

Abstract
The ROP5 family is a closely related set of polymorphic pseudokinases that are critical to the ability of Toxoplasma to cause disease. Polymorphisms in ROP5 also make it a major determinant of strain-specific differences in virulence. ROP5 possesses all of the major kinase motifs required for catalysis except for a substitution at the catalytic Asp. We show that this substitution in the catalytic loop of ROP5 is part of a motif conserved in other pseudokinases of both Toxoplasma and human origin, and that this motif is required for the full activity in vivo of ROP5. This suggests evolutionary selection at this site for a biochemical function, rather than simple drift away from catalysis. We present the crystal structures of a virulent isoform of ROP5 both in its ATP-bound and -unbound states and have demonstrated that despite maintaining the canonical ATP-binding motifs, ROP5 binds ATP in a distorted conformation mediated by unusual magnesium coordination sites that would not be predicted from the primary sequence. In addition, we have mapped the polymorphisms spread throughout the primary sequence of ROP5 to two major surfaces, including the activation segment of ROP5. This suggests that the pseudoactive site of this class of pseudokinases may have evolved to use the canonical ATP-binding motifs for non-catalytic signaling through allostery.
Keywords: Parasite, Protein Kinases, Protein Structure, Serine Threonine Protein Kinase, Signal Transduction, X-ray Crystallography, Toxoplasma, Pseudokinase, Virulence Factor
Introduction
Proteins involved in cellular signaling often have modular domain structure; whereas enzymatic domains catalyze the reactions that propagate a signal, the precise subcellular localization of a protein, and the recognition of partner molecules are usually accomplished by nonenzymatic domains (1, 2). Complicating this paradigm, noncatalytic enzymatic domains such as pseudokinases have been recognized as important regulators of signaling networks, for example, by acting as molecular scaffolds for an effector and its downstream targets, or by modulating the activity of a catalytically active enzyme (3).
The cellular signaling occurring at the interface between an intracellular pathogen and its host cell is particularly complex. Toxoplasma gondii is an obligate, intracellular parasite with the unusual ability to infect almost any nucleated cell of virtually any warm-blooded animal. During infection, Toxoplasma secretes a variety of effector molecules into its host cell to monitor and co-opt host signaling networks (4, 5). Although these effectors originate in the specialized secretory organelles of the parasite, called rhoptries (and thus the designation of these effectors as “ROPs”), they carry out their various functions in the cytosol of the host cell. Three ROPs have been identified as active kinases that drastically alter host signaling (6, 7) and disease outcome (6, 8, 9). Indeed, of the ∼160 predicted kinases of Toxoplasma, at least one-third are predicted to be secreted into the host cell (7), suggesting an active role in pathogenesis.
Intriguingly, the kinome of the Toxoplasma, especially its secreted effectors, is highly enriched in pseudokinases. We and others previously identified a closely related family of these secreted pseudokinases, the ROP5 family, as absolutely critical to pathogenesis in mice; ablation of the locus results in a complete loss of virulence in mice (10, 11). The ROP5 family is encoded by a locus of tandemly duplicated genes that are highly polymorphic (supplemental Fig. S1). Allelic variation of these isoforms is concentrated within the pseudokinase domains and is responsible for a >105 difference in virulence (LD50) in a mouse model of disease (10).
There is an abundance of structural information on active protein kinases, which has led to a detailed understanding both of the reaction mechanism of phosphoryl transfer and various methods of regulation by diverse actors such as small molecules (12) and both cis- (13) and trans- acting protein-protein interactions (14). Pseudokinases, however, remain something of a functional mystery. Recent structures of the pseudokinase domains of HER3 (15), ILK (16), and STRADα (17) have suggested these proteins act as “smart” scaffolds that allosterically regulate their binding partners. Other studies have indicated that some proteins originally predicted to be pseudokinases may catalyze phosphoryl transfer through atypical means (18–20).
Most studies of pseudokinases to date have concentrated on those residues that are missing from the canonical kinase motifs. The ROP5 family is a closely related family of proteins (up to 10 paralogous ROP5 genes exist within a given strain) under extraordinary selective pressure, and thus provides an opportunity to examine the functional relevance of conserved substitutions within the canonical kinase motifs (i.e. things conserved in pseudokinases that are divergent from an active kinase). Here, we present such an examination, guided by the crystal structure of the ROP5 pseudokinase domain, and demonstrate the in vivo relevance of a key substitution in the pseudoactive site to function for ROP5 in determining the outcome of Toxoplasma infection.
EXPERIMENTAL PROCEDURES
Parasite Culture and Genetic Manipulation
Human foreskin fibroblasts were grown in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum and 2 mm glutamine. Toxoplasma tachyzoites were maintained in confluent monolayers of human foreskin fibroblasts. Complemented RHΔrop5 strains were created by electroporating 15 μg of linearized plasmid for the targeted replacement of the UPRT locus (21) with the ROP5 wild-type or mutant of choice. Clonal parasites were grown from populations by limiting dilution.
Cloning and Plasmid Construction
DNA encoding residues 171–540 of ROP5BI was amplified by PCR using Phusion polymerase (New England Biolabs) from a plasmid encoding the Toxoplasma genomic locus for its gene, and cloned into the pET28a (Novagen) bacterial expression vector between the NdeI and XhoI sites. Mutagenesis was carried out according to the Phusion protocol. DNA encoding the R389D mutant ROP5AIII gene, including its native promoter (∼600 bp upstream from start) was cloned into a previously described vector (10) in-frame with a C-terminal HA tag and the Toxoplasma GRA2 3′-UTR, flanked by sequences for targeted recombination into the Toxoplasma uracil-phosphoribotransferase locus.
Immunofluorescence and Western Blot
For immunofluorescent analysis, cells were permeabilized and blocked for 1 h in PBS, 0.1% Triton X-100, 3% BSA. Cells were incubated for 2 h at room temperature with mouse monoclonal antibodies ROP2/4 (mAb T34A7, gift of J.-F. Dubremetz) or rat anti-HA (3F10, Roche Applied Science) at 1:200 and 1:500 dilution, respectively, in PBS, 3% BSA. Cells were washed 3 times with PBS and incubated for 1 h with anti-rat Alexa 488- and anti-mouse Alexa 594-conjugated secondary antibodies (Molecular Probes). Cells were again washed 3 times with PBS and mounted in VectaShield (Vector). Wide-field images were captured at ×100 on an Olympus BX60 and a Hamamatsu Orca100 CCD. For Western blot analysis, samples were separated by SDS-PAGE, transferred to a PVDF membrane (Millipore), and blocked in Tris-buffered saline, pH 7.5, with 0.01% Tween with 5% milk powder for 1 h. Membranes were then incubated for 2 h with HRP-conjugated rat anti-HA at a dilution of 1:500 in blocking buffer. Membranes were washed 3 times with TBST, and visualized with ECL reagent (Pierce). The membranes were then stripped, reblocked, and probed with primary mouse anti-SAG1 at 1:20,000 dilution for 2 h. After washing, membranes were probed for 1 h with HRP-conjugated secondary, washed again, and visualized as above.
Mouse Survival Analysis
10–12-Week-old female BALB/c (Jackson Labs) mice were infected by peritoneal injection with parasites diluted to the appropriate dose in phosphate-buffered saline, pH 7.4, and monitored daily for survival. Dosage was verified by plaque assay of inoculum. Infection of surviving mice was verified by testing serum for reactivity with Toxoplasma lysate. Data were analyzed in Prism (GraphPad Software) using the log-rank survival test.
Protein Expression and Purification
All recombinant protein was expressed in Escherichia coli Rosetta2(DE3) (EMD Biosciences). ROP5BI was purified on nickel-nitrilotriacetic acid resin (Qiagen), the His6 tag was removed by overnight thrombin cleavage at 4 °C and further purified by anion exchange and gel filtration chromatography. His6-ROP16-HA was purified as previously described (22).
