

1. Introduction
1.1. Discovery of the Phospholipase A2 Superfamily
Phospholipases represent one of the earliest enzyme activities to be identified and studied and the phospholipase A2 (PLA2) superfamily (see defining specificity1 in Figure 1) traces its roots to the identification of lytic actions of snake venom at the end of the 19th century. The enzyme was first purified and characterized from cobra venom and later from rattlesnake venom. As protein sequencing methodologies advanced in the 1970’s, it became apparent that these enzymes had an unusually large number of cysteines (over 10% of the amino acids) and as secreted enzymes, that they were all in the form of disulfide bonds. It was further recognized that in the case of PLA2, cobras and rattlesnakes had six disulfides in common, but one disulfide bond is located in distinctly different locations. This led to the designation of Type 1 and Type 2 for cobras (old world snakes) and rattlesnakes (new world snakes), respectively.2 During that same period, studies on the porcine pancreatic digestive enzyme that hydrolyzes phospholipids led to the determination that this mammalian enzyme (and also the human pancreatic enzyme) had the same disulfide bonding pattern as cobras and hence the designation as IB with the cobra enzyme as IA.
Figure 1.

The specific reaction catalyzed by phospholipase A2 at the sn-2 position of the glycerol backbone is shown. X, any of a number of polar headgroups; R1, fatty acids, or alkyl, or alkenyl groups and R2, fatty acids or acyl moieties.
A dramatic change in the phospholipase A2 field that attracted the attention of the broader scientific community occurred in July, 1988 when at the first FASEB Summer Conference on Phospholipases, Jeffery J. Seilhamer and Lorin K. Johnson from California Biotechnology Inc.3 and Ruth M. Kramer from Biogen Research Corporation4 independently and with much fanfare and excitement reported the purification, sequencing and cloning of the first human non-pancreatic secreted PLA2 which they each had isolated from the human synovial fluid of arthritic knee joints. Since the sequence revealed that the disulfide bond pattern was more like the rattlesnake than the human pancreatic enzyme, this new form of PLA2 was designated IIA. All of these enzymes then became known as secreted or sPLA2s.
It wasn’t until the late 1980’s that PLA2-like activities were reported in mammalian cells in contrast to extracellular secreted activities from venom and pancreas. In July, 1992, at the second FASEB Summer Conference on Phospholipases, James D. Clark from the Genetics Institute5 and Ruth M. Kramer (who had moved to Lilly Research Laboratories)6 independently reported the purification, sequencing, and cloning of the first human cytosolic PLA2 (cPLA2) from the U937 macrophage cell line. The sequence was unrelated to those of the secreted enzymes. To track this new enzyme and potentially additional PLA2s, a Group Numbering System7 was established utilizing the preexisting venom designation of I and II and expanding them to include subgroups IA, IB, and IIA (GIA, GIB, GIIA); adding Group III (GIII) for the clearly different PLA2 which had been purified from bee venom; and establishing the Group IV (GIV) designation for the new cytosolic PLA2 (cPLA2).
This was fortuitous because soon thereafter a new form of secreted PLA2 was discovered. It was produced by macrophages and it had the same six disulfide bonds as Group I and Group II, but lacked the seventh disulfide bond entirely. To make clear that this sPLA2 was neither GI nor GII, this enzyme was designated as Group V (GV). At the Third FASEB Summer Conference on Phospholipases held in July, 1995, Edward A. Dennis from the University of California, San Diego8 reported on another cystosolic PLA2 purified from macrophages that differed from Group IV cPLA2 in that its activity was not dependent on Ca2+ and Simon S. Jones from the Genetics Institute9 reported that the cloned form from CHO cells had a very different sequence than cPLA2. This new Ca2+-independent PLA2 (iPLA2) was designated as Group VI PLA2 (GVI).10
Earlier, investigators from the University of Utah11 had isolated an enzyme from human plasma which hydrolyzed platelet activating factor (PAF), a phosphatidylcholine containing an acetate at the sn-2 position, and in 1995 Larry W. Tjoelker from ICOS12 reported its cloning. This enzyme and other related PAF acetyl hydrolases (PAF-AH) were later recognized more broadly as PLA2s with a specificity for a short acyl chain on the sn-2 position and for the plasma one for oxidized lipids for which the same enzyme was independently named lipoproteinassociated phospholipase A2 (Lp-PLA2). These enzymes were designated Group VII and VIII (GVII and GVIII).13 As additional specific PLA2s were discovered, they were either designated by letters as subgroups of the original Groups indicated above or as additional Groups. Especially noteworthy was the discovery of a number of additional sPla2s in which the sequence and/or disulfide bonding pattern varied significantly from the traditional Groups I, II, III, and V sPla2s. These new forms led to the additional Groups IX, X, XI XII,XIII, and XIV sPla2s representing new human forms (especially Group X, which may have important functions) as well unique enzymes from snail venom, rice shoots, parvovirus, and fungi/bacteria. The only new type of PLA2 reported that did not naturally fit in the four types discussed above (secreted, cytosolic, Ca2+-independent, PAF acetylhydrolases) is the lysosomal PLA2 (LPLA2) which was designated as Group XV (GXV).14 Recently, a new PLA2 was isolated from adipose tissue and designated as Group XVI (GXVI);15 it appears to be a new type of PLA2 called adipose-PLA2 (AdPLA). The current designations are summarized in Table 1.
Table 1.
The Phospholipase A2 Superfamily
| Type | Group | Subgroup | Molecular Mass (kDa) |
Catalytic Residues |
|---|---|---|---|---|
| GI | A, B | 13–15 | ||
| GII | A, B, C, D, E, F | 13–17 | ||
| GIII | 15–18 | |||
| GV | 14 | |||
| sPLA2 | GIX | 14 | His/Asp | |
| GX | 14 | |||
| GXI | A, B | 12–13 | ||
| GXII | A, B | 19 | ||
| GXIII | <10 | |||
| GXIV | 13–19 | |||
| cPLA2 | GIV | A(α), B(β), C(γ), D(δ), E(ε), F(ζ) | 60–114 | Ser/Asp |
| iPLA2 | GVI | A(β), B(γ), C(δ), D(ε), E(ζ), F(η) | 84–90 | Ser/Asp |
| PAF-AH | GVII | A(Lp-PLA2), B(PAF-AH II) | 40–45 | Ser/His/Asp |
| GVIII | A (α1), B(α2), β | 26–40 | ||
| LPLA2 | GXV | 45 | Ser/His/Asp | |
| AdPLA | GXVI | 18 | His/Cys | |
In this review, we will discuss in turn each of the six types of PLA2. For each, we will first discuss the various forms, in terms of groups, subgroups and mechanism of action, their structure and interaction with membranes, their biological activities and role in disease, and the development of selective inhibitors. Of course the commonly used type designation has little meaning today since as we have learned more about these enzymes, it has been recognized that secreted, cytosolic, Ca2+-independent, PAF-AH, and lysosomal make little sense since all four of the later categories are actually intracellular (cytosolic) enzymes, that the secreted ones may occur intracellularly in various vesicles, and that the PAF-AHs, lysosomal and some forms of cPLA2 are also Ca2+-independent. Thus the Group Numbering System designation provides an unambiguous definition of each enzyme form. Over the years, numerous excellent reviews on either the broad family of PLA2s16 or specific types including sPLA2s,17 cPLA2s,18 iPLA2s,19 PAF-AHs20 and LPLA221 have appeared as well as several review articles summarizing the classes of PLA2 inhibitors and their potential role for the treatment of inflammatory diseases.22 We have employed all of these prior reviews heavily in preparing this up-to-date and comprehensive single review covering all aspects of the entire phospholipase A2 superfamily.
1.2 Accessing the In Vitro Activity of Phospholipase A2
Studying phospholipases has posed significant challenges because unlike classical water-soluble enzymes acting on water soluble substrates, phospholipases act on phospholipids which aggregate in aqueous solution to form structures termed micelles, vesicles, liposomes, etc. While most of the PLA2s studied in depth to date are water-soluble themselves, they catalyze hydrolysis of their water-insoluble substrates by catalytic action at the lipid-water interface. Since the substrate phospholipids are not monomeric, but rather are lined up in a two-dimensional interfaces, when the enzyme is associated with that interface, it binds its substrate phospholipid molecule in the interface where its substrate concentration can be best expressed in surface terms. Thus in kinetic experiments to determine activity, the observed activity depends on non-substrate lipids in the interface whether they be other non-substrate phospholipids, surface active detergents (surfactants), and even inhibitors that aggregate with the surface. The concept of “surface dilution kinetics”23 has been useful in measuring PLA2 activities, particularly in comparing the different enzyme forms and substrates as reviewed elsewhere24 and illustrated in Figure 2. Thus as one compares the specific activity, specificity, and especially the inhibition25 of each PLA2 Group, subgroup, and species, one must note the particular assay conditions and aggregated form of substrate used including the kinetic ramifications.
Figure 2.

Illustration of the application of “surface dilution kinetics” to the phospholipase A2-catalyzed hydrolysis of phospholipids contained in mixed micelles with nonionic surfactants such as Triton X-100. Two possibilities are shown: top, the “surface binding model” whereby the enzyme first associates nonspecifically with the micelle surface; and bottom, the “phospholipid binding model” whereby the enzyme first associates specifically with phospholipid in the micelle surface. In both cases, in a subsequent step, the enzyme associated with the micelle binds a phospholipid substrate molecule in the micelle in its catalytic site and carries out hydrolysis producing as products a lysophospholipid and a fatty acid, which may be released to solution or be retained in the micelle surface. Phospholipid molecules are depicted in red, detergent molecules in gold, and enzyme in blue. Adapted from Ref.24
2. Secreted Phospholipase A2 (sPLA2 Groups I, II, III, V, IX, X, XI, XII, XIII, XIV
2.1 Groups, Subgroups, Specificity and Mechanism
The sPLA2s are small secreted proteins of 14–18 kDa (except for Group III sPLA2) that usually contain 6 to 8 disulfide bonds (Table 2).14 A schematic presentation of the sequences (Figure 3) provides an overview of this PLA2 types. This group of enzymes uses an active site His/Asp dyad and requires mM Ca2+ for catalytic activity. Members of this type were first studied in phenomenological detail over 100 years ago using “poison” – venom from cobras.16c As various snake venom PLA2s were sequenced and disulfide bond patterns determined, those from old world snakes (cobras and kraits) were refered to as type I and those from new world snakes (rattlesnakes) were referred to as type II. The first non-venom PLA2, named GIB, was isolated from the pancreatic juices of cows, and was also found in many other mammals (similar disulfide bond pattern to GI snake venom). Later, another mammalian sPLA2, a non-pancreatic form (similar disulfide bond pattern to GII snake venom) named GIIA was found in the synovial fluid of patients with rheumatoid arthritis.3–4 To date, seventeen forms of sPLA2 (Table 2) have been identified in mammals, insects, mollusks, reptiles, plants and bacteria. The sPLA2s display a wide range of different tissue distribution patterns and distinct physiological functions.
Table 2.
Secreted Phospholipase A2s (sPLA2)
| Group | Source | Molecular Mass (kDa) |
Disulfide Bonds |
|---|---|---|---|
| IA | Cobras and Kraits | 13–15 | 7 |
| IB | Human/porcine pancreas | 13–15 | 7 |
| IIA | Rattlesnakes; human synovial | 13–15 | 7 |
| IIB | Gaboon viper | 13–15 | 6 |
| IIC | Rat/murine testis | 15 | 8 |
| IID | Human/murine pancreas/spleen | 14–15 | 7 |
| IIE | Human/murine brain/heart/uterus | 14–15 | 7 |
| IIF | Human/murine testis/embryo | 16–17 | 6 |
| III | Lizard/bee | 15–18 | 8 |
| Human/murine | 55 | 8 | |
| V | Human/murine heart/lung/macrophage | 14 | 6 |
| IX | Snail venom (conodipine-M) | 14 | 6 |
| X | Human spleen/thymus/leukocyte | 14 | 8 |
| XIA | Green rice shoots (PLA2-I) | 12.4 | 6 |
| XIB | Green rice shoots (PLA2-II) | 12.9 | 6 |
| XIIA | Human/murine | 19 | 7 |
| XIIB | Human/murine | 19 | 7 |
| XIII | Parvovirus | < 10 | 0 |
| XIV | Symbiotic fungus/bacteria | 13–19 | 2 |
This table has been adapted from.14
Figure 3.

Schematic presentation of secreted PLA2s. Calcium binding loops, active sites (red squares), N and C-terminal extension residues are shown.
Ten members of the sPLA2 family (group IB, IIA, IIC, IID, IIE, IIF, III, V, X and XII) have been identified in mammals; these are numbered and grouped according to their pattern of disulfide bonds and in order of their discovery.14 The human genome contains 9 sPLA2s and the mouse genome contains all ten, including GIIC sPLA2, which exists in the human genome only as a pseudogene.
Mammalian GIII sPLA2 is a multi-domain protein with a molecular weight of 55 kDa. It contains a central domain similar to that of bee venom GIII sPLA2, including a 130-amino acid N-terminal domain extension and a 219-amino acid C-terminal domain extension. GXIIB sPLA2 in humans and mice (a homologue of GXIIA sPLA2), has a natural mutation in the active site (H48L) and totally lacks enzymatic activity.26 GXIIB sPLA2 is highly expressed in the liver, small intestine and kidney, both in human and mouse, and the functions of GXIIB sPLA2 seem not to rely on enzymatic catalytic activity and might be related to its non-catalytic role in which the protein forms supermolecular aggregates with phospholipid vesicles and/or acts as a ligand for specific cellular targets.26 The prokaryotic sPLA2s have only two or zero disulfide bonds (GXIII and GXIV), which is strikingly different from the eukaryotic enzymes, which normally contain 6–8 disulfide bonds.
All of the sPLA2s display a characteristic increase in activity when the substrate concentration is changed from monomers to aggregates, which is refered as “interfacial activation”.24 The structural basis for the interfacial activation mechanism will be discussed in section 2.2. In contrast with cytosolic PLA2s (cPLA2s), which have a marked specificity for arachidonic acid at the sn-2 position of its phospholipid substrates, sPLA2s do not show distinct preference for the sn-2 position fatty acyl chains; there is, however, some specificity for certain head groups of the phospholipid substrates. In general, most of the sPLA2s show a higher activity with anionic phospholipids such as phosphatidylglycerol (PG), phosphatidylethanolamine (PE) and phosphatidylserine (PS). GIA and GXIV sPLA2s are more active against zwitterionic phosphatidylcholine (PC) vesicles. Mammalian GV and GX sPLA2s can hydrolyze both anionic phospholipid vesicles and zwitterionic PC-rich vesicles at comparable rates.27
2.2 Structural Characteristics and Interactions with Membranes
Almost all sPLA2s contain a highly conserved Ca2+ binding loop (XCGXGG) and a catalytic site (DXCCXXHD). Among the sPLA2s, crystal structures available are the cobra venom GIA, human, porcine and bovine pancreatic GIB, human GIIA, human GX, plant GXIB and prokaryotic GXIV sPLA2s. Although the amino acid identity level is low, all of these enzymes share a common protein fold (Figure 4 A) with slight differences and feature the same catalytic His/Asp dyad. GI, IIA and X sPLA2s have very similar structures, which contain three long α-helices, two-stranded β-sheets referred to as β-wings, and a conserved Ca2+ binding loop. The plant GXIB sPLA2 has been shown to lack β-wings.28 The prokaryotic GXIV sPLA2 shows a different, all α-helical, protein folding.29
Figure 4.

A. Overall structure of Group IA sPLA2. Helices are in magenta, β-strands are in green, calcium is shown as a red sphere and disulfide bonds are shown as yellow sticks. (PDB entry: 1PSH) B. The Group IA sPLA2 with phospholipid substrate modeled in the active site as a space filling model. The active site residues His-48 and Asp-93 and the bound Ca2+ are shown in purple. Ca2+ is bound by Asp-49 as well as the carbonyl oxygens of Tyr-28, Gly-30, and Gly-32. Aromatic residues are shown in white. Adapted from Dennis.7
Although the structures of other sPLA2 family members have not been reported to date, based on available structural data and sequence alignment, the GI, GII, GV and GX mammalian sPLA2s are expected to share a common three-dimensional structure. The human GIII sPLA2 would represent another class of fold. Its PLA2 domain is expected to be similar to that of bee venom GIII sPLA2, while its extra C- and N-terminal domains, as we mentioned above, are functionally unknown and structurally are not homologous to known proteins. GXIIA sPLA2 comprises the third structural class, which has an unusual Ca2+ binding loop.
The cobra venom GIA sPLA2 is one of the most extensively studied PLA2 enzymes and it has been considered an important model for phospholipase A2 enzymology. Therefore, we will use GIA sPLA2 as an example to show the unique structural characteristics of sPLA2 enzymes. GIA sPLA2 contains six conserved disulfide bonds with an additional disulfide bond between residue 11 and residue 71 (Figure 4 A). Calcium ions bind with the conserved aspartic acid residue as well as the carbonyl oxygens of the tyrosine and glycines from the calcium binding loop. Calcium is absolutely required for hydrolysis. The secreted sPLA2s shown do not form a classical acyl enzyme intermediate characteristic of serine proteases.7,30 Rather, they utilize the catalytic site His, assisted by an Asp, to polarize a bound H2O, which then attacks the carbonyl group. The calcium ion stabilizes the transition state by coordinating the carbonyl group and the negative charge from the phosphate oxygen.
Using the native and inhibitor-bound structure of GIA sPLA2 and a space-filling model for the phospholipid substrate, a model of how the substrate interacts with the active site was created (Figure 4B).7 In the model only about 9–10 carbons of the sn-2 acyl chain interact with the enzyme and the rest of the chains are buried in the lipid-water interface. This explains why sPLA2s fail to show specificity for the particular fatty acid in the sn-2 position. The hydrophobic residues, Leu-2, Phe-5, Trp-19, Tyr-52 and Tyr-69 wrap around the acyl chain of the lipid substrate.
The activity of most phospholipase A2 family members depends critically on the interaction of the protein with large lipid aggregates. The interfacial activation mechanism of PLA2 enzymes has long been an interesting topic in membrane protein enzymology.24,31 GIA sPLA2 can hydrolyze zwitterionic phospholipids. This is most likely due to the aromatic residues present on the interfacial binding surface. Recently hydrogen deuterium exchange mass spectrometry (DXMS) was used to study GIA sPLA2/phospholipid vesicles and calcium ion interaction in solution.32 The DXMS results show that the surface hydrophobic residues Tyr-3, Trp-61, Tyr-63 and Phe-64 penetrate into the lipid membrane phase to allow the enzyme to access the substrate from the lipid aggregates (Figure 5). A second calcium binding site was also indicated in the DXMS study, although there is only one calcium ion in the crystal structure. A second calcium binding site has also been found in other related sPLA2 structures.33 Among the aromatic amino acids, tryptophan was thought to be the most potent contributor to the interfacial binding. Replacing Try-31 causes human GV sPLA2 to lose its ability to bind to zwitterionic PC vesicles.34 A similar result was found in GX sPLA2, where replacing the only surface Try-67 residue with an alanine caused an 8-fold reduction in binding affinity to PC vesicles and also significantly decreased the hydrolysis activity of PC-rich vesicles.27b Human GIIA sPLA2, which binds poorly to PC-rich vesicles, exhibits much higher activity on DOPC membranes when a surface valine residue is mutated to tryptophan.27b,35
Figure 5.

Model of the lipid surface binding of the Group IA sPLA2 is shown with residues on the interfacial binding surface Tyr-3, Trp-19, Trp-61, and Phe-64 shown in blue stick form. Adapted from Burke et al.32
Both electrostatic and hydrophobic interactions contribute to the interfacial binding of sPLA2 to anionic phospholipid membranes. The interaction between basic residues on the binding surface with anionic vesicles plays an important role in interfacial binding.36 The large number of basic residues scattered over the GIIA sPLA2 surface could be the reason that this enzyme has highly selective binding to anionic vesicles. The recently defined crystal structure of GIB sPLA2 in a premicellar complex has shown that the enzyme uses the hydrophilic residues Arg-6 and Lys-10 to bind at the anionic interface.37 However, studies on the interfacial binding of bee venom GIII sPLA2 have shown that the interaction occurs predominantly through a nonelectrostatic mechanism.38 When the five basic residues on the bee venom sPLA2 binding surface are all changed to neutral glutamine residues, the resulting mutant does not show a significant decrease in binding to the anionic vesicles.38 However if the basic residues are mutated to charge-reversed glutamate residues, the mutant binds to PS/PC vesicles 3000-fold more weakly than the wild-type protein.38 This indicates that while the electrostatic interaction is not predominant between the enzyme and anionic phospholipids, the repulsion interaction will definitely destroy the binding. The interfacial binding surface of GX sPLA2 is charge neutral, which may explain why this enzyme is active on both zwitterionic and anionic phospholipids.39
The crystal structure of the trimeric form of human pancreatic pro-GIB sPLA2 was recently reported.40 The trimeric form shows a much more positively charged interfacial surface. The authors suggested that human GIB sPLA2 may use a different activation mechanism in which GIB sPLA2 switches from monomeric to trimeric form when it associates with the membrane using the highly positive charged trimer back side.40 The crystal structure of the trimeric form has also been shown in studies on cobra venom41 and Naja naja GIA sPLA2s.42 Anion-assisted dimer crystal structures were reported for other pancreatic sPLA2s.43 While the existence of dimeric and trimeric forms may be a functionally important feature of these sPLA2s, the higher oligomeric states of sPLA2 structures are found under high concentration crystallization conditions, so they may not be representative of the real physiological state. Whether the trimeric or dimmeric form is important for sPLA2 catalysis or not requires further investigation.
2.3 Biological Functions and Disease Implications
sPLA2s exhibit a large variety of cellular functions, though the specific function varies by group or subgroup. The major functions will be summarized below and include the ability to kill Gram-positive and Gram-negative bacteria thereby affecting host defense against bacterial infections.44 sPLA2s also show antiviral activity.45 sPLA2s are expressed and released by human inflammatory cells including macrophages, monocytes, T cells, mast cell and neutrophils and increased concentrations of different isoforms of sPLA2s have been detected in the blood of patients with inflammatory and autoimmune diseases.46 sPLA2s also play a role in the hydrolysis of oxidized lipids in low- and high-density lipoproteins ontributing to the development of atherosclerosis.47 Many experiments carried out both in vitro and in vivo, especially transgenic and gene knockout mice studies, have expanded our knowledge of the sPLA2 family. However, due to the large number of sPLA2 family members and the redundant expression in tissues, there is still much we do not understand about the functions and physiological roles of each individual sPLA2.
2.3.1 Antibacterial and Antiviral Functions of sPLA2s (sPLA2 Groups I, II, III, V, X)
There is a considerable body of evidence supporting the antibacterial functionality of sPLA2. GIIA sPLA2 has displayed antibacterial activity towards Gram-positive bacteria including Staphylococcus aureus48, Listeria monocytogenes,49 and others.17a The enzyme has also demonstrated antibacterial activity against some Gram-negative bacteria, such as E. coli and Salmonella typhimurium.44 High concentrations of GIIA sPLA2 are found in tears, where the majority of bactericidal action is due to GIIA sPLA2.50 The concentration of GIIA sPLA2 increases up to 500-fold in the serum samples of patients with severe acute diseases compared with healthy controls.49b High concentrations of GIIA sPLA2 have also been found in seminal plasma, inflammatory exudates, bronchoalveolar lavage and intestinal lumen.17e,51 Overexpression of GIIA sPLA2 in transgenic mice has resulted in decreased mortality in experimental Staphylococcus aureus infections and has improved clearance of bacteria from organs and body fluids of experimental animals.52 This enzyme also plays a protective role in vivo against experimental anthrax. Transgenic mice expressing human GIIA sPLA2 and mice that have had recombinant human GIIA sPLA2 administered in vivo are resistant to B. anthracis infection.53
The antibacterial activity of GIIA sPLA2 is calcium-dependent and is negated in the presence of EGTA.44,54 The antibacterial activity also depends on PLA2 activity to hydrolyze the cell membrane.55 To kill the bacteria, GIIA sPLA2 first penetrates the peptidoglycan envelope of Gram-positive bacteria, thus gaining access to the bacterial cell membrane phospholipids.56 The highly positively charged surface of GIIA sPLA2 also enables it to bind with the lipoteichonic acids of Gram-positive bacteria.57 The bactericidal effect of GIIA sPLA2 is highest against bacteria in the phase of logarithmic growth, which may be due to its ability to reach the bacterial plasma membrane through the dividing cell wall.58
In addition to GIIA other sPLA2s also have antibacterial activity.49a,56,59 The ranking of most to least potent sPLA2s against Gram-positive bacteria is GIIA > GX > GV > GXII > GIIE > GIB, GIIF for human, and GIIA > GIID > GV >GIIE > GIIC, GX > GIB, GIIF for murine.56 The antibacterial efficiency of a particular sPLA2 depends significantly on the highly positively charged protein surface. For example, the GIIA sPLA2 shows more antibacterial function than other sPLA2s-this may be related to its highly cationic nature (pI > 10.5). Antibacterial activity was also found in snake venom GIA sPLA2 against both Gram-positive and Gram-negative bacteria,60 however the mechanism is different. The catalytically inactive form (H49K), as well as a synthetic peptide containing residues 115–129, retained the bactericidal effect of the catalytically active intact protein,60 which indicates that the bactericidal effect is independent of the enzymatic activity.
The antibacterial mechanism against Gram-negative bacteria was thought to be different from the action against Gram-positive bacteria. In addition to the anionic peptidoglycan cell wall, Gram-negative bacteria have an outer layer of lipopolysacchride, which makes it difficult for the enzyme to access and hydrolyze the plasma membrane phospholipids. GIIA sPLA2 has been shown to be effective against bactericidal/permeability-increasing protein (BPI)-treated E. coli which depends on the presence of a cluster of basic residues within the surface region near the N-terminus.61 By mutating Ser-7 to a lysine residue, the pig pancreas GIB sPLA2 can be converted into an enzyme active against E. coli treated with BPI.62 GV sPLA2 was also found to hydrolyze phospholipids from E. coli in the presence of serum, but not as efficiently as GIIA sPLA2.59 GV sPLA2 has a lower pI (>7) and thus has lower intermediate activity on anionic cell wall-bound E. coli membranes.
In addition to their antibacterial functions, sPLA2s also display antivirus activity. GIII, GV and GX sPLA2s are capable of preventing host cells from being infected with adenovirus.63 The antivirus activity of GV and GX sPLA2s is due to the enzyme activity which converts PC to lysoPC in the host cell membranes. This hydrolysis of the cell membrane protects the host cells from adenovirus entry.63a GIII sPLA2 has a different antivirus mechanism, which requires the presence of both the catalytic domain and the N-terminal domain for the anti-adenovirus effect.63b By degrading the virus membrane and recognizing the virus envelop, human GX sPLA2 is capable of neutralizing HIV-1.64 Bee and snake venom sPLA2s and a peptide derived from bee venom sPLA2 can block HIV-1 entry into the host cells by steric inhibition of the chemokine receptor on the target cells without requiring enzyme catalytic activity.45,65
2.3.2 sPLA2 and Inflammation (sPLA2 Groups I, II, V, X)
The sPLA2s appear to play a role in several inflammatory diseases. The first evidence was from GIIA sPLA2, which is present at high concentrations in the synovial fluid of patients with rheumatoid arthritis.3 GIIA sPLA2 deficient mice have shown reduced signs of arthritis when compared with wild-type mice. Recently, GV sPLA2 has also been found in rheumatoid arthritis synovial fluid, but the expression was notably lower than GIIA sPLA2.66 However, GV sPLA2 may play an anti-inflammatory role rather than the normal pro-inflammatory role.66 GV sPLA2 deficient mice were protected from K/BxN arthritis when treated with exogenous recombinant GV sPLA2.66 Increased levels of sPLA2s have also been detected in the plasma or serum of patients with acute pancreatitis, septic shock, Crohn’s diseases and ulcerative colitis.47 Furthermore, sPLA2s seem to be involved in adult respiratory distress syndrome (ARDS) and inflammatory bowel disease.67
sPLA2s participate in inflammation through their enzymatic activity by releasing free fatty acids, including arachidonic acid (AA), thus initiating the biosynthesis of lipid mediators, including prostaglandins, thromboxanes and leukotrienes. In addition, the hydrolytic product, lysophospholipid, is also a proinflammatory lipid mediator. AA release by the PLA2 catalytic reaction is the initial and rate limiting step for the biosynthesis of eicosanoids.68 GIVA PLA2 has for a long time been considered the major PLA2 enzyme to release AA for eicosanoid production.68 However, more recent results have shown that sPLA2s may also be involved in AA release and eicosanoid biosynthesis. GIIA sPLA2s are capable of mediating cytokine-induced delayed AA release and ionophoreinduced immediate AA release.69 Exogenously added human GV sPLA2 could induce AA and leukotriene C4 (LTC4) release from unprimed human neutrophils.34 Peritoneal macrophages from GV sPLA2 knockout mice show reduced production of LTC4 upon stimulation with the yeast cell wall particle zymosan which amplifies the action of GIV cPLA2 in regulating eicosanoid biosynthesis.70 In mouse peritoneal macrophages stimulated with zymosan, GV sPLA2 can translocate to the phagosome and regulate phagocytosis through regulation of eicosanoid generation.71 The exogenously added human GV sPLA2 could also induce leukotriene B4 (LTB4) synthesis in human neutrophils by the activation of GIVA PLA2.72 A putative mechanism has been proposed whereby human GV sPLA2 first hydrolyzes the outer plasma membrane of neutrophils to release lysoPCs and fatty acids. The lysoPC and fatty acid then cause an increase in the calcium concentration and in turn activate membrane translocation of 5-lipoxygenase and cPLA2.72 In addition, exogenous GV sPLA2 could induce arachidonic acid release and LTC4 synthesis in isolated human peripheral blood eosinophils in a manner independent of activating cPLA2.73 Recombinant GX sPLA2 has been shown to induce a substantial release of AA independent of the action of GIVA PLA2, when added to human myeloid leukemia cells or adherent mammalian cells.74 A GIVA PLA2 knockout mice study further supports the results of the in vitro study which showed that the amount of AA released by GX sPLA2 from spleen cells was not significantly altered by GIVA PLA2 deficiency.75 GX sPLA2 knockout mice have shown a significant reduction in eicosanoid generation and this provides additional strong evidence for GX sPLA2 involvement in eicosanoid-mediated inflammation.76
sPLA2s can release AA by two mechanisms: an external plasma membrane pathway and a heparan sulfate proteoglycan (HSPG)-shuttling pathway.77 Among sPLA2s, GV and GX show high binding affinity and catalytic activity towards PCs. This would allow GV and GX sPLA2s to directly act on the outer leaflet of plasma membranes, to hydrolyze PCs to release fatty acids and lysophospholipids.77 Heparin-binding sPLA2s GIIA, GIID and GV may bind to the heparan sulfate chains of glypican, allowing the accumulation of the enzyme on the cell surface and promoting enzyme trafficking to intracellular compartments to release AA.77
In addition to eicosanoid production, sPLA2s also are able to activate inflammatory cells to induce the production of other proinflammatory mediators from macrophages, neutrophils, eocsinophils, monocytes and endothelia cells78 and the function may not require enzymatic activity. GIB, GIIA, GV and GX sPLA2s induce the production of proinflammatory cytokines and chemokines independent of their hydrolytic activity.79 GIIA and GIII sPLA2s are capable of upregulating the surface molecules on a variety of inflammatory cells.80
Adult respiratory distress syndrome is characterized by lung surfactant disorders that lead to increased surface tension, alveolar collapse, loss of liquid balance in the lung and severe disturbance of pulmonary gas exchange.67a Increased levels of sPLA2s have been detected in bronchoalveolar lavage fluids of ARDS patients at levels that correlate positively with the severity of ARDS.81 The involvement of sPLA2s in ARDS seems to depend on the enzymatic hydrolysis of surfactant phospholipids, since increased amounts of lyso-PC and decreased PC concentrations were found in bronchoalveolar lavage fluids of ARDS patients.82 In an animal model of acute lung injury inhibiting GIIA sPLA2 activity ameliorated lung dysfunction by protecting against surfactant degradation.83 Transgenic mice expressing human GV sPLA2, but not GIIA or GX sPLA2, show a significant reduction in the lung surfactant phospholipids phosphatidycholine and phosphatidylglycerol, a condition that leads to neonatal lethality due to lung dysfunction.84 The overexpression of human GX sPLA2 enzyme in transgenic mouse macrophages leads to massive degradation of surfactant phospholipids causing lethal lung inflammation.85
sPLA2 may be involved in the pathogensis of inflammatory bowel disease including Crohn’s disease and ulcerative colitis.86 GIIA sPLA2 protein and mRNA were detected in Paneth cells of the small intestinal mucosa in the intestine in Crohn’s disease patients.86b GIIA sPLA2 enzymatic activity was found to be significantly increased in actively inflamed colonic mucosa of Crohn’s disease patients and severely inflamed mucosa of ulcerative colitis patients compared with non-inflamed mucosa.86a GIIA sPLA2 was found to be localized to the Paneth’s cells at the site of active inflammation in the Crohn’s disease patients.86b
2.3.3 sPLA2s in Atherosclerosis (sPLA2 Groups II, III, V, X)
There is considerable evidence to show that sPLA2s play an important role in atherosclerosis. GIIA, GV and GX sPLA2s have been detected in human and/or mouse atherosclerotic lesions and are believed to enhance lipid accumulation in arterial intima.17c A study in patients with coronary artery disease shows that increased levels of plasma GIIA sPLA2 may be a significant risk indicator for coronary artery disease (CAD) and a predictor for clinical coronary events.87 Young adults with metabolic syndrome (MetS) were also found to have increased serum levels of sPLA2.88
Transgenic mice expressing human GIIA sPLA2 have exhibited a dramatic increase in atherosclerotic lesions compared with nontransgenic littermates, regardless of whether they were fed a high-fat, high-cholesterol diet or a low-fat chow diet.89 The transgenic mice also exhibit decreased levels of HDL-cholesterol and increased levels of LDL/VLDL-cholesterol.89 An animal model that transplanted bone marrow cells from transgenic mice to lethally irradiated LDL receptor knockout (LDLR−/−) mice indicates that the macrophage cells expressing human GIIA sPLA2 promote atherosclerotic lesion formation without plasma lipoprotein concentration changes.90 Additionally, the same mouse model shows 2.3 fold larger lesions compared with control mice when fed a high-fat diet and also shows enhanced collagen deposition independent of lesion size.91 This indicates that GIIA sPLA2 is a proatherogenic factor and it may regulate the collagen production in the plaque. The macrophage-specific expression of GIIA sPLA2 in transgenic mice induces increased mouse 12/15-lipoxygenase (12/15-LO), causing the generation of oxidative stress, thought to be a major contributing factor to atherogenesis.92 Studies of transgenic mice have found that the expression of recombinant apolipoprotein B100 (apoB100) and GIIA sPLA2 induces the formation of slightly smaller LDL particles, enriched with lysoPC.93 This is because GIIA sPLA2 modifies the LDL in circulation so that site A (residues 3148–3158) in apoB100 is exposed, increasing LDL/proteoglycan binding and making the LDL more proatherogenic.93
One possible mechanism for the role of sPLA2s in atherogenesis is the ability of sPLA2 to hydrolyze the phospholipids on LDL particles. The modified LDL could then promote lipid accumulation and lead to enhanced macrophage uptake. Among mammalian sPLA2s, GV and GX sPLA2 have shown higher activity in hydrolyzing LDL and HDL than other sPLA2s due to their high affinity binding to PCs.94 Thus, they may contribute more to atherosclerosis. GV sPLA2 was detected in both human and mouse atherosclerosis lesions.95 LDL particles modified by GV sPLA2 are significantly smaller than native LDL particles and promote foam cell formation through a mechanism that is independent of scavenger receptors SR-A and CD36.95–96 Using gain-of-function and loss-of-function approaches, Bostrom and coworkers have shown that macrophages are the major source of GV sPLA2 in mouse lesions and that overexpression of GV sPLA2 in bone marrow cells significantly increases collagen deposition in atherosclerotic lesions, providing the first in vivo data showing that GV sPLA2 promotes atherosclerosis.97
GX sPLA2 shows the highest activity towards PC, which is the major lipid component of both LDL and HDL. Elevated expression of GX sPLA2 was found in atherogenic lesions in the arterial intima of both human and apoE-deficient mice fed a high fat diet.98 LDL modified by GX sPLA2 enhances accumulation of cholesterol ester in macrophages,98 where it may promote the atherosclerotic process by hydrolyzing lipoproteins. Unlike GIIA and GV sPLA2, GX sPLA2 does not promote LDL particle aggregation and does not bind to proteoglycans.99 Recent research has shown GX sPLA2 may suppress macrophage expression of two transport proteins, ABCA1 and ABCG1, which efflux excess cholesterols to extracellular acceptors.100 This indicates that GX sPLA2 may negatively regulate the genes critical for cellular cholesterol efflux to affect atherosclerotic lipid accumulation.
In addition to GIIA, GV and GX, GIID, GIIE and GIII sPLA2s were also expressed in macrophage and smooth muscle cells (SMCs), and their expression increased with atherosclerosis development.101 Intimal SMCs are thought to contribute to the progression of atherosclerosis due to their ability to produce an extracellular matrix which binds to low-density lipoprotein (LDL) and leads to further intimal cholesterol deposition.102
Human GIII sPLA2, which is distinctive among mammalian sPLA2s due to its additional N- and C-terminal domains, may also play a role in atherosclerosis. Transgenic mice overexpressing human GIII sPLA2 have marked alterations in the levels of plasma lipoproteins and GIII sPLA2 modified LDL could promote the formation of foam cells.103
sPLA2 may also contribute to the atherosclerotic process by generating proinflammatory lipid mediators such as prostaglandins, thromboxanes and lysophospholipids. As we discussed in section 2.3.2, GIIA, GV and GX sPLA2 may be involved in AA release and thus promote eicosanoid biosynthesis. Moreover, as part of the release of fatty acids, another proinflammatory lipid mediator – lysophospholipid – is produced. Lysophosphatidylchonline, a highly atherogenic lipid, can induce multiple deleterious processes in the atherosclerotic plaque.104 In addition, GX sPLA2 can hydrolyze both platelet activating factor (PAF) in free form and PAF that has been partitioned into either large unilamellar PC vesicles or lipoproteins,105 which indicates that GX sPLA2 may function like GVIIA lipoprotein-associated PLA2 (Lp-PLA2), which regulates PAF in the inflammatory process.
sPLA2 may also exacerbate atherosclerosis through induction of proinflammatory cytokines. NFκB is considered to be a key regulator of inflammation in the atherosclerosis process.106 LDL containing GV sPLA2 incubated with J-774 macrophage-like cells showed a significant increased TNF-α and IL-6 mRNA expression and the induced cytokine secretion was promoted by activation of NFκB.107 The activation of NFκB was promoted by the lipid products generated by GV sPLA2 hydrolysis of LDL.107
2.3.4 Other Functions
Many of the biological functions of sPLA2s appear to be independent of their catalytic activity. A variety of sPLA2s exhibit potent anticoagulant activity.67b The mammalian GIIA, GIID and GV sPLA2s, in addition to several venomous sPLA2s, contain many basic residues on the protein surface which inhibit prothrombinase activity by binding to factor Xa. This effect is independent of phospholipid hydrolysis.108 However, at limiting concentrations of phosphatidylserine, sPLA2 enzymes inhibit the prothrombinase complex in a phospholipid-dependent manner either by hydrolyzing or binding to phospholipids and therefore inhibiting formation of coagulation complexes.108
Two mammalian cell surface sPLA2 receptors, N-type and M-type receptors, have been identified and found to bind to both sPLA2s from venom and mammalian.109 The N-type receptor is highly expressed in mammalian brain membranes. The M-type receptor is a 180 kDa protein, membrane-bound and soluble secreted receptor.17d The M-type receptor was cloned in various mammalian species and is expressed in various tissues. Several lines of evidence indicate that sPLA2s may exert physiological roles by acting in a cytokine-like fashion and independent of their catalytic function.79a,79c,110
GIB sPLA2, which is present at high levels in pancreatic juice, is the major enzyme responsible for the dietary phospholipid phosphatidycholine.111 GIB knockout mice fed on a chow diet show no difference in the absorption of dietary lipids compared with wild type mice111a. However, GIB knockout mice fed a high-fat diet are resistant to diet-induced obesity and obesity-related insulin resistance.112
Higher levels of sPLA2 activity were found in the bronchoalveolar lavage fluid of asthmatic patients than that of control groups.113 GIIA and GX sPLA2s were found in the airways of both asthmatic patients and controls. These two sPLA2s should be responsible for the majority of PLA2 enzymatic activity detected in the bronchoalveolar lavage fluid.114 GX sPLA2 is more highly expressed than GIIA sPLA2 in the airway epithelium of asthmatic patients.114 In a mouse asthma model, GX sPLA2 deficiency was shown to potentially reduce allergen-induced airway inflammation and remodeling.76 When transgenics expressing human GX sPLA2 in GX sPLA2 knockout mice were used in a recent study in a Th2 cytokine driven mouse asthma model, the mouse shown an induced airway inflammation and hyperresponsiveness which was not found in the GX sPLA2 knockout mouse asthma model.115 Altogether these studies suggest that GV sPLA2 may play an important role in asthma.
sPLA2s may also play a role in tumorigenesis.16e GIIA sPLA2 is overexpressed in almost all human prostate cancer specimens and increased levels correlate with advanced tumor grades.116 GIIA sPLA2 may stimulate tumor cell growth117 and be involved with the progression of prostate cancer, and could therefore potentially serve as a biomarker for prostate cancer.118 Increased expression of GIIA sPLA2 was also detected in colorectal adenomas of familial adenomatous polyposis patients, and the enzyme may also be involved in human colorectal tumor development and progression119. However the protective role of GIIA sPLA2 in colorectal cancer was also found in a mouse model. Transgenic Min (multiple intestinal neoplasia) mice carrying the functional PLA2G2AAKR allele have shown resistance to tumor development, including both reduced tumor multiplicity and size.120 In addition to GIIA, GIB, GX and GIII sPLA2s are expressed in various type of cancers and may also play a role in tumorigenesis.16e However the function of sPLA2s in cancer is a controversial issue, as it is not clear whether the enzyme is a tumor suppressor or tumor promoter.121
2.4 Chemical Inhibitors and Therapeutic Intervention
Numerous assays to evaluate the activity of inhibitors toward each PLA2 type exist,122 ranging from simple procedures to methods involving expensive instrumentation and from low sensitivity to highly sensitive procedures. As mentioned in section 1.2, it has to be emphasized that it makes no sense to compare the activity of various inhibitors (for example, the IC50 values) if they are measured in different assay systems, especially if assayed at different substrate concentrations. The IC50 is the inhibitor concentration that is required to reduce the activity of the enzyme in half. A review article in 2005 reported the various inhibitors of sPLA2s.22b The most recent review article on PLA2 inhibitors highlights the recent patent literature.22f
2.4.1 Early Attempts with Phospholipid Analogues
The early attempts to develop PLA2 inhibitors were focused on phospholipid analogues and started in earnest in the 1980’s. 1-Stearyl-2-stearoylaminodeoxy phosphatidylcholine (1) was studied and found to be a reversible inhibitor of PLA2 from cobra venom (Naja naja naja).123 At the same time, A series of long chain difluoro ketones was studied.124 Derivative 2a based on phosphatidylethanolamine was the most active in this series against cobra venom PLA2. Phospholipid analogues (3) containing a phosphonate group in place of the ester at the sn-2 position of the glycerol backbone were found to be tight-binding inhibitors of the same enzyme.125

A series of structurally modified phospholipids were used to delineate the structural features involved in the interaction between cobra venom PLA2 and its substrate.126 A very potent inhibitor (thioether amide PE, 4) was identified among them. At the same time, a class of acylamino analogues of phospholipids (5) was developed and studied as inhibitors of porcine pancreatic PLA2.127

Some of the above mentioned inhibitors, an acylamino and a phosphonate phospholipid analogue, were useful tools to resolve the structure of various sPLA2s by X-ray crystallography.30a,33,33,128 Thus, the interfacial catalytic mechanism of sPLA2 was proposed.33 The first structure of recombinant human synovial fluid PLA2 was reported in 1991128 and it was expected to be applied in structure based design.
A class of “suicide-inhibitory bifunctional linked substrates” (SIBLINKS, for example compound 6) has been reported.129 In addition, these invastigators alsot studied the effect of polar head groups on the interaction of cobra venom PLA2 with phosphonate transition-state analogues 7.130

The substrate specificity at the active site of recombinant human synovial fluid PLA2 was investigated by using a series of short-chain phospholipid analogues such as 8.131

2.4.2 Dicarboxylic Acids
In 1992, Bristol-Myers Squibb presented the dicarboxylic acid 9a (BMS-181162) as the first specific inhibitor of a 14 kDa PLA2 (specificly compared to other types of phospholipases, PLC, PLD and PLA1).132 BMS-181162 blocked the arachidonic acid release (IC50 10 µM) and the biosynthesis of LTB4 and PAF in calcium ionophore (A23187) stimulated human PMN’s.132b A similar derivative, BMS-188184, presented better stability and activity as a backup agent for BMS-181162 in clinical studies. BMS-181162 reached Phase II clinical trials as a cream for topical application for the treatment of psoriasis, but the results were disapointing as the drug could not penetrate beyond the outer layer of the skin. After the evaluation of these results, this inhibitor series was discontinued.

The mechanism of inhibition of GIIA sPLA2 (refered to by the authors as human nonpancreatic sPLA2) by the anti-inflammatory agent BMS-181162 was studied.132c BMS-181162 inhibited human platelet PLA2 with an IC50 40 µM and it was able to reduce mouse ear edema with an ED50 160 µg/ear in a phorbol-ester induced acute inflammation assay, while BMS-188184 inhibited human platelet PLA2 with an IC50 17 µM and reduced mouse ear edema with an ED50 9.37 µg/ear.132d It is ubclear whether the inhibtion observed in the mouse ear edema model reflected inhibtion of just GIIA sPLA2 or other PLA2s as well.
Among a series of biaryl diacid inhibitors of human sPLA2, biarylacetic acid derivatives were found to be more active than biaryl acids or biarylpropanoic acids.133 Compounds with larger hydrophobic groups were usually more potent inhibitors of the enzyme. Compounds 10a and 10b were found to possess significant anti-inflammatory activity in a phorbol ester induced mouse ear edema model of chronic inflammation (Table 3).
Table 3.
Group IIA PLA2 Inhibition by Diacid Inhibitors - Studies In Vitro and of Mouse Ear Edema
| IC50 (µM) | ED50 (µg/ear) | |
|---|---|---|
| 10a | 8 | 32 |
| 10b | 4 | 73 |

Through computer-assisted methods, Roche developed the very potent inhibitor 11 containing the iminodiacetic acid group (IC50 0.23 µM for human synovial fluid PLA2).134 Inhibitor 11 exhibited anti-inflammatory activity in two separate animal models of inflammation.

