

Abstract
The nuclear constitutive active/androstane receptor (CAR) is inactivated and sequestered in the cytoplasm when Thr-38 is phosphorylated. Here, we have demonstrated that activated ERK1/2 interacts with phosphorylated CAR to repress dephosphorylation of Thr-38. The phosphorylation-dependent interaction between CAR and ERK1/2 was examined by co-immunoprecipitation experiments of ectopically expressed FLAG-tagged CAR T38A and CAR T38D mutants with endogenous phospho-ERK1/2 in Huh-7 cells. Phospho-ERK1/2 coprecipitated only the phosphorylation-mimicking CAR T38D mutant; this coprecipitation was mediated by the interaction with the xenochemical response signal peptide near the C terminus of CAR. This interaction increased after EGF treatment and decreased after treatment with the MEK inhibitor U0126 as well as after knockdown of MEK1/2 by shRNA in Huh-7 cells. The phosphorylation levels of Thr-38 of CAR decreased in U0126-treated Huh-7 cells. Thus, activated ERK1/2 interacts with CAR and represses dephosphorylation of Thr-38, providing a cell signal-regulated mechanism for CAR activation.
Keywords: Cell Signaling, Cytochrome P450, Drug Action, Drug Metabolism, Gene Regulation, MAP Kinases (MAPKs), Metabolism, Nuclear Receptors
Introduction
The nuclear xenobiotic constitutive active/androstane receptor (CAR2; NR1I3) is an orphan member of the nuclear receptor superfamily. Phenobarbital was the first characterized activator of CAR, and CYP2B was the first CAR-targeted gene that was induced by phenobarbital to be identified (1–3). Whereas this nuclear receptor was first implicated as the key regulator in drug-induced drug metabolism, CAR is now known to regulate diverse liver functions ranging from energy metabolism to cell cycle and death, thus involving it in the development of diseases such as liver injury, diabetes, and hepatocellular carcinoma (4–13). Although the essential role of CAR as the target of phenobarbital in regulating various types of liver functions has now been well established, the molecular mechanism by which phenobarbital activates CAR is still not fully understood. The major hurdle for investigating this mechanism is the fact that phenobarbital indirectly activates CAR by apparently altering cell signaling. Here, we have investigated the cell signal mechanism that regulates CAR activation.
CAR is sequestered in the cytoplasm of hepatocytes and translocates into the nucleus in response to phenobarbital activation. Dephosphorylation was suggested to trigger this nuclear translocation (3). Our recent study identified Thr-38 of CAR as the target of dephosphorylation by protein phosphatase 2A, which activates CAR and translocates it into the nucleus (14). Regarding the cell signaling that regulates CAR, the growth factor-ERK1/2 pathway was shown to repress the activation and nuclear translocation of CAR in mouse primary hepatocytes (15). Moreover, the peptide sequence (residues 313–319) known as the xenochemical response signal (XRS), which resides near the C terminus of the CAR molecule, was determined previously to regulate nuclear translocation of CAR in mouse liver (16). However, at the same time, neither ERK1/2 nor the XRS has been linked to dephosphorylation of Thr-38 of CAR, which results in its activation with subsequent nuclear translocation.
To determine the role of ERK1/2 and the XRS in regulating the dephosphorylation of Thr-38 of CAR, we investigated the interaction between CAR and ERK1/2 upon growth factor treatment and how this interaction relates to dephosphorylation. CAR and its mutants were ectopically expressed in Huh-7 cells, and the interactions with endogenous ERK1/2 were examined by performing co-immunoprecipitation assays. An anti-phospho-Thr-38 peptide antibody that specifically detects phosphorylation of Thr-38 was utilized to determine whether or not the ERK1/2 interaction regulates dephosphorylation in response to growth factors. Here, we present experimental findings to support the hypothesis that the active (phosphorylated) form of ERK1/2 interacts with phosphorylated CAR via the XRS, thereby repressing dephosphorylation of Thr-38 and preventing CAR activation in Huh-7 cells. This growth factor-signaled ERK1/2 interaction provides a molecular basis for further investigations elucidating how CAR is indirectly activated not only by phenobarbital but also by many other therapeutics.
