

Abstract
Thrombogenic and inflammatory activity are two distinct aspects of platelet biology, which are sustained by the ability of activated platelets to interact with each other (homotypic aggregation) and to adhere to circulating leucocytes (heterotypic aggregation). These two events are regulated by distinct biomolecular mechanisms that are selectively activated in different pathophysiological settings. They can occur simultaneously, for example, as part of a pro-thrombotic/pro-inflammatory response induced by vascular damage, or independently, as in certain clinical conditions in which abnormal heterotypic aggregation has been observed in the absence of intravascular thrombosis. Current antiplatelet drugs have been developed to target specific molecular signalling pathways mainly implicated in thrombus formation, and their ever increasing clinical use has resulted in clear benefits in the treatment and prevention of arterial thrombotic events. However, the efficacy of currently available antiplatelet drugs remains suboptimal, most likely because their therapeutic action is limited to only few of the signalling pathways involved in platelet homotypic aggregation. In this context, modulation of heterotypic aggregation, which is believed to contribute importantly to acute thrombotic events, as well to the pathophysiology of atherosclerosis itself, may offer benefits over and above the classical antiplatelet approach. This review will focus on the distinct biomolecular pathways that, following platelet activation, underlie homotypic and heterotypic aggregation, aiming potentially to identify novel therapeutic targets.
Keywords: aspirin, heterotypic aggregation, homotypic aggregation, platelet activation, thienopyridines
Introduction
The classical model of platelet activation describes a series of phenotypic and morphological changes of resting platelets such that they acquire adhesiveness to the vascular wall in response to their exposure to pro-thrombotic molecules [e.g. collagen, von Willebrand factor (vWF)][1–3], forming platelet-to-platelet aggregates (homotypic aggregation) through interlinking by soluble adhesive proteins (fibrinogen) [4] and releasing a number of mediators that stabilize the initial aggregate and amplify thrombus formation [5]. The widespread view that vessel injury is the sole determinant of platelet activation has been largely abandoned in light of the evidence that a variety of pathophysiological stimuli, including pro-inflammatory cytokines and infective agents (recently reviewed by Semple & Freedman [6]) as well as shear stress [7], can activate platelets with no detectable vessel damage. Evidence has also accumulated that, in these and other diverse pathophysiological settings, platelets exhibit differential biological responses because of activation of distinct biomolecular pathways. Importantly, pro-inflammatory activity mediated by interaction with circulating leucocytes (heterotypic aggregation) and release of inflammatory mediators constitutes an important aspect of platelet biology and provides strong pathophysiological links between inflammation and thrombosis [6, 8]. As a consequence, platelet activation is not limited to the occurrence of a local thrombotic event triggered by vascular damage and mainly sustained by homotypic aggregation. On the contrary, it is often part of a systemic inflammatory response that can develop independently of, or in addition to, local injury-related factors. Indeed, formation of heterotypic aggregates in the peripheral circulation has been observed not only during acute thrombotic events [9–12], but also in those clinical conditions associated with high blood thrombogenicity in the absence of intravascular thrombosis, such as auto-immune disorders [13], haematological disease [14, 15] and in subjects with cardiovascular risk factors [16–18]. Given this, as well as the ongoing discovery of ever more biomolecular mechanisms underlying the platelet response to different agonists, it is likely that the signalling pathways that regulate thrombus formation and platelet–leucocyte interaction may operate either together or independently. In keeping with this, some antiplatelet drugs that effectively reduce platelet–platelet aggregation, such as aspirin, may have very little or no effect on heterotypic aggregation [19].
The increasing interest in pharmacological inhibition of heterotypic complex formation stems from the concept that platelet–leucocyte adhesion is not a simple epiphenomenon associated with thrombus development but represents a pro-thrombotic as well as a pro-atherogenic mechanism in itself. Indeed, interaction of activated platelets with white cells amplifies platelet activation and favours homotypic aggregation at sites of vascular injury [20], as well as exerting a damaging action on the vascular wall thereby predisposing to atherosclerotic disease [21, 22]. These observations raise the important question of whether inhibition of heterotypic aggregation might offer additional therapeutic benefit over and above the classical antithrombotic strategy, where the principal target and effect is platelet–platelet aggregation and thrombus formation.
We will here review the biomolecular mechanisms that are differentially implicated in homotypic and heterotypic aggregation, with a view to identifying, on the basis of recent data, potential therapeutic targets to prevent both the pro-thrombotic and pro-inflammatory effects of platelet activation. Table 1 compares some important features of homotypic and heterotypic aggregation.
Table 1.
Comparison between homotypic and heterotypic aggregation
| Homotypic aggregation | Heterotypic aggregation | |
|---|---|---|
| Localization | Confined to the vascular wall | Detectable in peripheral blood |
| Platelet activation required | Not under high shear rate | Yes |
| Mechanism of stabilization | Multiple ligand-receptor interactions with thrombogenic surface (demonstrated) | Multiple ligand-receptor interactions with leucocyte membrane (hypothesized) |
| Main effector molecule(s) | Varies depending on haemodynamic condition and lesion components | P-selectin, P-selectin glycoprotein ligand-1 |
| Platelet release reaction | Required for irreversible stabilization, mainly mediated by dense granule contents | Required for initiation, mediated by alpha granule contents |
Homotypic aggregation
Homotypic aggregation occurs at the site of a vascular lesion such as a ruptured atherosclerotic plaque or a traumatic injury, where exposed pro-thrombotic molecules provide an adhesive surface for recruitment of circulating platelets, followed by their activation and thrombotic plug formation [1–5]. In vivo and in vitro observations suggest that this phenomenon is a complex and dynamic multi-step process [20, 23, 24]. The initial phase of adhesion of platelets to the vascular wall as well as to each other (primary reversible aggregation) is followed by a second phase of stabilization and growth of the initial platelet plug (secondary irreversible aggregation). Platelet activation has long been assumed to have a dual role in this process, as an initiating factor in platelet arrest and as an essential mediator of the transition from reversible to irreversible aggregation [1, 25, 26]. Technical advances in intravital microscopy and real-time perfusion studies have demonstrated that primary aggregation can also occur without the need for platelet activation under conditions of elevated shear stress [24, 27]. However, when non-activated platelets adhere to the vessel wall they only form transient micro-aggregates that, in the absence of activation-dependent release and generation of soluble agonists [principally adenosine diphosphate (ADP), thrombin and thromboxane A2 (TxA2)], disaggregate with translocation of platelets in the direction of flow [24]. Central to homotypic aggregation is therefore the concept that platelets become activated in response to interaction with thrombogenic surfaces, and multiple ligand-receptor interactions are required to stabilize and amplify their adhesion and aggregation.
Biomolecular mechanisms of platelet activation leading to homotypic aggregation
Fibrinogen, vWF and collagen are able to initiate primary aggregation through the engagement of specific platelet integrins, namely glycoprotein (GP) IIb/IIIa (also designated α2bß3 integrin), GPIb and GPVI, respectively [25, 28, 29]. At low shear rate (<1000 s−1), the interaction between GPIIb/IIIa and fibrinogen has been demonstrated to constitute the predominant biomolecular event [1, 25, 30]. However, as GPIIb/IIIa is expressed in a low affinity state on the plasmalemma of quiescent platelets, initial stimulation of platelets by one or more soluble agonists in the vicinity of the lesion (e.g. ADP released from endothelial cells or thrombin locally produced) is required in order to activate downstream signalling pathways (inside-out signalling) that ultimately result in platelet shape change and activation of GPIIb/IIIa [4]. When the shear rate rises within the range 1000–10 000 s−1, platelet activation is not required to induce primary aggregation, as the synergistic action of GPIIb/IIIa and GPIb suffice in promoting tethering and transient aggregation of discoid-shaped quiescent platelets to the vascular wall. Nevertheless, the ensuing activation of platelets induced by integrin engagement leads to release of soluble agonists, mainly ADP, which is essential in stabilizing the initial aggregate [24]. At high shear rates (>10 000 s−1), Ruggeri et al. have shown both in vitro and in vivo that a thrombus can form efficiently through a mechanism independent of platelet activation, that is solely mediated by interaction between vWF and GPIb giving rise to stable local adhesion of platelets to a thrombogenic surface and homotypic aggregation [27].
The in vivo role of these ligand/receptor interactions has been evaluated in animal models selectively lacking one or more of the molecules involved in these pathways. In studies using vWF–/– mice, platelet accumulation and thrombus growth were markedly delayed but not absent in a model of ferric chloride-induced thrombosis [31], and the thrombogenic activity of platelets in laser-induced vessel wall injury was in fact comparable with that observed in wild-type mice [32], suggesting that platelet thrombus formation can occur in the absence of vWF. Fibrinogen/vWF knockout mice exhibit preserved platelet-to-platelet interaction [31], via a mechanism primarily triggered by thrombin and sustained by soluble agonist release (ADP) and different integrin signalling cascades [32, 33]. Activation of the coagulation cascade at the site of vessel injury, with consequent generation of thrombin through the tissue factor (TF) pathway, has been proposed as a major contributor to the thrombogenic component of atherothrombotic disease [34, 35]. However, recent evaluation of the dynamics of thrombus formation on atherosclerotic plaques has shown that TF has a predominant role only in the amplification phase of platelet aggregation, while the first key event of platelet arrest and aggregation is crucially regulated by engagement of the collagen receptor GPVI [36]. Indeed GPVI blockade, but not plaque TF suppression, significantly inhibits thrombus development. In similar experiments performed by Penz et al. [37], GPIb was found to be a crucial effector in plaque-induced thrombus, a finding in agreement with the work of Ruggeri et al. showing the importance of the vWF/GPIb axis in thrombus formation under conditions of high shear, and further confirmed in studies of interleukin 4-receptor/GPIb transgenic mice [38], in which lack of activity of GPIb gives rise to a severe bleeding phenotype. However, compared with the relatively mild effect on thrombogenic response to vascular injury observed in vWF-knockout mice, these results are strongly suggestive of an additional thrombogenic mechanism sustained by GPIb that may interact with ligands other than vWF.
Taken together, these findings imply that, independent of the initial thrombogenic stimulus and blood flow conditions, platelet-to-platelet interaction only results in irreversible aggregation once stable adhesion to the vascular wall, mediated by multiple receptor-ligand binding events, is established.
Heterotypic aggregation
The ability of platelets to interact with leucocytes was established in the late 19th century [39], from intravital microscopic studies of Bizzozero who first discovered platelets and reported the description of both homotypic and heterotypic aggregation at the site of vascular injury: ‘Blood platelets, swept along by the blood stream, are held up at the damaged spot as soon as they arrive at it. At first, one sees only two to four to six (platelets); very soon the number climbs to hundreds. Usually some white blood cells are held up amongst them.’ (Taken from a review by Brewer [40]).