ATP-binding Experiments
Purified recombinant protein was incubated at 4 °C with a 30-μl bed volume of γ-linked ATP-Sepharose (Innova Biosciences) in 10 mm Tris, pH 8.0, 150 mm NaCl, 2 mm DTT, 10 mm MgCl2, 0.1% Triton X-100. The beads were sedimented and briefly washed with 1 ml of buffer before protein was eluted in SDS loading buffer. Binding was analyzed by SDS-PAGE stained with Coomassie Blue.
Crystallization
The crystals of ROP5BI (apo form) were grown at 18 °C by mixing equal volumes of protein solution (∼0.5 mm; 10 mm HEPES, pH 7.0, 100 mm NaCl) and a reservoir containing 20% polyethylene glycol 3350, 10 mm MgCl2, and 0.3 m sodium malonate, pH 5.0. The ATP bound crystals were grown in a similar condition with the addition of 10 mm ATP and 20 mm MgCl2. To generate a platinum derivative, ROP5BI (apo) crystals were soaked with 10 mm K2PtCl4 for 2 h, washed quickly in mother liquor, and frozen. All crystals were flash frozen in a cryoprotectant of mother liquor with 30% ethylene glycol.
Data Collection, Structure Determination, and Refinement
The diffraction data for native crystals were collected at beamline 11.1 of SSRL (the Stanford Synchrotron Radiation Laboratory) at a wavelength of 1.00 Å and a temperature of 100 K. Data for the apo-ROP5BI platinum derivatives were collected in an inverse beam experiment at a wavelength of 0.979 Å. Integration, indexing, and scaling of the diffraction data were performed using the HKL2000 suite of programs (23). Initial phases at 2.7 Å were determined by single isomorphous replacement with anomalous scattering from two well occupied platinum sites and used to generate a starting model after density modification with the SOLVE/RESOLVE package (24, 25). The high resolution native data were incorporated for extension and map improvement in Phenix (26). Manual rebuilding in Coot (27) and refinement in Phenix led to a final 1.90-Å structure of apo-ROP5BI, which was deposited in the Protein Data Bank (PDB)3 (PDB accession 3Q5Z). Phases for the ATP-bound ROP5BI structure were determined by molecular replacement using Phaser (28) with apo-ROP5BI as a search model. Good density was observed for the bound ATP, two Mg2+ ions, and coordinating waters. The model was built and refined using Coot and Phenix, with a final resolution of 1.72 Å (PDB accession 3Q60). Both the apo- and ATP-bound models have excellent stereochemistry with no residues in disallowed regions (96.9 and 96.2% favored for the apo- and ATP-bound, respectively) from Ramachandran plots as validated by the program MOLPROBITY (29).
In Vitro Kinase Activity Assay
Either 10 ng of recombinant ROP16 or up to 10 μg of wild-type or H389D ROP5BI was incubated with 1 μg of substrate (either histone H1 or dephosphorylated myelin basic protein) in a 50-μl volume using protein kinase buffer (New England Biolabs) with 20 μCi of [γ-32P]ATP (PerkinElmer Life Sciences) for 1 h at 37 °C and halted by addition of SDS-loading buffer and boiling. The completed reactions were separated by SDS-PAGE, the gel was dried and exposed overnight to a phosphorscreen (GE Healthcare).
RESULTS
A Conserved Pseudokinase Pseudoactive Site Motif Is Required for Efficient ROP5 Function
As previously reported, polymorphisms between the various allelic isoforms of ROP5 are concentrated in the predicted pseudokinase domain (supplemental Fig. S1), which appears to be under strong, positive selection (10). It is striking, however, how well the residues comprising the former active site are conserved within the family, strongly suggesting a functional significance: save for the catalytic Asp (the D of the well studied “HRD” motif of active kinases), ROP5 possesses the canonical residues at each of the major positions required for ATP binding and catalysis. In ROP5, the catalytic Asp in the HRD motif is replaced with either an Arg or His depending on the ROP5 isoform. It is intriguing that these substitutions both replace the acidic Asp with a basic residue.
To determine how common such a substitution is in other pseudokinases, we examined the predicted sequences of the complete human and Toxoplasma kinomes. It is important to note that although Toxoplasma is genetically distant from mammals, many of its kinases are secreted into the cytosol of its mammalian hosts, where they exert their end function. As ∼30% of the predicted active kinases and pseudokinases of Toxoplasma are from a single family of homologous proteins specific to coccidian parasites (the so-called “rhoptry kinases” (7), which includes ROP5), similarities between these kinases and mammalian kinases are likely due to true convergent evolution rather than other mechanisms, such as horizontal gene transfer.
The Arg of the ROP5 HRD has been replaced with a Gly in each of the isoforms (Table 1 and supplemental Fig. S1). Although such a substitution would not, itself, be predicted to interfere with catalytic activity, it is, however, a remarkably rare substitution among human kinases and is highly predictive of a pseudokinase: only 27 of the ∼500 human proteins with kinase folds possess it, and of these, 19 are predicted pseudokinases (representing ∼40% of human pseudokinases; Table 1). The remaining 8 active kinases are enriched in proteins that are closely related to known pseudokinases (e.g. VRK2, IRAK1). Of these human “Gly” pseudokinases, very few residues appear as replacements for the catalytic Asp (Table 1), which can be clustered into two classes: short polar (Ser/Asn) and basic (His/Arg/Lys). Interestingly, this appears to be the case in Toxoplasma pseudokinases, as well (Table 1). Furthermore, the correlation is symmetric: of the 16 Toxoplasma and human pseudokinases with a basic residue substituted for the catalytic Asp, all but one are preceded by a Gly (Fig. 1A). It is important to note that even within the kinome of the organism, many of these pseudokinases appear to have evolved independently from one another; in both the human and Toxoplasma examples many of the members are more closely related to another, often active, kinase than to one another (Fig. 1A). Thus, such substitutions appear to identify a distinct subclass of pseudokinase, in which the HRD motif is replaced with an HGB tripeptide (where “B” denotes a basic residue).
TABLE 1.
Human and Toxoplasma kinases in which the Arg preceding the catalytic base has been substituted for a Gly
HRD motif and predicted activities are noted. HGB pseudokinases are boxed. Toxoplasma proteins predicted to be secreted into the host cell are starred.

FIGURE 1.

The noncanonical ROP5BI HRD motif contributes to its function in vivo. A, table of HGB pseudokinases, where B indicates a basic residue (His/Lys/Arg). Substitutions at other motifs are indicated, as is the closest relative of each pseudokinase, which are based on previously published alignments (7, 48). B, Western blot of lysates from Δrop5 parasites transgenic for ROP5AIII-HA or ROP5AIII(R389D)-HA. The blot was probed with antibodies recognizing the HA tag and the parasite SAG1 protein as a loading control. C, localization of the HA-tagged ROP5 proteins (green) in the parasites described above was compared with the rhoptry marker ROP2/4 (red). Scale bar is 5 μm. D, BALB/c mice were injected intraperitoneally with 106 parasites of either Δrop5 or Δrop5 complemented with either wild-type ROP5AIII-HA or ROP5AIII(R389D)-HA and monitored for mortality. n = 5 mice for Δrop5; n = 25 mice for complemented strains.
We next set out to determine whether this conserved but noncanonical motif in the ROP5 pseudoactive site was contributing to the function of ROP5. We had previously created a strain in which the entire ROP5 locus had been deleted (Δrop5), which resulted in the complete attenuation of an otherwise highly virulent strain (10). We also demonstrated that a single copy of the ROP5AIII isoform was able to partially complement the virulence phenotype. This provided the ideal background to address our current question. We reasoned that a charge swap returning the ROP5 HGB motif to an HGD was the most likely to affect ROP5 function without causing structural problems. Thus, we created a parasite line in which the only ROP5 expressed was ROP5AIII R389D. The gene was targeted to the identical locus as the wild-type copy, creating an isogenic parasite strain, except for the single point mutation. Both the wild-type and R389D ROP5A were expressed in-frame with a C-terminal HA tag, allowing us to validate the localization to the parasite rhoptries and equivalent protein expression of ROP5 in both strains (Fig. 1, B and C).