EXPERIMENTAL PROCEDURES
Reagents and Materials
U0126 was purchased from Promega (Madison, WI). U0124 and EGF were from Calbiochem. Phenobarbital was obtained from Sigma-Aldrich. Mouse monoclonal antibody to CAR was obtained from Perseus Proteomics Inc. (Tokyo, Japan). Mouse monoclonal antibody to FLAG was from Invitrogen. Goat polyclonal antibody to GFP was from Abcam (Cambridge, MA). Antibodies to phospho-ERK1/2 (p-ERK1/2; Thr-202/Tyr-204), ERK1/2, and MEK1/2 were from Cell Signaling Technology (Beverly, MA). Antibodies to α-tubulin and TATA-binding protein and normal rabbit IgG were from Santa Cruz Biotechnology (Santa Cruz, CA). Lab-TekTM Chamber SlideTM 2-well glass coverslips (Nunc) were obtained from Thermo Fisher Scientific. A rabbit anti-phospho-Thr-38 polyclonal antibody (made against the phospho-Thr-38 peptide of CAR) was produced in our previous work (14).
Plasmids
The pCR3-FLAG-hCAR, pCR3-FLAG-hCAR T38A, and pCR3-FLAG-hCAR T38D plasmids were constructed in our previous work (14). Polymerase chain amplification was performed using Pfu DNA polymerase and specific sets of primers bearing the newly created XhoI and EcoRI sites at the 5′- and 3′-ends of amplified DNAs, respectively. The QuikChange site-directed mutagenesis system (Stratagene) was utilized to mutate or internally delete nucleotides. Using pCR3-hCAR, pCR3-hCAR T38A, and pCR3-hCAR T38D plasmids as templates, cDNAs encoding hCAR T38A, hCAR T38D, hCAR(309–348), hCAR(309–328), and hCAR(329–348) were amplified and cloned into pEGFP-C1 (Clontech). Plasmids were prepared using a Qiagen plasmid maxi kit, and mutations and deletions were confirmed by nucleotide sequencing.
Cell Culture and Fractionation
Huh-7 cells were cultured on plastic dishes in minimal essential medium supplemented with 10% FBS, 2 mm l-glutamine, 100 units/ml penicillin, and 100 μg/ml streptomycin at 37 °C for 24 h prior to transfection. At ∼75% confluence, a given plasmid was transfected using Lipofectamine 2000 (Invitrogen) for 24 h. EGF (100 ng/ml) was then added to the transfected cells, which were precultured in serum-free medium for 30 min and then cultured for an additional 30 min. U0124 (20 μm) or U0126 (20 μm) was added 30 min prior to EGF treatment. Cells were scraped, collected by centrifugation, washed twice with PBS, and homogenized in 10 mm HEPES (pH 7.6) containing 10 mm KCl, 1.5 mm MgCl2, 0.3% Nonidet P-40, and Complete protease inhibitor mixture tablets (Roche Applied Science). The homogenates were centrifuged at 2000 × g for 5 min to obtain cytosolic extracts. The resulting pellets were resuspended and incubated at 4 °C for 30 min in 10 mm HEPES (pH 7.6) containing 0.4 m NaCl, 0.1 m KCl, 3 mm MgCl2, 0.1 mm EDTA, and Complete protease inhibitor mixture tablets to prepare nuclear extracts. Whole cell extracts were prepared by homogenizing Huh-7 cells in TBS containing 8 m urea, 0.1% SDS, and 0.05% Tween 20 and subsequently centrifuging homogenates at 15,000 rpm for 5 min.
Primary Hepatocytes
Primary hepatocytes were isolated from the livers of 6–8-week-old male C3H mice using a two-step collagenase perfusion as described previously (1, 3). Hepatocytes were suspended in Hanks' balanced salt solution containing 1.0 mg/ml collagenase and 10 mm HEPES (pH 7.4) by low speed centrifugation. The resulting pellet was suspended in prewarmed Williams' E medium supplemented with 10% FBS, 100 units/ml penicillin, and 100 μg/ml streptomycin and seeded on 10-cm dishes. One hour after seeding, the medium was changed to prewarmed Williams' E medium supplemented with or without a given chemical. Whole cell and nuclear extracts were prepared as described for Huh-7 cells.