More than a century of research in the field has provided multiple lines of evidence supporting the concept that, by contrast with homotypic aggregation, which is confined to the vascular wall, heterotypic complex formation occurs in circulating blood. The first detection of leucocyte–platelet aggregation in the human circulation was reported in subjects with coronary atherosclerosis [41], in whom heterotypic aggregates were identified within the coronary circulation, in close proximity to atherosclerotic lesions. The same study reported a higher level of heterotypic complexes in the vicinity of plaques with thrombotic complications as compared with uncomplicated ones. In further clinical studies, heterotypic complexes were observed in the systemic circulation of patients with coronary atherosclerosis [9–11], their levels being increased during acute thrombotic events. These findings suggest that circulating heterotypic aggregates form in parallel with thrombi, as a consequence of local platelet activation sustained by vascular damage. However, the presence of atherosclerotic lesions may not be mandatory for heterotypic aggregation, as raised levels of such aggregates have been described also in healthy subjects with cardiovascular risk factors [16–18], although such subjects may well have subclinical atherosclerosis, as well as in patients with other inflammatory conditions (reviewed by von Hundelshausen & Weber [42]). Adhesion between platelets and leucocytes is stable, does not require adhesion to pro-thrombotic surfaces, and is a systemic phenomenon whose extent depends on the degree of platelet activation. Early in vitro experiments performed on whole blood showed that platelet activation by a variety of agonists results initially in homotypic interaction mediated by fibrinogen-GPIIb/IIIa interaction, which is followed, in the absence of an adhesive vascular surface, by platelet disaggregation and subsequent platelet adhesion to leucocytes, with monocytes having a competitive advantage over other white cells in binding activated platelets [43]. Inhibition of fibrinogen binding to GPIIb/IIIa is even able to enhance the interaction of activated platelets with leucocytes, and the heterotypic complexes so formed, that are mainly composed of monocyte–platelet aggregates, display a strong pro-inflammatory phenotype mediated by surface-expressed molecules such as CD40L and P-selectin, along with pro-thrombotic activity sustained by their production of TF [44].
Biomolecular mechanisms of platelet activation leading to heterotypic aggregation
The pioneering work of Jungi et al. in 1986 [45] demonstrated that platelets acquire adhesiveness to leucocytes when stimulated with thrombin in a concentration-dependent manner, while non-activated platelets show little tendency to associate with white cells, any such association being explained by low-grade spontaneous activation of platelets undergoing handling in vitro. A number of other researchers confirmed these results [43, 46, 47], and it is now well established that platelet activation, independent of the initial stimulus, is required to trigger platelet-leucocyte interaction. As part of the underlying molecular mechanism, expression of the adhesion molecule P-selectin on the plasmalemma of activated platelets appears to be crucially involved [47, 48]. P-selectin, also designated CD62P and previously referred to as GMP-14O or PADGEM protein, is a transmembrane molecule stored in the alpha-granules of platelets. Upon stimulation, the granules fuse with the plasma membrane and P-selectin thus translocates from the cytosolic compartment to the extracellular surface, where it acts as a receptor for circulating white cells [48]. Its specific ligand is known as P-selectin glycoprotein ligand (PSGL)-1, and this is constitutively expressed on the plasmalemma of leucocytes [49]. Within the leucocyte population, the expression of PSGL-1 on the cellular surface of monocytes seems to be highest [50], and this could in part explain their elevated binding affinity to platelets over other white cells. Indeed, an in vivo study performed in apoE–/– mice [21] demonstrated that intravenous injection of activated platelets leads to sequestration of leucocytes within a few (approximately 5) minutes, persisting up to 80 min for polymorphonuclear cells (PMN) and 180 min for monocytes. No such effect was reported for lymphocytes, confirming once more the differential binding activity of activated platelets to different leucocyte sub-populations, as observed in early in vitro studies [43, 44]. A similar study in baboons showed similar results [9]: after injection of activated platelets, formation of circulating aggregates with monocytes and PMN occurred within 1 min, and the in vivo half-lives of circulating monocyte–platelet and PMN–platelet complexes were, respectively, 30 and 5 min; no heterotypic aggregates were detectable after 2 h.
The preferential binding of platelets to monocytes may also be ascribed to an additional role played by other ligands specifically present on the plasmalemma of monocytes, which may follow on from P-selectin/PSGL-1 interaction. Indeed, PSGL-1 is not only an adhesion but also a signalling molecule. PSGL-1 binding induces production of superoxide anion radicals from monocytes [51], and tyrosine phosphorylation of various cytoplasmic proteins, including pp125 focal adhesion kinase, ERK, Syk, Src kinase and paxillin [52–55]. Binding of PSGL-1 to P-selectin leads to integrin activation on monocytes, resulting in further stabilization of cellular adhesion in part mediated by additional bridging molecules. Further interactions are through CD40L/CD40, TREM-1 ligand/TREM-1 (which may also promote integrin expression) and CD36/CD36 via thrombospondin [56]. However, P-selectin/PSGL-1 linking clearly has a crucial role in heterotypic aggregation, as blocking monoclonal antibody to P-selectin [45] or PSGL-1 [16] completely abolishes the formation of leucocyte–platelet aggregates.
Platelet release reaction
Release of platelet granule contents represents a common biomolecular event in both homotypic and heterotypic aggregation (Table 1). In the former, it contributes to the amplification and stabilization of initial reversible aggregates, mainly through the release of platelet agonists. In the latter, it sustains the receptor-ligand interactions between platelets and white cells by promoting the expression of P-selectin. Platelet agonists and adhesion molecules are segregated into specific intracellular compartments [57, 58] and, for many decades, their release was considered to be directed by a structural re-organization of the cytoskeleton [59–61]. This conventional view of the platelet release reaction has been recently extended to include a more composite mechanism of fusion between the granule membrane and the plasma membrane, subtly regulated by N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) family proteins, chaperon and the composition of the lipid membrane (reviewed by Reed et al. [62] and Flaumenhaft [63]). The activity of this regulatory apparatus seems to be modulated by agonist-specific effector molecules, to ensure selectivity in dense and alpha granule content release as appropriate.
Platelet granules and selective regulation of release reaction
Immunocytochemical and electron microscopy studies over the years have given rise to the distinction between three main types of storage organelles in platelets, known as dense, alpha and lysosomal granules. Their content can be classified into three main groups on the basis of their principal function: platelet agonists [ADP, serotonin, epinephrine, 5-hydroxytryptamine (5-HT) and calcium], contained in the dense granules, adhesion molecules [P-selectin, fibrinogen, vWF, platelet factor 4 (PF4)], stored in alpha-granules and lysosomal enzymes (cathepsin, hexosaminidase) within the lysosomes [64]. Ultrastructural and morphological studies have revealed the intracellular organization of the different organelles, which distribute along an intricate network of membrane invaginations forming an open canalicular system (OCS) [64]. The process of actin polymerization in activated platelets has been generally considered to be the major determinant in organelle trafficking, because of the generation of contractile force with consequent expulsion of granules [59–65]. More recent studies have revealed that platelet shape changes, as sustained by actin re-organization following platelet stimulation, are not essential for the platelet release reaction [66–68]. Indeed, actin disruption has shown either inhibitory or stimulatory effects on platelet degranulation in different studies [66–70]. The discrepancy in results between these studies may be in part ascribed to the evidence, recently provided by Flaumenhaft et al. [71], that the cytoskeleton differentially regulates dense and alpha granule exocytosis; specifically, low grade actin disruption stimulates agonist-induced alpha granule release with only minimal effect on dense granule secretion, while high levels of actin disruption inhibit alpha granule release but promote dense granule secretion. The molecular mechanisms underlying the platelet release reaction are still poorly understood, but increasing evidence suggests the existence of distinct regulatory pathways for alpha and dense granule secretion (Figure 1). It has recently been reported that Munc13 proteins, which act in concert with the SNARE complex in mediating fusion between the granule/vesicle membrane and the plasma/target membrane [72], provide a single and common mechanism for the release reaction of all the different types of platelet granule. However, the residual secretion of alpha granule contents observed in response to thrombin in Munc13-4 knockout mice, in which dense granule release is totally abolished, suggests that secretion of alpha granules is less dependent on Munc13-4 activity than that of dense granules [72]. The scenario is complicated by recent observations that alpha organelles are composed of different subtypes of vesicle containing different adhesion molecules, which are released independently of each other [73–75].
Figure 1.
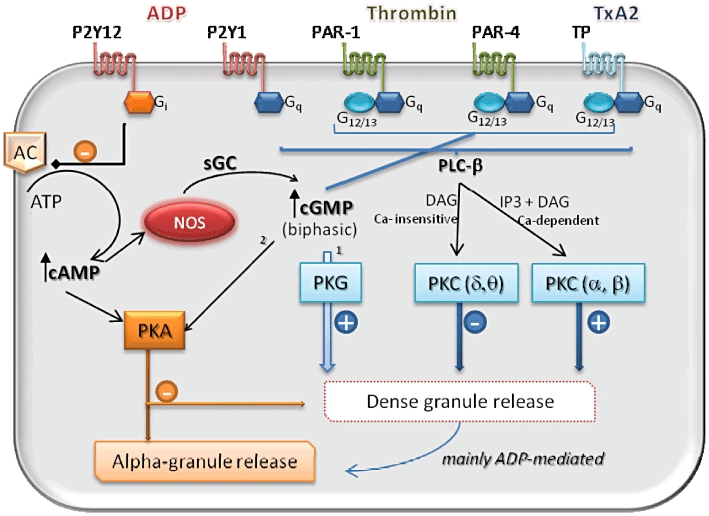
Model of agonist-selective modulation of alpha and dense granule release in human platelets
Given the fact that soluble molecules stored in dense organelles are predominantly involved in homotypic aggregation and thrombus formation, while P-selectin in alpha granules is mainly implicated in heterotypic complex formation, it is conceivable that, under different pathophysiological circumstances, different types of platelet release reaction occur. The distinct biomolecular pathways involved may differentially favour thrombus formation and/or platelet interaction with white cells.
Pharmacological modulation of homotypic and heterotypic aggregation
Aspirin and thienopyridine derivatives, including clopidogrel and prasugrel, are the most widely used anti-platelet drugs in clinical practice, especially in the chronic management of atherosclerotic disease. Clinical studies report a differential ability of these two classes of agent to modulate homotypic and heterotypic aggregation. In the following, we will examine the molecular mechanisms underlying their therapeutic action, in parallel with the molecular pathways activated in response to different platelet agonists, with a view to clarifying how the selective modulation of these different signalling pathways exerts differential effects on these two events which may allow identification of potential new therapeutic targets.
Aspirin
The antiplatelet effect of aspirin is mediated by the irreversible inhibition of thromboxane (Tx)A2 synthesis in platelets, because of acetylation of a serine hydroxyl group at position 529 in cyclo-oxygenase (COX)-1. This steric modification of the enzyme inhibits the access of arachidonic acid, the precursor of TxA2, to the active site of the enzyme. Blockade of COX-1 in platelets, as obtained either following in vivo treatment or in vitro incubation of platelets with aspirin, usually gives rise to effective inhibition of aggregation in response to a number of platelet agonists, although there is evidence of reduced aspirin response in some subjects, giving rise to the concept of aspirin resistance [76]. The antiplatelet effect of aspirin is mainly ascribed to negative modulation of dense granule release. Indeed, an early study reported that alpha granule exocytosis, as detected by P-selectin expression, vWF and beta-thromboglobulin secretion, was not at all influenced by COX-1 blockade in platelets stimulated with either ADP or thrombin [77]. These data imply that TxA2 signalling is primarily involved in dense granule but not alpha granule secretion.
In accordance with this, Li et al. reported no effect in healthy subjects of aspirin either on platelet P-selectin expression or on platelet–leucocyte aggregation, after stimulation with ADP, thrombin or platelet activating factor [78]. Similarly, Klinkhardt et al. found that aspirin did not affect the formation of leucocyte–platelet complexes in the circulation [19], and Moshfegh et al. demonstrated a reduced effect of aspirin when compared with other anti-platelet drugs in reducing platelet P-selectin expression [79]. As heterotypic aggregates are believed to promote atherosclerosis progression through sustaining a pro-inflammatory state, this relative lack of effect of aspirin on heterotypic aggregation may explain, at least in part, the lack of any clear effect on atherosclerosis progression as well as the poorer results on clinical outcome as compared with newer antiplatelet agents such as the thienopyridines, as discussed further below [80].
