging. Proc Natl Acad Sci. 1983;80:1521–1525. doi: 10.1073/pnas.80.6.1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goff SA, Goldberg AL. Production of abnormal proteins in E. coli stimulates transcription of lon and other heat shock genes. Cell. 1985;41:587–595. doi: 10.1016/s0092-8674(85)80031-3. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Flecha B, Demple B. Metabolic source of hydrogen peroxide in aerobically growing Escherichia coli. J Biol Chem. 1995;270:13681–13687. doi: 10.1074/jbc.270.23.13681. [DOI] [PubMed] [Google Scholar]
- Greenberg JT, Monach P, Chou JH, Josephy PD, Demple B. Positive control of a global antioxidant defense regulon activated by superoxide-generating agents in Escherichia coli. Proc Natl Acad Sci. 1990;87:6181–6185. doi: 10.1073/pnas.87.16.6181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross CA. In: Function and regulation of the heat shock proteins. In Escherichia coli and Salmonella: Cellular and molecular biology. Neidhardt FC, editor. Washington, D.C.: ASM Press; 1996. pp. 1382–1399. [Google Scholar]
- Halliwell B. Superoxide dismutase, catalase and glutathione peroxidase: Solutions to the problems of living with oxygen. New Phytol. 1974;73:1075–1086. [Google Scholar]
- Halliwell B, Gutteridge JMC. Oxygen toxicity, oxygen radicals, transition metals and disease. Biochem J. 1984;219:1–14. doi: 10.1042/bj2190001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hengge-Aronis R. Survival of hunger and stress: The role of rpoS in early stationary phase gene regulation in E. coli. Cell. 1993;72:165–168. doi: 10.1016/0092-8674(93)90655-a. [DOI] [PubMed] [Google Scholar]
- Ito K, Akiyama Y, Yura T, Shiba K. Diverse effects of the MalE-LacZ hybrid protein on Escherichia coli cell physiology. J Bacteriol. 1986;167:201–204. doi: 10.1128/jb.167.1.201-204.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins DE, Schultz JE, Matin A. Starvation-induced cross protection against heat or H2O2 challenge in Escherichia coli. J Bacteriol. 1988;170:3910–3914. doi: 10.1128/jb.170.9.3910-3914.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins DE, Auger EA, Matin A. Role of RpoH, a heat shock regulator protein, in Escherichia coli carbon starvation protein synthesis and survival. J Bacteriol. 1991;173:1992–1996. doi: 10.1128/jb.173.6.1992-1996.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolter R, Siegele DA, Tormo A. The stationary phase of the bacterial life cycle. Annu Rev Microbiol. 1993;47:855–874. doi: 10.1146/annurev.mi.47.100193.004231. [DOI] [PubMed] [Google Scholar]
- Lange R, Hengge-Aronis R. Identification of a central regulator of stationary phase gene expression in Escherichia coli. Mol Microbiol. 1991;5:49–59. doi: 10.1111/j.1365-2958.1991.tb01825.x. [DOI] [PubMed] [Google Scholar]
- Levine RL. Oxidative modification of glutamine synthetase. J Biol Chem. 1983;258:11823–11827. [PubMed] [Google Scholar]
- Levine RL, Mosoni L, Berlett BS, Stadtman ER. Methionine residues as endogenous antioxidants in proteins. Proc Natl Acad Sci. 1996;93:15036–15040. doi: 10.1073/pnas.93.26.15036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Clarke S. A protein methyltransferase specific for altered aspartyl residues is important in Escherichia coli stationary-phase survival and heat-shock resistance. Proc Natl Acad Sci. 1992;89:9885–9889. doi: 10.1073/pnas.89.20.9885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez A, Kolter R. Protection of DNA during oxidative stress by the nonspecific DNA-binding protein Dps. J Bacteriol. 1997;179:5188–5194. doi: 10.1128/jb.179.16.5188-5194.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCann MP, Kidwell JP, Matin A. The putative sigma factor KatF has a central role in development of starvation-mediated general resistance in Escherichia coli. J Bacteriol. 1991;173:4188–4194. doi: 10.1128/jb.173.13.4188-4194.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller J. Experiments in molecular genetics. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory; 1972. [Google Scholar]
- Moskovitz J, Rahman MA, Strassman J, Yancey SO, Kushner SR, Brot N, Weissbach H. Escherichia coli peptide methionine sulfoxide reductase gene: Regulation of expression and role in protecting against oxidative damage. J Bacteriol. 1995;177:502–507. doi: 10.1128/jb.177.3.502-507.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nossal NG, Heppel LA. The release of enzymes by osmotic shock from Escherichia coli in exponential phase. J Biol Chem. 1966;241:3055–3062. [PubMed] [Google Scholar]
- Nyström T. The glucose starvation stimulon of Escherichia coli: Induced and repressed synthesis of enzymes of central metabolic pathways and the role of acetyl phosphate in gene expression and starvation survival. Mol Microbiol. 1994;12:833–843. doi: 10.1111/j.1365-2958.1994.tb01069.x. [DOI] [PubMed] [Google Scholar]
