


Abstract
Annealing of the TAR RNA hairpin to the cTAR DNA hairpin is required for the minus-strand transfer step of HIV-1 reverse transcription. HIV-1 nucleocapsid protein (NC) plays a crucial role by facilitating annealing of the complementary hairpins. To gain insight into the mechanism of NC-mediated TAR RNA–DNA annealing, we used structural probes (nucleases and potassium permanganate), gel retardation assays, fluorescence anisotropy and cTAR mutants under conditions allowing strand transfer. In the absence of NC, cTAR DNA-TAR RNA annealing depends on nucleation through the apical loops. We show that the annealing intermediate of the kissing pathway is a loop–loop kissing complex involving six base-pairs and that the apical stems are not destabilized by this loop–loop interaction. Our data support a dynamic structure of the cTAR hairpin in the absence of NC, involving equilibrium between both the closed conformation and the partially open ‘Y’ conformation. This study is the first to show that the apical and internal loops of cTAR are weak and strong binding sites for NC, respectively. NC slightly destabilizes the lower stem that is adjacent to the internal loop and shifts the equilibrium toward the ‘Y’ conformation exhibiting at least 12 unpaired nucleotides in its lower part.
INTRODUCTION
Human immunodeficiency virus type 1 (HIV-1), like all retroviruses, replicates through reverse transcription, a complex process leading to the synthesis of double-stranded DNA from a single-stranded genomic RNA and requiring the HIV-1 nucleocapsid protein (NC) as well as at least two strand transfer reactions (1,2). The first strand transfer requires the repeat (R) sequences at both ends of the genome. During this reaction, which involves reverse transcriptase (RT) with its RNase H activity to promote degradation of the 5′-copied genomic RNA (donor RNA), the 3′-end of the minus-strand strong-stop DNA (ss-cDNA) is transferred to the 3′-end of the genomic RNA (acceptor RNA). This transfer is mediated by base pairing of the complementary R sequences at the 3′-ends of the RNA and DNA molecules. The 3′-R sequence of the genomic RNA folds into secondary structures corresponding to the transactivator response element (TAR) hairpin and the upper part of the poly(A) hairpin (3–6). The R sequence of ss-cDNA is predicted to fold into hairpins that are complementary to the TAR and poly(A) RNA sequences and are therefore named cTAR and cpoly(A), respectively (7,8). Mutational analysis of HIV-1 genomic RNA suggests that the great majority of first-strand transfers occur after completion of ss-cDNA synthesis, i.e. with the entire cTAR sequence (9). FRET-based assays performed with the cTAR sequence suggest that the majority (~70–80%) of the cTAR DNA molecules adopt the hairpin conformation (closed form) and the remaining (20–30%) are partially or totally melted (open forms) (8,10). Furthermore, single-molecule techniques (11,12) were used to show that the NC-coated cTAR hairpin exhibits a dynamic equilibrium between a ‘Y’-shaped and a closed conformation. In the ‘Y’ conformation, the lower stems are open and the upper stems are closed. Recently, using NMR and probing methods, we determined the structural and dynamic properties of the top half of the cTAR DNA (mini-cTAR, Figure 1A) (13). We showed that the upper stem located between the apical and the internal loops is stable, but that the lower stem of mini-cTAR is unstable. Note that the secondary structure of the full-length cTAR sequence has not been determined to date.
Figure 1.

(A) Predicted secondary structures for the TAR and cTAR sequences. Zuker’s folding programs (48) were used to predict the most stable secondary structures for RNAs and DNAs. The TAR RNA and cTAR DNA sequences are derived from the HIV-1 MAL isolate. Numbering is relative to the TAR and cTAR sequences. Mutations are shown as lower case letters in boxes. (B) Sequences of proteins used in this study.
Unfolding of the complementary hairpins is thought to be rate-limiting in the annealing process leading to the first-strand transfer (14). In vitro studies show that NC stimulates the first-strand transfer (15–18), due, at least in part, to the NC stimulatory effect on the rate of annealing of the complementary hairpins (14,19). NC is a nucleic acid chaperone that facilitates the rearrangement of nucleic acids into the most thermodynamically stable structures containing the maximum number of base pairs (20). This chaperone function of NC relies on two main components: duplex destabilization by the zinc fingers and nucleic acid aggregation, primarily by the basic N-terminal domain (Figure 1B) (19). NC induces a limited melting of the TAR RNA secondary structure (one base pair per molecule) (8), but largely enhances the fraying of the cTAR hairpin ends, shifting the distribution of hairpin conformations toward the more open structures (8,10,21). The cTAR sequence binds eight NC molecules at saturating protein concentrations under low ionic strength conditions (22), through both weak and strong binding sites (23), not yet identified.
Since mutations in the TAR apical loop decrease the first strand transfer in vitro, Berkhout et al. (7) suggested that this process involves a ‘kissing complex’ formed by the apical loops of TAR and cTAR. Studies using TAR RNA and cTAR DNA hairpins derived from the HIV-1 NL4-3 isolate, suggest that both the apical loops and the 3′/5′ termini of complementary hairpins are the initiation sites for the annealing reaction (24,25). Indeed, Vo et al. (24) reported that cTAR DNA–TAR RNA annealing in the absence of NC depends on nucleation through the apical loops, whereas cTAR DNA–TAR RNA annealing in the presence of saturated NC depends on nucleation through the 3′/5′ termini, resulting in the formation of a ‘zipper’ intermediate. Moreover, cTAR DNA and TAR RNA anneal via both ‘kissing’ and ‘zipper’ pathways under subsaturating concentrations of NC (24). Single-molecule FRET studies support the notion that the annealing process relies on two pathways (25). The annealing process has also been investigated using sequences derived from the HIV-1 MAL isolate (5,26). Thus, Godet et al. (26) reported that under low-salt concentrations, the TAR hairpins anneal almost exclusively via the ‘zipper’ pathway in the presence of NC(12–55), a truncated form of NC which lacks the N-terminal domain and is therefore a poor aggregating agent. Finally, we showed that efficient annealing of cTAR DNA to the 3′-end of the genomic RNA relies on sequence complementarities between TAR and cTAR apical loops under high-salt concentrations allowing cDNA synthesis by RT and strand transfer (5,17,27).
In this study, we investigated the annealing process using DNA and RNA sequences derived from the HIV-1 MAL isolate (Figure 1A) under high-salt concentrations. The ‘kissing complex’ was trapped using site-directed mutagenesis and investigated by fluorescence anisotropy (FA). Using chemical and enzymatic probes, and gel retardation assays, we determined the cTAR secondary structure and the NC binding sites within cTAR.
MATERIALS AND METHODS
Protein, DNA and RNA preparation
Full-length NC [NC(1–55)] and the truncated version of NC [NC(11–55)] were synthesized by the Fmoc/opfp chemical method and purified to homogeneity by HPLC (28). Proteins were dissolved at concentrations of 1 to 2 mg/ml in a buffer containing 25 mM HEPES (pH 6.5), 50 mM NaCl and 2.2 mol of ZnCl2 per mole of peptide. DNA oligonucleotides were purchased from Eurogentec and were 5′-end labeled using T4 polynucleotide kinase (New England Biolabs) and [γ-32P] ATP (Perkin Elmer). Full-length cTAR DNA was labeled at its 3′-end as follows. An oligonucleotide (5′-CTGGTCTCTCTTGTTAGACCAG-3′) was annealed to the 3′-end of cTAR. This oligonucleotide carries at its 5′-end a non-hybridizing tail of two nucleotides (5′-CT-3′). To add one labeled adenine residue to the 3′-end of cTAR, the annealed cTAR was incubated at 75°C with [α-32P] dATP and Taq DNA polymerase (New England Biolabs). The 5′- and 3′-end-labeled DNA oligonucleotides were purified by electrophoresis on 12% or 15 % denaturing polyacrylamide gels and isolated by elution followed by ethanol precipitation. DNA template for transcription of TAR RNA was prepared by PCR amplification of pHIVCG8.6 (29) using the expandTM high-fidelity PCR system (Roche Molecular Biochemicals) and oligonucleotides from Eurogentec. The upstream primer included the T7 promoter and its sequence was: 5′-GAGTAATACGACTCACTATAGGGTCTCTCTTGTTAG-3′. The sequence for the downstream primer was: 5′-GGGTTCCTTGCTAGCC-3′. The PCR product was purified as described previously (30). DNA template for transcription of mini-TAR RNA was prepared by annealing of two DNA oligonucleotides in the transcription buffer (31). The oligonucleotides contained the T7 promoter and the sequences were: 5′-TAATACGACTCACTATAG-3′ and 5′-GGCAGAGAGCTCCCGGGCTCGACCTGCCTATAGTGAGTCGTATTA-3′. RNAs were transcribed in vitro using the T7-MEGAshortscriptTM high yield transcription kit (Ambion). RNAs were purified by electrophoresis on 12% or 15% denaturing polyacrylamide gels as described previously (30). RNAs were treated with alkaline phosphatase from calf intestine (Roche Molecular Biochemicals) and 5′-end-labeled using T4 polynucleotide kinase (New England Biolabs) and [γ-32P] ATP (Perkin Elmer). The 5′-end-labeled RNAs were purified by electrophoresis on 12% or 15% denaturing polyacrylamide gels and isolated by elution followed by ethanol precipitation. The mini-TAR RNA labeled at its 5′-terminus by the 5/6-Rh6G was purchased from IBA GmbH Nucleic Acids Product Supply.