Thienopyridines
Ticlopidine, clopidogrel and prasugrel are selective, irreversible noncompetitive inhibitors of the P2Y12 receptor. All are prodrugs that require oxidation by hepatic cytochrome P450 to be converted into active metabolites [81]. Their effect on platelet aggregation has been investigated in many clinical trials, whose main end point was the reduction of fatal or non-fatal cardiovascular (CV) events [82–86]. Clear benefit from P2Y12 receptor blockade has been consistently found in the secondary prevention of CV disease. Compared with ticlopidine and clopidogrel, prasugrel provides faster and more uniform inhibition of P2Y12-dependent platelet activation [87], resulting in reportedly more effective reduction of major thrombotic events. However, the higher platelet inhibition achieved with prasugrel also gives rise to increased incidence of bleeding events compared with clopidogrel [88]. Reversible P2Y12 antagonists, such as cangrelor and ticagrelor, which belong to the cyclopentyl-triazolo-pyrimidine chemical class, have been recently developed [89] and offer a clinically useful alternative to thienopyridines. Unlike cangrelor, ticagrelor has shown superiority to clopidogrel in reducing CV events, as well as the advantage of shorter time of offset if surgery is contemplated (24–72 h) compared with thienopyridines which take at least 5 days to wear off [90].
Beyond a direct effect on homotypic aggregation, antagonism of P2Y12 receptor has demonstrated efficacy in reducing CD40L and P-selectin expression, as well as platelet–leucocyte aggregate formation [91]. In accordance with these findings, thienopyridines, but not aspirin, have been shown to reduce platelet P-selectin expression, which can be explained by their ability to inhibit ADP-induced alpha-granule release [19, 91]. Prasugrel has shown greater efficacy than clopidogrel in modulating heterotypic aggregation and P-selectin expression, in ADP-stimulated platelets obtained from subjects with atherosclerosis treated with the two different thienopyridines [92]. Thus, P2Y12 blockade may modulate vascular inflammation in addition to platelet thrombosis [93].
Gq protein-coupled receptors and the platelet release reaction
TxA2, ADP and thrombin all induce platelet activation through G-protein coupled receptors (GPCR). The TxA2 receptor (TP) couples to the Gq and G12/13 subtypes, as do the thrombin receptors protease activated receptor (PAR)-1 and PAR-4. G12/13-dependent signalling pathways positively feed back on the Gq signal-transduction cascade. ADP binds two main purinoceptors, namely P2Y1 and P2Y12, which, respectively, couple to Gq and Gi[94].
The main effector of Gq is phospholipase C (PLC), mainly the PLC-β isoform, which via hydrolysis of membrane phospholipids and consequent generation of two second messengers, inositol trisphosphate (IP3) and diacylglycerol (DAG), leads to the activation of protein kinase C (PKC) [5]. PKC comprises three groups of isoforms: the classical subgroup (α, β, γ), whose activation is calcium-dependent and mediated by the synergistic action of IP3 and DAG, the calcium-insensitive isoforms (δ, η, θ, ε), regulated by DAG and the IP3-sensitive atypical isoforms [95, 96]. The enzymatic activity of PKC has been demonstrated to have important implications in the regulation of the platelet release reaction [97, 98], as a consequence of its ability to phosphorylate and thereby activate the SNARE machinery [99]. Ablation of PKC-α in murine models gives rise to defective responsiveness of platelets to in vitro stimulation with thrombin, because of compromised release of dense granule contents. Indeed, platelets lacking expression of PKC-α show a decrease in ATP secretion compared with wild-type platelets, and the addition of exogenous ADP restores agonist-dependent aggregation [100]. The expression of P-selectin, an indicator of alpha-granule release, is also impaired in response to thrombin when PKC-α is not present. However, in these experiments, the ability of ADP to restore translocation of P-selectin to the platelet plasmalemma was not evaluated, so it is not known whether ADP also restores alpha-granule secretion in this model.
In separate in vitro experiments, the specific role of the PKC-δ isoform in granule content release was evaluated. Murugapan et al. [101] demonstrated that TxA2 and thrombin induce PKC-δ phosporylation, and that this effect is accompanied by dense granule exocytosis which is abolished by selective inhibition of PKC-δ activity. However, in collagen-stimulated platelets, PKC-δ was found to exert an inhibitory effect on platelet activation, implying that different stimuli may activate distinct transduction pathways. Similarly, ADP was not found to exert any modulatory action on PKC-δ. An individual comparison between the roles of different PKC subtypes has been recently reported by Gilio et al. [102], who used pharmacological approaches in human platelets as well as specific knockout mice to study platelet function under physiological flow conditions and in response to collagen. They demonstrated that activation of conventional Ca2+-dependent PKC isoforms stimulates dense granule release and platelet aggregation, while activation of Ca2+-insensitive isoforms inhibits these events (Figure 1). Moreover, although PKC-δ was found to regulate negatively dense granule release, no effect on alpha granule secretion was observed. However, in vivo studies indicate a co-adjuvant rather than essential role of PKC in thrombus formation. In PKC-α-deficient mice, no alteration of bleeding time or thrombus formation compared with wild-type mice is seen [100], despite a decrease in the volume of thrombus compared with wild-type controls in both PKC-α and PKC-β knockout mice secondary to compromised platelet-to-platelet adhesion [102].
The ability of ADP to correct the defect in platelet function in the presence of PKC deficiency demonstrates that its action on platelets is mediated by alternative signalling pathways. Its ability to activate Gi as well as Gq protein-dependent signalling seems to represent a crucial mechanism in ADP-mediated platelet activation, as specific inhibition of the former pathway has been demonstrated to compromise its co-adjuvant role in thrombin- or TxA2-mediated platelet stimulation [103, 104].
Gi protein-coupled receptors and adenylyl cyclase
The interaction of ADP with the P2Y12 receptor activates Gi-mediated signalling that, via negative modulation of adenylyl cyclase (AC) enzymatic activity, reduces intracellular concentrations of cyclic AMP (cAMP) [105]. AC represents an endogenous inhibitory mechanism of platelet activation, whose action is mediated by cAMP-dependent activation of protein kinase A (PKA). PKA-mediated inhibition of platelet activation involves interference with calcium release [106], phosphorylation of cytoskeletal proteins such actin binding protein (ABP) and myosin light chain kinase (MLCK) [107], as well as of the focal adhesion mediator vasodilator-stimulated phosphoprotein (VASP) [108]. The inhibition of AC, as caused by P2Y12 activation, therefore results in amplification of activator pathways triggered by different agonists. Indeed, P2Y12 stimulation potentiates the effect of thrombin and TxA2 on platelet aggregation as well as increasing P-selectin expression [103, 104]. Evidence for the essential role of Gi-dependent pathways in regulating the platelet release reaction has been provided by Li et al. [109], who studied signal-transduction regulated by TxA2 and thrombin in an animal model lacking expression of protein kinase G (PKG). They demonstrated that these two soluble agonists activate PKG, a cyclic GMP (cGMP)-dependent tyrosine kinase enzyme, through their specific Gq-protein coupled receptors. PKG activation plays an important role in dense granule release from platelets. Nevertheless, the impaired platelet response to thrombin and TxA2 observed in PKG knockout compared with wild-type animals was counteracted by addition of exogenous ADP, an effect abolished by selective Gi inhibition. The same findings were obtained in human platelets, in which blockade of Gi signalling nullified the potentiating effect of ADP on thrombin and TxA2 stimulation of platelets. Of note, recent work from Iyu et al. [110] has demonstrated that Gi blockade, as occurs in response to P2Y12 antagonists, converts the stimulatory activity of ADP on platelet activation into an inhibitory effect mediated by ADP-derived adenosine.
Other antagonists/agonists and pathways modulating adenylyl cyclase activity
Adenosine is a purine nucleoside that is formed from the sequential dephosphorylation of ATP. Adenosine modulates platelet function through the engagement of its specific receptors A2A and A2B, that couple to stimulatory Gs-proteins thereby activating AC. The resulting increase in cAMP gives rise to inhibition of platelet aggregation [110].
Epinephrine is contained in platelet dense granules and is released upon platelet activation [111, 112]. It acts on α2A-adrenergic receptors, coupled to Gz-proteins which belong to the Gi family [113, 114]. Epinephrine is considered a weak platelet agonist, as alone it does not sustain platelet aggregation in the absence of other agonists [94]; rather, it sensitizes platelets to the action of other agonists [115], and is able to induce P-selectin expression on platelets as well as to sustain platelet–leucocyte aggregation [116].
Prostaglandin E2 (PGE2) modulates platelet function via its binding to EP3 receptors, which are coupled to Gi protein [94] and EP4 receptors, which feed back on Gs-protein [117]. As with epinephrine, PGE2 is not an aggregating agent per se at low concentrations, but potentiates the effect of other agonists, as demonstrated in both in vitro experiments and in vivo animal models [118, 119]. The pro-aggregatory effect of PGE2 has been recently demonstrated to be secondary to cAMP reduction, as a consequence of EP3 receptor activation. On the other hand, PGE2 binding to EP4 induces an opposite inhibitory effect on platelet function, mediated by increased cAMP [117]. Similarly opposing effects on platelet activity have been described for prostaglandin E1 (PGE1) which, via binding to EP3 receptors, reduces cAMP thereby promoting platelet activation, while on the other hand, by interacting with IP receptors (as does prostacyclin), inhibiting platelet function [120].
All the chemokine receptors identified on the platelet plasmalemma, including CCR1, CCR2, CCR3, CCR4, CXCR4 and CX3CR1, are coupled to Gi. They all are considered weak platelet activators, but their role appears to be predominantly in the inflammatory response that accompanies atherosclerotic vascular disease and thrombosis [94].
Overall, the evidence suggests that negative modulation in platelets of the basal activity of AC, resulting in decreased cAMP, favours their aggregation, and even in the absence of effective homotypic aggregation, this mechanism suffices to induce platelet P-selectin expression and consequent heterotypic aggregation.
Pharmacological modulation of adenylyl cyclase: effect on heterotypic aggregation
Stimulation of AC with consequent cAMP elevation prevents platelet activation and adhesion. This effect has been observed in response to stimulation of Gs-coupled β-adrenoceptors in platelets that, via cAMP-dependent activation of nitric oxide (NO) synthase (NOS) type 3, primarily reduce platelet heterotypic adhesion with minimal, or no effect, on homotypic aggregation [121]. Similarly, in a study of healthy subjects, ex vivo treatment of platelets with exogenous NO was found to decrease their aggregation with monocytes [16]. In line with these results, Morell et al. [122] have shown that NO inhibits exocytosis of dense and alpha-granules, but the effect on the latter is more pronounced than on the former, as it is evident at concentrations as low as 10 nm compared with 1 µm required to modulate significantly dense granule release. Conversely, decreased NOS activity enhances monocyte–platelet complex formation [16, 123]. The effect of NO on platelet activation is mediated through soluble guanylyl cyclase (sGC) and consequent increase in cGMP. It has been reported by one group that cGMP plays a biphasic role on the platelet release reaction, with an early stimulatory action mediated through PKG and a late inhibitory effect occurring at higher cGMP concentrations and in a PKA-dependent manner [124, 125], although this is controversial.