Mice were then challenged with equal doses of either the Δrop5, or the strains complemented with either wild-type or R389D ROP5AIII (Fig. 1D). Interestingly, the mutant ROP5 protein maintained its ability to at least partially complement the virulence phenotype. However, whereas infection with parasites expressing wild-type ROP5AIII was uniformly lethal to mice, a portion of the mice infected with the R389D strain survived. In addition, the median survival time of the mice who succumbed to infection was significantly shifted (p < 0.0001 by log-rank analysis), strongly indicating that the mutant protein was not as efficient in mediating its effect on virulence as the wild-type protein.
These data are particularly intriguing in the context of the large amount of mutation data for a variety of active kinases. A partial attenuation of activity is contrary to what one would expect from the similar mutation of an active kinase; such mutations ablate the normal function of kinase, and are usually completely unable to complement a phenotype. In fact, mutation of the active site of the kinase often creates a dominant-negative phenotype that can disable the wild-type kinase in the same cell. Given that the mutation appears to not disrupt the stability or trafficking of the protein, our data suggest that the pseudoactive site of ROP5 is involved in its function, but is indeed not catalytic. It is, however, possible that the R389D mutation “rescued” ROP5 kinase activity, and it was this unintended kinase activity that was reducing the effect of ROP5 on virulence in vivo.
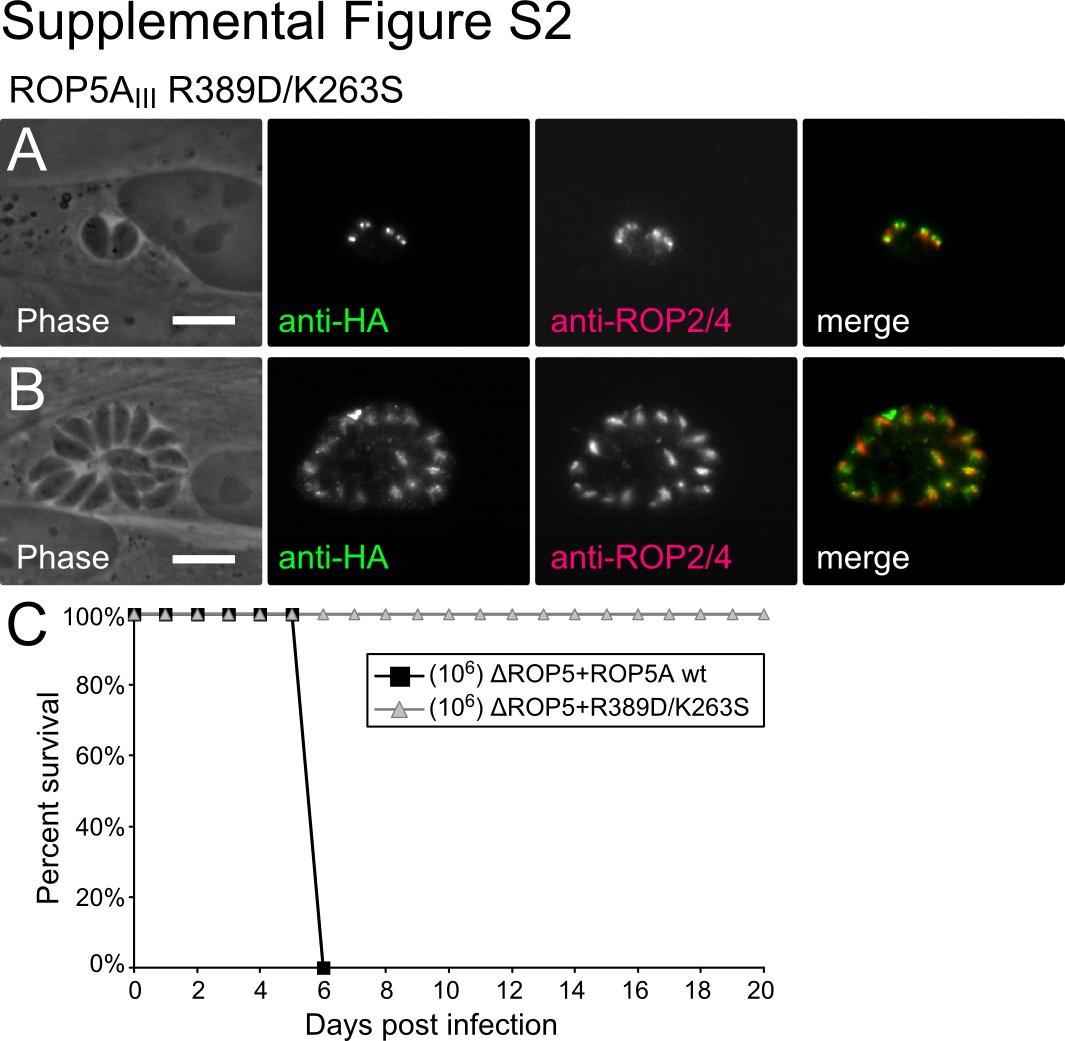
To rule this out, we created an additional Toxoplasma strain, in which the only ROP5 expressed was ROP5AIII carrying both R389D and K263S mutations. This latter mutation alters the Lys of the VAIK motif, which makes contacts with the β-phosphate of ATP and is required for catalysis in active kinases (30). It should be noted that among the human and Toxoplasma HGB pseudokinases, 11 of 16 have conserved the VAIK Lys, and 14 of 16 have conserved the DFG Asp, suggesting that these pseudokinases have conserved the ability to bind ATP. Although a portion of the ROP5A R389D/K263S mutant was correctly localized to the rhoptries, some of the protein appeared to be trapped in the endoplasmic reticulum or Golgi of the parasites (supplemental Fig. S2). This is consistent with protein that is either misfolded or is unable to make an interaction that is critical for its trafficking to the rhoptries. Not surprisingly, such mutant ROP5 was unable to complement the virulence of the ΔROP5 parasites (supplemental Fig. S2). We also observed either similar mislocalization or undetectable expression of ROP5 in multiple independent transfections to express protein with the single mutation K263S. This suggests that wild-type ROP5 binds ATP, and that this interaction with nucleotide promotes the maturation and trafficking of the protein. This interpretation is consistent with the behavior of active kinases, such as PKC, in which similar mutations that reduce the affinity of the protein for ATP disrupt the correct maturation of the protein (31). Due to the partial mislocalization of the ROP5 protein, these mutants were, however, otherwise uninformative as to the function of ROP5. We thus felt it necessary to move to in vitro studies of the biochemistry of ROP5.
ROP5 Binds ATP But Is Catalytically Inactive
To gain further insight into the function of ROP5 we sought to directly test whether wild-type or mutant protein had catalytic activity. Although we had difficulty obtaining suitable quantities of the pseudokinase domain of ROP5AIII, we were able to recombinantly express and purify ROP5BI, which we used in all of our in vitro assays. Although there are many residues that are polymorphic between the virulent alleles of the ROP5A and ROP5B isoforms (supplemental Fig. S1), the residues of the pseudoactive site are well conserved (aside from the catalytic base), suggesting that the biochemical properties of the individual purified proteins should be similar. As discussed above, ROP5 lacks the kinase catalytic base (the “HRD” Asp has been replaced by Arg-389 or His-389, depending on isoform; supplemental Fig. S1), resulting in its predicted catalytic inactivity. However, ROP5 has conserved residues that are known to coordinate both Mg2+ and ATP in active kinases (Asp-407 and Asn-394 in ROP5; supplemental Fig. S3). To test whether ROP5 was competent to bind ATP, we incubated recombinantly expressed and purified ROP5BI with γ-phosphate-linked ATP-Sepharose, washed the resin, eluted the bound protein with SDS-loading buffer and EDTA, and evaluated the binding by SDS-PAGE. As expected, ROP5 was efficiently pulled-down by the ATP resin in a Mg2+-dependent manner and excess soluble ATP competitively inhibited this interaction (Fig. 2A).
FIGURE 2.

