

Summary
The importance of proresolving mediators in the overall context of resolution of acute inflammation is accepted and prominent, though little is known on whether these anti-inflammatory and pro-resolving molecules act in concert. Here we focused on lipoxin A4 (LXA4) and annexin A1 (AnxA1) since these two very different mediators converge on a single receptor, formyl peptide receptor type 2 (acronym FPR2/ALX). Addition of LXA4 to human PMN provoked a concentration- and time-dependent mobilization of AnxA1 onto the plasma membrane, as determined by western blotting and flow cytometry analyses. This property was shared by another FPR2/ALX agonist, antiflammin-2 (AF2), and partly by fMLP or peptide Ac2-26 (an AnxA1 derivative which can activate all three members of the human FPR family). An FPR2/ALX antagonist blocked AnxA1 mobilization activated by LXA4 and AF2. Analysis of PMN degranulation patterns and phospho-AnxA1 status suggested a model where the two FPR2/ALX agonists mobilize the cytosolic (and not the granular) pool of AnxA1 through an intermediate phosphorylation step. Intravital microscopy investigations of the inflamed mesenteric microvasculature of wild type and AnxA1−/− mice revealed that LXA4 provoked leukocyte detachment from the post-capillary venule endothelium in the former (>50% within 10 min; P<0.05), but not the latter genotype (~15%; NS). Furthermore, recruitment of Gr1+ cells into dorsal air pouches, inflamed with IL-1β, was significantly attenuated by LXA4 in wild type, but not AnxA1−/− mice. Collectively, these data prompt us to propose the existence of an endogenous network in anti-inflammation centred on PMN AnxA1 and activated by selective FPR2/ALX agonists.
Keywords: Anti-inflammatory G-protein coupled receptor, Lipoxin A4, Antiflammin, Leukocyte Adhesion, Neutrophil Activation, Intravital Microscopy
Introduction
It is now appreciated that the process of acute inflammation relies on the active involvement of a series of pro-resolving mediators which assure temporal and spatial containment of the host reaction1. Therefore, a pro-inflammatory phase is followed by an anti-inflammatory and pro-resolving phase in line with “the beginning programmes the end” concept recently put forward2. However, whereas it is well accepted that pro-inflammatory mediators (eg. cytokines, chemokines, autacoids) often act in concert in a network-like fashion3, the significance of such network(s) to inflammatory resolution has been poorly appreciated and specific mediators/pathways have often been investigated in isolation.
Lipoxin A4 (LXA4)2 and Annexin A1 (AnxA1) are two effectors of endogenous anti-inflammation4 being able to halt leukocyte migration5, 6 and promote macrophage phagocytosis of infective agents as well as apoptotic leukocytes7,8,9. In 2002, we reported that this short-lived lipid, produced by trans-cellular synthesis and cooperation between lipoxygenases10, and the glucocorticoid-regulated protein both converged on a specific receptor target11, the formyl peptide receptor type 2 (the acronym FPR2/ALX is currently used to identify the human receptor12).
LXA4 and AnxA1 are very different not only chemically, but also with respect to their biosynthesis (produced on demand or stored in cell sources) and metabolic fate; we questioned whether there was a functional link between them, that justified a single receptor being the main transducer for their, essentially similar, pro-resolving properties. In addition, comparing the time course of generation of these two mediators during acute inflammation in mice and men reveals distinct windows for synthesis and secretion, with exudate LXA4 concentrations peaking at a much earlier time-point than AnxA1. In murine air-pouches, inflamed with the TLR2 agonist zymosan, LXA4 peaks at 4 h whereas exudate AnxA1 expression is highest at 24 h, coinciding with the peak of polymorphonuclear cells (PMN) influx11. The same holds true in the zymosan peritonitis model13,14. Furthermore, 15-epi-lipoxin induced by low-dose aspirin treatment inhibits leukocyte recruitment in a human skin blister model of acute inflammation15; in the same model, a delay in the onset of the inflammatory response, seen in ~40% of individuals, is associated with abnormal production of 15-epi-lipoxin16.
AnxA1 is very abundant in human PMN, representing between 2 and 4% of total intracellular proteins17. In resting PMN, a proportion (~60%) of intracellular AnxA1 is localized in gelatinase and azurophilic granules18,19, with the remaining being cytosolic in location. PMN adhesion to endothelial cell monolayers provokes a rapid mobilization of AnxA1 onto the cell surface20, where this agonist could encounter its receptor to activate juxtacrine and/or autocrine signals6, thus curtailing the extent of neutrophil trafficking21. Nonetheless, the cytosolic non-granular pool of the protein can also be mobilized, for instance in response to drug application22-24, and the latter process entails a phosphorylation step at Serine 27 (and possibly other residues) which seems to be a pre-requisite for secretion of the protein onto the cell surface25.
Antiflammin 2 (AF2) is a nonapeptide corresponding to region 246-254 of AnxA126. In experimental models of acute inflammation, AF2 produces inhibitory properties which, at variance from initial observations27, are not associated with inhibition of phospholipase A2 but rather to an interference with PMN activation, chemotaxis and adhesion to endothelial cells28, 29. In the AnxA1 protein structure, residues 246-254 are exposed on the outer side of the protein30 hence available for interaction, in conjunction with the N-terminal region31, with molecular targets. Congruent with this model, we found that AF2 binds to FPR2/ALX, eliciting genuine post-receptor signaling responses and inhibiting PMN interaction with endothelial monolayer under flow32.
The present study was undertaken to determine whether LXA4 and AF2 might affect AnxA1 localization in human PMNs. The data obtained indicate the existence of an anti-inflammatory network centred on FPR2/ALX that operates to regulate PMN activation and trafficking in the microcirculation.
Materials and methods
Human PMN isolation
Peripheral blood was collected from healthy volunteers by intravenous withdrawal in 3.2% sodium citrate solution (1:10). PMNs were isolated from blood by density centrifugation on Histopaque 1119/1077 (Sigma-Aldrich, Poole, UK) gradient according to the manufacturer’s instructions and suspended in PBS containing 0.5% BSA. Contaminating erythrocytes were removed by lysis with cold milliQ water. All healthy volunteers gave oral and written consent and cell separation was covered by ethical approval 05/Q0603/34 (East London and The City Research Ethics Committee 1).
PMN incubation with formyl-peptide receptor agonists
To monitor AnxA1 mobilization, peptide Ac2-26 (Ac-AMVSEFLKQAWFIENEEQEYVQTVK; Tocris, Bristol, UK), AF2 (HDMNKVLDL, Tocris, Bristol, UK) LXA4 (EMD chemicals, Darmstadt, Germany) or fMLP (Sigma-Aldrich, Dorset, UK) were added to freshly prepared PMNs (4×106 cells/well) for 10-60 min; incubations were then stopped by rapid centrifugation at 4°C and freezing. In some experiments, PMNs were stimulated with selected agonists in presence of the FPR1 antagonist cyclosporin H (CsH, −10min; Biomol GmbH, Hamburg, Germany), which was used at 10 μM as recently reported 33, 34. In other cases, the FPR2/ALX antagonist WRWWWW35 (WRW4, 10μM, −10 min; EMD chemicals, Darmstadt, Germany) was used. Altogether, these experiments aimed at investigating the involvement of FPR1 and FPR2/ALX in PMN AnxA1 mobilization after application of the four agonists used in the present study. Finally, in order to measure rapid formation of phospho-Serine27-AnxA1, PMNs were incubated with selected agonists for 10 min. In all cases, cell-free supernatants were collected with cells being processed for membrane and cytosolic fractions. AnxA1 localization and expression was monitored by Western blotting and FACS analysis (see below).
Isolation of PMN membrane (Mem) and cytosolic (Cyt) fractions
After treatment, cells (4×106) were re-suspended in lysis buffer (20 mM Tris-HCl, pH 7.5, with a protease and phosphatase inhibitor cocktail; Sigma-Aldrich, Dorset, UK) and processed as previously shown36. Briefly, 100μl of lysis buffer have been used and the cells were sheared through a 25-gauge needle ten times. Lysates were subsequently centrifuged for 2 min at 300xg, and supernatants centrifuged again for 45 min at 800 x g. The resultant supernatant (cytosolic fraction, Cyt) was collected and the remaining pellet re-suspended in 20 mM Tris-HCl, 1% Triton X-100, pH 7.5 (100 μl) for 15 min (membrane fraction, Mem). Membrane-bound AnxA1 was recovered by washing pelleted cells twice with 50 μl of 1 mM EDTA.