Co-immunoprecipitation and Western Blotting
Cytosolic extracts were incubated with an anti-p-ERK1/2 or anti-ERK1/2 antibody or anti-rabbit IgG for 2 h at 4 °C. Immunoprecipitates were prepared by incubation with protein G-Sepharose (Thermo Fisher Scientific) for 2 h at 4 °C. The precipitates were subjected to electrophoresis on 10% SDS-polyacrylamide gel and transferred to a PVDF membrane (Millipore). The membrane was blocked in 5% nonfat milk in 25 mm Tris-HCl (pH 7.5) containing 137 mm NaCl and 0.1% Tween 20 and then incubated for 1 h at room temperature with an HRP-conjugated anti-GFP, HRP-conjugated anti-FLAG, anti-p-ERK1/2, anti-ERK1/2, or anti-MEK1/2 primary antibody. HRP-conjugated anti-mouse or anti-rabbit IgG was used as the secondary antibody. Finally, protein bands were visualized using ECL Plus Western blotting detection reagent (GE Healthcare).
Knockdown by shRNA
Plasmids that contain shRNA constructs for human MEK1 (TRCN0000002330) and MEK2 (TRCN0000011062) were prepared from bacterial glycerol stocks (Sigma-Aldrich) and transfected with MISSION® Lentiviral Packaging Mix (Sigma-Aldrich) into HEK-293T cells to produce lentiviral particles according to the protocol provided. Huh-7 cells were infected with lentiviral control shRNA or a mixture of these lentiviral human MEK1 and MEK2 shRNAs (multiplicity of infection of 10 each) using hexadimethrine bromide (8 μg/ml) for 6 h and then subsequently transfected with an expression plasmid for FLAG-hCAR T38D for an additional 24 h.
Cytochemistry
Huh-7 cells (1 × 105 cells/well) were grown on 2-well glass coverslips and transfected with a given expression plasmid (GFP-hCAR T38A or GFP-hCAR T38D) for 24 h. These cells were fixed with methanol, and their nuclei were stained with bisbenzimide (Hoechst 33258, Invitrogen) and mounted with glycerol. Fluorescence was visualized using an LSM 510 Meta confocal microscope (Carl Zeiss).
Enzyme-linked Immunosorbent Assay
An anti-CAR antibody (0.1 μg in PBS/well) was coated on a 96-well polyvinyl chloride microtiter plate (Costar) overnight at 4 °C. After being blocked with PBS containing 5% BSA for 2 h at room temperature, the wells were washed three times with PBS buffer containing 0.05% Tween 20 (PBST buffer), incubated with cytosolic extracts for 2 h at room temperature to bind CAR to the anti-CAR antibody, and then washed three times with PBST buffer. To detect CAR phosphorylated at Thr-38, an anti-phospho-Thr-38 antibody was added to the wells for 2 h at room temperature, and the wells were washed with PBST buffer, incubated with HRP-conjugated anti-rabbit IgG in PBST buffer containing 0.5% BSA for 2 h at room temperature, and developed with 3,3′,5,5″-tetramethylbenzidine substrate (Cell Signaling Technology). The reaction was stopped by the addition of 1 m phosphoric acid, and the plates were read at 450 nm. In vitro phosphorylated GST-hCAR was used as a positive control to determine a standard correlation between phosphorylation and detected A values (supplemental Fig. 1).
RESULTS
Interaction between CAR T38D and p-ERK1/2 in Huh-7 Cells
CAR is known to be retained in the cytoplasm of liver cells; this cytoplasmic retention is mediated by phosphorylation of Thr-38 of CAR. Reflecting the role of this phosphorylation, the CAR T38D mutant, which mimics phosphorylated CAR, but not the CAR T38A mutant, which mimics non-phosphorylated CAR, is retained in the cytoplasm of mouse liver cells (14). However, WT CAR spontaneously accumulates in the nuclei of human hepatoma-derived Huh-7 cells. CAR T38D and CAR T38A were ectopically expressed to determine their localization in Huh-7 cells. Both Western blot analysis of FLAG-tagged CAR T38D and CAR T38A and fluorescent microscope analysis of GFP-tagged CAR T38D and CAR T38A revealed the exclusive cytoplasmic localization of CAR T38D, but not CAR T38A, in Huh-7 cells as observed in mouse liver cells (Fig. 1, A and B). Given the fact that Huh-7 cells retained CAR T38D in the cytoplasm and on the basis of our previous finding that ERK1/2 represses the nuclear accumulation of CAR in mouse primary hepatocytes (15), we employed co-immunoprecipitation assays to examine the interaction between CAR mutants and ERK1/2. FLAG-tagged CAR T38D or CAR T38A was transfected into Huh-7 cells, from which cytosolic extracts were prepared and precipitated by an antibody against active ERK1/2 (p-ERK1/2) for subsequent Western blot analysis with an anti-FLAG antibody. CAR T38D, but not CAR T38A, coprecipitated with endogenous p-ERK1/2 (Fig. 2).