Gel-shift annealing assay
For preparation of loose heteroduplexes, 100 pmol of mini-TAR 32P-RNA (2 × 102 cpm/pmol) or 120 pmol of mini-cTAR 32P-DNA (2 × 102 cpm/pmol) in 4 µl of water was heated at 90°C for 2 min and chilled for 2 min on ice. Then, 1 µl of strand transfer buffer was added (final concentrations: 75 mM KCl, 7 mM MgCl2 and 50 mM Tris–HCl, pH 7.8) and the sample was incubated at 37°C for 30 min. Unlabeled mini-cTAR DNA (120 pmol) or unlabeled mini-TAR RNA (100 pmol) underwent the same renaturation treatment and was then added to the labeled hairpins. The reaction mixture was then incubated at 4°C for 60 min. Then, 3.5 µl of loading buffer was added to the sample. The samples were loaded on 4% agarose (QA-AgaroseTM, Qbiogene) gels. Electrophoresis was carried out at 4°C in TBM buffer [45 mM Tris–borate (pH 8.3), 1 mM MgCl2] or at 25°C in TBE buffer [45 mM Tris–borate (pH 8.3), 1 mM EDTA]. After electrophoresis, the gel was fixed, dried and autoradiographed as described (32).
Gel retardation assays
Assays were carried out in a final volume of 10 µl. Mini-cTAR 32P-DNA (10 pmol) at 2 × 103 cpm/pmol was dissolved in 6 µl of water, heated at 90°C for 2 min and chilled for 2 min on ice. Then, 2 µl of strand transfer buffer was added (final concentrations: 75 mM KCl, 7 mM MgCl2 and 50 mM Tris–HCl, pH 7.8) and the sample was incubated for 15 min at 37°C in the absence or presence of protein at various concentrations. Gel loading buffer (final concentrations: 10 % w/v glycerol, 0.01 % w/v bromophenol blue, 0.01 % w/v xylene cyanol) was added and the samples were analyzed by electrophoresis on a 14% polyacrylamide gel [29:1 (w/w), acrylamide/bisacrylamide] at 4°C in 0.5× TBE buffer [45 mM Tris–borate (pH 8.3), 1 mM EDTA]. After electrophoresis, the gel was fixed, dried and autoradiographed. Free DNA and protein/DNA complexes were quantified using a PhosphorImager (Molecular Dynamics) and ImageQuant software. The fraction of bound mini-cTAR DNA (FR) was determined using the formula FR = 1−(IF/IB+IF), where IF and IB are the band intensities of free and bound mini-cTAR DNAs, respectively.
Structural probing of cTAR DNA and loose heteroduplexes
Potassium permanganate and piperidine were purchased from Sigma-Aldrich. Mung bean nuclease, DNase I and RNase T1, were purchased from New England Biolabs, Promega and Life Technologies, respectively. Structural probing of cTAR DNA was carried out in a final volume of 10 µl. cTAR 32P-DNA (1 pmol at 5×104 cpm/pmol) in 5.5 µl of water was heated at 90°C for 2 min and chilled for 2 min on ice. Then, 2.5 µl of renaturation buffer (final concentrations: 75 mM KCl, 7 mM MgCl2 and 50 mM Tris–HCl, pH 7.8 for probing with KMnO4 or DNase I; 75 mM KCl, 7 mM MgCl2 and 50 mM sodium cacodylate, pH 6.5 for probing with mung bean nuclease) was added and the sample was incubated for 30 min at 37°C. At the end of this incubation, the samples were incubated for 15 min at 37°C in the absence or presence of NC at various concentrations. The samples were then incubated with KMnO4 or an enzyme as follows: 1.5, 3 or 6 U of mung bean nuclease for 15 min at 37°C; or 0.05, 0.1 or 0.2 U of DNase I for 7 min at 37°C. These cleavage reactions were stopped by phenol–chloroform extraction followed by ethanol precipitation. The dried pellets were resuspended in 10 µl of loading buffer (7 M urea, 0.03 w/v% bromophenol blue and 0.03 w/v% xylene cyanol). For potassium permanganate probing, cTAR DNA was treated with 0.25, 0.5 or 1 mM of KMnO4 for 1 min at 37°C. The treatment was stopped by adding 40 µl of the termination buffer [0.7 M β-mercaptoethanol, 0.4 M NaOAc (pH 7.0), 10 mM EDTA, 25 µg/ml tRNA]. DNA was then extracted with phenol–chloroform, ethanol precipitated and dried. DNA was subjected to piperidine cleavage by resuspension of the dried pellet in 100 µl of freshly diluted 1 M piperidine and heating at 90°C for 30 min. The samples were then lyophilized, resuspended in 20 µl of water and lyophilized again. After a second lyophilization from 15 µl of water, the samples were resuspended in 10 µl of loading buffer. G and T+C sequence markers of the labeled cTAR were produced by the Maxam–Gilbert method (33). All samples were analyzed by short and long migration times on denaturing 14% polyacrylamide gels.
Structural probing of loose heteroduplexes was carried out in a final volume of 10 µl. For RNase T1 probing, 100 pmol of mini-TAR 32P-RNA (3 × 102 cpm/pmol) in 4 µl of water was heated at 90°C for 2 min and chilled for 2 min on ice. Then, 1 µl of strand transfer buffer was added (final concentrations: 75 mM KCl, 7 mM MgCl2 and 50 mM Tris–HCl, pH 7.8) and the sample was incubated at 37°C for 30 min. Unlabeled mini-cTARS3 DNA (50, 100 or 200 pmol) underwent the same renaturation treatment and was then added to refolded mini-TAR 32P-RNA. The reaction mixture was incubated at 4°C for 60 min. Then, 0.2 U of RNase T1 was added and the sample was incubated at 4°C for 10 min. This cleavage reaction was stopped by phenol–chloroform extraction followed by ethanol precipitation. The dried pellet was resuspended in 10 µl of loading buffer. For mung bean nuclease probing, 100 pmol of mini-cTARS3 32P-DNA (3 × 102 cpm/pmol) in 4 µl of water was heated at 90°C for 2 min and chilled for 2 min on ice. Then, 1 µl of renaturation buffer was added (final concentrations: 75 mM KCl, 7 mM MgCl2 and 50 mM sodium cacodylate pH 6.5) and the sample was incubated at 37°C for 30 min. Unlabeled mini-TAR RNA (50, 100 or 200 pmol) underwent the same renaturation treatment and was then added to refolded mini-cTARS3 32P-DNA. The reaction mixture was incubated at 4°C for 60 min. Then, 30 U of mung bean nuclease were added and the sample was incubated at 4°C for 20 min. This cleavage reaction was stopped by phenol-chloroform extraction followed by ethanol precipitation. The dried pellet was resuspended in 10 µl of loading buffer. All samples were analyzed by short and long migration times on denaturing 17% polyacrylamide gels.
Fluorescence anisotropy
Binding of the mini-cTAR DNA sequences to 5′ 5/6-Rh6G mini-TAR RNA was investigated using FA. Measurements were performed in 50 µl quartz cuvettes using a Fluorolog spectrophotometer (Jobin Yvon) equipped with a Peltier thermostated cell holding the sample at 4°C. FA values were obtained by recording the IVV, IVH, IHV and IHH intensities (V and H characterizing the vertical and horizontal light polarisation for the excitation and the emission paths, respectively) of each solution. The steady-state anisotropy r was calculated according to: r = (IVV−G.IVH)/(IVV+2 G.IVH) in which G = IHV/IHH. Excitation and emission wavelengths were set at 520 nm and 560 nm, respectively. FA data are the result of two independent experiments performed in duplicate. Apparent equilibrium dissociation constants were determined from the FA values plotted as a function of DNA concentrations. The binding curves were fitted according to:
| (1) |
where r0 and rf are the anisotropy of mini-TAR RNA in the free and in fully bound form, respectively, and R corresponds to the fluorescence quantum yield ratio of bound mini-TAR relative to free mini-TAR. Assuming a 1:1 stoichiometry, the molar fraction ν of bound mini-cTAR DNA can be expressed as:
in which N and D stand for the concentrations of mini-TAR RNA and mini-cTAR DNA, respectively.