2.4.3 Sulfonamides
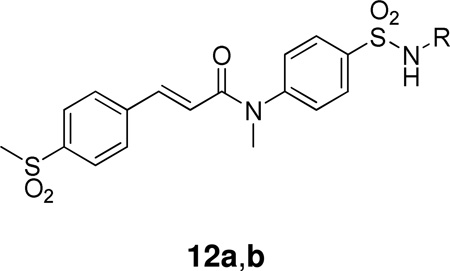
A novel series of benzenesulfonamides were prepared and evaluated as membrane-bound PLA2 inhibitors.135 Several compounds (12a,b), which proved to be potent inhibitors in vitro, significantly reduced the size of myocardial infarction in coronary occluded rats by iv administrations prior to the ligation. Compound 12a (ER-3826), which showed the protective in vivo effects at doses higher than 0.3 mg/kg iv (Table 4), was finally chosen as a leading candidate.135
Table 4.
Intracellular Membrane Bound PLA2 Inhibition by Benzenesulfonamides
 | ||
|---|---|---|
| Compound | R | IC30 (µM) |
| 12a | 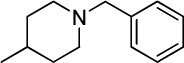 |
0.028 ± 0.012 |
| 12b | 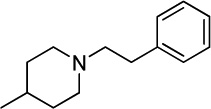 |
0.009 ± 0.004 |
Sulfonamide 13 (SB-203347) was used to understand the contribution of GII sPLA2 to prostaglandin formation.136 Computer-assisted methods contributed to the rational design of the sulfonamide inhibitor 14.137

2.4.4 Amides
Taking into consideration that ionic phospholipid analogues would not be cell permeable and therefore not useful in determining the roles of PLA2s in cellular processes, primary amides of long unsaturated acids were synthesized and studied. Two of them (15a,b) presented interesting inhibition of porcine pancreatic and human synovial fluid PLA2 (Table 5).138
Table 5.
Group IB and Group IIA PLA2 Inhibition by Primary Amides of Long Chain Unsaturated Fatty Acids.a
|
Compound |
XI(50) | |
|---|---|---|
| pGIB PLA2 | hGIIA PLA2 | |
| 15a | 0.0008 | 0.002 |
| 15b | 0.0003 | 0.004 |
XI(50) is the molar fraction of an inhibitor in the lipid layer that is required to reduce the activity of the enzyme in half.

Acylamino phospholipid analogues were synthesized and evaluated as pancreatic PLA2 inhibitors (Table 6).139 The mode of binding of these inhibitors to the active site of the enzyme was determined using two-dimensional NMR and molecular modeling techniques.
Table 6.
Group IB PLA2 Inhibition by Acylamino Phospholipid Analogues
| Compound | IC50 (µM) |
|---|---|
| 16a | 1.4 |
| 16b | 0.23 |
| 16c | 4.5 |

A continuation of this work reported non-phospholipid inhibitors of sPLA2, where a simple carboxylic group replaced the phosphocholine moiety.140 Structure-activity relationship studies led to interesting results (Table 7). The most potent inhibitor in this series (FPL67047XX) presented an IC50 value against the human platelet sPLA2 of 21 ± 4 nM. The precise binding interactions of this inhibitor with the human nonpancreatic sPLA2 were determined by high-resolution X-ray crystallography.141
Table 7.
Group IB and Group IIA PLA2 Inhibition by Non-phospholipid Amides
|
Compound |
IC50 (µM) | |
|---|---|---|
| pGIB PLA2 | hGIIA PLA2 | |
| 17a | 0.19 | NT |
| 17b | 0.023 | NT |
| 17c | 0.016 | 1.85 |
| 18 | 0.015 | 0.021 |

Most recently, new GIIA sPLA2 inhibitors were designed based on docking calculations by modifying the pharmacophore segments of the FPL67047XX inhibitor.142
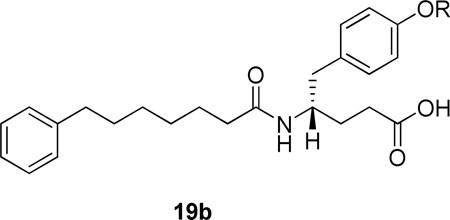
Another similar series of potent inhibitors was created by derivatization of D-tyrosine.143 The activities of various derivatives are summarized in Table 8. Inhibitor 19b (R=benzyl) was co-crystallized with hGIIA PLA2 and the crystal structure revealed a chelation to a Ca2+ ion through carboxylate and amide oxygen atoms, H-bonding through an amide NH group to His48, multiple hydrophobic contacts and a T-shaped aromatic group – His6 interaction.143
Table 8.
Group IIA PLA2 Inhibition by Amides Derived from D-tyrosine
 | |
|---|---|
| R | IC50 (µM) |
| benzyl- | 0.029 |
| 2-picolyl- | 0.214 |
| cyclopentylmethyl- | 0.057 |
| 1-napthylmethyl- | 0.019 |
| 2-napthylmethyl- | 0.039 |
| cinnamyl- | 0.116 |
| iso-butyl | 0.170 |
| n-heptyl | 0.086 |
| H | 2.57 |

Inhibitor 19b (R=benzyl) was found to protect the rat small intestine from I/R injury after oral or intravenous administration.144 In addition, this inhibitor of GIIA sPLA2 protected rats from TNBS-induced colitis,145 exhibited antifibrotic activity in young spontaneously hypertensive rats,146 and preserved bone architecture following ovariectomy in adult rats.147
A recent study on natural and non-natural amino acid-based amide and 2-oxoamide inhibitors of human PLA2 enzymes showed that amide 20, based on (R)-γ-norleucine, is a selective inhibitor of GV sPLA2 (XI(50) 0.003 ± 0.0004) not affecting the activities of intracellular GIVA PLA2 and GVIA PLA2.148

2.4.5 Indoles
In 1995, researchers at Lilly reported a highly potent sPLA2 inhibitor having a novel indole structure by using computer-aided drug design and chemical modification of a lead compound, which was discovered in the course of high-volume screening. Inhibitor 21a was co-crystallized with human recombinant GIIA PLA2 and the three dimensional structure showed that the inhibitor was in fact located in the active site.149 The replacement of the carboxylic acid functionality with an amide one and the methyl group by an ethyl group led to considerable increase of activity (Table 9).150
Table 9.
Group IIA PLA2 Inhibition by Indoles Using the Chromogenic Assay
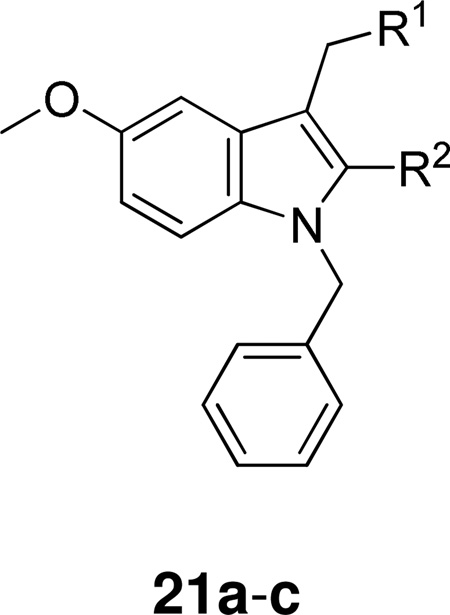 | |||
|---|---|---|---|
| Compound | R1 | R2 | IC50 (µM) |
| 21a | COOH | CH3 | 13.6 ± 4.2 |
| 21b | CONH2 | CH3 | 0.84 ± 0.17 |
| 21c | CONH2 | CH2CH3 | 0.26 ± 0.11 |
Further implementation of this structure-based design strategy and continued SAR development led to indole-3-acetamides with additional functionalities which provide increased interaction with important residues within the enzyme active site. These inhibitors 22 presented substantially enhanced potency and selectivity (Table 10).151
Table 10.
Group IIA and Group IB PLA2 Inhibition by Indoles Developed by SAR Study Using the Chromogenic Assay.
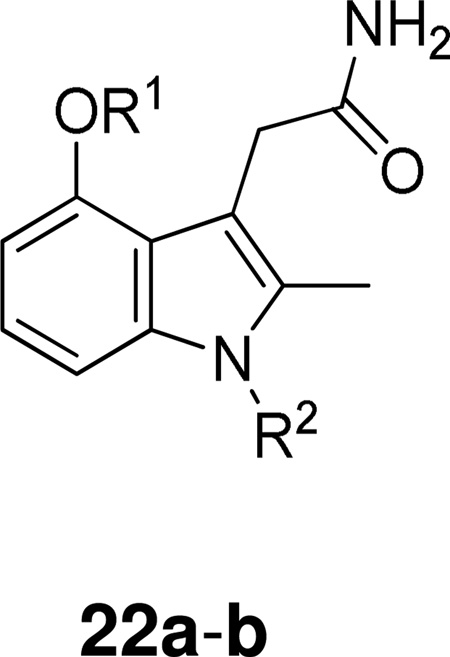 | |||||
|---|---|---|---|---|---|
| Compound | R1 | R2 | IC50 (µM) | ||
| hGIIA PLA2 | hGIB PLA2 | PLA2 pGIB PLA2 | |||
| 22a | CH2COONa |  |
0.010 ± 0.001 | 4.09 | 0.014 |
| 22b | CH2COOH |  |
0.052 ± 0.010 | 1.4 | 0.15 |
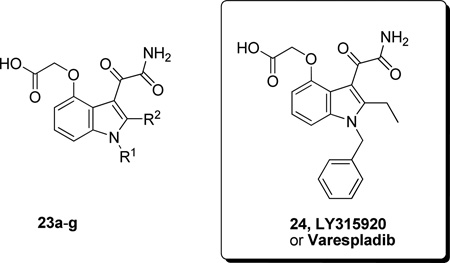
Structure-activity relationship studies were extended to include a series of indole-3-glyoxamide derivatives. Functionalized indole-3-glyoxamides with an acidic substituent appended to the 4- or 5-position of the indole ring were prepared and studied. Indole-3-glyoxamides with a 4-oxyacetic acid substituent had optimal inhibitory activity. These inhibitors exhibited an improvement in potency over the best of the indole-3-acetamides (Table 11), and LY315920 or Varespladib (24) was selected for evaluation clinically as a hGIIA PLA2 inhibitor.152
Table 11.
Comparison of GIIA and Group IB PLA2 Inhibition by Varespladib and Analogues Using the Chromogenic Assay
 | |||||
|---|---|---|---|---|---|
| Compound | R1 | R2 | IC50 (µM) | ||
| hGIIA PLA2 | hGIB PLA2 | pGIB PLA2 | |||
| 23a | C6H5CH2 | CH3 | 0.011 ± 0.004 | 0.761 | 0.015 |
| 23b | 2-(C6H5)C6H4CH2 | CH3 | 0.006 ± 0.001 | 0.364 | 0.097 |
| 23c | 3-(C6H5)C6H4CH2 | CH3 | 0.009 ± 0.001 | 0.57 | 0.007 |
| 23d | 1-naphthylCH2 | CH3 | 0.009 ± 0.004 | 1.2 | |
| 23e | n-C8H17 | CH3 | 0.008 ± 0.003 | 0.78 | |
| 23f | 2-(C6H5CH2)C6H4CH2 | CH2CH3 | 0.004 ± 0.001 | 0.062 | |
| 23g | 3-ClC6H4CH2 | CH2CH3 | 0.007 ± 0.002 | 0.390 | 0.003 |
| 24 | C6H5CH2 | CH2CH3 | 0.009 ± 0.001 | 0.228 | 0.048 |
LY315920 was 40-fold less active against human GIB pancreatic PLA2 and was inactive against cPLA2 and the constitutive and inducible forms of cycloxygenase.153 LY315920Na showed prophylactic effects on the high mortality, severe pancreas tissue damage, and blood biochemical changes in a lipolytic enzyme-related severe pancreatitis model.154 Varespladib was advanced in clinical trials as an intravenously-administered therapy for sepsis-induced systemic inflammatory response syndrome.155 At the end of the Phase I study, Varespladib was found to have an acceptable safety profile in patients with severe sepsis.156 However, the development of Varespladib for the treatment of severe sepsis was terminated because the Phase II study showed poorer than expected efficacy.
Lilly also synthesized methyl varespladib 25, LY333013, which functions as a prodrug and is rapidly converted in vivo to Varespladib. Using inhibitor 25 the role of GIIA PLA2 in rat colitis induced by dextran sulfate sodium was studied.157 A randomized, double-blinded, placebo-controlled clinical trial of LY333013 showed that the treatment for 12 weeks was well tolerated, but ineffective as an adjunct to disease modifying antirheumatic drugs.158 LY333013 was also used to study the possible role of GII sPLA2 in asthma, however it had no impact on the primary outcome variables of the areas under the FEV1 response curve early (0–3 hours) (AUCearly) and late (3 –8 hours) (AUClate) following inhaled allergen challenge.159

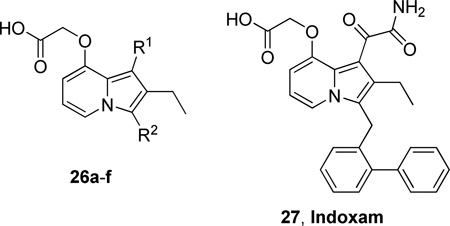
In 1996, Shionogi reported the synthesis, structure-activity relationship, and inhibitory activities of indolizine and indene derivatives. 1-(Carbamoylmethyl) indolizine derivatives were potent inhibitors, but were not stable to air oxidation. Introduction of an oxamoyl group to the C-1 position made the derivative stable and highly potent. By chemical modification at the C-3 position with various hydrophobic substituents and at the C-1 or C-8 position with hydrophilic substituents, some compounds approached the stoichiometric limit of the chromogenic assay (Table 12).160
Table 12.
GIIA PLA2 Inhibition by Indoxam and Analogues Using the Chromogenic Assay and a Deoxycholate Assay
 | ||||
|---|---|---|---|---|
| Compound | R1 | R2 | IC50 (µM) | |
| chromogenic assay |
PC/DOC assay |
|||
| 26a | CH2CONH2 |  |
0.014 | 0.014 |
| 26b | CH2CONH2 |  |
0.013 | 0.028 |
| 26c | COCONH2 |  |
0.008 | 0.005 |
| 26d | COCONH2 |  |
0.005 | 0.024 |
| 26e | COCONH2 |  |
0.006 | 0.0013 |
| 26f | COCONH2 |  |
0.009 | 0.0042 |
| 27 – Indoxam | COCONH2 |  |
0.006 | 0.003 |
The effect of indoxam on murine endotoxic shock was studied and the results suggested that indoxam blocks the production of proinflammatory cytokines during endotoxemia through PLA2-IIA independent mechanisms, possibly via blockade of the PLA2R function.161
In 2002, the expression of the full set of human and mouse groups I, II, V, X, and XII sPLA2s in Escherichia coli and insect cells provided pure recombinant enzymes for detailed comparative interfacial kinetic and binding studies. Analysis of the inhibition by a set of 12 active site-directed, competitive inhibitors revealed a large variation in the potency among the mammalian sPLA2s, with Me-Indoxam being the most generally potent sPLA2 inhibitor.27a

A structure-guided design was employed in a search for potent and selective inhibitors of mammalian sPLA2s. Although no compounds were found to be highly specific for a single human or mouse sPLA2, combinations of Me-Indoxam analogues were discovered that could be used to distinguish the action of various sPLA2s in cellular events.162
Using the X-ray structure of human GX sPLA2, the first potent inhibitor of this enzyme was developed.163 In addition, a series of inhibitors of sPLA2s based on substituted indoles, 6,7-benzoindoles, and indolizines derived from LY315920 were reported.164 Compound 29a was found to be selectively potent against hGX over all other human and mouse sPLA2 enzymes, while the substituted 6,7-benzoindole 29b inhibited nearly all human and mouse sPLA2s in the low nanomolar range.

Most recently, molecular docking and 3D Quantitative Structure-Activity Relationship Comparative Molecular Field Analysis (3D-QSAR CoMFA) studies on indole inhibitors of GIIA sPLA2 led to a model which was used for the design and evaluation of new compounds.165
A series of bis-indole compounds were designed and synthesized on the basis of the enzyme structure of human nonpancreatic sPLA2. Their inhibition activities were improved compared to that of the monofunctional protocompound. The potent compound 30 (IC50 24 nM) revealed that cooperative binding interactions between the two enzyme molecules also contributed to the stability of the ternary complex.166

Recently, Anthera Pharmaceuticals disclosed the sPLA2 inhibitors Varespladib (A-001, previously known as LY315920) and Varespladib methyl (A-002, previously known as LY333013) for the treatment of cardiovascular diseases.167 A-002 was shown to lower levels of GIIAs PLA2 by >90%, LDL-C by 12% to 18% and high-sensitivity CRP by 20% to 40% in stable CHD patients.168 A-002 acts synergistically with pravastatin to decrease atherosclerosis in the heart and proximal aorta of apoE−/− mice, possibly through decreased levels of systemic inflammation or decreased lipid levels.169 The FRANCIS (Fewer Recurrent Acute Coronary Events With Near-Term Cardiovascular Inflammation Suppression, http://clinicaltrials.gov/, Identifier: NCT00743925) study demonstrated that treatment with Varespladib methyl reduced concentrations of LDL-C, hsCRP and sPLA2 in ACS patients treated with evidence-based therapies inclusive of high-dose atorvastatin.170 Enrollment has commenced in the Phase 3 clinical study named VISTA-16 (Vascular Inflammation Suppression to Treat Acute Coronary Syndrome for 16 Weeks, http://clinicaltrials.gov/, Identifier: NCT01130246). The primary objective of this study is to determine whether 16 weeks of treatment with A-002 plus atorvastatin and standard of care is superior to placebo plus atorvastatin and standard of care for reducing the hazard of the first occurrence of the combined endpoint of cardiovascular death, non-fatal myocardial infarction, non-fatal stroke, or documented unstable angina with objective evidence of ischemia requiring hospitalization.
2.4.6 Oxadiazolones
A series of 4-alkoxybenzamidines was synthesized and their inhibitory potency against sPLA2 was evaluated.171 4-Tetradecyloxybenzamidine (31a, PMS815) was shown to exert an anti-inflammatory effect in vivo on the carrageenan-induced rat paw edema. Starting from PMS815, a series of oxadiazolones was synthesized and studied.172 The leading compound 31b (PMS1062) exhibited a micromolar IC50 towards three GII PLA2s, while inactive towards four GI and one GIII enzymes in two in vitro enzymatic assay conditions.

In a continuation, glycerol-containing derivatives of PMS1062 were synthesized.173 Among them, compound 32 was as potent as Me-Indoxam for hGIIA PLA2.

Replacement of the long chains by substituted piperazine derivatives led to inhibitors 33.174 Compound 33a inhibited hGIIA sPLA2 and in a carrageenan-induced edema test in rats showed to be as potent as indomethacin (Table 13).
Table 13.
Group IB and Group IIA PLA2 Inhibition by Oxadiazolones In Vitro and of Mouse Ear Edema
| Compound | IC50 (µM) | Ear Edema | |
|---|---|---|---|
| pGIB PLA2 | hGIIA PLA2 | ||
| 31b | >100 | 4.0 ± 0.9 | |
| 32 | >100 | 0.28 ± 0.02 | |
| 33a | 9 | 47.83 ± 12.34 (1.0 mg/ear) | |
| 33b | 0.1 | ||
| 34a | 7.41 | 0.03 | |
| 34b | >50 | 0.05 | |
| Indomethacin | 43.11 ± 8.79 (0.5 mg/ear) | ||

Most recently, further SAR towards the change of the rigidity of the piperazine region produced the more potent inhibitors 34 (Table 13).175

2.4.7 In Silico-Guided Identification of Inhibitors
A collection of 2150 druggable active sites from the Protein Data Bank was screened by high-throughput docking to identify putative targets for five representative molecules of a combinatorial library sharing a 1,3,5-triazepan-2,6-dione scaffold.176 Out of the five proposed proteins, sPLA2 was shown to be a true target for a panel of 1,3,5-triazepan-2,6-diones which exhibited micromolar affinities toward two human sPLA2 members (Table 14).
Table 14.
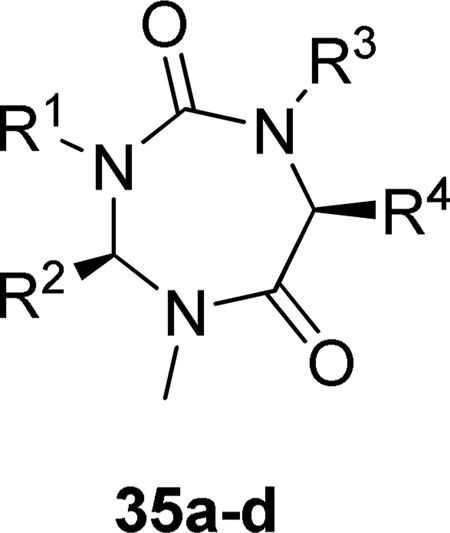
Group V and Group X PLA2 Inhibition by 1,3,5-Triazepan-2,6-diones
 | ||||||
|---|---|---|---|---|---|---|
| Compound | R1 | R2 | R3 | R4 | IC50 (µM) | |
| hGV PLA2 |
hGX PLA2 |
|||||
| 35a | CH2CH=CHCOOH | H | H | CH2C6H5 | 12 | 10 |
| 35b | CH2COOC(CH3)3 | H | H | CH(CH3)2 | 12 | 11 |
| 35c | H | CH2C6H5 | H | CH2C6H5 | 11 | 13 |
| 35d | CH2COOC(CH3)3 | H | CH2COOC(CH3)3 | CH2C6H5 | 11 | 11 |
2.4.8 Aptamers and Peptides
A family of sequence-related 2’-aminopyrimidine, 2’-hydroxylpurine aptamers, developed by oligonucleotide-based combinatorial chemistry, SELEX (systematic evolution of ligand by exponential enrichment) technology, was found to bind human nonpancreatic sPLA2 with nanomolar affinities and inhibit enzymatic activity.177
A number of synthetic peptides were designed and screened for sPLA2 inhibition.178 The linear peptide 36 (PIP-18) inhibited the recombinant human synovial sPLA2 activity with an IC50 of 1.19 µM. The peptide interfered with the function of sPLA2, but it also appeared to inhibit mRNA expression of sPLA2 and various MMPs in IL-1β-stimulated RA synovial fibroblast (RASF) cultures and thereby the production of the corresponding proteins (>80% inhibition).

2.4.9 Natural Products

In the mid 1980s, the natural product manoalide 37 was reported to be the first inhibitor of cobra venom PLA2.179 Manoalide is a sesterpene, which was isolated in the early 1980s from the sponge Luffariella variabilis and it was found to have anti-inflammatory activity in vivo. Manoalide inhibits also bee venom PLA2180 and human synovial fluid PLA2.181 Mechanistic studies on manoalide and analogues182 revealed that two specific lysine residues are responsible for the inhibition of the enzyme.183,184 Manoalide was licensed to Allergan Pharmaceuticals and reached Phase II clinical trials as a topical antipsoriatic, its development was, however, discontinued due to formulation problems.185
A number of sesterpenes of marine origin that contain the γ-hydroxybutenolide moiety have been studied for their anti-inflammatory activity and inhibition of PLA2. Petrosaspongiolide M (38) displayed a potent inhibitory activity toward GII and GIII sPLA2 and the molecular basis of the inhibition of this product as well as petrosaspongiolides M-R was studied.186 More recently, the binding mode of petrosaspongiolide to human GIIA PLA2 was analyzed in detail.187 Scalaradial (39) is another marine metabolite, which inhibits GII and GIII PLA2 and presents in vivo anti-inflammatory activity.188

Thielocin B3 is a very potent naturally occurring inhibitor of human nonpancreatic sPLA2 (GII). Structure-activity relationship studies led to a number of analogues with potency comparable to the parent natural product.189,190

YM-26567 (41), a natural product isolated from the fruit of Horsfieldia amygdaline, is a micromolar inhibitor of rabbit platelet sPLA2.191 Further studies led to YM-26734 (42) which has increased potency against the enzyme (IC50 85 nM)192 and to simplified analogues.193

Flavonoids are widely produced polyphenolic plant secondary metabolites. Some flavonoids have demonstrated inhibition of PLA2194 and the mechanism of inhibition of human sPLA2 has been investigated.195
2.4.10 Summary Status of sPLA2 Inhibitors
As presented above, a wide variety of structurally different inhibitors has been studied for their inhibition of several sPLA2s. Starting in the mid 1980s, before crystal structures were widely available, the first studied inhibitors of sPLA2 were substrate analogues and marine natural products. The first potent and specific (when compared to PLC, PLD and PLA1) dicarboxylic acid inhibitor of sPLA2 was presented by Bristol-Myers Squibb in 1992. It reached Phase II clinical trials for the topical treatment of psoriasis, but since it could not reach the inner layer of the skin, the studies were discontinued. Sulfonamide and various amide inhibitors developed over the years proved useful tools for mechanistic studies. Indole inhibitors have attracted a great deal of attention as drug candidates and they constitute the most comprehensively studied class of sPLA2 inhibitors. Lilly Research Laboratory presented a highly potent sPLA2 inhibitor (LY315920) which was developed via molecular modeling techniques combined with chemical modification of a lead compound. This inhibitor, which is selective for GIIA sPLA2, reached Phase II studies as a treatment for severe sepsis, but the trials were discontinued when results did not meet expectations. One of the reasons that indole inhibitors may not have shown the expected efficacy in clinical studies may be that they are cell impermeable and therefore incapable of blocking intercellular effects. Even though sPLA2 is a secreted enzyme, inhibitors that are both potent and cell permeable may show greater effects clinically. Furthermore, similar properties in inhibitors may affect tissue distribution and permeability as well but this has not been extensively investigated.
A prodrug of the Lilly inhibitor was also used to study the possible role of GII sPLA2 in asthma. At the same time, a structurally similar inhibitor was introduced by Shionogi, namely Me-Indoxam. A few years later, when the full set of human sPLA2s was expressed, indole-type inhibitors were developed that could somewhat distinguish the action of various sPLA2s in cellular events, even though no compound was highly specific for a single sPLA2. Currently, the previously mentioned LY315920, also called Varespladib or A-001, together with Varespladib methyl or A-002 are in Phase III trials by Anthera Pharmaceuticals to treat cardiovascular diseases. In the quest for sPLA2 inhibitors, one should keep in mind that while selective sPLA2 inhibitors can help us study the biological activity of specific sPLA2s in vitro and in vivo, they are also essential for helping us to distinguish the activities of the different groups of sPLA2.
3. Cytosolic Phospholipase A2 [Group IV cPLA2]
3.1 Groups, Subgroups, Specificity and Mechanism
The first cytosolic PLA2, now attributed to GIVA PLA2 (or cPLA2α), was reported as an activity by Christina Leslie and Ruth Kramer in human neutrophils196 and platelets197 respectively in 1986. The enzyme was purified, sequenced and cloned by James Clark at the Genetics Institute and Ruth Kramer at Lilly Research Laboratories in 1991.5–6 GIVB PLA2 (cPLA2β)198 and GIVC PLA2 (cPLA2γ)198a,199 were subsequently reported in 1998-99 by Lilly Research Laboratories and Genetics Institute investigators. GIVD (cPLA2δ), GIVE (cPLA2ε) and GIVF (cPLA2ζ) PLA2 were identified in mice by the Shimizu’s group in Japan in 2004–2005.200 Currently, the group IV PLA2 is comprised of these six known phospholipases.14 A summary of the characteristics of each member of the GIV PLA2 family (Table 15) and a schematic presentation of the sequences (Figure 6) provide an overview of this PLA2 group.
Table 15.
Group GIV Cytosolic Phospholipase A2s (cPLA2)
| Subgroup | Initial/common sources |
Residues /Molecular Mass |
Domain | Activation Factor |
Substrate | Activity | Post Translational Modification |
Human chromosome |
Swiss- Prot |
|---|---|---|---|---|---|---|---|---|---|
| GIVA (cPLA2α) |
Human macrophage–like U937 cells/ platelets/RAW 264.7/rat kidney, Ubiquitous |
749/ 85 Kda |
C2 α/ β hydrolase Cap |
Ca2+ PIP2 C1P Phosphor y-lation |
PC, PE, PI High sn- 2AA specificity |
PLA2, PLA1, Lyso-PLA transacylase |
Phosphorylation | 1q25 | P47712 |
| GIVB (cPLA2β) |
Human pancreas/ liver/heart/brain, Ubiquitous |
1012 /100− 114 Kda |
JmjC insert C2 α/ β hydrolase |
Ca2+ | PC, PE No sn-2 specificity |
PLA1, PLA2, Lyso-PLA transacylase |
15q11.2 - q21.3 |
AAD321 35 |
|
| GIVC (cPLA2γ) |
Human heart /skeletal muscle |
541/ 61 Kda |
α/β hydrolase |
PC Low sn-2 AA specificity |
PLA1, PLA2 Lyso-PLA |
Farnesylation | 19q13.3 | AAC328 23 |
|
| GIVD (cPLA2δ) |
Murine placenta | 818/ 91 Kda |
C2 α/ β hydrolase |
Ca2+ | PC, PE | PLA1, PLA2 Lyso-PLA |
15q15.1 |
Q86XP0 .2 |
|
| GIVE (cPLA2ε) |
Murine heart /skeletal muscle/testis/thyroid |
838/ 95 Kda |
C2 α/ β hydrolase |
Ca2+ | PC, PE | PLA1,PLA2 Lyso-PLA |
15q15.1 |
Q3MJ16 .2 |
|
| GIVF (cPLA2ζ) |
Murine thyroid /stomach |
849/ 95 Kda |
C2 α/ β hydrolase |
Ca2+ | PC, PE | PLA1,PLA2 Lyso-PLA |
15q15.1 |
Q68DD2 .2 |
Figure 6.

Schematic presentation of Group IV cPLA2s. Calcium binding loops (CBL), active sites (red squares), and the 242 residue- and 120 residue-inserts of GIVB PLA2 are shown.
3.1.1 Group IVA Phospholipase A2 (cPLA2α)
After the identification of cytosolic PLA2 in neutrophils and platelets by cell-free assay,196–197 the human GIVA PLA2 was cloned and sequenced in 1991 from U937 cells.5–6 The GIVA PLA2 gene has been mapped to human chromosome 1q25 and is ubiquitously expressed in most human tissues (Table 15).201 This enzyme contains 749 amino acids and was shown to be a 85 kDa protein using SDS-PAGE.5–6 GIVA PLA2 consists of an N-terminal C2 domain and a C-terminal catalytic domain (Figure 6),5,202 and the structural details of this enzyme can be seen in the X-ray crystal structure produced by Dessen et al in 1999.202a GIVA PLA2 is regulated by intracellular calcium, and calcium binding to the C2 domain of GIVA PLA2 can activate the enzyme resulting in the localization of the enzyme to the phospholipid membrane.5,203 After localization to the membrane, the catalytic domain of GIVA PLA2 utilizes an active site dyad Ser-228/Asp-549, located in the α/β hydrolase domain, to catalyze the hydrolysis.202a,204 In addition, MAP kinase phosphorylation205 and the lipid mediators ceramide-1-phosphate (C1P)206 and phosphatidylinositol 4,5-bis phosphate (PIP2)207 were shown to further increase GIVA PLA2 activity.
Most phospholipids, such as phosphatidylcholine (PC), phosphatidylethanoamine (PE) and phosphatidylinositol (PI), are substrates of GIVA PLA2.208 Depending on the experimental setup, the preference for these phospholipids may vary slightly. But, PC is a relatively good substrate and is commonly used to determine the enzymatic activity of GIVA PLA2 since its discovery.5–6 GIVA PLA2 hydrolysis of phospholipid substrates has high substrate specificity for arachidonic acid at the sn-2 position.5,18a In addition to phospholipase activity, GIVA PLA2 also possesses lysophospholipase activity and transacylase activity.204b In contrast to the calcium dependent phospholipase activity, the lysophospholipase activity toward micelle substrates was found to be calcium-independent.204b Recently, a comprehensive interfacial kinetic study compared the activities of all six GIV PLA2 isoforms and showed that they all exhibit phospholipase A1/A2 and lysophospholipase activities.209
3.1.2 Group IVB Phospholipase A2 (cPLA2β)
The full length human GIVB PLA2 was initially cloned in 1999 and the recombinant protein was then expressed in Sf-9 insect cells.198 The GIVB PLA2 gene is encoded by chromosome 15q11.2–q21.3 and the mRNA is expressed ubiquitously, but found at especially high levels in the pancreas and cerebellum (Table 15).198 The enzyme contains 1012 amino acids and its size is reported at 100–114 kDa using SDS-PAGE (Figure 6).198 GIVB PLA2 contains a C2 domain and a catalytic domain that are similar to those in GIVA PLA2, with the primary sequences sharing 30% identical residues.18a The major difference is that the GIVB PLA2 has two insertions (before and after the C2 domain). The first is a 242-residue insertion at the N-terminus and the second is a 120-residue insertion between the C2 domain and the catalytic domain (Figure 6). The 242-residue insertion is part of a JmjC domain, which is often found to regulate chromatin stability in nuclear proteins.210 Interestingly, the JmjC domain is in the gene upstream from that of GIVB PLA2. The α/β hydrolase domain is highly homologous to GIVA PLA2 and the active site dyad Ser-538 and Asp-566 is conserved. When we ran a multiple sequence alignment of all six GIV PLA2s, the alignment shows that GIVB PLA2 does not have the phosphorylation sites, C1P binding sites, or PIP2 binding sites, and has a significantly different cap and lid region (Figure 6). Three GIVB PLA2 splice variants, b1, b2 and b3, have been observed.211 The alternative splicing on the catalytic domain results in the splice variants b2 and b3, which are shorter than the original b1 variant. GIVB PLA2 preserves the conserved calcium binding sites on the C2 domain in the sequence alignment and thus demonstrates calcium-dependent PLA2 activity. Compared to GIVA PLA2, its phospholipase activity is low and it has low substrate specificity for sn-2 fatty acids.14,212 It also has low lysophospholipase A1/A2 and transacylase activities.198b,204b,213
3.1.3 GIVC PLA2 (cPLA2γ)
The human GIVC PLA2 gene, located at chromosome 19q13.3, was cloned in 1998 (Table 15).198a,199 Its transcript is expressed highly in the heart and skeletal muscle.198a,199 The GIVC PLA2 is a 61 kDa protein and has 541 amino acid residues, showing 30% homology to the catalytic domain of GIVA PLA2 (Figure 6). The active site dyad in the catalytic domain is conserved. Because this enzyme does not contain a C2 domain, GIVC PLA2 has no calcium-dependent phospholipase activities.198a,199 Neither the C1P nor the PIP2 binding sites nor the phosphorylation sites are observed. Cys-538 has shown to be farnesylated in insect cells.214 GIVC PLA2 is also able to hydrolyze both sn-1 and sn-2 acyl chains from a phosphatidylcholine substrate with PLA2 and PLA1 activity.198b,199 It has low sn-2 specificity for arachidonic acid, less than the GIVA PLA2.199
3.1.4 GIVD (cPLA2δ), GIVE (cPLA2ε) and GIVF (cPLA2ζ)
The human GIVD PLA2 gene was found to be differentially expressed between normal and psoriatic skin, and was then cloned in 2004.200b The Shimizu laboratory later performed the cloning of murine GIVD, GIVE and GIVF in 2005.200a All three genes form a cluster with GIVB PLA2 at human chromosome 15q15.1 (Table 15). The murine transcripts of all three genes are tissue specific. GIVD PLA2 was detected in the placenta, GIVE PLA2 was detected in the thyroid, heart and skeletal muscles, and GIVF PLA2 was detected in the thyroid.200a The sequence lengths of GIVD, E and F PLA2 are 818 amino acids/91 kDa, 838 amino acids/95 kDa and 849 amino acids/95 kDa respectively (Figure 6). All three proteins contain a conserved C2 domain and a phospholipase domain. The calcium binding sites are conserved in the C2 domain and the active site dyad is also conserved in the catalytic domain, but no C1P, PIP2 or phosphorylation sites are observed. All GIVD, E and F PLA2s exhibit calcium-dependent PLA2 activities.200a GIVF PLA2 has higher activity toward phosphatidylethanolamine (PE) than phosphatidylcholine (PC), which is the opposite of GIVA PLA2, suggesting preference for charged substrates. A comparison of the three enzymes shows GIVF PLA2 has higher PLA2 activity than GIVD PLA2 and GIVE PLA2. GIVF PLA2 shows high phospholipase A2 and lysophospholipase activities.215 In response to ionomycin, GIVF PLA2 translocates to ruffles and dynamic vesicular structures, which is different from how GIVA PLA2 translocates to the Golgi and endoplasmic reticulum.215 The biological functions of these enzymes have not been well studied to date.
3.2 Structural Characteristics and Activation Mechanisms
The six members of GIV PLA2 show some structural similarities in the C2 and the α/β hydrolase domains, but the differences among them are significant. Because our understanding of GIVB, C, D, E and F PLA2 are still limited, we utilize the well-studied GIVA PLA2 enzyme to represent the structural characteristics and activation mechanisms of the entire GIV PLA2 type in this section. The early work showed that GIVA PLA2 is activated by calcium, and determined that the C2 domain is responsible for the calcium binding and GIVA PLA2 translocation.202b,203 The crystal structure and the NMR structure of the C2 domains were reported in 1998, and showed calcium binding to the C2 domain.202b,216 Dessen et al. (1999) solved the crystal structure of the whole GIVA PLA2202a, including the C2 domain which is linked to a catalytic domain (Figure 7). The C2 domain structure of the intact enzyme is similar to the C2 domain structure alone, showing two calcium ions binding to the anion hole at the tip of the C2 domain.
Figure 7.

Features of the Group IVA cPLA2 crystal structure. The C2 domain is in orange, the α/β hydrolase domain is in green, the cap region is yellow, and the lid is in pink. The C1P binding site is in magenta, the PIP2 binding site is in blue and the active site residues are in red. Adapted from Ref.218
The catalytic domain is tethered to the C2 domain by a single peptide strand consisting of residues 139–143. The catalytic domain is composed of an α/β hydrolase core and a cap region. The α/β hydrolase core folds similarly to many lipases, but the sequence similarity is low.202a The active site is located on the top of the α/β hydrolase core, which is also similar to other lipases. The novel cap region (residues 370–548) is found only in GIVA PLA2, and is not conserved in other members of the GIV PLA2 family. The homologies among GIVA, B and C PLA2 in the cap region are very low. The homologies in the cap region among GIVD, E and F PLA2 are higher than the rest of GIV PLA2s. The cap region of GIVA PLA2 sits on top of the α/β hydrolase core and so the active site dyad is buried within a tunnel. The cap region seems to be very flexible, so that a wide range of residues cannot provide enough electron density for crystallization.
Within the cap region, a segment of peptide (415–432) has been crystallized, characteristically surrounded by low-electron density residues. This segment is right on top of the active site and is defined as a lid. The presence of a lid region blocks the phospholipid substrate from approaching the active site. The region of the lid that faces away from the enzymes contains three Glu residues, Glu-418, Glu-419, and Glu-420. In addition, one of the hinges that hold the lid in place also contains four negatively charged groups, i.e., Asp-436, Asp-438, Asp-39, and Asp-440. It appears that they interact with the lipid interface and that the presence or absence of negatively-charged lipids in the surface affects binding due to their interactions with these groups. At the same time the negative interactions may facilitate the movement of the lid away from the catalytic site.
This crystal structure defined the catalysis pocket, which is composed of Ser-228 in the consensus sequence GXSXS and Asp-549. These two residues act as an active site dyad (Figure 7) for both PLA2 and lysophospholipase activities. Arg-200 was shown to be critical for phospholipase activity204a and is in the proximity of the pocket. It may bind to the charged headgroups of the phospholipid substrate and may therefore be responsible for substrate affinity and the release of lysophsopholipid.202a,217 Although the determination of the crystal structure has led to an understanding of the GIVA PLA2 catalytic activity, due to the lack of electron density in some regions of the crystal structure, the C2 domain and the catalytic domain do not show any contact or interaction. This disparity with the deuterium exhchange results suggests that the unstructured linker region is the only region that could bring the two domains together. The hydrogen/deuterium exchange experiments on the intact GIVA PLA2 enzyme allowed the determination of the rates of exchange in regions that lack electron density. Additionally, the exchange results of the intact enzyme were compared with the C2 domain deletion mutant and the catalytic domain alone. The hydrogen/deuterium exchange experiments indicated that there are extensive intradomain interactions between the C2 domain and the catalytic domain.218 The cap region shows a significant increase in deuterium exchange in the C2 domain deletion mutant, suggesting the intradomain interaction may play a role in the enzymatic activity.
The activation mechanism of this enzyme includes many steps. The enzyme is first recruited to the membrane by a calcium-dependent translocation of the C2 domain. The lipid second messengers ceramide-1-phosphate219 and phosphatidylinositol (4, 5) bisphosphate220 can further activate the enzyme, at least in vitro. The phosphorylation status has also been shown to regulate the enzymatic activities.205,221
3.2.1 Calcium Activation
In response to extracellular stimuli, the intracellular Ca2+ concentration increases, and the GIVA PLA2 then translocates from the cytosol to the perinuclear membrane region.203 Calcium binding to the C2 domain is crucial for membrane localization, but is not directly involved in the catalytic activity of the GIVA PLA2.202a The GIVA PLA2 C2 domain has three calcium binding loops, CBL1, CBL2, and CBL3, which form an anion hole on the tip of the C2 domain. Two calcium ions coordinate with the calcium-binding loops, CBL1, CBL2, and CBL3, in the C2 domain, as illustrated in Figure 7.202,216 These two Ca2+ ions neutralize the negative charge in the anion hole to facilitate the C2 domain's hydrophobic interaction with phospholipid membranes.202,216 We utilized hydrogen/deuterium (H/D) experiments to show that calcium binds to a low H/D exchange region, indicating that the anion hole is a stable region before calcium binding.218 Calcium binding also causes a conformational change in the C2 domain to stabilize the C2 domain structure, as shown by a decreased rate of H/D exchange.218 This rigidified conformation of the C2 domain is crucial for proper orientation of the stabilized C2 domain binding to the phospholipid membrane, leading to enzyme interfacial activation.
The calcium-bound C2 domain is also important for phospholipid hydrolysis by GIVA PLA2. The catalytic domain of GIVA PLA2 alone does not have phospholipase activity.218,220 We have shown that the intradomain interactions between the C2 domain and the catalytic domain stabilize both domains218. This intradomain interaction changed our understanding of the relative position between the C2 domain and the catalytic domain as illustrated in the crystal structure.218 Membrane binding to the calcium-activated GIVA PLA2 has shown an increased H/D exchange on the cap region, including the part of the region involved in the intradomain interaction.222 Active site targeting inhibitors also shows changes in the H/D exchange rate in the same cap region.217 While the calcium binding effects are mainly seen in the C2 domain, calcium binding may extend its effects and have implications for the catalytic domain as well through the contact residues in the cap region.
3.2.2 PIP2/C1P Activation
GIVA PLA2 is also activated by binding to the lipid second messenger phosphatidylinositol-4,5-bis phosphate (PIP2) in a Ca2+ independent manner.207b,220 An increase of intracellular PIP2 levels caused the increase of arachidonic acid release by the calcium-independent activation of GIVA PLA2.223 GIVA PLA2 has a high binding affinity and specificity toward PIP2.207b The lysine cluster, Lys-488, Lys-541, Lys-543, and Lys-544, have been identified as PIP2 binding sites and critical for PIP2-mediated GIVA PLA2 activation220,224 and translocation of the enzyme to phagosomes.225 The lysine cluster is located in the intradomain contact region (Figure 7).218 PIP2 activation requires the presence of the C2 domain, indicating that the orientation of the C2 domain and the catalytic domain is critical for phospholipase activity.220
Ceramide 1-phosphate (C1P) is a phosphorylated bioactive sphingolipid involved in inflammation.226 C1P is also a lipid second messenger involved in cell signaling.227 A C1P analog was shown to inhibit GIVA PLA2 and increase cell toxicity.228 Ceramide kinase, which phosphorylates cermide to synthesize C1P, was also found to be an activator of GIVA PLA2.226,229 Ceramide kinase is also involved in the biogenesis of lipid droplets through GIVA PLA2 activation.230 C1P binds directly to the C2 domain of GIVA PLA2 at Arg-57, Lys-58, and Arg-59 (Figure 7).219 It is also a required bioactive lipid for GIVA PLA2 to translocate to intracellular membranes in response to inflammatory agonists.231 Unlike PIP2 activation, the C1P activation mechanism is Ca2+ dependent. Interestingly, recent research showed that C1P activates GIVA PLA2 by decreasing the dissociation constant to increase the residence time on membranes, while PIP2 activates GIVA PLA2 by increasing membrane penetration for higher catalytic efficiency.232 More recently, GIVA PLA2 was also shown to be inhibited by sphingomyelin at the biomembrane interface.233
3.2.3 Phosphorylation
GIVA PLA2 phosphorylation regulates the enzymatic activity at the phospholipid membrane and its cellular functions. GIVA PLA2 was initially found to be phosphorylated at Ser-505 and activated by p42-MAP kinase and PKC in 1993.205,234 Ser-515, and Ser-727 were also found to be phosphorylated by mitogen activated protein kinases (MAPKs), calmodulin kinase II (CamKII), and mitogen activated protein kinase interacting kinase (MNK1).221 The phosphorylation sites were later reported at Ser-505, Ser-437, Ser-454, Ser-515, and Ser-727 in Sf9 cells.235 Since then, the level of phosphorylation, especially the level of Ser-505 phosphorylation by MAP kinase, has been implicated in the activation of GIVA PLA2 in response to various cellular stimuli.203b,205,235–236
The phosphorylation sites are all located in the low electron density areas in the crystal structure.202a Among them, Ser-437, Ser-454, Ser-505 and Ser-515 are in the cap region, indicating the critical role of phosphorylation in affecting the conformation of the cap or the flexibility around the phosphorylated region. DXMS studies have revealed these regions are flexible and significantly increase the deuteration level after activation.218,222 When GIVA PLA2 is already activated by calcium, Ser-505 phosphorylation increases activity by 30–60%.237 In low (2.5 µM) Ca2+ concentration, membrane binding showed a 60-fold increase in membrane affinity.238 Interestingly, this effect was also observed in the PIP2 activation mechanism.220 Both effects indicate a conformational change in the cap region to facilitate interfacial activation. The heterotetramer (A2t) of p11 and annexin A2 has been shown to bind to GIVA PLA2 and inhibit the membrane binding. Phosphorylation of Ser-727 has been shown to disrupt the binding of GIVA PLA2 to heterotetramer (A2t) to activate GIVA PLA2.239 To date, Ser-505, Ser-515 and Ser-727 are considered as the three phosphorylation sites involved in cellular functions.
3.2.4 Membrane Interaction
Interfacial activation, showing higher activity against large phospholipid aggregates (or a cellular phospholipid membrane), is required for GIVA PLA2 activity.240 For the enzyme to be active, it must be sequestered to a phospholipid interface. As we have described, the binding of the GIVA PLA2 to the membrane is mediated through three mechanisms: Ca2+-mediated translocation, binding of secondary lipid messengers, and phosphorylation. Each of these mechanisms increases GIVA PLA2 catalytic efficiency in a different way. Calcium binding assists in membrane penetration of the C2 domain. We found that the presence of the phospholipid membrane decreases the H/D exchange rate on CBL1 and CBL3 in the C2 domain, which are composed of amino acids 35–39, and 96–98 (Figure 8).222
Figure 8.