Western blot analysis
Samples boiled in 6x Laemmli buffer were subjected to standard SDS-polyacrylamide gel electrophoresis (12%) and electrophoretically blotted onto polyvinylidene difluoride membranes (PVDF, Millipore, Watford, UK). Membranes were incubated with mouse monoclonal antibodies (mAb) anti-human AnxA1 (clone 1B; dilution 1:1,00037) and anti-human actin (1:10,000; Sigma-Aldrich) in Tris-buffer saline solution containing 0.1% Tween 20 (TBST) and 5% (w/v) non-fat dry milk overnight at 4°C. Membranes were washed for 30 min with TBST with the solution being changed at 10-min intervals; membranes were then incubated with secondary antibody (HRP-conjugated goat anti-mouse 1:5000, Dako, Cambridge, UK), for 2 h at room temperature. Phospho-Ser27-AnxA1 was detected with rabbit Ser27 phosphospecific antibody (kind gift from Dr. Egle Solito, 1:1000 in TBST, 1% BSA solution23, 25). Proteins were then detected using the ECL detection kit and visualized on Hyperfilm (Amersham Biosciences, Amersham, UK).
FACS Analysis
Granule markers and FPR receptors
PMN were treated with the different FPR agonists, as described above, and incubated with mAb against the different granule/vesicle markers CD35, CD66b, CD63 (all FITC-conjugated monoclonal antibodies, 1:10 final dilution; Serotec, Abingdon, UK). CD35 recognizes secretory vesicles, CD66b is a marker for specific granules and CD63 is a specific marker for azurophilic granules. These intracellular organelles are mobilized in a sequential manner by activated PMNs38,39.
FPR1 and FPR2/ALX expression
FPR1 and FPR2/ALX cell surface expression was measured by incubation with anti-FPR1 or anti-FPR2 mAb (1:10 dilution in both cases; from R&D System, Abingdon, UK and Genovac, Brussels, Belgium, respectively); a final staining with a rabbit anti-mouse IgG (1:200 dilution, clone STAR9B; Serotec) was then conducted.
Flow cytometry analysis was performed analyzing ≥10,000 events using a FACScalibur flow cytometer (Becton Dickinson, San José, CA) using CellQuest software (Becton Dickinson). Results are reported as median fluorescence intensity (MFI) units.
In vivo analyses in AnxA1 deficient mice
AnxA1 deficient (AnxA1−/−)40 and wild type C57BL/6J (AnxA1+/+) mice were obtained from B&K Limited (Hull, UK). Male age-matched 5-8 weeks old mice were used for all experiments, approved and performed under the guidelines of the Ethical Committee for the Use of Animals, Barts and The London School of Medicine and Home Office regulations (Scientific Procedures Act, 1986).
PMN recruitment of post-capillary venules: the detachment effect
Intravital microscopy was performed as previously reported41,42. Briefly, mice were anesthetized and the left jugular vein was cannulated with polyethylene tubing (PE 10, internal diameter 0.28mm). A cautery incision was made along the abdominal region, the superior mesenteric artery (SMA) exposed and clamped with a microaneurysm clip to induce ischemia in the mesentery for 30 min, followed by a 45-min reperfusion phase. Toward the end of the reperfusion period (ie, after approximately 35 min), the mesenteric vascular bed was exteriorized, placed on a viewing Plexiglas stage, and mounted on a Zeiss Axioskop “FS” with a water-immersion objective lens (magnification 40; Carl Zeiss, Welwyn Garden City, United Kingdom) and an eyepiece (magnification x10; Carl Zeiss). Tissue preparations were transilluminated with a 12 V, 100W halogen light source. A Hitachi charge-coupled device color camera (model KPC571; Tokyo, Japan) acquired images that were displayed onto a Sony Trinitron color video monitor (model PVM 1440QM; Tokyo, Japan) and recorded on a Sony super-VHS videocassette recorder (model SVO-9500 MDP) for subsequent offline analysis. A video time-date generator (FOR.A video timer, model VTG-33, Tokyo, Japan) projected the time, date, and stopwatch function onto the monitor. Mesenteries were superfused with thermostated (37°C) bicarbonate-buffered solution (g/L: NaCl, 7.71; KCl, 0.25; MgSO4, 0.14; NaHCO3, 1.51; and CaCl2, 0.22, pH 7.4, gassed with 5% CO2/95%N2) at a rate of 2 mL/min. When a suitable post-capillary venule was selected (diameter 20 - 40 μm; with equivalent numbers of 5-8 adherent leukocytes per 100 μm vessel length), PBS (100 μl), LXA4 or AF2 were administered (1 μg dose) via the jugular vein, and the fate of the adherent leukocytes was monitored for up to 10 min. Offline analysis monitored i) the number of adherent cells that rejoined the bloodstream and ii) the number of newly adherent leukocytes, as described previously41,42.
PMN trafficking into inflamed air-pouches
Air-pouches were formed on the dorsal area of C57Bla6 mice by injection of 2.5 ml air on day 0 and 3. On day 6, 10ng of mouse IL-1b (Peprotech, London, UK) were injected in 0.5 ml of 0.5% carboxymethyl-cellulose as described43. Four hours later, air-pouches were lavaged and exudate fluids stained with Turk’s solution for total cell counting in light microscopy. The extent of PMN influx was determined by FACS analysis by using a FITC-conjugated mAb for Ly-6G/Gr1 (clone RB6-8C5; 10 μg/ml final concentration; eBioscience, San Diego, CA) or isotype controls (eBR2a; eBioscience). FACS analysis was conducted as above, determining the percentage of Gr1 positive (Gr1+) cells and calculating the total number of PMN per air-pouch taking into account total cell number.
Data analysis
Western blot data are reported as mean ± SEM, expressing them as percentage of vehicle-treated cytosolic AnxA1 expression from at least three experiments performed in triplicate and plotted as X(time)/Y(expression) graph. The n number of mice has been reported for the in vivo experiments. Differences were evaluated using the Mann-Whitney U test for non-parametric data (in vitro analyses) or by ANOVA followed by Dunnett post-test (in vivo analyses). In all cases, a probability value <0.05 was taken as threshold for rejecting the null hypothesis.
Results
Agonist-dependent mobilization of intracellular AnxA1 in human PMN
Resting PMN contained large amounts of AnxA1, which was largely intracellularly located, with a small proportion in the membrane pool. This can be seen in Figure 1A and the following figures, with densitometric values of cumulative experiments showing a ratio of ~80±12% intracellular AnxA1 vs. 20±12% membrane AnxA1 (10 experiments, performed in triplicate). This is congruent with the predominant intracellular localization of the protein in resting PMN20. However, cell activation with soluble stimuli would mobilize the protein onto the cell surface in an EDTA-labile pool, where partial cleavage would occur, hence the appearance of the doublet36.
Figure 1. AnxA1 disposition in human PMN upon agonist stimulation.
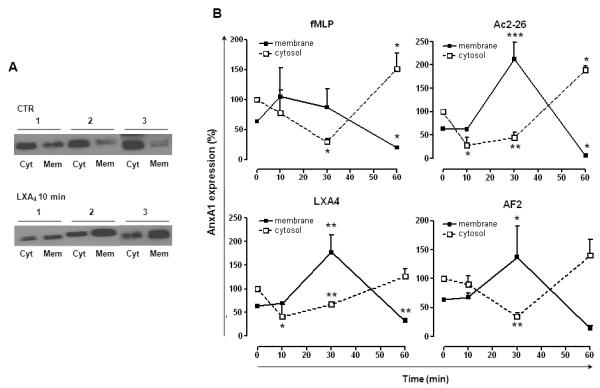
A, Representative blots showing AnxA1 localization in unstimulated and LXA4 treated human PMN; AnxA1, mainly present in cytosol (Cyt), is mobilized towards the membrane surface (Mem) upon stimulation with agonists (LXA4). Data are from 3 distinct PMN preparations. B, Human PMN were treated with vehicle (CTR), AF2 (10 μM), peptide Ac2-26 (10 μM), LXA4 (100nM) or fMLP (1 μM) for 10-60 min and then expression of AnxA1 in cytosolic and membrane fraction was determined by Western blotting. Blots are showed as line graphs reporting expression vs. time. Data, mean ± SEM, are calculated as percentage of vehicle-treated cytosolic AnxA1 expression taken as 100 (*p<0.05; **p<0.01; ***p<0.001 vs. CTR; n=3).