Cilastozol, which is a phosphodiesterase (PDE) 3 inhibitor, can increase platelet cAMP [126], as well as efficiently suppress P-selectin expression and platelet–leucocyte aggregate formation induced by a variety of agonists [126, 127]. Addition of cilastozol to standard dual antiplatelet therapy (aspirin plus clopidogrel) in patients undergoing coronary artery stent implantation has been reported to result in increased inhibition of platelet aggregation and P-selectin expression, in ex vivo functional assays [128].
Pharmacological modulation of the serotoninergic system can modify platelet activation status and heterotypic complex formation. Indeed, selective serotonin re-uptake inhibitors, commonly used for the treatment of depression, have been shown to inhibit platelet–leucocyte aggregation [129, 130], most likely as a consequence of their demonstrated ability to activate AC in platelets [131]. Similar findings have been reported in rodents treated with herbal medicines such as magnolol and honokiol from Magnolia officinalis[132], used for treatment of depression in traditional Chinese medicine, as well as curcumin from Curcuma longa[133], used by alternative practitioners for improving cognitive function and for its purported anti-inflammatory and antitumour effects, all of which enhance platelet AC activity and thus intracellular cAMP.
From all of these data, Gi-dependent signalling appears to be crucially implicated in alpha granule release and consequent heterotypic aggregation, independently of other biomolecular events activated during platelet adhesion to the vascular wall and thrombus formation. At the same time, it exerts a modulatory effect on platelet activation and homotypic aggregation induced by agonists whose direct action is Gq-mediated.
Table 2 summarizes the effects of different agents that modulate intraplatelet cAMP on homotypic and heterotypic aggregation.
Table 2.
Effects of platelet agonists/inhibitors on intraplatelet cAMP and consequent effects on homotypic vs. heterotypic aggregation
| cAMP modulation | Effect on homotypic aggregation | Effect on heterotypic aggregation | |
|---|---|---|---|
| ADP | ↓ | Stimulatory, requires activation of both P2Y1 and P2Y12 | Stimulatory, mainly sustained by P2Y12 |
| Adenosine | ↑ | Inhibitory | Inhibitory |
| Epinephrine | ↑ | Very little | Stimulatory |
| Prostaglandin E2 | via EP3 receptor ↓ | None alone | Stimulatory |
| Prostaglandin E2 | via EP4 receptor ↑ | Inhibitory | Inhibitory |
| Prostaglandin E1 | via EP3 receptor ↓ | None alone | Stimulatory |
| Prostaglandin E1 | via IP receptor ↑ | Inhibitory | Inhibitory |
| Chemokines | ↓ | None alone | Likely stimulatory |
| β-adrenoceptors | ↑ | Inhibitory | Inhibitory |
| Cilostazol | ↑ | Inhibitory | Inhibitory |
| SSRI | ↑ | Inhibitory | Inhibitory |
| Magnolol, homokiol, curcumin | ↑ | Inhibitory | Inhibitory |
Novel therapeutic approaches to modulation of homotypic and heterotypic aggregation
The development of new drugs which target the above described molecular mechanisms underlying adhesion molecule expression on activated platelets, as well as that of agents which interfere with the multiple receptor-ligand interactions involved in homotypic or heterotypic aggregation, represent exciting and promising strategies for future antithrombotic therapies. GPIIb/IIIa blockers are an emblematic example of this pharmacological approach. They have been demonstrated to prevent thrombotic events when given acutely and intravenously in patients with acute coronary syndromes and in those undergoing percutaneous coronary intervention, albeit at a price of increased risk of bleeding complications [134]. However, their counterpart oral GPIIb/IIIa inhibitors have shown no benefit or even increased mortality in clinical trials [135–137].
Interference in GPVI interaction with collagen has been also proposed as a viable antithrombotic strategy, and GPVI blocking agents are under development. Indeed, soluble GPVI dimers and anti-GPVI antibodies have been produced and tested in a variety of animal models [138–140]. However those tested to date appear to lack potency and efficacy and consequently in vivo data have not demonstrated a convincing antithrombotic effect. Fab fragment of anti-GPVI monoclonal antibody [141] and a novel GPVI-binding peptide [142] are currently undergoing evaluation in terms of their GPVI affinity and antithrombotic effectiveness in different species.
A novel and potentially attractive area of therapeutic development in this field is represented by so-called aptamers. Aptamers are protein-binding oligonucleotides that offer potential advantages over standard methods, in particular the possibility to combine a selective action against a specific protein–protein interaction with lack of antigenic activity (extensively reviewed by Keefe & Shaub [143]). Amongst the different antithrombotic aptamers that have been developed, inhibitors of vWF have shown good efficacy as antiplatelet agents in animal models, including non-human primates [144, 145], and have moved into the clinical setting with a first phase I trial [146] which has shown promising results.
Despite these considerations, the complexity of homotypic aggregation, whose main effector molecules vary depending on the haemodynamic conditions and specific features of the vascular lesions, may render it difficult to select a single ligand/receptor axis on which pharmacological modulation might result in a clear therapeutic anti-thrombotic action. On the other hand, interference with the adhesive interaction sustaining heterotypic complex formation may be more straightforward, given the critical role of P-selectin/PSGL-1 binding in mediating platelet-leucocyte aggregation. A number of different molecules have been identified or chemically synthesized with demonstrated ability to interfere with this binding. Heparin and heparin sulphate are ligands for P-selectin and block its binding to sialylated, fucosylated carbohydrate antigen related to sialyl LewisX (SLeX), which is expressed on its ligands including PSGL-1. Several chemically-modified heparin derivatives (heparinoids) have been developed and have shown in vivo anti-inflammatory activity because of their blocking activity on P-selectin as well as L-selectin, the latter being expressed on leucocytes and endothelial cells [147]. More specific inhibitors of P-selectin such as monoclonal antibodies [148], SLeX mimetics [149, 150] and recombinant PSGL-1 [151] have been produced and have demonstrated in vivo antithrombotic as well as anti-inflammatory effects in different animal models. However, the development of these compounds into effective drugs for clinical use has been greatly limited because of their relatively low selectivity amongst the different selectins, short circulating half-life, expense and potential antigenicity. In this context, apatmers offer a good therapeutic alternative. Indeed, P-selectin inhibitor aptamers are now available and have demonstrated higher binding affinity for P-selectin compared with SLeX, and consequently a selective effect on PSGL-1 engagement. Moreover, little cross-interaction appears to occur with other selectins, including the L- and E-isoforms. It now needs to be established whether the in vitro data showing inhibition of heterotypic aggregation mediated by P-selectin aptamers [152] will be supported by antithrombotic and anti-inflammatory effects in vivo.
Conclusions
We have here reviewed how the two main biological functions of platelet activation, namely thrombotic and inflammatory activity, can be differentially regulated in platelets. Examination of Gq- and Gi-dependent signalling pathways reveals that the combined activation of these two transduction cascades is necessary for effective thrombus formation, which requires the full platelet release reaction. However, regulation of Gi signalling alone, as occurs with pro-inflammatory cytokines, ADP and molecules involved in redox regulation, such as NO, appears to be crucially involved in alpha granule release, even in the absence of full platelet activation secondary to vascular damage. Pharmacological intervention on this molecular pathway, as provided by thienopyridine derivatives or cilastozol, as well as NO-potentiating agents, reduces heterotypic aggregate formation. Additionally, aptamers represent a novel and potentially effective means to inhibit platelet aggregation, especially of the heterotypic kind. However, in spite of rapid advances in our understanding of the molecular mechanisms regulating platelet function, further studies are required to define better the role of the different molecular effectors so far identified, as well as to identify novel potential molecular targets in order to develop more effective therapeutic strategies to correct platelet disorders.
Competing Interests
There are no competing interests to declare.
REFERENCES
- 1.Savage B, Almus-Jacobs F, Ruggeri ZM. Specific synergy of multiple substrate-receptor interactions in platelet thrombus formation under flow. Cell. 1998;94:657–66. doi: 10.1016/s0092-8674(00)81607-4. [DOI] [PubMed] [Google Scholar]
- 2.Tschopp TB, Weiss HJ, Baumgartner HR. Decreased adhesion of platelet to subendothelium in von Willebrand's disease. J Lab Clin Med. 1973;83:296–300. [PubMed] [Google Scholar]
- 3.Weiss HJ, Turitto VT, Baumgartner HR. Effect of shear rate on platelet interaction with subendothelium in citrated and native blood. I. Shear rate-dependent decrease of adhesion in von Willebrand's disease and the Bernard-Soulier syndrome. J Lab Clin Med. 1978;92:750–64. [PubMed] [Google Scholar]