ROP5 can bind ATP, but is catalytically inactive. A, ROP5BI was incubated with γ-linked ATP-Sepharose in the presence of EDTA, MgCl2, or MgCl2 and 1 mm ATP. Protein was separated by SDS-PAGE and visualized with Coomassie Blue. Control indicates Sepharose without cross-linked ATP in the presence of MgCl2. B, representative in vitro radioactive kinase assay data using bacterially expressed, purified recombinant protein. Either 10 ng of the catalytically active Toxoplasma kinase ROP16 or 10 μg of wild-type or mutant ROP5BI (H389D) were incubated with substrate in the presence of [γ-32P]ATP. Substrates used were recombinant histone H1 (H1) or myelin basic protein (MBP). The multiple bands in the ROP16 lane likely correspond to multiple autophosphorylated species. C, the active sites of ATP-unbound ROP5BI (black) is overlaid on that of the ATP-bound structure of PKA (light blue, PDB code 1ATP). Residues important for ATP binding and catalysis in PKA are represented as sticks and numbered according to their position in ROP5BI and PKA, respectively. The PKA ATP and alternate conformation of ROP5BI His-389 are not shown for clarity.
We tested the same recombinant protein for in vitro kinase activity. We incubated purified, recombinantly expressed ROP5BI with two standard substrates and [γ-32P]ATP and measured the level of phosphorylation by autoradiogram (Fig. 2B). Although ∼0.01 μg of recombinantly expressed ROP16, a paralog that we have previously demonstrated to be an active kinase (22), was efficiently autophosphorylated, even 10 μg of ROP5 produced no detectable autophosphorylation nor phosphorylation of myelin basic protein or histone H1. This is consistent with current understanding of the protein kinase reaction mechanism, in which the invariant catalytic Asp is thought to accept the proton from the substrate hydroxyl (32, 33).
As ROP5 appears to differ from active kinases only in that it lacks the catalytic Asp, we reasoned that by mutating His-389 of ROP5BI to Asp, we might “rescue” the kinase activity of ROP5. ROP5BI H389D expressed well and was similarly soluble to the wild-type protein. As expected, the mutant protein maintained its ability to bind ATP (Fig. 2A). Importantly, however, we were unable to detect in vitro kinase activity from ROP5BI H389D (Fig. 2B), suggesting that the conformation of the ATP-bound ROP5 active site was somehow distorted from that required for phosphoryl transfer. These data also strongly suggest the difference in virulence we observed in vivo with our ROP5AIII R389D mutant was not due to aberrant kinase activity.
To gain insight into the structural basis for the lack of activity of ROP5, we solved the crystal structure of the apo-ROP5BI pseudokinase domain using the single isomorphous replacement with anomalous scattering method with native data to 1.9 Å and a platinum derivative with data to 2.1 Å. The final model was completed using native data to 1.9 Å (crystallographic information in Table 2). We then compared our crystal structure of ROP5 to that of PKA. Save for its missing catalytic Asp, the active site of ROP5 appears structurally similar to an active kinase; all of the major motifs involved in ATP binding overlaid with those of ATP-bound PKA (Fig. 2C), suggesting ROP5 should bind ATP in a similar mode.
TABLE 2.
Data collection and refinement statistics
| Data collection | Apo-ROP5BI (platinum) | Apo-ROP5BI native | ROP5BI:ATP |
|---|---|---|---|
| Space group | P21212 | P21212 | P21212 |
| Cell dimensions | |||
| a, b, c (Å) | 116.98, 69.83, 43.30 | 117.34, 70.55, 43.01 | 123.36, 69.33, 43.39 |
| α, β, γ (°) | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 |
| Resolution (Å) | 2.05 | 1.90 | 1.72 |
| Rsym | 8.5 (53.2) | 5.4 (48.6) | 7.2 (54.0) |
| I/σI | 24.8 (2.1) | 42.9 (3.4) | 34.2 (2.9) |
| Completeness (%) | 97.8 (85.0) | 98.7 (88.7) | 98.8 (89.0) |
| Redundancy | 12.5 (5.1) | 12.8 (6.5) | 13.9 (9.3) |
| Refinement | |||
| Resolution (Å) | 36.7–1.90 | 30.2–1.72 | |
| No. reflections | 27,733 | 39,069 | |
| Rwork/Rfree | 0.209/0.261 | 0.179/0.210 | |
| No. atoms | |||
| Protein | 2675 | 2741 | |
| Ligand/ion | 16 (4 EDO) | 40 (1 ATP, 2 Mg, 1 MLI) | |
| Water | 160 | 276 | |
| B-factors | |||
| Protein | 41.7 | 29.9 | |
| Ligand/ion | 46.3 | 21.4 | |
| Water | 49.3 | 39.2 | |
| Root mean square deviations | |||
| Bond lengths (Å) | 0.019 | 0.017 | |
| Bond angles (°) | 1.585 | 1.707 | |
ROP5 Uses Canonical Residues to Bind ATP in a Conformation Unsuitable for Catalysis
We next sought to determine the structure of the ATP-bound conformation of ROP5. Crystals of ROP5BI were grown in the presence of ATP and MgCl2 and a native dataset were collected to 1.72 Å. The structure was solved by molecular replacement using the apo-ROP5BI structure as a search model. Consistent with the lack of catalytic activity of ROP5, the nucleotide was completely unhydrolyzed during crystallization. Strong electron density was found for all atoms of ATP, its two coordinated Mg2+ cations, and their associated water molecules. Surprisingly, the structure of ROP5BI was unchanged upon binding ATP; the two structures have a root mean square deviation of 0.8 Å for 313 of 351 Cα atoms, and neither the P-loop nor the activation segment show significant movement in the two structures (Fig. 3A and supplemental Fig. S4) This is consistent with the recently solved structures of other pseudokinases that lack a Gly-rich P-loop: Toxoplasma ROP2 (34, 35), and mammalian ILK (16) and VRK3 (36) each appear to be locked in the closed conformation typical of active, ATP-bound kinases. This is in contrast to STRADα, which despite its degraded catalytic core, has maintained the Gly-rich P-loop, and appears to use the dynamics typically thought to be inherent in the kinase fold to allosterically regulate its binding partners (17, 37).
FIGURE 3.

ROP5 binds ATP in an unusual conformation. A, a stereo view of the pseudoactive site of ROP5BI is shown comparing the ATP-bound (gray) and unbound (blue) structures. Residues that make contact with ATP are shown as sticks. The ATP and its associated Mg2+ and waters are superposed with the 1.72-Å 2Fo − Fc electron density map contoured at 2.5 σ. The PKA (B) and ROP5BI (C) active sites are shown with bound ATP in an orientation to highlight the magnesium conformation. D, the ATP molecules bound to ROP5 (black) or PKA (green) have been overlaid to highlight triphosphate conformation.
Surprisingly, given the conservation of the ATP-binding motifs, and despite being coordinated by two Mg2+ in the active site, the ROP5-bound ATP triphosphate is in a substantially different conformation than found in PKA and other active kinases (Fig. 3, B–D). The PKA conformation has been confirmed by simulation to be that competent for catalysis (33, 38). Simulation studies have also shown that the efficiency of phosphoryl transfer by a protein kinase is sensitive to motions on the order of 0.1 Å (39). As each of the phosphorous atoms of the ATP are displaced by ∼2 Å in the ROP5 structure from their positions in that of PKA, we would predict these differences to have a significant deleterious effect on the phosphoryl transfer activity of ROP5. This is consistent with the lack of detectable catalytic activity of either the wild-type or H389D ROP5BI.