Incubation of PMN alone over a 60-min time-course provoked only mild changes in AnxA1 expression in the absence of receptor agonists. Cell treatment with peptide Ac2-26 (10 μM), AF2 (10 μM), LXA4 (100 nM) or fMLP (1 μM) provoked significant changes in AnxA1 intracellular location though they occurred to a different extent and pattern (Fig. 1B). Over the time course, LXA4 rapidly (10 min) reduced AnxA1 cytosolic content and this was mirrored by an increase in the membrane pool at 30 min (Fig. 1A-B). AF2 provoked qualitatively similar – yet slower - effects, with significance only being reached at the 30 min time-point. Peptide Ac2-26 seemed to provoke changes similar to LXA4 but its effects were lost by 60 min, with the pool of intracellular AnxA1 apparently replenished. The FPR1 agonist fMLP mobilized AnxA1 with kinetics similar to those produced by peptide Ac2-26: at 30 min there was significant reduction of intracellular AnxA1, which was partially replenished by 60 min, possibly consequence to a reduction of the membrane pool by this time-point (Fig.1B).
Changes in AnxA1 concentrations in cell supernatants were detected only after 30 min treatment (data not shown) implying some delay in the completion of the AnxA1 externalization process after its exposure on the plasma membrane20, 36; normally AnxA1 is not detectable in the culture medium of resting PMN (data not shown). From this set of experiments, we selected the 30 min time-point and the two FPR2/ALX agonists AF2 and LXA4, for further studies.
Characterization of AF2 and LXA4 induced AnxA1 mobilization
Incubation of human PMN with AF2 (1-10μM) provoked a reduction of cytosolic AnxA1 and a simultaneous increase in either membrane fraction and cell supernatant (Fig 2A) AnxA1. This effect was evident at 1μM AF2, and robustly observed at 3-10 μM concentrations. Treatment of cells with LXA4 (10-100 nM) induced changes in AnxA1 location similar to AF2; notably, these effects were concentration-dependent and optimal at ~10 nM (Fig. 2B).
Figure 2. AnxA1 mobilization in human PMN: concentration-response studies.

Treatment of human PMN either with AF2 (1-10 μM, A) or LXA4 (10-100 nM, B) for 30 min provoked a decrease in cytosolic AnxA1 levels and a parallel increase in AnxA1 membrane surface expression. Blots are representative of three different experiments. Data, mean ± SEM, are calculated as percentage of vehicle-treated cytosolic AnxA1 expression taken as 100 (*p<0.05; **p<0.01; ***p<0.001 vs. vehicle, CTR).
Pre-treatment of human PMN with the antagonist WRW4 (10μM, 10 min pre-incubation) affected both LXA4 and AF2-induced AnxA1 mobilization. In particular, the marked mobilization of AnxA1 on the cell surface induced by either FPR2/ALX agonists was essentially abolished by WRW4; representative blots are shown (Fig. 3A) with cumulative data for cytosolic, membrane and supernatant pools of AnxA1, respectively (Fig. 3B-D). Peptide Ac2-26 was also tested in these experiments however its modulation of PMN AnxA1 localization was not affected by WRW4 (Fig. 3B-D).
Figure 3. WRW4 prevents LXA4 and AF2-elicited mobilization of AnxA1 in human PMN.

Human PMN were pre-treated with WRW4 (10 μM, 10 min), prior addition of vehicle (CTR), Ac2-26 (10 μM), LXA4 (100 nM) or AF2 (10 μM) for further 30 min. Subcellular fractions were then processed and analyzed by Western blotting. A, Representative blots from three different experiments in the presence (+) or absence (−) of WRW4. B-D, Densitometric analysis for AnxA1 expression in cytosol (B), membrane (C) and supernatant (D), respectively. Data, mean ± SEM, are calculated as percentage of vehicle-treated cytosolic AnxA1 expression taken as 100 (*p<0.05; **p<0.01; ***p<0.001 vs. CTR, i.e. no WRW4; §p<0.05; §§p<0.01; §§§p<0.001 vs. vehicle, i.e. in presence of WRW4 only).
Experiments done in presence of the selective FPR1 antagonist CsH, showed that AnxA1 mobilization induced by fMLP was genuinely due to engagement of this receptor (Fig. 4). A different scenario emerged for peptide Ac2-26 which retained its ability to mobilize AnxA1 (Fig. 4). Of interest, even when a combination of CsH and WRW4 was tested, peptide Ac2-26 used at 10 μM, yielded marked mobilization of AnxA1 (Fig. 1S).
Figure 4. CsH affects fMLP-elicited mobilization of AnxA1 in human PMN.
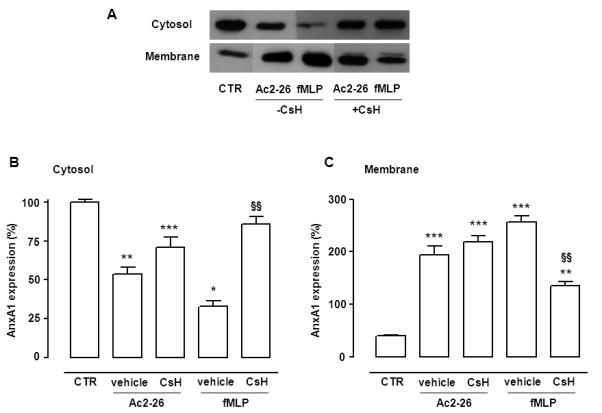
Human PMN were pre-treated with CsH (10 μM, 10 min), prior addition of vehicle (CTR), Ac2-26 (10 μM) or fMLP (1μM) for further 30 min. Subcellular fractions were then processed and analyzed by Western blotting. A, Representative blots from three different experiments in the presence (+) or absence (−) of CsH. B-C, Densitometric analysis for AnxA1 expression in cytosol (B) and membrane (C) respectively. Data, mean ± SEM, are calculated as percentage of vehicle-treated cytosolic AnxA1 expression taken as 100 (*p<0.05; **p<0.01; ***p<0.001 vs. CTR, i.e. no CsH; §p<0.05; §§p<0.01; §§§p<0.001 vs. vehicle, i.e. in presence of CsH).
To determine whether AnxA1 mobilization was secondary to, or accompanied by, granule mobilization, antibodies specific for PMN granules, such as CD35 (secretory vesicles), CD66b (specific granules) and CD63 (azurophil granules) were used. Flow cytometry analysis showed that, following 30 min incubation, unlike PMA (positive control) neither LXA4 nor AF2 induced changes in the PMN cell surface expression of any of these markers (Fig. 5A). Interestingly, both agonists caused internalization of FPR2/ALX over the 30 min incubation time, with no effect on FPR1 (Fig. 5B). In these experiments, fMLP (1 μM) provoked significant up-regulation of CD66b (+60%; Fig. 5A).
Figure 5. FACS analysis on human PMN after treatment with LXA4 and AF2.
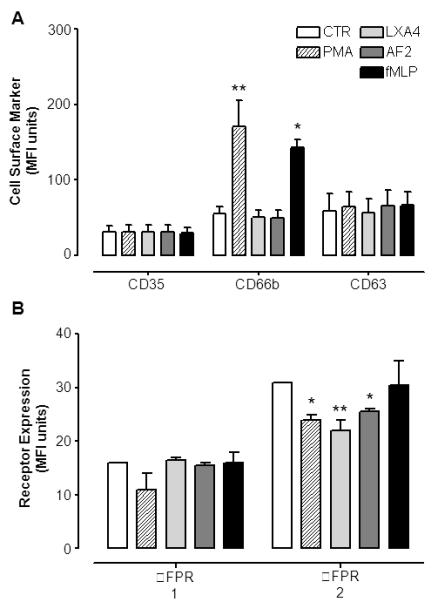
Human PMN were treated with vehicle (CTR), AF2 (10 μM), LXA4 (100nM) or fMLP (1 μM) for 30 min prior staining with a specific mAbs. PMA (100 nM) was as positive control for PMN degranulation. A, Mean fluorescence intensity (MFI) determination for cell surface staining for PMN granule markers CD35, CD66b and CD63. B, Mean fluorescence intensity for FPR1 and FPR2/ALX receptors surface expression upon treatment with FPR2/ALX agonists LXA4 and AF2. Data, reported as MFI unites, are mean ± SEM from three different experiments (*p<0.05; **p<0.01 vs. CTR).