RESULTS
Analysis of the loop–loop interaction in the absence of NC
The mini-TAR and mini-cTAR hairpins (Figure 1) are appropriate substrates to investigate the kissing pathway in the absence of NC (24,34). To better understand the nucleation through the apical loops, we investigated the mini-TAR RNA–DNA annealing process in the strand transfer buffer, i.e. under high-salt conditions previously shown to promote annealing of cTAR DNA to the 3′-end of the genomic RNA (5). The apical loop of mini-cTAR is probably constituted of six but not four residues, since the T.G base-pair predicted by mfold (Figure 1) was not detected by NMR methods (13). An intermolecular loop–loop interaction can be characterized by the formation of loose duplexes that are stable upon gel migration at 4°C in Tris–borate magnesium (TBM) but not in TBE at 25°C (35–37). In contrast, the tight duplexes can be seen under both electrophoretic conditions (36–38). Despite numerous attempts, we could not detect loose duplexes using the wild-type and mutant sequences at low concentrations (data not shown). Therefore, annealing assays were performed at 4°C and at high concentrations of complementary hairpin molecules, i.e. under conditions allowing observation of RNA–DNA kissing complexes (39,40). To identify the annealing products, either mini-TAR 32P RNA (Figure 2A, lanes 1–6) or mini-cTAR 32P DNAs (Figure 2A, lanes 7–12) were incubated with or without the unlabeled complementary sequences. After renaturation and incubation at 4°C in the strand transfer buffer, mini-TAR RNA and mini-cTAR DNAs were mostly monomeric (Figure 2A, lanes 2 and 10–12). Addition of wild-type mini-cTAR to the wild-type mini-TAR strongly increased the band intensity that migrated at a rate expected for the heteroduplex formed by mini-TAR and mini-cTAR (Figure 2A, lanes 3). Since the band intensity did not decrease in the TBE gel, the heteroduplex was stable. These results suggest that the loop–loop interaction was converted into an extended duplex under our experimental conditions. Consistent with an annealing mechanism involving nucleation through a loop–loop interaction, mini-cTAR2CA and mini-cTARS1 (Figure 1A) did not form tight heteroduplexes with mini-TAR (Figure 2B). In addition, the results obtained with mini-cTARS1 show that a loose heteroduplex cannot be formed through a loop-loop interaction involving four base-pairs. It has been reported that the apical loop of mini-TAR RNA and the terminal loop of a DNA aptamer form a DNA–RNA loop–loop complex involving 5 bp (39,40). Interestingly, this complex and the RNA–RNA loop–loop complexes formed by the dimerization sequences of HIV-1 and avian leukosis virus are flanked by at least one unpaired nucleotide (30,41). The mini-cTARTA, mini-cTARGT and mini-cTARGC mutants were designed so that they have the potential to form a loop–loop complex involving 5 bp and one unpaired nucleotide (Figure 1). These mutants did not form loose heteroduplexes (Figure 2B), suggesting that the loop–loop complex was not formed or was very unstable. To trap the loop–loop interaction involving 6 bp, the mini-cTARS2 and mini-cTARS3 mutants were designed (Figure 1). Both mutants formed loose heteroduplexes that were observed on the TBM gel but not on the TBE gel (Figure 2A, lanes 5–8). Since the mini-cTARS4 mutant formed loose heteroduplexes (Figure 2A, lanes 4 and 9), we can exclude the possibility that the mini-cTAR mutants formed loose heteroduplexes through the 5′-/3′-terminal ends.
Figure 2.

Mutational analysis of mini-TAR RNA–DNA annealing in the absence of NC. Annealing was performed as described in ‘Materials and Methods’ section. (A) Each sample was divided in two aliquots which were analyzed by 4% agarose gel electrophoresis at 4°C in TBM buffer and at 25°C in TBE buffer. Nucleic acids were visualized by ethidium bromide staining (top) and autoradiography (bottom). Lane 1, heat-denatured mini-TAR 32P-RNA. Mini-TAR 32P-RNA was incubated in the absence (lane 2) or presence of unlabeled mini-cTAR DNAs (lanes 3–6). Mini-cTAR 32P-DNAs were incubated in the absence (lanes 10–12) or in the presence of unlabeled TAR RNA (lanes 7–9). Monomeric and dimeric forms of mini-TAR RNA are indicated by m1 and d1, respectively. Monomeric and dimeric forms of mini-cTAR DNAs are indicated by m2 and d2, respectively. Loose and tight heteroduplexes are indicated by h1 and h2, respectively. (B) The table summarizes the results of the mutational analysis.
To confirm that formation of loose heteroduplexes relies on an intermolecular loop–loop interaction, probing experiments were performed with mini-TAR RNA and mini-cTARS3 (Figure 3). First, we probed the structure of mini-TAR RNA with RNase T1 (specific for unpaired guanines) in the absence and presence of mini-cTARS3 (Figure 3A). In the absence of mini-cTARS3, there were strong and moderate T1 cleavages in the apical loop (Figure 3A, lane 1). As expected, the guanine residues in the predicted stems were not cleaved by RNase T1. The guanine residues in the apical loop became markedly unreactive to RNase T1 in the presence of increasing concentrations of mini-cTARS3 (Figure 3A, lanes 2–4). Second, we probed the structure of mini-cTARS3 DNA with mung bean nuclease (MB) in the absence and presence of mini-TAR RNA (Figure 3B). MB is highly selective for single-stranded nucleic acids and single-stranded regions in double-stranded nucleic acids (42). Note that single-base mismatches in double-stranded DNA are poor substrates for MB cleavage at 37°C (42,43). In the absence of mini-TAR RNA, there were moderate and strong MB cleavages in the apical loop and within the sequence between positions 34 and 38 (Figure 3B, lane 1). These results are consistent with our study showing that the lower stem of mini-cTAR does not form a stable double-stranded structure (13). The residues in the apical loop became markedly unreactive to MB in the presence of increasing concentrations of mini-TAR RNA (Figure 3B, lanes 2–4). In contrast, the cleavage rate for MB in the lower part of mini-cTARS3 was not significantly reduced in the presence of increasing concentrations of mini-TAR RNA. Taken together, our results show that the linkage structure of the loose heteroduplex is the loop–loop complex. To determine the apparent dissociation constants (Kd) of loop–loop complexes, 5′ 5/6-Rh6G mini-TAR RNA was mixed with increasing amounts of mini-cTARS2 or mini-cTARS3. Heteroduplexes were allowed to form for one hour at 4°C in the strand transfer buffer, before the fluorescence anisotropy of the mixture was measured. Adding the mini-cTAR mutants to 5′ 5/6-Rh6G mini-TAR RNA induced a slight but significant increase of the anisotropy. Since no change in the steady-state fluorescence spectra were observed when DNA sequences were added, the anisotropy increase indicated that mini-cTAR DNAs interacted with mini-TAR RNA. Using Equation (1) to fit the binding curves shown in Figure 4, Kd values of 3.5 (±2) × 106 M−1 were obtained with both mini-cTARS2/mini-TAR and mini-cTARS3/mini-TAR.
Figure 3.

Probing of mini-TAR:mini-cTARS3 duplex. (A) Enzymatic probing with RNase T1. Mini-TAR 32P-RNA alone (lanes C and 1) or mixed with mini-cTARS3 (lanes 2–4) was incubated in the absence (lane C) or in the presence of RNase T1 (lanes 1–4). Lanes 2–4, the RNA:DNA ratios were 1:0.5, 1:1 and 1:2, respectively. Lanes T1 and L refer to RNAse T1 sequencing and to alkaline ladder, respectively. (B) Enzymatic probing with mung bean nuclease. Mini-cTARS3 32P-DNA alone (lanes C and 1) or mixed with mini-TAR (lanes 2–4) was incubated in the absence (lane C) or in the presence of mung bean nuclease (lanes 1–4). Lanes 2–4, the DNA:RNA ratios were 1:0.5, 1:1 and 1:2, respectively. Lanes G and T+C refer to Maxam–Gilbert sequence markers. Samples were analyzed by electrophoresis on 17% denaturing polyacrylamide gels. Arrows indicate the cleavage sites. Closed, gray and open symbols indicate strong, medium and weak cleavage sites, respectively. The strong protections induced by mini-cTARS3 (A) or mini-TAR (B) are indicated by asterisks.
Figure 4.

Characterization of loose heteroduplexes by fluorescence anisotropy. 5′ 5/6-Rh6G mini-TAR RNA was mixed with increased amounts of mini-cTARS2 (triangles) or mini-cTARS3 (squares) in the absence of NC and incubated at 4°C for 60 min as described in ‘Materials and Methods’ section. The fluorescence anisotropy of the mixture was measured at the end of incubation.
Analysis of cTAR secondary structure in the absence of NC
The secondary structure of cTAR DNA was investigated at 37°C in the strand transfer buffer, i.e. under conditions previously shown to promote strand transfer and annealing of cTAR DNA to the 3′-end of the genomic RNA (5,17,27). Using gel-shift annealing assays, we checked that the cTAR DNA hairpin anneals to the TAR RNA hairpin under our high-salt conditions (Supplementary Figure S1). Consistent with previous reports (24,26), the TAR RNA–DNA annealing reaction is extremely slow in the absence of NC (Supplementary Figure S1). In addition, the full-length cTAR was monomeric after incubation as assessed by native agarose gel electrophoresis in the TBM and TBE buffers (data not shown). Therefore, the structural analysis was not complicated by the presence of homoduplexes. The secondary structure of cTAR DNA was probed with potassium permanganate (KMnO4), MB and DNase I. DNase I is a double-strand-specific endonuclease that produces single-strand nicks (44). KMnO4 can be used to detect regions of DNA that are unpaired or distorted (45,46): it is an oxidizing agent that preferentially attacks the 5,6 double bond of thymine. In B-DNA, this bond is shielded by base stacking interactions and, thus, the T residues in such DNA duplexes are relatively resistant to oxidation. After treatment of cTAR DNA with piperidine, the DNA backbone was cleaved at the site of the modified thymines. The cleavage fragments generated by the nucleases and the KMnO4/piperidine treatment were analyzed by electrophoresis on denaturing 14% polyacrylamide gels. Running Maxam–Gilbert sequence markers of cTAR on the same gels in parallel identified the cleavage sites. Representative examples of probing experiments are shown in Figures 5 and 6 and the results of a series of independent experiments are summarized in Figure 7. Note that the nucleases cleave the phosphodiester bond and generate a 3′-hydroxyl terminus on 5′-end-labeled DNA. In contrast, the KMnO4/piperidine treatment and Maxam–Gilbert reactions generate a 3′-phosphorylated terminus on 5′-end-labeled DNA (33,47). The electrophoretic mobility of Maxam–Gilbert sequence markers is therefore slightly greater than that of fragments produced by nucleases. To determine the precise position of cleavage at some sites, the 3′-end-labeled cTAR was used (examples in Figure 5).
Figure 5.