- ————— The glucose-starvation stimulon of Escherichia coli: Role of integration host factor in starvation survival and growth-phase-dependent proteins synthesis. J Bacteriol. 1995;177:5707–5710. doi: 10.1128/jb.177.19.5707-5710.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyström T, Neidhardt FC. Cloning, mapping and nucleotide sequencing of a gene encoding a universal stress protein in Escherichia coli. Mol Microbiol. 1992;6:3187–3198. doi: 10.1111/j.1365-2958.1992.tb01774.x. [DOI] [PubMed] [Google Scholar]
- Nyström T, Larsson C, Gustafsson L. Bacterial defense against aging: Role of the Escherichia coli ArcA regulator in gene expression, readjusted energy flux and survival during stasis. EMBO J. 1996;15:3219–3228. [PMC free article] [PubMed] [Google Scholar]
- O’Farrel PH. High resolution two-dimensional electrophoresis of proteins. J Biol Chem. 1975;250:4007–4021. [PMC free article] [PubMed] [Google Scholar]
- Parsell DA, Sauer RT. Induction of a heat shock-like response by unfolded protein in Escherichia coli: Dependence on protein level not protein degradation. Genes & Dev. 1989;3:1226–1232. doi: 10.1101/gad.3.8.1226. [DOI] [PubMed] [Google Scholar]
- Pedersen S, Reeh SV, Parker J, Watson RJ, Friesen JD, Fiil NP. Analysis of the proteins synthesized in ultraviolet light-irradiated Escherichia coli following infection with the bacteriophage λdrifd18 and λdfus-3. Mol & Gen Genet. 1976;144:2205–2213. doi: 10.1007/BF00341733. [DOI] [PubMed] [Google Scholar]
- Petasne RG, Zika RG. Fate of superoxide in coastal sea water. Nature. 1987;325:516–518. [Google Scholar]
- Prinz WA, Åslund F, Holmgren A, Beckwith J. The role of the thioredoxin and glutaredoxin pathways in reducing protein disulfide bonds in the Escherichia coli cytoplasm. J Biol Chem. 1997;272:15661–15667. doi: 10.1074/jbc.272.25.15661. [DOI] [PubMed] [Google Scholar]
- Reeve CA, Bockman AT, Matin A. Role of protein synthesis in the survival of carbon-starved Escherichia coli K-12. J Bacteriol. 1984;160:1041–1046. doi: 10.1128/jb.160.3.1041-1046.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivett AJ, Levine RL. Metal-catalysed oxidation of Escherichia coli glutamine synthetase: Correlation of structural and functional changes. Arch Biochem Biophys. 1990;278:26–34. doi: 10.1016/0003-9861(90)90226-o. [DOI] [PubMed] [Google Scholar]
- Rockabrand D, Livers K, Austin T, Kaiser R, Jensen D, Burgess R, Blum P. Roles of DnaK and RpoS in starvation-induced thermotolerance of Escherichia coli. J Bacteriol. 1998;180:846–854. doi: 10.1128/jb.180.4.846-854.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sak BD, Eisenstark A, Touati D. Exonuclease III and the catalase hydroperoxidase II in Escherichia coli are both regulated by the katF gene product. Proc Natl Acad Sci. 1989;86:3271–3275. doi: 10.1073/pnas.86.9.3271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: A laboratory manual. 2nd ed. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- Spence J, Cegielska A, Georgopoulos C. Role of Escherichia coli heat shock proteins DnaK and HtpG (C62.5) in response to nutritional deprivation. J Bacteriol. 1990;172:7157–7166. doi: 10.1128/jb.172.12.7157-7166.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadtman ER. Protein oxidation and aging. Science. 1992;257:1220–1224. doi: 10.1126/science.1355616. [DOI] [PubMed] [Google Scholar]
- Tamarit J, Cabiscol E, Ros J. Identification of the major oxidatively damaged proteins in Escherichia coli cells exposed to oxidative stress. J Biol Chem. 1998;273:3027–3032. doi: 10.1074/jbc.273.5.3027. [DOI] [PubMed] [Google Scholar]
- Tsaneva IR, Weiss B. soxR, a locus governing a superoxide response regulon in Escherichia coli K-12. J Bacteriol. 1990;172:4197–4205. doi: 10.1128/jb.172.8.4197-4205.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyler RH, Bar H, Singh M, Latorre A, Graves JL, Mueller LD, Rose MR, Ayala FJ. The effect of superoxide dismutase alleles on aging in Drosophila. Genetica. 1993;91:143–149. doi: 10.1007/BF01435994. [DOI] [PubMed] [Google Scholar]
- VanBogelen RA, Neidhardt FC. Ribosomes as sensors of heat and cold shock in Escherichia coli. Proc Natl Acad Sci. 1990;87:5589–5593. doi: 10.1073/pnas.87.15.5589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanBogelen RA, Hutton ME, Neidhardt FC. Gene–protein database of Escherichia coli K-12: Edition 3. Electrophoresis. 1990;11:1131–1166. doi: 10.1002/elps.1150111205. [DOI] [PubMed] [Google Scholar]
- Von Ossowski I, Melvey MR, Leco PA, Borys A, Loewen PC. Nucleotide sequence of Escherichia coli katE, which encodes catalase HPII. J Bacteriol. 1991;173:514–520. doi: 10.1128/jb.173.2.514-520.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warner HR. Superoxide dismutase, aging, and degenerative disease. Free Radicals Biol Med. 1994;17:249–258. doi: 10.1016/0891-5849(94)90080-9. [DOI] [PubMed] [Google Scholar]
- Yan LJ, Levine RL, Sohal RS. Oxidative damage during aging targets mitochondrial aconitase. Proc Natl Acad Sci. 1997;94:11168–11172. doi: 10.1073/pnas.94.21.11168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng M, Åslund F, Storz G. Activation of the OxyR transcription factor by reversible disulfide bond formation. Science. 1998;279:1718–1721. doi: 10.1126/science.279.5357.1718. [DOI] [PubMed] [Google Scholar]