Since membrane AnxA1 did not appear to derive from intracellular granules, we monitored AnxA1 phosphorylation status in the cytosolic pool25,24, using an antibody against phospho-Ser27-AnxA125. Shorter time-points were tested here, with Fig. 6 reporting data obtained at 10 min. In resting cells there was little membrane phosphorylated AnxA1; addition of LXA4 and AF2 provoked rapid increase in this species (Fig. 6A). Incubation with fMLP augmented AnxA1 on the membrane surface (Fig. 6B), but this did not appear to be phosphorylated.
Figure 6. Phospho-Serine 27-AnxA1 detection in PMN membrane fractions.
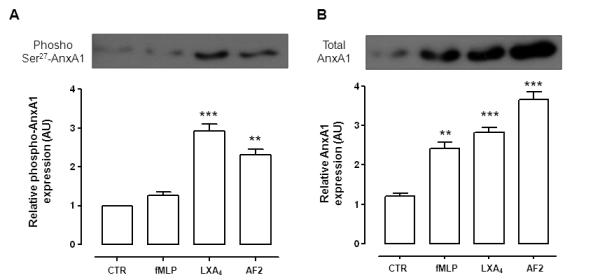
Human PMN were treated with vehicle (CTR) AF2 (10 μM), LXA4 (100nM) or fMLP (1 μM) for 10 min prior to recovery of membrane-bound AnxA1 by EDTA. A, Phospho Serine 27-AnxA1 with representative western blotting and densitometric cumulative values. B, total AnxA1 in the membrane pool with representative western blotting and densitometric cumulative values, Data reported as relative optical density arbitrary unit (AU), are mean ± SEM from three experiments (**p<0.01; ***p<0.001 vs. CTR).
Cell-surface AnxA1 in stimulated PMN could also be quantified by flow cytometry. At the optimal 30 min incubation time, both LXA4 and AF2 augmented AnxA1 expression at the single cell level (>2-fold increase in MFI Units; Fig. 7A-B) and there was a marked increase in AnxA1 positive PMN, with values close to 70% versus control (resting PMN) levels of ~15%. A similar pattern was seen following incubation with fMLP.
Figure 7. Detection of cell surface AnxA1 by flow cytometry.
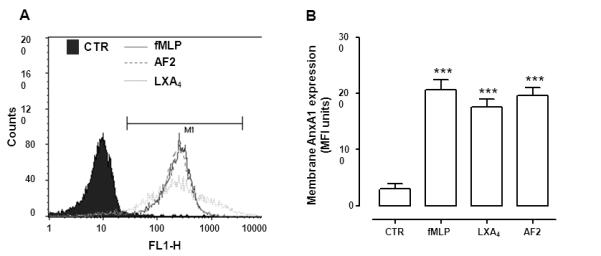
Human PMN were treated with vehicle (CTR), AF2 (10 μM), LXA4 (100nM) or fMLP (1 μM) for 30 min prior to staining with a specific anti-AnxA1 mAb. A, Representative histograms depicting the increase in fluorescence intensity as AnxA1 becomes externalized. B, Cumulative data for AnxA1 expression. Data, reported as MFI unites, are mean ± SEM from three different experiments (***p<0.001 vs. CTR).
Anti-inflammatory properties of LXA4 and AF2 are tightly coupled to AnxA1
Next, we tested if the rapid non-genomic modulation of PMN AnxA1 might be of relevance to PMN trafficking in inflammation. To this end, we used a validated protocol whereby LXA442 and AnxA1 itself exquisitely modulate the fate of adherent leukocytes, a biological effect referred to as ‘the detachment phenomenon’41. The mesenteric microcirculation of AnxA1+/+ mice was inflamed with an ischaemia/reperfusion procedure, allowing the selection of post-capillary venules with a ~6-8 adherent leukocytes, in contrast to the minimal leukocyte interaction with the post-capillary venule endothelium observed in sham operated animals (0-2 cells per vessel) (see Figure 2S).
Injection of a single bolus of LXA4 (1μg i.v., corresponding to 2.8nmol) to AnxA1+/+ mice provoked a marked (>50%) detachment of PMN from the endothelium within the 10 min observation period (Fig. 8A). This effect was drastically reduced in AnxA1−/− mice, with no significant alteration in the fate of the adherent leukocytes (~15%; Fig. 8C). A similar result was obtained with AF2 (1μg i.v., corresponding to 1 nmol) which afforded significant detachment of adherent leukocytes in AnxA1+/+, but not AnxA1−/−, mice (Fig. 8B-C).
Figure 8. LXA4 and AF2 provoke leukocyte detachment by mobilizing endogenous AnxA1.
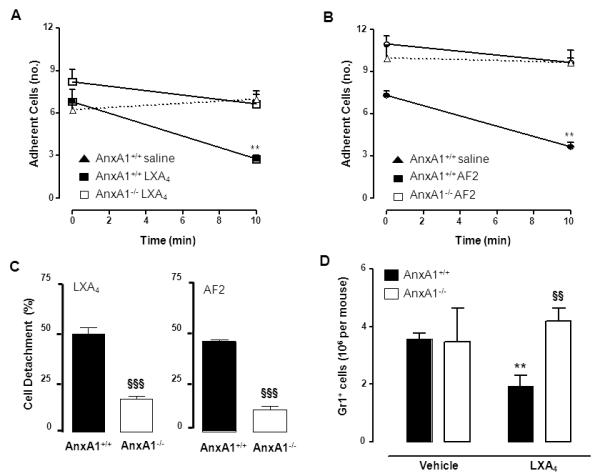
Intravital microscopy analyses of AnxA1+/+ and AnxA1−/− mouse mesenteric microcirculation inflamed by an ischaemia-reperfusion procedure. A-B, Administration of LXA4 and AF2 (1 μg i.v. at time 0, that is once the preparation is on stage and post-capillary venule selected) induced detachment of adherent leukocytes in AnxA1+/+ but not in AnxA1−/− mice (**p<0.01 vs. time 0). C, Cumulative modulation of the detachment process, as recorded at the end of the 10 min observation period. Data are Mean ± SEM of 6 mice per group (§§§p<0.001 vs. AnxA1+/+). D, Dorsal air pouches of AnxA1+/+ and AnxA−/− mice were injected with IL-1β (10 ng in 0.5 ml) at time 0; vehicle (100 μl) or LXA4 (1 μg/100μl) were injected i.v. via the tail vein immediately after. In all cases, air-pouches were washed 4 h later and cells were stained to determine amount of recruited PMN (Gr1+ cells). Data are mean ± SEM of 6 mice per group (**p<0.01 vs. vehicle; §§p<0.01 vs. AnxA1+/+).
It should be noted that in line with the reported alterations within the microcirculation of in AnxA1−/− 43 absence of AnxA1 was associated with a slightly higher degree of adherent cells in both sets of experiments (e.g. Fig. 8A-B).
Finally, we investigated whether the crucial role of endogenous AnxA1 in LXA4-induced effects was also evident in a more severe type of inflammation. IL-1β injection into mice dorsal air-pouches provoked an intense recruitment of Gr1+ cells at 4h time-point, which was inhibited by treatment with LXA4 in AnxA1+/+, but not AnxA1−/−, mice (Fig. 8D).
Discussion
In this study we provide strong evidence that engagement of FPR2/ALX by selective agonists would induce AnxA1 phosphorylation and mobilization in human PMN. We propose this cellular response operates to control PMN recruitment to post-capillary venules and migration to inflamed sites, as demonstrated with proof-of-concept experiments in AnxA1−/− mice, in which LXA4 and AF2 lost their anti-migratory properties.