Enzymatic probing of cTAR. Enzymatic probing experiments were performed as described in ‘Materials and Methods’ section. (A and B) In the absence of NC, the 3′-end-labeled cTAR DNA was incubated with DNase I (0.05, 0.1 and 0.2 U) or mung bean nuclease (MB) (1.5, 3 and 6 U). (C) In the absence of NC, the 5′-end-labeled cTAR DNA was incubated with MB (1.5, 3 and 6 U). Lanes C are controls without enzyme. G and T+C refer to Maxam–Gilbert sequence markers. Arrows indicate the cleavage sites. (D) 5′-end-labeled cTAR DNA was incubated with MB (3 U) in the absence (lane 1) or in the presence of NC (lanes 2–5). The protein to nucleotide molar ratios were 1:8 (lane 2), 1:4 (lane 3), 1:2 (lane 4) and 1:1 (lane 5). The strong protections induced by NC at the level of MB cleavage sites are indicated by asterisks. (E) Secondary structures for the partially melted and closed forms of cTAR. Delta G-values were predicted by mfold. Closed, gray and open symbols indicate strong, medium and weak cleavage sites, respectively, for the various enzymes (triangle for DNase I and circle for mung bean nuclease).
Figure 6.

KMnO4 probing of cTAR. KMnO4 probing experiments were performed as described in ‘Materials and Methods’ section. (A) In the absence of NC, the 5′-end-labeled cTAR DNA was incubated with KMnO4 (0.25, 0.5 and 1 mM). (B) In the absence (lane 1) or in the presence of NC (lanes 2–5), the 5′-end-labeled cTAR DNA was incubated with KMnO4 (0.25 mM). Lanes C are controls without NC and any chemical treatment. Lane Cpip is the control without NC and KMnO4 treatment but with piperidine treatment. The protein to nucleotide molar ratios were 1:8 (lane 2), 1:4 (lane 3), 1:2 (lane 4) and 1:1 (lane 5). Arrows indicate the reactive thymine residues. The strong protection induced by NC at the level of T40 is indicated by an asterisk. The thymine residues where the reactivity to KMnO4 is increased by NC are indicated by the stars. G and T+C refer to Maxam-Gilbert sequence markers. (C) Secondary structures for the partially melted and closed forms of cTAR. Delta G values were predicted by mfold. The color codes used for the reactivity of thymine residues are indicated in the inset.
Figure 7.
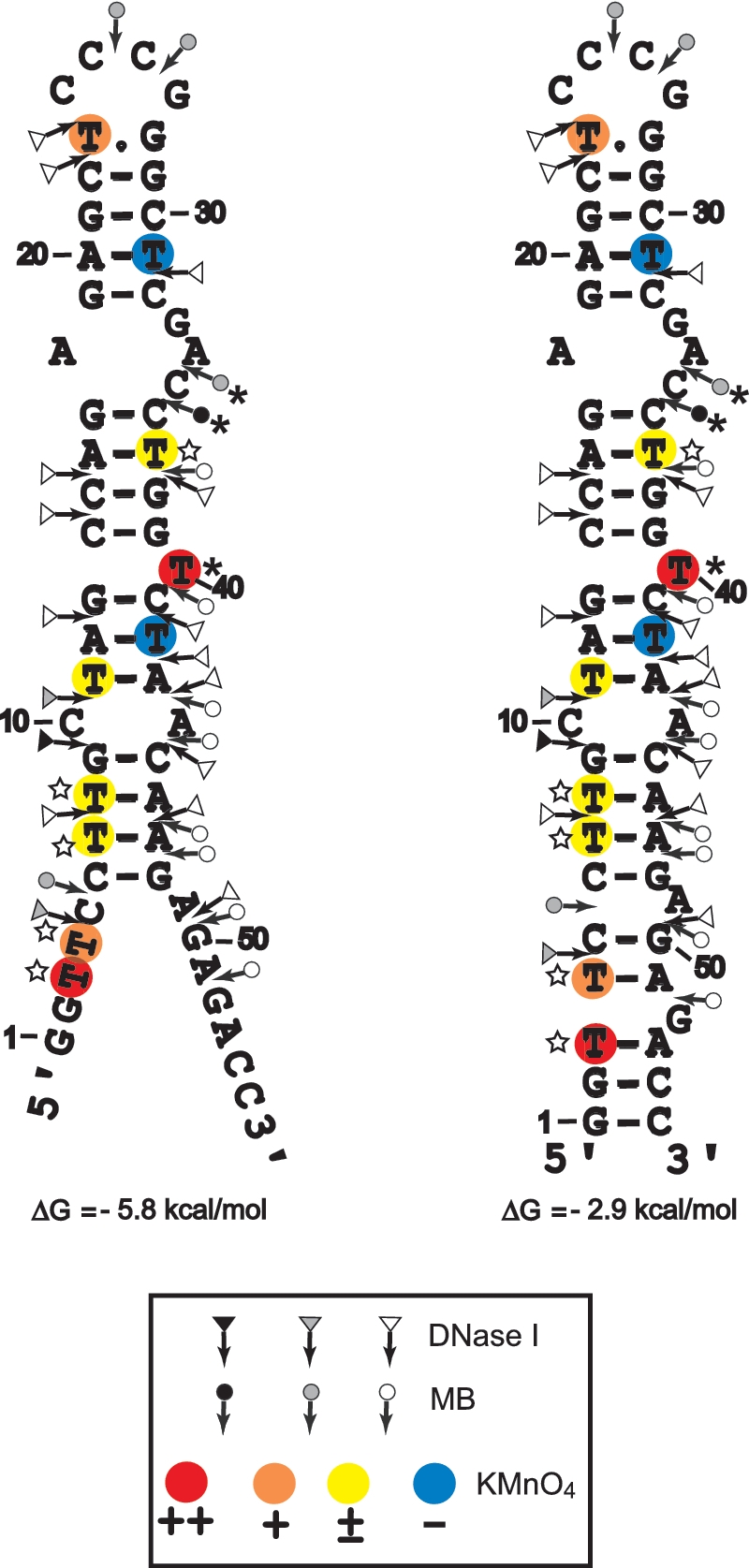
Secondary structures for the partially melted and closed forms of cTAR. Delta G-values were predicted by mfold. Closed, gray and open symbols indicate strong, medium and weak cleavage sites, respectively, for the various enzymes (triangle for DNase I and circle for mung bean nuclease). The color codes used for the reactivity of thymine residues are indicated in the inset. The strong protections induced by NC at the level of MB cleavage sites and thymine residues are indicated by asterisks. The thymine residues where the reactivity to KMnO4 is increased by NC are indicated by the stars.
The partially melted ‘Y’ conformation is the most stable cTAR structure predicted by the Mfold program (48). It has been proposed that the cTAR sequence forms the closed conformation (12,24,26). Therefore, the probing data were superimposed on these two conformations (Figure 7). In both conformations, the 6–48 sequence forms the same stem–loop structure. There were strong and moderate DNase I cleavages between G9–C10 and C10–T11, all of which are predicted to lie at the ends of stems. There were moderate and strong MB cleavages between C25–C26, C26–G27, A34–C35 and C35–C36 that are predicted to be in the apical and internal loops. The moderate and high sensitivities of T23 and T40 to KMnO4 (Figure 6A), respectively, indicate that at least one side of the plane of the heterocyclic ring of these thymine residues is exposed. These data suggest that the predicted T.G mismatched base pair is not involved in strong stacking interactions with the residues of the apical loop and T40 adopts the looped-out bulge conformation. Consistent with the formation of stems, T31 and T42 were unreactive to KMnO4 and the sensitivity of T8, T11 and T37 to KMnO4 was very low. It is likely that the C–A mismatch slightly destabilizes the adjacent base-pairs and the internal loop destabilizes the G17–C36 base pair (13), since T8, T11 and T37 were not totally protected by the surrounding base pairs. Fraying of the C6–G48 base pair is probably responsible for the low sensitivity of T7. The high and moderate sensitivities of T3 and T4 to KMnO4, respectively and the moderate MB cleavage between C5 and C6 are consistent with the partially melted conformation of cTAR. However, the 3′-/5′-terminal ends may be involved in transient interactions to form the closed conformation since there was moderate DNase I cleavage between T4 and C5. Taken together, our probing data support a structural heterogeneity for the 3′-/5′-terminal ends of cTAR.
Analysis of cTAR secondary structure in the presence of NC
We checked that the cTAR DNA hairpin anneals to the TAR RNA hairpin in the presence of NC under structural analysis conditions (Supplementary Figure S1). Consistent with previous reports (24,26), the TAR RNA–DNA annealing reaction is dramatically accelerated by NC (Supplementary Figure S1). The full-length cTAR was monomeric after incubation with NC as assessed by native polyacrylamide gel electrophoresis in the TBE buffer (data not shown). Therefore, the structural analysis was not complicated by the presence of homoduplexes. To identify destabilized regions and protections induced by NC in the cTAR hairpin, we compared the enzymatic and KMnO4 probing patterns of cTAR in the absence or presence of increasing concentrations of NC (Figures 5 and 6). Consistent with the formation of stable stems, T11, T31 and T42 did not become more reactive to KMnO4 in the presence of NC (Figure 6B). The sensitivity of T3, T4, T7 and T8 to KMnO4 was slightly increased with NC at protein to nucleotide molar ratios of 1:8, 1:4 and 1:2 (Figure 6B, lanes 2–4), suggesting that the population of cTAR in the ‘Y’ conformation increases in the presence of NC. It is likely that the stem containing T37 is slightly destabilized by NC, since the sensitivity of this thymine residue was slightly increased with NC at protein to nucleotide molar ratios of 1:8, 1:4 and 1:2 (Figure 6B, lanes 2–4). In the presence of NC at protein to nucleotide molar ratios higher than 1:2, the efficiency of DNA recovery after phenol–chloroform extraction was reduced for KMnO4 assays. Therefore, the sensitivity of thymine residues to KMnO4 seems to decrease with NC at a protein to nucleotide molar ratio of 1:1 (Figure 6B, lane 5). The double-stranded region containing the T bulge is probably a binding site for NC since the protein induced a significant decrease in KMnO4 sensitivity for T40. In the presence of increasing concentrations of NC, DNase I cleavage at the level of all sites decreased at the same rate, i.e. NC did not induce specific protection against DNase I (data not shown). These results indicate that the double-stranded regions of cTAR do not contain preferential binding sites for NC. Interestingly, NC induced a strong decrease in MB sensitivity for A34 and C35 (Figure 5D) that are located in the internal loop (Figure 7). PhosphorImager quantification indicated that the cleavage rate for MB in the internal loop at a protein to nucleotide molar ratio of 1:4 corresponded to 52% of that observed in the absence of NC. In contrast, at the same ratio, the cleavage rate for MB in the apical loop (residues C25 and C26) corresponded to 96% of that observed in the absence of NC. These data indicate that the internal loop is a strong binding site for NC, and that, if NC binds the apical loop, its binding is too weak to be detected at a protein to nucleotide molar ratio of 1:4. However, phosphorImager quantification indicated that the cleavage rate for MB in the apical loop at a protein to nucleotide molar ratio of 1:2 corresponded to 55% of that observed in the absence of NC, i.e. NC binds the apical loop at protein to nucleotide molar ratios equal to or greater than 1:2 (Figure 5D, lanes 4 and 5). Taken together the data indicate that the apical and internal loops are weak and strong binding sites for NC, respectively. Finally, our results show that NC preferentially binds the internal loop, slightly destabilizes the lower stem that is adjacent to this loop, and shifts the equilibrium toward the ‘Y’ conformation.