- 4.Isenberg WM, McEver RP, Phillips DR, Shuman MA, Bainton DF. The platelet fibrinogen receptor: an immunogold-surface replica study of agonist-induced ligand binding and receptor clustering. J Cell Biol. 1987;104:1655–63. doi: 10.1083/jcb.104.6.1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Siess W. Molecular mechanisms of platelet activation. Physiol Rev. 1989;69:58–178. doi: 10.1152/physrev.1989.69.1.58. [DOI] [PubMed] [Google Scholar]
- 6.Semple JW, Freedman JE. Platelets and innate immunity. Cell Mol Life Sci. 2010;67:499–511. doi: 10.1007/s00018-009-0205-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kroll MH, Hellums JD, McIntire LV, Schafer AI, Moake JL. Platelets and shear stress. Blood. 1996;88:1525–41. [PubMed] [Google Scholar]
- 8.Freedeman JE, Loscalzo J. Platelet–monocyte aggregates: bridging thrombosis and inflammation. Circulation. 2002;105:2130–2. doi: 10.1161/01.cir.0000017140.26466.f5. [DOI] [PubMed] [Google Scholar]
- 9.Michelson D, Barnard MR, Krueger LA, Valeri CR, Furman MI. Circulating monocyte–platelet aggregates are a more sensitive marker of in vivo platelet activation than platelet P-selectin: studies in baboons, human coronary intervention, and human myocardial infarction. Circulation. 2001;104:1533–7. doi: 10.1161/hc3801.095588. [DOI] [PubMed] [Google Scholar]
- 10.Sarma J, Laan CA, Alam S, Jha A, Fox KA, Dransfield I. Increased platelet binding to circulating monocytes in acute coronary syndromes. Circulation. 2002;105:2166–71. doi: 10.1161/01.cir.0000015700.27754.6f. [DOI] [PubMed] [Google Scholar]
- 11.Furman MI, Barnard MR, Krueger LA, Fox ML, Shilale EA, Lessard DM, Marchese P, Frelinger A. L, 3rd, Goldberg RJ, Michelson AD. Circulating monocyte–platelet aggregates are an early marker of acute myocardial infarction. J Am Coll Cardiol. 2001;38:1002–6. doi: 10.1016/s0735-1097(01)01485-1. [DOI] [PubMed] [Google Scholar]
- 12.Marquardta L, Andersb C, Bugglea F, Palma F, Hellsternb P, Graua AJ. Leukocyte–platelet aggregates in acute and subacute ischemic stroke. Cerebrovasc Dis. 2009;28:276–82. doi: 10.1159/000228710. [DOI] [PubMed] [Google Scholar]
- 13.Joseph JE, Harrison P, Mackie IJ, Isenberg DA, Machin SJ. Increased circulating platelet–leucocyte complexes and platelet activation in patients with antiphospholipid syndrome, systemic lupus erythematosus and rheumatoid arthritis. Br J Haematol. 2001;115:451–9. doi: 10.1046/j.1365-2141.2001.03101.x. [DOI] [PubMed] [Google Scholar]
- 14.Jensen MK, de Nully Brown P, Lund BV, Nielsen OJ, Hasselbalch HC. Increased circulating platelet–leukocyte aggregates in myeloproliferative disorders is correlated to previous thrombosis, platelet activation and platelet count. Eur J Haematol. 2001;66:143–51. doi: 10.1034/j.1600-0609.2001.00359.x. [DOI] [PubMed] [Google Scholar]
- 15.Wun T, Cordoba M, Rangaswami A, Cheung AW, Paglieroni T. Activated monocytes and platelet–monocyte aggregates in patients with sickle cell disease. Clin Lab Haematol. 2002;24:81–8. doi: 10.1046/j.1365-2257.2002.00433.x. [DOI] [PubMed] [Google Scholar]
- 16.Gkaliagkousi E, Corrigall V, Becker S, de Winter P, Shah A, Zamboulis C, Ritter J, Ferro A. Decreased platelet nitric oxide contributes to increased circulating monocyte–platelet aggregates in hypertension. Eur Heart J. 2009;30:3048–54. doi: 10.1093/eurheartj/ehp330. [DOI] [PubMed] [Google Scholar]
- 17.Harding SA, Sarma J, Josephs DH, Cruden NL, Din JN, Twomey PJ, Fox KA, Newby DE. Upregulation of the CD40/CD40 ligand dyad and platelet–monocyte aggregation in cigarette smokers. Circulation. 2004;109:1926–9. doi: 10.1161/01.CIR.0000127128.52679.E4. [DOI] [PubMed] [Google Scholar]
- 18.Harding SA, Sommerfield AJ, Sarma J, Twomey PJ, Newby DE, Frier BM, Fox KA. Increased CD40 ligand and platelet–monocyte aggregates in patients with type 1 diabetes mellitus. Atherosclerosis. 2004;176:321–5. doi: 10.1016/j.atherosclerosis.2004.05.008. [DOI] [PubMed] [Google Scholar]
- 19.Klinkhardt U, Bauersachs R, Adams J, Graff J, Lindhoff-Last E, Harder S. Clopidogrel but not aspirin reduces P-selectin expression and formation of platelet–leukocyte aggregates in patients with atherosclerotic vascular disease. Clin Pharmacol Ther. 2003;73:232–41. doi: 10.1067/mcp.2003.13. [DOI] [PubMed] [Google Scholar]
- 20.Furie B, Furie BC. Thrombus formation in vivo. J Clin Invest. 2005;115:3355–62. doi: 10.1172/JCI26987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huo Y, Schober A, Forlow SB, Smith DF, Hyman MC, Jung S, Littman DR, Weber C, Ley K. Circulating activated platelets exacerbate atherosclerosis in mice deficient in apolipoprotein E. Nat Med. 2003;9:61–7. doi: 10.1038/nm810. [DOI] [PubMed] [Google Scholar]
- 22.van Gils JM, Zwaginga JJ, Hordijk PL. Molecular and functional interactions among monocytes, platelets, and endothelial cells and their relevance for cardiovascular diseases. J Leukoc Biol. 2009;85:195–204. doi: 10.1189/jlb.0708400. [DOI] [PubMed] [Google Scholar]
- 23.Kulkarni S, Dopheide SM, Yap CL, Ravanat C, Freund M, Mangin P, Heel KA, Street A, Harper IS, Lanza F, Jackson SP. A revised model of platelet aggregation. J Clin Invest. 2000;105:783–91. doi: 10.1172/JCI7569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maxwell MJ, Westein E, Nesbitt WS, Giuliano S, Dopheide SM, Jackson SP. Identification of a 2-stage platelet aggregation process mediating shear-dependent thrombus formation. Blood. 2007;109:566–76. doi: 10.1182/blood-2006-07-028282. [DOI] [PubMed] [Google Scholar]
- 25.Savage B, Saldivar E, Ruggeri ZM. Initiation of platelet adhesion by arrest onto fibrinogen or translocation on von Willebrand factor. Cell. 1996;84:289–97. doi: 10.1016/s0092-8674(00)80983-6. [DOI] [PubMed] [Google Scholar]
- 26.Packham MA, Mustard JF. Platelet adhesion. Prog Hemost Thromb. 1984;7:211–88. [PubMed] [Google Scholar]
- 27.Ruggeri ZM, Orje JN, Habermann R, Federici AB, Reininger AJ. Activation-independent platelet adhesion and aggregation under elevated shear stress. Blood. 2006;108:1903–10. doi: 10.1182/blood-2006-04-011551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nachman RL, Leung LL. Complex formation of platelet membrane glycoproteins IIb and IIIa with fibrinogen. J Clin Invest. 1982;69:263–9. doi: 10.1172/JCI110448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Emsley J, Knight CG, Farndale RW, Barnes MJ, Liddington RC. Structural basis of collagen recognition by integrin alpha2beta1. Cell. 2000;101:47–56. doi: 10.1016/S0092-8674(00)80622-4. [DOI] [PubMed] [Google Scholar]
- 30.Ruggeri ZM. Mechanisms initiating platelet thrombus formation. Thromb Haemost. 1997;78:611–6. [PubMed] [Google Scholar]
- 31.Ni H, Denis CV, Subbarao S, Degen JL, Sato TN, Hynes RO, Wagner DD. Persistence of platelet thrombus formation in arterioles of mice lacking both von Willebrand factor and fibrinogen. J Clin Invest. 2000;106:385–92. doi: 10.1172/JCI9896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dubois C, Panicot-Dubois L, Gainor JF, Furie BC, Furie B. Thrombin-initiated platelet activation in vivo is vWF independent during thrombus formation in a laser injury model. J Clin Invest. 2007;117:953–60. doi: 10.1172/JCI30537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang H, Reheman A, Chen P, Zhu G, Hynes RO, Freedman J, Wagner DD, Ni H. Fibrinogen and von Willebrand factor-independent platelet aggregation in vitro and in vivo. J Thromb Haemost. 2006;4:2230–7. doi: 10.1111/j.1538-7836.2006.02116.x. [DOI] [PubMed] [Google Scholar]
- 34.Corti R, Farkouh ME, Badimon JJ. The vulnerable plaque and acute coronary syndromes. Am J Med. 2002;113:668–80. doi: 10.1016/s0002-9343(02)01344-x. [DOI] [PubMed] [Google Scholar]
- 35.Libby P. Inflammation in atherosclerosis. Nature. 2002;420:868–74. doi: 10.1038/nature01323. [DOI] [PubMed] [Google Scholar]
- 36.Reininger AJ, Bernlochner I, Penz SM, Ravanat C, Smethurst P, Farndale RW, Gachet C, Brandl R, Siess W. A 2-step mechanism of arterial thrombus formation induced by human atherosclerotic plaques. J Am Coll Cardiol. 2010;55:1147–58. doi: 10.1016/j.jacc.2009.11.051. [DOI] [PubMed] [Google Scholar]
- 37.Penz SM, Reininger AJ, Toth O, Deckmyn H, Brandl R, Siess W. Glycoprotein Ibα inhibition and ADP receptor antagonists, but not aspirin, reduce platelet thrombus formation in flowing blood exposed to atherosclerotic plaques. Thromb Haemost. 2007;97:435–43. [PubMed] [Google Scholar]
- 38.Bergmeier W, Piffath CL, Goerge T, Cifuni SM, Ruggeri ZM, Ware J, Wagner DD. The role of platelet adhesion receptor GPIbalpha far exceeds that of its main ligand, von Willebrand factor, in arterial thrombosis. Proc Natl Acad Sci USA. 2007;103:16900–5. doi: 10.1073/pnas.0608207103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bizzozero J. Über einen neuen Formbestandteil des Blutes und dessen Rolle bei der Thrombose und Blutgerinnung. Virchow's Arch Path Anat Physiol Klin Med. 1882;90:261–332. [Google Scholar]
- 40.Brewer DB. Max Schultze (1865), G. Bizzozero (1882) and the discovery of the platelet. Br J Haematol. 2006;133:251–8. doi: 10.1111/j.1365-2141.2006.06036.x. [DOI] [PubMed] [Google Scholar]
- 41.Mickelson JK, Lakkis NM, Villarreal-Levy G, Hughes BJ, Smith CW. Leukocyte activation with platelet adhesion after coronary angioplasty: a mechanism for recurrent disease. J Am Coll Cardiol. 1996;28:345–53. doi: 10.1016/0735-1097(96)00164-7. [DOI] [PubMed] [Google Scholar]
- 42.von Hundelshausen P, Weber C. Platelets as immune cells: bridging inflammation and cardiovascular disease. Circ Res. 2007;100:27–40. doi: 10.1161/01.RES.0000252802.25497.b7. [DOI] [PubMed] [Google Scholar]
- 43.Rinder HM, Bonan JL, Rinder CS, Ault KA, Smith BR. Dynamics of leukocyte–platelet adhesion in whole blood. Blood. 1991;78:1730–7. [PubMed] [Google Scholar]
- 44.Zhao L, Bath PM, May J, Lösche W, Heptinstall S. P-selectin, tissue factor and CD40 ligand expression on platelet–leucocyte conjugates in the presence of a GPIIb/IIIa antagonist. Platelets. 2003;14:473–80. doi: 10.1080/09537100310001638562. [DOI] [PubMed] [Google Scholar]