Model of the lipid-binding surface of Group IVA cPLA2. Residues interacting with the lipid membrane are based on the experiments of hydrogen/deuterium exchange in the presence of phospholipid vesicles. Adapted from Burke et al.222
The C2 domain anchors GIVA PLA2 to the membrane and causes the catalytic domain to approach the membrane.202b,203 The catalytic sites in GIVA PLA2 must maintain the correct orientation toward the membrane, although calcium activated, C1P/PIP2 bound, or phosphorylated GIVA PLA2 may have slightly different phospholipid interactions. The intradomain interaction may also be affected by the conformational change in the C2 domain, phosphorylation, or C1P/PIP2 binding, and further changes the membrane interaction of the catalytic domain. Two helices in the regions 268–279 and 466–470 in the cap region have been shown to interact with the phospholipid membrane, in addition to the C2 domain (Figure 8).222
Once GIVA PLA2 has localized to the membrane, the active site is in the correct orientation to allow substrate molecules to enter the active site. We have mentioned that within the cap region, there is a lid region that prevents the phospholipid substrate from binding to the active site.202a A substrate mimicking the inhibitor MAFP was used to examine the substrate binding effects. Recent work using a lipid substrate and a covalent inhibitor bound to the active site has indeed shown a conformational change of the lid region in the presence of the substrate.222 Further experiments using the 2-oxoamide inhibitor (see Section 3.4.7) show that the head group is binding to the Arg-200.217 Based on the model developed, the phospholipid molecule must interact with the residues near the cap region and be extracted from the membrane interface.217–218,222 The molecule will then go into the tunnel or push away the lid and bind to the active sites.
3.3 Biological Functions and Disease Implications
3.3.1 Biological Functions
3.3.1.1 Phospholipid Hydrolysis
GIVA PLA2 specifically favors hydrolyzing the phospholipid membrane at the sn-2 position of unsaturated arachidonic acid in response to cellular stimuli.18a,241 Free arachidonic acid may be metabolized along the cyclooxygenase (COX) pathway or lipoxygenase pathway (LOX). COX-1, COX-2 and other terminal synthases convert arachidonic acid and generate prostaglandins and thromboxanes.242 On the other hand, arachidonic acid can be oxidized by 5-lipoxygenase or 12/15-lipoxygenase and other downstream enzymes leukotriene A4 hydrolase and LTC4 synthase to produce leukotrienes and lipoxins.242a,243 The eicosanoids are important in intracellular immunity244 and implicated in several diseases, including thrombosis, cancer, atherosclerosis, asthma, arthritis and rhinitis.242a,245
GIVA PLA2 deficiency in patients and knockout mouse models have shown decreased eicosanoid production and an easing in the effects of inflammatory diseases.241,246 Because arachidonic acid is the precursor of these eicosanoids, it has been proposed that GIVA PLA2 plays a major role in inflammatory diseases. GIVA PLA2 is now considered a central enzyme for mediating eicosanoid production. GIVA PLA2 has also been implicated in apoptosis triggered by the hydrolysis product arachidonic acid and its metabolites.247 The inhibition of arachidonyl-CoA transferase results in accumulation of arachidonic acid, which triggers apoptosis.248 Arachidonic acid metabolites, including 12-EET and 19-HETE, have been shown to trigger apoptosis.245a,249
Lysophospholipids represent another product of phospholipid hydrolysis by GIVA PLA2. This biological function is not specific to GIVA PLA2, since the lysophospholipid can be potentially generated by all PLA2 members. But it has been suggested that lysophospholipids act as second messengers for GPCR signaling and are strongly associated with cancer.250
3.3.1.2 Golgi Function Regulation
The Golgi complex and the trans-Golgi network are cellular organelles involved in the trafficking of cell lipids and proteins as part of the endocytic and biosynthetic pathways.251 Lipids are not only the main component of the cell membrane, but are also implicated in the regulation of membrane-protein trafficking, vesicular fusion, and signaling.242a,251 When GIVA PLA2 is localized in the Golgi complex in epithelial cells, it functions as a Golgi regulatory enzyme instead of inducing cell proliferation.252 GIVA PLA2 also regulates the junction protein transports from the Golgi complex to endothelial cell contacts.253 It was recently shown that GIVA PLA2 associates with the Golgi complex and is involved in regulation of the Golgi complex structure, tubule formation and intra-Golgi transport.18c,254
3.3.1.3 Regulation of NADPH Oxidase
GIVA PLA2 regulates the stimulation of NADPH oxidase to produce superoxides.255 Correlations have been demonstrated between PGE2 production and GIVA PLA2 translocation to the nuclear membranes and between superoxide production and GIVA PLA2 translocation to the plasma membranes.255–256 While GIVA PLA2 binds to PC-enriched nuclear membranes in a calcium-dependent process using its C2 domain, which is correlated with its phospholipid-binding specificity, it is anchored to the plasma membranes by the assembled NADPH oxidase which serves as an adaptor protein for GIVA PLA2.255–257 Under oxidative stress, down-regulation of GIVA PLA2 can suppress the NADPH oxidasemediated reactive oxygen species production in astrocytes.258 GIVA PLA2 is targeted to the p47phox-PX domain of the assembled NADPH oxidase via a novel binding site in its C2 domain.259
3.3.2 Disease Implications
3.3.2.1 Human Mutation and Knockout Mice
A patient with two heterozygous single base pair mutations (Ser111Pro; Arg485His) in the coding regions of his GIVA PLA2 gene has been identified.246b,260 The Arg485His mutation is located in a critical region, and is involved in intradomain interaction and membrane interaction.218,222 Because PLA2 activity in platelets was not observed in the patient, the mutation sites must be critical for GIVA PLA2 activity. The patient showed small intestinal ulcerations without COX deficiency. Inherited human GIVA PLA2 deficiency is associated with impaired eicosanoid biosynthesis.
Genetic knockout mice were developed in 1997 by two different groups.246a,261 The normal phenotype of GIVA PLA2-null mice suggested that this enzyme is not crucial for development.246a,261 However, when the mice were tested for various disease models, especially those involving inflammation, the signs of illness were much milder than wild-type mice and the mice demonstrated a great reduction in lipid mediator production. In particular, GIVA PLA2-null mice have been shown to be resistant to ischemia-reperfusion injury,262 anaphylactic responses,263 acute respiratory distress syndrome caused by acid or endotoxin,264 bleomycin-induced pulmonary fibrosis,265 collagen-induced autoimmune arthritis,266 experimental allergic encephalomyelitis,267 fatty liver damage,268 and autoimmune diabetes.269 Concerning normal physiology, the loss-of- GIVA PLA2 causes some defects to the renal concentrating function.270 The most notable defects caused by the loss-of- GIVA PLA2 are found in the female reproduction system.261 These female knockout mice showed a decreased level of eicosanoids and platelet-activating factor in peritoneal macrophages, gained less weight and had a smaller litter size. Ultimately, they failed to carry to term and deliver offspring. However, these effects could be prevented by injection of a progesterone-receptor antagonist during pregnancy.
3.3.2.2 Cancer
Alterations in the levels and functional activity of GIVA PLA2 have also been associated with cancer pathogenesis.245a GIVA PLA2 activation was shown to mediate estrogen-dependent breast cancer cell growth.271 Deletion of GIVA PLA2 in the APC716 mouse of the human FAP model showed a decrease in the number of and repressed the growth of polyps in the small intestine.272 GIVA PLA2 plays an important organ-specific role in modulating intestinal tumorigenesis. Administering azoxymethane (AOM) triggered the formation of colon tumors in studies of GIVA PLA2-null mice, showing significant increases in the number of tumors over wild type mice.273 GIVA PLA2 null mice also developed 43% less tumors than wild type mice after exposure to the lung carcinogen urethane.274 In analysis of adenocarcinoma tumors, GIVA PLA2 expression was detected in 18% of Barrett's oesophagus patients and is inversely associated with depth of tumor infiltration.275 Additionally, the GIVA PLA2 null mice also show reduction of tumor progression in the glioblastoma (GL261) and Lewis lung carcinoma tumor models.276
3.4 Chemical Inhibitors and Therapeutic Intervention
The most recent review on inhibitors of the four major PLA2 types focused on the recent patent literature.22f The GIVA PLA2 has been considered to be the PLA2 enzyme that plays the most central role in inflammatory diseases, thus numerous inhibitors have been reported.22c,d
3.4.1 Fatty Acid Trifluoromethyl Ketones and Tricarbonyls
The first inhibitor of GIVA PLA2 was the activated ketone, arachidonoyl trifluoromethyl ketone (43).277 AACOCF3 was shown to be a slow tight binding inhibitor of GIVA PLA2, 4 orders of magnitude more potent for this enzyme than for sPLA2. 19F and 13C-NMR experiments elucidated the structure of the GIVA PLA2•AACOCF3 complex and that the mole ratio of AACOCF3 with respect to the enzyme was 1:1.278 The concentration-dependent inhibition of the enzyme by 43 and the palmitoyl trifluoromethyl ketone (44) was also studied.279 The trifluoromethyl ketone analogues of γ-linolenic (45) and linoleic acid (46) were also found to be GIVA PLA2 inhibitors.280

A series of fatty acid trifluoromethyl ketones were analyzed with phospholipid vesicle-, detergent-phospholipid mixed micelle-, and natural membrane-based assays.281 With few exceptions, the relative inhibitor potencies measured with the three assays were similar (Table 16). Note that some trifluoromethylketones may also inhibit GVIA iPLA2 (see, Section 4.4.1).
Table 16.
Group IVA PLA2 Inhibition by Fatty Acid Trifluoromethyl Ketones
| R-COCF3 | XI(50) | ||
|---|---|---|---|
| DOPM/GLU | mixed-micelles | natural membrane | |
| 20:4 | 0.0050±0.001 | 0.036±0.006 | 0.05±0.02 |
| 11, 14, 17–20:3 | 0.0085±0.002 | 0.020±0.002 | 0.10±0.03 |
| 16:1 | 0.0083±0.001 | 0.010±0.003 | 0.07±0.02 |
| 16:0 | 0.025±0.002 | 0.022±0.006 | 0.20±0.03 |
AACOCF3 was also able to inhibit the lysophospholipase activity of GIVA PLA2 at a similar level to PLA2 activity.213b Fatty acid tricarbonyl derivatives, like compound 47, were found to be inhibitors of GIVA PLA2.279 Their potency was approximately the same as that of the corresponding trifluoromethyl ketones.
AACOCF3 has been used in various experiments in cells and in vivo; however, the results obtained with this inhibitor have to be viewed with some caution, because it is not a selective GIVA PLA2 inhibitor and it may inhibit other enzymes, for example cycloxygenases.282 AACOCF3 blocked the production of arachidonic acid and 12-hydroxyeicosatetraenoic acid (12-HETE) in platelets283 and in human monocytes causing a dose dependent inhibition of GIVA PLA2 activity and LDL lipid oxidation.284 Daily treatment of prion-infected cell lines with AACOCF3 for seven days prevented the accumulation of protease-resistant PrP (PrPres) indicating a pivotal role for GIVA PLA2 in prion disease.285
The role of GIVA PLA2 in allodynia has been studied. Intracerebroventricular injection of AACOCF3 significantly reduced responses to von Frey hair stimulation at 8 h and 1 day after facial carrageenan injection.286 Intrathecal administration of AACOCF3 dose-dependently prevented thermal hyperalgesia induced by intrapolar carrageenan as well as formalin-induced flinching.287 Using AACOCF3, Kalyvas and David disclosed that cPLA2 plays an important role in the pathogenesis of experimental autoimmune encephalomyelitis (EA), the animal model of multiple sclerosis.288 More recently, AACOCF3 has been used in a study reporting that GIVA PLA2 reduction ameliorates cognitive deficits in a mouse model of Alzheimer’s disease.289
3.4.2 Methyl Arachidonyl Fluorophosphonate
Methyl arachidonyl fluorophosphonate (48, MAFP) was reported to be a potent time-dependent irreversible inhibitor of GIVA PLA2 but not of the human sPLA2. The inactivation of the enzyme by some additional alkyl methyl fluorophosphonates was also studied.281 Like AACOCF3, intrathecal administration of MAFP dose-dependently prevented thermal hyperalgesia induced by intraplantar carrageenan as well as formalin-induced flinching.290

3.4.3 Trifluoromethyl Ketones
Bristol-Myers Squibb reported in several US patents a series of α- and β-substituted trifluoromethyl ketones,291 such as compounds 49 and 50, as GIVA PLA2 inhibitors for the treatment of inflammatory diseases. These inhibitors presented IC50 values in the 1–50 µM range.

BMS-229724 is a tight-binding inhibitor of GIVA PLA2 that acts at the lipid/water interface and possesses anti-inflammatory activity in skin inflammation models.292 In a UVB-induced skin erythema model in hairless guinea pigs, BMS-229724 was orally active.

3.4.4 Pyrrolidines
In the search for low molecular weight inhibitors of GIVA PLA2, Shionogi identified compounds 52 (IC50 1.5 µM) and 53 (IC50 1.7 µM) as lead candidates. When these two structures were combined and studied in SAR experiments, a series of even more potent inhibitors were identified.293 Compounds 54 and 55 were the most promising (Table 17). Compound 55 inhibited the arachidonic acid release in A23187 stimulated THP-1- cells with an IC50 value of 22 nM.293 Compound 54 inhibited GIVA PLA2 by 50%, when present at approximately 0.002 mole fraction in the interface in a number of in vitro assays.294
Table 17.
Group IVA PLA2 Inhibition by Pyrrolidines In Vitro and in THP-1 Cells
| Compound | IC50 (µM) | |
|---|---|---|
| PC/DOG | THP-1 | |
| 54 | 0.031 ± 0.0006 | 0.052 ± 0.009 |
| 55 | 0.0018 ± 0.0005 | 0.022 ± 0.001 |


In a continuation of the previous studies, pyrrophenone (56) proved to be an excellent inhibitor of human GIVA PLA2 (IC50 4.2 nM).295,296 Pyrrophenone strongly inhibited arachidonic acid release, prostaglandin E2, thromboxane B2 and leukotriene B4 formation in human whole blood. The magnitudes of PGE2 and thromboxane B2 inhibition were the same as those of indomethacine. Pyrrophenone showed reversible inhibition of GIVA PLA2, not displaying characteristics of slow-binding inhibition. Pyrrophenone potently inhibited LT, PGE2 and PAF biosynthesis in human neutrophils with IC50s in the range of 1–20 nM.297 A structurally related inhibitor, pyrroxyphene (57), displayed anti-arthritic and anti-bone destructive action in a murine arthritis model.298


3.4.5 Pyrroles
A series of substituted pyrroles have been developed by Lehr.299 Inhibitor 58 (IC50 13 µM) displayed activity similar to that of the reference inhibitor AACOCF3 (11 µM).299b

To define the structural requirements for improved cPLA2 inhibition, Lehr varied systematically the alkanoic acid group, the acyl residue, the nature of the substituents of the pyrrole ring, as well as the pyrrole nitrogen relative to the pyrrole ring substituents. The best inhibitor of this class of compounds was compound 59 (IC50 3.3 µM).299d When several of these pyrrole derivatives were tested in intact bovine and human platelets using the calcium ionophore A23187 as stimulant and the results were similar for both cell types.299e
3.4.6 Indoles
Lehr also developed a series of indoles. The first potent inhibitor of this series, compound 60, presented inhibition of GIVA PLA2 with an IC50 value of 8 µM.300

He later reported a series of 3-acylindole-2-carboxylic acid derivatives where the influence of the carboxylic acid group, the acyl residue, the moiety in position 1 and the substituent of the benzoic acid residue over the inhibition was investigated, along with the introduction of substituents in the phenyl ring of the indole. Compounds 61a and 61b showed the best inhibitory activity of the series with IC50 values of 0.5 µM and 0.52 µM, respectively, in a cPLA2-mediated arachidonic acid release assay from human platelets stimulated with calcium ionophore A23187.301 However, compounds 61a and b were not found to inhibit isolated GIVA PLA2, a fact that indicates that they do not directly interact with the enzyme.302
Genetic Institute developed several series of indoles as inhibitors of GIVA PLA2.303 Compounds 62 and 63 inhibited the enzyme in both an isolated enzyme assay and in cell-based assays. In addition, they proved to be active in the rat carrageenan paw edema test.

Wyeth (which acquired Genetics Institute) has undertaken a tremendous effort aimed toward the discovery of novel GIVA PLA2 inhibitors. The exploration of their application as novel pharmaceutical agents for inflammatory diseases led to their study in clinical trials. Using a substrate mimetic approach, a SAR study led to inhibitors 64a,b.304 Extensive SAR data led to the conclusion that increasing the distance between the indole and the benzoic acid moieties provided more potency as illustrated with compound 65 (Table 18).305 A continuation of this research resulted in the discovery of ecopladib (66).306 Ecopladib displayed oral efficacy in the rat carrageenan air pouch and rat carrageenan-induced paw edema models and advanced to Phase I clinical trials.
Table 18.
Group IVA PLA2 Inhibition by Indoles
| Compound | IC50 (µM) | |
|---|---|---|
| GLU assay | RWB assay | |
| 65 | 0.5 | 0.8 |
| 66 | 0.15 | 0.11 |
| 67 | 0.04 | 0.07 |
| 68 | 0.01 | 0.03 |
| 70 | 0.065 | 0.10 |
| 71 | 0.013 | 0.17 |

Another study on the importance of the substituent at C3 and the substitution pattern of the phenylmethane sulfonamide region led to efipladib (67) and WAY-196025 (68).307 These two compounds have shown efficacy when dosed orally in multiple acute and chronic prostaglandin and leukotriene dependent in vivo models.

The efficacy of a similar indole inhibitor, giripladib (69), in two different mouse models of rheumatoid arthritis has been reported.308 Giripladib, also known as PLA-695, was advanced into a phase II clinical trial for osteroarthritis, but the trial was terminated due to a failure to differentiate from the standard of care with naproxan because of gastroenterologic effects.

Further studies aimed at the optimization of in vitro potency and rat pharmacokinetics for oral efficacy309 and lowering the lipophilicity of the inhibitors.310 Examples are structures 70 and 71.

Wyeth has also reported 1,2,4-oxadiazolidin-3,5-diones and 1,3,5-triazin-2,4,6-triones311 as well as quinazoline-2,4(1H,3H)-dione GIVA PLA2 inhibitors (for example, compound 72) with reduced lipophilicity and improved aqueous solubility.312

3.4.7 2-Oxoamides
In 2002, a novel class of GIVA PLA2 inhibitors was reported, including AX006 (73a) and AX007 (74a), designed to contain the 2-oxoamide functionality and a free carboxyl group.313

Structure-activity relationship studies showed that long chain 2-oxoamides based on γ- or δ-amino acids, for example compounds 74a and 74b, are potent selective inhibitors of GIVA PLA2, while the corresponding esters may inhibit both GIVA PLA2 and GVIA PLA2 (Table 19).314 AX007 and AX109 presented a potent in vivo effect in the carrageenan paw edema test.314c Inhibitor AX048 (73b) showed a potent anti-hyperalgesic affect, being able to block spinal PGE2 release.315
Table 19.
Group IVA and GVIA PLA2 Inhibition by 2-Oxoamides
| Compound | XI(50) | |
|---|---|---|
| GIVA PLA2 | GVIA PLA2 | |
| AX006 | 0.024 ± 0.015 | N.D. |
| AX007 | 0.009 ± 0.004 | N.D. |
| AX048 | 0.022 ± 0.009 | 0.027 ± 0.009 |
| AX109 | 0.005 ± 0.002 | N.D. |
Recently, the location of the 2-oxoamide inhibitor AX007 (74a), as well as of pyrrophenone (56), in the active site of GIVA PLA2 was determined by a combination of deuterium exchange mass spectrometry with molecular dynamics.217 The models showed that both inhibitors interact with key residues that also exhibit changes in deuterium exchange upon inhibitor binding. Pyrrophenone was bound to the protein through numerous hydrophobic residues located distal from the active site, while the oxoamide was bound mainly through contacts near the active site as illustrated in Figure 9.
Figure 9.

A. Group IVA cPLA2 residues involved in binding pyrrophenone. B. Group IVA cPLA2 residues involved in binding oxoamide AX007. The residues that have contact with pyrrophenone or AX007 greater than 90% of the time in the molecular dynamics simulation are represented as red sticks and labeled in the figure. The inhibitor is shown in the licorice representation, with carbon, hydrogen, oxygen, nitrogen, and phosphorus atoms colored cyan, white, red, blue, and yellow, respectively. Adapted from Ref.217
Further studies,316 revealed that the sulfonamide group constitues a bioisosteric group suitable to replace the carboxyl group in 2-oxoamide inhibitors (for example, compound 75),316b while the pseudodipeptide-based inhibitor 76 inhibited the arachidonic acid release (IC50 2 µM) in RAW 264.7 macrophages.317

3.4.8 1,3-Disubstituted Propan-2-ones
AstraZeneca reported a series of heterocyclic compounds, such as compound 77, able to inhibit the GIVA PLA2-mediated arachidonic acid release from HL60 cells.318

In 2002, Conolly et al. presented a series of novel potent inhibitors of GIVA PLA2 based on a 1,3-disubstituted propan-2-one skeleton, where the electrophilicity of the carbonyl group could be altered over a wide range of structural modifications.319 Compound 78 (AR-C70484XX), which contains a decyloxy lipophilic side chain and a benzoic acid group, inhibited the enzyme presenting an IC50 value of 0.008 µM in a bilayer assay, 0.03 µM in a soluble assay and 2.8 µM in a whole cell assay. In an effort to reduce the lipophilicity, modifications of the decyloxyphenyl chain led to inhibitor 79 (IC50 0.56 µM in a soluble assay and 1.0 µM in a HL60 cell assay).320

In 2006, Lehr presented several 1-indol-1-yl-3-phenoxypropan-2-ones.321 Their activity was evaluated in a vesicle assay with isolated enzyme as well as in cellular assays with intact human platelets. Compound 80 presented an IC50 value of 8 µM, similar to some of the indole inhibitors Lehr had presented in the past.300,301a

Structure-activity relationship studies on the influence of the position of the carboxylic acid group, the nature of the substituent of the indole ring, the introduction of a second substituent in position 3 of the indole ring and the substitution of the octyl chain by a decyloxy chain led to compounds 81a and 81b (Table 20).321
Table 20.
Group IVA PLA2 Inhibition by 1,3-Disubstituted Propan-2-ones In Vitro and in Cellular Assay
| Compound | IC50 (µM) | ||
|---|---|---|---|
| vesicle assay with the isolated enzyme |
cellular assay with human platelets stimulant A23187 |
cellular assay with human platelets stimulant TPA |
|
| 81a | 0.020 | 0.21 | 0.0078 |
| 81b | 0.0043 | 0.57 | 0.0009 |

Replacement of the carboxylic acid and carboxamide moiety by other bioisosteric groups did not lead to more potent inhibitors.322 To enhance the water solubility and metabolic stability of this class of compounds, an additional series was evaluated. Compound 82 presented interesting aqueous solubility, but also a 10-fold decrease in the inhibitory potency.323 The indole ring of the above inhibitors was replaced by benzimidazole, benzotriazole and indazole rings and the inhibitory activity, the metabolic stability and the water solubility were evaluated.324 Compound 83 bearing an indazole ring (IC50 value of 5 nM in the vesicle assay) proved to be the most stable metabolically. Most of the above compounds were also tested for their activity against fatty acid amide hydrolase (FAAH), in order to evaluate their dual inhibitory potency.325 Compounds 83 and AR-C70484XX were found to be the best dual inhibitors for cPLA2 and FAAH.

In an attempt to develop clinically active GIVA PLA2 inhibitors, a series of structurally related indole-5-carboxylic acids with reduced lipophilicity was synthesized.326 The cPLA2 inhibition, the thermodynamic solubility and the metabolic stability of the new compounds was evaluated. Compound 84 was the most potent inhibitor (IC50 value of 12 nM against the isolated enzyme), and also possessed the highest water solubility (212 µg/mL at pH 7.4). Unfortunately, the po application of 84 (100 mg/kg) in mice only led to low concentrations of the substance in blood plasma and a very high plasma clearance was observed after intravenous administration (10 mg/kg). In a topical murine model of contact dermatitis, compound 84 showed a pronounced anti-inflammatory in vivo activity.326

In the most recent article by Lehr et al., the effect of the substituents in position 3 of the indole ring was evaluated.327 The most potent inhibitor was compound 85 bearing a 3-methyl-1,2,4-oxadiazol-5-yl-moiety with an IC50 value of 2.1 nM and excellent metabolic stability (81%); however, this compound presented poor aqueous solubility. In addition, compound 86 presented inhibition of cPLA2 with an IC50 value of 22 nM, excellent metabolic stability (93%) and aqueous solubility (194 µg/mL) making this compound interesting for further investigation. Compound 86 did not inhibit at the high concentration of 10 µM calcium-independent PLA2 (iPLA2) from rat brain cytosol and group IB secretory PLA2 (sPLA2) from porcine pancreas. The bioavailability of compound 86 was disappointing, but its concentration in intestine after po administration made this compound interesting for in vivo testing in animal models of inflammatory bowel diseases.327

3.4.9 Other Inhibitors
Annexin V belongs to a family of proteins that interact with phospholipids in a Ca2+-dependent manner. Both recombinant and human placental purified annexin V inhibited GIVA PLA2 activity whatever the stimulus used.328,329 The inhibition of cPLA2 by Annexin A6 was found to be linked to caveolin-1 export from the Golgi.330
Arylsulfonamides, such as compound 87 were reported to inhibit GIVA PLA2 and cytokine release.331 9,10-Dihydro-9,10-ethanoanthracene derivatives were also reported to inhibit GIVA PLA2 without affecting sPLA2 activity.332 Compound 88 inhibited paw swelling in the carrageenan edema model after i.p. administration with an ED50 of 16 mg/kg. A series of pyrimidines, such as compounds 89, were reported to inhibit the enzyme with IC50 values <100 nM.333

FTY720 is a novel immunosuppressive agent that was derived from myriocin, a sphingosine-like fungal metabolite. FTY720 inhibits the egress of lymphocytes from secondary lymphoid tissues and thymus and it has recently been approved by the FDA as a first-line therapy for multiple sclerosis. It has been shown that FTY720 inhibited cPLA2 independently of sphingosine-1-phosphate receptors.334

3.4.10 Summary Status of cPLA2 Inhibitors
A variety of different classes of GIVA cPLA2 inhibitors have been developed in the last 20 years. The first potent inhibitor was the trifluoromethyl ketone of arachidonic acid, which has been widely used to study the role of GIVA cPLA2 in vitro, in cells and in vivo. Even though this inhibitor does not inhibit sPLA2s, it does inhibit GVIA iPLA2, as well as other enzymes, so the interpretation of each studies should be taken with caution. More complex structures that also include a trifluoromethyl ketone group were presented by Bristol-Mayers Squibb as potent inhibitors of GIVA cPLA2. Shionogi has introduced a series of pyrrolidines that are excellent inhibitors of GIVA cPLA2, e.g., pyrrophenone, which can be used in ex vivo experiments. Although the above mentioned inhibitors are excellent tools for studying the role of the enzyme, none of them seems to be appropriate as a drug candidate. One of the most well studied class of inhibitors are the indoles bearing various simple or complex side chains. Indoles bearing a carboxylic acid group have been introduced as GIVA cPLA2 inhibitors by Lehr and Genetics Institute, which was later acquired by Wyeth. Ecopladib and later efipladib, WAY-196025 and giripladib are some of these indole inhibitors that have been used in in vivo models of inflammation, rheumatoid arthritis, etc. Another important class of GIVA cPLA2 inhibitors are 2-oxoamides such as AX007 and AX048 that have been shown to bind in the active site of GIVA cPLA2 and to present potent anti-hyperalgesic activity. Finally, another significant and well studied class of GIVA cPLA2 inhibitors are the 1,3-disubstituted propan-2-ones first introduced by Astrazeneca and later expanded upon by Lehr. A series of these inhibitors has been tested in several in vitro and in vivo experiments. To conclude, the scientific community awaits the results of the clinical trials using cPLA2 inhibitors to determine the importance of cPLA2 inhibition on inflammatory diseases.
4 Calcium Independent Phospholipase A2 [Group VI iPLA2]
4.1 Groups, Subgroups, Specificity and Mechanism
Group VIA phospholipase A2 is a member of the phospholipase A2 superfamily13–14 and is characterized by its calcium-independent phospholipase A2 activity. Although the activities of GIVC, GVII, GVIII and lysosomal PLA2s are all independent of calcium, the common name of “calcium independent” PLA2 (iPLA2) applies only to GVI PLA2. The first member of this family, GVIA PLA2, was purified and characterized from macrophages in 1994.8 To date, the Group VI calcium independent PLA2 (iPLA2) includes six different members: GVIA (iPLA2β; PNPLA9), GVIB (iPLA2γ; PNPLA8), GVIC (iPLA2δ; PNPLA6), GVID (iPLA2ε; PNPLA3), GVIE (iPLA2ζ; PNPLA2), and GVIF (iPLA2η; PNPLA4) (Table 21).14 All of these enzymes function through a catalytic serine at the active site in a patatin-like α/β-hydrolase domain (Figure 10). Because of the homology with patatin, GVI A, B, C, D, E and F are also included in the patatin-like protein family and named PNPLA9, 8, 6, 3, 2 and 4 respectively.335
Table 21.
Group VI Calcium Independent Phospholipase A2s (iPLA2)
| Subgroup | Alternative name |
Sources | Residues /Molecular Mass |
Domain | Alt Splice |
Activity | Post Translational Modification |
Human chromosome |
Swiss- Prot |
|---|---|---|---|---|---|---|---|---|---|
| GVIA | iPLA2β PNPLA9 |
Human/ Murine |
750/85 kDa 806/87 kDa |
7–8 Ankyrin Repeats α/ β hydrolase (Patatin like Lipase) |
5 | PLA2, Lyso-PLA transacylase acyl-CoA thioesterase |
22q13. 1 |
O60733 | |
| GVIB | iPLA2γ PNPLA8 |
Human/ Murine |
782/90 kDa | α/ β hydrolase (Patatin like Lipase) |
2 | PLA1, PLA2, Lyso-PLA transacylase |
7q31 | Q9NP8 0 |
|
| GVIC | iPLA2δ PNPLA6 NTE |
Human/ Murine |
1366/146 kDa |
3 cNMP α/ β hydrolase (Patatin like Lipase) |
3 | PLA2 Lyso-PLA |
Phosphorylatio n (5) |
19p13. 3-13.2 |
Q8IY17 |
| GVID | iPLA2ε PNPLA3 ADPN |
Human | 481/52 kDa | α/ β hydrolase (Patatin like Lipase) |
2 | PLA2, TG hydrolase, transacylase |
22q13. 31 |
Q9NST 1 |
|
| GVIE | iPLA2ζ PNPLA2 ATGL |
Human | 504/55 kDa | α/ β hydrolase (Patatin like Lipase) |
2 | PLA2 TG hydrolase, transacylase |
Phosphorylatio n (1) |
11p15. 5 |
Q96AD 5 |
| GVIF | iPLA2η PNPLA4 GS2 |
Human | 253/27 kDa | α/ β hydrolase (Patatin like Lipase) |
PLA2, TG hydrolase, retinylester hydrolase acylglycerol and retinol transacylase |
Phosphorylatio n (1) |
xp22.3 | P41247 |
Figure 10.

Schematic presentation of Group VI iPLA2. Ankyrin repeats (ANK), nucleotide phosphate binding domain (cNMP), active site residues in red squares, and patatin-like lipase domains are indicated in green.
4.1.1 Group VIA PLA2 (iPLA2β; PNPLA9)
Calcium-independent PLA2 was so named in order to differentiate it from sPLA2 and cPLA2, whose activities are calcium-dependent. Its gene, located at 22q13.1, expresses GVIA-1 PLA2 as a 752 amino acid protein with a molecular mass of 85 kDa that contains eight ankyrin repeats and a catalytic domain.9,336 The initial reports of Ca2+-independent PLA2 activity referred to a 40-kDa enzyme described as iPLA2337. Subsequently, an 85-kDa iPLA2 was purified and well characterized which is now known as GVIA iPLA2 (also iPLA2β)8. The same iPLA2 was isolated from CHO cells, sequenced, cloned and expressed9,336a. The size, sequence and properties of the 85-kDa enzyme were subsequently confirmed by studies in many laboratories.338 This enzyme showed potent phospholipase, lysophospholipase and transacylase activities which could be inhibited by a variety of inhibitors including bromoenol lactone (BEL) and various fluoroketones.240,339 The active site contains a lipase consensus sequence (GXS465XG) located in the catalytic domain (Figure 10).9 GVIA-1 PLA2 was shown to be active as an oligomer through radiation inactivation studies.336a ATP binding protected GVIA-1 PLA2 from cysteine oxidation and prevented loss of activity.240,336a,340
The human Group VIA PLA2 gene was found to express multiple splice variants, including GVIA-1, GVIA-2, GVIA-3 PLA2, GVIA Ankyrin-1 and GVIA Ankyrin-2,338a,b,341 in tissue-dependent patterns,342 At least two of these isoforms, GVIA-1 and GVIA-2 PLA2 are active. GIVA-2 is the longer (88 kDa) splice variant and is composed of 7 ankyrin repeats, a linker region and a catalytic domain (Figure 10). The eighth ankyrin repeat is disrupted by a 54 amino acid insert.338b GVIA-1 PLA2 activity was reported to be unaffected by ATP, while GVIA-2 PLA2 activity is enhanced by ATP.337c,341,343 GVIA-2 PLA2 is membrane-associated when overexpressed in COS-7 cells, rat vascular smooth muscle cells,338a,341 and Sf-9 insect cells.344 The Ankyrin-1 and Ankyrin-2 splice variants are mainly the ankyrin repeats without phospholipase activity, and it has been suggested that they inhibit GVIA-2 PLA2 by forming hetero-oligomers.338b
GVI PLA2 and GIV PLA2 both use a serine active site to catalyze the cleavage of the sn-2 ester bond, but GVI PLA2 does not show arachidonic acid specificity in the sn-2 position, while GIV PLA2 does.240 GVIA PLA2 also exhibits lysophospholipase activity, transacylase activity and acyl-CoA thioesterase activity.342,345 It has been suggested that the activity of GVIA PLA2 is regulated through many different mechanisms, including ATP binding, caspase cleavage, calmodulin, and possible ankyrin repeat mediated protein aggregation.16c
4.1.2 Group VIB PLA2 (iPLA2γ; PNPLA8)
The GVIB PLA2 gene at 7q31 was cloned in 2000 from heart, skeletal muscle,346 and lymphocyte cDNA347 and the transcript was detected in heart, placenta, kidney, liver, brain, and skeletal muscle.346–347 GVIB PLA2 is a 90 kDa protein with 782 amino acids.347 The region 445–640 is a patatin-like lipase domain and the GXSXG motif implies that the active site is at Ser-483 (Figure 10).348 The catalytic core shares approximately 25% identity with GVIA-1 PLA2.13 GVIB PLA2 protein activity has been identified in membrane fractions346–347 and was later found to associate with mitochondria and peroxisomes.349 Typical localization motifs of mitochondria and peroxisomes were identified.349 GVIB PLA2 exhibits both PLA1 and PLA2 activity and shows low specificity to sn-2 fatty acids for PC hydrolysis.346,350 Research in GVIB PLA2-transgenic and GVIB PLA2-deficient mice show that GVIB PLA2 is critical to cardiac phospholipid homeostasis and mitochondrial function.349,351 This enzyme may be responsible for cardiolipin remodeling in mitochondria.351
4.1.3 Group VIC PLA2 (iPLA2δ; PNPLA6)
Group VIC PLA2 is commonly known as a neuropathy target esterase (NTE), since its discovery in 2002.352 The human Group VIC PLA2 gene is located at chromosome 19p13.3-13.2. It is expressed in neurons and is localized to the endoplasmic reticulum (ER) and Golgi apparatus.352 The expressed enzyme is a membrane protein with 1366 amino acids (146 kDa) and exhibits esterase activity in phenyl valerate substrate. The patatin-like catalytic domain is in the region 933–1100 and the active site Ser-1005 is in the GXSXG motif (Figure 10).348 The recombinant esterase domain (residues 727–1216) also demonstrates calcium-independent phospholipase and lysophospholipase activities.352 Inhibition of NTE lysophospholipase activity may be responsible for the accumulation of its physiological substrate lysolecithin, leading to delayed toxicity.353 Inhibition of NTE by organophosphorus esters can initiate axonal degeneration syndrome. NTE knockout mice are embryonically lethal and the NTE inhibitor caused hyperactivity in NTE(+/−) mice.354 NTE deletion mice have exhibited neuronal pathology in the hippocampus and thalamus, cerebellum defects, and abnormal cellular features, including disruption of the ER, vacuolation of nerve cell bodies, and abnormal reticular aggregates.355 Human patients with NTE mutations were found to suffer from the severe NTE-related motor neuron disease.356
4.1.4 Group VID PLA2 (iPLA2ε; PNPLA3)
GVID PLA2 was identified in adipocytes in 2001, and is also called adiponutrin because of its transcript responses to a nutritional diet.357 Human adiponutrin is a transmembrane protein that is expressed in adipocytes.357 Its gene is located at 22q13.31 and the protein has 481 amino acids/ 52 kDa. It contains a patatin-like lipase domain in residues 10–180 and Ser-47, within this domain, is predicted to be the active site (Figure 10).335 The phylogenetic tree shows the closest relation as GVIE PLA2 among the GVI PLA2s.335 Human GVID PLA2 has exhibited triacylglycerol hydrolase, transacylase, and calcium-independent PLA2 activity.358 The adiponutrin gene has been strongly associated with nonalcoholic fatty liver disease,359 liver dysfunction,360 insulin secretion and obesity.361
4.1.5 Group VIE PLA2 (iPLA2ζ; PNPLA2)
PNPLA2 was identified in 2004358a,362 and was named adipose triglyceride lipase (ATGL), iPLA2ζ, GVIE PLA2 and PNPLA2, due to its functions and structural homology.14,358a,362 The gene is located at chromosome 11p15.5 and translates to a 55 kDa protein with 504 amino acids.335b,358a,362 The protein is expressed mostly in white and brown adipocytes and is localized to the membrane and lipid droplets.362–363 Similar to GVID PLA2, it also contains a patatin-like lipase domain in residues 10–180 and Ser47 is expected to be an active site residue (Figure 10).335 ATGL exhibits triglyceride lipase, transacylase and phospholipase A2 activity in vitro.358a,362b,363a,364 The phospholipase activity is independent of calcium and ATP.358a An ATGL knockout mouse study has confirmed its importance in triglyceride hydrolysis.365 Human mutations in the ATGL gene cause accumulation of triacylglycerol in multiple tissues and neutral lipid storage disease in patients, along with skeletal- and cardiomyopathy.366 The polymorphisms in PNPLA2 have also been associated with decreased plasma fatty acid and triacylglycerol levels and increased risk for type 2 diabetes.367
4.1.6 Group VIF PLA2 (iPLA2η; PNPLA4)
Human PNPLA4 was identified in 1994 and was named gene sequence-2 (GS2)368. The gene at xp22.3 expresses ubiquitously in all tissues examined.348,358b,368–369 GVIF PLA2 is a 27 kDa protein comprised of 253 amino acids. The region 6–177 contains a patatin-like catalytic domain and Ser-43 is the expected active site in the GXSXG motif (Figure 10).348 GS2 has shown retinylester hydrolase,369 acylglycerol and retinol transacylase,358a,369 TG hydrolase,358b,369 and PLA2 activity.358a
4.2 Structural Characteristics and Activation Mechanisms
There are currently six forms of the GVI PLA2. Among them, GVIA is the most well studied and recognized iPLA2. In this section, we will use the long form splice variant GVIA-2 PLA2 as our main example for discussing the structural characteristics of iPLA2. As described in section 4.1.1, all GVI PLA2 isoforms contain a patatin-like lipase domain. GVID, E and F PLA2s are all small enzymes primarily composed of only the patatin-like domain.335,348 GVIB PLA2 shows the highest homology to GVIA PLA2, but does not contain the ankyrin repeats.335,348 GVIC PLA2 is a transmembrane protein and also the largest protein in GVI.352 It shares low similarity with all of the other GVI PLA2s, except for the patatin-like lipase domain. The 85 kDa human GVIA-2 PLA2 (806 amino acids) contains seven ankyrin repeats (residues 152–382), a linker region (residues 383–474) with the eighth repeat disrupted by a 54-amino acid insert, and a catalytic domain (residues 475–806) (Figure 11). Definition of the segments of GVIA PLA2 is still vague. To avoid confusion, the regions that show high homology to the patatin protein were defined as the catalytic domain. And the rest of the region was defined as a regulatory domain, which contains the N-terminal region, ankyrin repeats and the linker region. A computational homology model based on homology structures was constructed.344 This model was validated by comparing the deuterium exchange results with the predicted structure.
Figure 11.

Features of Group VIA PLA2 homology models. The domains and binding sites are differentiated by colors. Adapted from Ref.344
The regulatory domain is specific for GVIA PLA2 only. The region with a disrupted ankyrin repeat becomes the linker region in splice variant 2. The ankyrin repeats show a recurring 30–40 residue repeat with a helix-turn-helix-loop structure and has sequence homology with the human Ankyrin-R D34 protein.240,370 Although no crystal structure of the ankyrin repeats in GVIA PLA2 had been obtained, the identified ankyrin repeats seem to all fold in a similar pattern. A homology model of the ankyrin repeats has been built based on the human Ankyrin-R D34 protein (Figure 11).344,370c The ankyrin repeats are packed side by side and form a curved structure. While two additional ankyrin repeats in the n-terminal region fit in the model, they do not have a conventional ankyrin repeat sequence identity. We have shown that the linker region is a highly flexible region in our H/D exchange experiments.344
The catalytic domain is also called a patatin-like lipase domain. The published homology model shows 40% homology to patatin (Figure 11) and is obviously larger than the 170 residue conserved core.344,371 Comparison of our GVIA PLA2 model to the GIVA PLA2 crystal structure shows similar folding of the α/β-hydrolase domain to GIVA PLA2 without the cap region, but the overall sequence homology is low.202a,344,371 The active site serine of the GVIA PLA2 lies within a lipase consensus sequence (Gly-X-Ser519-X-Gly) on top of the catalytic domain (Figure 11). Unlike the GIVA PLA2 active site, which is under the cap region,202a the active GVIA PLA2 site, Ser519, is quite exposed to solvent.344 This suggests that the cap region contributes to the GIVA PLA2 AA specificity and that the exposed serine active site results in low substrate specificity. We further found that GVIA PLA2 also has Asp-652 near the active site, which has been identified as the active site dyad in patatin.344,371 Moreover, there is no histidine near the active site Ser-519. The homology model strongly suggests GVIA PLA2 is also a Ser519/Asp652 catalytic dyad, similar to the dyad in GIVA PLA2 and patatin.
This enzyme is known to be regulated by ATP binding, caspase cleavage, oligomerization and calmodulin binding. They all take part in modulating GVIA PLA2 activity and function. We will further discuss GVIA PLA2 regulation mechanisms in the following section.
4.2.1 ATP Activation
Regulation of ATP is critical for various cellular functions, especially in mitochondria.372 GVIA PLA2 has been shown to localize in mitochondria in various cell types.373 GVIA PLA2 is the only PLA2 reported to be regulated by ATP and ADP binding.240 GVIA PLA2 can be activated by ATP, but ATP is not a substrate or cofactor for GVIA PLA2.240 The binding of ATP by the GVIA enzyme either stabilizes the enzyme’s structure or activates the enzyme.8,240,341 Its activation likely results from a conformational change in the enzyme. ATP has also been shown to prevent loss of activity in GVIA PLA2.240 Research has shown that ATP binding can prevent the dimerization and cysteine oxidation of GVIA PLA2, enabling it to maintain its activity over time.340 Because of GVIA PLA2’s homology to protein kinases, it has been suggested that the binding site is in the G485XGXXG motif,374 close to the active site serine within a lipase consensus sequence (Gly-X-Ser519-X-Gly) in the α/β hydrolase domain. However, this proposed ATP binding site is in a highly negatively charged environment in the homology model of the catalytic domain of GVIA PLA2, based on the crystal structure of patatin.344 Although the homology model may not provide accurate electrostatic potentials, it is unlikely that ATP would bind to a region surrounded by negatively charged residues. In addition, the proposed ATP binding sequences may not be similar to the glycine rich loop of protein kinases. Recently, the crystal structure of the ankyrin repeats of the transient receptor potential cation channel TRPV1 showed that ATP can bind to ankyrin repeats and regulate the calcium channels.375 This process utilized a completely different ATP binding mechanism than that found in protein kinases, which does not require metal ions.
4.2.2 Caspase Cleavage
GVIA PLA2 plays an important role in homeostasis, maintaining a constant level of lysoPC in resting P388D1 macrophages cells.342,376 Imbalances in the homeostasis can seriously damage a cell. Caspase proteolysis of the GVIA enzyme in apoptosis produces a truncated protein in which the first of the ankyrin repeats has been clipped, resulting in a hyperactive form of the protein.377 In the U937 system, both the GIVA and GVIA PLA2s are targets of caspase-3 proteolysis, but only the GVIA enzyme remains active following cleavage.377 Three forms of the caspase cleaved GVIA PLA2 have been identified, 70 kDa, 26 kDa and 32 kDa19a The preferred cleavage site is DVTD183, while DLFD513 and MVVD733 are minor cleavage sites.377–378 Thapsigargin-induced apoptosis results in caspase-3 cleavage and generate an active 62-kDa protein localized to the nucleus.379 During apoptosis, active GVIA PLA2 may participate in membrane damage and provide bioactive lipid metabolites, such as lysoPC, for phagocytosis.19a,380
4.2.3 Calmodulin Inhibition
Calmodulin is activated by binding to four Ca2+ ions, which induces a conformational change.381 Activated calmodulin can interact with calmodulin binding proteins to change their subcellular localization or enzymatic activity.381 Calmodulin binding proteins that are not directly regulated by Ca2+ can respond to intracellular Ca2+ through calmodulin. Calmodulin associates with GVIA PLA2 and was used to purify GVIA PLA2.338c No evidence of Ca2+ binding to GVIA PLA2 has ever been reported, and we have shown membrane penetration by GVIA PLA2 in the absence of Ca2+.344 Although GVIA PLA2 activity is independent of Ca2+, Ca2+-activated calmodulin has been reported to bind to the IQ motif in the tryptic C-terminal 15 kDa region of GVIA PLA2, inhibiting its activity.382 The IQ motif is located in residues 694–705 of the catalytic domain of GVIA-1 PLA2.
4.2.4 Oligomerization
Radiation inactivation studies of murine GVIA PLA2 suggest that it is active as an oligomer of 340 kDa.8 Early work suggested that the 340 kDa complex was a tetramer of the 85 kDa GVIA enzyme.8 It was assumed that the eight ankyrin repeats near the N-terminus of the protein played a role in this aggregation. The requirement of the ankyrin repeats for GVIA PLA2 activity is identified by the truncation mutation.338b The splice variant GVI Ankyrin-1 was proposed to be a potential negative regulator of GVIA PLA2 by blocking oligomerization.338b GVIA PLA2 may also associate with other regulatory proteins to form active complexes.377 GVIA PLA2 has been shown to form a signaling complex with the calcium/calmodulin-dependent protein kinase IIβ that is expressed in pancreatic islet beta-cells.383 Additionally, the ankyrin repeats are reported to be involved in protein-protein interactions, such as 53BP2-p53, GA-binding protein α-GAbinding protein β, p16INK4a-CDK6, and IκBα-NFκB.336b
4.2.5 Membrane Interaction
The activity of phospholipases depends critically on the interaction of the protein with phospholipid membranes. GVIA-2 PLA2 is composed of seven consecutive N-terminal ankyrin repeats, a linker region, and a C-terminal phospholipase catalytic domain.8,240,341 No crystal structure of GVIA PLA2 has ever been published, and no information is known about the membrane binding surface. Deuterium exchange experiments on GVIA-2 PLA2 in the presence of phospholipid substrate (PAPC) was carried out to locate regions in the protein that change upon lipid binding (Figure 12A).344 The region with the greatest change was region 708–730, which showed a >70% decrease in deuteration levels at numerous time points. No decreases in exchange due to phospholipid binding were seen in the ankyrin repeat domain of the protein. The homology model, combined with the deuterium exchange results in the presence of lipid substrate, has led to the first structural model of GVIA-2 PLA2 as well as the interfacial lipid binding region (Figure 12B).
Figure 12.