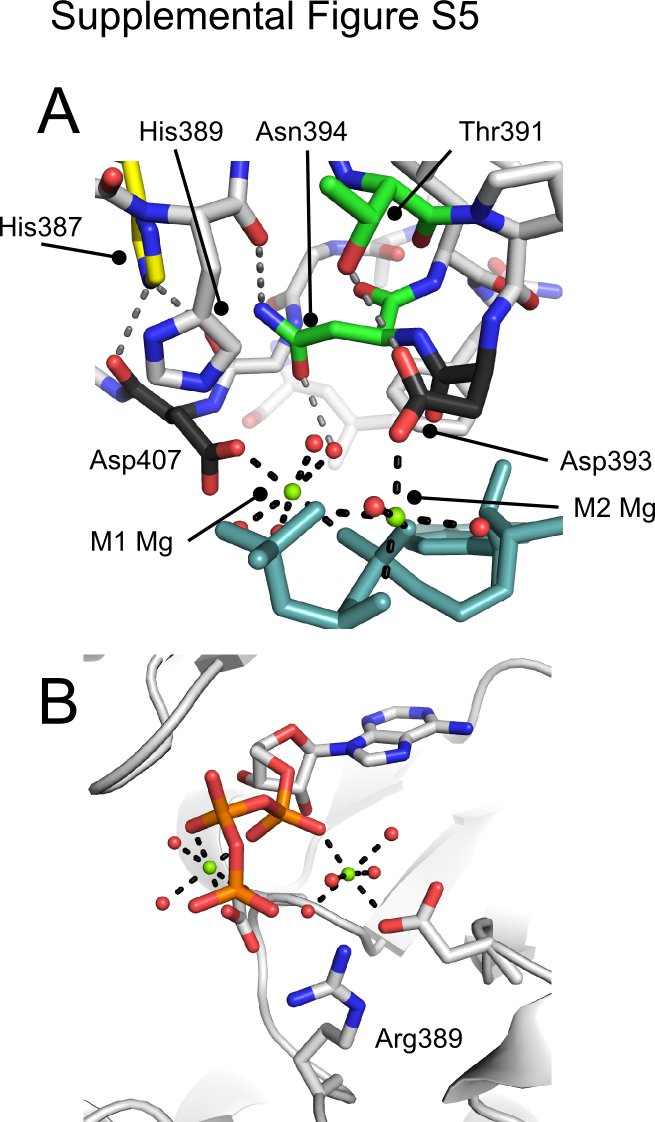
These distortions in the ATP conformation bound in the ROP5 pseudoactive site are mediated by an unusual coordination of Mg2+ cations. Despite having conserved both the canonical M1 (the “DFG” motif, Asp-184 in PKA, Asp-407 in ROP5) and M2 (Asn-171 in PKA, Asn-394 in ROP5) Mg2+-binding residues, the M2 site in ROP5 has been shifted. Although PKA Asp-184 contributes to the coordination of both Mg2+ ions, ROP5 Asp-407 coordinates only the M1 Mg2+. Also, in ROP5 the Asn-394 side chain is orthogonal to the orientation found in PKA and, instead, Asp-393 of ROP5 is coordinating the M2 Mg2+. ROP5 Asn-394, however, plays a supporting role, hydrogen bonding both with one of the waters that coordinates the M2 Mg2+ and with the His-389 backbone carbonyl, assisting in orienting it toward the ATP γ-phosphate (supplemental Fig. S5). Together, these differences result in a more splayed-out conformation of ATP than found in PKA (Fig. 3D). It is important to note that neither the displaced Mg2+ coordination nor the distorted ATP conformation would be predicted from either the primary sequence of ROP5 or its apo-structure.
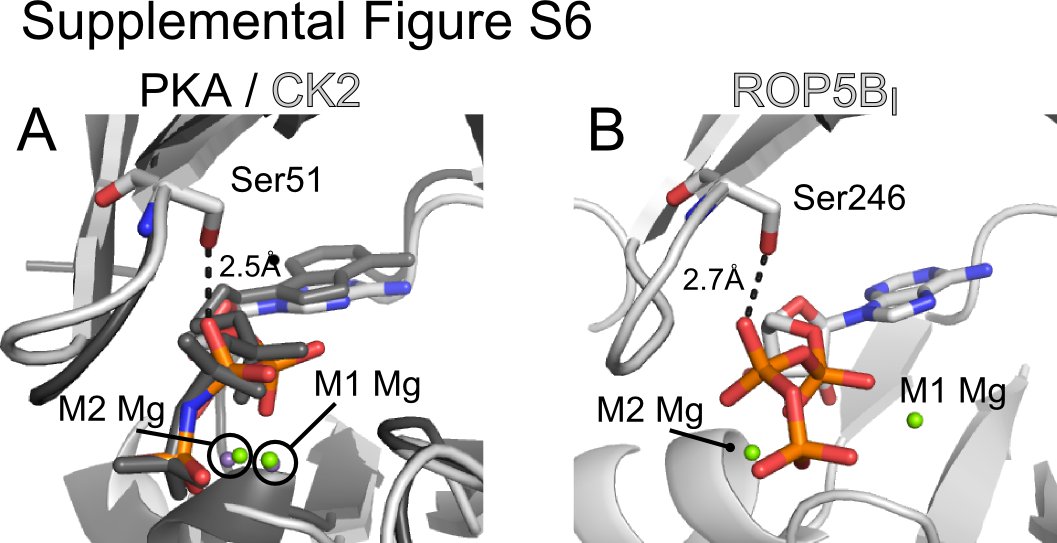
In addition to these major differences in Mg2+ coordination, ROP5 makes several other unusual contacts with its bound ATP. In many kinases, a Lys two residues C-terminal to the catalytic Asp (Lys-168 in PKA) assists in orienting the triphosphate (Fig. 3B). In ROP5, this residue is Thr-391 that is hydrogen bonding with Asp-393 (supplemental Fig. S5A). In apparent compensation, ROP5 Arg-245 (a bulky hydrophobic in most kinases; Phe-54 in PKA) swings down from the P-loop above the ATP to salt bridge with the γ-phosphate (Fig. 3A). In addition, ROP5 Ser-246 (Gly-55 in PKA) hydrogen bonds with the ATP β-phosphate. This substitution in the P-loop, however, is unlikely to participate in distorting the ATP conformation, as other, active kinases have equivalent substitutions at this position. For example, casein kinase 2 from both mammals and plants has a Ser at the equivalent position to ROP5 Ser-246, which makes similar contacts with the nucleotide β-phosphate but does not perturb the nucleotide conformation (PDB code 1DAW, supplemental Fig. S6), nor does it affect catalytic activity.
Although ROP5 His-389 (the catalytic Asp-166 in PKA) is oriented toward the ATP γ-phosphate, it is relatively distant (4–5 Å) to form an effective salt bridge. However, in three of the allelic isoforms of ROP5, His-389 is replaced by an Arg (supplemental Fig. S1). Although we do not have structural information on these isoforms, modeling predicts that with its extended reach, such an Arg would directly coordinate the ATP γ-phosphate (supplemental Fig. S5B).