An effective inflammatory reaction relies on the concerted action of several pathways which attract white blood cells from the vasculature into specific tissue sites. A classical example is that of the synovial joint; here TNF-α initiates a cascade of mediator generation and release (e.g. IL-1 or IL-6)44,45 culminating in the recruitment of blood borne leukocytes into the synovial tissue and joint space. Blocking TNF-α activity is therefore a very successful therapeutic strategy 44,46. In recent years an innovative approach to anti-inflammatory therapeutic development has emerged, which aims to capitalize on the action of endogenous anti-inflammatory and pro-resolving mediators47, 48. The evidence that the pro-inflammatory phase is required for an appropriate induction of the endogenous anti-inflammatory phase is now well accepted2. As an example, pro-inflammatory prostanoids induce LXA4 synthesis in, what has been proposed to be, a ‘class switching’ from the pro-inflammatory to the pro-resolving phase of an acute experimental inflammation49. However, the option that a network might be operative within anti-inflammatory mediators themselves has been poorly explored: we demonstrate here a functional link between LXA4 and endogenous AnxA1. Incubation of human PMN with LXA4 induced a rapid translocation of AnxA1 to the cell surface with a time profile different from that provoked by peptide Ac2-26 or fMLP. The effect of LXA4 was mimicked by the anti-inflammatory synthetic nonapeptide AF2.
It is intriguing that both LXA4 and AF2 inhibit neutrophil activation in vitro as well as recruitment of these cells in vivo. AF2 reduces HL-60 cells adhesion to endothelial cells, suggesting that it dampens inflammation by blocking leukocyte trafficking and the subsequent eicosanoid production29. Furthermore, AF2 is effective in suppressing endotoxin-induced uveitis in rats50. The data presented here indicate that such an effect might occur at least partly via endogenous AnxA1. Our new data and ensuing hypothesis may provide a mechanistic explanation for the observation that endogenous LXA4 and AnxA1 are both operative in germ-free mice for optimal generation of IL-10 in the gut51.
It should also be noted that very few examples of crosstalk in resolution are beginning to emerge, with resolvin E1 promoting exudate expression of LXA4, although the specific cell target(s) of this mediator were not identified52. In addition, there is indication that endogenous epi-LXA4, synthesized upon aspirin unique blockade of cyclo-oxygenase 2, can mobilize nitric oxide from the endothelium (constitutive nitric oxide synthase activation) and macrophages (inducible nitric oxide synthase activation) to elicit anti-inflammatory effects in a model of pleurisy as well as in the microcirculation53. We cannot exclude that an involvement of endogenous nitric oxide might be occurring also in our settings, though the experiments of leukocyte detachment reported here took place within 10-min. In any case, the recent involvement of rapidly produced endothelial nitric oxide and prostacyclin in the anti-migratory effects of resolvin D254 would suggest to keep this option viable also upon the acute application of LXA4 and AF2. Finally, the positive association between endogenous AnxA1 and expression of mouse Fpr2 during resolving colitis should be reported55.
Most of the studies summarized above begin to propose the existence of positive non-genomic loops in resolution, however, there is also emerging evidence for genomic associations. One classical example is the one linking glucocorticoids to over-expression of AnxA1, with increments in gene promoter activity being measured with a reporter gene14. In addition, LXA4 could induce two specific signaling molecules, NGFI-A binding protein 1 (NAB1)56 and suppressor of cytokine signaling 257 to re-programme the phenotype of PMN and dendritic cells, respectively. Among the biological effects elicited by LXA4, its analogs and other pro-resolving lipid mediators, it is worth highlighting the augmented expression of CCR5 on apoptotic leukocytes, a response that facilitate removal of migrated cells and tissue resolution58. De novo gene synthesis has, clearly, been ascribed to glucocorticoids; in this context it must be noted that they will not only augment AnxA1 synthesis and release, but would also increase expression of anti-inflammatory FPR2/ALX59,60.
The observation that exogenously applied LXA4 mobilizes AnxA1 and that this contributes, at least in part, to its inhibitory properties on PMN trafficking, adds another layer of complexity to the molecular and functional inter-relationship between these two endogenous effectors of anti-inflammation and resolution. Moreover, it is intriguing how a pattern of distinct inducers of AnxA1 externalization is emerging. Processes such as cell adhesion and activation, which induce re-arrangement of intracellular organelles within the PMN, would mobilize preferentially the pool of AnxA1 contained in granules and vesicles20,61 (see Fig. 9 for a schematic summary). On the other hand, the typical mobilization induced by glucocorticoids utilizing a receptor-dependent non-genomic pathway 62,63,25 seems to be replicated by other classes of drugs including cromones 23, 24 and, as described here, LXA4 and AF2. Mobilization of this cytosolic pool of AnxA1 is downstream PKC activation and phosphorylation at Ser27 on the AnxA1 N-terminus25 (Fig. 9). It is unclear whether PKC isoform(s) - advocated for dexamethasone and nedocromil action 23, 25 – also operates downstream FPR2/ALX activation by LXA4 and AF2. In any case, the involvement of this specific GPCR is unambiguous, as shown by the effects produced with the receptor antagonist and the observation of FPR2/ALX internalization after agonist application. It should be noted that a recent study indicated selective internalization of the receptor upon application of LXA4 as a prerequisite for stimulation of phagocytosis by this anti-inflammatory lipid64.
Figure 9. Schematic model for AnxA1 mobilization in human PMN.
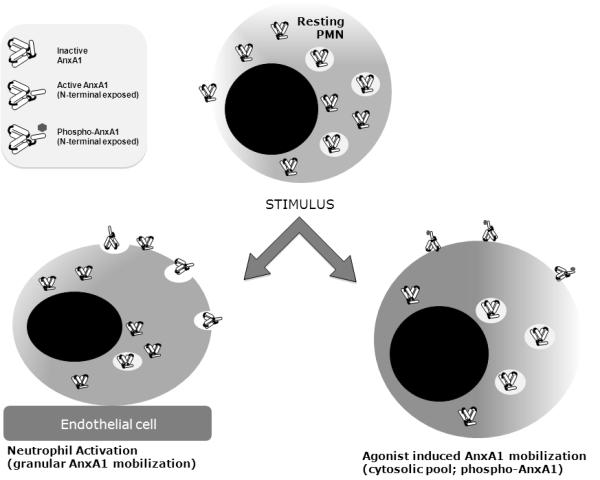
Proposed scheme of AnxA1 mobilization whereby cell-to-cell interaction (e.g. adhesion to endothelial cells) would externalize AnxA1 by mobilizing predominantly the granular pool18, 19,20 , whereas the non-granular (cytosolic) pool seems to be mobilized in response to soluble compound addition, e.g. glucocorticoids, cromones and inhibitors of the cystic fibrosis transducer factor22-24. The present study adds FPR2/ALX to the list of compounds known to ‘activate’ the AnxA1 pathway in the neutrophil. The latter mode of mobilization seems to have, as a consistent pre-requisite, phosphorylation of Ser27 on the AnxA1 N-terminus. It is yet unclear if PMN adhesion to endothelial cells20 leads to externalization of phosphorylated AnxA1 species.
As stated above, LXA4 is a potent regulator of PMN trafficking in experimental inflammation, with its ability to inhibit recruitment of this cell type to a variety of tissue sites and in response to several distinct inflammogens6. The same is partially true for AF2, though this nonapeptide has been used more often as an inhibitor of PMN activation in vitro 27,29 rather than of cell recruitment in vivo65. For instance, AF2 inhibits PMN adhesion to endothelial cells28 and leukocyte chemotaxis66. Both compounds were tested in the IVM protocol which we reported being sensitive to LXA4 treatment42. The results we present here indicate a crucial role for endogenous AnxA1 in the detachment phenomenon promoted by either FPR2/ALX agonist.