Phospholipid binding of the Group VIA iPLA2. A. Phospholipid membrane binding effects on H/D exchange of the Group VIA iPLA2 mapped onto the catalytic domain model. B. Model of the lipid-binding surface of the Group VIA iPLA2 based on interactions with lipid membrane. Adapted from Ref.344
Only one of these regions was located in the regulatory domain. Region 378–389, located in the middle of the linker region, had a 2.2 deuteron increase in exchange at 5 min of on-exchange.344 When deuterium exchange experiments were carried out with the isolated ankyrin repeat linker construct, the entire linker region had increased exchange compared with the intact enzyme. This implies that the linker region is in contact with the catalytic domain. The increase in exchange at 378–389 may imply that upon lipid binding there is a conformational change in the orientation of the catalytic domain relative to the ankyrin repeat linker region. GVIA-2 PLA2 was found to be membrane-associated when overexpressed in COS-7 cells, and this was further confirmed in rat vascular smooth muscle cells.338a,341 The other active splice variant, GVIA-1, is cytosolic and not specific in targeting membrane surfaces,338a,341 indicating that these two splice variants use two different regulatory mechanisms. The 54-residue insertion in the eighth ankyrin repeat alters the property of GVIA-2 PLA2 for membrane association.
Four of the regions that showed changes in deuterium exchange are localized to the catalytic domain, and all showed decreases in exchange (Figure 12A).344 The decreases in exchange on the catalytic domain occurred in areas that have numerous different hydrophobic residues. The region with the greatest decrease in exchange is 708–730. This region had a 13.2 deuteron decrease in exchange at the 10s time point but only a 3.5 deuteron decrease at 166 min. This region is the most highly solvent-exposed region in the protein in the absence of vesicles. The almost total absence of exchange at 10s of on-exchange in the presence of lipid vesicles implies that this region is penetrating into the membrane surface. This region contains hydrophobic residues Val-708, Phe-709, Trp-715, Leu-717, Val-721, Phe-722, and Leu-727 that may mediate penetration into the lipid surface.
The negatively charged region 773–778 also shows a decrease in exchange (Figure 12A).344 This region consists of an α-helix that is part of the catalytic core, which explains the low rates of H/D exchange. There are no hydrophobic residues in this region, and it is most likely that decreases here are due to either electrostatic interactions between the charged membrane head group and the charged residues Asp-771, Glu-772, and Asp-775 or conformational changes induced by substrate binding. Regions 631–655 and 658–664 are both spatially very close to the active site Ser-519 and are on the same face of the enzyme as the proposed membrane penetration region, 708–730. Neither of these regions has hydrophobic regions and are also most likely interacting with the charged surface of the membrane rather than mediating penetration.
The model of GIVA PLA2 membrane binding suggests penetration of the 708–730 region into the membrane surface with regions 631–655, 658–664, and 773–778 interacting with the charged head group of the phospholipid substrate (Figure 12B).
4.3 Biological Functions and Disease Implications
4.3.1 Biological Functions
The Group VI enzymes are diverse in terms of their structure and function, even though they all have calcium-independent phospholipase A2 activity and a patatin-like lipase domain.13–14,335a Most of these enzymes have multiple catalytic activities and localize to different organelles. The GVID, GVIE and GVIF are all shorter forms of GVI PLA2.14,335a,358a All of them were found to localize to adipose cells.358a It is suggested that fatty acid acylation is the main cellular function of these enzymes.358a GVIA and GVIB have closer relationships than the other GVI enzymes in the phylogenic tree and both have been shown to localize to mitochondria.335b,348 GVIC, neuropathy target esterase, is quite different from the rest of the GVI enzymes.352 We have briefly described the general functions of all GVI enzymes in the previous sections. Here we focus on representative GVIA enzymes to discuss their biological functions and disease implications.
GVIA-1 has been shown to be important in membrane homeostasis and remodeling376b, and it appears that this enzyme is the primary PLA2 for day to day metabolic functions within the cell. GVIA PLA2 is involved in cell proliferation,384 cell cycle progression,385 apoptosis,376b,386 bone formation,387 sperm development,388 glucose-induced insulin secretion,389 cardiolipin acetylation,390 and monocyte recruitment,384a,391 which also shows that its function may vary by cell and tissue. Interestingly, GVIA PLA2 has been involved in both cell growth and cell death in various cell types. On one hand, GVIA PLA2 promotes cell cycle progression and proliferation by GVIA PLA2-generated lipid mediators, including free fatty acids, eicosanoids and LPA.317,384c,d,385,392 On the other hand, GVIA PLA2 has been shown to be involved in various stimuli-induced apoptosis, including Fas-ligand, thapsigargin, H2O2, ROS generation, and chemotherapeutic drugs.19b,377,393 Several studies of insulin secretion, cell proliferation and apoptosis in beta-cells have established the involvement of GVIA PLA2 with diabetes.394
Several lines of evidence have shown that GVI PLA2 is involved in regulating store-operated calcium channels in glial cells, astrocytes, endothelial cells, epithelial cells and smooth muscle cells, and also mediates store-operated calcium entry.395 GVI PLA2 has also been shown to be activated during ischemia in rabbit myocardium and human coronary artery endothelial cells396. Although the GVI PLA2 knockout mice do not exhibit the myocardial function defect, they are relatively refractory to ischemia-induced cardiac arrhythmias, which can be completely suppressed by the administration of GVI PLA2 inhibitor BEL19e,388.
Several lines of evidence also show the significant role of GVIA PLA2 in muscle functions in muscle degeneration,395b skeletal muscle contractility,397 cardiomyopathy,398 drip formation in muscle,399 skeletal muscle fatty acid oxidation,345b vascular smooth muscle cell contraction400 and Barth syndrome with cardinal disorder characteristics. GVIA PLA2 function is also strongly related to function and dysfunction in the nerve system, such as neurotransmission and nerve degeneration.401 Researchers have shown a correlation of GVIA PLA2 activity with several diseases including neurodegeneration with brain iron accumulation (NBIA) disorders,402 infantile neuroaxonal dystrophy,403 and memory loss.404 GVIA PLA2 also plays an important role in Wallerian degeneration and axon regeneration in nerve injury in mouse models.165,405
4.3.2 Diabetes
GVIA PLA2 has been related to diabetes due to its participation in β-cell apoptosis, superoxide production and glucose-induced RhoA/Rho kinase activation. GVIA PLA2 genetically-modified mice and various cellular studies suggest that GVIA PLA2 participates in β-cell apoptosis.379,406 β-cell apoptosis and ceramide production may be responsible for the loss of beta-cell mass that is associated with the onset and progression of type 1 and type 2 diabetes mellitus.406a In diabetes mellitus patients, GVIA PLA2 enhanced superoxide generation in neutrophils and siRNA knockdown inhibited superoxide generation.407 GVIA PLA2-deficient cell and inhibitor studies have found that GVIA PLA2 participates in the high glucose-induced RhoA/Rho kinase/CPI-17 activation pathway, which has been shown to contribute to diabetes-associated vascular smooth muscle hypercontractility.408
4.3.3 Barth Syndrome
Barth syndrome is associated with mutations of the X-linked tafazzin gene (TAZ).409 Cardiolipin is a distinct phospholipid component of the mitochondrial membrane.410 Tafazzin is responsible for cardiolipin homeostasis in mitochondria.390a,411 GVIA PLA2 localizes in mitochondria, maintains the integrity of the mitochondrial membrane and prevents the rupture of mitochondria and the release of cytochorme c under oxidative stress.373 Recent research suggests that GVIA PLA2 participates in cardiolipin remodeling in cardiomyocytes.390a The other isoform, GVIB PLA2, shows alterations to hippocampal cardiolipin content in transgenic and deficient mice412 and a role in cardiac phospholipid homeostasis and mitochondrial function.349,351 This enzyme may be responsible for cardiolipin remodeling in mitochondria.351 GVIA PLA2 is also responsible for cardiolipin deacylation and monolyso cardiolipin accumulation in Barth syndrome.390b Genetic inactivation of GVIA PLA2 can suppress the phenotype of tafazzin knockout drosophila.390b GVIA PLA2 inhibition is a possible treatment for Barth syndrome patients.
4.3.4 NBIA/Neuroaxonal Dystrophy
Recent clinical studies have shown that GVIA PLA2 mutations are associated with “neurodegeneration with brain iron accumulation” (NBIA) disorders and infantile neuroaxonal dystrophy.389b,402,413 Neither the active site, Ser-519, nor the potential active site, Asp-652, was found to be mutated in the more than 40 mutation sites occurred in patients. These GVIA PLA2 mutants may retain different level of activities, except for some truncation mutants. Double mutations of GVIA PLA2 tend to correlate to earlier age of disease onset, suggesting that GVIA PLA2 activity is critical in development.389b,402
4.4 Chemical Inhibitors and Therapeutic Intervention
In comparison to the secreted and cytosolic Ca2+-dependent PLA2 enzymes, the research on inhibitors of GVI PLA2 is much more limited. However, a summary of GVIA PLA2 inhibitors is included in two review articles.22c,22f
4.4.1 Fatty Acid Trifluoromethyl Ketones and Tricarbonyls
In 1995, the inhibition of macrophage Ca2+-independent PLA2 by AACOCF3 (43) and palmitoyl trifluoromethyl ketone (44) were reported.414 In contrast to the case with GIVA PLA2, for GVIA PLA2 the saturated derivative was found to be 4-fold more potent than AACOCF3 (Table 22). Fatty acyl tricarbonyls 47 and 91 also inhibited GVIA PLA2 but appeared to be much poorer inhibitors than the corresponding trifluoromethyl ketones.279
Table 22.
Group VIA PLA2 Inhibition by Fatty Acyl Trifluoromethyl Ketones
| Compound | IC50 (µM) | XI(50) |
|---|---|---|
| 43 | 15 | 0.028 |
| 44 | 3.8 | 0.0075 |
| 95 | 0.0043 |

4.4.2. Methyl fluorophosphonates
MAFP (48) was found to irreversibly inhibit GVIA PLA2.415 In addition, the saturated fluorophosphonates 92 and 93 inhibited GVIA PLA2 showing similar potencies and considerably higher potency than that of MAFP.

4.4.3. Bromoenol lactone

Bromoenol lactone (94, BEL) is an irreversible, covalent inhibitor of GVIA PLA2,414,376b inhibiting the enzyme at concentrations far lower than those required to inhibit sPLA2 or cPLA2 family members. As a result, BEL is commonly used to selectively inhibit GVIA PLA2 in cellular systems. However, although BEL can distinguish GVIA PLA2 among other PLA2s, it may also inhibit other enzymes, for example the magnesium-dependent phosphatidate phosphohydrolase-1,416 and was first identified as a serine protease inhibitor.417 Thus, studies involving iPLA2 inhibition by BEL are ambiguous and require confirmation by other experiments.
Initially, BEL had been characterized as a suicide inhibitor of canine myocardial calcium-independent PLA2.418 Using BEL it has been suggested that PGE2 generation upon stimulation may be partially mediated by iPLA2 in addition to sPLA2.419 In a number of studies, Turk et al. showed that GVIA PLA2 (iPLA2β) played a signalling role in β-cells that differed from housekeeping functions in PC biosynthesis and degradation in P388D1 and CHO cells.379 BEL was found to decrease arachidonic acid release and PGE2 production in 3T6 fibroplast cultures stimulated by fetal calf serum.420 Prosynaptic injection of BEL selectively increased AMPA receptor-mediated synaptic transmission.421 Intracerebroventricular injection of BEL significantly reduced responses to von Frey hair stimulation after facial carrageenan injection in both C57BL/6J (B6) and BALB/c mice.286
The effect of inhibition of iPLA2β on chemotherapeutic-induced death and phospholipid profiles in renal cells was studied.393c Inhibition of iPLA2 by BEL decreased prostate cancer cell growth by p53-dependent and independent mechanisms.422 Alterations in Mdm2 and epidermal growth factor receptor activation following BEL exposure suggested novel roles for iPLA2 in prostate cancer cell signaling. Most recently, it was shown that iPLA2 inhibition by BEL activated p38 MAPK signaling pathways during cytostasis in prostate cancer cells.423
Interestingly, in a number of studies it has been demonstrated that (R)- and (S)-enantiomers of BEL present different inhibitory properties.424 GVIA PLA2 (iPLA2β) and GVIB PLA2 (iPLA2γ) have been found to be selectively inhibited by (S)- and (R)-enantiomers of BEL, respectively.424a,b Turk et al. have reported that BEL inactivates GVIA PLA2 by generating a diffusible bromomethyl keto acid that alkylates cysteine thiols, rather than creating an acyl-enzyme intermediate with the active-site serine.425
4.4.4. Polyfluoroketones
In 1999, a variety of trifluoromethyl ketones were studied for the inhibition of GVIA PLA2 in a mixed-micelle assay.269 Trifluoromethyl ketone 95 was found to be a potent inhibitor of GVIA PLA2 (Table 22), 10-fold more potent in comparison to GIVA PLA2. In 2008, a series of polyfluoro ketones for the selective inhibition of GVIA PLA2 was synthesized.405b Such a task is very challenging, because both the intracellular GVIA PLA2 and GIVA PLA2 share the same catalytic mechanism and cross-reactivity is expected for inhibitors targeting the active site serine.

It was found that the pentafluoroethyl ketone functionality favored selective inhibition of GVIA PLA2. FKGK11 (96) was found to be a selective GVIA PLA2 inhibitor, while the trifluoromethyl ketone FKGK2 (97) can be considered as a pan-inhibitor inhibiting GVIA PLA2, GIVA PLA2, and even GV sPLA2 (Table 23).
Table 23.
Comparison of Group VIA, Group IVA and Group V PLA2 Inhibition by Polyfluoroalkyl Ketonesa
| Compound | GVIA PLA2 | GIVA PLA2 | GV sPLA2 |
|---|---|---|---|
| 96 | >90% 0.0073 ± 0.0007 |
N.D. | 28 ± 1% |
| 97 | >90% 0.0169 ± 0.021 |
>90% 0.0098 ± 0.0006 |
86 ± 2 % |
Average percent inhibition at 0.091 mol fraction and XI(50) and standard error (n = 3) are reported for each compound. N.D. signifies compounds with less than 25% inhibition (or no detectable inhibition).

In a continuation of SAR studies on polyfluoro ketones and using an improved assay for GVIA PLA2, compound FKGK18 (98) was identified as the most potent GVIA PLA2 inhibitor yet reported (Table 24).405c Being 195 and >455 times more potent for GVIA PLA2 than for GIVA PLA2 and GV sPLA2, respectively, makes it a valuable tool to explore the role of GVIA PLA2 in cells and in vivo models. Heptafluoro derivative 99 also presented interesting inhibition of GVIA PLA2, inhibiting GIVA PLA2 and GV sPLA2 at least 90 times less potently (Table 24).
Table 24.
Comparison of Group VIA, Group IVA and Group V PLA2 Inhibition by Polyfluoroalkyl Ketonesa
| Compound | GVIA PLA2 XI(50) |
GIVA PLA2 % Inhibition |
GV sPLA2 % Inhibition |
|---|---|---|---|
| 98 | 0.0002 ±0.0000 | 80.8 ± 1.5 % | 36.8 ± 7.9 % |
| 99 | 0.0010 ± 0.0001 | 55.8 ± 2.1 % | 46.3 ±10.0 % |
Average percent inhibition at 0.091 mol fraction and standard error (n = 3) are reported for each compound or XI(50) and standard error (n = 3) for those with >90% inhibition.

Selective PLA2 inhibitors contributed to the clarification of the role of each PLA2 class in neurological disorders.405a,426 Using the selective GVIA PLA2 inhibitor FKGK11, the selective GIVA PLA2 AX059 and the pan-inhibitor FKGK2, the role of the various classes of PLA2 in EAE, the animal model of multiple sclerosis, was studied.426 The results suggested that GIVA PLA2 plays a role in the onset of the disease, while GVIA PLA2 plays a key-role both on the onset and the progression of the disease. Thus, it seems that GVIA PLA2 is a target enzyme for the development of novel therapies for multiple sclerosis.426
4.4.5. 2-Oxoamides
As discussed in the section on GIVA PLA2 inhibitors, 2-oxoamides based on esters of amino acids may inhibit both GVIA PLA2 and GIVA PLA2. Recently, two 2-oxoamides based on esters of dipeptides or pseudodipeptides (100 and 101) were reported to preferentially inhibit GVIA PLA2 (Table 25).317
Table 25.
Comparison of Group VIA, Group IVA and Group V PLA2 Inhibition by 2-Oxoamidesa
| Compound | GVIA PLA2 XI(50) |
GIVA PLA2 % Inhibition |
GV PLA2 % Inhibition |
|---|---|---|---|
| 100 | 0.011 ± 0.001 | 72 | 59 |
| 101 | 0.017 ± 0.002 | 52 | 81 |
Average percent inhibition at 0.091 mol fraction are reported for each compound or XI(50) and standard error (n = 3) for those with >90% inhibition.

4.4.6 Summary Status of iPLA2 Inhibitors
Research on GVIA iPLA2 inhibitors is relatively limited, due partially to its lack of a crystal structure, but also to the fact that only recently has it emerged that GVIA iPLA2 plays a significant role in a number of medical conditions. As with GIVA cPLA2, the first series of potent reversible GVIA iPLA2 inhibitors were trifluoromethyl ketones of fatty acids. Bromoenol lactone (BEL) is the most important irreversible inhibitor of GVIA iPLA2 and it has been used in a number of in vitro, ex vivo and in vivo studies to elucidate the role of GVIA iPLA2. One should note that even though BEL is selective against GVIA iPLA2 when compared to the other PLA2 groups, it also inhibits other serine enzymes and therefore the data obtained from ex vivo and in vivo studies of its inhibitory activity should be carefully considered. The most potent, selective and reversible inhibitors of GVIA iPLA2 are polyfluoroketones bearing an aromatic ring and a small aliphatic chain as a spacer between the two funcional groups. The most potent and selective polyfluoroketone inhibitor, namely FKGK11, has been used to show the important role that GVIA iPLA2 plays in EAE, the animal model of multiple sclerosis. Most recently, it has been reported that a combination therapy of GVIA iPLA2 inhibitors (BEL as well as FKGK11) with the anticancer drug paclitaxel is highly effective at blocking development of ovarian cancer.427 As the significance of GVIA iPLA2 emerges and efforts to acquire its crystal structure advance, it seems essential to persist in developing potent and selective GVIA iPLA2 inhibitors.
5. PAF Acetylhydrolases (GVII and GVIII PAF-AH PLA2s)
5.1 Groups, Subgroups, Specificity and Mechanism
Platelet-activating factor (PAF) is a potent phospholipid activator and mediator of many leukocyte functions, particularly as a mediator of inflammation.428 Two groups of PLA2s, designated GVII and GVIII (Table 26), can catalyze the hydrolysis of the acetyl group from the sn-2 position of PAF to produce lyso-PAF and acetate, which is why the enzymes were originally named as PAF acetylhydrolases (PAF-AH).14 GVIIA PLA2 is a secreted enzyme with a molecular weight of 45 kDa that associates with both low-density lipoprotein (LDL) and high-density lipoprotein (HDL) in human plasma429. Therefore, the enzyme is also known as plasma PAF-AH (pPAF-AH) or lipoprotein-associated PLA2 (Lp-PLA2). GVIIB PLA2, also referred to as PAF-AH II, is an intracellular protein with a molecular weight of 40 kDa that has an N-terminal myristoylation site and which shares 41% amino acid sequence identity with GVIIA PLA2.430 GVIII PLA2 is a brain intracellular heterotrimeric protein complex that is also referred to as PAF-AH Ib.431 GVIII PLA2 consists of two 26 kDa catalytic subunits (α subunits) and one 45 kDa non-catalytic regulatory subunit (β subunit or LIS1).432 The catalytic subunits GVIIIA and GVIIIB PLA2, also termed α1 and α2, form catalytically active homo- and hetero-dimers. The α1 and α2 subunits share 69% amino acid sequence identity with each other and do not show significant amino acid sequence identity with GVII PLA2s.
Table 26.
Group VII and Group VIII Phospholipase A2s (PAF-AH)
| Group | Source | Molecular mass (kDa) |
Features | Alternate names |
|---|---|---|---|---|
| VIIA | Human, murine, porcine, bovine | 45 | Secreted α/βhydrolase N-linked glycosylation |
Lipoprotein-associated PLA2 (Lp-PLA2) Plasma PAF-AH (pPAF-AH) |
| VIIB | Human bovine | 40 | Intracellular, myristoylated, α/βhydrolase | PAF-AH II |
| VIIIA | Human | 26 | Intracellular, Homodimer or heterodimer with GVIIIB associates with regulatory β subnunit |
PAF-AH Ib (α1 subunit) |
| VIIIB | Human | 26 | Intracellular, Homodimer or heterodimer with GVIIIA associates with regulatory β subnunit |
PAF-AH Ib (α2 subunit) |
PAF acetylhydrolases are calcium independent PLA2s. These enzymes all feature the same catalytic triad, Ser/His/Asp.20a The secreted GVIIA Lp-PLA2 shows broad substrate specificity. In addition to PAF, GVIIA Lp-PLA2 can also hydrolyze phosphatidylcholines with short chain sn-2 residues.433 Lp-PLA2 hydrolyzes PCs with decreased efficiency when the sn-2 residue is lengthened. The C5 homologue was 60% as efficient as PAF, the C6 homologue was 20% as efficient, and the C9 homologue was only 2% as efficient as PAF.433 However, the enzyme activity is dramatically increased when the ω-end contains an oxidized functional group, such as an aldehyde or a carboxyl.433 When the sn-2 residue terminates with an aldehydic functional group, residues up to C9 are effective substrates for Lp-PLA2. Several studies indicate that Lp-PLA2 can use oxidized phospholipids as substrates.433–434 A mass spectrometry based analysis of the effects of in vitro oxidation in the absence and presence of an irreversible Lp-PLA2 inhibitor on the PC compositions of human LDL has shown that oxidized PCs are recognized as substrates by Lp-PLA2 during LDL oxidation.434b The study shows that oxidatively modified di-unsaturated and poly-unsaturated fatty acid-containing PC species are efficient substrates for Lp-PLA2 and that truncated oxidized PCs are major substrates.434b In a recent study, both Lp-PLA2 and the intracellular GVIIB PAF-AH II have been shown to have the ability to release F2-isoprostanes from esterified phospholipids, though at a much slower rate than they hydrolyze PAF or POVPC.435 Interestingly, both GVII PLA2s show about a 200- to 1000-fold higher affinity for esterified F2-isoprostanes than for PAF and POVPC.435 GVIIB PAF-AH II shows very similar substrate specificity to the plasma form of Lp-PLA2.436 Both forms of GVII PLA2 display PLA1 activity and transacetylase activity that transfers acetic acid from PAF to lysophospholipids.436–437 Neither enzyme distinguishes between an ester and an ether at the sn-1 position of PAF or PAF analogues.20a
Compared with GVII PLA2s, GVIII PLA2 is more restricted at the sn-2 position, selecting only for acetyl groups, but not selective at all for the head groups at the sn-3 position.438 GVIII catalytic homodimers and heterodimers show different activities towards PAF and PAF analogues. The α2/α2 homodimer hydrolyzes PAF and 1-O-alkyl-2-acetyl-sn-glycerol-3-phosphorylethanolamine more efficiently,438 while the α1/α1 homodimer and α1/α2 heterodimer exhibit higher activity towards 1-O-alkyl-2-acetyl-sn-glycerol-3-phosphoric acid.438 The β subunit possesses a regulatory role for catalytic activity in a catalytic dimer composition-dependent manner. The β subunit accelerates PAF hydrolysis of α2/α2 homodimers up to 4-fold, slightly suppresses the activity of α1/α1 homodimers, and has little effect on the activity of the α1/α2 heterodimers.438
5.2 Structural Characteristics and Interaction with Membranes
5.2.1 GVIIA PLA2 (Lp-PLA2)
Lp-PLA2 is a 45 kDa, Ca2+-independent PLA2 that contains a GXSXG motif, which is a characteristic fingerprint for neutral lipases and serine esterases.429 The first 20 residues comprise a hydrophobic signal peptide12. The N-terminus of this protein was found to be heterogeneous in human blood (it can be Ser-35, Ile-42 or Lys-55); the C-terminus is Asn-441.429 While removal of 21 amino acids from the C-terminal residues caused a slight loss of activity, a 30 residue deletion reduced catalysis to below the limit of detection.429 Lp-PLA2 may contain heterogeneous asparagine-conjugated sugar chains at residues Asp-423 and/or Asp-433.439 Though N-linked glycosylation does not affect secretion of the enzyme or its catalytic activity, it may hinder the enzyme’s ability to associate with HDL particles.439a
The crystal structure shows that Lp-PLA2 contains a classic lipase/esterase α/β hydrolase fold and features a catalytic serine/histidine/aspartic acid triad (Figure 13 A).440 A mutagenesis study has shown that residues Trp-115, Leu-116 and Tyr-205 are important for enzyme binding with LDL particles.441 An Lp-PLA2/LDL binding assay using LDL from transgenic mice expressing truncated apoB-100 lipoproteins has demonstrated that the C-terminus of apoB-100 (residues 4119–4279) could be important for LDL binding to Lp-PLA2.441 In fact, in humans who are apoB-100 deficient, Lp-PLA2 was found to be entirely associated with HDL442. A recent study has identified a domain (residues 367–370) that mediates Lp-PLA2/HDL association.443 The data also shows that residues Met-368 and Leu-369 are necessary for binding to HDL and His-367 and Lys-370 may participate in the association as well.443 Based on the above experiments and the solved crystal structure, two surface exposed hydrophobic α helices (residues 114–126 and residues 362–369) (Figure 13A) have been suggested as important for mediating the enzyme’s association with lipoproteins and/or lipid membranes.440 However it’s still possible that other regions may also participate in the lipoprotein association. There is a large patch of carboxylate residues together with three basic residues on the protein surface that may play a role in LDL/HDL lipoprotein partitioning.440
Figure 13.

A. The α/β hydrolase fold of the Group VIIA PLA2 (PAF-AH/Lp-PLA2) crystal structure (PDB entry:3D59). Helixes are shown in purple, β strands in green and loops in light gray. The predicted LDL (residues 114–126) and HDL(residues 362–369) binding surface are shown as indicated. B. Hypothetical model of Lp-PLA2 association with the DMPC lipid membrane surface. The Lp-PLA2 region implicated for Lp-PLA2/liposome association (residues 113–120), is shown in blue and the proposed key residues for Lp-PLA2/liposome association, Trp-115 and Leu-116 are shown in red, as are the catalytic triad residues, Ser-273, Asp-296 and His-351.446
Although it has been earlier suggested that Lp-PLA2 acts on the substrate PAF by a non-interfacial mechanism,444 considering that the surfaces of both LDL and HDL are enriched with phospholipids, it has been expected that under physiological conditions Lp-PLA2 would bind to its substrate(s) from the lipid membrane phase, as is the case for all classic membrane-associating PLA2 enzymes. A recent study by Pande445 has shown that the lipid composition of membrane vesicles affects Lp-PLA2 activity and that membrane binding of Lp-PLA2 increases enzyme activity, which suggests that Lp-PLA2 may also operate by an interfacial mechanism. Recently peptide amide hydrogen-deuterium exchange mass spectrometry (DXMS) was employed to characterize, at the molecular level, the association of Lp-PLA2 with lipid membranes.446 It was found that specific residues 113–120 in one of the enzyme’s surface-exposed hydrophobic α-helices likely mediate liposome binding (Figure 13 B).446 In the resulting model, the active site opens to the solvent, but faces the interfacial surface that accesses substrates from the lipoprotein particles. The active site would theoretically allow substrates to enter from the aqueous phase as well as substrates partitioning into the lipoproteins.
The distribution and location of Lp-PLA2 in LDL/HDL lipoproteins have been suggested to affect Lp-PLA2 function and/or its physiological role.447 An abnormal distribution of the enzyme may correlate with diseases.447 The enzyme localized in LDL was shown to be more active than the same enzyme localized on HDL.442 In vitro study using assays at low PAF concentrations that mimic physiological levels showed that the enzyme associated with HDL particles is inactive.448 Thus the enzyme localized in LDL and HDL may have different functions. One suggestion is that the enzyme associated with HDL could serve as a reservoir for plasma Lp-PLA2 under the circumstance that excess enzyme is present.449 This suggests that the Lp-PLA2 interaction with HDL could be different for the same enzyme interacting with LDL and the enzyme associated with HDL may form a catalyticly unactive conformation. In order to access the substrate from the lipid membrane, Lp-PLA2 would have to either undergo a conformational change or dissociate from the HDL.
Additionally, Lp-PLA2 shows preferential association with dense LDL and with the very high density lipoprotein-1 (VHDL-1) subfraction in human plasma.450 The electronegative LDL subfraction [LDL(−)] has shown 5-fold higher PAF-AH activity than the nonelectronegative LDL subfraction; Lp-PLA2 is mainly associated with the LDL(−) subfraction.451 Lipoprotein(a) [Lp(a)] also accounts for Lp-PLA2 activity to a small extent.20d In addition to lipoproteins, Lp-PLA2 may also associate with microparticles in human plasma.452 Furthermore, different lipoprotein carriers of Lp-PLA2 may result in different roles of the enzyme in atherosclerosis.447
5.2.2 GVIII PLA2 (PAF-AH Ib)
Several three-dimensional crystal structures of GVIII PAF-AH Ib were determined, including the α1/α1 homodimer, the α1/α2 heterodimer and the α2/α2 homodimer complex with LIS1.431,453 PAF-AH Ib contains a single α/β domain with a similar fold to that found in GTPases431. The side chains of Thr-103, Leu-48 and Leu-194, which are all conserved in α1 and α2 isoforms, form a hydrophobic pocket which could only fit a substrate’s acetyl moiety (Figure 14).431 Mutants replacing Thr-103 or Leu-48 or Leu-194 with an alanine residue showed higher relative activity against phospholipids with an sn-2 acyl chain longer than an acetyl453a. This structural characteristic can explain why PAF-AH Ib shows much more strict substrate specificity than other PAF-AHs.
Figure 14.

The crystal structure of Group VIII PLA2 (PAF-AH IB) α1 subunit (PDB entry 1WAB). Helixes are shown in light orange, β strands in purple and loops in light gray. The catalytic triad residues Ser-47, Asp-192 and His-195 are shown in red sticks. The specific pocket is comprised of residues Leu-48, Thr-103 and Leu-194, which defines the enzyme substrate specificity, are shown in blue sticks.
The crystal structure of the LIS1 complex with the α2/α2 homodimer shows that one LIS1 homodimer binds symmetrically to one α2/α2 homodimer via the highly conserved top faces of the LIS1 β propellers.453c A site-directed mutagenesis study has shown that Glu-39 of the murine PAF-AH Ib α2 subunit is crucial for LIS1 binding and the E39D mutation results in a complete loss of LIS1 binding.454 Comparing the α2/α2 structure complexed with LIS1 with α1/α1 and α 1/α2 dimer structures, no major changes were found.453c This indicates that the α2/α2 dimeric structure is not impacted significantly when it associates with LIS1. The mechanism of the β subunit’s regulation of PAF-AH Ib activity requires further investigation.
5.3 Biological Functions and Disease Implications
5.3.1 Lp-PLA2 in Atherosclerosis
Lp-PLA2 is secreted predominantly by macrophages.11,455 Its expression and secretion increase significantly as human monocytes differentiate into macrophages, and increase even more dramatically during activation of macrophages in the atherosclerotic lesion.456 Thus, Lp-PLA2 is considered a very important enzyme for atherosclerotic progression. However, since its discovery, the role of Lp-PLA2 in atherosclerosis has always been a controversial issue. By inactivating PAF and PAF-like lipid mediators and hydrolyzing oxPC in the oxidized LDL particles, Lp-PLA2 could act as a potent anti-atherogenic enzyme. However, growing evidence has shown that Lp-PLA2 may also play a pro-atherosclerotic role. This is because Lp-PLA2 generates the pro-inflammatory and proapoptotic lipid mediators lyso-PC and oxidized nonesterified fatty acids, which play an important role in the development of atherosclerotic necrotic cores. In the current review, we summarize progress in understanding the anti-atherosclerotic and pro-atherosclerotic roles of Lp-PLA2.
Lp-PLA2 was initially considered to be an anti-inflammatory enzyme. Recombinant Lp-PLA2 markedly decreases vascular leakage in pleurisy and paw edema and blocks inflammation.12 Probably the strongest evidence is from a loss of function mutation study. A missense mutation (V279F) found in 4% of the Japanese population leads to a complete loss of enzyme activity457 and Lp-PLA2 deficiency is shown to be an independent risk factor for cardiovascular disease and stroke458. Minimally oxidized LDL obtained from Japanese subjects with this mutation consistently induced greater monocyte adhesion.455 Several other experimental data from either in vitro or animal models also support the protective role of Lp-PLA2. Lp-PLA2-treated mildly oxidized low density lipoprotein (MM-LDL) lost the ability to induce endothelial cells to bind to monocytes.459 Lp-PLA2 destroys the action of MM-LDL by facilitating hydrolysis of bioactive oxidized phospholipids to lysophospholipids.459 Macrophages in both human and rabbit atherosclerotic lesions express Lp-PLA2 and modulation of Lp-PLA2 activity could lead to anti-atherogenic effects in the vessel wall.455 Transgenic apoE deficient mice expressing human Lp-PLA2 have shown increased Lp-PLA2 enzyme activity, decreased oxidized lipoprotein accumulation in the injured vessels, and reduced macrophage homing.460 The same mouse model has also shown that gene transfer of Lp-PLA2 inhibits injury-induced neointima formation and reduces spontaneous atherosclerosis in the absence of mechanical injury.461 In nonhyperlipidemic rabbits the expression of Lp-PLA2 reduces oxLDL accumulation in arteries and exerts anti-inflammatory, antithrombotic, and antiproliferative effects.462 All together, the potential anti-atherosclerotic role of Lp-PLA2 may be attributed to hydrolysis of the oxidized phospholipids and therefore a reduction in the accumulation of oxidized lipoproteins. However, Lp-PLA2 associates mainly with HDL in either mice or rabbits; even in pigs, which have a similar lipoprotein profile to that of humans, 90% of the Lp-PLA2 is associated with HDL and only 5% with LDL.463 In fact, humans are the only mammals where Lp-PLA2 predominantly associates with LDL. So the results from animal models may not be the case in humans and the human enzyme may not protect against atherosclerosis.
Increasing evidence suggests that Lp-PLA2 plays a critical role in the development and progression of atherosclerosis. Epidemiological studies that began in 2000 with about 80,000 participants have shown that increased Lp-PLA2 activity and mass in plasma are associated with increased risk of coronary disease, stroke, and mortality.20e,464 Both the lyso-PC and nonesterified fatty acids that are produced by hydrolyzing oxidized LDL are pro-inflammatory and atherogenic, and they play a critical role in atherosclerosis. Increased levels of Lp-PLA2 and lyso-PC were found in symptomatic carotid artery plaques and the increase is correlated with markers of tissue oxidative stress, inflammation, and instability.465 A study in hypercholesterolemic pigs also shows increased Lp-PLA2 activity is associated with increased levels of lyso-PC, oxidized LDL and inflammation.463 Thus increased Lp-PLA2 levels further accelerate atherosclerosis in the hypercholesterolemic minipig model.463 A recent study has shown that the ratio of Lp-PLA2 to oxLDL is higher in carotid atherosclerotic tissue and plasma than it is in normal tissue and plasma.466 The oxPC/apoB 100 ratio has been shown to be a significant risk factor for cardiovascular disease (CVD) and when associated with high levels of Lp-PLA2 activity, the risk for CVD is increased.456 More evidence suggesting a pro-atherogenic role for Lp-PLA2 comes from an inhibitor study. In vitro experiments using GlaxoSmithKline (GSK) inhibitors SB-222657 (see section 5.4) and SB-677116 demonstrate reduced generation of lyso-PC and oxidized nonesterified fatty acids.434a,467 Lp-PLA2 activity inhibited by SB-222657 showed reduced atherosclerotic plaque development in a 3-month rabbit model.468 A more specific and efficient inhibitor from GSK, SB-480848 (Darapladid), can selectively inhibit Lp-PLA2 and hence reduce the development of advance coronary atherosclerosis in diabetic and hypercholesterolemic swine.469 Thus Lp-PLA2 is now considered a risk factor, a potential biomarker, and a target of therapy in the treatment of cardiovascular disease.
A non-functional V279F allele was first discovered in the Japanese population and was later found in other East Asian populations.470 This null allele is rarely found in Middle Eastern populations and is almost absent in Europeans.471 A recent study of the V279F null mutation population in Japan has shown that there is no reduced risk of Alzheimer’s disease with genetic deficiency of Lp-PLA2.472 However, another study in South Korean men has shown that the V279F null allele carriers are protected from coronary artery disease.473 In addition to V279F, another inactive mutant Q281R is also found in humans.474 Residue 281, near the active site Ser-273, may affect the active site folding and/or substrate binding and therefore causes a loss of enzymatic activity. Other polymorphisms, such as R92H, I198T and A379V, have also been identified, and their presence may correlate with CVD.475 Details regarding these mutations and polymorphisms are in a recent published review.476
5.3.2 Lp-PLA2 and Neonatal Necrotizing Enterocolitis
Several studies have indicated that Lp-PLA2 may play an important role in the pathogenesis of neonatal necrotizing enterocolitis (NEC), which afflicts premature newborn infants and is characterized by an acute onset of intestinal inflammatory necrosis. Human infants with NEC have systemic accumulation of PAF and decreased serum Lp-PLA2 levels.477 A neonatal rat model treated with human recombinant Lp-PLA2 shows a reduced incidence of NEC compared with controls.478 Enteric Lp-PLA2 administration resulted in significant intestinal Lp-PLA2 activity but no serum Lp-PLA2 activity.478 Lp-PLA2 knockout mice show lower mortality rates before 24 h of life compared with wild type controls in response to bacterial exposure, formula feeding, and asphyxia.479 However, the knockout mice have a significantly higher incidence of NEC after 24 h and showed increased expression of intestinal pro-inflammatory mediators compared with wild type controls.479 Therefore, Lp-PLA2 may play a protective role in the development of NEC and exogenous Lp-PLA2 supplementation may help to reduce the incidence of NEC in premature infants.
In addtion to atherosclerosis and neonatal necrotizing enterocolitis, Lp-PLA2 may also be related to severe anaphylaxis. The PAF hydrolysis activity of Lp-PLA2 was found to be significantly lower in patients with fatal anaphylactic reaction to peanuts than control groups and Lp-PLA2 enzymatic activity was inversely correlated with the severity of the anaphylactic response.480
5.3.3 GVIIB PLA2 (PAF-AH II)
The intracellular form of PAF-AH II is predominantly expressed in epithelial cells, such as kidney proximal and distal tubules, intestinal column epithelium, and hepatocytes.481 PAF-AH II is thought to have an antioxidant function. During oxidative stress, PAF-AH II translocates from cytosol to the membrane and protects the cell against oxidative stress induced cell death.482 Overexpression of PAF-AH II suppresses the oxidative stress-induced cell death.482 The antioxidant function may depend on the enzyme’s ability to hydrolyze oxidized phospholipids. The N-myristoylated property of PAF-AH II makes it possible for it to be present both in the cytosol and membranes.
PAF-AH II knockout mice are not phenotypically distinguishable from wild-type mice, although PAF-AH activity was almost abolished in the liver and kidney of knockout mice.481 However, the knockout mice showed a delay in hepatic injury recovery when injected with carbon tetrachloride.481 Moreover, the levels of F2-isoprostane esterified phospholipids in the liver are higher in knockout mice than wild type mice after the injection of carbon tetrachloride.481 As discussed above, plasma containing both PAF-AH and PAF-AH II can efficiently hydrolyze F2-isoprostane esterified phospholipids in vitro.435 Therefore, the accumulation of F2-isoprostane esterified phospholipids in knockout mice should account for the loss of PAF-AH II expression. PAF-AH II may be involved in the metabolism of esterified 8-isoprostaglandin F2α and may protect tissues from oxidative stress-induced injury.481
Overexpression of PAF-AH II also shows protective effects on neurons in a transgenic mouse model of focal cerebral ischemia.483 Overexpression of PAF-AH II in neurons may protect the central nervous system neurons against ischemic damage by hydrolyzing PAF, PAF like lipids and oxidized phospholipids.483
In addition to mammals, PAF-AH II has also been found in C. elegans and it is important for epithelial morphogenesis.484 However, PAF is not present in C. elegans and therefore PAF-AH II seems not to function in a PAF metabolism pathway in C. elegans. There’s really not much evidence to support the idea that GVII PAF-AHs are involved in the PAF metabolism pathway, although the enzyme was first thought to regulate PAF. Additional work must be done to determine the relevant substrate of GVII PLA2s in vivo.
5.3.4 GVIII PLA2 (PAF-AH Ib)
The β subunit of GVIII PAF-AH Ib is a product of the LIS1 gene for type I lissencephaly, a severe developmental brain disorder caused by abnormal neuronal migration. In addition to forming a complex with PAF-AH Ib catalytic subunits α1 and α2, LIS1 interacts with a number of other proteins, such as cytoplasmic dynein, tubulin and NudE. Both PAF-AH Ib catalytic subunits α1 and α2 and LIS1 are expressed at high levels in the brain and testis.485 Haplo insufficiency of LIS1 results in neuronal migration defects in mice and mice that are homozygous null for LIS1 experience early embryonic lethality after implantation.486 Thus, PAF-AH Ib was thought to be important for brain development. However, none of the α1, α2 knockout mice or the α1/α2 double knockout mice exhibit brain abnormalities.485,487 α1 Knockout mice are indistinguishable from wild type mice and neither double knockout mice exhibit brain α2 nor LIS1 levels are changed.485 α2 Knockout male mice show a significant reduction in testis size and both α1 and LIS1 levels are significantly reduced compared with wild type mice.485 Unexpectedly, α1/α2 double knockout mice exhibit severe impairment in spermatogenesis with no significant reduction of LIS1 levels.485,487 These data shows that the PAF-AH Ib catalytic subunits may not be required for brain development and that the catalytic units may mediate the signaling pathway by interacting with LIS1.
In addition to interacting with LIS1, the PAF-AH Ib catalytic αsubunits are shown to interact with the Reelin very low density lipoprotein receptor (VLDLR), binding to the C terminal of VLDLR, but not with the apolipoprotein E receptor 2.488 The binding of α subunits to VLDLR is very specific and requires the NPxY domain and the presence of a leucine residue immediately following the sequence in the VLDLR.488 PAF-AH Ib may have a functional role in Reelin signaling during brain development.
PAF-AH Ib subunits expression levels are shown to be proportional to the expression levels of α-tublin.485 Recently, Bechler and coworkers have shown that the PAF-AH Ib complex can regulate the functional organization of the Golgi complex.489 PAF-AH Ib can stimulate membrane tubules from Golgi complexes in vitro, and the catalytic activity is required.489 But the catalytic activity and LIS1 binding are not required for PAF-AH Ib α1 and α2 to associate with Golgi membranes489. Both PLA2 enzymatic activity and LIS1 are important for maintaining the Golgi structure. Knockdown of either PAF-AH Ib α1 and α2 or LIS1 results in the formation of mini-stacks and inhibits tubule-mediated Golgi assembly and reduces anterograde trafficking.489
5.4 Chemical Inhibitors and Therapeutic Intervention
Although a limited number of synthetic inhibitor classes of GVIIA Lp-PLA2 has been reported, one of them, darapladib, has reached the most advanced step of clinical trials (Phase III). The recent developments on Lp-PLA2 inhibitors are summarized in a number of review articles.22e–i
5.4.1. Azetidinones
In 1998, SmithKline Beecham presented a novel series of monocyclic β-lactams (or azetidinones) as inhibitors of Lp-PLA2.490 Even though these compounds presented only modest inhibition, they gave way to the further investigation on Lp-PLA2 inhibitors and to the first potent azetidinone inhibitor of Lp-PLA2, SB-222657 (102).434a This inhibitor was used to investigate the role of the enzyme in the oxidative modification of lipoproteins, and it was found that the inhibition was stereoselective since SB-222657 presented a Ki of 40 ± 3 nM, while its enantiomer had a Ki of 6.3 ± 0.5 µM.491

Another β-lactam, SB-245713 (103a), also presented inhibitory potency with an IC50 of 5.2 nM, while its ethyl ester (103b), acting as an effective prodrug, was used in a 3 month proof of concept study in Watanabe hereditable hyperlipidaemic rabbits (WHHL rabbits).468,492 Histological analysis of aortic segments showed a decrease in both lesion cross-sectional area and thickness, particularly in segments with the most complex, raised plaque. These results supported the theory that Lp-PLA2 plays a significant role in the development of atherosclerotic plaque and that Lp-PLA2 inhibitors would be effective in blocking the later stages of plaque progression, including stability.468,492 β-Lactams acted as covalent inhibitors and presented a poor pharmacokinetic profile.