Binding of NC to mini-cTAR
To confirm that the apical loop is not a strong binding site for NC, we carried out gel retardation assays with wild-type and mutant mini-cTAR DNAs (Figure 1A). Since NC binds nucleic acids with a preference for sequences containing unpaired guanine residues (49–55), we altered the apical and internal loops containing guanine residues. The LS mutant was designed so that four T bases replaced the CCCG sequence in the apical loop. The internal loop sequence was deleted in the IN2 mutant. Protein:DNA complexes were formed under the same salt conditions and at protein to nucleotide molar ratios as those used in probing experiments (see ‘Materials and Methods’ section). Heat-denatured mini-cTAR DNAs (Figure 8A and B, lanes 2) were used to locate the position of monomeric mini-cTAR DNAs. To identify the positions of bands corresponding to dimeric mini-cTAR DNAs, the proteins were removed before gel analysis (Figure 8A and B, lanes 1). After renaturation and incubation at a protein to nucleotide molar ratio of 1:1, the majority of mini-cTAR DNAs remained monomeric (Figure 8A and B, lanes 1). Addition of increasing amounts of NC resulted in the appearance of two bands (Figure 8A): band CI migrating at a rate expected for the NC:mini-cTAR complex and band CII corresponding to high-molecular-weight protein:mini-cTAR complexes (aggregates) that stuck in the wells. The LS mutation did not lead to reduction in NC binding (Figure 8C). Interestingly, deletion of the internal loop significantly reduced the binding of NC to the IN2 mutant (Figure 8C).
Figure 8.

Gel retardation assays of protein:mini-cTAR DNA complexes formed in vitro. Mini-cTAR 32P-DNAs were incubated in presence of NC (A) or NC(11-55) (B) and analyzed by electrophoresis on a 14% polyacrylamide gel as described in ‘Materials and Methods’ section. Lanes 1, controls mini-cTAR dimerization induced by NC or NC(11-55) at a protein to nucleotide molar ratio of 1:1 [NC and NC(11-55) were removed by phenol/chloroform before gel electrophoresis]. Lanes 2, heat-denatured mini-cTAR DNAs. Lanes 3, controls without protein; lanes 4–7, protein to nucleotide molar ratios were 1:8, 1:4, 1:2 and 1:1. Monomeric and dimeric forms of free mini-cTAR DNAs are indicated by fm and fd, respectively. CI indicates the protein:mini-cTAR complexes. CII indicates the high molecular mass protein:mini-cTAR complexes (aggregates). (C) The graph was derived from the experiments shown in (A). (D) The graph was derived from the experiments shown in (B). Symbols: filled circles, wt; open circles, LS; open squares, IN2.
The nucleic acid aggregating activity of NC resides primarily in the basic N-terminal domain (29,56–58), and the duplex destabilizing activity of NC has been mapped to its zinc finger structures (22,59–62). NC exhibits sequence-specific binding to single-stranded regions through interactions that involve the zinc fingers (51,53,63). To characterize specific interactions between the zinc-finger structures and the mini-cTAR hairpin, we used NC(11–55) which lacks the basic N-terminal domain (Figure 1B). As expected, NC(11–55) did not form aggregates with mini-cTAR DNAs (Figure 8B). Addition of increasing amounts of NC(11–55) resulted in the appearance of band CI migrating at a rate expected for the NC(11–55):mini-cTAR complex. Binding of NC(11–55) to the LS mutant was not impaired by mutations in the apical loop (Figure 8B and D). Interestingly, NC(11–55) did not bind to the IN2 mutant. Taken together, these results show that the internal loop sequence is important for tight binding of NC(11–55) to cTAR and that the NC zinc fingers do not bind the apical loop under conditions allowing strand transfer.
DISCUSSION
The full-length cTAR adopts two alternative conformations in the absence of NC
FRET-based assays have been developed to study the folding of the cTAR DNA molecule derived from the MAL (8) or NL4-3 isolate (10). These studies suggest that the majority (~70–80%) of the cTAR DNA molecules adopt the hairpin conformation (closed form) and the remaining (20–30%) are partially or totally melted (open forms) in the absence of NC. Using single-molecule spectroscopy (SMS) techniques and cTAR DNA molecules derived from the NL4-3 isolate, it has been reported that the partially open ‘Y’ conformation is the dominant form for the cTAR hairpin in the NC-mediated annealing process (12,64). These studies suggest that the 5′ and 3′ unpaired termini of the ‘Y’ conformation are accessible for TAR RNA–DNA annealing. Note that the double-stranded and single-stranded regions of the ‘Y’ conformation were deduced from the analysis of hairpin mutants. Here, we used structural probes (nucleases and potassium permanganate) to directly determine the secondary structure of the full-length cTAR derived from the MAL isolate. The probing experiments were performed under high-salt conditions previously shown to promote strand transfer and annealing (5,17,27). It is unlikely that a population of cTAR DNA molecules is totally melted in the absence of NC under our conditions, since T31 and T42 were unreactive to potassium permanganate and the sensitivity of T8, T11 and T37 to this reagent was very low (Figures 6 and 7). The 6–48 sequence forms a stem–loop structure containing a C–A mismatch, a T bulge in the looped-out conformation and an internal loop (Figure 7). Consistent with the stem involving nucleotides 6–9/45–48, the sensitivity of T8 to potassium permanganate was very low and there was a strong DNase I cleavage between G9 and C10. Nucleotides 1–5 and 50–55 are paired in the closed conformation and are unpaired in the ‘Y’ conformation. In summary, our probing data are consistent with the equilibrium between the closed and ‘Y’ conformations of the cTAR hairpin in the absence of NC.
NC’ effects on the structure of the cTAR DNA hairpin
FRET-based assays suggest that the populations of the partially or totally melted forms of the cTAR hairpin are moderately increased in the presence of NC or NC(12–55) (8,10). The regions opened by NC are expected not to become accessible to MB because they probably bind the nucleocapsid protein. Consistent with this notion, MB cleavage did not increase in the presence of NC (Figure 5D). Studies performed with DNA oligonucleotides showed that NC binds to the single-stranded regions with the following preference: TG > TNG, GNG > G, where N corresponds to either A or C (50,52,65). Recently, NMR studies performed with DNA oligonucleotides containing TG and TNG sequences showed that the nucleobase of the thymine residue is partly inserted in the N-terminal zinc finger of NC and is involved in stacking interactions with the Phe16 side chain (63,66). Residues T3, T4, T23, T40 and T42 are predicted not to be involved in stacking interactions with the Phe16 side chain of NC, since they are not present in TG and TNG sequences. In addition, a close examination of the structures of the NC:DNA complexes (63,66) shows that the 5,6 double bond of the nucleobase of the thymine residue is accessible to potassium permanganate (KMnO4) that is a small probe. Therefore, KMnO4 is expected to interact with the nucleobases of unpaired thymine residues that interact with the N-terminal zinc finger of NC. Since T31 and T42 did not become sensitive to KMnO4 in the presence of NC (Figure 6B), it is likely that the 12A–T42 and 20A–T31 base pairs are stable. Therefore, there is no evidence that a population of cTAR DNA molecules is totally melted in the presence of NC under our experimental conditions. Our probing data are consistent with the SMS study that does not report the observation of totally melted forms (64). NC induced a slight increase in KMnO4 sensitivity for residues T3, T4, T7 and T8 (Figure 6B), suggesting that the population of cTAR in the ‘Y’ conformation (Figure 7) increases in the presence of NC. Our results are in agreement with the hypothesis that NC shifts the equilibrium toward the O1 and O2 partially open forms (8). In the lower part of the cTAR hairpin, the O1 and O2 forms exhibit 12 and 22 unpaired nucleotides, respectively. The sensitivity of T37 to KMnO4 was slightly increased with NC (Figure 6B), suggesting that the stem involving nucleotides 14CCAG17/36CTGG39 is slightly destabilized by NC. For the full-length cTAR hairpin in the absence of NC, this stem is stable since the sensitivity of T37 to KMnO4 was barely detectable (Figure 6A). However, NMR investigation of mini-cTAR in the absence of NC suggests that this stem may be destabilized by alternative possibilities of base pairing involving the residues of the internal loop and the 17G–C36 base pair (13). An attractive hypothesis is that binding of NC to the internal loop facilitates these conformational exchange phenomena. Finally, our data suggest that NC destabilizes the lower stem that is adjacent to the internal loop and shifts the equilibrium toward the ‘Y’ conformation.