The ROP5 Pseudokinase Domain Exists in a Conformation That Likely Precludes Catalysis
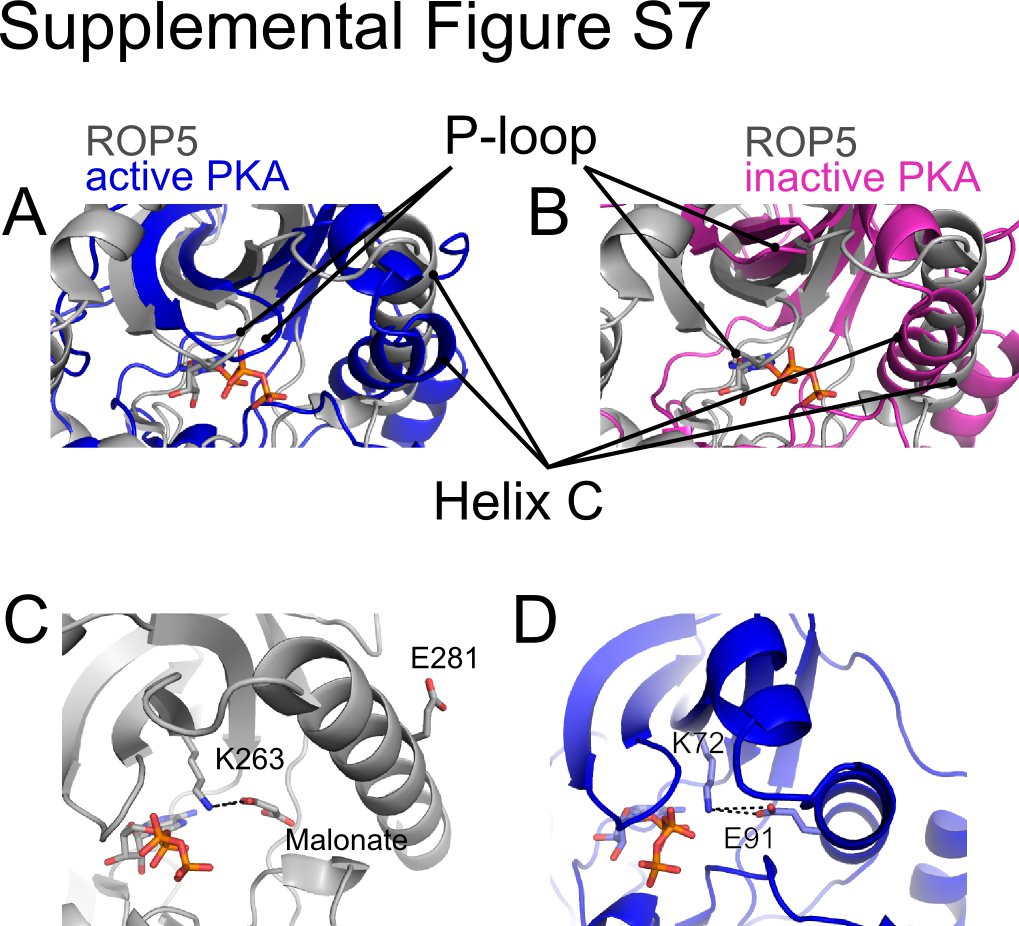
Although the binding of ATP in a conformation suitable for catalysis is critical for phosphoryl transfer, the global conformation of the kinase fold plays an important role in stabilizing additional conserved intramolecular contacts that facilitate catalysis. Given the lack of conformational change between the ATP-bound and unbound structures of ROP5 (supplemental Fig. S4), it is difficult to assess whether the domain is in a truly “active” conformation. One of the hallmarks of the active conformation of a protein kinase is the clamping down of the P-loop onto the bound nucleotide. Regardless of nucleotide occupancy, the P-loop of ROP5 aligns more closely with that of active PKA (PDB code 1ATP; 3.1 Å root mean square deviation for 217 of 351 Cα atoms) as compared with an inactive conformation (PDB code 1SYK (40); 3.9 Å root mean square deviation for 224 of 351 Cα atoms; supplemental Fig. S7, A and B). On the other hand, in the ROP5 structures, helix C is in a substantially different conformation than in PKA (supplemental Fig. S7). The relative position of this helix can, however, vary greatly among the structures of different kinases, and even between structures of the same kinase (e.g. p38; PDB codes 1CM8 and 1YWR (41, 42)). The conserved Glu in the center of helix C makes a critical hydrogen bond with the catalytic Lys (30). In ROP5, Glu-281 may be the conserved glutamate in question. However, the ROP5 helix C is in an orientation that flips Glu-281 out and creates an empty cavity where the conserved Glu would normally sit during the catalytic cycle. Interestingly, in the ATP-bound structure of ROP5, this space is filled by a crystallographic malonate, whose carboxylate interacts with Lys-263 in a manner analogous to the helix C Glu (supplemental Fig. S7, C and D). It is also important to note that this cavity near ROP5 helix C creates a gap in the “regulatory spine,” a conserved series of contacts that appear to be required for a catalysis in an active enzyme (43). Although the present structures do not suggest it, we cannot exclude the possibility that, under other conditions, the ROP5 helix C reorients to adopt a conformation more similar to that observed in other, active kinases.
Structurally Conserved Regulatory Elements Bridge Polymorphic Surfaces on ROP5
Many protein kinases are activated by the structural transition of their activation segments from disordered to ordered. The activation segments comprise the sequence from the conserved DFG to APE motifs (44) (Asp-407 to Glu-431 in ROP5). This transition can be caused both by interaction with regulatory molecules and by phosphorylation of residues in the activation loop. The activation segment of ROP5, however, appears locked in an ordered conformation, as it is well ordered in each of our structures, independent of the ATP-binding state (supplemental Figs. S4 and S8). In ROP5, the DFG Gly has been substituted for Ser (Ser-409) and two prolines align with both the activation loop phosphorylation site and GT motif Thr of PKA (supplemental Fig. S8), which may help rigidify the ROP5 loop.
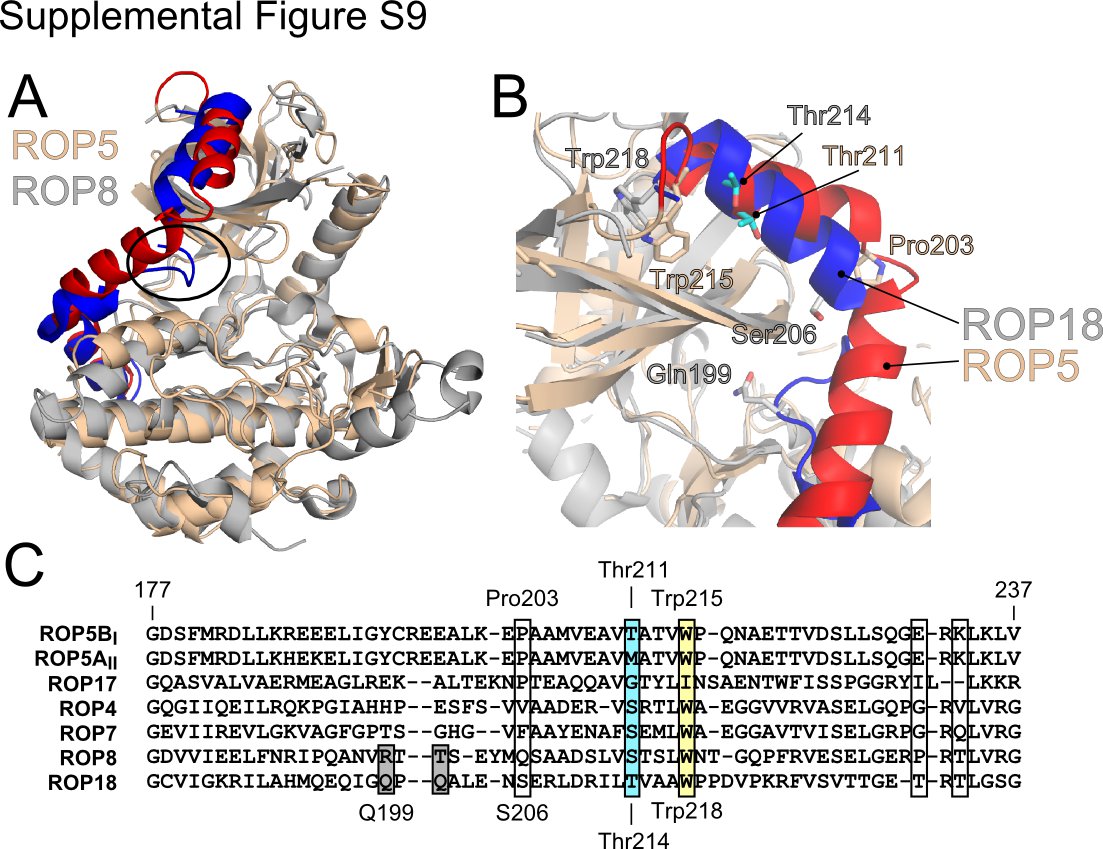
The ROP5 pseudokinase domain, like other members of the rhoptry kinase family has an N-terminal extension (NTE) in primary sequence that plays a major role in allowing profiling methods to distinguish the members of this family from other kinases (7). In the structures of two of closest relatives of ROP5, the non-ATP-binding pseudokinases ROP2 and ROP8, the center of the NTE has an extended conformation, with a loop whose side chains tuck between the N- and C-lobes and occlude the ATP-binding site (34, 35) (supplemental Fig. S9). In ROP5, however, the NTE forms three α-helices that wrap around the structure, tethering the N- and C-lobes (Fig. 4A and supplemental Fig. S9). Although these helices pack tightly against the N-lobe, none of the residues occlude the ATP-binding site. Interestingly, the C-terminal linker of the AGC family (which includes PKA and PKC) occupies the same three-dimensional space as the NTE, but arises from the opposite side of the primary sequence (Fig. 4 and supplemental Fig. S3). The C-terminal region of the AGC family terminates in a hydrophobic motif, whose two Phe are buried in the kinase N-lobe. Analogously, the ROP5 NTE terminates in the conserved Trp-215, which is also buried in the N-lobe (supplemental Fig. S9B). In the AGC family, divergence of this C-terminal span has been suggested as a major mechanism of diversification of regulatory elements in the family (13). Strengthening the analogy, the sequences of the rhoptry kinase NTE are quite divergent, even among close relatives (supplemental Fig. S9C). The space occupied by the ROP5 NTE has been suggested to be a critical hinge region that can alter the interdomain motions of the N- and C-lobes of a kinase (13). Thus the varied rhoptry kinase NTE and AGC C-terminal sequences may represent convergent solutions to the problem of regulation.