FIGURE 1.

Cytoplasmic localization of hCAR T38D in Huh-7 cells. A, ectopically expressed GFP-hCAR T38A and GFP-hCAR T38D were visualized using a confocal microscope. Hoechst DNA staining was used to visualize the nuclei. Scale bars = 10 μm. B, ectopically expressed FLAG-hCAR T38A or FLAG-hCAR T38D in cytosolic and nuclear extracts was subjected to Western blot analysis with an anti-FLAG antibody. Blots were subsequently stripped and restained with anti-α-tubulin or anti-TATA-binding protein antibody as a loading control. IB, immunoblot.
FIGURE 2.

Specific interaction between hCAR T38D and p-ERK1/2 in Huh-7 cells. As described under “Experimental Procedures,” FLAG-hCAR T38A and FLAG-hCAR T38D were ectopically expressed in Huh-7 cells, from which cell extracts were prepared and incubated with an anti-p-ERK1/2 antibody. The resultant precipitates were analyzed by Western blotting with an anti-FLAG antibody for co-immunoprecipitation (IP) of CAR with p-ERK1/2. Cytosolic extracts were also blotted with an anti-FLAG antibody to confirm expression levels of CAR mutants (2% input). IB, immunoblot.
Various deletion mutants of CAR were generated to delineate the interaction between CAR and p-ERK1/2 (Fig. 3A). To study this interaction, we first focused on the C-terminal region of CAR that contains the peptide sequence (residues 313–319) known as the XRS, which regulates the nuclear translocation of CAR in mouse liver (16). The GFP-tagged C-terminal fragment from residues 309 to 348 of CAR, which contains both the XRS and AF2 domain, was coprecipitated by the anti-p-ERK1/2 antibody (Fig. 3B). Removal of the XRS from the C-terminal fragment (residues 329–348), leaving only the AF2 domain, abolished this co-immunoprecipitation, whereas the fragment that retained the XRS but not the AF2 domain (residues 309–328) was coprecipitated. To confirm further the involvement of the XRS in the interaction between CAR T38D and p-ERK1/2, the XRS was internally deleted within the context of CAR T38D. When the C-terminal half of XRS (Δ316/321) was deleted, the resulting mutant was coprecipitated; however, when the N-terminal half of the XRS (Δ312/316) was removed, coprecipitation with p-ERK1/2 no longer occurred (Fig. 3C), thereby indicating that the N-terminal region is required for interaction with p-ERK1/2. There are four leucine residues within this N-terminal region (312LLGLL316), and they form multiple nested LLXL, LXXL, and LXL motifs for a potential interacting site with p-ERK1/2. Leu-316 is not required for the interaction because the Δ316/321 mutant interacted with p-ERK1/2. Because any single mutation of the first three leucine residues did not produce reproducible data, all three residues were simultaneously mutated to alanine. The resultant mutant (312AAGA315) no longer coprecipitated with p-ERK1/2 (Fig. 3D). These results indicate that the p-ERK1/2-interacting peptide overlaps with the N-terminal region of the XRS. This potential p-ERK1/2-interacting site (312LLGL315) is colored green and located on helix 11 within the ligand-binding domain of the CAR molecule in supplemental Fig. 2.
FIGURE 3.

Interaction between p-ERK1/2 and the XRS of hCAR T38D. A, schematic representation of the domains and motifs of the CAR molecule and GFP-tagged deletion constructs. XRS+AF2 contains the XRS and AF2 domain. XRS and AF2 contain only the XRS or AF2 domain. ΔN has an internal deletion of the N-terminal half of XRS (Δ312/316) within the context of GFP-hCAR T38D, whereas the C-terminal half of XRS (Δ316/321) was internally deleted in ΔC. In the AAGA construct, three leucines (residues 312, 313, and 315) were simultaneously mutated to alanines within the context of the GFP-hCAR T38D mutant. DBD, DNA-binding domain; LBD, ligand-binding domain. B–D, these GFP-tagged mutants were expressed in Huh-7 cells for 24 h, and cytosolic extracts were prepared and incubated with an anti-p-ERK1/2 antibody. The resultant precipitates were analyzed by Western blotting with an anti-GFP antibody for co-immunoprecipitation (IP) of these hCAR mutants with p-ERK1/2. Cytosolic extracts were also blotted with an anti-GFP antibody to confirm the expression of CAR mutants (2% input). IB, immunoblot.