NC binding sites in the cTAR hairpin
A recent study suggests that the cTAR sequence contains weak and strong binding sites for NC (23). Identification of preferred binding sites in the cTAR molecule is necessary to propose models for the annealing mechanism mediated by NC. Our study is the first to identify the NC binding sites using full-length NC and full-length cTAR DNA under salt conditions previously shown to promote strand transfer and annealing (5,17,27). The DNase I probing data indicate that the double-stranded regions of cTAR bind NC but do not contain preferential binding sites for this protein. In a previous report, we showed that the N-terminal basic domain of NC is required for providing protection against RNase V1 that cleaves double-stranded RNA (5). These results are consistent with the notion that NC binds the double-stranded regions of nucleic acids non-specifically through electrostatic interactions of the basic residues with the phosphodiester backbone (29,67).
Binding of NC to the double-stranded region containing the T bulge is probably responsible for the significant decrease in KMnO4 sensitivity for T40 (Figure 6B). A likely explanation is that this thymine residue is stacked between the flanking bases in the presence of NC while it was in looped out conformation in the absence of NC. Interestingly, Kalnik et al. (68) reported an equilibrium between the stacked and looped-out conformations of a T bulge. Since NC can bend short segments of double-stranded nucleic acids (67), an attractive hypothesis is that the NC-induced bending of cTAR shifts the equilibrium towards the stacked conformation of the T bulge. Using mung bean nuclease (MB), we showed that the apical and internal loops are weak and strong binding sites for NC, respectively. Moreover, since mutations in the apical loop had no influence on the binding of the mini-cTAR to NC and NC(11–55) (Figure 8) and since NC(11–55) did not bind the mini-cTARIN2 mutant displaying the apical loop but not the internal loop, it is likely that binding of NC to the apical loop relies mainly on non-specific interactions involving the N-terminal basic domain. Moreover, NMR experiments performed with mini-cTAR in low ionic strength buffer showed that there is no significant NC(11–55) binding to the apical loop (66). It is likely that the NC zinc fingers do not interact with the two unpaired guanine residues of the apical loop because these residues are involved in stacking or transient hydrogen bonding interactions with neighbor nucleotides (13).
Mechanisms of annealing and roles of NC
Our probing data support the equilibrium between the closed and ‘Y’ conformations of the cTAR hairpin, i.e. the existence of two sites (the single-stranded ends and the apical loop) for initiation of the annealing reaction. The secondary structure of the ‘Y’ conformation (Figure 7) suggests that nucleation through the 3′/5′ termini results in formation of a zipper intermediate involving at least 12 bp which is subsequently converted into the final extended duplex. Since the apical loop of the cTAR DNA hairpin is a weak binding site for NC, it can probably interact with the apical loop of the TAR RNA hairpin in the presence of NC. Using mini-cTAR mutants, gel-shift annealing assays, probing experiments and fluorescence anisotropy we showed that the annealing intermediate of the kissing pathway is the loop–loop interaction involving 6 intermolecular base pairs (Figures 2–4). The enzymatic and chemical probing data suggest that the apical stems are not destabilized by the loop–loop interaction (Figure 3 and data not shown). In their preceding work, Vo et al. (34) used a mini-cTAR mutant named Inv-1 that contains inverted sequences for the internal loop and the apical stem. This mutant designed to trap the 6 bp kissing complex, did not form heteroduplex with mini-TAR RNA (34). From these results, Vo et al. (34) suggested that the annealing intermediate involves 17 intermolecular base pairs, including the nucleotides from the apical stem–loop and from the internal loop of cTAR. The apparent discrepancy between our findings and this previous work can be explained by the fact that the secondary structure of our mutants is similar to that of the wild-type mini-cTAR, whereas this is not the case for the Inv-1 mutant. Indeed, the configuration of the internal loop in our mutants is similar to that of the wild-type mini-cTAR hairpin (the internal loop is located near the 3′-end), whereas it is not the case for the Inv-1 mutant (the internal loop is located near the 5′-end). In addition, we used experimental conditions (low temperature, high concentrations of nucleic acids) that were different from those used in the previous study. Our findings in conjunction with previous works (24,26) are consistent with annealing mechanisms that involve the fast formation of unstable kissing and zipper intermediates, followed by a rate-limiting strand exchange between the stems of hairpins. TAR RNA–DNA annealing via the zipper intermediate is favored in the presence of saturated NC (24). This finding is consistent with our probing data showing that the stem end is less stable than the apical stem which is not destabilized by NC. In addition, NC shifts the equilibrium toward the ‘Y’ conformation and slightly destabilizes the lower stem that is adjacent to the internal loop. Preferential binding of NC zinc fingers to the internal loop is probably responsible for destabilization of the adjacent stem.
CONCLUSIONS
Here, we investigated the TAR RNA–DNA annealing mechanism in the absence or presence of NC under high-salt conditions previously shown to promote strand transfer (17,27). Our study is the first to show that the annealing intermediate of the kissing pathway is the loop–loop complex involving 6 intermolecular base pairs. Our results support a model for the NC–cTAR interactions in which one molecule of NC binds the internal loop tightly via its zinc fingers, whereas other NC molecules bind the apical loop and the double-stranded regions weakly via the basic residues scattered along the protein. The timing of HIV-1 core disassembly is essential for viral DNA synthesis, but there is still no consensus on exactly when uncoating occurs (69). Dissociation of the capsid shell surrounding the nucleoprotein complex is believed to take place during reverse transcription. The NC molecules are probably diluted by the disassembly of viral core. Dilution of NC is expected to induce a significant dissociation of NC molecules that are weakly bound to the minus-strand strong stop DNA. An attractive hypothesis, based on our results, is that the initiation sites for the annealing reaction (the apical loop and the 3′/5′ termini of cTAR) are not bound to NC and that NC binds only its strong binding site (the cTAR internal loop) when uncoating and the first strand occur simultaneously. Binding of NC to the internal loop of the cTAR DNA hairpin would be essential for destabilization of this hairpin, and therefore for the annealing step of first strand transfer.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Agence Nationale de Recherche sur le Sida; Institut national de la santé et de la recherche médicale; Postdoctoral fellowship from Agence Nationale de Recherche sur le Sida (to I.K.); PhD fellowship from Ministère des Affaires Etrangères (to Y.C.); Funding for open access charge: Centre National de la Recherche Scientifique (CNRS LBPA UMR 8113).
Conflict of interest statement. None declared.
Supplementary Material
REFERENCES
- 1.Basu VP, Song M, Gao L, Rigby ST, Hanson MN, Bambara RA. Strand transfer events during HIV-1 reverse transcription. Virus Res. 2008;134:19–38. doi: 10.1016/j.virusres.2007.12.017. [DOI] [PubMed] [Google Scholar]
- 2.Mougel M, Houzet L, Darlix JL. When is it time for reverse transcription to start and go? Retrovirology. 2009;6:24. doi: 10.1186/1742-4690-6-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Feng S, Holland EC. HIV-1 tat trans-activation requires the loop sequence within tar. Nature. 1988;334:165–167. doi: 10.1038/334165a0. [DOI] [PubMed] [Google Scholar]
- 4.Berkhout B, Klaver B, Das AT. A conserved hairpin structure predicted for the poly(A) signal of human and simian immunodeficiency viruses. Virology. 1995;207:276–281. doi: 10.1006/viro.1995.1077. [DOI] [PubMed] [Google Scholar]
- 5.Kanevsky I, Chaminade F, Ficheux D, Moumen A, Gorelick R, Negroni M, Darlix JL, Fosse P. Specific interactions between HIV-1 nucleocapsid protein and the TAR element. J. Mol. Biol. 2005;348:1059–1077. doi: 10.1016/j.jmb.2005.03.046. [DOI] [PubMed] [Google Scholar]