- 45.Jungi TW, Spycher MO, Nydegger UE, Barandun S. Platelet–leukocyte interaction: selective binding of thrombin-stimulated platelets to human monocytes, polymorphonuclear leukocytes, and related cell lines. Blood. 1986;67:629–36. [PubMed] [Google Scholar]
- 46.Rinder HM, Bonan JL, Rinder CS, Ault KA, Smith BR. Activated and unactivated platelet adhesion to monocytes and neutrophils. Blood. 1991;78:1760–9. [PubMed] [Google Scholar]
- 47.Hamburger SA, McEver RP. GMP-140 mediates adhesion of stimulated platelets to neutrophils. Blood. 1990;75:550–4. [PubMed] [Google Scholar]
- 48.Larsent E, Celi A, Gilbert GE, Furie BC, Erban JK, Bonfanti R, Wagner DD, Furie' B. PADGEM protein: a receptor that mediates the interaction of activated platelets with neutrophils and monocytes. Cell. 1989;59:305–12. doi: 10.1016/0092-8674(89)90292-4. [DOI] [PubMed] [Google Scholar]
- 49.Yang J, Furie BC, Furie B. The biology of P-selectin glycoprotein ligand-1: its role as a selectin counterreceptor in leukocyte–endothelial and leukocyte–platelet interaction. Thromb Haemost. 1999;81:1–7. [PubMed] [Google Scholar]
- 50.Kappelmayer J, Kiss A, Karaaszi E, Veszpreami A, Jako J, Kiss C. Identification of P-selectin glycoprotein ligand-1 as a useful marker in acute myeloid leukaemias. Br J Haematol. 2001;115:903–9. doi: 10.1046/j.1365-2141.2001.03179.x. [DOI] [PubMed] [Google Scholar]
- 51.Tsuji T, Nagata K, Koike J, Todoroki N, Irimura T. Induction of superoxide anion production from monocytes and neutrophils by activated platelets through the P-selectin-sialyl Lewis X interaction. J Leukoc Biol. 1994;56:583–7. doi: 10.1002/jlb.56.5.583. [DOI] [PubMed] [Google Scholar]
- 52.Hidari KI, Weyrich AS, Zimmerman GA, McEver RP. Engagement of P-selectin glycoprotein ligand-1 enhances tyrosine phosphorylation and activates mitogen-activated protein kinases in human neutrophils. J Biol Chem. 1997;272:28750–6. doi: 10.1074/jbc.272.45.28750. [DOI] [PubMed] [Google Scholar]
- 53.Urzainqui A, Serrador JM, Viedma F, Yanez-Mo M, Rodriguez A, Corbi AL, Alonso-Lebrero JL, Luque A, Deckert M, Vazquez J, Sanchez-Madrid FITA. M-based interaction of ERM proteins with Syk mediates signaling by the leukocyte adhesion receptor PSGL-1. Immunity. 2002;17:401–12. doi: 10.1016/s1074-7613(02)00420-x. [DOI] [PubMed] [Google Scholar]
- 54.Haller H, Kunzendorf U, Sacherer K, Lindschau C, Walz G, Distler A, Luft FC. T cell adhesion to P-selectin induces tyrosine phosphorylation of pp125 focal adhesion kinase and other substrates. J Immunol. 1997;158:1061–7. [PubMed] [Google Scholar]
- 55.Wang HB, Wang JT, Zhang L, Geng ZH, Xu WL, Xu T, Huo Y, Zhu X, Plow EF, Chen M, Geng JG. P-selectin primes leukocyte integrin activation during inflammation. Nat Immunol. 2007;8:882–92. doi: 10.1038/ni1491. [DOI] [PubMed] [Google Scholar]
- 56.van Gils JM, Zwaginga JJ, Hordijk PL. Molecular and functional interactions among monocytes, platelets, and endothelial cells and their relevance for cardiovascular diseases. J Leukoc Biol. 2009;85:195–204. doi: 10.1189/jlb.0708400. [DOI] [PubMed] [Google Scholar]
- 57.Heijnen HF, Debili N, Vainchencker W, Breton-Gorius J, Geuze HJ, Sixma JJ. Multivesicular bodies are an intermediate stage in the formation of platelet alpha-granules. Blood. 1998;91:2313–25. [PubMed] [Google Scholar]
- 58.Sixma JJ, Slot J-W, Geuze HJ. Immunocytochemical localization of platelet granule proteins. Methods Enzymology. 1989;169:301–11. doi: 10.1016/0076-6879(89)69070-2. [DOI] [PubMed] [Google Scholar]
- 59.Pollard TD, Fujiwara K, Handin R, Weiss G. Contractile proteins in platelet activation and contraction. Ann N Y Acad Sci. 1977;283:218–36. [Google Scholar]
- 60.White JG, Gerrard JM. Interaction of microtubules and microfilaments in platelet contractile physiology. Methods Achiev Exp Pathol. 1979;9:1–39. [PubMed] [Google Scholar]
- 61.Loftus JC, Choate J, Albrecht RM. Platelet activation and cytoskeletal reorganization : high voltage electron microscopic examination of intact and Triton-extracted whole mounts. J Cell Biol. 1989;98:2019–25. doi: 10.1083/jcb.98.6.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Reed GL, Fitzgerald ML, Polga J. Molecular mechanisms of platelet exocytosis: insights into the ‘secrete’ life of thrombocytes. Blood. 2000;96:3334–42. [PubMed] [Google Scholar]
- 63.Flaumenhaft R. Molecular Basis of Platelet Granule Secretion. Arterioscler Thromb Vasc Biol. 2003;23:1152–60. doi: 10.1161/01.ATV.0000075965.88456.48. [DOI] [PubMed] [Google Scholar]
- 64.Ren Q, Ye S, Whiteheart SW. The Platelet Release Reaction: Just when you thought platelet secretion was simple. Curr Opin Hematol. 2008;15:537–41. doi: 10.1097/MOH.0b013e328309ec74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Painter RG, Ginsberg MH. Centripetal myosin redistribution in thrombinstimulated platelets: relationship to platelet factor 4 secretion. Exp Cell Res. 1984;155:198–212. doi: 10.1016/0014-4827(84)90781-x. [DOI] [PubMed] [Google Scholar]
- 66.Kirkpatrick JP, McIntire LV, Moake JL, Cimo PL. Differential effects of cytochalasin B on platelet release, aggregation and contractility: evidence against a contractile mechanism for the release of platelet granular contents. Thromb Haemost. 1980;42:1483–9. [PubMed] [Google Scholar]
- 67.Lefebvre P, White JG, Krumwiede MD, Cohen I. Role of actin in platelet function. Eur J Cell Biol. 1993;62:194–204. [PubMed] [Google Scholar]
- 68.White JG, Rao GH. Effects of a microtubule stabilizing agent on the response of platelets to vincristine. Blood. 1982;60:474–83. [PubMed] [Google Scholar]
- 69.Cox AC. Cytochalasin E enhances the protein kinase C-dependent process of secretion. Biochem Biophys Res Commun. 1988;150:745–51. doi: 10.1016/0006-291x(88)90454-8. [DOI] [PubMed] [Google Scholar]
- 70.Yatomi Y, Higashihara M, Tanabe A, Ohashi T, Takahata K, Kariya T, Kume S. Separable function of platelet release reaction and clot retraction. Biochem Biophys Res Commun. 1986;140:329–34. doi: 10.1016/0006-291x(86)91094-6. [DOI] [PubMed] [Google Scholar]
- 71.Flaumenhaft R, Dilks JR, Rozenvayn N, Monahan-Earley RA, Feng D, Dvorak AM. The actin cytoskeleton differentially regulates platelet α-granule and dense-granule secretion. Blood. 2005;105:3879–87. doi: 10.1182/blood-2004-04-1392. [DOI] [PubMed] [Google Scholar]
- 72.Ren Q, Wimmer C, Chicka MC, Ye S, Ren Y, Hughson FM, Whiteheart SW. Munc13-4 is a limiting factor in the pathway required for platelet granule release and hemostasis. Blood. 2010;116:869–77. doi: 10.1182/blood-2010-02-270934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.van Nispen tot Pannerden H, de Haas F, Geerts W, Posthuma G, van Dijk S, Heijnen HFG. The platelet interior revisited: electron tomography reveals tubular alpha granule subtypes. Blood. 2010;116:1147–56. doi: 10.1182/blood-2010-02-268680. [DOI] [PubMed] [Google Scholar]
- 74.Sehgal S, Storrie B. Evidence that differential packaging of the major platelet granule proteins von Willebrand factor and fibrinogen can support their differential release. J Thromb Haemost. 2007;5:2009–16. doi: 10.1111/j.1538-7836.2007.02698.x. [DOI] [PubMed] [Google Scholar]
- 75.Italiano JE, Jr, Richardson JL, Patel-Hett S, Battinelli E, Zaslavsky A, Short S, Ryeom S, Folkman J, Klement GL. Angiogenesis is regulated by a novel mechanism: pro- and antiangiogenic proteins are organized into separate platelet {alpha} granules and differentially released. Blood. 2008;111:1227–33. doi: 10.1182/blood-2007-09-113837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Maree AO, Fitzgerald DJ. Variable platelet response to aspirin and clopidogrel in atherothrombotic disease. Circulation. 2007;115:2196–207. doi: 10.1161/CIRCULATIONAHA.106.675991. [DOI] [PubMed] [Google Scholar]
- 77.Rinder CS, Student LA, Bonan JL, Rinder HM, Smith BI. Aspirin does not inhibit adenosine diphosphate-induced platelet a-granule release. Blood. 1993;82:505–12. [PubMed] [Google Scholar]
- 78.Li N, Hu H, Hjemdahl P. Aspirin treatment does not attenuate platelet or leukocyte activation as monitored by whole blood flow cytometry. Thromb Res. 2003;111:165–70. doi: 10.1016/j.thromres.2003.08.026. [DOI] [PubMed] [Google Scholar]
- 79.Moshfegh K, Redondo M, Julmy F, Wuillemin WA, Gebauer MU, Haeberli A, Meyer BJ. Antiplatelet effects of clopidogrel compared with aspirin after myocardial infarction: enhanced inhibitory effects of combination therapy. J Am Coll Cardiol. 2000;36:699–705. doi: 10.1016/s0735-1097(00)00817-2. [DOI] [PubMed] [Google Scholar]
- 80.Frelinger AL, Li Y, Linden MD, Barnard MR, Fox ML, Christie DJ, Furman MI, Michelson AD. Association of cyclooxygenase-1-dependent and -independent platelet function assays with adverse clinical outcomes in aspirin-treated patients presenting for cardiac catheterization. Circulation. 2009;120:2586–96. doi: 10.1161/CIRCULATIONAHA.109.900589. [DOI] [PubMed] [Google Scholar]
- 81.Levine GN, Berger PB, Cohen DJ, Maree AO, Rosenfield K, Wiggins BS, Spinler SA. Newer pharmacotherapy in patients undergoing percutaneous coronary interventions: a guide for pharmacists and other health care professionals. Pharmacotherapy. 2006;26:1537–56. doi: 10.1592/phco.26.11.1537. [DOI] [PubMed] [Google Scholar]
- 82.CAPRIE Steering Committee. A randomized, blinded, trial of clopidogrel versus aspirin in patients at risk of ischemic events (CAPRIE) Lancet. 1996;348:1329–39. doi: 10.1016/s0140-6736(96)09457-3. [DOI] [PubMed] [Google Scholar]
- 83.Sabatine MS, Cannon CP, Gibson CM, Lopez-Sendon JL, Montalescot G, Theroux P, Claeys MJ, Cools F, Hill KA, Skene AM, McCabe CH, Braunwald E. Addition of clopidogrel to aspirin and fibrinolytic therapy for myocardial infarction with ST-segment elevation. N Engl J Med. 2005;352:1179–89. doi: 10.1056/NEJMoa050522. [DOI] [PubMed] [Google Scholar]