FIGURE 4.

Structurally conserved regulatory elements in ROP5 bridge polymorphic surfaces. A, the structures of ROP5BI (gray with NTE in blue) are overlaid PKA (PDB code 1ATP; tan with C-terminal linker in red). The hydrophobic α-helix F is highlighted purple for ROP5 and pink for PKA. The ROP5 ATP is shown as sticks. B, the ATP-bound ROP5BI structure colored as in A, and polymorphic residues are indicated in green (surface exposed) or as a yellow surface (near active site). A disulfide bond at the C terminus of helix F, conserved throughout the rhoptry kinase family, is shown as orange sticks. The ROP5 APE motif is indicated as red space filling, and the conserved Trp-447 of helix F against which the APE motif packs is indicated in purple space filling.
A recently proposed model suggests the various regulatory elements of protein kinases are structurally anchored on the conserved hydrophobic α-helix F (43), which serves as a central point of allosteric cross-talk between the regulatory elements. In ROP5, as with other kinases, the center of helix F packs against the activation segment (Fig. 4). Interestingly, the activation segment of ROP5 has several substitutions that are typical of differences between isoforms (rather than strains; Fig. 4B and supplemental Fig. S1). The C terminus of the ROP5 helix F has a disulfide bridge (Cys-458 to Cys-492), conserved in many rhoptry kinases (45), that directly anchors it to the polymorphic patch from which the NTE extends (Fig. 4B). The high selective pressure on this surface strongly implicates it as a binding site for the proteins with which ROP5 may interact to affect virulence. As our in vivo mutagenesis data demonstrate the ROP5 pseudoactive site is required for its full function (Fig. 1D), it is tempting to hypothesize that the NTE and hydrophobic helix F serve to allosterically link the ROP5 pseudoactive site to the two polymorphic regions on its surface, which may represent sites at which ROP5 interacts with its downstream targets. Such a model is consistent with recent structural studies that demonstrate that STRADα (17) and ILK (16) regulate their binding partners through interaction with their activation segments and pseudoactive sites.
DISCUSSION
We have solved the structure of the pseudokinase domain of ROP5, a protein that is critical for the ability of Toxoplasma to cause disease and is the major determinant of strain-specific variation in virulence (10, 11). The rhoptry kinase family is specific to coccidian parasites, and is highly enriched in proteins that are predicted to be secreted (7), implicating the family as a focal point in the interaction between Toxoplasma and its varied hosts. Recent work demonstrated that the ROP5 pseudokinase domain contains many polymorphic residues that appear to be under strong positive selection (10). Building on this, we have shown that these polymorphic residues throughout the primary sequence are concentrated within a major surface. Our results indicate a strong selective pressure has operated on this region of the protein, which is adjacent to the hinge between the N- and C-lobes of the kinase fold. Taken together, these data suggest that ROP5 mediates its effect on virulence through interaction with and dysregulation of its target or targets using this surface. Although we cannot yet know whether the binding partners are of host or parasite origin, the fact that the ROP5 proteins reside on the parasitophorous vacuolar membrane and are exposed to the host cytosol (45–47), suggests that the potential binding partner(s) derive from the host.
The structure of ROP5 is especially interesting in its relationship with current thinking of both kinase and pseudokinase function. Analysis of proteins with kinase folds often invokes the conservation of canonical motifs (30), whose contribution to catalysis has been exhaustively studied structurally, biochemically, and through simulation. Of the pseudokinases whose structures are known, ROP5 has among the greatest conservation of the canonical kinase motifs, apparently lacking only the catalytic Asp, suggesting that ROP5 should be competent to bind ATP in a canonical conformation competent for catalysis. Remarkably, in the structure of ROP5BI bound to ATP, the canonical ATP-binding residues in the ROP5 pseudoactive site have evolved to stabilize a surprisingly non-canonical conformation of ATP, which appears protected from hydrolysis. As shown here, this ATP conformation appears incompatible with catalytic activity, even with the re-introduction of the catalytic Asp by mutagenesis. Thus, it is possible that even proteins with apparently complete canonical kinase motifs may not be catalytically active, which provides an interesting corollary to the recent discovery that several proteins originally thought to be pseudokinases appear to have maintained their catalytic activities even with a non-canonical active site (19, 20). For instance, Wnk1 originally appeared to lack a catalytically active Lys, but instead has adapted to use a non-canonical Lys from a different position in its primary sequence (18). CASK, on the other hand, whereas missing the Mg2+-coordinating DFG motif, has adapted a new set of residues to form a magnesium-independent active site (19). Interestingly, in CASK, mutagenesis of the canonical catalytic Asp and Lys did not alter its catalytic activity (19), indicating that its atypical active site truly acts independently from many of the canonical motifs. This is in contrast to our mutagenesis data of ROP5, in which substitution to restore the canonical HRD motif Asp resulted in a less functional (but, biologically, not inactive) protein in vivo. Particular care should be taken when ascribing function or non-function to novel sequences; nevertheless, and although we cannot prove unequivocally that the ROP5 proteins described here lack catalytic activity, the presence of a basic residue in place of the catalytic Asp, the altered conformation of the bound ATP, and our failure to detect any activity in biochemical assays, even using a mutant form engineered to reintroduce the Asp, makes the possibility of ROP5 being a functional kinase extremely unlikely.
By examining the kinomes of both humans and Toxoplasma, we have identified a potential subclass of pseudokinases based on their convergent evolution of the substitution of the canonical HRD motif with an HGB motif. We confirmed the importance of this motif by testing the in vivo activity of ROP5 in which a charge-swap mutation had replaced its basic Arg to restore the canonical Asp. In vitro, such protein maintained its ability to bind ATP, although it was still not catalytically active. In vivo, R389D mutant ROP5 was able to partially complement the virulence of a Δrop5 phenotype, but to a significantly lesser extent as compared with complementation with the wild-type ROP5. This is consistent with the proposed role of ROP5 as a molecular scaffold rather than as an atypical, active enzyme. By extension, our data implicate the conserved non-canonical HGB motif as important to the function of multiple other pseudokinases in a variety of organisms. Hence, pseudokinases may not be the outliers as they first appeared; just as diligent research has elucidated general principles that govern a catalytic activity of the kinase, with further study the principles that govern the functioning of specific families of pseudokinases should soon be determinable.
Supplementary Material
Acknowledgments
We thank Arvin Dar for suggesting the ATP-Sepharose pull-down experiment, Mark Wilson and Tim Fenn for crystallographic software assistance, the Brünger lab for crystallization space, members of the Boothroyd lab for thoughtful suggestions, and Eva LaDow, Kara Norman, Selena Sagan, Mark Wilson, and Rich Yu for critical comments on the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grant AI73756.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S9.