Signal-mediated Dephosphorylation of Thr-38
It was demonstrated previously that treatment with a growth factor represses drug-induced activation and nuclear translocation of CAR in mouse primary hepatocytes (15). We have now examined whether or not Huh-7 cells retain any ability to dephosphorylate Thr-38 of CAR and whether or not p-ERK1/2 regulates dephosphorylation of CAR in Huh-7 cells. Following expression of FLAG-CAR, Huh-7 cells were treated with either EGF or PBS, and cytosolic extracts were prepared for subsequent co-immunoprecipitation with an anti-ERK1/2 antibody. EGF treatment increased the amounts of co-immunoprecipitated CAR, which correlated with the increased levels of p-ERK1/2 in Huh-7 cells (Fig. 4A). Conversely, treatment with the MEK1/2 inhibitor U0126, but not U0124, decreased coprecipitated CAR as p-ERK1/2 levels decreased (Fig. 4B). To substantiate further the role of MEK1/2 in this interaction between p-ERK1/2 and CAR, a lentivirus-based shRNA was employed to knock down the function of ERK1/2 as an alternative of U0126 (Fig. 4C). Huh-7 cells were first infected with lentiviral MEK1/2 shRNA and then the transfected plasmid for ectopic expression of FLAG-CAR T38D. This infection resulted in decreased levels of MEK1/2 and p-ERK1/2 in Huh-7 cells, from which cytosolic extracts were prepared for co-immunoprecipitation assays. Although an anti-ERK1/2 antibody precipitated equal amounts of ERK1/2 from the cytosolic extracts prepared from lentiviral control shRNA- and MEK1/2 shRNA-infected Huh-7 cells, the precipitation of p-ERK1/2 decreased in the MEK1/2 shRNA-infected cells. Correlating with the decrease in p-ERK1/2, co-immunoprecipitation of CAR T38D decreased as well (Fig. 4C). Thus, these results confirm that MEK1/2 regulates the interaction between CAR T38D and p-ERK1/2 through phosphorylation of ERK1/2.
FIGURE 4.

p-ERK1/2-CAR interaction is regulated by growth factors. FLAG-hCAR-expressing Huh-7 cells were treated with EGF (A) or U0126 or U0124 (B) as described under “Experimental Procedures.” Cytosolic extracts were prepared for immunoprecipitation (IP) with an anti-ERK1/2 antibody and subsequent Western blot analysis with antibodies against FLAG, p-ERK1/2, and ERK1/2. IB, immunoblot. C, cytosolic extracts were prepared from shRNA-treated Huh-7 cells and subjected to immunoprecipitation with an anti-ERK1/2 antibody and Western blot analysis with antibodies against FLAG, p-ERK1/2, ERK1/2, and α-tubulin. Input (2%) shows the expression levels of a given protein. Cont, control; shMEK, MEK1/2 shRNA.
Because only the phosphorylation-mimicking CAR T38D mutant interacted with p-ERK1/2 (Fig. 2), and EGF treatment increased this interaction (Fig. 4A), Thr-38 of CAR may be phosphorylated at detectable levels in Huh-7 cells. An anti-phospho-Thr-38 peptide antibody was employed to provide further evidence for this dephosphorylation. Upon Western blot analysis, this antibody detected the phosphorylation of Thr-38 of CAR ectopically expressed in U0124-treated Huh-7 cells and the dephosphorylation of Thr-38 after treatment with U0126 (Fig. 5A). In contrast, the CAR T38D mutant was neither phosphorylated nor dephosphorylated in these Huh-7 cells. ELISA further confirmed the U0126-dependent dephosphorylation of Thr-38 of CAR in Huh-7 cells; WT CAR, but not CAR T38D, was dephosphorylated in Huh-7 cells after treatment with U0126 (Fig. 5B). Similar to this observation in Huh-7 cells, U0126 treatment decreased the phosphorylation levels of Thr-48 (corresponding to Thr-38 in human CAR) of endogenous CAR in mouse primary hepatocytes, as did phenobarbital, whereas phenobarbital treatment decreased the p-ERK1/2 levels (Fig. 6). In addition, siRNA was employed in Huh-7 cells to support the role of ERK1/2 in dephosphorylation and CAR-mediated transactivation of the NR1 luciferase reporter construct (supplemental Fig. 3). siRNA knockdown of both ERK1 and ERK2 resulted in increased dephosphorylation of Thr-38 and enhancement of the CAR-mediated activation of NR1. Thus, these results link the p-ERK1/2 signaling to the repression of dephosphorylation of Thr-38 and CAR activation.