- 84.Sabatine MS, Cannon CP, Gibson CM, Lopez-Sendon JL, Montalescot G, Theroux P, Lewis BS, Murphy SA, McCabe CH, Braunwald E. Effect of clopidogrel pretreatment before percutaneous coronary intervention in patients with ST-elevation myocardial infarction treated with fibrinolytics: the PCI-CLARITY study. JAMA. 2005;294:1224–32. doi: 10.1001/jama.294.10.1224. [DOI] [PubMed] [Google Scholar]
- 85.Diener HC, Bogousslavsky J, Brass LM, Cimminiello C, Csiba L, Kaste M, Leys D, Matias-Guiu J, Rupprecht HJ, MATCH investigators Aspirin and clopidogrel compared with clopidogrel alone after recent ischaemic stroke or transient ischaemic attack in high-risk patients (MATCH): randomised, double-blind, placebo-controlled trial. Lancet. 2004;364:331–7. doi: 10.1016/S0140-6736(04)16721-4. [DOI] [PubMed] [Google Scholar]
- 86.Leon C, Alex M, Klocke A, Morgenstern E, Moosbauer C, Eckly A, Spannagl M, Gachet C, Engelmann B. Platelet ADP receptors contribute to the initiation of intravascular coagulation. Blood. 2004;103:594–600. doi: 10.1182/blood-2003-05-1385. [DOI] [PubMed] [Google Scholar]
- 87.Farid NA, Payne CD, Small DS, Winters KJ, Ernest CS, 2nd, Brandt JT, Darstein CD, Jakubowski JA, Salazar DE. Cytochrome P450 3A inhibition by ketonazole affects prasugrel and clopidogrel pharmacokinetics and pharmacodynamics differently. Clin Pharmacol Ther. 2007;81:735–41. doi: 10.1038/sj.clpt.6100139. [DOI] [PubMed] [Google Scholar]
- 88.Wiviott SD, Braunwald E, McCabe CH, Montalescot G, Ruzyllo W, Gottlieb S, Neuman FJ, Ardissino D, De Servi S, Murphy SA, Riesmeyer J, Weerakkody G, Gibson CM, Antman EM, TRITON-TIMI 38 Investigators Prasugrel versus clopidogrel in patients with acute coronary syndromes. N Engl J Med. 2007;357:2001–5. doi: 10.1056/NEJMoa0706482. [DOI] [PubMed] [Google Scholar]
- 89.Van Giezen JJJ, Humphries RG. Preclinical and clinical studies with selective reversible direct P2Y12 antagonists. Sem Thromb Hemost. 2005;31:195–204. doi: 10.1055/s-2005-869525. [DOI] [PubMed] [Google Scholar]
- 90.Cattaneo M, Podda GM. State of the art of new P2Y12 antagonists. Intern Emerg Med. 2010;5:385–91. doi: 10.1007/s11739-010-0363-z. [DOI] [PubMed] [Google Scholar]
- 91.Storey RF, Judge HM, Wilcox RG, Heptinstall S. Inhibition of ADP-induced P-selectin expression and platelet–leukocyte conjugate formation by clopidogrel and the P2Y12 receptor antagonist AR-C69931MX but not aspirin. Thromb Haemost. 2002;88:488–94. [PubMed] [Google Scholar]
- 92.Braun OO, Johnell M, Varenhorst C, James S, Brandt JT, Jakubowski JA, Winters KJ, Wallentin L, Erlinge D, Siegbahn A. Greater reduction of platelet activation markers and platelet–monocyte aggregates by prasugrel compared to clopidogrel in stable coronary artery disease. Thromb Haemost. 2008;100:626–33. [PubMed] [Google Scholar]
- 93.Antonino MJ, Mahla E, Bliden KP, Tantry US, Gurbel PA. Effect of long-term clopidogrel treatment on platelet function and inflammation in patients undergoing coronary arterial stenting. Am J Cardiol. 2009;103:1546–50. doi: 10.1016/j.amjcard.2009.01.367. [DOI] [PubMed] [Google Scholar]
- 94.Offermanns S. Activation of platelet function through G protein–coupled receptors. Circ Res. 2006;99:1293–304. doi: 10.1161/01.RES.0000251742.71301.16. [DOI] [PubMed] [Google Scholar]
- 95.Newton AC. Regulation of protein kinase C. Curr Opin Cell Biol. 1997;9:161–67. doi: 10.1016/s0955-0674(97)80058-0. [DOI] [PubMed] [Google Scholar]
- 96.Mellor H, Parker PJ. The extended protein kinase C superfamily. Biochem J. 1998;332:281–92. doi: 10.1042/bj3320281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Paul BZ, Jin J, Kunapuli SP. Molecular mechanism of thromboxane A(2)-induced platelet aggregation. Essential role for p2t(ac) and alpha(2a) receptors. J Bio Chem. 1999;274:29108–14. doi: 10.1074/jbc.274.41.29108. [DOI] [PubMed] [Google Scholar]
- 98.Quinton TM, Kim S, Dangelmaier C, Dorsam RT, Jin J, Daniel JL, Kunapuli SP. Protein kinase C- and calcium-regulated pathways independently synergize with Gi pathways in agonist-induced fibrinogen receptor activation. Biochem J. 2002;368:535–53. doi: 10.1042/BJ20020226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Chung SH, Polgar J, Reed GL. Protein kinase C phosphorylation of syntaxin 4 in thrombin-activated human platelets. J Biol Chem. 2000;275:25286–91. doi: 10.1074/jbc.M004204200. [DOI] [PubMed] [Google Scholar]
- 100.Konopatskaya O, Gilio K, Harper MT, Zhao Y, Cosemans JM, Karim ZA, Whiteheart SW, Molkentin JD, Verkade P, Watson SP, Heemskerk JW, Poole AW. PKCalpha regulates platelet granule secretion and thrombus formation in mice. J Clin Invest. 2009;119:399–407. doi: 10.1172/JCI34665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Murugappan S, Tuluc F, Dorsam RT, Shankar H, Kunapuli SP. Differential role of protein kinase C delta isoform in agonist-induced dense granule secretion in human platelets. J Biol Chem. 2004;279:2360–7. doi: 10.1074/jbc.M306960200. [DOI] [PubMed] [Google Scholar]
- 102.Gilio K, Harper MT, Cosemans JM, Konopatskaya O, Munnix IC, Prinzen L, Leitges M, Liu Q, Molkentin JD, Heemskerk JW, Poole AW. Functional divergence of platelet protein kinase C (PKC) isoforms in thrombus formation on collagen. J Biol Chem. 2010;285:23410–9. doi: 10.1074/jbc.M110.136176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Li Z, Zhang G, Le Breton GC, Gao X, Malik AB, Du X. Two waves of platelet secretion induced by thromboxane A2 receptor and a critical role for phosphoinositide 3-kinases. J Biol Chem. 2003;278:30725–31. doi: 10.1074/jbc.M301838200. [DOI] [PubMed] [Google Scholar]
- 104.Paul BZ, Jin J, Kunapuli SP. Molecular mechanism of thromboxane A(2)-induced platelet aggregation. Essential role for p2t(ac) and alpha(2a) receptors. J Biol Chem. 1999;274:29108–14. doi: 10.1074/jbc.274.41.29108. [DOI] [PubMed] [Google Scholar]
- 105.Hollopeter G, Jantzen HM, Vincent D, Li G, England L, Ramakrishnan V, Yang RB, Nurden P, Nurden A, Julius D, Conley PB. Identification of the platelet ADP receptor targeted by antithrombotic drugs. Nature. 2001;409:202–7. doi: 10.1038/35051599. [DOI] [PubMed] [Google Scholar]
- 106.Dean WL, Chen D, Brandt PC, Vanaman TC. Regulation of platelet plasmalemma Ca2+-ATPase by cAMP-dependent and tyrosin phosphorylation. J Biol Chem. 1997;272:15113–9. doi: 10.1074/jbc.272.24.15113. [DOI] [PubMed] [Google Scholar]
- 107.Hallam TJ, Daniel JL, Kendrick Jones J, Rink TJ. Relationship between cytosplasmic free calcium and myosin light chain phosphorylation in intact platelets. Biochem J. 1985;232:373–7. doi: 10.1042/bj2320373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Butt E, Abel K, Krieger M, Palm D, Hoppe V, Hoppe J, Walter J. cAMP and cGMP-dependent protein kinase [hosphorylation sites of the focal vasodilator-stimulated phosphoprotein (VASP) in vitro in intact human plateltes. J Biol Chem. 1994;269:14509–17. [PubMed] [Google Scholar]
- 109.Li Z, Zhang G, Marjanovic JA, Ruan C, Du X. A platelet secretion pathway mediated by cGMP-dependent protein kinase. J Biol Chem. 2004;279:42469–75. doi: 10.1074/jbc.M401532200. [DOI] [PubMed] [Google Scholar]
- 110.Iyú D, Glenn GR, White AE, Fox SC, Heptinstall S. Adenosine derived from ADP can contribute to inhibition of platelet. Arterioscler Thromb Vasc Biol. 2010;31:416–22. doi: 10.1161/ATVBAHA.110.219501. [DOI] [PubMed] [Google Scholar]
- 111.Born GV, Smith JB. Uptake, metabolism and release of (3H)-adrenalin by human platelets. Br J Pharmacol. 1970;39:765–78. doi: 10.1111/j.1476-5381.1970.tb09903.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Smith CC. Evidence for separate serotonin and catecholamine compartments in human platelets. Biochim Biophys Acta. 1996;1291:1–4. doi: 10.1016/0304-4165(96)00035-9. [DOI] [PubMed] [Google Scholar]
- 113.Kaywin P, McDonough M, Insel PA, Shattil SJ. Platelet function is essential in thrmbocythemia: decreased epinephrine responsiveness of platelets associated with a deficiency in alpha-adrenergic receptors. N Engl J Med. 1978;299:505–9. doi: 10.1056/NEJM197809072991002. [DOI] [PubMed] [Google Scholar]
- 114.Woulfe D, Jiang H, Mortensen R, Yang J, Brass LF. Activation of Rap1B by Gi family members in platelets. J Biol Chem. 2002;277:23382–90. doi: 10.1074/jbc.M202212200. [DOI] [PubMed] [Google Scholar]
- 115.Hjemdahl P, Chronos NA, Wilson DJ, Bouloux P, Goodall AH. Epinephrine sensitizes human platelets in vivo and in vitro as studied by fibrinogen binding and P-selectin expression. Arterioscler Thromb Vasc Biol. 1994;14:77–84. doi: 10.1161/01.atv.14.1.77. [DOI] [PubMed] [Google Scholar]
- 116.Horn NA, Anastase DM, Hecker KE, Baumert JH, Robitzsch T, Rossaint R. Epinephrine enhances platelet–neutrophil adhesion in whole blood in vitro. Anesth Analg. 2005;100:520–6. doi: 10.1213/01.ANE.0000141527.60441.B7. [DOI] [PubMed] [Google Scholar]
- 117.Iyú D, Glenn JR, White AE, Johnson AJ, Fox SC, Heptinstall S. The role of prostanoid receptors in mediating the effects of PGE(2) on human platelet function. Platelets. 2010;21:329–42. doi: 10.3109/09537101003718065. [DOI] [PubMed] [Google Scholar]
- 118.Fabre JE, Nguyen M, Athirakul K, Coggins K, McNeish JD, Austin JD, Austin S, Parisse LK, FitzGerald GA, Coffman TM, Koller BH. Activation of the murine EP3 receptor for PGE2 inhibits cAMP production and promotes platelet aggregation. J Clin Invest. 2001;107:603–10. doi: 10.1172/JCI10881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Ma H, Hara A, Xiao CY, Okada Y, Takahata O, Nakaya K, Sugimoto Y, Ichikawa A, Narumiya S, Ushikubi F. Increased bleeding tendency and decrease susceptibility to thromboembolism in mice lacking the prostaglandin E2 receptor subtype EP(3) Circulation. 2001;104:1176–80. doi: 10.1161/hc3601.094003. [DOI] [PubMed] [Google Scholar]