5.4.2. Pyrimidones
High throughput screening of a broad compound bank led to the identification of pyrimidones that were reversible inhibitors of Lp-PLA2. For example, compounds 104 and 105 inhibited the enzyme with IC50 values of 54 nM and 1.1 µM, respectively.493

In a series of communications, various modifications were undertaken in those first pyrimidone structures in order to find the most potent inhibitor that would at the same time present excellent in vivo activity and oral bioavailability.494 Based on this series, novel 1-((amidolinked)-alkyl)-pyrimidones were designed as nanomolar inhibitors of human Lp-PLA2. These compounds showed greatly improved activity in isolated plasma, while compounds 106a and 106b (Table 27) were orally active with a good duration of action.495,494a
Table 27.
Group VIIA PLA2 Inhibition by Pyrimidones
 | ||
|---|---|---|
| Compound | R | IC50 (nM) |
| 106a | (E)-(CH2)3CONH(CH2)8CH=CHC8H17 | 0.7 |
| 106b | (Z)-(CH2)3CONH(CH2)8CH=CHC8H17 | 0.4 |
One of the most promising inhibitors was 1-(arylpiperazinylamidoalkyl)-pyrimidone 107 that presented an IC50 value of 20 nM, 77% inhibition in human plasma at 100 nM and was orally active, properties that suggested that it would be an excellent lead.494c In 2002, GlaxoSmithKline presented a series of 1-(biphenylmethylamidoalkyl)-pyrimidones that were highly potent inhibitors of Lp-PLA2 and showed excellent activity in the WHHL rabbit.494d

Based on this series, they discovered a more potent, orally active inhibitor of Lp-PLA2, SB-435495 (108) with an IC50 of 0.06 nM and a suitable profile for evaluation in man.496

Simplification of the pyrimidone 5-substituent of compound 108 led to the inhibitor, SB-480848 (109, Darapladib), that demonstrated excellent in vitro and in vivo profiles and was selected for progression to man.497 SB-480848 presented IC50 0.25 nM against rhLp-PLA2, showed prolonged inhibition of plasma Lp-PLA2 and a good correlation of pharmacodynamic and pharmacokinetic effects. Mechanistic studies indicated this compound to be a freely reversible, non-covalently-bound inhibitor of rhLp-PLA2. Furthermore, the presence of SB-480848 during the copper catalyzed oxidation of human LDL prevented the production of lysoPC (IC50 4 ± 3 nM). Additional in vivo studies showed that SB-480848 had good oral bioavailability, but also presented excellent inhibition of Lp-PLA2 within atherosclerotic plaque after oral administration of 109 to the WHHL rabbit.497

The results of a large case-control study provided strong evidence for an independent and clinically relevant relationship between elevated concentrations of Lp-PLA2 and risk of stable coronary artery disease (CAD) and thus further support to the hypothesis that Lp-PLA2 may be considered as a novel risk marker for CAD.464f The effect of darapladib on plasma Lp-PLA2 activity and cardiovascular biomarkers in patients with stable coronary heart disease or coronary heart disease risk equivalent was studied.498 Darapladib produced sustained inhibition of plasma Lp-PLA2 activity in patients receiving intensive atorvastatin therapy and caused changes in IL-6 and hs-CRP after 12 weeks of darapladib 160 mg suggesting a possible reduction in inflammatory burden. A study compared the effects of 12-month treatment with darapladib or placebo on coronary atheroma deformability and plasma high-sensitivity C-reactive protein in 330 patients with angiographically documented coronary disease.499 The necrotic core volume continued to expand among patients receiving placebo, while Lp-PLA2 inhibition with darapladib prevented necrotic core expansion, a key determinant of plaque vulnerability. In another study, selective inhibition of Lp-PLA2 with darapladib inhibited progression to advanced coronary atherosclerotic lesions and confirmed a crucial role of vascular inflammation independent from hypercholesterolemia in the development of lesions implicated in the pathogenesis of myocardial infarction and stroke.469
A number of review articles published in 2010 summarize the completed preclinical and early phase clinical studies with darapladib.22g–i,500 Two phase III clinical studies are in progress and are expected to be completed by the end of 2012. The Stabilization of Atherosclerotic Plaque by Inhibition of Darapladib Therapy Trial (STABILITY, http://clinicaltrials.gov/, Identifier: NCT00799903) is a phase III randomized, double-blind, parallel-assignment, safety/efficacy study. The Stabilization of Plaques using Darapladib – Thrombolysis in Myocardial Infarction 52 (SOLID-TIMI 52, http://clinicaltrials.gov/, Identifier: NCT01000727) trial will test whether daily administration of darapladib (160 mg po) versus placebo when treatment is initiated within 30 days after an ACS will reduce the risk of CVD death, nonfatal MI or nonfatal stroke.
5.4.3 Summary Status of Lp-PLA2 Inhibitors
Darapladib is a potent, selective inhibitor which is in a phase III trial and should also be useful for mechanistic studies.
6. Lysosomal Phospholipase A2 [Group XV LPLA2]
6.1 Groups, Subgroups, Specificity and Mechanism
GXV PLA2 was first identified in the soluble fraction of Madin-Darby canine kidney (MDCK) cells501 and was subsequently purified from bovine brain.502 The genes encoding this enzyme were also identified in mouse, rat, cow and human (Table 28).503 GXV LPLA2 has high protein sequence identity to a human lecithin: cholesterol acyltransferase-like lysophospholipase (LLPL).504 However GXV LPLA2s do not show significant lecithin cholesterol acyltransferase or lysophospholipase activity under acidic or neutral conditions.504 Instead, the protein possesses Ca2+ independent PLA2 and transacylase activity as well as 1-O-acylceramide synthase (ACS) activity, which esterifies an acyl group with the hydroxyl group in the C-1 position of ceramide using phospholipids as the acyl group donor.502 Therefore, this enzyme was also named ACS. Hiraoka et. al. proposed that the hydrolyzed acyl group is transferred through an enzyme-acyl intermediate to ceramide or water, resulting either in the production of 1-O-acylceramide (ACS activity) or the release of free fatty acids (PLA2 activity).504–505 Thus, ACS activity may be related to PLA2 activity. GXV PLA2 shows optimal enzymatic activity at pH 4.5 and the protein co-localizes with betahexosaminidase, suggesting that the enzyme localizes to lysosomes. Hence, the enzyme is known as a lysosomal PLA2 (LPLA2). In addition, LPLA2 also shows PLA1 activity.
Table 28.
Group XV Lysosomal Phospholipase A2 (LPLA2)
| Group | Source | Molecular mass (kDa) |
Features | Alternate names |
|---|---|---|---|---|
| XV | Human murine bovine | 45 (deglycosylated) | Ser/His/Asp triad, glycosylated, N-terminal signal sequence | ACS, LLPL |
GXV LPLA2 is a water-soluble glycoprotein with a molecular mass of 45 kDa. GXV LPLA2 has a signal sequence cleavage site and several N-linked glycosylation sites.504 Although the enzymatic activity of GXV LPLA2 does not require divalent cations, such as Ca2+ and Mg2+, milimolar Ca2+ or Mg2+ does enhance the activity 502. LPLA2 has shown specificity toward phosphatidylcholine (PC) and phosphatidylethanolamine (PE) and also has positional specificity. LPLA2 is able to hydrolyze the acyl chain at both the sn-1 and sn-2 positions of PC or PE. LPLA2 has a higher preference for the sn-2 position of 1-O-palmitoyl-2-unsaturated PCs and PEs.506 However this is not true if the substrate is PAPC or PAPE. LPLA2 has demonstrated a higher specificity for the sn-1 position over the sn-2 position of PAPC or PAPE.506 The authors suggested that the polyunsaturated acyl chains affect the lipid bilayer packing structure and hence weaken the sn-2 preference of LPLA2. In addition, LPLA2 has shown preference for an unsaturated acyl group over a saturated acyl group of PC. In the cases of 1-stearoyl-2-oleoyl-sn-glycero-3-phosphocholine (SOPC) or 1-oleoyl-2-steraroyl-sn-glycero-3-phosphocholine (OSPC) and 1-oleoyl-2-palmitoyl-sn-glycero-3-phosphocholine (POPC) or 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (OPPC) as substrates, LPLA2 favors the oleoyl group at the sn-2 position of SOPC and POPC or the sn-1 position of OSPC and OPPC.506
LPLA2 contains a GXSXG motif, which is a characteristic fingerprint of neutral lipases and serine esterases. LPLA2 belongs to the α/β hydrolase superfamily and contains the Ser-198, Asp-360 and His-392 catalytic triad. All three residues are required for enzymatic activity and replacement of the catalytic triad with alanine residues totally eliminated the transacylase activity.507 Four cysteine residues (Cys-65, Cys-89, Cys-330, and Cys-371) are conserved between LPLA2 and lecithin cholesterol acyltransferase (LCAT)507. A site-directed mutagensis study has shown that there is one disulfide bond between Cys-65 and Cys-89 and that there are free cysteine residues at Cys-330 and Cys-371, which are required for full expression of LPLA2 activity. Quadruple mutations at all four cysteine residues and double mutations at Cys-65 and Cys-89 and a single mutation at Cys-65 or Cys-89 have been shown to cause total loss of LPLA2 activity.507 However, the double mutations at Cys-330 and Cys-371 and the signal mutations at Cys-330 or Cys-371 only show partially reduced activity.507
LPLA2 shows increased activity towards zwitterionic phospholipids that contain negatively charged lipids, e.g., PA, PE and PS, under acidic conditions.505 This may be due to the electrostatic interaction between LPLA2 and the negatively charged lipid, and the interaction could promote the association of LPLA2 with lipid vesicles. LPLA2 operates using the same interfacial mechanism as the classic membrane-associating PLA2s where substrate exclusively originates from the lipid vesicle. Adding NaCl or increasing the pH can markedly reduce the increase in LPLA2 activity due to negatively charged lipids.505 The increased Na+ concentration could destroy the electrostatic interaction between LPLA2 and the lipid-water interfacial surface.
6.2 Biological Functions and Disease Implications
LPLA2 plays an important role in lysosomal phospholipid degradation. LPLA2 knockout mice (LPLA2−/−) showed an accumulation of PC and PE in alveolar macrophages as well as peritoneal macrophages and spleen, compared with wild type mice.508 This is consistent with the preference of the enzyme for hydrolysis of PC and PE.508 Recombinant LPLA2 protein from HEK293 cells was added to LPLA2−/− mice alveolar macrophages. The uptake of exogenous LPLA2 significantly decreased phospholipid accumulation.509 In contrast, the catalytically inactive LPLA2-protein treated LPLA2−/− mice alveolar macrophages do not show a decrease in phospholipid accumulation, which suggests that LPLA2 enzymatic activity is responsible for the reduction in phospholipid.509
LPLA2 may be involved in surfactant phospholipid catabolism in alveolar macrophages. LPLA2 is highly expressed in alveolar macrophages.503 Granulocyte macrophage colony stimulating factor (GM-CSF) knockout mice, a model of impaired surfactant catabolism, were found to have six times lower LPLA2 activity than wild type mice.503 One year old LPLA2−/− mice showed marked splenomegaly, foam cell formation, and increased lung surfactant phospholipid levels.508 Thus, LPLA2 may be a major enzyme that is responsible for pulmonary surfactant phospholipid degradation.
Several lines of evidence suggest that LPLA2 is involved in phospholipidosis. In addition to the accumulation of phospholipid, LPLA2 knockout mice show the formation of foam cells with lamellar inclusion bodies, which is a hallmark of cellular phospholipidosis.508 LPLA2 has also been shown to be involved in the phospholipidosis induced by cationic amphiphilic drugs (CAD).510 Treatment of MDCK cells with two CADs, 2-{4-[(2-butyl-1-benzofuran-3-yl) carbonyl]-2,6-diiodophenoxy} ethyl) diethylamine (AMIOD) and D-threo-1-phenyl-2-decanoylamino-3-morpholino-1-propanol (PDMP), shows the formation of numerous multi-lamellar inclusion bodies.510 AMIOD and PDMP both inhibited the transacylase activity of the soluble fraction from LPLA2-overexpressed COS-7 cells in a concentration dependent manner.510 CAD induced phospholipidosis may be due to the decreased phospholipid catabolism caused by inhibition of LPLA2 activity.
In apolipoprotein E (apoE) knockout mice, LPLA2 was found in foam cells in severe atherosclerotic lesions.511 LPLA2 and apoE double knockout mice showed increased lesion formation but little effect on the plasma-lipid profile when fed on a normal diet. However the double knockout mice did not show a significant difference in the extent of atherogenesis when fed an atherogenic, Western-style diet.511 Thioglycolate-elicited peritoneal macrophages from the double knockout mice were more sensitive to apoptosis induced by oxLDL.511 All together, this suggests that LPLA2 may play an important role in atherogenesis.
7. Adipose-Specific Phospholipase A2 [GXVI AdPLA]
7.1 Groups, Subgroups, Specificity and Mechanism
Adipose-specific phospholipase A2 (AdPLA) was initially cloned and characterized as a tumor suppressor.512 The enzyme has been considered to be a member of the lecithin retinol acyltransferase (LRAT) family.513 Recently, it was found to exhibit phospholipase A2 activity15a,513–514 and was designated as GXVI PLA2.15a Human AdPLA is expressed ubiquitously in various tissues and is highly expressed in adipose tissue.15 The enzyme contains 162 amino acids and is detected as an 18 kDa protein in an immunoblot.15b,514 In 3T3-L1 adipocytes, AdPLA showed perinuclear localization and was partially co-localized with the endoplasmic reticulum.15a
AdPLA exhibits calcium-independent phospholipase A1 and A2 activities toward PC and PE, but lacks acyltransferase activity.15a,514 Its PLA1 activity is higher than its PLA2 activity.514 The 18kDa AdPLA is distinct from iPLA2 and cPLA2 in size. The GXSXG or GXSGS motif in the active sites of iPLA2 and cPLA2 is not found in any of the LRAT family members. Instead, the LRAT members adopted a conserved His23/Cys113 dyad as the catalytic site.15a,514 Although it uses His in the active site, as does sPLA2, AdPLA does not contain the highly disulfide bonded features or calcium dependency of sPLA2s. Note that other members of the LRAT family, such as the calcium-independent N-acyltransferase (iNAT), also exhibit phospholipase A1/A2 activity.514
7.2 Biological Functions and Disease Implications
AdPLA has been categorized as a Class II tumor suppressor.512 Researchers have found down-regulation of H-Rev107 in human ovarian carcinomas and involvement of H-Rev107 in interferon-dependent cell death.515 AdPLA is also involved in eicosanoid production in adipose tissue. AdPLA-null mice showed reduced adipose tissue mass, triglyceride content and adipose PGE2 levels.15b These knock-out mice exhibit a high rate of lipolysis and increased fatty acid oxidation in adipocytes.15b Studies of AdPLA ablation in mice have indicated the importance of AdPLA in the development of obesity.15b
8. Other Lipases Expressing Phospholipase A2 Activities
The phospholipase A2 superfamily had earlier been categorized into three major types, sPLA2, cPLA2 and iPLA2.13–14 The diversity and growing number of enzymes, whose activity is independent of Ca2+ in addition to GVI iPLA2, including the GVII and GVIII PAF-AHs and GXV lysosomal PLA2, have complicated the classification and there are still some enzymes that display PLA2 functionality or demonstrate homologous sequences to current PLA2 enzymes that are not included in the naming system. We will discuss particularly the Otoconin-90-containing homology domain to sPLA2, the phospholipase B1 containing homology domain to cPLA2, the patatin domain containing proteins containing a homology domain to iPLA2 and peridoxin-6, which is also an acidic lysosomal phospholipase (Table 29).
Table 29.
Summery of Human Proteins Not Traditionally Categorized as Phospholipase A2s, but which Express Phospholipase A2 Activity
| Type | Name | Alternative name |
Homology | Identification | Residues /Molecular Mass(Kd a) |
PLA2 Activity | Other activity | Domain | Swiss-Prot |
|---|---|---|---|---|---|---|---|---|---|
| sPLA2 | Otoconin-90 | PLA2L | sPLA2 | Protein (mouse) | 493/53 | Predicted | sPLA2-like (2) | Q02509 | |
| cPLA2 | PLB1 | Phospholipase B1 | Protein | 1458/163 | Verified | Lyso-lecithin acylhydrolas e | Glycosylation | Q6P1J6 | |
| iPLA2 | PNPLA7 | NTE-related esterase (NRE) | PNPLA6 | Protein | 1317/145 | Lyso- | Patatin-Like | Q6ZV29 | |
| PNPLA5 | GS2-like (GS2L) | Protein | 429/47 | TG-hydrolase | Patatin-Like | Q7Z6Z6 | |||
| PNPLA1 | Transcript | 532/57 | Patatin-Like | Q8N8W4 | |||||
| aiPLA2 | Peroxiredoxin-6 | 1-Cys peroxiredoxin aiPLA2 | Protein | 224/25 | Verified | peroxiredoxi n activity | Thioredoxin domain | P30041 |
8.1 Otoconin-90 (sPLA2 homology)
sPLA2 has been characterized by the properties of its extracellular secretion, small size, highly disulfide bonded and His/Asp dyad active site. One major characteristic that stands out is the highly disulfide bonded PLA2 domain. Otoconin-90/95 (OC90), also named as PLA2-like (PLA2L), contains a similar domain to Groups I, II, V, and X sPLA2s. The human otoconin-90 was identified with 493 amino acids.516 The partial murine otoconin-90 containing 453 residues and two sPLA2 homologous domains were cloned in 1998.516a
Otoconin is the major component of otoconia, which are protein-calcium carbonate crystals of the vestibular system that are indispensable for the perception of gravity in mammals.516 The predominant mammalian otoconin, otoconin-90, is essential for formation of the organic matrix of otoconia by recruiting and associating with other matrix components, which includes otolin and cerebellin-1.517 The arrest of otoconia genesis by NADPH oxidase organizer 1 (Noxo1) inactivation can result in an accumulation of otoconial protein, otoconin-90.518 Otoconin-90 deletion leads to abnormal otoconia formation and physical imbalance but normal hearing in mouse models.519 In vitro, the recombinant Otoconin 90 can facilitate nucleation and inhibit calcite crystal growth in a concentration-dependent manner and induce morphologic changes in native otoconia.520 However, PLA2 activity has not yet been demonstrated in this protein and it has not been categorized as a PLA2.
8.2 Phospholipase B (cPLA2 homolog)
cPLA2 was initially characterized by the cytosolic property and calcium dependent activity. The current members of the cPLA2 type contain a C2 domain, except for the GIVC PLA2, which has high homology in the catalytic domain.14 Phospholipase B (PLB) was mentioned in association with the PLA2 family in our previous review.13 Because its PLA2 activity in a mammalian form has still not been clearly studied to date, and these enzymes are well known in the literature as PLB, we have still not included them as part of the GVI PLA2 type. The PLB family contains PLB1, PLB2 and PLB3. By definition, these enzymes can hydrolyze both sn-1 and sn-2 acyl chains in phospholipid substrates. All PLBs contains the GXSXG serine lipase consensus sequence similar to the cPLA2 group, as well as the other critical Asp active site dyad. Mutations of the Ser146 or Asp392 of the dyad abolish catalytic activity of PLB1 in cryptococcus neoformans.521
PLB’s studied from yeast, PLB1, PLB2 and PLB3 show significant PLB/lysoPL activity and PLB1 was shown to be responsible for much of the PLB and lysoPL activity.522 The P. notatum PLB was shown to be highly glycosylated.523 Although PLB1, PLB2 and PLB3 have been identified in various species, PLB1 is the only human phospholipase B cloned and identified.524 The gene is encoded in chromosome 2p23.2 and found to be expressed in epidermis.524
8.3 PNPLA (iPLA2 homolog)
iPLA2 is currently defined as a calcium independent PLA2. However, there are more and more groups of PLA2 having calcium independent activity. Now, iPLA2 is referred to as the GVI PLA2. In GVI PLA2, the key element of these enzymes is the patatin-like lipase domain and they are also included in the PNPLA family.335,348 Currently, there are 9 members PNPLA1-9 in this family and GVI PLA2s are included in 6 of them.14,335a The PLA2 activities and functions of the other three enzymes, PNPLA1, 5 and 7 are not yet clearly determined.
Patatin is a protein from potatoes and other plants with confirmed PLA1 and PLA2 activity.525 Patatins have the lipase consensus sequence (Gly-Thr-Ser-Thr-Gly) as does the GVIA PLA2. Its crystal structure was determined and showed the same catalytic dyad as cPLA2.371 The sequence alignment and structure modeling showed that patatin has high homology to iPLA2.344 Current studies on PNPLA1 are very limited and are only at the transcript level.335a The mRNA of PNPLA5 has been detected in both mouse and human tissues.335a,348,358b PNPLA5, also named as GS2-like, shows TG hydrolase activity,358b but the results are not consistent and need to be further confirmed.335a The phylogenetic tree shows the PNPLA6 (NTE) and PNPLA7, NTE-related esterase (NRE), are closely related. Based on the sequence homology to NTE, NRE contains three neucleotide binding domains, a patatin-like lipase domain and a GXSXG motif. NRE exhibits lysophospholipase activity, but no phospholipase activity was detected.526 NRE transcript levels are strongly regulated by the nutritional diet and downregulated by indulin.526
8.4 aiPLA2 (Peroxiredoxin-6)
Peroxiredoxin 6 in mammals (Prx6) contains a conserved cysteine at the active site to catalyze the reduction of hydrogen peroxide and alkyl hydroperoxides.527 This 25 kDa enzyme has been suggested to be a bifunctional enzyme that shows both peroxidase activity528 and phospholipase A2 activity.529 It was shown to be a lysosomal protein and has an optimal Ca2+-independent PLA2 activity at pH 4.0,529–530 and also named as acidic calcium-independent PLA2 (aiPLA2) in 1997. It is structurally and functionally different from the GXV LPLA2. Its function of hydrogen peroxide peroxidase activity was identified and named as 1-cysteine peroxiredoxin.528
Because this enzyme is a non-selenium glutathione peroxidase that can reduce oxidized phospholipid hydroperoxides with glutathione as an electron donor,531 the role of the active site serine in a phospholipase catalytic process is questionable.13 However, other researches support the idea that the active site serine provides a phospholipase activity.532
9. Concluding Remarks
In summary, phospholipase A2 has been studied for over a century, first from the venoms of a variety of snakes and later from mammalian pancreatic extracts. However, the emphasis was quite academic in understanding the biological function of PLA2s, mainly as digestive enzymes, and then eventually as the enzymes were purified, the emphasis turned to their structure and function as proteins and their interaction with membranes and micelles. It wasn’t until the mid-1980s that scientists began to appreciate the broader role of PLA2 in inflammatory and other diseases and that they were not just digestive enzymes. By the late 1980’s, there began an explosion of interest in PLA2s due to the isolation, characterization, cloning, and general availability of the pure human recombinant enzymes. With time the specialized role of each type of PLA2 in metabolism has become more and more appreciated. Although prior to the 1990’s, many laboratories worked on developing inhibitors of the pancreatic and venom enzymes, once the pure cloned human non-pancreatic PLA2s were available, numerous academic and industrial laboratories focused on the development of potent and selective inhibitors of these enzymes for specific disease applications.
The first comprehensively focused papers on inhibitors of sPLA2, the oldest type of PLA2 enzyme, appeared in 1985 and at that time the attempts were focused on synthetic phospholipid analogues and on marine natural products. Ten years later, Lilly Research Laboratories developed a class of indole inhibitors and one of them, Varespladib Methyl, was entered into clinical trials for the treatment of severe sepsis. However, the trials terminated at Phase II because the results were not robust. Years later, in 2008, Anthera Pharmaceuticals pursued the same inhibitor, now named A-002 for the treatment of cardiovascular diseases and this inhibitor is currently in Phase III trials. Apart from sPLA2 inhibitors, much effort has been devoted to the discovery of inhibitors for cPLA2. It has to be noted that for both sPLA2 and cPLA2 many structurally different classes of synthetic inhibitors have been reported. However, although cPLA2 is considered to play the major role in inflammatory diseases, only in the mid 2000’s did an inhibitor reach Phase II trials, an indole inhibitor developed by Wyeth (now part of Pfizer) for rheumatoid arthritis. Unfornately, this trial (http://clinicaltrials.gov/, Identifier: NCT00396955) was terminated because of an “imbalance of gastrointestinal and lipase effects”.
Although Lp-PLA2 is the most recently recognized enzyme among the major PLA2 types and although only two chemical classes of inhibitors have been reported for this enzyme, the pyrimidinone derivative Darapladib is at the most advanced clinical trials stage (Phase III). iPLA2 has received little attention up to now as a therapeutic target. However, recent studies on animal models have demonstrated the importance of this PLA2 type in a large variety of pathological conditions, for example, in neurological disorders. In conclusion, the results of the Phase III clinical trials on Darapladib and Varespladib Methyl within the next year should demonstrate whether or not PLA2 inhibitors will become useful in clinical practice for cardiovascular diseases. In addition, we anticipate that the continuing research efforts on cPLA2 and iPLA2 inhibitors may provide new chemical entities as potential novel investigational drugs that may eventually reach evaluation in clinical trials.
Identifying selective inhibitors for the various human PLA2 groups is of paramount importance in the effort to develop PLA2 inhibitors as phamaceutical agents. Selective inhibitors for the major groups that have been especially useful for in vitro mechanistic studies have been reported, for example pyrrophenone for cPLA2, varespladib for sPLA2, pentafluoroketone FKGK11 for iPLA2. However, inhibitors able to selectively inhibit the various sPLA2 groups and subgroups (IIA, V, X) and the various cPLA2 and iPLA2 subgroups are still needed for use in animal and human studies.
Of course, the availability of the pure, cloned human PLA2s covering a variety of types has opened up a large yield of basic research as to their structure and function and how they interact with substrate in the lipid-water interface presented to them as micelles or bilayer membranes. Much is still to be learned in the coming decades about the phospholipase A2 superfamily!
Acknowledgment
We would like to thank the National Institutes of Health RO1-GM20501(EAD) for support of our work on phospholipase A2 enzymes.
Biographies

Edward A. Dennis is Distinguished Professor in the Department of Chemistry & Biochemistry and Department of Pharmacology at the School of Medicine of the University of California, San Diego. He received his BA from Yale University in 1963 and his PhD from Harvard University in 1968. Since completing his postdoctoral fellowship at Harvard Medical School in 1969, Dr. Dennis has been a professor at UCSD, also serving as Chair of the Department of Chemistry and Biochemistry, Chair of the Faculty Academic Senate, and on the Board of Overseers. He has authored over 330 original research publications and edited 13 books. He is currently Editor-in-Chief of the J. Lipid Res. and Director of the LIPID MAPS Consortium (www.LIPIDMAPS.org). He was named an inaugural Fellow of the AAAS in 1984 and was the recipient of the American Society of Biochemistry and Molecular Biology’s Avanti Award in Lipid Enzymology in 2000, the European Federation for Lipid Science and Technology’s European Lipid Science Award in 2008, and the Yale Medal from Yale University in 2008. Dr. Dennis’ career research focus has been on the structure, function, mechanism, and inhibition of the enzyme phospholipase A2 as well as on signal transduction, inflammation, lipid metabolism, eicosanoid action and lipidomics.

Jian Cao received his BS in applied chemistry (2000) and his MS in polymer chemistry (2003) from Jilin University, China and his PhD in biological chemistry (2008) under the supervision of Prof. Debra Dunaway-Mariano from the University of New Mexico. In 2009 he joined the Edward A. Dennis group at the University of California, San Diego as a postdoctoral fellow to study the interaction mechanisms of Group VII phospholipase A2 (PAF-AH) with lipid membranes as well as high density and low density lipoproteins. His research interests include protein structure and function analysis, phospholipase A2 enzymology, and hydrogen/deuterium exchange mass spectrometry.

Yuan-Hao Hsu received his BS from National Chung-Shin University in 1994 and his MS from National Taiwan University in 1996. He moved across the Pacific Ocean to the University of California, Riverside and received an MS in 2001 and a PhD in 2006, both in Biochemistry, with Dr. Jolinda A. Traugh. He joined the Edward A. Dennis group at the University of California, San Diego in 2006 to study the function, activation mechanisms and membrane interactions of cytosolic phospholipase A2 (cPLA2) and calcium independent phospholipase A2 (iPLA2).

Victoria Magrioti studied Chemistry at the University of Athens. In 2003, she obtained her Ph.D. degree in Organic Chemistry under the supervision of Prof. Violetta Constantinou at the Agricultural University of Athens. During her Ph.D. studies she joined for a few months the group of Dr. Robert Verger at the CNRS, Marseille where she studied assays for lipase inhibitors. She continued her postdoctoral studies in the group of Prof. George Kokotos at the University of Athens and in the group of Prof. Alexandros Makriyannis at the Center for Drug Discovery of Northeastern University in Boston. She is currently a Lecturer in the Department of Chemistry, University of Athens. Her research is mainly focused on the design and development of novel inhibitors of lipolytic enzymes, such as human digestive lipases and phospholipase A2s. She is also working on new synthetic methodologies for medicinally interesting enzyme inhibitors.