FIGURE 5.

Dephosphorylation of Thr-38 of hCAR in U0126-treated Huh-7 cells. A, whole cell extracts were prepared from drug-treated Huh-7 cells ectopically expressing FLAG-hCAR or FLAG-hCAR T38D as described under “Experimental Procedures.” Western blotting was performed using anti-phospho-Thr-38, anti-FLAG, anti-p-ERK1/2, and anti-ERK1/2 antibodies. IB, immunoblot. B, ELISA with an anti-phospho-Thr-38 antibody was used for the cytosolic extracts, which were prepared from drug-treated Huh-7 cells ectopically expressing FLAG-hCAR or FLAG-hCAR T38D as described under “Experimental Procedures.” *, significantly different (p < 0.05) from WT CAR treated with U0124 (unpaired t test). ±Error bars represent the mean ± S.E.
FIGURE 6.
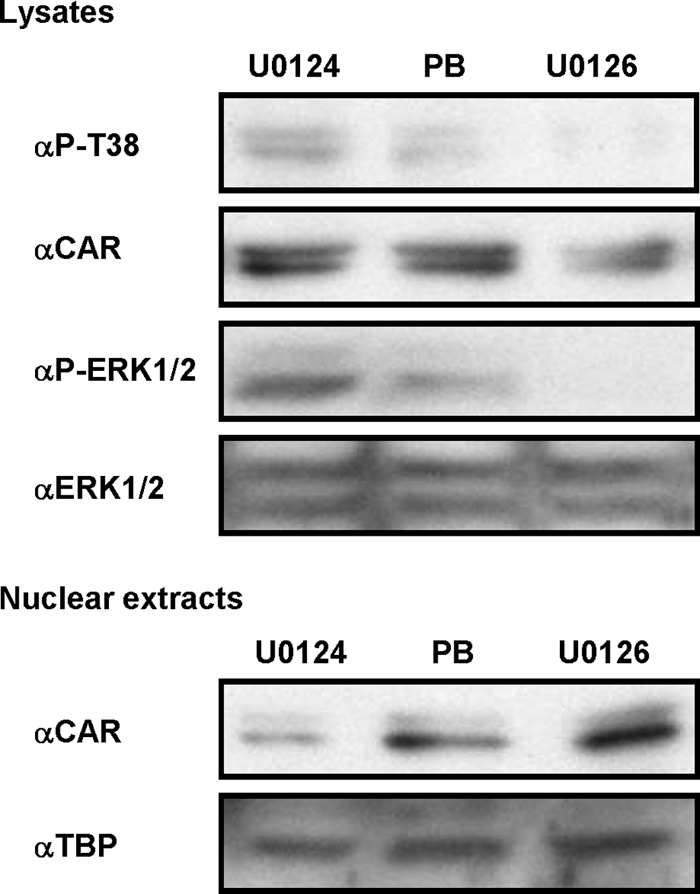
Dephosphorylation of CAR in mouse primary hepatocytes after treatment with U0126. Primary hepatocytes were prepared from male C3H mice and treated with phenobarbital (PB; 2 mm), U0124 (20 μm), or U0126 (20 μm) for 1 h. Whole cell and nuclear extracts were then prepared for Western blot analysis with anti-CAR, anti-ERK1/2, anti-p-ERK1/2, and anti-TATA-binding protein antibodies. The TATA-binding protein levels were used as a loading control.