Peptide Ac2-26 was not tested in the leukocyte detachment phenomenon here, in view of the in vitro profile we obtained with human PMNs. However, it has been previously shown that administration of this peptide to mice can activate this process in an Fpr1-independent fashion42 and peptide Ac2-26 also retains its anti-migratory effects in AnxA1-null mice21. In the present in vitro settings, this AnxA1 mimetic induces AnxA1 mobilization in a different manner with respect to LXA4 and AF2 (and fMLP) since it does not appear to evoke involvement of either FPR1 and FPR2, as shown by experiments performed with FPR1 or FPR2/ALX selective antagonists. It should be noted that PMN activating effects associated to AnxA1 N-terminal derived peptides (Ac9-25) do not require activation of FPR1 or FPR2/ALX67.
Finally, it should be noted that the inhibitory properties exhibited by LXA4 in the air-pouch model were lost in AnxA1−/− mice. Coupled with the data recent observation of LXA4 lack of effect in Fpr2−/− mice, using the same experimental model and inflammogen68, we propose a model whereby intravascular activation of PMN Fpr2, in response to the agonists tested here, would bring about AnxA1 mobilization. This event would in turn act in a juxtacrine/autocrine fashion6, reinforcing the overall attenuation of the process of leukocyte recruitment. Clearly, other intracellular pathways 69,70 and mediators might also act downstream LXA4 application 5,66, nonetheless the data presented and discussed here point to the non-genomic mobilization of AnxA1 as one of the major downstream effectors of the LXA4, and AF2, actions that negatively modulate the cell trafficking process. Networks of pro-resolving mediators and receptors are an emerging feature of the endogenous homeostatic response; once more loops are discovered and dissected, these ‘anti-inflammatory networks’ are likely to become an accepted paradigm in the area of inflammatory resolution.
Supplementary Material
Acknowledgements
We would like to thank Dr Egle Solito for providing the AnxA1-Ser27 phospho-specific antibody.
1 Funding: Wellcome Trust (programme 086867/Z/08). This work forms part of the research themes contributing to the translational research portfolio of Barts and the London Cardiovascular Biomedical Research Unit, which is supported and funded by the National Institutes of Health Research.
Footnotes
Abbreviations: AF2, antiflammin 2; AnxA1, Annexin A1; CsH, cyclosporin H; Cyt, cytosolic pool; FPR2/ALX, formyl peptide receptor 2/Lipoxin A4 receptor; GPCR, G-protein coupled receptor; LXA4, lipoxin A4; Mem, membrane pool; PMN, polymorphonuclear leukocytes; WRW4; peptide WRWWWW.
References
- 1.Nathan C. Neutrophils and immunity: challenges and opportunities. Nat Rev Immunol. 2006;6:173–182. doi: 10.1038/nri1785. [DOI] [PubMed] [Google Scholar]
- 2.Serhan CN, Savill J. Resolution of inflammation: the beginning programs the end. Nat Immunol. 2005;6:1191–1197. doi: 10.1038/ni1276. [DOI] [PubMed] [Google Scholar]
- 3.Brennan FM, McInnes IB. Evidence that cytokines play a role in rheumatoid arthritis. J Clin Invest. 2008;118:3537–3545. doi: 10.1172/JCI36389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Serhan CN, Brain SD, Buckley CD, Gilroy DW, Haslett C, O’Neill LA, Perretti M, Rossi AG, Wallace JL. Resolution of inflammation: state of the art, definitions and terms. Faseb J. 2007;21:325–332. doi: 10.1096/fj.06-7227rev. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chiang N, Serhan CN, Dahlen SE, Drazen JM, Hay DW, Rovati GE, Shimizu T, Yokomizo T, Brink C. The lipoxin receptor ALX: potent ligand-specific and stereoselective actions in vivo. Pharmacol Rev. 2006;58:463–487. doi: 10.1124/pr.58.3.4. [DOI] [PubMed] [Google Scholar]
- 6.Perretti M, D’Acquisto F. Annexin A1 and glucocorticoids as effectors of the resolution of inflammation. Nat Rev Immunol. 2009;9:62–70. doi: 10.1038/nri2470. [DOI] [PubMed] [Google Scholar]
- 7.Maderna P, Yona S, Perretti M, Godson C. Modulation of phagocytosis of apoptotic neutrophils by supernatant from dexamethasone-treated macrophages and annexin-derived peptide Ac2-26. J Immunol. 2005;174:3727–3733. doi: 10.4049/jimmunol.174.6.3727. [DOI] [PubMed] [Google Scholar]
- 8.Schwab JM, Chiang N, Arita M, Serhan CN. Resolvin E1 and protectin D1 activate inflammation-resolution programmes. Nature. 2007;447:869–874. doi: 10.1038/nature05877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Godson C, Mitchell S, Harvey K, Petasis NA, Hogg N, Brady HR. Cutting edge: lipoxins rapidly stimulate nonphlogistic phagocytosis of apoptotic neutrophils by monocyte-derived macrophages. J Immunol. 2000;164:1663–1667. doi: 10.4049/jimmunol.164.4.1663. [DOI] [PubMed] [Google Scholar]
- 10.Serhan CN, Yacoubian S, Yang R. Anti-inflammatory and proresolving lipid mediators. Annu Rev Pathol. 2008;3:279–312. doi: 10.1146/annurev.pathmechdis.3.121806.151409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Perretti M, Chiang N, La M, Fierro IM, Marullo S, Getting SJ, Solito E, Serhan CN. Endogenous lipid- and peptide-derived anti-inflammatory pathways generated with glucocorticoid and aspirin treatment activate the lipoxin A(4) receptor. Nat Med. 2002;8:1296–1302. doi: 10.1038/nm786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ye RD, Boulay F, Wang JM, Dahlgren C, Gerard C, Parmentier M, Serhan CN, Murphy AP. International Union of Basic and Clinical Pharmacology. LXXIII. Nomenclature for the Formyl Peptide Receptor (FPR) Family. Pharmacol Rev. 2009 doi: 10.1124/pr.109.001578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bannenberg GL, Chiang N, Ariel A, Arita M, Tjonahen E, Gotlinger KH, Hong S, Serhan CN. Molecular circuits of resolution: formation and actions of resolvins and protectins. J Immunol. 2005;174:4345–4355. doi: 10.4049/jimmunol.174.7.4345. [DOI] [PubMed] [Google Scholar]
- 14.Damazo AS, Yona S, Flower RJ, Perretti M, Oliani SM. Spatial and Temporal Profiles for Anti-Inflammatory Gene Expression in Leukocytes during a Resolving Model of Peritonitis. J Immunol. 2006;176:4410–4418. doi: 10.4049/jimmunol.176.7.4410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Morris T, Stables M, Hobbs A, de Souza P, Colville-Nash P, Warner T, Newson J, Bellingan G, Gilroy DW. Effects of low-dose aspirin on acute inflammatory responses in humans. J Immunol. 2009;183:2089–2096. doi: 10.4049/jimmunol.0900477. [DOI] [PubMed] [Google Scholar]
- 16.Morris T, Stables M, Colville-Nash P, Newson J, Bellingan G, de Souza PM, Gilroy DW. Dichotomy in duration and severity of acute inflammatory responses in humans arising from differentially expressed proresolution pathways. Proc Natl Acad Sci U S A. 2010;107:8842–8847. doi: 10.1073/pnas.1000373107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rosales JL, Ernst JD. Calcium-dependent neutrophil secretion: characterization and regulation by annexins. J Immunol. 1997;159:6195–6202. [PubMed] [Google Scholar]