George Kokotos is Professor of Organic Chemistry and Director of the Organic Chemistry Laboratory at the University of Athens, Greece. He studied chemistry at the University of Athens where he also obtained his Ph.D. (1984). He then conducted postdoctoral work in the Department of Pharmaceutical and Biological Chemistry at the University of London. He has spent a sabbatical leave as a visiting Professor in the Department of Chemistry and Biochemistry at the University of California, San Diego. He has authored over 110 publications in peer-reviewed journals and edited two books on Bioactive Lipids and Lipases. He is also co-inventor of more than 10 international patents. He is currently the Chairman of the Division of Organic and Medicinal Chemistry (The Association of Greek Chemists) and a member of the European Committee of the Division of Organic Chemistry (European Association of Chemical and Molecular Sciences). His research interests include the design and synthesis of bioactive compounds, in particular enzyme inhibitors, amino acid and peptide chemistry, development of new organocatalysts, and applications of enzymes in Organic Chemistry.
References
- 1.Dennis EA. In: The Enzymes. Boyer P, editor. Vol. 16. New York: Academic Press; 1983. [Google Scholar]
- 2.(a) Davidson FF, Dennis EA. J. Mol. Evol. 1990;31:228. doi: 10.1007/BF02109500. [DOI] [PubMed] [Google Scholar]; (b) Davidson FF, Dennis EA. In: Handbook of Natural Toxins. Tu AT, editor. Vol. 5. New York: Marcel Dekker; 1991. [Google Scholar]
- 3.Seilhamer JJ, Pruzanski W, Vadas P, Plant S, Miller JA, Kloss J, Johnson LK. J. Biol. Chem. 1989;264:5335. [PubMed] [Google Scholar]
- 4.Kramer RM, Hession C, Johansen B, Hayes G, McGray P, Chow EP, Tizard R, Pepinsky RB. J. Biol. Chem. 1989;264:5768. [PubMed] [Google Scholar]
- 5.Clark JD, Lin LL, Kriz RW, Ramesha CS, Sultzman LA, Lin AY, Milona N, Knopf JL. Cell. 1991;65:1043. doi: 10.1016/0092-8674(91)90556-e. [DOI] [PubMed] [Google Scholar]
- 6.Kramer RM, Roberts EF, Manetta J, Putnam JE. J. Biol. Chem. 1991;266:5268. [PubMed] [Google Scholar]
- 7.Dennis EA. J. Biol. Chem. 1994;269:13057. [PubMed] [Google Scholar]
- 8.Ackermann EJ, Kempner ES, Dennis EA. J. Biol. Chem. 1994;269:9227. [PubMed] [Google Scholar]
- 9.Tang J, Kriz RW, Wolfman N, Shaffer M, Seehra J, Jones SS. J. Biol. Chem. 1997;272:8567. doi: 10.1074/jbc.272.13.8567. [DOI] [PubMed] [Google Scholar]
- 10.Dennis EA. Trends Biochem. Sci. 1997;22:1. doi: 10.1016/s0968-0004(96)20031-3. [DOI] [PubMed] [Google Scholar]
- 11.Stafforini DM, Elstad MR, McIntyre TM, Zimmerman GA, Prescott SM. J. Biol. Chem. 1990;265:9682. [PubMed] [Google Scholar]
- 12.Tjoelker LW, Wilder C, Eberhardt C, Stafforini DM, Dietsch G, Schimpf B, Hooper S, Le Trong H, Cousens LS, Zimmerman GA, Yamada Y, McIntyre TM, Prescott SM, Gray PM. Nature. 1995;374:549. doi: 10.1038/374549a0. [DOI] [PubMed] [Google Scholar]
- 13.Six DA, Dennis EA. Biochim. Biophys. Acta. 2000;1488:1. doi: 10.1016/s1388-1981(00)00105-0. [DOI] [PubMed] [Google Scholar]
- 14.Schaloske RH, Dennis EA. Biochim. Biophys. Acta. 2006;1761:1246. doi: 10.1016/j.bbalip.2006.07.011. [DOI] [PubMed] [Google Scholar]
- 15.(a) Duncan RE, Sarkadi-Nagy E, Jaworski K, Ahmadian M, Sul HS. J. Biol. Chem. 2008;283:25428. doi: 10.1074/jbc.M804146200. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Jaworski K, Ahmadian M, Duncan RE, Sarkadi-Nagy E, Varady KA, Hellerstein MK, Lee HY, Samuel VT, Shulman GI, Kim KH, de Val S, Kang C, Sul HS. Nat. Med. 2009;15:159. doi: 10.1038/nm.1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.(a) Kudo I, Murakami M. Prostaglandins Other Lipid Mediators. 2002;68–69:3. doi: 10.1016/s0090-6980(02)00020-5. [DOI] [PubMed] [Google Scholar]; (b) Murakami M, Kudo I. J. Biochem. 2002;131:285. doi: 10.1093/oxfordjournals.jbchem.a003101. [DOI] [PubMed] [Google Scholar]; (c) Burke JE, Dennis EA. Cardiovasc. Drugs Ther. 2009;23:49. doi: 10.1007/s10557-008-6132-9. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Burke JE, Dennis EA. J. Lipid Res. 2009;50 Suppl:S237. doi: 10.1194/jlr.R800033-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Scott KF, Sajinovic M, Hein J, Nixdorf S, Galettis P, Liauw W, de Souza P, Dong Q, Graham GG, Russell PJ. Biochimie. 2010;92:601. doi: 10.1016/j.biochi.2010.03.019. [DOI] [PubMed] [Google Scholar]
- 17.(a) Menschikowski M, Hagelgans A, Siegert G. Prostaglandins Other Lipid Mediators. 2006;79:1. doi: 10.1016/j.prostaglandins.2005.10.005. [DOI] [PubMed] [Google Scholar]; (b) Nevalainen TJ, Graham GG, Scott KF. Biochim. Biophys. Acta. 2008;1781:1. doi: 10.1016/j.bbalip.2007.12.001. [DOI] [PubMed] [Google Scholar]; (c) Rosenson RS, Gelb MH. Curr. Cardiol. Rep. 2009;11:445. doi: 10.1007/s11886-009-0064-2. [DOI] [PubMed] [Google Scholar]; (d) Lambeau G, Gelb MH. Annu. Rev. Biochem. 2008;77:495. doi: 10.1146/annurev.biochem.76.062405.154007. [DOI] [PubMed] [Google Scholar]; (e) Boyanovsky BB, Webb NR. Cardiovasc. Drugs Ther. 2009;23:61. doi: 10.1007/s10557-008-6134-7. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Karabina SA, Gora S, Atout R, Ninio E. Biochimie. 2010;92:594. doi: 10.1016/j.biochi.2010.02.002. [DOI] [PubMed] [Google Scholar]; (g) Murakami M, Taketomi Y, Girard C, Yamamoto K, Lambeau G. Biochimie. 2010;92:561. doi: 10.1016/j.biochi.2010.03.015. [DOI] [PubMed] [Google Scholar]
- 18.(a) Ghosh M, Tucker DE, Burchett SA, Leslie CC. Prog. Lipid Res. 2006;45:487. doi: 10.1016/j.plipres.2006.05.003. [DOI] [PubMed] [Google Scholar]; (b) Kita Y, Ohto T, Uozumi N, Shimizu T. Biochim. Biophys. Acta. 2006;1761:1317. doi: 10.1016/j.bbalip.2006.08.001. [DOI] [PubMed] [Google Scholar]; (c) Leslie CC, Gangelhoff TA, Gelb MH. Biochimie. 2010;92:620. doi: 10.1016/j.biochi.2010.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.(a) Balsinde J, Perez R, Balboa MA. Biochim. Biophys. Acta. 2006;1761:1344. doi: 10.1016/j.bbalip.2006.07.013. [DOI] [PubMed] [Google Scholar]; (b) Lei X, Barbour SE, Ramanadham S. Biochimie. 2010;92:627. doi: 10.1016/j.biochi.2010.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Hooks SB, Cummings BS. Biochem. Pharmacol. 2008;76:1059. doi: 10.1016/j.bcp.2008.07.044. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Green JT, Orr SK, Bazinet RP. J. Lipid Res. 2008;49:939. doi: 10.1194/jlr.R700017-JLR200. [DOI] [PubMed] [Google Scholar]; (e) Cedars A, Jenkins CM, Mancuso DJ, Gross RW. J. Cardiovasc. Pharmacol. 2009;53:277. [PMC free article] [PubMed] [Google Scholar]
- 20.(a) Arai H, Koizumi H, Aoki J, Inoue K. J. Biochem. 2002;131:635. doi: 10.1093/oxfordjournals.jbchem.a003145. [DOI] [PubMed] [Google Scholar]; (b) Karasawa K, Harada A, Satoh N, Inoue K, Setaka M. Prog. Lipid Res. 2003;42:93. doi: 10.1016/s0163-7827(02)00049-8. [DOI] [PubMed] [Google Scholar]; (c) McIntyre TM, Prescott SM, Stafforini DM. J. Lipid Res. 2009;50 Suppl:S255. doi: 10.1194/jlr.R800024-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Stafforini DM. Cardiovasc. Drugs Ther. 2009;23:73. doi: 10.1007/s10557-008-6133-8. [DOI] [PubMed] [Google Scholar]; (e) Wilensky RL, Macphee CH. Curr. Opin. Lipidol. 2009;20:415. doi: 10.1097/MOL.0b013e3283307c16. [DOI] [PubMed] [Google Scholar]
- 21.Shayman JA, Kelly R, Kollmeyer J, He Y, Abe A. Prog. Lipid Res. 2011;50:1. doi: 10.1016/j.plipres.2010.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.(a) Lehr M. Expert Opin. Ther. Pat. 2001;11:1123. [Google Scholar]; (b) Reid RC. Curr. Med. Chem. 2005;12:3011. doi: 10.2174/092986705774462860. [DOI] [PubMed] [Google Scholar]; (c) Magrioti V, Kokotos G. Anti-Inflammatory Anti-Allergy Agents Med. Chem. 2006;5:189. [Google Scholar]; (d) Lehr M. Anti-Inflammatory Anti-Allergy Agents Med. Chem. 2006;5:149. [Google Scholar]; (e) Rosenson RS. Cardiovasc. Drugs Ther. 2009;23:93. doi: 10.1007/s10557-008-6148-1. [DOI] [PubMed] [Google Scholar]; (f) Magrioti V, Kokotos G. Expert Opin. Ther. Pat. 2010;20:1. doi: 10.1517/13543770903463905. [DOI] [PubMed] [Google Scholar]; (g) Suckling K. Atherosclerosis. 2010;212:357. doi: 10.1016/j.atherosclerosis.2010.03.011. [DOI] [PubMed] [Google Scholar]; (h) Chauffe RJ, Wilensky RL, Mohler ER., Iii Curr. Atheroscler. Rep. 2010;12:43. doi: 10.1007/s11883-009-0076-9. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Rosenson RS. Curr. Opin. Lipid. 2010;21:473. doi: 10.1097/MOL.0b013e32833eb581. [DOI] [PubMed] [Google Scholar]
- 23.Deems RA, Eaton BR, Dennis EA. J. Biol. Chem. 1975;250:9013. [PubMed] [Google Scholar]
- 24.Carman GM, Deems RA, Dennis EA. J. Biol. Chem. 1995;270:18711. doi: 10.1074/jbc.270.32.18711. [DOI] [PubMed] [Google Scholar]
- 25.Balsinde J, Balboa MA, Insel PA, Dennis EA. Annu. Rev. Pharmacol. Toxicol. 1999;39:175. doi: 10.1146/annurev.pharmtox.39.1.175. [DOI] [PubMed] [Google Scholar]
- 26.Rouault M, Bollinger JG, Lazdunski M, Gelb MH, Lambeau G. Biochemistry. 2003;42:11494. doi: 10.1021/bi0349930. [DOI] [PubMed] [Google Scholar]
- 27.(a) Singer AG, Ghomashchi F, Le Calvez C, Bollinger J, Bezzine S, Rouault M, Sadilek M, Nguyen E, Lazdunski M, Lambeau G, Gelb MH. J. Biol. Chem. 2002;277:48535. doi: 10.1074/jbc.M205855200. [DOI] [PubMed] [Google Scholar]; (b) Bezzine S, Bollinger JG, Singer AG, Veatch SL, Keller SL, Gelb MH. J. Biol. Chem. 2002;277:48523. doi: 10.1074/jbc.M203137200. [DOI] [PubMed] [Google Scholar]
- 28.Guy JE, Stahl U, Lindqvist Y. J. Biol. Chem. 2009;284:19371. doi: 10.1074/jbc.M109.008466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Matoba Y, Katsube Y, Sugiyama M. J. Biol. Chem. 2002;277:20059. doi: 10.1074/jbc.M200263200. [DOI] [PubMed] [Google Scholar]
- 30.(a) Thunnissen MM, Ab E, Kalk KH, Drenth J, Dijkstra BW, Kuipers OP, Dijkman R, de Haas GH, Verheij HM. Nature. 1990;347:689. doi: 10.1038/347689a0. [DOI] [PubMed] [Google Scholar]; (b) Yu L, Dennis EA. Proc. Natl. Acad. Sci. U. S. A. 1991;88:9325. doi: 10.1073/pnas.88.20.9325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Berg OG, Gelb MH, Tsai MD, Jain MK. Chem. Rev. 2001;101:2613. doi: 10.1021/cr990139w. [DOI] [PubMed] [Google Scholar]
- 32.Burke JE, Karbarz MJ, Deems RA, Li S, Woods VL, Jr, Dennis EA. Biochemistry. 2008;47:6451. doi: 10.1021/bi8000962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Scott DL, White SP, Otwinowski Z, Yuan W, Gelb MH, Sigler PB. Science. 1990;250:1541. doi: 10.1126/science.2274785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Han SK, Kim KP, Koduri R, Bittova L, Munoz NM, Leff AR, Wilton DC, Gelb MH, Cho W. J. Biol. Chem. 1999;274:11881. doi: 10.1074/jbc.274.17.11881. [DOI] [PubMed] [Google Scholar]
- 35.Beers SA, Buckland AG, Giles N, Gelb MH, Wilton DC. Biochemistry. 2003;42:7326. doi: 10.1021/bi0343222. [DOI] [PubMed] [Google Scholar]
- 36.Winget JM, Pan YH, Bahnson BJ. Biochim. Biophys. Acta. 2006;1761:1260. doi: 10.1016/j.bbalip.2006.08.002. [DOI] [PubMed] [Google Scholar]
- 37.Pan YH, Bahnson BJ. Biochim. Biophys. Acta. 2010;1804:1443. doi: 10.1016/j.bbapap.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bollinger JG, Diraviyam K, Ghomashchi F, Murray D, Gelb MH. Biochemistry. 2004;43:13293. doi: 10.1021/bi049390i. [DOI] [PubMed] [Google Scholar]
- 39.Pan YH, Yu BZ, Singer AG, Ghomashchi F, Lambeau G, Gelb MH, Jain MK, Bahnson BJ. J. Biol. Chem. 2002;277:29086. doi: 10.1074/jbc.M202531200. [DOI] [PubMed] [Google Scholar]
- 40.Xu W, Yi L, Feng Y, Chen L, Liu J. J. Biol. Chem. 2009;284:16659. doi: 10.1074/jbc.M808029200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fremont DH, Anderson DH, Wilson IA, Dennis EA, Xuong NH. Proc. Natl. Acad. Sci. U. S. A. 1993;90:342. doi: 10.1073/pnas.90.1.342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Segelke BW, Nguyen D, Chee R, Xuong NH, Dennis EA. J. Mol. Biol. 1998;279:223. doi: 10.1006/jmbi.1998.1759. [DOI] [PubMed] [Google Scholar]
- 43.Bahnson BJ. Arch. Biochem. Biophys. 2005;433:96. doi: 10.1016/j.abb.2004.08.013. [DOI] [PubMed] [Google Scholar]
- 44.Harwig SS, Tan L, Qu XD, Cho Y, Eisenhauer PB, Lehrer RI. J. Clin. Invest. 1995;95:603. doi: 10.1172/JCI117704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fenard D, Lambeau G, Valentin E, Lefebvre JC, Lazdunski M, Doglio A. J. Clin. Invest. 1999;104:611. doi: 10.1172/JCI6915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Triggiani M, Granata F, Frattini A, Marone G. Biochim. Biophys. Acta. 2006;1761:1289. doi: 10.1016/j.bbalip.2006.07.003. [DOI] [PubMed] [Google Scholar]
- 47.Sartipy P, Camejo G, Svensson L, Hurt-Camejo E. Adv. Exp. Med. Biol. 2002;507:3. doi: 10.1007/978-1-4615-0193-0_1. [DOI] [PubMed] [Google Scholar]
- 48.Weinrauch Y, Elsbach P, Madsen LM, Foreman A, Weiss J. J. Clin. Invest. 1996;97:250. doi: 10.1172/JCI118399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.(a) Gronroos JO, Laine VJ, Janssen MJ, Egmond MR, Nevalainen TJ. J. Immunol. 2001;166:4029. doi: 10.4049/jimmunol.166.6.4029. [DOI] [PubMed] [Google Scholar]; (b) Gronroos JO, Laine VJ, Nevalainen TJ. J. Infect. Dis. 2002;185:1767. doi: 10.1086/340821. [DOI] [PubMed] [Google Scholar]
- 50.(a) Nevalainen TJ, Aho HJ, Peuravuori H. Invest. Ophthalmol. Vis. Sci. 1994;35:417. [PubMed] [Google Scholar]; (b) Saari KM, Aho V, Paavilainen V, Nevalainen TJ. Invest. Ophthalmol. Vis. Sci. 2001;42:318. [PubMed] [Google Scholar]
- 51.Gronroos JO, Salonen JH, Viander M, Nevalainen TJ, Laine VJ. Scand. J. Immunol. 2005;62:413. doi: 10.1111/j.1365-3083.2005.01678.x. [DOI] [PubMed] [Google Scholar]
- 52.(a) Grass DS, Felkner RH, Chiang MY, Wallace RE, Nevalainen TJ, Bennett CF, Swanson ME. J. Clin. Invest. 1996;97:2233. doi: 10.1172/JCI118664. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Laine VJ, Grass DS, Nevalainen TJ. J. Immunol. 1999;162:7402. [PubMed] [Google Scholar]
- 53.Piris-Gimenez A, Paya M, Lambeau G, Chignard M, Mock M, Touqui L, Goossens PL. J. Immunol. 2005;175:6786. doi: 10.4049/jimmunol.175.10.6786. [DOI] [PubMed] [Google Scholar]
- 54.(a) Weinrauch Y, Abad C, Liang NS, Lowry SF, Weiss J. J. Clin. Invest. 1998;102:633. doi: 10.1172/JCI3121. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Qu XD, Lehrer RI. Infect. Immun. 1998;66:2791. doi: 10.1128/iai.66.6.2791-2797.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Buckland AG, Heeley EL, Wilton DC. Biochim. Biophys. Acta. 2000;1484:195. doi: 10.1016/s1388-1981(00)00018-4. [DOI] [PubMed] [Google Scholar]
- 56.Koduri RS, Gronroos JO, Laine VJ, Le Calvez C, Lambeau G, Nevalainen TJ, Gelb MH. J. Biol. Chem. 2002;277:5849. doi: 10.1074/jbc.M109699200. [DOI] [PubMed] [Google Scholar]
- 57.Beers SA, Buckland AG, Koduri RS, Cho W, Gelb MH, Wilton DC. J. Biol. Chem. 2002;277:1788. doi: 10.1074/jbc.M109777200. [DOI] [PubMed] [Google Scholar]
- 58.Foreman-Wykert AK, Weinrauch Y, Elsbach P, Weiss J. J. Clin. Invest. 1999;103:715. doi: 10.1172/JCI5468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Degousee N, Ghomashchi F, Stefanski E, Singer A, Smart BP, Borregaard N, Reithmeier R, Lindsay TF, Lichtenberger C, Reinisch W, Lambeau G, Arm J, Tischfield J, Gelb MH, Rubin BB. J. Biol. Chem. 2002;277:5061. doi: 10.1074/jbc.M109083200. [DOI] [PubMed] [Google Scholar]
- 60.(a) Paramo L, Lomonte B, Pizarro-Cerda J, Bengoechea JA, Gorvel JP, Moreno E. Eur J. Biochem. 1998;253:452. doi: 10.1046/j.1432-1327.1998.2530452.x. [DOI] [PubMed] [Google Scholar]; (b) Chioato L, Aragao EA, Lopes Ferreira T, Medeiros AI, Faccioli LH, Ward RJ. Biochim. Biophys. Acta. 2007;1768:1247. doi: 10.1016/j.bbamem.2007.01.023. [DOI] [PubMed] [Google Scholar]
- 61.Weiss J, Inada M, Elsbach P, Crowl RM. J. Biol. Chem. 1994;269:26331. [PubMed] [Google Scholar]
- 62.Weiss J, Wright G, Bekkers AC, van den Bergh CJ, Verheij HM. J. Biol. Chem. 1991;266:4162. [PubMed] [Google Scholar]
- 63.(a) Mitsuishi M, Masuda S, Kudo I, Murakami M. Biochem. J. 2006;393:97. doi: 10.1042/BJ20050781. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Mitsuishi M, Masuda S, Kudo I, Murakami M. Biochim. Biophys. Acta. 2007;1771:1389. doi: 10.1016/j.bbalip.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 64.Kim JO, Chakrabarti BK, Guha-Niyogi A, Louder MK, Mascola JR, Ganesh L, Nabel GJ. J. Virol. 2007;81:1444. doi: 10.1128/JVI.01790-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fenard D, Lambeau G, Maurin T, Lefebvre JC, Doglio A. Mol. Pharmacol. 2001;60:341. doi: 10.1124/mol.60.2.341. [DOI] [PubMed] [Google Scholar]
- 66.Boilard E, Lai Y, Larabee K, Balestrieri B, Ghomashchi F, Fujioka D, Gobezie R, Coblyn JS, Weinblatt ME, Massarotti EM, Thornhill TS, Divangahi M, Remold H, Lambeau G, Gelb MH, Arm JP, Lee DM. EMBO Mol. Med. 2010;2:172. doi: 10.1002/emmm.201000072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.(a) Kitsiouli E, Nakos G, Lekka ME. Biochim. Biophys. Acta. 2009;1792:941. doi: 10.1016/j.bbadis.2009.06.007. [DOI] [PubMed] [Google Scholar]; (b) Nevalainen TJ, Haapamaki MM, Gronroos JM. Biochim. Biophys. Acta. 2000;1488:83. doi: 10.1016/s1388-1981(00)00112-8. [DOI] [PubMed] [Google Scholar]
- 68.Dennis EA. Am. J. Respir. Crit. Care Med. 2000;161:S32. doi: 10.1164/ajrccm.161.supplement_1.ltta-7. [DOI] [PubMed] [Google Scholar]
- 69.Murakami M, Shimbara S, Kambe T, Kuwata H, Winstead MV, Tischfield JA, Kudo I. J. Biol. Chem. 1998;273:14411. doi: 10.1074/jbc.273.23.14411. [DOI] [PubMed] [Google Scholar]
- 70.Satake Y, Diaz BL, Balestrieri B, Lam BK, Kanaoka Y, Grusby MJ, Arm JP. J. Biol. Chem. 2004;279:16488. doi: 10.1074/jbc.M313748200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Balestrieri B, Hsu VW, Gilbert H, Leslie CC, Han WK, Bonventre JV, Arm JP. J. Biol. Chem. 2006;281:6691. doi: 10.1074/jbc.M508314200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kim YJ, Kim KP, Han SK, Munoz NM, Zhu X, Sano H, Leff AR, Cho W. J. Biol. Chem. 2002;277:36479. doi: 10.1074/jbc.M205399200. [DOI] [PubMed] [Google Scholar]
- 73.Munoz NM, Kim YJ, Meliton AY, Kim KP, Han SK, Boetticher E, O'Leary E, Myou S, Zhu X, Bonventre JV, Leff AR, Cho W. J. Biol. Chem. 2003;278:38813. doi: 10.1074/jbc.M302476200. [DOI] [PubMed] [Google Scholar]
- 74.(a) Hanasaki K, Ono T, Saiga A, Morioka Y, Ikeda M, Kawamoto K, Higashino K, Nakano K, Yamada K, Ishizaki J, Arita H. J. Biol. Chem. 1999;274:34203. doi: 10.1074/jbc.274.48.34203. [DOI] [PubMed] [Google Scholar]; (b) Bezzine S, Koduri RS, Valentin E, Murakami M, Kudo I, Ghomashchi F, Sadilek M, Lambeau G, Gelb MH. J. Biol. Chem. 2000;275:3179. doi: 10.1074/jbc.275.5.3179. [DOI] [PubMed] [Google Scholar]
- 75.Saiga A, Uozumi N, Ono T, Seno K, Ishimoto Y, Arita H, Shimizu T, Hanasaki K. Prostaglandins Other Lipid Mediators. 2005;75:79. doi: 10.1016/j.prostaglandins.2004.10.001. [DOI] [PubMed] [Google Scholar]
- 76.Henderson WR, Jr, Chi EY, Bollinger JG, Tien YT, Ye X, Castelli L, Rubtsov YP, Singer AG, Chiang GK, Nevalainen T, Rudensky AY, Gelb MH. J. Exp. Med. 2007;204:865. doi: 10.1084/jem.20070029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Murakami M, Koduri RS, Enomoto A, Shimbara S, Seki M, Yoshihara K, Singer A, Valentin E, Ghomashchi F, Lambeau G, Gelb MH, Kudo I. J. Biol. Chem. 2001;276:10083. doi: 10.1074/jbc.M007877200. [DOI] [PubMed] [Google Scholar]
- 78.Granata F, Nardicchi V, Loffredo S, Frattini A, Ilaria Staiano R, Agostini C, Triggiani M. Immunobiology. 2009;214:811. doi: 10.1016/j.imbio.2009.06.006. [DOI] [PubMed] [Google Scholar]
- 79.(a) Granata F, Petraroli A, Boilard E, Bezzine S, Bollinger J, Del Vecchio L, Gelb MH, Lambeau G, Marone G, Triggiani M. J. Immunol. 2005;174:464. doi: 10.4049/jimmunol.174.1.464. [DOI] [PubMed] [Google Scholar]; (b) Triggiani M, Granata F, Oriente A, De Marino V, Gentile M, Calabrese C, Palumbo C, Marone G. J. Immunol. 2000;164:4908. doi: 10.4049/jimmunol.164.9.4908. [DOI] [PubMed] [Google Scholar]; (b) Triggiani M, Granata F, Balestrieri B, Petraroli A, Scalia G, Del Vecchio L, Marone G. J. Immunol. 2003;170:3279. doi: 10.4049/jimmunol.170.6.3279. [DOI] [PubMed] [Google Scholar]; (d) Jo EJ, Lee HY, Lee YN, Kim JI, Kang HK, Park DW, Baek SH, Kwak JY, Bae YS. J. Immunol. 2004;173:6433. doi: 10.4049/jimmunol.173.10.6433. [DOI] [PubMed] [Google Scholar]
- 80.(a) Takasaki J, Kawauchi Y, Yasunaga T, Masuho Y. J. Leukocyte Biol. 1996;60:174. doi: 10.1002/jlb.60.2.174. [DOI] [PubMed] [Google Scholar]; (b) Fuentes L, Hernandez M, Fernandez-Aviles FJ, Crespo MS, Nieto ML. Circ Res. 2002;91:681. doi: 10.1161/01.res.0000038341.34243.64. [DOI] [PubMed] [Google Scholar]
- 81.(a) Kim DK, Fukuda T, Thompson BT, Cockrill B, Hales C, Bonventre JV. Am. J. Physiol. 1995;269:L109. doi: 10.1152/ajplung.1995.269.1.L109. [DOI] [PubMed] [Google Scholar]; (b) Kitsiouli EI, Nakos G, Lekka ME. J. Lipid Res. 1999;40:2346. [PubMed] [Google Scholar]
- 82.Touqui L, Wu YZ. Acta Pharmacol. Sin. 2003;24:1292. [PubMed] [Google Scholar]
- 83.Furue S, Mikawa K, Nishina K, Shiga M, Ueno M, Tomita Y, Kuwabara K, Teshirogi I, Ono T, Hori Y, Matsukawa A, Yoshinaga M, Obara H. Crit. Care Med. 2001;29:719. doi: 10.1097/00003246-200104000-00004. [DOI] [PubMed] [Google Scholar]
- 84.Ohtsuki M, Taketomi Y, Arata S, Masuda S, Ishikawa Y, Ishii T, Takanezawa Y, Aoki J, Arai H, Yamamoto K, Kudo I, Murakami M. J. Biol. Chem. 2006;281:36420. doi: 10.1074/jbc.M607975200. [DOI] [PubMed] [Google Scholar]
- 85.Curfs DM, Ghesquiere SA, Vergouwe MN, van der Made I, Gijbels MJ, Greaves DR, Verbeek JS, Hofker MH, de Winther MP. J. Biol. Chem. 2008;283:21640. doi: 10.1074/jbc.M710584200. [DOI] [PubMed] [Google Scholar]
- 86.(a) Minami T, Tojo H, Shinomura Y, Matsuzawa Y, Okamoto M. Gut. 1994;35:1593. doi: 10.1136/gut.35.11.1593. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Haapamaki MM, Gronroos JM, Nurmi H, Alanen K, Nevalainen TJ. Am. J. Gastroenterol. 1999;94:713. doi: 10.1111/j.1572-0241.1999.00941.x. [DOI] [PubMed] [Google Scholar]
- 87.Kugiyama K, Ota Y, Takazoe K, Moriyama Y, Kawano H, Miyao Y, Sakamoto T, Soejima H, Ogawa H, Doi H, Sugiyama S, Yasue H. Circulation. 1999;100:1280. doi: 10.1161/01.cir.100.12.1280. [DOI] [PubMed] [Google Scholar]
- 88.Mattsson N, Magnussen CG, Ronnemaa T, Mallat Z, Benessiano J, Jula A, Taittonen L, Kahonen M, Juonala M, Viikari JS, Raitakari OT. Arterioscler. Thromb. Vasc. Biol. 2010;30:1861. doi: 10.1161/ATVBAHA.110.204669. [DOI] [PubMed] [Google Scholar]
- 89.Ivandic B, Castellani LW, Wang XP, Qiao JH, Mehrabian M, Navab M, Fogelman AM, Grass DS, Swanson ME, de Beer MC, de Beer F, Lusis AJ. Arterioscler. Thromb. Vasc. Biol. 1999;19:1284. doi: 10.1161/01.atv.19.5.1284. [DOI] [PubMed] [Google Scholar]
- 90.Webb NR, Bostrom MA, Szilvassy SJ, van der Westhuyzen DR, Daugherty A, de Beer FC. Arterioscler. Thromb. Vasc. Biol. 2003;23:263. doi: 10.1161/01.atv.0000051701.90972.e5. [DOI] [PubMed] [Google Scholar]
- 91.Ghesquiere SA, Gijbels MJ, Anthonsen M, van Gorp PJ, van der Made I, Johansen B, Hofker MH, de Winther MP. J. Lipid Res. 2005;46:201. doi: 10.1194/jlr.M400253-JLR200. [DOI] [PubMed] [Google Scholar]
- 92.Tietge UJ, Pratico D, Ding T, Funk CD, Hildebrand RB, Van Berkel T, Van Eck M. J. Lipid Res. 2005;46:1604. doi: 10.1194/jlr.M400469-JLR200. [DOI] [PubMed] [Google Scholar]
- 93.Flood C, Gustafsson M, Pitas RE, Arnaboldi L, Walzem RL, Boren J. Arterioscler. Thromb. Vasc. Biol. 2004;24:564. doi: 10.1161/01.ATV.0000117174.19078.85. [DOI] [PubMed] [Google Scholar]
- 94.(a) Gesquiere L, Cho W, Subbaiah PV. Biochemistry. 2002;41:4911. doi: 10.1021/bi015757x. [DOI] [PubMed] [Google Scholar]; (b) Pruzanski W, Lambeau L, Lazdunsky M, Cho W, Kopilov J, Kuksis A. Biochim. Biophys. Acta. 2005;1736:38. doi: 10.1016/j.bbalip.2005.07.005. [DOI] [PubMed] [Google Scholar]
- 95.Wooton-Kee CR, Boyanovsky BB, Nasser MS, de Villiers WJ, Webb NR. Arterioscler., Thromb., Vasc. Biol. 2004;24:762. doi: 10.1161/01.ATV.0000122363.02961.c1. [DOI] [PubMed] [Google Scholar]
- 96.Boyanovsky BB, van der Westhuyzen DR, Webb NR. J. Biol. Chem. 2005;280:32746. doi: 10.1074/jbc.M502067200. [DOI] [PubMed] [Google Scholar]
- 97.Bostrom MA, Boyanovsky BB, Jordan CT, Wadsworth MP, Taatjes DJ, de Beer FC, Webb NR. Arterioscler., Thromb., Vasc. Biol. 2007;27:600. doi: 10.1161/01.ATV.0000257133.60884.44. [DOI] [PubMed] [Google Scholar]
- 98.Hanasaki K, Yamada K, Yamamoto S, Ishimoto Y, Saiga A, Ono T, Ikeda M, Notoya M, Kamitani S, Arita H. J. Biol. Chem. 2002;277:29116. doi: 10.1074/jbc.M202867200. [DOI] [PubMed] [Google Scholar]
- 99.Karabina SA, Brocheriou I, Le Naour G, Agrapart M, Durand H, Gelb M, Lambeau G, Ninio E. FASEB J. 2006;20:2547. doi: 10.1096/fj.06-6018fje. [DOI] [PubMed] [Google Scholar]
- 100.Shridas P, Bailey WM, Gizard F, Oslund RC, Gelb MH, Bruemmer D, Webb NR. Arterioscler., Thromb., Vasc. Biol. 2010;30:2014. doi: 10.1161/ATVBAHA.110.210237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kimura-Matsumoto M, Ishikawa Y, Komiyama K, Tsuruta T, Murakami M, Masuda S, Akasaka Y, Ito K, Ishiguro S, Morita H, Sato S, Ishii T. Atherosclerosis. 2008;196:81. doi: 10.1016/j.atherosclerosis.2006.08.062. [DOI] [PubMed] [Google Scholar]
- 102.Camejo G, Hurt-Camejo E, Wiklund O, Bondjers G. Atherosclerosis. 1998;139:205. doi: 10.1016/s0021-9150(98)00107-5. [DOI] [PubMed] [Google Scholar]
- 103.Sato H, Kato R, Isogai Y, Saka G, Ohtsuki M, Taketomi Y, Yamamoto K, Tsutsumi K, Yamada J, Masuda S, Ishikawa Y, Ishii T, Kobayashi T, Ikeda K, Taguchi R, Hatakeyama S, Hara S, Kudo I, Itabe H, Murakami M. J. Biol. Chem. 2008;283:33483. doi: 10.1074/jbc.M804628200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hsieh CC, Yen MH, Liu HW, Lau YT. Atherosclerosis. 2000;151:481. doi: 10.1016/s0021-9150(00)00453-6. [DOI] [PubMed] [Google Scholar]
- 105.Gora S, Lambeau G, Bollinger JG, Gelb M, Ninio E, Karabina SA. Biochim. Biophys. Acta. 2006;1761:1093. doi: 10.1016/j.bbalip.2006.08.004. [DOI] [PubMed] [Google Scholar]
- 106.de Winther MP, Kanters E, Kraal G, Hofker MH. Arterioscler., Thromb., Vasc. Biol. 2005;25:904. doi: 10.1161/01.ATV.0000160340.72641.87. [DOI] [PubMed] [Google Scholar]
- 107.Boyanovsky BB, Li X, Shridas P, Sunkara M, Morris AJ, Webb NR. Cytokine. 2010;50:50. doi: 10.1016/j.cyto.2009.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kini RM. Toxicon. 2005;45:1147. doi: 10.1016/j.toxicon.2005.02.018. [DOI] [PubMed] [Google Scholar]
- 109.Valentin E, Lambeau G. Biochim. Biophys. Acta. 2000;1488:59. doi: 10.1016/s1388-1981(00)00110-4. [DOI] [PubMed] [Google Scholar]
- 110.Mandal AK, Zhang Z, Chou JY, Mukherjee AB. FASEB J. 2001;15:1834. doi: 10.1096/fj.00-0831fje. [DOI] [PubMed] [Google Scholar]
- 111.(a) Richmond BL, Boileau AC, Zheng S, Huggins KW, Granholm NA, Tso P, Hui DY. Gastroenterology. 2001;120:1193. doi: 10.1053/gast.2001.23254. [DOI] [PubMed] [Google Scholar]; (b) Eerola LI, Surrel F, Nevalainen TJ, Gelb MH, Lambeau G, Laine VJ. Biochim. Biophys. Acta. 2006;1761:745. doi: 10.1016/j.bbalip.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 112.Huggins KW, Boileau AC, Hui DY. Am. J. Physiol. Endocrinol. Metab. 2002;283:E994. doi: 10.1152/ajpendo.00110.2002. [DOI] [PubMed] [Google Scholar]
- 113.Triggiani M, Giannattasio G, Calabrese C, Loffredo S, Granata F, Fiorello A, Santini M, Gelb MH, Marone G. J. Allergy Clin. Immunol. 2009;124:558. doi: 10.1016/j.jaci.2009.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Hallstrand TS, Lai Y, Ni Z, Oslund RC, Henderson WR, Jr, Gelb MH, Wenzel SE. Clin. Exp. Allergy. 2011;41:801. doi: 10.1111/j.1365-2222.2010.03676.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Hendersen WR, Jr, Oslund RC, Bollinger JG, Ye X, Tien YT, Xue J, Gelb MH. J. Biol. Chem. 2011 doi: 10.1074/jbc.M111.235812. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.(a) Kallajoki M, Alanen KA, Nevalainen M, Nevalainen TJ. Prostate. 1998;35:263. doi: 10.1002/(sici)1097-0045(19980601)35:4<263::aid-pros5>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]; (b) Jiang J, Neubauer BL, Graff JR, Chedid M, Thomas JE, Roehm NW, Zhang S, Eckert GJ, Koch MO, Eble JN, Cheng L. Am. J. Pathol. 2002;160:667. doi: 10.1016/S0002-9440(10)64886-9. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Graff JR, Konicek BW, Deddens JA, Chedid M, Hurst BM, Colligan B, Neubauer BL, Carter HW, Carter JH. Clin. Cancer Res. 2001;7:3857. [PubMed] [Google Scholar]
- 117.Sved P, Scott KF, McLeod D, King NJ, Singh J, Tsatralis T, Nikolov B, Boulas J, Nallan L, Gelb MH, Sajinovic M, Graham GG, Russell PJ, Dong Q. Cancer Res. 2004;64:6934. doi: 10.1158/0008-5472.CAN-03-3018. [DOI] [PubMed] [Google Scholar]
- 118.Dong Z, Liu Y, Scott KF, Levin L, Gaitonde K, Bracken RB, Burke B, Zhai QJ, Wang J, Oleksowicz L, Lu S. Carcinogenesis. 2010;31:1948. doi: 10.1093/carcin/bgq188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.(a) Cormier RT, Hong KH, Halberg RB, Hawkins TL, Richardson P, Mulherkar R, Dove WF, Lander ES. Nat. Genet. 1997;17:88. doi: 10.1038/ng0997-88. [DOI] [PubMed] [Google Scholar]; (b) Belinsky GS, Rajan TV, Saria EA, Giardina C, Rosenberg DW. Mol. Carcinog. 2007;46:106. doi: 10.1002/mc.20271. [DOI] [PubMed] [Google Scholar]; (c) Buhmeida A, Bendardaf R, Hilska M, Laine J, Collan Y, Laato M, Syrjanen K, Pyrhonen S. Ann. Oncol. 2009;20:1230. doi: 10.1093/annonc/mdn783. [DOI] [PubMed] [Google Scholar]
- 120.Cormier RT, Hong KH, Halberg RB, Hawkins TL, Richardson P, Mulherkar R, Dove WF, Lander ES. Nat. Genet. 1997;17:88. doi: 10.1038/ng0997-88. [DOI] [PubMed] [Google Scholar]
- 121.Laye JP, Gill JH. Drug Discov Today. 2003;8:710. doi: 10.1016/s1359-6446(03)02754-5. [DOI] [PubMed] [Google Scholar]
- 122.Reynolds SD, Washburn WN, Deems RA, Dennis EA. In: Methods in Enzymology. Dennis EA, editor. Vol. 197. San Diego: Academic Press; 1991. [Google Scholar]
- 123.Davidson FF, Hajdu J, Dennis EA. Biochem. Biophys. Res. Commun. 1986;137:587. doi: 10.1016/0006-291x(86)91118-6. [DOI] [PubMed] [Google Scholar]
- 124.(a) Gelb MH. J. Am. Chem. Soc. 1986;108:3146. [Google Scholar]; (b) Yuan W, Berman RJ, Gelb MH. J. Am. Chem. Soc. 1987;109:8071. [Google Scholar]
- 125.(a) Yuan W, Gelb MH. J. Am. Chem. Soc. 1988;110:2665. [Google Scholar]; (b) Yuan W, Quinn DM, Sigler PB, Gelb MH. Biochemistry. 1990;29:6082. doi: 10.1021/bi00477a028. [DOI] [PubMed] [Google Scholar]
- 126.(a) Yu L, Deems RA, Hajdu J, Dennis EA. J. Biol. Chem. 1990;265:2657. [PubMed] [Google Scholar]; (b) Yu L, Dennis EA. J. Am. Chem. Soc. 1992;114:8757. [Google Scholar]
- 127.(a) De Haas GH, Van Oort MG, Dijkman R, Verger R. Biochem. Soc. Trans. 1989;17:274. doi: 10.1042/bst0170274. [DOI] [PubMed] [Google Scholar]; (b) De Haas GH, Dijkman R, Van Oort MG, Verger R. Biochim. Biophys. Acta. 1990;1043:75. doi: 10.1016/0005-2760(90)90112-b. [DOI] [PubMed] [Google Scholar]; (c) De Haas GH, Dijkman R, Ransac S, Verger R. Biochim. Biophys. Acta. 1990;1046:249. doi: 10.1016/0005-2760(90)90238-s. [DOI] [PubMed] [Google Scholar]; (d) Ransac S, Deveer AMTJ, Riviere C, Slotboom AJ, Gancet C, Verger R, De Haas GH. Biochim. Biophys Acta. 1992;1123:92. doi: 10.1016/0005-2760(92)90175-u. [DOI] [PubMed] [Google Scholar]
- 128.Wery JP, Schevitz RW, Clawson DK, Bobbitt JL, Dow ER, Gamboa G, Goodson T, Jr, Hermann RB, Kramer RM, McClure DB, Mihelich ED, Putnam JE, Sharp JD, Stark DH, Teater C, Warrick MW, Jones ND. Nature. 1991;352:79. doi: 10.1038/352079a0. [DOI] [PubMed] [Google Scholar]
- 129.(a) Washburn WN, Dennis EA. J. Am. Chem. Soc. 1990;112:2040. [Google Scholar]; (b) Washburn WN, Dennis EA. J. Am. Chem. Soc. 1990;112:2042. [Google Scholar]
- 130.Yu L, Dennis EA. Biochemistry. 1993;32:10185. doi: 10.1021/bi00089a039. [DOI] [PubMed] [Google Scholar]
- 131.Wheeler TN, Blanchard SG, Andrews RC, Fang F, Gray-Nunez Y, Harris CO, Lambert MH, Mehrotra MM, Parks DJ, Ray, Andrew RC. J. Med. Chem. 1994;37:4118. doi: 10.1021/jm00050a009. [DOI] [PubMed] [Google Scholar]
- 132.(a) Tramposch KM, Steiner SA, Stanley PL, Nettleton DO, Franson RC, Lewin AH, Ivy Carroll F. Biochem. Biophys. Res. Commun. 1992;189:272. doi: 10.1016/0006-291x(92)91554-4. [DOI] [PubMed] [Google Scholar]; (b) Tramposch KM, Chilton FH, Stanley PL, Franson RC, Havens MB, Nettleton DO, Davern LB, Darling IM, Bonney RJ. J. Pharmacol. Exp. Ther. 1994;271:852. [PubMed] [Google Scholar]; (c) Burke JR, Gregor KR, Tramposch KM. J. Biol. Chem. 1995;270:274. doi: 10.1074/jbc.270.1.274. [DOI] [PubMed] [Google Scholar]; (d) Springer DM, Luh B-Y, D'Andrea SV, Bronson JJ, Mansuri MM, Burke JR, Gregor KR, Stanley PL, Tramposch KM. Bioorg. Med. Chem. Lett. 1997;7:793. [Google Scholar]
- 133.Springer DM, Luh B-Y, Bronson JJ, McElhone KE, Mansuri MM, Gregor KR, Nettleton DO, Stanley PL, Tramposch KM. Bioorg. Med.l Chem. 2000;8:1087. doi: 10.1016/s0968-0896(00)00047-x. [DOI] [PubMed] [Google Scholar]
- 134.LeMahieu RA, Carson M, Han RJ, Madison VS, Hope WC, Chen T, Morgan DW, Hendrickson HS. J. Med. Chem. 1993;36:3029. doi: 10.1021/jm00072a025. [DOI] [PubMed] [Google Scholar]
- 135.Oinuma H, Takamura T, Hasegawa T, Nomoto K, Naitoh T, Daiku Y, Hamano S, Kakisawa H, Minami N. J. Med. Chem. 1991;34:2260. doi: 10.1021/jm00111a048. [DOI] [PubMed] [Google Scholar]
- 136.(a) Marshall LA, Hall RH, Winkler JD, Badger A, Bolognese B, Roshak A, Flamberg PL, Sung CM, Chabot-Fletcher M, Adams JL. J. Pharmacol. Exp. Ther. 1995;274:1254. [PubMed] [Google Scholar]; (b) Munns MJ, King RG, Rice GE. Prostaglandins Other Lipid Mediators. 1999;57:361. doi: 10.1016/s0090-6980(99)00012-x. [DOI] [PubMed] [Google Scholar]
- 137.Pisabarro MT, Ortiz AR, Palomer A, Cabre F, Garcia L, Wade RC, Gago F, Mauleon D, Carganico G. J. Med. Chem. 1994;37:337. doi: 10.1021/jm00029a004. [DOI] [PubMed] [Google Scholar]
- 138.Jain MK, Ghomashchi F, Yu BZ, Bayburt T, Murphy D, Houck D, Brownell J, Reid JC, Solowiej JE. J. Med. Chem. 1992;35:3584. doi: 10.1021/jm00097a018. [DOI] [PubMed] [Google Scholar]
- 139.Bennion C, Connolly S, Gensmantel NP, Hallam C, Jackson CG, Primrose WU, Roberts GCK, Robinson DH, Slaich PK. J. Med. Chem. 1992;35:2939. doi: 10.1021/jm00094a003. [DOI] [PubMed] [Google Scholar]
- 140.Beaton HG, Bennion C, Connolly S, Cook AR, Gensmantel NP, Hallam C, Hardy K, Hitchin B, Jackson CG, Robinson DH. J. Med. Chem. 1994;37:557. doi: 10.1021/jm00031a001. [DOI] [PubMed] [Google Scholar]
- 141.Cha SS, Lee D, Adams J, Kurdyla JT, Jones CS, Marshall LA, Bolognese B, AbdelMeguid SS, Oh BH. J. Med. Chem. 1996;39:3878. doi: 10.1021/jm960502g. [DOI] [PubMed] [Google Scholar]
- 142.Mouchlis VD, Mavromoustakos TM, Kokotos G. J. Comput.-Aided Mol. Des. 2010;24:107. doi: 10.1007/s10822-010-9319-7. [DOI] [PubMed] [Google Scholar]
- 143.Hansford KA, Reid RC, Clark CI, Tyndall JDA, Whitehouse MW, Guthrie T, McGeary RP, Schafer K, Martin JL, Fairlie DP. ChemBioChem. 2003;4:181. doi: 10.1002/cbic.200390029. [DOI] [PubMed] [Google Scholar]
- 144.Arumugam TV, Arnold N, Proctor LM, Newman M, Reid RC, Hansford KA, Fairlie DP, Shiels IA, Taylor SM. Br. J. Pharmacol. 2003;140:71. doi: 10.1038/sj.bjp.0705402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Woodruff TM, Arumugam TV, Shiels IA, Newman ML, Ross PA, Reid RC, Fairlie DP, Taylor SM. Int. Immunopharmacology. 2005;5:883. doi: 10.1016/j.intimp.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 146.Levick S, Loch D, Rolfe B, Reid RC, Fairlie DP, Taylor SM, Brown L. J. Immunol. 2006;176:7000. doi: 10.4049/jimmunol.176.11.7000. [DOI] [PubMed] [Google Scholar]
- 147.Gregory LS, Kelly WL, Reid RC, Fairlie DP, Forwood MR. Bone. 2006;39:134. doi: 10.1016/j.bone.2005.12.017. [DOI] [PubMed] [Google Scholar]
- 148.Antonopoulou G, Barbayianni E, Magrioti V, Cotton N, Stephens D, Constantinou-Kokotou V, Dennis EA, Kokotos G. Bioorg. Med. Chem. 2008;16:10257. doi: 10.1016/j.bmc.2008.10.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Schevitz RW, Bach NJ, Carlson DG, Chirgadze NY, Clawson DK, Dillard RD, Draheim SE, Hartley LW, Jones ND, Mihelich ED, Olkowski JL, Snyder DW, Sommers C, Wery JP. Nat. Struct. Mol. Biol. 1995;2:458. doi: 10.1038/nsb0695-458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Dillard RD, Bach NJ, Draheim SE, Berry DR, Carlson DG, Chirgadze NY, Clawson DK, Hartley LW, Johnson LM, Jones ND, McKinney ER, Mihelich ED, Olkowski JL, Schevitz RW, Smith AC, Snyder DW, Sommers CD, Wery J-P. J. Med. Chem. 1996;39:5119. doi: 10.1021/jm960485v. [DOI] [PubMed] [Google Scholar]
- 151.Dillard RD, Bach NJ, Draheim SE, Berry DR, Carlson DG, Chirgadze NY, Clawson DK, Hartley LW, Johnson LM, Jones ND, McKinney ER, Mihelich ED, Olkowski JL, Schevitz RW, Smith AC, Snyder DW, Sommers CD, Wery J-P. J. Med. Chem. 1996;39:5137. doi: 10.1021/jm960486n. [DOI] [PubMed] [Google Scholar]
- 152.Draheim SE, Bach NJ, Dillard RD, Berry DR, Carlson DG, Chirgadze NY, Clawson DK, Hartley LW, Johnson LM, Jones ND, McKinney ER, Mihelich ED, Olkowski JL, Schevitz RW, Smith AC, Snyder DW, Sommers CD, Wery J-P. J. Med. Chem. 1996;39:5159. doi: 10.1021/jm960487f. [DOI] [PubMed] [Google Scholar]
- 153.Snyder DW, Bach NJ, Dillard RD, Draheim SE, Carlson DG, Fox N, Roehm NW, Armstrong CT, Chang CH, Hartley LW, Johnson LM, Roman CR, Smith AC, Song M, Fleisch JH. J. Pharmacol. Exp. Ther. 1999;288:1117. [PubMed] [Google Scholar]
- 154.Tomita Y, Kuwabara K, Furue S, Tanaka K, Yamada K, Ueno M, Ono T, Maruyama T, Ajiki T, Onoyama H, Yamamoto M, Hori Y. J. Pharmacol. Sci. 2004;96:144. doi: 10.1254/jphs.fp0040314. [DOI] [PubMed] [Google Scholar]
- 155.Loh A, Macias W, Skerjanec S Eli Lilly & Co. Method for treating sepsis. WO 0205796. Patent. 2002
- 156.Abraham E, Naum C, Bandi V, Gervich D, Lowry SF, Wunderink R, Schein RM, Macias W, Skerjanec S, Dmitrienko A, Farid N, Forgue ST, Jiang F. Crit. Care Med. 2003;31:718. doi: 10.1097/01.CCM.0000053648.42884.89. [DOI] [PubMed] [Google Scholar]
- 157.Tomita Y, Jyoyama H, Kobayashi M, Kuwabara K, Furue S, Ueno M, Yamada K, Ono T, Teshirogi I, Nomura K, Arita H, Okayasu I, Hori Y. Eur. J. Pharmacol. 2003;472:147. doi: 10.1016/s0014-2999(03)01859-4. [DOI] [PubMed] [Google Scholar]