DISCUSSION
CAR is sequestered in the cytoplasm of mouse hepatocytes due to phosphorylation of its Thr-38 residue (14). Huh-7 cells do not effectively phosphorylate CAR as well as mouse hepatocytes do, as evidenced by the fact that ectopically expressed WT CAR spontaneously accumulates in their nuclei. In this study, the phosphorylation-mimicking CAR T38D mutant was found to remain in the cytoplasm of Huh-7 cells, suggesting that the Thr-38 phosphorylation can sequester CAR in the cytoplasm. Moreover, our newly developed ELISA detected the phosphorylation of Thr-38 in ectopically expressed WT CAR in Huh-7 cells, therefore demonstrating that these cells retain the capability to keep CAR phosphorylated to some degree. Given these findings, we have now determined the molecular mechanism by which growth factors repress CAR activation. p-ERK1/2 interacts with the XRS region residing near the C terminus of the CAR T38D (but not CAR T38A) molecule in a growth factor-dependent manner. This growth factor-regulated interaction prevents dephosphorylation of Thr-38 of CAR in Huh-7 cells, thereby sequestering CAR in the cytoplasm and repressing its transactivation.
Ever since the characterization of CAR as a phenobarbital-activated nuclear receptor, the activation mechanism of CAR by drugs has remained elusive (1–3). Because phenobarbital indirectly activates CAR, it has been suggested that this activation could be cell signal-regulated. Two different cell signaling pathways have been suggested to regulate CAR activation: the AMP-activated protein kinase signal (18, 19) and growth factor-ERK1/2 (15, 20) pathways. Nuclear translocation is the initial step of CAR activation, yet phenobarbital was still able to induce nuclear translocation in primary hepatocytes prepared from AMP-activated protein kinase knock-out mice (18). Treatment with growth factors repressed nuclear translocation of CAR in mouse primary hepatocytes, whereas treatment with U0126, a MEK inhibitor, spontaneously accumulated CAR in the nucleus, with subsequent gene activation (15). Thr-38 of CAR can be phosphorylated by protein kinase C, and this phosphorylated Thr-38 residue is then dephosphorylated following phenobarbital treatment in mouse primary hepatocytes (14). Consistent with previous findings that okadaic acid inhibits the phenobarbital-induced nuclear accumulation of CAR (3), protein phosphatase 2A was identified recently as the enzyme that dephosphorylates this Thr-38 residue.3 Therefore, growth factor-MEK1/2-ERK1/2 signaling appears to be a negative regulator that prevents dephosphorylation of Thr-38 of CAR by protein phosphatase 2A, thereby repressing CAR activation in the absence of CAR activators such as phenobarbital. This negative signaling may present a mechanism through which phenobarbital indirectly activates CAR because phenobarbital treatment resulted in a decrease in p-ERK1/2 in mouse primary hepatocytes. The biological implication of growth signal-mediated regulation of CAR in the absence of exogenous CAR activators can only be speculated on at the present time. Upon drug activation, CAR is known to repress TNFα-induced death of mouse primary hepatocytes (12) and also appears to regulate cell cycles (8, 13). Through these and additional unknown functions, CAR activation may cause various effects on growth factor-mediated signals.
The XRS (313LGLLAEL319) is a functional motif defined by the inability of the alanine mutants to translocate into the nuclei of hepatocytes in mouse liver after treatment with exogenous CAR activators (16). Each of these leucines was mutated to alanine within the context of GFP-tagged CAR. The resultant GFP-tagged CAR mutants were directly expressed in mouse livers by injecting their expression plasmids via tail veins, and their intracellular localizations were determined using a confocal microscope; these alanine mutants no longer accumulated in the nucleus after treatments with phenobarbital (16). The XRS has now been found to interact with p-ERK1/2, and this interaction represses dephosphorylation of Thr-38 of CAR, providing the molecular basis for further investigations to decipher the growth factor-mediated regulation of CAR activation. Phenobarbital induces this dephosphorylation to translocate CAR into the nucleus (14). Although this study with mouse primary hepatocytes revealed that phenobarbital treatment decreased the phosphorylation levels of ERK1/2, whether or not this XRS-p-ERK1/2 interaction is directly involved in the regulation of phenobarbital-induced dephosphorylation and nuclear translocation of CAR remains only a possibility at the present time. The XRS is essential for CAR activators for the nuclear translocation of CAR regardless of the type of CAR activator (i.e. either indirect or direct) because not only phenobarbital but also the CAR ligand 1,4-bis[2-(3,5-dichloropyridyloxy)] benzene requires the XRS to translocate CAR into the nuclei of mouse livers (16). The XRS sequence appears to be conserved in various nuclear receptors such as the vitamin D, estrogen, retinoid X, and peroxisome proliferator-activated receptors, so the role of the XRS may go beyond that of phenobarbital and CAR.