- 18.Perretti M, Christian H, Wheller SK, Aiello I, Mugridge KG, Morris JF, Flower RJ, Goulding NJ. Annexin I is stored within gelatinase granules of human neutrophils and mobilised on the cell surface upon adhesion but not phagocytosis. Cell Biol Int. 2000;24:163–174. doi: 10.1006/cbir.1999.0468. [DOI] [PubMed] [Google Scholar]
- 19.Lominadze G, Powell DW, Luerman GC, Link AJ, Ward RA, McLeish KR. Proteomic analysis of human neutrophil granules. Mol Cell Proteomics. 2005;4:1503–1521. doi: 10.1074/mcp.M500143-MCP200. [DOI] [PubMed] [Google Scholar]
- 20.Perretti M, Croxtall JD, Wheller SK, Goulding NJ, Hannon R, Flower RJ. Mobilizing lipocortin 1 in adherent human leukocytes downregulates their transmigration. Nat Med. 1996;2:1259–1262. doi: 10.1038/nm1196-1259. [DOI] [PubMed] [Google Scholar]
- 21.Gastardelo TS, Damazo AS, Dalli J, Flower RJ, Perretti M, Oliani SM. Functional and ultrastructural analysis of annexin A1 and its receptor in extravasating neutrophils during acute inflammation. Am J Pathol. 2009;174:177–183. doi: 10.2353/ajpath.2009.080342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dalli J, Rosignoli G, Hayhoe RP, Edelman A, Perretti M. CFTR inhibition provokes an inflammatory response associated with an imbalance of the annexin A1 pathway. Am J Pathol. 2010;177:176–186. doi: 10.2353/ajpath.2010.091149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yazid S, Solito E, Christian H, McArthur S, Goulding N, Flower R. Cromoglycate drugs suppress eicosanoid generation in U937 cells by promoting the release of Anx-A1. Biochem Pharmacol. 2009;77:1814–1826. doi: 10.1016/j.bcp.2009.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yazid S, Leoni G, Getting SJ, Cooper D, Solito E, Perretti M, Flower RJ. Antiallergic cromones inhibit neutrophil recruitment onto vascular endothelium via annexin-A1 mobilization. Arterioscler Thromb Vasc Biol. 2010;30:1718–1724. doi: 10.1161/ATVBAHA.110.209536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Solito E, Christian HC, Festa M, Mulla A, Tierney T, Flower RJ, Buckingham JC. Post-translational modification plays an essential role in the translocation of annexin A1 from the cytoplasm to the cell surface. Faseb J. 2006;20:1498–1500. doi: 10.1096/fj.05-5319fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miele L, Cordella-Miele E, Facchiano A, Mukherjee AB. Novel anti-inflammatory peptides from the region of highest similarity between uteroglobin and lipocortin I. Nature. 1988;335:726–730. doi: 10.1038/335726a0. [DOI] [PubMed] [Google Scholar]
- 27.Camussi G, Tetta C, Bussolino F, Baglioni C. Antiinflammatory peptides (antiflammins) inhibit synthesis of platelet-activating factor, neutrophil aggregation and chemotaxix, and intradermal inflammatory reactions. Journal of Experimental Medicine. 1990;171:913–927. doi: 10.1084/jem.171.3.913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zouki C, Ouellet S, Filep JG. The anti-inflammatory peptides, antiflammins, regulate the expression of adhesion molecules on human leukocytes and prevent neutrophil adhesion to endothelial cells. Faseb J. 2000;14:572–580. doi: 10.1096/fasebj.14.3.572. [DOI] [PubMed] [Google Scholar]
- 29.Moreno JJ. Antiflammin-2 prevents HL-60 adhesion to endothelial cells and prostanoid production induced by lipopolysaccharides. J Pharmacol Exp Ther. 2001;296:884–889. [PubMed] [Google Scholar]
- 30.Rosengarth A, Gerke V, Luecke H. X-ray Structure of Full-length Annexin 1 and Implications for Membrane Aggregation. J Mol Biol. 2001;306:489–498. doi: 10.1006/jmbi.2000.4423. [DOI] [PubMed] [Google Scholar]
- 31.Rosengarth A, Luecke H. A Calcium-driven Conformational Switch of the N-terminal and Core Domains of Annexin A1. J Mol Biol. 2003;326:1317–1325. doi: 10.1016/s0022-2836(03)00027-5. [DOI] [PubMed] [Google Scholar]
- 32.Kamal AM, Hayhoe RP, Paramasivam A, Cooper D, Flower RJ, Solito E, Perretti M. Antiflammin-2 activates the human formyl-peptide receptor like 1. ScientificWorldJournal. 2006;6:1375–1384. doi: 10.1100/tsw.2006.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, Brohi K, Itagaki K, Hauser CJ. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. 2010;464:104–107. doi: 10.1038/nature08780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McDonald B, Pittman K, Menezes GB, Hirota SA, Slaba I, Waterhouse CC, Beck PL, Muruve DA, Kubes P. Intravascular danger signals guide neutrophils to sites of sterile inflammation. Science. 2010;330:362–366. doi: 10.1126/science.1195491. [DOI] [PubMed] [Google Scholar]
- 35.Bae YS, Lee HY, Jo EJ, Kim JI, Kang HK, Ye RD, Kwak JY, Ryu SH. Identification of peptides that antagonize formyl peptide receptor-like 1-mediated signaling. J Immunol. 2004;173:607–614. doi: 10.4049/jimmunol.173.1.607. [DOI] [PubMed] [Google Scholar]
- 36.Vong L, D’Acquisto F, Pederzoli-Ribeil M, Lavagno L, Flower RJ, Witko-Sarsat V, Perretti M. Annexin 1 cleavage in activated neutrophils: a pivotal role for proteinase 3. J Biol Chem. 2007;282:29998–30004. doi: 10.1074/jbc.M702876200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pepinsky RB, Sinclair LK, Dougas I, Liang C-M, Lawton P, Browning JL. Monoclonal antibodies to lipocortin-1 as probes for biological functions. FEBS Letters. 1990;261:247–252. doi: 10.1016/0014-5793(90)80564-y. [DOI] [PubMed] [Google Scholar]
- 38.Borregaard N, Cowland JB. Granules of the human neutrophilic polymorphonuclear leukocyte. Blood. 1997;89:3503–3521. [PubMed] [Google Scholar]
- 39.Witko-Sarsat V, Rieu P, Descamps-Latscha B, Lesavre P, Halbwachs-Mecarelli L. Neutrophils: molecules, functions and pathophysiological aspects. Lab Invest. 2000;80:617–653. doi: 10.1038/labinvest.3780067. [DOI] [PubMed] [Google Scholar]
- 40.Hannon R, Croxtall JD, Getting SJ, Roviezzo F, Yona S, Paul-Clark MJ, Gavins FN, Perretti M, Morris JF, Buckingham JC, Flower RJ. Aberrant inflammation and resistance to glucocorticoids in annexin 1−/− mouse. Faseb J. 2003;17:253–255. doi: 10.1096/fj.02-0239fje. [DOI] [PubMed] [Google Scholar]
- 41.Lim LH, Solito E, Russo-Marie F, Flower RJ, Perretti M. Promoting detachment of neutrophils adherent to murine postcapillary venules to control inflammation: effect of lipocortin 1. Proc Natl Acad Sci U S A. 1998;95:14535–14539. doi: 10.1073/pnas.95.24.14535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gavins FN, Yona S, Kamal AM, Flower RJ, Perretti M. Leukocyte antiadhesive actions of annexin 1: ALXR- and FPR-related anti-inflammatory mechanisms. Blood. 2003;101:4140–4147. doi: 10.1182/blood-2002-11-3411. [DOI] [PubMed] [Google Scholar]
- 43.Chatterjee BE, Yona S, Rosignoli G, Young RE, Nourshargh S, Flower RJ, Perretti M. Annexin 1 deficient neutrophils exhibit enhanced transmigration in vivo and increased responsiveness in vitro. J Leukoc Biol. 2005;78:639–646. doi: 10.1189/jlb.0405206. [DOI] [PubMed] [Google Scholar]
- 44.Brennan FM, Chantry D, Jackson A, Maini R, Feldmann M. Inhibitory effect of TNF alpha antibodies on synovial cell interleukin-1 production in rheumatoid arthritis. Lancet. 1989;2:244–247. doi: 10.1016/s0140-6736(89)90430-3. [DOI] [PubMed] [Google Scholar]