- 158.Bradley JD, Dmitrienko AA, Kivitz AJ, Gluck OS, Weaver AL, Wiesenhutter C, Myers SL, Sides GD. J. Rheumatol. 2005;32:417. [PubMed] [Google Scholar]
- 159.Bowton DL, Dmitrienko AA, Israel E, Zeiher BG, Sides GD. J. Asthma. 2005;42:65. doi: 10.1081/jas-200044748. [DOI] [PubMed] [Google Scholar]
- 160.Hagishita S, Yamada M, Shirahase K, Okada T, Murakami Y, Ito Y, Matsuura T, Wada M, Kato T, Ueno M, Chikazawa Y, Yamada K, Ono T, Teshirogi I, Ohtani M. J. Med. Chem. 1996;39:3636. doi: 10.1021/jm960395q. [DOI] [PubMed] [Google Scholar]
- 161.Yokota Y, Hanasaki K, Ono T, Nakazato H, Kobayashi T, Arita H. Biochim. Biophys. Acta. 1999;1438:213. doi: 10.1016/s1388-1981(99)00053-0. [DOI] [PubMed] [Google Scholar]
- 162.Smart BP, Pan YH, Weeks AK, Bollinger JG, Bahnson BJ, Gelb MH. Bioorg. Med. Chem. 2004;12:1737. doi: 10.1016/j.bmc.2004.01.022. [DOI] [PubMed] [Google Scholar]
- 163.Smart BP, Oslund RC, Walsh LA, Gelb MH. J. Med. Chem. 2006;49:2858. doi: 10.1021/jm060136t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Oslund RC, Cermak N, Gelb MH. J. Med. Chem. 2008;51:4708. doi: 10.1021/jm800422v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Mouchlis VD, Mavromoustakos TM, Kokotos G. J. Chem. Inf. Mod. 2010;50:1589. doi: 10.1021/ci100217k. [DOI] [PubMed] [Google Scholar]
- 166.Zhou L, Fang C, Wei P, Liu S, Liu Y, Lai L. J. Med. Chem. 2008;51:3360. doi: 10.1021/jm7010707. [DOI] [PubMed] [Google Scholar]
- 167.Trias J, Hislop C, Truex P, Fraser H, Odink D, Chadwick S, Gould K Anthera Pharmaceuticals Inc. Treatment of cardiovascular disease and dyslipidemia using secretory phospholipase A2 (sPLA2) inhibitors and sPLA2 inhibitor combination therapies. WO 2008137803. Patent. 2008
- 168.Rosenson RS, Hislop C, McConnell D, Elliott M, Stasiv Y, Wang N, Waters DD. Lancet. 2009;373:649. doi: 10.1016/S0140-6736(09)60403-7. [DOI] [PubMed] [Google Scholar]
- 169.Shaposhnik Z, Wang X, Trias J, Fraser H, Lusis AJ. J. Lipid Res. 2009;50:623. doi: 10.1194/jlr.M800361-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Rosenson RS, Hislop C, Elliott M, Stasiv Y, Goulder M, Waters D. J. Am. Coll. Cardiol. 2010;56:1079. doi: 10.1016/j.jacc.2010.06.015. [DOI] [PubMed] [Google Scholar]
- 171.Aitdafoun M, Mounier C, Heymans F, Binisti C, Cassian B, Godfroid JJ. Biochem. Pharmacol. 1996;51:737. doi: 10.1016/0006-2952(95)02172-8. [DOI] [PubMed] [Google Scholar]
- 172.Assogba L, Ahamada-Himidi A, Habich NMB, Aoun D, Boukli L, Massicot F, Mounier CM, Huet J, Lamouri A, Ombetta JE, Godfroid JJ, Dong CZ, Heymans F. Eur. J. Med. Chem. 2005;40:850. doi: 10.1016/j.ejmech.2005.03.027. [DOI] [PubMed] [Google Scholar]
- 173.Touaibia M, Djimdé A, Cao F, Boilard E, Bezzine S, Lambeau G, Redeuilh C, Lamouri A, Massicot F, Chau F, Dong CZ, Heymans F. J. Med. Chem. 2007;50:1618. doi: 10.1021/jm060082n. [DOI] [PubMed] [Google Scholar]
- 174.Boukli L, Touaibia M, Meddad-Belhabich N, Djimdé A, Park CH, Kim JJ, Yoon JH, Lamouri A, Heymans F. Bioorg. Med. Chem. 2008;16:1242. doi: 10.1016/j.bmc.2007.10.077. [DOI] [PubMed] [Google Scholar]
- 175.Meddad-Belhabich N, Aoun D, Djimdé A, Redeuilh C, Dive G, Massicot F, Chau F, Heymans F, Lamouri A. Bioorg. Med. Chem. 2010;18:3588. doi: 10.1016/j.bmc.2010.03.049. [DOI] [PubMed] [Google Scholar]
- 176.Muller P, Lena G, Boilard E, Bezzine S, Lambeau G, Guichard G, Rognan D. J. Med. Chem. 2006;49:6768. doi: 10.1021/jm0606589. [DOI] [PubMed] [Google Scholar]
- 177.Bridonneau P, Chang YF, O'Connell D, Gill SC, Snyder DW, Johnson L, Goodson T, Jr, Herron DK, Parma DH. J. Med. Chem. 1998;41:778. doi: 10.1021/jm970579k. [DOI] [PubMed] [Google Scholar]
- 178.Thwin MM, Satyanarayanajois SD, Nagarajarao LM, Sato K, Arjunan P, Ramapatna SL, Kumar PV, Gopalakrishnakone P. J. Med. Chem. 2007;50:5938. doi: 10.1021/jm070385x. [DOI] [PubMed] [Google Scholar]
- 179.Lombardo D, Dennis EA. J. Biol. Chem. 1985;260:7234. [PubMed] [Google Scholar]
- 180.Glaser KB, Jacobs RS. Biochem. Pharmacol. 1986;35:449. doi: 10.1016/0006-2952(86)90218-2. [DOI] [PubMed] [Google Scholar]
- 181.Jacobson PB, Marshall LA, sung A, Jacobs RS. Biochem. Pharmacol. 1990;39:1557. doi: 10.1016/0006-2952(90)90521-l. [DOI] [PubMed] [Google Scholar]
- 182.Reynolds LJ, Morgan BP, Hite GA, Mihelich ED, Dennis EA. J. Am. Chem. Soc. 1988;110:5172. [Google Scholar]
- 183.Potts BCM, Faulkner DJ, Decarvalho MS, Jacobs RS. J. Am. Chem. Soc. 1992;114:5093. [Google Scholar]
- 184.Bianco ID, Kelley MJ, Crowl RM, Dennis EA. Biochim. Biophys. Acta. 1995;1250:197. doi: 10.1016/0167-4838(95)00051-u. [DOI] [PubMed] [Google Scholar]
- 185.Haefner B. Drug Discovery Today. 2003;8:536. doi: 10.1016/s1359-6446(03)02713-2. [DOI] [PubMed] [Google Scholar]
- 186.(a) Dal Piaz F, Casapullo A, Randazzo A, Riccio R, Pucci P, Marino G, Gomez-Paloma L. ChemBioChem. 2002;3:664. doi: 10.1002/1439-7633(20020703)3:7<664::AID-CBIC664>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]; (b) Monti MC, Casapullo A, Riccio R, Gomez-Paloma L. Bioorg. Med. Chem. 2004;12:1467. doi: 10.1016/j.bmc.2003.12.038. [DOI] [PubMed] [Google Scholar]
- 187.Monti MC, Casapullo A, Cavasotto CN, Tosco A, Dal Piaz F, Ziemys A, Margarucci L, Riccio R. Chem.-Eur. J. 2009;15:1155. doi: 10.1002/chem.200801512. [DOI] [PubMed] [Google Scholar]
- 188.Monti MC, Casapullo A, Cavasotto CN, Napolitano A, Riccio R. ChemBioChem. 2007;8:1585. doi: 10.1002/cbic.200700217. [DOI] [PubMed] [Google Scholar]
- 189.Tanaka K, Matsutani S, Kanda A, Kato T, Yoshida T. J. Antibiot. 1994;47:631. doi: 10.7164/antibiotics.47.631. [DOI] [PubMed] [Google Scholar]
- 190.Teshirogi I, Matsutani S, Shirahase K, Fujii Y, Yoshida T, Tanaka K, Ohtani M. J. Med. Chem. 1996;39:5183. doi: 10.1021/jm960437a. [DOI] [PubMed] [Google Scholar]
- 191.Miyake A, Yamamoto H, Takebayashi Y, Imai H, Honda K. J. Pharmacol. Exp. Ther. 1992;263:1302. [PubMed] [Google Scholar]
- 192.Miyake A, Yamamoto H, Kubota E, Hamaguchi K, Kouda A, Honda K, Kawashima H. Br. J. Pharmacol. 1993;110:447. doi: 10.1111/j.1476-5381.1993.tb13831.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Oslund RC, Cermak N, Verlinde CLMJ, Gelb MH. Bioorg. Med. Chem. Lett. 2008;18:5415. doi: 10.1016/j.bmcl.2008.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Lindahl M, Tagesson C. Inflammation. 1997;21:347. doi: 10.1023/a:1027306118026. [DOI] [PubMed] [Google Scholar]
- 195.Lattig J, Bohl M, Fischer P, Tischer S, Tietbohl C, Menschikowski M, Gutzeit HO, Metz P, Pisabarro MT. J. Comput.-Aided Mol. Des. 2007;21:473. doi: 10.1007/s10822-007-9129-8. [DOI] [PubMed] [Google Scholar]
- 196.Alonso F, Henson PM, Leslie CC. Biochim. Biophys. Acta. 1986;878:273. doi: 10.1016/0005-2760(86)90156-6. [DOI] [PubMed] [Google Scholar]
- 197.Kramer RM, Checani GC, Deykin A, Pritzker CR, Deykin D. Biochim. Biophys. Acta. 1986;878:394. doi: 10.1016/0005-2760(86)90248-1. [DOI] [PubMed] [Google Scholar]
- 198.(a) Pickard RT, Strifler BA, Kramer RM, Sharp JD. J. Biol. Chem. 1999;274:8823. doi: 10.1074/jbc.274.13.8823. [DOI] [PubMed] [Google Scholar]; (b) Song C, Chang XJ, Bean KM, Proia MS, Knopf JL, Kriz RW. J. Biol. Chem. 1999;274:17063. doi: 10.1074/jbc.274.24.17063. [DOI] [PubMed] [Google Scholar]
- 199.Underwood KW, Song C, Kriz RW, Chang XJ, Knopf JL, Lin LL. J. Biol. Chem. 1998;273:21926. doi: 10.1074/jbc.273.34.21926. [DOI] [PubMed] [Google Scholar]
- 200.(a) Ohto T, Uozumi N, Hirabayashi T, Shimizu T. J. Biol. Chem. 2005;280:24576. doi: 10.1074/jbc.M413711200. [DOI] [PubMed] [Google Scholar]; (b) Chiba H, Michibata H, Wakimoto K, Seishima M, Kawasaki S, Okubo K, Mitsui H, Torii H, Imai Y. J. Biol. Chem. 2004;279:12890. doi: 10.1074/jbc.M305801200. [DOI] [PubMed] [Google Scholar]
- 201.Tay A, Simon JS, Squire J, Hamel K, Jacob HJ, Skorecki K. Genomics. 1995;26:138. doi: 10.1016/0888-7543(95)80093-2. [DOI] [PubMed] [Google Scholar]
- 202.(a) Dessen A, Tang J, Schmidt H, Stahl M, Clark JD, Seehra J, Somers WS. Cell. 1999;97:349. doi: 10.1016/s0092-8674(00)80744-8. [DOI] [PubMed] [Google Scholar]; (b) Perisic O, Fong S, Lynch DE, Bycroft M, Williams RL. J. Biol. Chem. 1998;273:1596. doi: 10.1074/jbc.273.3.1596. [DOI] [PubMed] [Google Scholar]
- 203.(a) Channon JY, Leslie CC. J. Biol. Chem. 1990;265:5409. [PubMed] [Google Scholar]; (b) Gijon MA, Spencer DM, Kaiser AL, Leslie CC. J. Cell. Biol. 1999;145:1219. doi: 10.1083/jcb.145.6.1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.(a) Pickard RT, Chiou XG, Strifler BA, DeFelippis MR, Hyslop PA, Tebbe AL, Yee YK, Reynolds LJ, Dennis EA, Kramer RM, Sharp JD. J. Biol. Chem. 1996;271:19225. doi: 10.1074/jbc.271.32.19225. [DOI] [PubMed] [Google Scholar]; (b) Reynolds LJ, Hughes LL, Louis AI, Kramer RM, Dennis EA. Biochim. Biophys. Acta. 1993;1167:272. doi: 10.1016/0005-2760(93)90229-3. [DOI] [PubMed] [Google Scholar]; (c) Sharp JD, Pickard RT, Chiou XG, Manetta JV, Kovacevic S, Miller JR, Varshavsky AD, Roberts EF, Strifler BA, Brems DN, Kramer RM. J. Biol. Chem. 1994;269:23250. [PubMed] [Google Scholar]
- 205.Lin LL, Wartmann M, Lin AY, Knopf JL, Seth A, Davis RJ. Cell. 1993;72:269. doi: 10.1016/0092-8674(93)90666-e. [DOI] [PubMed] [Google Scholar]
- 206.Nakamura H, Hirabayashi T, Shimizu M, Murayama T. Biochem. Pharmacol. 2006;71:850. doi: 10.1016/j.bcp.2005.12.027. [DOI] [PubMed] [Google Scholar]
- 207.(a) Leslie CC, Channon JY. Biochim. Biophys. Acta. 1990;1045:261. doi: 10.1016/0005-2760(90)90129-l. [DOI] [PubMed] [Google Scholar]; (b) Mosior M, Six DA, Dennis EA. J. Biol. Chem. 1998;273:2184. doi: 10.1074/jbc.273.4.2184. [DOI] [PubMed] [Google Scholar]; (c) Tamiya-Koizumi K, Umekawa H, Yoshida S, Ishihara H, Kojima K. Biochim. Biophys. Acta. 1989;1002:182. doi: 10.1016/0005-2760(89)90285-3. [DOI] [PubMed] [Google Scholar]
- 208.Leslie CC, Voelker DR, Channon JY, Wall MM, Zelarney PT. Biochim. Biophys. Acta. 1988;963:476. doi: 10.1016/0005-2760(88)90316-5. [DOI] [PubMed] [Google Scholar]
- 209.Ghomashchi F, Naika GS, Bollinger JG, Aloulou A, Lehr M, Leslie CC, Gelb MH. J. Biol. Chem. 2010;285:36100. doi: 10.1074/jbc.M110.165647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Clissold PM, Ponting CP. Trends Biochem. Sci. 2001;26:7. doi: 10.1016/s0968-0004(00)01700-x. [DOI] [PubMed] [Google Scholar]
- 211.Ghosh M, Loper R, Gelb MH, Leslie CC. J. Biol. Chem. 2006;281:16615. doi: 10.1074/jbc.M601770200. [DOI] [PubMed] [Google Scholar]
- 212.Lucas KK, Dennis EA. Biochim. Biophys. Acta. 2004;1636:213. doi: 10.1016/j.bbalip.2003.12.009. [DOI] [PubMed] [Google Scholar]
- 213.(a) Leslie CC. J. Biol. Chem. 1991;266:11366. [PubMed] [Google Scholar]; (b) Loo RW, Conde-Frieboes K, Reynolds LJ, Dennis EA. J. Biol. Chem. 1997;272:19214. doi: 10.1074/jbc.272.31.19214. [DOI] [PubMed] [Google Scholar]
- 214.Jenkins CM, Han X, Yang J, Mancuso DJ, Sims HF, Muslin AJ, Gross RW. Biochemistry. 2003;42:11798. doi: 10.1021/bi034611q. [DOI] [PubMed] [Google Scholar]
- 215.Ghosh M, Loper R, Ghomashchi F, Tucker DE, Bonventre JV, Gelb MH, Leslie CC. J. Biol. Chem. 2007;282:11676. doi: 10.1074/jbc.M608458200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Xu GY, McDonagh T, Yu HA, Nalefski EA, Clark JD, Cumming DA. J. Mol. Biol. 1998;280:485. doi: 10.1006/jmbi.1998.1874. [DOI] [PubMed] [Google Scholar]
- 217.Burke JE, Babakhani A, Gorfe AA, Kokotos G, Li S, Woods VL, Jr, McCammon JA, Dennis EA. J. Am. Chem. Soc. 2009;131:8083. doi: 10.1021/ja900098y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Hsu YH, Burke JE, Stephens DL, Deems RA, Li S, Asmus KM, Woods VL, Jr, Dennis EA. J. Biol. Chem. 2008;283:9820. doi: 10.1074/jbc.M708143200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Stahelin RV, Subramanian P, Vora M, Cho W, Chalfant CE. J. Biol. Chem. 2007;282:20467. doi: 10.1074/jbc.M701396200. [DOI] [PubMed] [Google Scholar]
- 220.Six DA, Dennis EA. J. Biol. Chem. 2003;278:23842. doi: 10.1074/jbc.M301386200. [DOI] [PubMed] [Google Scholar]
- 221.(a) Borsch-Haubold AG, Bartoli F, Asselin J, Dudler T, Kramer RM, Apitz-Castro R, Watson SP, Gelb MH. J. Biol. Chem. 1998;273:4449. doi: 10.1074/jbc.273.8.4449. [DOI] [PubMed] [Google Scholar]; (b) Hefner Y, Borsch-Haubold AG, Murakami M, Wilde JI, Pasquet S, Schieltz D, Ghomashchi F, Yates JR, 3rd, Armstrong CG, Paterson A, Cohen P, Fukunaga R, Hunter T, Kudo I, Watson SP, Gelb MH. J. Biol. Chem. 2000;275:37542. doi: 10.1074/jbc.M003395200. [DOI] [PubMed] [Google Scholar]; (c) Muthalif MM, Hefner Y, Canaan S, Harper J, Zhou H, Parmentier JH, Aebersold R, Gelb MH, Malik KU. J. Biol. Chem. 2001;276:39653. doi: 10.1074/jbc.M103136200. [DOI] [PubMed] [Google Scholar]
- 222.Burke JE, Hsu YH, Deems RA, Li S, Woods VL, Jr, Dennis EA. J. Biol. Chem. 2008;283:31227. doi: 10.1074/jbc.M804492200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Balsinde J, Balboa MA, Li WH, Llopis J, Dennis EA. J. Immunol. 2000;164:5398. doi: 10.4049/jimmunol.164.10.5398. [DOI] [PubMed] [Google Scholar]
- 224.Das S, Cho W. J. Biol. Chem. 2002;277:23838. doi: 10.1074/jbc.M202322200. [DOI] [PubMed] [Google Scholar]
- 225.(a) Casas J, Valdearcos M, Pindado J, Balsinde J, Balboa MA. J. Lipid Res. 2010;51:388. doi: 10.1194/jlr.M001461. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Casas J, Meana C, Esquinas E, Valdearcos M, Pindado J, Balsinde J, Balboa MA. J. Immunol. 2009;183:2767. doi: 10.4049/jimmunol.0901530. [DOI] [PubMed] [Google Scholar]
- 226.Lamour NF, Chalfant CE. Mol. Interv. 2005;5:358. doi: 10.1124/mi.5.6.8. [DOI] [PubMed] [Google Scholar]
- 227.Chalfant CE, Spiegel S. J. Cell. Sci. 2005;118:4605. doi: 10.1242/jcs.02637. [DOI] [PubMed] [Google Scholar]
- 228.Makiyama T, Nagasaka N, Houjyo Y, Yamaura E, Nakamura H, Koide Y, Nishida A, Murayama T. Biochem. Pharmacol. 2010;80:1396. doi: 10.1016/j.bcp.2010.07.028. [DOI] [PubMed] [Google Scholar]
- 229.(a) Pettus BJ, Bielawska A, Subramanian P, Wijesinghe DS, Maceyka M, Leslie CC, Evans JH, Freiberg J, Roddy P, Hannun YA, Chalfant CE. J. Biol. Chem. 2004;279:11320. doi: 10.1074/jbc.M309262200. [DOI] [PubMed] [Google Scholar]; (b) Subramanian P, Stahelin RV, Szulc Z, Bielawska A, Cho W, Chalfant CE. J. Biol. Chem. 2005;280:17601. doi: 10.1074/jbc.M414173200. [DOI] [PubMed] [Google Scholar]
- 230.Gubern A, Barcelo-Torns M, Barneda D, Lopez JM, Masgrau R, Picatoste F, Chalfant CE, Balsinde J, Balboa MA, Claro E. J. Biol. Chem. 2009;284:32359. doi: 10.1074/jbc.M109.061515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Lamour NF, Subramanian P, Wijesinghe DS, Stahelin RV, Bonventre JV, Chalfant CE. J. Biol. Chem. 2009;284:26897. doi: 10.1074/jbc.M109.001677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Subramanian P, Vora M, Gentile LB, Stahelin RV, Chalfant CE. J. Lipid Res. 2007;48:2701. doi: 10.1194/jlr.M700356-JLR200. [DOI] [PubMed] [Google Scholar]
- 233.Nakamura H, Wakita S, Suganami A, Tamura Y, Hanada K, Murayama T. J. Lipid Res. 2010;51:720. doi: 10.1194/jlr.M002428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Nemenoff RA, Winitz S, Qian NX, Van Putten V, Johnson GL, Heasley LE. J. Biol. Chem. 1993;268:1960. [PubMed] [Google Scholar]
- 235.de Carvalho MG, McCormack AL, Olson E, Ghomashchi F, Gelb MH, Yates JR, 3rd, Leslie CC. J. Biol. Chem. 1996;271:6987. doi: 10.1074/jbc.271.12.6987. [DOI] [PubMed] [Google Scholar]
- 236.Tucker DE, Ghosh M, Ghomashchi F, Loper R, Suram S, John BS, Girotti M, Bollinger JG, Gelb MH, Leslie CC. J. Biol. Chem. 2009;284:9596. doi: 10.1074/jbc.M807299200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.(a) Bayburt T, Gelb MH. Biochemistry. 1997;36:3216. doi: 10.1021/bi961659d. [DOI] [PubMed] [Google Scholar]; (b) de Carvalho MG, Garritano J, Leslie CC. J. Biol. Chem. 1995;270:20439. doi: 10.1074/jbc.270.35.20439. [DOI] [PubMed] [Google Scholar]
- 238.Das S, Rafter JD, Kim KP, Gygi SP, Cho W. J. Biol. Chem. 2003;278:41431. doi: 10.1074/jbc.M304897200. [DOI] [PubMed] [Google Scholar]
- 239.Tian W, Wijewickrama GT, Kim JH, Das S, Tun MP, Gokhale N, Jung JW, Kim KP, Cho W. J. Biol. Chem. 2008;283:3960. doi: 10.1074/jbc.M707345200. [DOI] [PubMed] [Google Scholar]
- 240.Lio YC, Dennis EA. Biochim. Biophys. Acta. 1998;1392:320. doi: 10.1016/s0005-2760(98)00049-6. [DOI] [PubMed] [Google Scholar]
- 241.Uozumi N, Shimizu T. Prostaglandins Other Lipid Mediators. 2002;68–69:59. doi: 10.1016/s0090-6980(02)00021-7. [DOI] [PubMed] [Google Scholar]
- 242.(a) Buczynski MW, Dumlao DS, Dennis EA. J. Lipid Res. 2009;50:1015. doi: 10.1194/jlr.R900004-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Rouzer CA, Marnett LJ. J. Lipid Res. 2009;50 Suppl:S29. doi: 10.1194/jlr.R800042-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Simmons DL, Botting RM, Hla T. Pharmacol. Rev. 2004;56:387. doi: 10.1124/pr.56.3.3. [DOI] [PubMed] [Google Scholar]; (d) Smith WL, DeWitt DL, Garavito RM. Annu. Rev. Biochem. 2000;69:145. doi: 10.1146/annurev.biochem.69.1.145. [DOI] [PubMed] [Google Scholar]
- 243.(a) Murphy RC, Hammarstrom S, Samuelsson B. Proc. Natl. Acad. Sci. U. S. A. 1979;76:4275. [PubMed] [Google Scholar]; (b) Sigal E, Craik CS, Highland E, Grunberger D, Costello LL, Dixon RA, Nadel JA. Biochem. Biophys. Res. Commun. 1988;157:457. doi: 10.1016/s0006-291x(88)80271-7. [DOI] [PubMed] [Google Scholar]
- 244.Vignola MJ, Kashatus DF, Taylor GA, Counter CM, Valdivia RH. J. Biol. Chem. 2010;285:21625. doi: 10.1074/jbc.M110.103010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.(a) Nakanishi M, Rosenberg DW. Biochim. Biophys. Acta. 2006;1761:1335. doi: 10.1016/j.bbalip.2006.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Vanhoutte PM. Clin. Pharmacol. Ther. 2009;86:212. doi: 10.1038/clpt.2009.108. [DOI] [PubMed] [Google Scholar]; (c) Wang YX, Ulu A, Zhang LN, Hammock B. Curr. Atheroscler. Rep. 2010;12:174. doi: 10.1007/s11883-010-0108-5. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Raichel L, Berger S, Hadad N, Kachko L, Karter M, Szaingurten-Solodkin I, Williams RO, Feldmann M, Levy R. Eur J. Immunol. 2008;38:2905. doi: 10.1002/eji.200838545. [DOI] [PubMed] [Google Scholar]
- 246.(a) Bonventre JV, Huang Z, Taheri MR, O'Leary E, Li E, Moskowitz MA, Sapirstein A. Nature. 1997;390:622. doi: 10.1038/37635. [DOI] [PubMed] [Google Scholar]; (b) Adler DH, Cogan JD, Phillips JA, 3rd, Schnetz-Boutaud N, Milne GL, Iverson T, Stein JA, Brenner DA, Morrow JD, Boutaud O, Oates JA. J. Clin. Invest. 2008;118:2121. doi: 10.1172/JCI30473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Cao Y, Pearman AT, Zimmerman GA, McIntyre TM, Prescott SM. Proc. Natl. Acad. Sci. U. S. A. 2000;97:11280. doi: 10.1073/pnas.200367597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Perez R, Matabosch X, Llebaria A, Balboa MA, Balsinde J. J. Lipid Res. 2006;47:484. doi: 10.1194/jlr.M500397-JLR200. [DOI] [PubMed] [Google Scholar]
- 249.Caro AA, Cederbaum AI. Free Radical Biol. Med. 2006;40:364. doi: 10.1016/j.freeradbiomed.2005.10.044. [DOI] [PubMed] [Google Scholar]
- 250.Im DS. Acta Pharmacol. Sin. 2010;31:1213. doi: 10.1038/aps.2010.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.De Matteis MA, Luini A. Nat. Rev. Mol. Cell Biol. 2008;9:273. doi: 10.1038/nrm2378. [DOI] [PubMed] [Google Scholar]
- 252.Herbert SP, Ponnambalam S, Walker JH. Mol. Biol. Cell. 2005;16:3800. doi: 10.1091/mbc.E05-02-0164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Regan-Klapisz E, Krouwer V, Langelaar-Makkinje M, Nallan L, Gelb M, Gerritsen H, Verkleij AJ, Post JA. Mol. Biol. Cell. 2009;20:4225. doi: 10.1091/mbc.E08-02-0210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.(a) Grewal S, Ponnambalam S, Walker JH. J. Cell Sci. 2003;116:2303. doi: 10.1242/jcs.00446. [DOI] [PubMed] [Google Scholar]; (b) Choukroun GJ, Marshansky V, Gustafson CE, McKee M, Hajjar RJ, Rosenzweig A, Brown D, Bonventre JV. J. Clin. Invest. 2000;106:983. doi: 10.1172/JCI8914. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) San Pietro E, Capestrano M, Polishchuk EV, DiPentima A, Trucco A, Zizza P, Mariggio S, Pulvirenti T, Sallese M, Tete S, Mironov AA, Leslie CC, Corda D, Luini A, Polishchuk RS. PLoS Biol. 2009;7 doi: 10.1371/journal.pbio.1000194. e1000194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Levy R. Biochim. Biophys. Acta. 2006;1761:1323. doi: 10.1016/j.bbalip.2006.09.004. [DOI] [PubMed] [Google Scholar]
- 256.Shmelzer Z, Haddad N, Admon E, Pessach I, Leto TL, Eitan-Hazan Z, Hershfinkel M, Levy R. J. Cell Biol. 2003;162:683. doi: 10.1083/jcb.200211056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Dana R, Leto TL, Malech HL, Levy R. J. Biol. Chem. 1998;273:441. doi: 10.1074/jbc.273.1.441. [DOI] [PubMed] [Google Scholar]
- 258.Zhu D, Hu C, Sheng W, Tan KS, Haidekker MA, Sun AY, Sun GY, Lee JC. Biochem. J. 2009;421:201. doi: 10.1042/BJ20090356. [DOI] [PubMed] [Google Scholar]
- 259.Shmelzer Z, Karter M, Eisenstein M, Leto TL, Hadad N, Ben-Menahem D, Gitler D, Banani S, Wolach B, Rotem M, Levy R. J. Biol. Chem. 2008;283:31898. doi: 10.1074/jbc.M804674200. [DOI] [PubMed] [Google Scholar]
- 260.Adler DH, Phillips JA, 3rd, Cogan JD, Iverson TM, Schnetz-Boutaud N, Stein JA, Brenner DA, Milne GL, Morrow JD, Boutaud O, Oates JA. J. Gastroenterol. 2009;44 Suppl 19:1. doi: 10.1007/s00535-008-2253-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Uozumi N, Kume K, Nagase T, Nakatani N, Ishii S, Tashiro F, Komagata Y, Maki K, Ikuta K, Ouchi Y, Miyazaki J, Shimizu T. Nature. 1997;390:618. doi: 10.1038/37622. [DOI] [PubMed] [Google Scholar]
- 262.Tabuchi S, Uozumi N, Ishii S, Shimizu Y, Watanabe T, Shimizu T. Acta Neurochir. Suppl. 2003;86:169. doi: 10.1007/978-3-7091-0651-8_36. [DOI] [PubMed] [Google Scholar]
- 263.Nakatani N, Uozumi N, Kume K, Murakami M, Kudo I, Shimizu T. Biochem. J. 2000;352(Pt 2):311. [PMC free article] [PubMed] [Google Scholar]
- 264.(a) Nagase T, Uozumi N, Aoki-Nagase T, Terawaki K, Ishii S, Tomita T, Yamamoto H, Hashizume K, Ouchi Y, Shimizu T. Am. J. Physiol. Lung Cell Mol. Physiol. 2003;284:L720. doi: 10.1152/ajplung.00396.2002. [DOI] [PubMed] [Google Scholar]; (b) Nagase T, Uozumi N, Ishii S, Kume K, Izumi T, Ouchi Y, Shimizu T. Nat. Immunol. 2000;1:42. doi: 10.1038/76897. [DOI] [PubMed] [Google Scholar]
- 265.Nagase T, Uozumi N, Ishii S, Kita Y, Yamamoto H, Ohga E, Ouchi Y, Shimizu T. Nat. Med. 2002;8:480. doi: 10.1038/nm0502-480. [DOI] [PubMed] [Google Scholar]
- 266.Hegen M, Sun L, Uozumi N, Kume K, Goad ME, Nickerson-Nutter CL, Shimizu T, Clark JD. J. Exp. Med. 2003;197:1297. doi: 10.1084/jem.20030016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Marusic S, Leach MW, Pelker JW, Azoitei ML, Uozumi N, Cui J, Shen MW, DeClercq CM, Miyashiro JS, Carito BA, Thakker P, Simmons DL, Leonard JP, Shimizu T, Clark JD. J. Exp. Med. 2005;202:841. doi: 10.1084/jem.20050665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Ii H, Yokoyama N, Yoshida S, Tsutsumi K, Hatakeyama S, Sato T, Ishihara K, Akiba S. PLoS One. 2009;4:e8089. doi: 10.1371/journal.pone.0008089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Oikawa Y, Yamato E, Tashiro F, Yamamoto M, Uozumi N, Shimada A, Shimizu T, Miyazaki J. FEBS Lett. 2005;579:3975. doi: 10.1016/j.febslet.2005.06.024. [DOI] [PubMed] [Google Scholar]
- 270.Downey P, Sapirstein A, O'Leary E, Sun TX, Brown D, Bonventre JV. Am. J. Physiol. Renal. Physiol. 2001;280:F607. doi: 10.1152/ajprenal.2001.280.4.F607. [DOI] [PubMed] [Google Scholar]
- 271.Caiazza F, Harvey BJ, Thomas W. Mol. Endocrinol. 2010;24:953. doi: 10.1210/me.2009-0293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.(a) Takaku K, Sonoshita M, Sasaki N, Uozumi N, Doi Y, Shimizu T, Taketo MM. J. Biol. Chem. 2000;275:34013. doi: 10.1074/jbc.C000585200. [DOI] [PubMed] [Google Scholar]; (b) Hong KH, Bonventre JC, O'Leary E, Bonventre JV, Lander ES. Proc. Natl. Acad. Sci. U. S. A. 2001;98:3935. doi: 10.1073/pnas.051635898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Ilsley JN, Nakanishi M, Flynn C, Belinsky GS, De Guise S, Adib JN, Dobrowsky RT, Bonventre JV, Rosenberg DW. Cancer Res. 2005;65:2636. doi: 10.1158/0008-5472.CAN-04-3446. [DOI] [PubMed] [Google Scholar]
- 274.Meyer AM, Dwyer-Nield LD, Hurteau GJ, Keith RL, O'Leary E, You M, Bonventre JV, Nemenoff RA, Malkinson AM. Carcinogenesis. 2004;25:1517. doi: 10.1093/carcin/bgh150. [DOI] [PubMed] [Google Scholar]
- 275.Lagorce-Pages C, Paraf F, Wendum D, Martin A, Flejou JF. Virchows Arch. 2004;444:426. doi: 10.1007/s00428-004-1003-7. [DOI] [PubMed] [Google Scholar]
- 276.Linkous AG, Yazlovitskaya EM, Hallahan DE. J. Natl. Cancer Inst. 2010;102:1398. doi: 10.1093/jnci/djq290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Street IP, Lin HK, Laliberte F, Ghomashchi F, Wang Z, Perrier H, Tremblay NM, Huang Z, Weech PK, Gelb MH. Biochemistry. 1993;32:5935. doi: 10.1021/bi00074a003. [DOI] [PubMed] [Google Scholar]
- 278.Trimble LA, Street IP, Perrier H, Tremblay NM, Weech PK, Bernstein MA. Biochemistry. 1993;32:12560. doi: 10.1021/bi00210a002. [DOI] [PubMed] [Google Scholar]
- 279.CondeFrieboes K, Reynolds LJ, Lio YC, Hale MR, Wasserman HH, Dennis EA. J. Am. Chem. Soc. 1996;118:5519. [Google Scholar]
- 280.AmandiBurgermeister E, Tibes U, Kaiser BM, Friebe WG, Scheuer WV. Eur. J. Pharmacol. 1997;326:237. doi: 10.1016/s0014-2999(97)85419-2. [DOI] [PubMed] [Google Scholar]
- 281.Ghomashchi F, Loo R, Balsinde J, Bartoli F, Apitz-Castro R, Clark JD, Dennis EA, Gelb MH. Biochim. Biophys. Acta. 1999;1420:45. doi: 10.1016/s0005-2736(99)00056-5. [DOI] [PubMed] [Google Scholar]
- 282.Leis HJ, Windischhofer W. Br. J. Pharmacol. 2008;155:731. doi: 10.1038/bjp.2008.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Riendeau D, Guay J, Weech PK, Laliberte F, Yergey J, Li C, Desmarais S, Perrier H, Liu S, Nicoll-Griffith D. J. Biol. Chem. 1994;269:15619. [PubMed] [Google Scholar]
- 284.Li Q, Cathcart MK. J. Biol. Chem. 1997;272:2404. doi: 10.1074/jbc.272.4.2404. [DOI] [PubMed] [Google Scholar]
- 285.Bate C, Reid S, Williams A. J. Biol. Chem. 2004;279:36405. doi: 10.1074/jbc.M404086200. [DOI] [PubMed] [Google Scholar]
- 286.Yeo JF, Ong WY, Ling SF, Farooqui AA. Pain. 2004;112:148. doi: 10.1016/j.pain.2004.08.009. [DOI] [PubMed] [Google Scholar]
- 287.Lucas KK, Svensson CI, Hua XY, Yaksh TL, Dennis EA. Br. J. Pharmacol. 2005;144:940. doi: 10.1038/sj.bjp.0706116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Kalyvas A, David S. Neuron. 2004;41:323. doi: 10.1016/s0896-6273(04)00003-0. [DOI] [PubMed] [Google Scholar]
- 289.Sanchez-Mejia RO, Newman JW, Toh S, Yu GQ, Zhou YU, Halabisky B, Cisse M, Scearce-Levie K, Cheng IH, Gan L, Palop JJ, Bonventre JV, Mucke L. Nat. Neurosci. 2008;11:1311. doi: 10.1038/nn.2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Svensson CI, Lucas KK, Hua XY, Powell HC, Dennis EA, Yaksh TL. Neuroscience. 2005;133:543. doi: 10.1016/j.neuroscience.2005.01.024. [DOI] [PubMed] [Google Scholar]
- 291.(a) Banville J, Remillard R, Balasubramanian N, Bouthillier G, Martel A Bristol-Myers Squibb Company. Alpha-amino,-thio,-oxo substituted ketones as phospholipase inhibitors. 37875. US Patent. 2002; (b) Banville J, Plamondon S, Gai Y, Balasubramanian N Bristol-Myers Squibb Company. Alpha-substituted thio,-oxo trifluoromethylketones as phospholipase inhibitors. 68722. US Patent. 2002; (c) Banville J, Marinier A, Gai Y, Plamondon S, Roy S, Balasubramanian N Bristol-Myers Squibb Co. Alpha-and beta- substituted trifluoromethyl ketones as phospholipase inhibitors. 65246. US Patent. 2002
- 292.Burke JR, Davern LB, Stanley PL, Gregor KR, Banville J, Remillard R, Russell JW, Brassil PJ, Witmer MR, Johnson G, Tredup JA, Tramposch KM. J. Pharmacol. Exp. Ther. 2001;298:376. [PubMed] [Google Scholar]
- 293.Seno K, Okuno T, Nishi K, Murakami Y, Watanabe F, Matsuura T, Wada M, Fujii Y, Yamada M, Ogawa T, Okada T, Hashizume H, Kii M, Hara S, Hagishita S, Nakamoto S, Yamada K, Chikazawa Y, Ueno M, Teshirogi I, Ono T, Ohtani M. J. Med. Chem. 2000;43:1041. doi: 10.1021/jm9905155. [DOI] [PubMed] [Google Scholar]
- 294.Ghomashchi F, Stewart A, Hefner Y, Ramanadham S, Turk J, Leslie CC, Gelb MH. Biochim. Biophys. Acta. 2001;1513:160. doi: 10.1016/s0005-2736(01)00349-2. [DOI] [PubMed] [Google Scholar]
- 295.Seno K, Okuno T, Nishi K, Murakami Y, Yamada K, Nakamoto S, Ono T. Bioorg. Med. Chem. Lett. 2001;11:587. doi: 10.1016/s0960-894x(01)00003-8. [DOI] [PubMed] [Google Scholar]
- 296.Ono T, Yamada K, Chikazawa Y, Ueno M, Nakamoto S, Okuno T, Seno K. Biochem. J. 2002;363:727. doi: 10.1042/0264-6021:3630727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Flamand N, Picard S, Lemieux L, Pouliot M, Bourgoin SG, Borgeat P. Br. J. Pharmacol. 2006;149:385. doi: 10.1038/sj.bjp.0706879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Tai N, Kuwabara K, Kobayashi M, Yamada K, Ono T, Seno K, Gahara Y, Ishizaki J, Hori Y. Inflammation Res. 2010;59:53. doi: 10.1007/s00011-009-0069-8. [DOI] [PubMed] [Google Scholar]
- 299.(a) Lehr M. Eur. J. Pharm. Sci. 1994;2:91. [Google Scholar]; (b) Lehr M. Arch. Pharm. 1996;329:483. doi: 10.1002/ardp.19963291103. [DOI] [PubMed] [Google Scholar]; (c) Lehr M. Eur. J. Med. Chem. 1997;32:805. [Google Scholar]; (d) Lehr M. J. Med. Chem. 1997;40:3381. doi: 10.1021/jm970045j. [DOI] [PubMed] [Google Scholar]; (e) Lehr M, Elfringhoff AS. Arch. Pharm. 2000;333:312. doi: 10.1002/1521-4184(20009)333:9<312::aid-ardp312>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 300.Lehr M. Arch. Pharm. 1996;329:386. doi: 10.1002/ardp.19963290803. [DOI] [PubMed] [Google Scholar]
- 301.(a) Lehr M. J. Med. Chem. 1997;40:2694. doi: 10.1021/jm960863w. [DOI] [PubMed] [Google Scholar]; (b) Lehr M, Klimt M, Elfringhoff AS. Bioorg. Med. Chem. Lett. 2001;11:2569. doi: 10.1016/s0960-894x(01)00472-3. [DOI] [PubMed] [Google Scholar]
- 302.Ghasemi A, Schulze Elfringhoff A, Lehr M. J. Enzyme Inhib. Med. Chem. 2005;20:429. doi: 10.1080/14756360500228338. [DOI] [PubMed] [Google Scholar]
- 303.(a) Seehra JS, McKew JC, Lovering F, Bemis JE, Xiang Y, Chen L, Knopf JL Genetics Institute, INC. Inhibitors of phospholipase enzymes. WO 9943654. Patent. 1999; (b) Seehra JS, Xiang Y, Bemis JE, McKew JC, Kaila N, Chen L Genetics Institute, INC. Inhibitors of phospholipase A2. WO 9943672. Patent. 1999
- 304.McKew JC, Lovering F, Clark JD, Bemis J, Xiang Y, Shen M, Zhang W, Alvarez JC, Joseph-McCarthy D. Bioorg. Med. Chem. Lett. 2003;13:4501. doi: 10.1016/j.bmcl.2003.08.070. [DOI] [PubMed] [Google Scholar]
- 305.McKew JC, Foley MA, Thakker P, Behnke ML, Lovering FE, Sum FW, Tam S, Wu K, Shen MWH, Zhang W, Gonzalez M, Liu S, Mahadevan A, Sard H, Khor SP, Clark JD. J. Med. Chem. 2006;49:135. doi: 10.1021/jm0507882. [DOI] [PubMed] [Google Scholar]
- 306.Lee KL, Foley MA, Chen L, Behnke ML, Lovering FE, Kirincich SJ, Wang W, Shim J, Tam S, Shen MWH, Khor S, Xu X, Goodwin DG, Ramarao MK, Nickerson-Nutter C, Donahue F, Ku MS, Clark JD, McKew JC. J. Med. Chem. 2007;50:1380. doi: 10.1021/jm061131z. [DOI] [PubMed] [Google Scholar]
- 307.McKew JC, Lee KL, Shen MWH, Thakker P, Foley MA, Behnke ML, Hu B, Sum FW, Tam S, Hu Y, Chen L, Kirincich SJ, Michalak R, Thomason J, Ipek M, Wu K, Wooder L, Ramarao MK, Murphy EA, Goodwin DG, Albert L, Xu X, Donahue F, Ku MS, Keith J, Nickerson-Nutter CL, Abraham WM, Williams C, Hegen M, Clark JD. J. Med. Chem. 2008;51:3388. doi: 10.1021/jm701467e. [DOI] [PubMed] [Google Scholar]
- 308.Lamothe J, Lee K, Schelling S, Stedman N, Leach M, McKew J, Clark J, Nickerson-Nutter C, Hegen M. Clin. Immun. 2008;127:S89. [Google Scholar]
- 309.Lee KL, Behnke ML, Foley MA, Chen L, Wang W, Vargas R, Nunez J, Tam S, Mollova N, Xu X, Shen MWH, Ramarao MK, Goodwin DG, Nickerson-Nutter CL, Abraham WM, Williams C, Clark JD, McKew JC. Bioorg. Med. Chem. 2008;16:1345. doi: 10.1016/j.bmc.2007.10.060. [DOI] [PubMed] [Google Scholar]
- 310.Chen L, Wang W, Lee KL, Shen MWH, Murphy EA, Zhang W, Xu X, Tam S, Nickerson-Nutter C, Goodwin DG, Clark JD, McKew JC. J. Med. Chem. 2009;52:1156. doi: 10.1021/jm8009876. [DOI] [PubMed] [Google Scholar]
- 311.Gopalsamy A, Yang H, Ellingboe JW, McKew JC, Tam S, Joseph-McCarthy D, Zhang W, Shen M, Clark JD. Bioorg. Med. Chem. Lett. 2006;16:2978. doi: 10.1016/j.bmcl.2006.02.067. [DOI] [PubMed] [Google Scholar]
- 312.Kirincich SJ, Xiang J, Green N, Tam S, Yang HY, Shim J, Shen MWH, Clark JD, McKew JC. Bioorg. Med. Chem. 2009;17:4383. doi: 10.1016/j.bmc.2009.05.027. [DOI] [PubMed] [Google Scholar]
- 313.(a) Kokotos G, Kotsovolou S, Six DA, Constantinou-Kokotou V, Beltzner CC, Dennis EA. J. Med. Chem. 2002;45:2891. doi: 10.1021/jm025538p. [DOI] [PubMed] [Google Scholar]; (b) Kokotos G, Six DA, Loukas V, Smith T, Constantinou-Kokotou V, Hadjipavlou-Litina D, Kotsovolou S, Chiou A, Beltzner CC, Dennis EA. J. Med. Chem. 2004;47:3615. doi: 10.1021/jm030485c. [DOI] [PubMed] [Google Scholar]
- 314.(a) Constantinou-Kokotou V, Peristeraki A, Kokotos CG, Six DA, Dennis EA. J. Pept. Sci. 2005;11:431. doi: 10.1002/psc.628. [DOI] [PubMed] [Google Scholar]; (b) Stephens D, Barbayianni E, Constantinou-Kokotou V, Peristeraki A, Six DA, Cooper J, Harkewicz R, Deems RA, Dennis EA, Kokotos G. J. Med. Chem. 2006;49:2821. doi: 10.1021/jm050993h. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Six DA, Barbayianni E, Loukas V, Constantinou-Kokotou V, Hadjipavlou-Litina D, Stephens D, Wong AC, Magrioti V, Moutevelis-Minakakis P, Baker SF, Dennis EA, Kokotos G. J. Med. Chem. 2007;50:4222. doi: 10.1021/jm0613673. [DOI] [PubMed] [Google Scholar]
- 315.Yaksh TL, Kokotos G, Svensson CI, Stephens D, Kokotos CG, Fitzsimmons B, Hadjipavlou-Litina D, Hua XY, Dennis EA. J. Pharmacol. Exp. Ther. 2006;316:466. doi: 10.1124/jpet.105.091686. [DOI] [PubMed] [Google Scholar]
- 316.(a) Moutevelis-Minakakis P, Neokosmidi A, Filippakou M, Stephens D, Dennis EA, Kokotos G. J. Pept. Sci. 2007;13:634. doi: 10.1002/psc.889. [DOI] [PubMed] [Google Scholar]; (b) Antonopoulou G, Magrioti V, Stephens D, Constantinou-Kokotou V, Dennis EA, Kokotos G. J. Pept. Sci. 2008;14:1111. doi: 10.1002/psc.1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Barbayianni E, Stephens D, Grkovich A, Magrioti V, Hsu YH, Dolatzas P, Kalogiannidis D, Dennis EA, Kokotos G. Bioorg. Med. Chem. 2009;17:4833. doi: 10.1016/j.bmc.2009.03.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Conolly S, Mete A. ASTRAZENECA UK LTD. Novel compounds. WO 0034254. Patent. 2000
- 319.Connolly S, Bennion C, Botterell S, Croshaw PJ, Hallam C, Hardy K, Hartopp P, Jackson CG, King SJ, Lawrence L, Mete A, Murray D, Robinson DH, Smith GM, Stein L, Walters I, Wells E, Withnall WJ. J. Med. Chem. 2002;45:1348. doi: 10.1021/jm011050x. [DOI] [PubMed] [Google Scholar]
- 320.Walters I, Bennion C, Connolly S, Croshaw PJ, Hardy K, Hartopp P, Jackson CG, King SJ, Lawrence L, Mete A, Murray D, Robinson DH, Stein L, Wells E, Withnall WJ. Bioorg. Med. Chem. Lett. 2004;14:3645. doi: 10.1016/j.bmcl.2004.05.024. [DOI] [PubMed] [Google Scholar]
- 321.Ludwig J, Bovens S, Brauch C, Elfringhoff AS, Lehr M. J. Med. Chem. 2006;49:2611. doi: 10.1021/jm051243a. [DOI] [PubMed] [Google Scholar]
- 322.Hess M, Schulze Elfringhoff A, Lehr M. Bioorg. Med. Chem. 2007;15:2883. doi: 10.1016/j.bmc.2007.02.016. [DOI] [PubMed] [Google Scholar]
- 323.Fritsche A, Elfringhoff AS, Fabian J, Lehr M. Bioorg. Med. Chem. 2008;16:3489. doi: 10.1016/j.bmc.2008.02.019. [DOI] [PubMed] [Google Scholar]
- 324.Bovens S, Kaptur M, Elfringhoff AS, Lehr M. Bioorg. Med. Chem. Lett. 2009;19:2107. doi: 10.1016/j.bmcl.2009.03.019. [DOI] [PubMed] [Google Scholar]
- 325.Forster L, Ludwig J, Kaptur M, Bovens S, Elfringhoff AS, Holtfrerich A, Lehr M. Bioorg. Med. Chem. 2010;18:945. doi: 10.1016/j.bmc.2009.11.028. [DOI] [PubMed] [Google Scholar]
- 326.Drews A, Bovens S, Roebrock K, Sunderkötter C, Reinhardt D, Schäfers M, Van Der Velde A, Schulze Elfringhoff A, Fabian J, Lehr M. J. Med. Chem. 2010;53:5165. doi: 10.1021/jm1001088. [DOI] [PubMed] [Google Scholar]
- 327.Bovens S, Schulze Elfringhoff A, Kaptur M, Reinhardt D, Schäfers M, Lehr M. J. Med. Chem. 2010;53:8298. doi: 10.1021/jm101094p. [DOI] [PubMed] [Google Scholar]
- 328.Mira JP, Dubois T, Oudinet JP, Lukowski S, RussoMarie F, Geny B. J. Biol. Chem. 1997;272:10474. doi: 10.1074/jbc.272.16.10474. [DOI] [PubMed] [Google Scholar]
- 329.Buckland AG, Wilton DC. Biochem. J. 1998;329:369. doi: 10.1042/bj3290369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.Cubells L, de Muga SV, Tebar F, Bonventre JV, Balsinde J, Pol A, Grewal T, Enrich C. J. Biol. Chem. 2008;283:10174. doi: 10.1074/jbc.M706618200. [DOI] [PubMed] [Google Scholar]
- 331.Varghese J, Rydel R, Thorsett E. Athena Neurosciences, Inc. Arylsulfonamides as phospholipase A2 inhibitors. WO 9825893. Patent. 1998
- 332.Friebe W, Tibes U, Scheuer W. Roche Diagnostics GmbH. 9,10-Dihydro-9,10-ethanoanthracene derivatives as phospholipase inhibitors. WO 9915493. Patent. 1999
- 333.Varghese J, Rydel R, Dappen M, Thorsett E. ELAN Pharmaceuticals. Substituted pyrimidine compositions and methods of use. WO 0027824. Patent. 2000
- 334.Payne SG, Oskeritzian CA, Griffiths R, Subramanian P, Barbour SE, Chalfant CE, Milstien S, Spiegel S. Blood. 2007;109:1077. doi: 10.1182/blood-2006-03-011437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.(a) Kienesberger PC, Oberer M, Lass A, Zechner R. J. Lipid Res. 2009;50 Suppl:S63. doi: 10.1194/jlr.R800082-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Saarela J, Jung G, Hermann M, Nimpf J, Schneider WJ. BMC Genomics. 2008;9:281. doi: 10.1186/1471-2164-9-281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336.(a) Balboa MA, Balsinde J, Jones SS, Dennis EA. J. Biol. Chem. 1997;272:8576. doi: 10.1074/jbc.272.13.8576. [DOI] [PubMed] [Google Scholar]; (b) Sedgwick SG, Smerdon SJ. Trends Biochem. Sci. 1999;24:311. doi: 10.1016/s0968-0004(99)01426-7. [DOI] [PubMed] [Google Scholar]