In conclusion, growth factors repress dephosphorylation of Thr-38 of CAR in Huh-7 cells. The XRS of CAR has now been characterized as the peptide that regulates this dephosphorylation through its interaction with active p-ERK1/2. Phenobarbital may target the growth factor-ERK1/2 pathway to dephosphorylate Thr-38 of CAR, thus activating it in mouse livers.
Supplementary Material
Acknowledgments
We thank all members of the Pharmacogenetics Section for valuable discussion. We express our gratitude to Lee Pedersen and Lalith Perera, who simulated the model structure of the CAR molecule.
This work was supported, in whole or in part, by the National Institutes of Health Intramural Research Program and National Institutes of Health Grant Z01ES1005-01 from NIEHS.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. 1–3.
S. Mutoh and M. Negishi, unpublished data.
- CAR
- constitutive active/androstane receptor
- XRS
- xenochemical response signal
- p-ERK1/2
- phospho-ERK1/2
- hCAR
- human CAR.
REFERENCES
- 1. Honkakoski P., Zelko I., Sueyoshi T., Negishi M. (1998) Mol. Cell. Biol. 18, 5652–5658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sueyoshi T., Kawamoto T., Zelko I., Honkakoski P., Negishi M. (1999) J. Biol. Chem. 274, 6043–6046 [DOI] [PubMed] [Google Scholar]
- 3. Kawamoto T., Sueyoshi T., Zelko I., Moore R., Washburn K., Negishi M. (1999) Mol. Cell. Biol. 19, 6318–6322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sueyoshi T., Negishi M. (2001) Annu. Rev. Pharmacol. Toxicol. 41, 123–143 [DOI] [PubMed] [Google Scholar]
- 5. Ueda A., Kakizaki S., Negishi M., Sueyoshi T. (2002) Mol. Pharmacol. 61, 1284–1288 [DOI] [PubMed] [Google Scholar]
- 6. Kodama S., Koike C., Negishi M., Yamamoto Y. (2004) Mol. Cell. Biol. 24, 7931–7940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yamamoto Y., Moore R., Goldsworthy T. L., Negishi M., Maronpot R. R. (2004) Cancer Res. 64, 7197–7200 [DOI] [PubMed] [Google Scholar]
- 8. Osabe M., Sugatani J., Takemura A., Yamazaki Y., Ikari A., Kitamura N., Negishi M., Miwa M. (2008) Biochem. Biophys. Res. Commun. 369, 1027–1033 [DOI] [PubMed] [Google Scholar]
- 9. Dong B., Saha P. K., Huang W., Chen W., Abu-Elheiga L. A., Wakil S. J., Stevens R. D., Ilkayeva O, Newgard C. B., Chan L., Moore D. D. (2009) Proc. Natl. Acad. Sci. U.S.A. 106, 18831–18836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Konno Y., Negishi M., Kodama S. (2008) Drug Metab. Pharmacokinet. 23, 8–13 [DOI] [PubMed] [Google Scholar]
- 11. Wada T., Gao J., Xie W. (2009) Trends Endocrinol. Metab. 20, 273–279 [DOI] [PubMed] [Google Scholar]
- 12. Yamamoto Y., Moore R., Flavell R. A., Lu B., Negishi M. (2010) PLoS ONE 5, e10121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kamino H., Negishi M. (May 6, 2011) Mol. Carcinog. 10.1002/mc.20783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mutoh S., Osabe M., Inoue K., Moore R., Pedersen L., Perera L., Rebolloso Y., Sueyoshi T., Negishi M. (2009) J. Biol. Chem. 284, 34785–34792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Koike C., Moore R., Negishi M. (2007) Mol. Pharmacol. 71, 1217–1221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zelko I., Sueyoshi T., Kawamoto T., Moore R., Negishi M. (2001) Mol. Cell. Biol. 21, 2838–2846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Deleted in proof.
- 18. Rencurel F., Foretz M., Kaufmann M. R., Stroka D., Looser R., Leclerc I., da Silva Xavier G., Rutter G. A., Viollet B., Meyer U. A. (2006) Mol. Pharmacol. 70, 1925–1934 [DOI] [PubMed] [Google Scholar]
- 19. Shindo S., Numazawa S., Yoshida T. (2007) Biochem. J. 401, 735–741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bauer D., Wolfram N., Kahl G. F., Hirsch-Ernst K. I. (2004) Mol. Pharmacol. 65, 172–180 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.