- PDB
- Protein Data Bank
- NTE
- N-terminal extension.
REFERENCES
- 1. Kuriyan J., Cowburn D. (1997) Annu. Rev. Biophys. Biomol. Struct. 26, 259–288 [DOI] [PubMed] [Google Scholar]
- 2. Pawson T. (2007) Curr. Opin. Cell Biol. 19, 112–116 [DOI] [PubMed] [Google Scholar]
- 3. Boudeau J., Miranda-Saavedra D., Barton G. J., Alessi D. R. (2006) Trends Cell. Biol. 16, 443–452 [DOI] [PubMed] [Google Scholar]
- 4. Boothroyd J. C., Dubremetz J. F. (2008) Nat. Rev. Microbiol. 6, 79–88 [DOI] [PubMed] [Google Scholar]
- 5. Bradley P. J., Sibley L. D. (2007) Curr. Opin. Microbiol. 10, 582–587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Saeij J. P., Coller S., Boyle J. P., Jerome M. E., White M. W., Boothroyd J. C. (2007) Nature 445, 324–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Peixoto L., Chen F., Harb O. S., Davis P. H., Beiting D. P., Brownback C. S., Ouloguem D., Roos D. S. (2010) Cell Host Microbe 8, 208–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Saeij J. P., Boyle J. P., Coller S., Taylor S., Sibley L. D., Brooke-Powell E. T., Ajioka J. W., Boothroyd J. C. (2006) Science 314, 1780–1783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Taylor S., Barragan A., Su C., Fux B., Fentress S. J., Tang K., Beatty W. L., Hajj H. E., Jerome M., Behnke M. S., White M., Wootton J. C., Sibley L. D. (2006) Science 314, 1776–1780 [DOI] [PubMed] [Google Scholar]
- 10. Reese M. L., Zeiner G. M., Saeij J. P., Boothroyd J. C., Boyle J. P. (2011) Proc. Natl. Acad. Sci. U.S.A. 108, 9625–9630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Behnke M. S., Khan A., Wootton J. C., Dubey J. P., Tang K., Sibley L. D. (2011) Proc. Natl. Acad. Sci. U.S.A. 108, 9631–9636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Knight Z. A., Shokat K. M. (2005) Chem. Biol. 12, 621–637 [DOI] [PubMed] [Google Scholar]
- 13. Kannan N., Haste N., Taylor S. S., Neuwald A. F. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 1272–1277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pellicena P., Kuriyan J. (2006) Curr. Opin. Struct. Biol. 16, 702–709 [DOI] [PubMed] [Google Scholar]
- 15. Jura N., Shan Y., Cao X., Shaw D. E., Kuriyan J. (2009) Proc. Natl. Acad. Sci. U.S.A. 106, 21608–21613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fukuda K., Gupta S., Chen K., Wu C., Qin J. (2009) Mol. Cell 36, 819–830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zeqiraj E., Filippi B. M., Deak M., Alessi D. R., van Aalten D. M. (2009) Science 326, 1707–1711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Min X., Lee B. H., Cobb M. H., Goldsmith E. J. (2004) Structure 12, 1303–1311 [DOI] [PubMed] [Google Scholar]
- 19. Mukherjee K., Sharma M., Urlaub H., Bourenkov G. P., Jahn R., Südhof T. C., Wahl M. C. (2008) Cell 133, 328–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Shi F., Telesco S. E., Liu Y., Radhakrishnan R., Lemmon M. A. (2010) Proc. Natl. Acad. Sci. U.S.A. 107, 7692–7697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Donald R. G., Roos D. S. (1995) Proc. Natl. Acad. Sci. U.S.A. 92, 5749–5753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ong Y. C., Reese M. L., Boothroyd J. C. (2010) J. Biol. Chem. 285, 28731–28740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Otwinowski Z., Minor W. (1997) in Methods in Enzymology: Macromolecular Crystallography, Part A (Carter C. W., Jr., Sweet R. M. eds) Vol. 276, pp. 307–326, Academic Press, New York: [DOI] [PubMed] [Google Scholar]
- 24. Terwilliger T. (2004) J. Synchrotron Radiat. 11, 49–52 [DOI] [PubMed] [Google Scholar]
- 25. Terwilliger T. C., Berendzen J. (1999) Acta Crystallogr. D Biol. Crystallogr. 55, 849–861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Emsley P., Lohkamp B., Scott W. G., Cowtan K. (2010) Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chen V. B., Arendall W. B., 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., Richardson D. C. (2010) Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hanks S. K., Hunter T. (1995) FASEB J. 9, 576–596 [PubMed] [Google Scholar]
- 31. Cameron A. J., Escribano C., Saurin A. T., Kostelecky B., Parker P. J. (2009) Nat. Struct. Mol. Biol. 16, 624–630 [DOI] [PubMed] [Google Scholar]
- 32. Valiev M., Kawai R., Adams J. A., Weare J. H. (2003) J. Am. Chem. Soc. 125, 9926–9927 [DOI] [PubMed] [Google Scholar]
- 33. Turjanski A. G., Hummer G., Gutkind J. S. (2009) J. Am. Chem. Soc. 131, 6141–6148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Qiu W., Wernimont A., Tang K., Taylor S., Lunin V., Schapira M., Fentress S., Hui R., Sibley L. D. (2009) EMBO J. 28, 969–979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Labesse G., Gelin M., Bessin Y., Lebrun M., Papoin J., Cerdan R., Arold S. T., Dubremetz J. F. (2009) Structure 17, 139–146 [DOI] [PubMed] [Google Scholar]
- 36. Scheeff E. D., Eswaran J., Bunkoczi G., Knapp S., Manning G. (2009) Structure 17, 128–138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zeqiraj E., Filippi B. M., Goldie S., Navratilova I., Boudeau J., Deak M., Alessi D. R., van Aalten D. M. (2009) PLoS Biol. 7, e1000126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Khavrutskii I. V., Grant B., Taylor S. S., McCammon J. A. (2009) Biochemistry 48, 11532–11545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Henkelman G., LaBute M. X., Tung C. S., Fenimore P. W., McMahon B. H. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 15347–15351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wu J., Yang J., Kannan N., Madhusudan, Xuong N. H., Ten Eyck L. F., Taylor S. S. (2005) Protein Sci. 14, 2871–2879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bellon S., Fitzgibbon M. J., Fox T., Hsiao H. M., Wilson K. P. (1999) Structure 7, 1057–1065 [DOI] [PubMed] [Google Scholar]
- 42. Golebiowski A., Townes J. A., Laufersweiler M. J., Brugel T. A., Clark M. P., Clark C. M., Djung J. F., Laughlin S. K., Sabat M. P., Bookland R. G., VanRens J. C., De B., Hsieh L. C., Janusz M. J., Walter R. L., Webster M. E., Mekel M. J. (2005) Bioorg. Med. Chem. Lett. 15, 2285–2289 [DOI] [PubMed] [Google Scholar]
- 43. Kornev A. P., Taylor S. S., Ten Eyck L. F. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 14377–14382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Nolen B., Taylor S., Ghosh G. (2004) Mol. Cell 15, 661–675 [DOI] [PubMed] [Google Scholar]
- 45. El Hajj H., Demey E., Poncet J., Lebrun M., Wu B., Galéotti N., Fourmaux M. N., Mercereau-Puijalon O., Vial H., Labesse G., Dubremetz J. F. (2006) Proteomics 6, 5773–5784 [DOI] [PubMed] [Google Scholar]
- 46. Reese M. L., Boothroyd J. C. (2009) Traffic 10, 1458–1470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. El Hajj H., Lebrun M., Fourmaux M. N., Vial H., Dubremetz J. F. (2007) Cell. Microbiol. 9, 54–64 [DOI] [PubMed] [Google Scholar]
- 48. Manning G., Whyte D. B., Martinez R., Hunter T., Sudarsanam S. (2002) Science 298, 1912–1934 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}