- 6.Watts JM, Dang KK, Gorelick RJ, Leonard CW, Bess JW, Jr, Swanstrom R, Burch CL, Weeks KM. Architecture and secondary structure of an entire HIV-1 RNA genome. Nature. 2009;460:711–716. doi: 10.1038/nature08237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Berkhout B, Vastenhouw NL, Klasens BI, Huthoff H. Structural features in the HIV-1 repeat region facilitate strand transfer during reverse transcription. RNA. 2001;7:1097–1114. doi: 10.1017/s1355838201002035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bernacchi S, Stoylov S, Piemont E, Ficheux D, Roques BP, Darlix JL, Mely Y. HIV-1 nucleocapsid protein activates transient melting of least stable parts of the secondary structure of TAR and its complementary sequence. J. Mol. Biol. 2002;317:385–399. doi: 10.1006/jmbi.2002.5429. [DOI] [PubMed] [Google Scholar]
- 9.Ohi Y, Clever JL. Sequences in the 5' and 3' R elements of human immunodeficiency virus type 1 critical for efficient reverse transcription. J. Virol. 2000;74:8324–8334. doi: 10.1128/jvi.74.18.8324-8334.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hong MK, Harbron EJ, O'Connor DB, Guo J, Barbara PF, Levin JG, Musier-Forsyth K. Nucleic acid conformational changes essential for HIV-1 nucleocapsid protein-mediated inhibition of self-priming in minus-strand transfer. J. Mol. Biol. 2003;325:1–10. doi: 10.1016/s0022-2836(02)01177-4. [DOI] [PubMed] [Google Scholar]
- 11.Azoulay J, Clamme JP, Darlix JL, Roques BP, Mely Y. Destabilization of the HIV-1 complementary sequence of TAR by the nucleocapsid protein through activation of conformational fluctuations. J. Mol. Biol. 2003;326:691–700. doi: 10.1016/s0022-2836(02)01430-4. [DOI] [PubMed] [Google Scholar]
- 12.Liu HW, Zeng Y, Landes CF, Kim YJ, Zhu Y, Ma X, Vo MN, Musier-Forsyth K, Barbara PF. Insights on the role of nucleic acid/protein interactions in chaperoned nucleic acid rearrangements of HIV-1 reverse transcription. Proc. Natl Acad. Sci. USA. 2007;104:5261–5267. doi: 10.1073/pnas.0700166104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zargarian L, Kanevsky I, Bazzi A, Boynard J, Chaminade F, Fosse P, Mauffret O. Structural and dynamic characterization of the upper part of the HIV-1 cTAR DNA hairpin. Nucleic Acids Res. 2009;37:4043–4054. doi: 10.1093/nar/gkp297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.You JC, McHenry CS. Human immunodeficiency virus nucleocapsid protein accelerates strand transfer of the terminally redundant sequences involved in reverse transcription. J. Biol. Chem. 1994;269:31491–31495. [PubMed] [Google Scholar]
- 15.Darlix JL, Vincent A, Gabus C, de RH, Roques B. Trans-activation of the 5' to 3' viral DNA strand transfer by nucleocapsid protein during reverse transcription of HIV1 RNA. C. R. Acad. Sci. III. 1993;316:763–771. [PubMed] [Google Scholar]
- 16.Peliska JA, Balasubramanian S, Giedroc DP, Benkovic SJ. Recombinant HIV-1 nucleocapsid protein accelerates HIV-1 reverse transcriptase catalyzed DNA strand transfer reactions and modulates RNase H activity. Biochemistry. 1994;33:13817–13823. doi: 10.1021/bi00250a036. [DOI] [PubMed] [Google Scholar]
- 17.Guo J, Henderson LE, Bess J, Kane B, Levin JG. Human immunodeficiency virus type 1 nucleocapsid protein promotes efficient strand transfer and specific viral DNA synthesis by inhibiting TAR-dependent self-priming from minus-strand strong-stop DNA. J. Virol. 1997;71:5178–5188. doi: 10.1128/jvi.71.7.5178-5188.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen Y, Balakrishnan M, Roques BP, Bambara RA. Steps of the acceptor invasion mechanism for HIV-1 minus strand strong stop transfer. J. Biol. Chem. 2003;278:38368–38375. doi: 10.1074/jbc.M305700200. [DOI] [PubMed] [Google Scholar]
- 19.Levin JG, Guo J, Rouzina I, Musier-Forsyth K. Nucleic acid chaperone activity of HIV-1 nucleocapsid protein: critical role in reverse transcription and molecular mechanism. Prog. Nucleic Acid Res. Mol. Biol. 2005;80:217–286. doi: 10.1016/S0079-6603(05)80006-6. [DOI] [PubMed] [Google Scholar]
- 20.Thomas JA, Gorelick RJ. Nucleocapsid protein function in early infection processes. Virus Res. 2008;134:39–63. doi: 10.1016/j.virusres.2007.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Beltz H, Azoulay J, Bernacchi S, Clamme JP, Ficheux D, Roques B, Darlix JL, Mely Y. Impact of the terminal bulges of HIV-1 cTAR DNA on its stability and the destabilizing activity of the nucleocapsid protein NCp7. J. Mol. Biol. 2003;328:95–108. doi: 10.1016/s0022-2836(03)00244-4. [DOI] [PubMed] [Google Scholar]
- 22.Beltz H, Clauss C, Piemont E, Ficheux D, Gorelick RJ, Roques B, Gabus C, Darlix JL, de Rocquigny H, Mely Y. Structural determinants of HIV-1 nucleocapsid protein for cTAR DNA binding and destabilization, and correlation with inhibition of self-primed DNA synthesis. J. Mol. Biol. 2005;348:1113–1126. doi: 10.1016/j.jmb.2005.02.042. [DOI] [PubMed] [Google Scholar]
- 23.Shvadchak VV, Klymchenko AS, de RH, Mely Y. Sensing peptide-oligonucleotide interactions by a two-color fluorescence label: application to the HIV-1 nucleocapsid protein. Nucleic Acids Res. 2009;37:e25. doi: 10.1093/nar/gkn1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vo MN, Barany G, Rouzina I, Musier-Forsyth K. HIV-1 nucleocapsid protein switches the pathway of transactivation response element RNA/DNA annealing from loop-loop “kissing” to “zipper”. J. Mol. Biol. 2009;386:789–801. doi: 10.1016/j.jmb.2008.12.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu HW, Cosa G, Landes CF, Zeng Y, Kovaleski BJ, Mullen DG, Barany G, Musier-Forsyth K, Barbara PF. Single-molecule FRET studies of important intermediates in the nucleocapsid-protein-chaperoned minus-strand transfer step in HIV-1 reverse transcription. Biophys. J. 2005;89:3470–3479. doi: 10.1529/biophysj.105.065326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Godet J, de RH, Raja C, Glasser N, Ficheux D, Darlix JL, Mely Y. During the early phase of HIV-1 DNA synthesis, nucleocapsid protein directs hybridization of the TAR complementary sequences via the ends of their double-stranded stem. J. Mol. Biol. 2006;356:1180–1192. doi: 10.1016/j.jmb.2005.12.038. [DOI] [PubMed] [Google Scholar]
- 27.Moumen A, Polomack L, Roques B, Buc H, Negroni M. The HIV-1 repeated sequence R as a robust hot-spot for copy-choice recombination. Nucleic Acids Res. 2001;29:3814–3821. doi: 10.1093/nar/29.18.3814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.de Rocquigny H, Ficheux D, Gabus C, Fournie-Zaluski MC, Darlix JL, Roques BP. First large scale chemical synthesis of the 72 amino acid HIV-1 nucleocapsid protein NCp7 in an active form. Biochem. Biophys. Res. Commun. 1991;180:1010–1018. doi: 10.1016/s0006-291x(05)81166-0. [DOI] [PubMed] [Google Scholar]
- 29.Lapadat-Tapolsky M, de RH, Van GD, Roques B, Plasterk R, Darlix JL. Interactions between HIV-1 nucleocapsid protein and viral DNA may have important functions in the viral life cycle. Nucleic Acids Res. 1993;21:831–839. doi: 10.1093/nar/21.4.831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Polge E, Darlix JL, Paoletti J, Fosse P. Characterization of loose and tight dimer forms of avian leukosis virus RNA. J. Mol. Biol. 2000;300:41–56. doi: 10.1006/jmbi.2000.3832. [DOI] [PubMed] [Google Scholar]
- 31.Milligan JF, Groebe DR, Witherell GW, Uhlenbeck OC. Oligoribonucleotide synthesis using T7 RNA polymerase and synthetic DNA templates. Nucleic Acids Res. 1987;15:8783–8798. doi: 10.1093/nar/15.21.8783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fosse P, Motte N, Roumier A, Gabus C, Muriaux D, Darlix JL, Paoletti J. A short autocomplementary sequence plays an essential role in avian sarcoma-leukosis virus RNA dimerization. Biochemistry. 1996;35:16601–16609. doi: 10.1021/bi9613786. [DOI] [PubMed] [Google Scholar]
- 33.Maxam AM, Gilbert W. Sequencing end-labeled DNA with base-specific chemical cleavages. Methods Enzymol. 1980;65:499–560. doi: 10.1016/s0076-6879(80)65059-9. [DOI] [PubMed] [Google Scholar]
- 34.Vo MN, Barany G, Rouzina I, Musier-Forsyth K. Mechanistic studies of mini-TAR RNA/DNA annealing in the absence and presence of HIV-1 nucleocapsid protein. J. Mol. Biol. 2006;363:244–261. doi: 10.1016/j.jmb.2006.08.039. [DOI] [PubMed] [Google Scholar]
- 35.Skripkin E, Paillart JC, Marquet R, Ehresmann B, Ehresmann C. Identification of the primary site of the human immunodeficiency virus type 1 RNA dimerization in vitro. Proc. Natl Acad. Sci. USA. 1994;91:4945–4949. doi: 10.1073/pnas.91.11.4945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Laughrea M, Jette L. Kissing-loop model of HIV-1 genome dimerization: HIV-1 RNAs can assume alternative dimeric forms, and all sequences upstream or downstream of hairpin 248-271 are dispensable for dimer formation. Biochemistry. 1996;35:1589–1598. doi: 10.1021/bi951838f. [DOI] [PubMed] [Google Scholar]