- 120.Iyú D, Jüttner M, Glenn JR, White AE, Johnson AJ, Fox SC, Heptinstall S. PGE(1) and PGE(2) modify platelet function through different prostanoid receptors. Prostaglandins Other Lipid Mediat. 2010 doi: 10.1016/j.prostaglandins.2010.11.001. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 121.Queen LR, Xu B, Horinouchi K, Fisher I, Ferro A. B2-adrenoreceptors activate nitric oxide synthase in human platelets. Circ Res. 2000;87:39–44. doi: 10.1161/01.res.87.1.39. [DOI] [PubMed] [Google Scholar]
- 122.Morell AN, Matsushita K, Chiles K, Scharp RB, Yamakuchi M, Mason RJA, Bergmeier W, Mankowski JL, Baldwin W, Farady N, Lowenstein CJ. Regulation of platelet granule exocytosis by S-nitrosylation. Proc N Acad Sci. 2005;102:3782–7. doi: 10.1073/pnas.0408310102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Goubareva I, Gkaliagkousi E, Shah A, Queen L, Ferro A. Age decreases nitric oxide synthesis and responsiveness in human platelets and increases formation of monocyte–platelet aggregates. Cardiovasc Res. 2007;75:793–802. doi: 10.1016/j.cardiores.2007.05.021. [DOI] [PubMed] [Google Scholar]
- 124.Li Z, Xi X, Gu M, Feil R, Ye RD, Eigenthaler M, Hofman F, Du X. A stimulatory role for cGMP-dependent protein kinase in platelet activation. Cell. 2003;112:77–86. doi: 10.1016/s0092-8674(02)01254-0. [DOI] [PubMed] [Google Scholar]
- 125.Li Z, Ajdic J, Eigenthaler M, Du X. A predominant role for cAMP-dependent protein kinase in cGMP-induced phosphorylation of vasodilator-stimulated phosphoprotein and platelet inhibition in humans. Blood. 2003;101:4423–9. doi: 10.1182/blood-2002-10-3210. [DOI] [PubMed] [Google Scholar]
- 126.Kariyazono H, Nakamra K, Shinkawa T, Yamaguchi T, Sakata R, Yamada K. Inhibition of platelet aggregation and the release of P-selectin from platelets by cilostazol. Thrombosis Res. 2001;101:445–53. doi: 10.1016/s0049-3848(00)00415-1. [DOI] [PubMed] [Google Scholar]
- 127.Ito H, Miyakoda G, Mori T. Cilostazol inhibits platelet leukocyte interaction by suppression of platelet activation. Platelets. 2004;15:293–301. doi: 10.1080/09537100410001715583. [DOI] [PubMed] [Google Scholar]
- 128.Lee SW, Park SW, Kim YH. Comparison of triple versus dual anti-platelet therapy after drug-eluting stent implantation (from the DECLARE-Long trial) Am J Cardiol. 2007;100:1103–8. doi: 10.1016/j.amjcard.2007.05.032. [DOI] [PubMed] [Google Scholar]
- 129.Serebruany VL, Glassman AH, Malinin AI, Atar D, Sane DC, Oshrine BR, Ferguson JJ, O'Connor CM. Selective serotonin reuptake inhibitors yield additional antiplatelet protection in patients with congestive heart failure treated with antecedent aspirin. Eur J Heart Fail. 2003;5:517–21. doi: 10.1016/s1388-9842(03)00005-9. [DOI] [PubMed] [Google Scholar]
- 130.Morel-Kopp MC, McLean L, Chen Q, Tofler GH, Tennant C, Maddison V, Ward CM. The association of depression with platelet activation: evidence for a treatment effect. J Thromb Haemos. 2009;7:573–81. doi: 10.1111/j.1538-7836.2009.03278.x. [DOI] [PubMed] [Google Scholar]
- 131.Duman RS. Novel therapeutic approaches beyond the serotonin receptor. Biol Psychiatry. 1998;44:324–35. doi: 10.1016/s0006-3223(98)00031-6. [DOI] [PubMed] [Google Scholar]
- 132.Xu Q, Yi LT, Pan Y, Wang X, Li YC, Li JM, Wang CP, Kong LD. Antidepressant-like effects of the mixture of honokiol and magnolol from the barks of Magnolia officinalis in stressed rodents. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32:715–25. doi: 10.1016/j.pnpbp.2007.11.020. [DOI] [PubMed] [Google Scholar]
- 133.Li YC, Wang FM, Pan Y, Qiang LQ, Cheng G, Zhang WY, Kong LD. Antidepressant-like effects of curcumin on serotonergic receptor-coupled AC-cAMP pathway in chronic unpredictable mild stress of rats. Prog Neuropsychopharmacol Biol Psychiatry. 2009;33:435–4. doi: 10.1016/j.pnpbp.2009.01.006. [DOI] [PubMed] [Google Scholar]
- 134.Bosch X, Marrugat J, Sanchis J. Platelet glycoprotein IIb/IIIa blockers during percutaneous coronary intervention and as the initial medical treatment of non-ST segment elevation acute coronary syndromes. Cochrane Database Syst Rev. 2010;(9) doi: 10.1002/14651858.CD002130.pub2. CD002130. [DOI] [PubMed] [Google Scholar]
- 135.O'Neill WW, Serruys P, Knudtson M, van Es GA, Timmis GC, van der Zwaan C, Kleiman J, Gong J, Roecker EB, Dreiling R, Alexander J, Anders R. Long-term treatment with a platelet glycoprotein-receptor antagonist after percutaneous coronary revascularization. EXCITE Trial Investigators. Evaluation of Oral Xemilofiban in Controlling Thrombotic Events. N Engl J Med. 2000;342:1316–24. doi: 10.1056/NEJM200005043421803. [DOI] [PubMed] [Google Scholar]
- 136.Comparison of sibrafiban with aspirin for prevention of cardiovascular events after acute coronary syndromes: a randomised trial. The SYMPHONY Investigators. Sibrafiban versus Aspirin to Yield Maximum Protection from Ischemic Heart Events Post-acute Coronary Syndromes. Lancet. 2000;355:337–45. [PubMed] [Google Scholar]
- 137.SYMPHONY II Investigators. Randomized trial of aspirin, sibrafiban, or both for secondary prevention after acute coronary syndromes. Circulation. 2001;103:1727–33. doi: 10.1161/01.cir.103.13.1727. [DOI] [PubMed] [Google Scholar]
- 138.Grüner S, Prostredna M, Koch M, Miura Y, Schulte V, Jung SM, Moroi M, Nieswandt B. Relative antithrombotic effect of soluble GPVI dimer compared with anti-GPVI antibodies in mice. Blood. 2005;105:1492–9. doi: 10.1182/blood-2004-06-2391. [DOI] [PubMed] [Google Scholar]
- 139.Schulte V, Rabie T, Prostredna M, Aktas B, Gruner S, Nieswandt B. Targeting of the collagen-binding site on glycoprotein VI is not essential for in vivo depletion of the receptor. Blood. 2003;101:3948–52. doi: 10.1182/blood-2002-10-3242. [DOI] [PubMed] [Google Scholar]
- 140.Smethurst PA, Joutsi-Korhonen L, O'Connor MN, Wilson E, Jennings NS, Garner SF, Zhang Y, Knight CG, Dafforn TR, Buckle A. IJsseldijk MJ, De Groot PG, Watkins NA, Farndale RW, Ouwehand WH. Identification of the primary collagen binding surface on human glycoprotein VI by site-directed mutagenesis and by a blocking phage antibody. Blood. 2004;103:903–11. doi: 10.1182/blood-2003-01-0308. [DOI] [PubMed] [Google Scholar]
- 141.Matsumoto Y, Takizawa H, Gong X, Le S, Lockyer S, Okuyama K, Tanaka M, Yoshitake M, Tandon NN, Kambayashi J. Highly potent anti-human GPVI monoclonal antibodies derived from GPVI knockout mouse immunization. Thromb Res. 2007;119:319–29. doi: 10.1016/j.thromres.2006.01.023. [DOI] [PubMed] [Google Scholar]
- 142.Kato-Takagaki K, Mizukoshi Y, Yoshizawa Y, Akazawa D, Torii Y, Ono K, Tanimura R, Shimada I, Takahashi H. Structural and interaction analysis of glycoprotein VI-binding peptide selected from a phage display library. J Biol Chem. 2009;284:10720–27. doi: 10.1074/jbc.M808563200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Keefe AD, Shaub RG. Aptamers as candidate therapeutics for cardiovascular indications. Curr Opin Pharm. 2008;8:147–52. doi: 10.1016/j.coph.2007.12.005. [DOI] [PubMed] [Google Scholar]
- 144.McGhie AI, McNatt J, Ezov N, Cui K, Lowell K, Mower LK, Hagay Y, Buja LM, Garfinkel LI, Gorecki M, Willerson JT. Abolition of cyclic flow variations in stenosed, endothelium-injured coronary arteries in nonhuman primates with a peptide fragment (VCL) derived from human plasma von Willebrand factor–glycoprotein Ib binding domain. Circulation. 1994;90:2976–81. doi: 10.1161/01.cir.90.6.2976. [DOI] [PubMed] [Google Scholar]
- 145.Diener JL, Daniel Lagassé HA, Duerschmied D, Merhi Y, Tanguay JF, Hutabarat R, Gilbert J, Wagner DD, Schaub R. Inhibition of von Willebrand factor-mediated platelet activation and thrombosis by the anti-von Willebrand factor A1-domain aptamer ARC1779. J Thromb Haemost. 2009;7:1155–62. doi: 10.1111/j.1538-7836.2009.03459.x. [DOI] [PubMed] [Google Scholar]
- 146.Gilbert JC, DeFeo-Fraulini T, Hutabarat RM, Horvath CJ, Merlino PG, Marsh HN, Healy JM, Boufakhreddine S, Holohan TV, Schaub RG. First-in-human evaluation of anti von Willebrand factor therapeutic aptamer ARC1779 in healthy volunteers. Circulation. 2007;116:2678–86. doi: 10.1161/CIRCULATIONAHA.107.724864. [DOI] [PubMed] [Google Scholar]
- 147.Wang L, Brown JR, Varki A, Esko JD. Heparin's anti-inflammatory effects require glucosamine 6-O-sulfation and are mediated by blockade of L- and P-selectins. J Clin Invest. 2002;110:127–36. doi: 10.1172/JCI14996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Oostingh GJ, Pozgajova M, Ludwig RJ, Krahn T, Boehncke WH, Nieswandt B, Schön MP. Diminished thrombus formation and alleviation of myocardial infarction and reperfusion injury through antibody- or small-molecule-mediated inhibition of selectin-dependent platelet functions. Haematologica. 2007;92:502–12. doi: 10.3324/haematol.10741. [DOI] [PubMed] [Google Scholar]
- 149.Skurk C, Buerke M, Guo JP, Paulson J, Lefer AM. Sialyl Lewisx-containing oligosaccharide exerts beneficial effects in murine traumatic shock. Am J Physiol Heart Circ Physiol. 1994;267:H2124–31. doi: 10.1152/ajpheart.1994.267.6.H2124. [DOI] [PubMed] [Google Scholar]
- 150.Sanders WJ, Gordon EJ, Dwir O, Beck PJ, Alon R, Kiessling LL. Inhibition of L-selectin-mediated leukocyte rolling by synthetic glycoprotein mimics. J Biol Chem. 1999;274:5271–8. doi: 10.1074/jbc.274.9.5271. [DOI] [PubMed] [Google Scholar]
- 151.Hayward R, Campbell B, Shin YK, Scalia R, Lefer AM. Recombinant soluble P-selectin glycoprotein ligand-1 protects against myocardial ischemic reperfusion injury in cats. Cardiovasc Res. 1999;41:65–76. doi: 10.1016/s0008-6363(98)00266-1. [DOI] [PubMed] [Google Scholar]
- 152.Jenison RD, Jennings SD, Walker DW, Bargatze RF, Parma D. Oligonucleotide inhibitors of P-selectin-dependent neutrophil–platelet adhesion. Antisense Nucleic Acid Drug Dev. 1998;8:265–79. doi: 10.1089/oli.1.1998.8.265. [DOI] [PubMed] [Google Scholar]