- 45.Hata H, Sakaguchi N, Yoshitomi H, Iwakura Y, Sekikawa K, Azuma Y, Kanai C, Moriizumi E, Nomura T, Nakamura T, Sakaguchi S. Distinct contribution of IL-6, TNF-alpha, IL-1, and IL-10 to T cell-mediated spontaneous autoimmune arthritis in mice. J Clin Invest. 2004;114:582–588. doi: 10.1172/JCI21795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Arif S, Cox P, Afzali B, Lombardi G, Lechler RI, Peakman M, Mirenda V. Anti-TNFalpha therapy--killing two birds with one stone? Lancet. 2010;375:2278. doi: 10.1016/S0140-6736(10)60394-7. [DOI] [PubMed] [Google Scholar]
- 47.Gilroy DW, Lawrence T, Perretti M, Rossi AG. Inflammatory resolution: new opportunities for drug discovery. Nat Rev Drug Discov. 2004;3:401–416. doi: 10.1038/nrd1383. [DOI] [PubMed] [Google Scholar]
- 48.Serhan CN. Resolution phase of inflammation: novel endogenous anti-inflammatory and proresolving lipid mediators and pathways. Annu Rev Immunol. 2007;25:101–137. doi: 10.1146/annurev.immunol.25.022106.141647. [DOI] [PubMed] [Google Scholar]
- 49.Levy BD, Clish CB, Schmidt B, Gronert K, Serhan CN. Lipid mediator class switching during acute inflammation: signals in resolution. Nat Immunol. 2001;2:612–619. doi: 10.1038/89759. [DOI] [PubMed] [Google Scholar]
- 50.Chan CC, Ni M, Miele L, Cordella-Miele E, Ferrick M, Mukherjee AB, Nussenblatt RB. Effects of antiflammins on endotoxin-induced uveitis in rats. Arch Ophthalmol. 1991;109:278–281. doi: 10.1001/archopht.1991.01080020124058. [DOI] [PubMed] [Google Scholar]
- 51.Souza DG, Fagundes CT, Amaral FA, Cisalpino D, Sousa LP, Vieira AT, Pinho V, Nicoli JR, Vieira LQ, Fierro IM, Teixeira MM. The required role of endogenously produced lipoxin A4 and annexin-1 for the production of IL-10 and inflammatory hyporesponsiveness in mice. J Immunol. 2007;179:8533–8543. doi: 10.4049/jimmunol.179.12.8533. [DOI] [PubMed] [Google Scholar]
- 52.Haworth O, Cernadas M, Yang R, Serhan CN, Levy BD. Resolvin E1 regulates interleukin 23, interferon-gamma and lipoxin A4 to promote the resolution of allergic airway inflammation. Nat Immunol. 2008;9:873–879. doi: 10.1038/ni.1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Paul-Clark MJ, Van Cao T, Moradi-Bidhendi N, Cooper D, Gilroy DW. 15-epi-lipoxin A4-mediated induction of nitric oxide explains how aspirin inhibits acute inflammation. J Exp Med. 2004;200:69–78. doi: 10.1084/jem.20040566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Spite M, Norling LV, Summers L, Yang R, Cooper D, Petasis NA, Flower RJ, Perretti M, Serhan CN. Resolvin D2 is a potent regulator of leukocytes and controls microbial sepsis. Nature. 2009;461:1287–1291. doi: 10.1038/nature08541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Babbin BA, Laukoetter MG, Nava P, Koch S, Lee WY, Capaldo CT, Peatman E, Severson EA, Flower RJ, Perretti M, Parkos CA, Nusrat A. Annexin A1 regulates intestinal mucosal injury, inflammation, and repair. J Immunol. 2008;181:5035–5044. doi: 10.4049/jimmunol.181.7.5035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Qiu FH, Devchand PR, Wada K, Serhan CN. Aspirin-triggered lipoxin A4 and lipoxin A4 up-regulate transcriptional corepressor NAB1 in human neutrophils. Faseb J. 2001 doi: 10.1096/fj.01-0576fje. 10.1096/fj.01-0576fje ( www.fasebj.org) [DOI] [PubMed] [Google Scholar]
- 57.Machado FS, Johndrow JE, Esper L, Dias A, Bafica A, Serhan CN, Aliberti J. Anti-inflammatory actions of lipoxin A4 and aspirin-triggered lipoxin are SOCS-2 dependent. Nat Med. 2006;12:330–334. doi: 10.1038/nm1355. [DOI] [PubMed] [Google Scholar]
- 58.Ariel A, Fredman G, Sun YP, Kantarci A, Van Dyke TE, Luster AD, Serhan CN. Apoptotic neutrophils and T cells sequester chemokines during immune response resolution through modulation of CCR5 expression. Nat Immunol. 2006;7:1209–1216. doi: 10.1038/ni1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sawmynaden P, Perretti M. Glucocorticoid upregulation of the annexin-A1 receptor in leukocytes. Biochem Biophys Res Commun. 2006;349:1351–1355. doi: 10.1016/j.bbrc.2006.08.179. [DOI] [PubMed] [Google Scholar]
- 60.Hashimoto A, Murakami Y, Kitasato H, Hayashi I, Endo H. Glucocorticoids co-interact with lipoxin A4 via lipoxin A4 receptor (ALX) up-regulation. Biomed Pharmacother. 2007;61:81–85. doi: 10.1016/j.biopha.2006.06.023. [DOI] [PubMed] [Google Scholar]
- 61.Gerke V, Creutz CE, Moss SE. Annexins: linking Ca2+ signalling to membrane dynamics. Nat Rev Mol Cell Biol. 2005;6:449–461. doi: 10.1038/nrm1661. [DOI] [PubMed] [Google Scholar]
- 62.Croxtall JD, van Hal PT, Choudhury Q, Gilroy DW, Flower RJ. Different glucocorticoids vary in their genomic and non-genomic mechanism of action in A549 cells. Br J Pharmacol. 2002;135:511–519. doi: 10.1038/sj.bjp.0704474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Castro-Caldas M, Duarte CB, Carvalho AP, Lopes MC. Dexamethasone induces the secretion of annexin I in immature lymphoblastic cells by a calcium-dependent mechanism. Mol Cell Biochem. 2002;237:31–38. doi: 10.1023/a:1016502120139. [DOI] [PubMed] [Google Scholar]
- 64.Maderna P, Cottell DC, Toivonen T, Dufton N, Dalli J, Perretti M, Godson C. FPR2/ALX receptor expression and internalization are critical for lipoxin A4 and annexin-derived peptide-stimulated phagocytosis. FASEB J. 2010;24:4240–4249. doi: 10.1096/fj.10-159913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lloret S, Moreno JJ. In vitro and in vivo effects of the anti-inflammatory peptides, antiflammins. Biochemical Pharmacology. 1992;44:1437–1441. doi: 10.1016/0006-2952(92)90546-u. [DOI] [PubMed] [Google Scholar]
- 66.Moreno JJ. Antiflammin-2, a nonapeptide of lipocortin-1, inhibits leukocyte chemotaxis but not arachidonic acid mobilization. Eur J Pharmacol. 1996;314:129–135. doi: 10.1016/s0014-2999(96)00521-3. [DOI] [PubMed] [Google Scholar]
- 67.Karlsson J, Fu H, Boulay F, Dahlgren C, Hellstrand K, Movitz C. Neutrophil NADPH-oxidase activation by an annexin AI peptide is transduced by the formyl peptide receptor (FPR), whereas an inhibitory signal is generated independently of the FPR family receptors. J Leukoc Biol. 2005;78:762–771. doi: 10.1189/jlb.0305153. [DOI] [PubMed] [Google Scholar]
- 68.Dufton N, Hannon R, Brancaleone V, Dalli J, Patel HB, Gray M, D’Acquisto F, Buckingham JC, Perretti M, Flower RJ. Anti-inflammatory role of the murine formyl-peptide receptor 2: ligand-specific effects on leukocyte responses and experimental inflammation. J Immunol. 2010;184:2611–2619. doi: 10.4049/jimmunol.0903526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kieran NE, Doran PP, Connolly SB, Greenan MC, Higgins DF, Leonard M, Godson C, Taylor CT, Henger A, Kretzler M, Burne MJ, Rabb H, Brady HR. Modification of the transcriptomic response to renal ischemia/reperfusion injury by lipoxin analog. Kidney Int. 2003;64:480–492. doi: 10.1046/j.1523-1755.2003.00106.x. [DOI] [PubMed] [Google Scholar]
- 70.Mitchell D, Rodgers K, Hanly J, McMahon B, Brady HR, Martin F, Godson C. Lipoxins inhibit Akt/PKB activation and cell cycle progression in human mesangial cells. Am J Pathol. 2004;164:937–946. doi: 10.1016/S0002-9440(10)63181-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.