- 37.Ben Ali M, Chaminade F, Kanevsky I, Ennifar E, Josset L, Ficheux D, Darlix JL, Fosse P. Structural requirements for nucleocapsid protein-mediated dimerization of avian leukosis virus RNA. J. Mol. Biol. 2007;372:1082–1096. doi: 10.1016/j.jmb.2007.07.026. [DOI] [PubMed] [Google Scholar]
- 38.Lanchy JM, Rentz CA, Ivanovitch JD, Lodmell JS. Elements located upstream and downstream of the major splice donor site influence the ability of HIV-2 leader RNA to dimerize in vitro. Biochemistry. 2003;42:2634–2642. doi: 10.1021/bi0271190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Boiziau C, Dausse E, Yurchenko L, Toulme JJ. DNA aptamers selected against the HIV-1 trans-activation-responsive RNA element form RNA-DNA kissing complexes. J. Biol. Chem. 1999;274:12730–12737. doi: 10.1074/jbc.274.18.12730. [DOI] [PubMed] [Google Scholar]
- 40.Collin D, van HC, Boiziau C, Toulme JJ, Guittet E. NMR characterization of a kissing complex formed between the TAR RNA element of HIV-1 and a DNA aptamer. Nucleic Acids Res. 2000;28:3386–3391. doi: 10.1093/nar/28.17.3386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Paillart JC, Shehu-Xhilaga M, Marquet R, Mak J. Dimerization of retroviral RNA genomes: an inseparable pair. Nat. Rev. Microbiol. 2004;2:461–472. doi: 10.1038/nrmicro903. [DOI] [PubMed] [Google Scholar]
- 42.Desai NA, Shankar V. Single-strand-specific nucleases. FEMS Microbiol. Rev. 2003;26:457–491. doi: 10.1111/j.1574-6976.2003.tb00626.x. [DOI] [PubMed] [Google Scholar]
- 43.Till BJ, Burtner C, Comai L, Henikoff S. Mismatch cleavage by single-strand specific nucleases. Nucleic Acids Res. 2004;32:2632–2641. doi: 10.1093/nar/gkh599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hampshire AJ, Rusling DA, Broughton-Head VJ, Fox KR. Footprinting: a method for determining the sequence selectivity, affinity and kinetics of DNA-binding ligands. Methods. 2007;42:128–140. doi: 10.1016/j.ymeth.2007.01.002. [DOI] [PubMed] [Google Scholar]
- 45.Holstege FC, Timmers HT. Analysis of open complex formation during RNA polymerase II transcription initiation using heteroduplex templates and potassium permanganate probing. Methods. 1997;12:203–211. doi: 10.1006/meth.1997.0472. [DOI] [PubMed] [Google Scholar]
- 46.Craig ML, Tsodikov OV, McQuade KL, Schlax PE, Jr, Capp MW, Saecker RM, Record MT., Jr DNA footprints of the two kinetically significant intermediates in formation of an RNA polymerase-promoter open complex: evidence that interactions with start site and downstream DNA induce sequential conformational changes in polymerase and DNA. J. Mol. Biol. 1998;283:741–756. doi: 10.1006/jmbi.1998.2129. [DOI] [PubMed] [Google Scholar]
- 47.Rubin CM, Schmid CW. Pyrimidine-specific chemical reactions useful for DNA sequencing. Nucleic Acids Res. 1980;8:4613–4619. doi: 10.1093/nar/8.20.4613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Morellet N, Demene H, Teilleux V, Huynh-Dinh T, de RH, Fournie-Zaluski MC, Roques BP. Structure of the complex between the HIV-1 nucleocapsid protein NCp7 and the single-stranded pentanucleotide d(ACGCC) J. Mol. Biol. 1998;283:419–434. doi: 10.1006/jmbi.1998.2098. [DOI] [PubMed] [Google Scholar]
- 50.Fisher RJ, Rein A, Fivash M, Urbaneja MA, Casas-Finet JR, Medaglia M, Henderson LE. Sequence-specific binding of human immunodeficiency virus type 1 nucleocapsid protein to short oligonucleotides. J. Virol. 1998;72:1902–1909. doi: 10.1128/jvi.72.3.1902-1909.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.De Guzman RN, Wu ZR, Stalling CC, Pappalardo L, Borer PN, Summers MF. Structure of the HIV-1 nucleocapsid protein bound to the SL3 psi-RNA recognition element. Science. 1998;279:384–388. doi: 10.1126/science.279.5349.384. [DOI] [PubMed] [Google Scholar]
- 52.Vuilleumier C, Bombarda E, Morellet N, Gerard D, Roques BP, Mely Y. Nucleic acid sequence discrimination by the HIV-1 nucleocapsid protein NCp7: a fluorescence study. Biochemistry. 1999;38:16816–16825. doi: 10.1021/bi991145p. [DOI] [PubMed] [Google Scholar]
- 53.Amarasinghe GK, De Guzman RN, Turner RB, Chancellor KJ, Wu ZR, Summers MF. NMR structure of the HIV-1 nucleocapsid protein bound to stem-loop SL2 of the psi-RNA packaging signal. Implications for genome recognition. J. Mol. Biol. 2000;301:491–511. doi: 10.1006/jmbi.2000.3979. [DOI] [PubMed] [Google Scholar]
- 54.Shubsda MF, Paoletti AC, Hudson BS, Borer PN. Affinities of packaging domain loops in HIV-1 RNA for the nucleocapsid protein. Biochemistry. 2002;41:5276–5282. doi: 10.1021/bi016045+. [DOI] [PubMed] [Google Scholar]
- 55.Paoletti AC, Shubsda MF, Hudson BS, Borer PN. Affinities of the nucleocapsid protein for variants of SL3 RNA in HIV-1. Biochemistry. 2002;41:15423–15428. doi: 10.1021/bi026307n. [DOI] [PubMed] [Google Scholar]
- 56.Lapadat-Tapolsky M, Pernelle C, Borie C, Darlix JL. Analysis of the nucleic acid annealing activities of nucleocapsid protein from HIV-1. Nucleic Acids Res. 1995;23:2434–2441. doi: 10.1093/nar/23.13.2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Stoylov SP, Vuilleumier C, Stoylova E, de RH, Roques BP, Gerard D, Mely Y. Ordered aggregation of ribonucleic acids by the human immunodeficiency virus type 1 nucleocapsid protein. Biopolymers. 1997;41:301–312. doi: 10.1002/(SICI)1097-0282(199703)41:3<301::AID-BIP5>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 58.Cruceanu M, Gorelick RJ, Musier-Forsyth K, Rouzina I, Williams MC. Rapid kinetics of protein-nucleic acid interaction is a major component of HIV-1 nucleocapsid protein's nucleic acid chaperone function. J. Mol. Biol. 2006;363:867–877. doi: 10.1016/j.jmb.2006.08.070. [DOI] [PubMed] [Google Scholar]
- 59.Guo J, Wu T, Anderson J, Kane BF, Johnson DG, Gorelick RJ, Henderson LE, Levin JG. Zinc finger structures in the human immunodeficiency virus type 1 nucleocapsid protein facilitate efficient minus- and plus-strand transfer. J. Virol. 2000;74:8980–8988. doi: 10.1128/jvi.74.19.8980-8988.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Williams MC, Gorelick RJ, Musier-Forsyth K. Specific zinc-finger architecture required for HIV-1 nucleocapsid protein's nucleic acid chaperone function. Proc. Natl Acad. Sci. USA. 2002;99:8614–8619. doi: 10.1073/pnas.132128999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Heath MJ, Derebail SS, Gorelick RJ, DeStefano JJ. Differing roles of the N- and C-terminal zinc fingers in human immunodeficiency virus nucleocapsid protein-enhanced nucleic acid annealing. J. Biol. Chem. 2003;278:30755–30763. doi: 10.1074/jbc.M303819200. [DOI] [PubMed] [Google Scholar]
- 62.Narayanan N, Gorelick RJ, DeStefano JJ. Structure/function mapping of amino acids in the N-terminal zinc finger of the human immunodeficiency virus type 1 nucleocapsid protein: residues responsible for nucleic acid helix destabilizing activity. Biochemistry. 2006;45:12617–12628. doi: 10.1021/bi060925c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bourbigot S, Ramalanjaona N, Boudier C, Salgado GF, Roques BP, Mely Y, Bouaziz S, Morellet N. How the HIV-1 nucleocapsid protein binds and destabilises the (-)primer binding site during reverse transcription. J. Mol. Biol. 2008;383:1112–1128. doi: 10.1016/j.jmb.2008.08.046. [DOI] [PubMed] [Google Scholar]
- 64.Cosa G, Harbron EJ, Zeng Y, Liu HW, O'Connor DB, Eta-Hosokawa C, Musier-Forsyth K, Barbara PF. Secondary structure and secondary structure dynamics of DNA hairpins complexed with HIV-1 NC protein. Biophys. J. 2004;87:2759–2767. doi: 10.1529/biophysj.104.043083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Avilov SV, Godet J, Piemont E, Mely Y. Site-specific characterization of HIV-1 nucleocapsid protein binding to oligonucleotides with two binding sites. Biochemistry. 2009;48:2422–2430. doi: 10.1021/bi8022366. [DOI] [PubMed] [Google Scholar]
- 66.Bazzi A, Zargarian L, Chaminade F, Boudier C, de RH, Rene B, Mely Y, Fosse P, Mauffret O. Structural insights into the cTAR DNA recognition by the HIV-1 nucleocapsid protein: role of sugar deoxyriboses in the binding polarity of NC. Nucleic Acids Res. 2011;39:3903–3916. doi: 10.1093/nar/gkq1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wang H, Yeh YS, Barbara PF. HIV-1 nucleocapsid protein bends double-stranded nucleic acids. J. Am. Chem. Soc. 2009;131:15534–15543. doi: 10.1021/ja9070046. [DOI] [PubMed] [Google Scholar]
- 68.Kalnik MW, Norman DG, Li BF, Swann PF, Patel DJ. Conformational transitions in thymidine bulge-containing deoxytridecanucleotide duplexes. Role of flanking sequence and temperature in modulating the equilibrium between looped out and stacked thymidine bulge states. J. Biol. Chem. 1990;265:636–647. [PubMed] [Google Scholar]
- 69.Levin JG, Mitra M, Mascarenhas A, Musier-Forsyth K. Role of HIV-1 nucleocapsid protein in HIV-1 reverse transcription. RNA. Biol. 2010;7:754–774. doi: 10.4161/rna.7.6.14115. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.