- 337.(a) Hazen SL, Stuppy RJ, Gross RW. J. Biol. Chem. 1990;265:10622. [PubMed] [Google Scholar]; (b) Hazen SL, Gross RW. Biochem. J. 1991;280(Pt 3):581. doi: 10.1042/bj2800581. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Ramanadham S, Wolf MJ, Jett PA, Gross RW, Turk J. Biochemistry. 1994;33:7442. doi: 10.1021/bi00189a052. [DOI] [PubMed] [Google Scholar]
- 338.(a) Larsson Forsell PK, Kennedy BP, Claesson HE. Eur. J. Biochem. 1999;262:575. doi: 10.1046/j.1432-1327.1999.00418.x. [DOI] [PubMed] [Google Scholar]; (b) Larsson PK, Claesson HE, Kennedy BP. J. Biol. Chem. 1998;273:207. doi: 10.1074/jbc.273.1.207. [DOI] [PubMed] [Google Scholar]; (c) Wolf MJ, Gross RW. J. Biol. Chem. 1996;271:30879. doi: 10.1074/jbc.271.48.30879. [DOI] [PubMed] [Google Scholar]; (d) Wolf MJ, Gross RW. J. Biol. Chem. 1996;271:20989. doi: 10.1074/jbc.271.35.20989. [DOI] [PubMed] [Google Scholar]
- 339.(a) Ackermann EJ, Conde-Frieboes K, Dennis EA. J. Biol. Chem. 1995;270:445. doi: 10.1074/jbc.270.1.445. [DOI] [PubMed] [Google Scholar]; (b) Lio YC, Reynolds LJ, Balsinde J, Dennis EA. Biochim. Biophys. Acta. 1996;1302:55. doi: 10.1016/0005-2760(96)00002-1. [DOI] [PubMed] [Google Scholar]; (c) Hazen SL, Zupan LA, Weiss RH, Getman DP, Gross RW. J. Biol. Chem. 1991;266:7227. [PubMed] [Google Scholar]
- 340.Song H, Bao S, Ramanadham S, Turk J. Biochemistry. 2006;45:6392. doi: 10.1021/bi060502a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341.Ma Z, Wang X, Nowatzke W, Ramanadham S, Turk J. J. Biol. Chem. 1999;274:9607. doi: 10.1074/jbc.274.14.9607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342.Winstead MV, Balsinde J, Dennis EA. Biochim. Biophys. Acta. 2000;1488:28. doi: 10.1016/s1388-1981(00)00107-4. [DOI] [PubMed] [Google Scholar]
- 343.Hazen SL, Gross RW. Biochem. J. 1991;280(Pt 3):581. doi: 10.1042/bj2800581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344.Hsu YH, Burke JE, Li S, Woods VL, Jr, Dennis EA. J. Biol. Chem. 2009;284:23652. doi: 10.1074/jbc.M109.021857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345.(a) Jenkins CM, Yan W, Mancuso DJ, Gross RW. J. Biol. Chem. 2006;281:15615. doi: 10.1074/jbc.M511623200. [DOI] [PubMed] [Google Scholar]; (b) Carper MJ, Zhang S, Turk J, Ramanadham S. Biochemistry. 2008;47:12241. doi: 10.1021/bi800923s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346.Mancuso DJ, Jenkins CM, Gross RW. J. Biol. Chem. 2000;275:9937. doi: 10.1074/jbc.275.14.9937. [DOI] [PubMed] [Google Scholar]
- 347.Tanaka H, Takeya R, Sumimoto H. Biochem. Biophys. Res. Commun. 2000;272:320. doi: 10.1006/bbrc.2000.2776. [DOI] [PubMed] [Google Scholar]
- 348.Wilson PA, Gardner SD, Lambie NM, Commans SA, Crowther DJ. J. Lipid Res. 2006;47:1940. doi: 10.1194/jlr.M600185-JLR200. [DOI] [PubMed] [Google Scholar]
- 349.Mancuso DJ, Han X, Jenkins CM, Lehman JJ, Sambandam N, Sims HF, Yang J, Yan W, Yang K, Green K, Abendschein DR, Saffitz JE, Gross RW. J. Biol. Chem. 2007;282:9216. doi: 10.1074/jbc.M607307200. [DOI] [PubMed] [Google Scholar]
- 350.Yan W, Jenkins CM, Han X, Mancuso DJ, Sims HF, Yang K, Gross RW. J. Biol. Chem. 2005;280:26669. doi: 10.1074/jbc.M502358200. [DOI] [PubMed] [Google Scholar]
- 351.Mancuso DJ, Sims HF, Han X, Jenkins CM, Guan SP, Yang K, Moon SH, Pietka T, Abumrad NA, Schlesinger PH, Gross RW. J. Biol. Chem. 2007;282:34611. doi: 10.1074/jbc.M707795200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352.van Tienhoven M, Atkins J, Li Y, Glynn P. J. Biol. Chem. 2002;277:20942. doi: 10.1074/jbc.M200330200. [DOI] [PubMed] [Google Scholar]
- 353.Quistad GB, Barlow C, Winrow CJ, Sparks SE, Casida JE. Proc. Natl. Acad. Sci. U. S. A. 2003;100:7983. doi: 10.1073/pnas.1232473100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354.Winrow CJ, Hemming ML, Allen DM, Quistad GB, Casida JE, Barlow C. Nat. Genet. 2003;33:477. doi: 10.1038/ng1131. [DOI] [PubMed] [Google Scholar]
- 355.Akassoglou K, Malester B, Xu J, Tessarollo L, Rosenbluth J, Chao MV. Proc. Natl. Acad. Sci. U. S. A. 2004;101:5075. doi: 10.1073/pnas.0401030101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 356.Rainier S, Bui M, Mark E, Thomas D, Tokarz D, Ming L, Delaney C, Richardson RJ, Albers JW, Matsunami N, Stevens J, Coon H, Leppert M, Fink JK. Am. J. Hum. Genet. 2008;82:780. doi: 10.1016/j.ajhg.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 357.Baulande S, Lasnier F, Lucas M, Pairault J. J. Biol. Chem. 2001;276:33336. doi: 10.1074/jbc.M105193200. [DOI] [PubMed] [Google Scholar]
- 358.(a) Jenkins CM, Mancuso DJ, Yan W, Sims HF, Gibson B, Gross RW. J. Biol. Chem. 2004;279:48968. doi: 10.1074/jbc.M407841200. [DOI] [PubMed] [Google Scholar]; (b) Lake AC, Sun Y, Li JL, Kim JE, Johnson JW, Li D, Revett T, Shih HH, Liu W, Paulsen JE, Gimeno RE. J. Lipid Res. 2005;46:2477. doi: 10.1194/jlr.M500290-JLR200. [DOI] [PubMed] [Google Scholar]
- 359.Romeo S, Kozlitina J, Xing C, Pertsemlidis A, Cox D, Pennacchio LA, Boerwinkle E, Cohen JC, Hobbs HH. Nat. Genet. 2008;40:1461. doi: 10.1038/ng.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 360.Yuan X, Waterworth D, Perry JR, Lim N, Song K, Chambers JC, Zhang W, Vollenweider P, Stirnadel H, Johnson T, Bergmann S, Beckmann ND, Li Y, Ferrucci L, Melzer D, Hernandez D, Singleton A, Scott J, Elliott P, Waeber G, Cardon L, Frayling TM, Kooner JS, Mooser V. Am. J. Hum. Genet. 2008;83:520. doi: 10.1016/j.ajhg.2008.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 361.Johansson LE, Lindblad U, Larsson CA, Rastam L, Ridderstrale M. Eur. J. Endocrinol. 2008;159:577. doi: 10.1530/EJE-08-0426. [DOI] [PubMed] [Google Scholar]
- 362.(a) Villena JA, Roy S, Sarkadi-Nagy E, Kim KH, Sul HS. J. Biol. Chem. 2004;279:47066. doi: 10.1074/jbc.M403855200. [DOI] [PubMed] [Google Scholar]; (b) Zimmermann R, Strauss JG, Haemmerle G, Schoiswohl G, Birner-Gruenberger R, Riederer M, Lass A, Neuberger G, Eisenhaber F, Hermetter A, Zechner R. Science. 2004;306:1383. doi: 10.1126/science.1100747. [DOI] [PubMed] [Google Scholar]
- 363.(a) Notari L, Baladron V, Aroca-Aguilar JD, Balko N, Heredia R, Meyer C, Notario PM, Saravanamuthu S, Nueda ML, Sanchez-Sanchez F, Escribano J, Laborda J, Becerra SP. J. Biol. Chem. 2006;281:38022. doi: 10.1074/jbc.M600353200. [DOI] [PubMed] [Google Scholar]; (b) Kershaw EE, Hamm JK, Verhagen LA, Peroni O, Katic M, Flier JS. Diabetes. 2006;55:148. [PMC free article] [PubMed] [Google Scholar]
- 364.Smirnova E, Goldberg EB, Makarova KS, Lin L, Brown WJ, Jackson CL. EMBO Rep. 2006;7:106. doi: 10.1038/sj.embor.7400559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 365.Haemmerle G, Lass A, Zimmermann R, Gorkiewicz G, Meyer C, Rozman J, Heldmaier G, Maier R, Theussl C, Eder S, Kratky D, Wagner EF, Klingenspor M, Hoefler G, Zechner R. Science. 2006;312:734. doi: 10.1126/science.1123965. [DOI] [PubMed] [Google Scholar]
- 366.(a) Igal RA, Rhoads JM, Coleman RA. J. Pediatr. Gastroenterol. Nutr. 1997;25:541. doi: 10.1097/00005176-199711000-00011. [DOI] [PubMed] [Google Scholar]; (b) Fischer J, Lefevre C, Morava E, Mussini JM, Laforet P, Negre-Salvayre A, Lathrop M, Salvayre R. Nat. Genet. 2007;39:28. doi: 10.1038/ng1951. [DOI] [PubMed] [Google Scholar]
- 367.Schoenborn V, Heid IM, Vollmert C, Lingenhel A, Adams TD, Hopkins PN, Illig T, Zimmermann R, Zechner R, Hunt SC, Kronenberg F. Diabetes. 2006;55:1270. doi: 10.2337/db05-1498. [DOI] [PubMed] [Google Scholar]
- 368.Lee WC, Salido E, Yen PH. Genomics. 1994;22:372. doi: 10.1006/geno.1994.1397. [DOI] [PubMed] [Google Scholar]
- 369.Gao JG, Simon M. Biochem. Biophys. Res. Commun. 2007;360:501. doi: 10.1016/j.bbrc.2007.06.089. [DOI] [PubMed] [Google Scholar]
- 370.(a) Gaudet R. Mol Biosyst. 2008;4:372. doi: 10.1039/b801481g. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Li J, Mahajan A, Tsai MD. Biochemistry. 2006;45:15168. doi: 10.1021/bi062188q. [DOI] [PubMed] [Google Scholar]; (c) Michaely P, Tomchick DR, Machius M, Anderson RG. EMBO J. 2002;21:6387. doi: 10.1093/emboj/cdf651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 371.Rydel TJ, Williams JM, Krieger E, Moshiri F, Stallings WC, Brown SM, Pershing JC, Purcell JP, Alibhai MF. Biochemistry. 2003;42:6696. doi: 10.1021/bi027156r. [DOI] [PubMed] [Google Scholar]
- 372.Brookes PS, Yoon Y, Robotham JL, Anders MW, Sheu SS. Am. J. Physiol. Cell Physiol. 2004;287:C817. doi: 10.1152/ajpcell.00139.2004. [DOI] [PubMed] [Google Scholar]
- 373.(a) Gadd ME, Broekemeier KM, Crouser ED, Kumar J, Graff G, Pfeiffer DR. J. Biol. Chem. 2006;281:6931. doi: 10.1074/jbc.M510845200. [DOI] [PubMed] [Google Scholar]; (b) Liou JY, Aleksic N, Chen SF, Han TJ, Shyue SK, Wu KK. Exp. Cell. Res. 2005;306:75. doi: 10.1016/j.yexcr.2005.01.011. [DOI] [PubMed] [Google Scholar]; (c) Seleznev K, Zhao C, Zhang XH, Song K, Ma ZA. J. Biol. Chem. 2006;281:22275. doi: 10.1074/jbc.M604330200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 374.Hemmer W, McGlone M, Tsigelny I, Taylor SS. J. Biol. Chem. 1997;272:16946. doi: 10.1074/jbc.272.27.16946. [DOI] [PubMed] [Google Scholar]
- 375.Lishko PV, Procko E, Jin X, Phelps CB, Gaudet R. Neuron. 2007;54:905. doi: 10.1016/j.neuron.2007.05.027. [DOI] [PubMed] [Google Scholar]
- 376.(a) Balsinde J, Dennis EA. J. Biol. Chem. 1997;272:16069. doi: 10.1074/jbc.272.26.16069. [DOI] [PubMed] [Google Scholar]; (b) Balsinde J, Bianco ID, Ackermann EJ, Conde-Frieboes K, Dennis EA. Proc. Natl. Acad. Sci. U. S. A. 1995;92:8527. doi: 10.1073/pnas.92.18.8527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 377.(a) Atsumi G, Murakami M, Kojima K, Hadano A, Tajima M, Kudo I. J. Biol. Chem. 2000;275:18248. doi: 10.1074/jbc.M000271200. [DOI] [PubMed] [Google Scholar]; (b) Atsumi G, Tajima M, Hadano A, Nakatani Y, Murakami M, Kudo I. J. Biol. Chem. 1998;273:13870. doi: 10.1074/jbc.273.22.13870. [DOI] [PubMed] [Google Scholar]
- 378.Lauber K, Bohn E, Krober SM, Xiao YJ, Blumenthal SG, Lindemann RK, Marini P, Wiedig C, Zobywalski A, Baksh S, Xu Y, Autenrieth IB, Schulze-Osthoff K, Belka C, Stuhler G, Wesselborg S. Cell. 2003;113:717. doi: 10.1016/s0092-8674(03)00422-7. [DOI] [PubMed] [Google Scholar]
- 379.Ramanadham S, Hsu FF, Zhang S, Jin C, Bohrer A, Song H, Bao S, Ma Z, Turk J. Biochemistry. 2004;43:918. doi: 10.1021/bi035536m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 380.Kim SJ, Gershov D, Ma X, Brot N, Elkon KB. J. Exp. Med. 2002;196:655. doi: 10.1084/jem.20020542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 381.Chin D, Means AR. Trends Cell Biol. 2000;10:322. doi: 10.1016/s0962-8924(00)01800-6. [DOI] [PubMed] [Google Scholar]
- 382.Jenkins CM, Wolf MJ, Mancuso DJ, Gross RW. J. Biol. Chem. 2001;276:7129. doi: 10.1074/jbc.M010439200. [DOI] [PubMed] [Google Scholar]
- 383.Wang Z, Ramanadham S, Ma ZA, Bao S, Mancuso DJ, Gross RW, Turk J. J. Biol. Chem. 2005;280:6840. doi: 10.1074/jbc.M405287200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 384.(a) Balboa MA, Perez R, Balsinde J. FEBS J. 2008;275:1915. doi: 10.1111/j.1742-4658.2008.06350.x. [DOI] [PubMed] [Google Scholar]; (b) Roshak AK, Capper EA, Stevenson C, Eichman C, Marshall LA. J. Biol. Chem. 2000;275:35692. doi: 10.1074/jbc.M002273200. [DOI] [PubMed] [Google Scholar]; (c) Sanchez T, Moreno JJ. J. Cell Physiol. 2002;193:293. doi: 10.1002/jcp.10162. [DOI] [PubMed] [Google Scholar]; (d) Song Y, Wilkins P, Hu W, Murthy KS, Chen J, Lee Z, Oyesanya R, Wu J, Barbour SE, Fang X. Biochem. J. 2007;406:427. doi: 10.1042/BJ20070631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 385.Herbert SP, Walker JH. J. Biol. Chem. 2006;281:35709. doi: 10.1074/jbc.M600699200. [DOI] [PubMed] [Google Scholar]
- 386.(a) Bao S, Li Y, Lei X, Wohltmann M, Jin W, Bohrer A, Semenkovich CF, Ramanadham S, Tabas I, Turk J. J. Biol. Chem. 2007;282:27100. doi: 10.1074/jbc.M701316200. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Perez R, Balboa MA, Balsinde J. J. Immunol. 2006;176:2555. doi: 10.4049/jimmunol.176.4.2555. [DOI] [PubMed] [Google Scholar]
- 387.Ramanadham S, Yarasheski KE, Silva MJ, Wohltmann M, Novack DV, Christiansen B, Tu X, Zhang S, Lei X, Turk J. Am. J. Pathol. 2008;172:868. doi: 10.2353/ajpath.2008.070756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 388.Bao S, Miller DJ, Ma Z, Wohltmann M, Eng G, Ramanadham S, Moley K, Turk J. J. Biol. Chem. 2004;279:38194. doi: 10.1074/jbc.M406489200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 389.(a) Bao S, Song H, Wohltmann M, Ramanadham S, Jin W, Bohrer A, Turk J. J. Biol. Chem. 2006;281:20958. doi: 10.1074/jbc.M600075200. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Gregory A, Westaway SK, Holm IE, Kotzbauer PT, Hogarth P, Sonek S, Coryell JC, Nguyen TM, Nardocci N, Zorzi G, Rodriguez D, Desguerre I, Bertini E, Simonati A, Levinson B, Dias C, Barbot C, Carrilho I, Santos M, Malik I, Gitschier J, Hayflick SJ. Neurology. 2008;71:1402. doi: 10.1212/01.wnl.0000327094.67726.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 390.(a) Zachman DK, Chicco AJ, McCune SA, Murphy RC, Moore RL, Sparagna GC. J. Lipid Res. 2010;51:525. doi: 10.1194/jlr.M000646. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Malhotra A, Edelman-Novemsky I, Xu Y, Plesken H, Ma J, Schlame M, Ren M. Proc. Natl. Acad. Sci. U. S. A. 2009;106:2337. doi: 10.1073/pnas.0811224106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 391.(a) Mishra RS, Carnevale KA, Cathcart MK. J. Exp. Med. 2008;205:347. doi: 10.1084/jem.20071243. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Balgoma D, Montero O, Balboa MA, Balsinde J. FEBS J. 2008;275:6180. doi: 10.1111/j.1742-4658.2008.06742.x. [DOI] [PubMed] [Google Scholar]
- 392.Sengupta S, Xiao YJ, Xu Y. FASEB J. 2003;17:1570. doi: 10.1096/fj.02-1145fje. [DOI] [PubMed] [Google Scholar]
- 393.(a) Perez R, Melero R, Balboa MA, Balsinde J. J. Biol. Chem. 2004;279:40385. doi: 10.1074/jbc.M402562200. [DOI] [PubMed] [Google Scholar]; (b) Brustovetsky T, Antonsson B, Jemmerson R, Dubinsky JM, Brustovetsky N. J Neurochem. 2005;94:980. doi: 10.1111/j.1471-4159.2005.03248.x. [DOI] [PubMed] [Google Scholar]; (c) Zhang L, Peterson BL, Cummings BS. Biochem. Pharmacol. 2005;70:1697. doi: 10.1016/j.bcp.2005.09.008. [DOI] [PubMed] [Google Scholar]
- 394.Su X, Han X, Mancuso DJ, Abendschein DR, Gross RW. Biochemistry. 2005;44:5234. doi: 10.1021/bi047773a. [DOI] [PubMed] [Google Scholar]
- 395.(a) Boittin FX, Gribi F, Serir K, Beny JL. Am. J. Physiol. Heart Circ. Physiol. 2008;295:H2466. doi: 10.1152/ajpheart.00639.2008. [DOI] [PubMed] [Google Scholar]; (b) Boittin FX, Petermann O, Hirn C, Mittaud P, Dorchies OM, Roulet E, Ruegg UT. J. Cell Sci. 2006;119:3733. doi: 10.1242/jcs.03184. [DOI] [PubMed] [Google Scholar]; (c) Ross K, Whitaker M, Reynolds NJ. J. Cell Physiol. 2007;211:569. doi: 10.1002/jcp.20993. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Singaravelu K, Lohr C, Deitmer JW. J. Neurosci. 2006;26:9579. doi: 10.1523/JNEUROSCI.2604-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Smani T, Zakharov SI, Leno E, Csutora P, Trepakova ES, Bolotina VM. J. Biol. Chem. 2003;278:11909. doi: 10.1074/jbc.M210878200. [DOI] [PubMed] [Google Scholar]; (f) Vanden Abeele F, Lemonnier L, Thebault S, Lepage G, Parys JB, Shuba Y, Skryma R, Prevarskaya N. J. Biol. Chem. 2004;279:30326. doi: 10.1074/jbc.M400106200. [DOI] [PubMed] [Google Scholar]
- 396.(a) Hazen SL, Ford DA, Gross RW. J. Biol. Chem. 1991;266:5629. [PubMed] [Google Scholar]; (b) Ford DA, Hazen SL, Saffitz JE, Gross RW. J. Clin. Invest. 1991;88:331. doi: 10.1172/JCI115296. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Meyer MC, McHowat J. Am. J. Physiol. Cell Physiol. 2007;292:C251. doi: 10.1152/ajpcell.00120.2006. [DOI] [PubMed] [Google Scholar]; (d) Poulsen KA, Pedersen SF, Kolko M, Lambert IH. Am. J. Physiol. Cell Physiol. 2007;293:C1605. doi: 10.1152/ajpcell.00012.2007. [DOI] [PubMed] [Google Scholar]
- 397.Gong MC, Arbogast S, Guo Z, Mathenia J, Su W, Reid MB. J. Appl. Physiol. 2006;100:399. doi: 10.1152/japplphysiol.00873.2005. [DOI] [PubMed] [Google Scholar]
- 398.Kan H, Xie Z, Finkel MS. J. Mol. Cell. Cardiol. 2006;40:131. doi: 10.1016/j.yjmcc.2005.10.006. [DOI] [PubMed] [Google Scholar]
- 399.Poulsen KA, Young JF, Theil P, Kolko M, Oksbjerg N, Lambert IH. J. Agric. Food Chem. 2007;55:1970. doi: 10.1021/jf062341n. [DOI] [PubMed] [Google Scholar]
- 400.Ratz PH, Miner AS, Barbour SE. Cell Calcium. 2009;46:65. doi: 10.1016/j.ceca.2009.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 401.(a) Berti-Mattera LN, Harwalkar S, Hughes B, Wilkins PL, Almhanna K. J Neurochem. 2001;79:1136. doi: 10.1046/j.1471-4159.2001.00642.x. [DOI] [PubMed] [Google Scholar]; (b) Mendes CT, Gattaz WF, Schaeffer EL, Forlenza OV. J. Neural. Transm. 2005;112:1297. doi: 10.1007/s00702-004-0271-3. [DOI] [PubMed] [Google Scholar]
- 402.Kurian MA, Morgan NV, MacPherson L, Foster K, Peake D, Gupta R, Philip SG, Hendriksz C, Morton JE, Kingston HM, Rosser EM, Wassmer E, Gissen P, Maher ER. Neurology. 2008;70:1623. doi: 10.1212/01.wnl.0000310986.48286.8e. [DOI] [PubMed] [Google Scholar]
- 403.(a) Biancheri R, Rossi A, Alpigiani G, Filocamo M, Gandolfo C, Lorini R, Minetti C. Eur. J. Paediatr. Neurol. 2007;11:175. doi: 10.1016/j.ejpn.2006.11.013. [DOI] [PubMed] [Google Scholar]; (b) Westaway SK, Gregory A, Hayflick SJ. J. Med. Genet. 2007;44:e64. doi: 10.1136/jmg.2006.044966. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Malik I, Turk J, Mancuso DJ, Montier L, Wohltmann M, Wozniak DF, Schmidt RE, Gross RW, Kotzbauer PT. Am. J. Pathol. 2008;172:406. doi: 10.2353/ajpath.2008.070823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 404.(a) Schaeffer EL, Gattaz WF. Psychopharmacology (Berl) 2005;181:392. doi: 10.1007/s00213-005-2256-9. [DOI] [PubMed] [Google Scholar]; (b) Schaeffer EL, Forlenza OV, Gattaz WF. Psychopharmacology (Berl) 2009;202:37. doi: 10.1007/s00213-008-1351-0. [DOI] [PubMed] [Google Scholar]
- 405.(a) Lopez-Vales R, Navarro X, Shimizu T, Baskakis C, Kokotos G, Constantinou-Kokotou V, Stephens D, Dennis EA, David S. Brain. 2008;131:2620. doi: 10.1093/brain/awn188. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Baskakis C, Magrioti V, Cotton N, Stephens D, Constantinou-Kokotou V, Dennis EA, Kokotos G. J. Med. Chem. 2008;51:8027. doi: 10.1021/jm800649q. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Kokotos G, Hsu YH, Burke JE, Baskakis C, Kokotos CG, Magrioti V, Dennis EA. J. Med. Chem. 2010;53:3602. doi: 10.1021/jm901872v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 406.(a) Lei X, Barbour SE, Ramanadham S. Biochimie. 2010;92:627. doi: 10.1016/j.biochi.2010.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lei X, Zhang S, Bohrer A, Bao S, Song H, Ramanadham S. Biochemistry. 2007;46:10170. doi: 10.1021/bi700017z. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Lei X, Zhang S, Bohrer A, Ramanadham S. J. Biol. Chem. 2008;283:34819. doi: 10.1074/jbc.M807409200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 407.Ayilavarapu S, Kantarci A, Fredman G, Turkoglu O, Omori K, Liu H, Iwata T, Yagi M, Hasturk H, Van Dyke TE. J. Immunol. 2010;184:1507. doi: 10.4049/jimmunol.0901219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 408.Xie Z, Gong MC, Su W, Xie D, Turk J, Guo Z. J. Biol. Chem. 2010;285:8628. doi: 10.1074/jbc.M109.057711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 409.Barth PG, Scholte HR, Berden JA, Van der Klei-Van Moorsel JM, Luyt-Houwen IE, Van 't Veer-Korthof ET, Van der Harten JJ, Sobotka-Plojhar MA. J. Neurol. Sci. 1983;62:327. doi: 10.1016/0022-510x(83)90209-5. [DOI] [PubMed] [Google Scholar]
- 410.Newman HA, Gordesky SE, Hoppel C, Cooper C. Biochem. J. 1968;107:381. doi: 10.1042/bj1070381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 411.Houtkooper RH, Turkenburg M, Poll-The BT, Karall D, Perez-Cerda C, Morrone A, Malvagia S, Wanders RJ, Kulik W, Vaz FM. Biochim. Biophys. Acta. 2009;1788:2003. doi: 10.1016/j.bbamem.2009.07.009. [DOI] [PubMed] [Google Scholar]
- 412.Mancuso DJ, Kotzbauer P, Wozniak DF, Sims HF, Jenkins CM, Guan S, Han X, Yang K, Sun G, Malik I, Conyers S, Green KG, Schmidt RE, Gross RW. J. Biol. Chem. 2009;284:35632. doi: 10.1074/jbc.M109.055194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 413.(a) Tonelli A, Romaniello R, Grasso R, Cavallini A, Righini A, Bresolin N, Borgatti R, Bassi MT. Clin. Genet. 2010;78:432. doi: 10.1111/j.1399-0004.2010.01417.x. [DOI] [PubMed] [Google Scholar]; (b) Morgan NV, Westaway SK, Morton JE, Gregory A, Gissen P, Sonek S, Cangul H, Coryell J, Canham N, Nardocci N, Zorzi G, Pasha S, Rodriguez D, Desguerre I, Mubaidin A, Bertini E, Trembath RC, Simonati A, Schanen C, Johnson CA, Levinson B, Woods CG, Wilmot B, Kramer P, Gitschier J, Maher ER, Hayflick SJ. Nat. Genet. 2006;38:752. doi: 10.1038/ng1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 414.Ackermann EJ, Condefrieboes K, Dennis EA. J. Biol. Chem. 1995;270:445. doi: 10.1074/jbc.270.1.445. [DOI] [PubMed] [Google Scholar]
- 415.Lio YC, Reynolds LJ, Balsinde J, Dennis EA. Biochim. Biophys.Acta. 1996;1302:55. doi: 10.1016/0005-2760(96)00002-1. [DOI] [PubMed] [Google Scholar]
- 416.Balsinde J, Dennis EA. J. Biol. Chem. 1996;271:31937. doi: 10.1074/jbc.271.50.31937. [DOI] [PubMed] [Google Scholar]
- 417.Daniels SB, Cooney E, Sofia MJ, Chakravarty PK, Katzenellenbogen JA. J. Biol. Chem. 1983;258:15046. [PubMed] [Google Scholar]
- 418.(a) Hazen SL, Zupan LA, Weiss RH, Getman DP, Gross RW. J. Biol. Chem. 1991;266:7227. [PubMed] [Google Scholar]; (b) Zupan LA, Weiss RH, Hazen SL, Parnas BL, Aston KW, Lennon PJ, Getman DP, Gross RW. J. Med. Chem. 1993;36:95. doi: 10.1021/jm00053a012. [DOI] [PubMed] [Google Scholar]
- 419.Akiba S, Hayama M, Sato T. FEBS Lett. 1998;437:225. doi: 10.1016/s0014-5793(98)01236-8. [DOI] [PubMed] [Google Scholar]
- 420.Sanchez T, Moreno JJ. Biochem. Pharmacol. 2001;61:811. doi: 10.1016/s0006-2952(01)00555-x. [DOI] [PubMed] [Google Scholar]
- 421.St-Gelais F, Ménard C, Congar P, Trudeau LE, Massicotte G. Hippocampus. 2004;14:319. doi: 10.1002/hipo.10176. [DOI] [PubMed] [Google Scholar]
- 422.Sun B, Zhang X, Talathi S, Cummings BS. J. Pharmacol. Exp. Ther. 2008;326:59. doi: 10.1124/jpet.108.138958. [DOI] [PubMed] [Google Scholar]
- 423.Sun B, Zhang X, Yonz C, Cummings BS. Biochem. Pharmacol. 2010;79:1727. doi: 10.1016/j.bcp.2010.02.005. [DOI] [PubMed] [Google Scholar]
- 424.(a) Jenkins CM, Han X, Mancuso DJ, Gross RW. J. Biol. Chem. 2002;277:32807. doi: 10.1074/jbc.M202568200. [DOI] [PubMed] [Google Scholar]; (b) Saavedra G, Zhang W, Peterson B, Cummings BS. J. Pharmacol. Exp. Ther. 2006;318:1211. doi: 10.1124/jpet.106.105650. [DOI] [PubMed] [Google Scholar]; (c) Kinsey GR, Cummings BS, Beckett CS, Saavedra G, Zhang W, McHowat J, Schnellmann RG. Biochem. Biophys. Res. Commun. 2005;327:287. doi: 10.1016/j.bbrc.2004.12.016. [DOI] [PubMed] [Google Scholar]
- 425.Song H, Ramanadham S, Bao S, Hsu FF, Turk J. Biochemistry. 2006;45:1061. doi: 10.1021/bi052065q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 426.Kalyvas A, Baskakis C, Magrioti V, Constantinou-Kokotou V, Stephens D, Lpez-Vales R, Lu JQ, Yong VW, Dennis EA, Kokotos G, David S. Brain. 2009;132:1221. doi: 10.1093/brain/awp002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 427.Li J, Zhao Z, Antalis C, Zhao Z, Emerson R, Wei G, Zhang S, Zhang Z-Y, Xu Y. Am. J. Pathol. 2011;179:452. doi: 10.1016/j.ajpath.2011.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 428.Prescott SM, Zimmerman GA, Stafforini DM, McIntyre TM. Annu. Rev. Biochem. 2000;69:419. doi: 10.1146/annurev.biochem.69.1.419. [DOI] [PubMed] [Google Scholar]
- 429.Tjoelker LW, Eberhardt C, Unger J, Trong HL, Zimmerman GA, McIntyre TM, Stafforini DM, Prescott SM, Gray PW. J. Biol. Chem. 1995;270:25481. doi: 10.1074/jbc.270.43.25481. [DOI] [PubMed] [Google Scholar]
- 430.Hattori K, Adachi H, Matsuzawa A, Yamamoto K, Tsujimoto M, Aoki J, Hattori M, Arai H, Inoue K. J. Biol. Chem. 1996;271:33032. doi: 10.1074/jbc.271.51.33032. [DOI] [PubMed] [Google Scholar]
- 431.Ho YS, Swenson L, Derewenda U, Serre L, Wei Y, Dauter Z, Hattori M, Adachi T, Aoki J, Arai H, Inoue K, Derewenda ZS. Nature. 1997;385:89. doi: 10.1038/385089a0. [DOI] [PubMed] [Google Scholar]
- 432.Hattori M, Adachi H, Tsujimoto M, Arai H, Inoue K. Nature. 1994;370:216. doi: 10.1038/370216a0. [DOI] [PubMed] [Google Scholar]
- 433.Stremler KE, Stafforini DM, Prescott SM, McIntyre TM. J. Biol. Chem. 1991;266:11095. [PubMed] [Google Scholar]
- 434.(a) MacPhee CH, Moores KE, Boyd HF, Dhanak D, Ife RJ, Leach CA, Leake DS, Milliner KJ, Patterson RA, Suckling KE, Tew DG, Hickey DM. Biochem. J. 1999;338(Pt 2):479. [PMC free article] [PubMed] [Google Scholar]; (b) Davis B, Koster G, Douet LJ, Scigelova M, Woffendin G, Ward JM, Smith A, Humphries J, Burnand KG, Macphee CH, Postle AD. J. Biol. Chem. 2008;283:6428. doi: 10.1074/jbc.M709970200. [DOI] [PubMed] [Google Scholar]
- 435.Stafforini DM, Sheller JR, Blackwell TS, Sapirstein A, Yull FE, McIntyre TM, Bonventre JV, Prescott SM, Roberts LJ., 2nd J. Biol. Chem. 2006;281:4616. doi: 10.1074/jbc.M507340200. [DOI] [PubMed] [Google Scholar]
- 436.Min JH, Wilder C, Aoki J, Arai H, Inoue K, Paul L, Gelb MH. Biochemistry. 2001;40:4539. doi: 10.1021/bi002600g. [DOI] [PubMed] [Google Scholar]
- 437.(a) Tsoukatos DC, Liapikos TA, Tselepis AD, Chapman MJ, Ninio E. Biochem. J. 2001;357:457. doi: 10.1042/0264-6021:3570457. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Tsoukatos DC, Brocheriou I, Moussis V, Panopoulou CP, Christofidou ED, Koussissis S, Sismanidis S, Ninio E, Siminelakis S. J. Lipid Res. 2008;49:2240. doi: 10.1194/jlr.M800188-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 438.Manya H, Aoki J, Kato H, Ishii J, Hino S, Arai H, Inoue K. J. Biol. Chem. 1999;274:31827. doi: 10.1074/jbc.274.45.31827. [DOI] [PubMed] [Google Scholar]
- 439.(a) Tselepis AD, Karabina SA, Stengel D, Piedagnel R, Chapman MJ, Ninio E. J. Lipid Res. 2001;42:1645. [PubMed] [Google Scholar]; (b) Akiyama M, Sugatani J, Suzuki T, Suzuki Y, Miwa M. J. Biochem. 1998;123:786. doi: 10.1093/oxfordjournals.jbchem.a022005. [DOI] [PubMed] [Google Scholar]
- 440.Samanta U, Bahnson BJ. J. Biol. Chem. 2008;283:31617. doi: 10.1074/jbc.M804750200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 441.Stafforini DM, Tjoelker LW, McCormick SP, Vaitkus D, McIntyre TM, Gray PW, Young SG, Prescott SM. J. Biol. Chem. 1999;274:7018. doi: 10.1074/jbc.274.11.7018. [DOI] [PubMed] [Google Scholar]
- 442.Stafforini DM, Carter ME, Zimmerman GA, McIntyre TM, Prescott SM. Proc. Natl. Acad. Sci. U. S. A. 1989;86:2393. doi: 10.1073/pnas.86.7.2393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 443.Gardner AA, Reichert EC, Topham MK, Stafforini DM. J. Biol. Chem. 2008;283:17099. doi: 10.1074/jbc.M802394200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 444.Min JH, Jain MK, Wilder C, Paul L, Apitz-Castro R, Aspleaf DC, Gelb MH. Biochemistry. 1999;38:12935. doi: 10.1021/bi991149u. [DOI] [PubMed] [Google Scholar]
- 445.Pande AH, Tillu VA. Biochim. Biophys. Acta. 2011;1811:46. doi: 10.1016/j.bbalip.2010.09.002. [DOI] [PubMed] [Google Scholar]
- 446.Cao J, Hsu YH, Li S, Woods VL, Dennis EA. Biochemistry. 2011;50:5314. doi: 10.1021/bi101916w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 447.Tellis CC, Tselepis AD. Biochim. Biophys. Acta. 2009;1791:327. doi: 10.1016/j.bbalip.2009.02.015. [DOI] [PubMed] [Google Scholar]
- 448.Stafforini DM, McIntyre TM, Carter ME, Prescott SM. J. Biol. Chem. 1987;262:4215. [PubMed] [Google Scholar]
- 449.Stafforini DM, Prescott SM, Zimmerman GA, McIntyre TM. Biochim. Biophys. Acta. 1996;1301:161. doi: 10.1016/0005-2760(96)00040-9. [DOI] [PubMed] [Google Scholar]
- 450.Tselepis AD, Dentan C, Karabina SA, Chapman MJ, Ninio E. Arterioscler., Thromb., Vasc. Biol. 1995;15:1764. doi: 10.1161/01.atv.15.10.1764. [DOI] [PubMed] [Google Scholar]
- 451.Benitez S, Sanchez-Quesada JL, Ribas V, Jorba O, Blanco-Vaca F, Gonzalez-Sastre F, Ordonez-Llanos J. Circulation. 2003;108:92. doi: 10.1161/01.CIR.0000072791.40232.8F. [DOI] [PubMed] [Google Scholar]
- 452.Mitsios JV, Vini MP, Stengel D, Ninio E, Tselepis AD. Arterioscler., Thromb., Vasc. Biol. 2006;26:1907. doi: 10.1161/01.ATV.0000228821.79588.ef. [DOI] [PubMed] [Google Scholar]
- 453.(a) Ho YS, Sheffield PJ, Masuyama J, Arai H, Li J, Aoki J, Inoue K, Derewenda U, Derewenda ZS. Protein Eng. 1999;12:693. doi: 10.1093/protein/12.8.693. [DOI] [PubMed] [Google Scholar]; (b) Sheffield PJ, McMullen TW, Li J, Ho YS, Garrard SM, Derewenda U, Derewenda ZS. Protein Eng. 2001;14:513. doi: 10.1093/protein/14.7.513. [DOI] [PubMed] [Google Scholar]; (c) Tarricone C, Perrina F, Monzani S, Massimiliano L, Kim MH, Derewenda ZS, Knapp S, Tsai LH, Musacchio A. Neuron. 2004;44:809. doi: 10.1016/j.neuron.2004.11.019. [DOI] [PubMed] [Google Scholar]
- 454.Yamaguchi N, Koizumi H, Aoki J, Natori Y, Nishikawa K, Takanezawa Y, Arai H. Genes Cells. 2007;12:1153. doi: 10.1111/j.1365-2443.2007.01126.x. [DOI] [PubMed] [Google Scholar]
- 455.Hakkinen T, Luoma JS, Hiltunen MO, Macphee CH, Milliner KJ, Patel L, Rice SQ, Tew DG, Karkola K, Yla-Herttuala S. Arterioscler., Thromb., Vasc. Biol. 1999;19:2909. doi: 10.1161/01.atv.19.12.2909. [DOI] [PubMed] [Google Scholar]
- 456.Elstad MR, Stafforini DM, McIntyre TM, Prescott SM, Zimmerman GA. J. Biol. Chem. 1989;264:8467. [PubMed] [Google Scholar]
- 457.Stafforini DM, Satoh K, Atkinson DL, Tjoelker LW, Eberhardt C, Yoshida H, Imaizumi T, Takamatsu S, Zimmerman GA, McIntyre TM, Gray PW, Prescott SM. J. Clin. Invest. 1996;97:2784. doi: 10.1172/JCI118733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 458.(a) Yamada Y, Ichihara S, Fujimura T, Yokota M. Metabolism. 1998;47:177. doi: 10.1016/s0026-0495(98)90216-5. [DOI] [PubMed] [Google Scholar]; (b) Hiramoto M, Yoshida H, Imaizumi T, Yoshimizu N, Satoh K. Stroke. 1997;28:2417. doi: 10.1161/01.str.28.12.2417. [DOI] [PubMed] [Google Scholar]
- 459.Watson AD, Navab M, Hama SY, Sevanian A, Prescott SM, Stafforini DM, McIntyre TM, Du BN, Fogelman AM, Berliner JA. J. Clin. Invest. 1995;95:774. doi: 10.1172/JCI117726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 460.Theilmeier G, De Geest B, Van Veldhoven PP, Stengel D, Michiels C, Lox M, Landeloos M, Chapman MJ, Ninio E, Collen D, Himpens B, Holvoet P. FASEB J. 2000;14:2032. doi: 10.1096/fj.99-1029com. [DOI] [PubMed] [Google Scholar]
- 461.Quarck R, De Geest B, Stengel D, Mertens A, Lox M, Theilmeier G, Michiels C, Raes M, Bult H, Collen D, Van Veldhoven P, Ninio E, Holvoet P. Circulation. 2001;103:2495. doi: 10.1161/01.cir.103.20.2495. [DOI] [PubMed] [Google Scholar]
- 462.Arakawa H, Qian JY, Baatar D, Karasawa K, Asada Y, Sasaguri Y, Miller ER, Witztum JL, Ueno H. Circulation. 2005;111:3302. doi: 10.1161/CIRCULATIONAHA.104.476242. [DOI] [PubMed] [Google Scholar]
- 463.De Keyzer D, Karabina SA, Wei W, Geeraert B, Stengel D, Marsillach J, Camps J, Holvoet P, Ninio E. Arterioscler., Thromb., Vasc. Biol. 2009;29:2041. doi: 10.1161/ATVBAHA.109.196592. [DOI] [PubMed] [Google Scholar]
- 464.(a) Caslake MJ, Packard CJ, Suckling KE, Holmes SD, Chamberlain P, Macphee CH. Atherosclerosis. 2000;150:413. doi: 10.1016/s0021-9150(99)00406-2. [DOI] [PubMed] [Google Scholar]; (b) Blake GJ, Dada N, Fox JC, Manson JE, Ridker PM. J. Am. Coll. Cardiol. 2001;38:1302. doi: 10.1016/s0735-1097(01)01554-6. [DOI] [PubMed] [Google Scholar]; (c) Gazi I, Lourida ES, Filippatos T, Tsimihodimos V, Elisaf M, Tselepis AD. Clin. Chem. 2005;51:2264. doi: 10.1373/clinchem.2005.058404. [DOI] [PubMed] [Google Scholar]; (d) Anderson JL. Am. J. Cardiol. 2008;101:23F. doi: 10.1016/j.amjcard.2008.04.015. [DOI] [PubMed] [Google Scholar]; (e) Winkler K, Hoffmann MM, Winkelmann BR, Friedrich I, Schafer G, Seelhorst U, Wellnitz B, Wieland H, Boehm BO, Marz W. Clin. Chem. 2007;53:1440. doi: 10.1373/clinchem.2007.086298. [DOI] [PubMed] [Google Scholar]; (f) Thompson A, Gao P, Orfei L, Watson S, Di Angelantonio E, Kaptoge S, Ballantyne C, Cannon CP, Criqui M, Cushman M, Hofman A, Packard C, Thompson SG, Collins R, Danesh J. Lancet. 2010;375:1536. doi: 10.1016/S0140-6736(10)60319-4. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Casas JP, Ninio E, Panayiotou A, Palmen J, Cooper JA, Ricketts SL, Sofat R, Nicolaides AN, Corsetti JP, Fowkes FG, Tzoulaki I, Kumari M, Brunner EJ, Kivimaki M, Marmot MG, Hoffmann MM, Winkler K, Marz W, Ye S, Stirnadel HA, Khaw KT, Humphries SE, Sandhu MS, Hingorani AD, Talmud PJ. Circulation. 2010;121:2284. doi: 10.1161/CIRCULATIONAHA.109.923383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 465.Mannheim D, Herrmann J, Versari D, Gossl M, Meyer FB, McConnell JP, Lerman LO, Lerman A. Stroke. 2008;39:1448. doi: 10.1161/STROKEAHA.107.503193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 466.Vickers KC, Maguire CT, Wolfert R, Burns AR, Reardon M, Geis R, Holvoet P, Morrisett JD. J. Lipid Res. 2009;50:1735. doi: 10.1194/jlr.M800342-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 467.(a) Carpenter KL, Dennis IF, Challis IR, Osborn DP, Macphee CH, Leake DS, Arends MJ, Mitchinson MJ. FEBS Lett. 2001;505:357. doi: 10.1016/s0014-5793(01)02840-x. [DOI] [PubMed] [Google Scholar]; (b) Rosenson RS, Vracar-Grabar M, Helenowski I. Cardiovasc. Drugs Ther. 2008;22:55. doi: 10.1007/s10557-008-6080-4. [DOI] [PubMed] [Google Scholar]
- 468.Leach CA, Hickey DM, Ife RJ, Macphee CH, Smith SA, Tew DG. Farmaco. 2001;56:45. doi: 10.1016/s0014-827x(01)01011-4. [DOI] [PubMed] [Google Scholar]
- 469.Wilensky RL, Shi Y, Mohler ER, 3rd, Hamamdzic D, Burgert ME, Li J, Postle A, Fenning RS, Bollinger JG, Hoffman BE, Pelchovitz DJ, Yang J, Mirabile RC, Webb CL, Zhang L, Zhang P, Gelb MH, Walker MC, Zalewski A, Macphee CH. Nat. Med. 2008;14:1059. doi: 10.1038/nm.1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 470.Jang Y, Kim OY, Koh SJ, Chae JS, Ko YG, Kim JY, Cho H, Jeong TS, Lee WS, Ordovas JM, Lee JH. J. Clin. Endocrinol. Metab. 2006;91:3521. doi: 10.1210/jc.2006-0116. [DOI] [PubMed] [Google Scholar]
- 471.Song KNM, Aponte J, Manas ES, Bacanu SA, Yuan X, Kong X, Cardon L, Mooser VE, Whittaker JC, Waterworth DM. Pharmacogenomics J. 2011 doi: 10.1038/tpj.2011.20. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 472.Koshy B, Miyashita A, St Jean P, Stirnadel H, Kaise T, Rubio JP, Mooser V, Kuwano R, Irizarry MC. J. Alzheimers Dis. 2010;21:775. doi: 10.3233/JAD-2010-100513. [DOI] [PubMed] [Google Scholar]
- 473.Jang Y, Waterworth D, Lee JE, Song K, Kim S, Kim HS, Park KW, Cho HJ, Oh IY, Park JE, Lee BS, Ku HJ, Shin DJ, Lee JH, Jee SH, Han BG, Jang HY, Cho EY, Vallance P, Whittaker J, Cardon L, Mooser V. PloS one. 2011;6 doi: 10.1371/journal.pone.0018208. e18208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 474.Stafforini DM, Numao T, Tsodikov A, Vaitkus D, Fukuda T, Watanabe N, Fueki N, McIntyre TM, Zimmerman GA, Makino S, Prescott SM. J. Clin. Invest. 1999;103:989. doi: 10.1172/JCI5574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 475.Kruse S, Mao XQ, Heinzmann A, Blattmann S, Roberts MH, Braun S, Gao PS, Forster J, Kuehr J, Hopkin JM, Shirakawa T, Deichmann KA. Am. J. Hum. Genet. 2000;66:1522. doi: 10.1086/302901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 476.Stafforini DM. Pharmaceuticals. 2009;2:94. doi: 10.3390/ph2030094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 477.Caplan M, Hsueh W, Kelly A, Donovan M. Prostaglandins. 1990;39:705. doi: 10.1016/0090-6980(90)90030-y. [DOI] [PubMed] [Google Scholar]
- 478.Caplan MS, Lickerman M, Adler L, Dietsch GN, Yu A. Pediatr. Res. 1997;42:779. doi: 10.1203/00006450-199712000-00010. [DOI] [PubMed] [Google Scholar]
- 479.Lu J, Pierce M, Franklin A, Jilling T, Stafforini DM, Caplan M. Pediatr. Res. 2010;68:225. doi: 10.1203/PDR.0b013e3181eb2efe. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 480.Vadas P, Gold M, Perelman B, Liss GM, Lack G, Blyth T, Simons FE, Simons KJ, Cass D, Yeung J. N. Eng. J. Med. 2008;358:28. doi: 10.1056/NEJMoa070030. [DOI] [PubMed] [Google Scholar]
- 481.Kono N, Inoue T, Yoshida Y, Sato H, Matsusue T, Itabe H, Niki E, Aoki J, Arai H. J. Biol. Chem. 2008;283:1628. doi: 10.1074/jbc.M708622200. [DOI] [PubMed] [Google Scholar]
- 482.Matsuzawa A, Hattori K, Aoki J, Arai H, Inoue K. J. Biol. Chem. 1997;272:32315. doi: 10.1074/jbc.272.51.32315. [DOI] [PubMed] [Google Scholar]
- 483.Umemura K, Kato I, Hirashima Y, Ishii Y, Inoue T, Aoki J, Kono N, Oya T, Hayashi N, Hamada H, Endo S, Oda M, Arai H, Kinouchi H, Hiraga K. Stroke. 2007;38:1063. doi: 10.1161/01.STR.0000257981.09329.d2. [DOI] [PubMed] [Google Scholar]
- 484.Inoue T, Sugimoto A, Suzuki Y, Yamamoto M, Tsujimoto M, Inoue K, Aoki J, Arai H. Proc. Natl. Acad. Sci. U. S. A. 2004;101:13233. doi: 10.1073/pnas.0405507101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 485.Koizumi H, Yamaguchi N, Hattori M, Ishikawa TO, Aoki J, Taketo MM, Inoue K, Arai H. J. Biol. Chem. 2003;278:12489. doi: 10.1074/jbc.M211836200. [DOI] [PubMed] [Google Scholar]
- 486.Hirotsune S, Fleck MW, Gambello MJ, Bix GJ, Chen A, Clark GD, Ledbetter DH, McBain CJ, Wynshaw-Boris A. Nat. Genet. 1998;19:333. doi: 10.1038/1221. [DOI] [PubMed] [Google Scholar]
- 487.Yan W, Assadi AH, Wynshaw-Boris A, Eichele G, Matzuk MM, Clark GD. Proc. Natl. Acad. Sci. U. S. A. 2003;100:7189. doi: 10.1073/pnas.1236145100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 488.Zhang G. PLoS One. 2007;2:1. doi: 10.1371/journal.pone.0000164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 489.Bechler ME, Doody AM, Racoosin E, Lin L, Lee KH, Brown WJ. J. Cell Biol. 2010;190:45. doi: 10.1083/jcb.200908105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 490.Tew DG, Boyd HF, Ashman S, Theobald C, Leach CA. Biochemistry. 1998;37:10087. doi: 10.1021/bi9801412. [DOI] [PubMed] [Google Scholar]
- 491.Macphee CH, Moores KE, Boyd HF, Dhanak D, Ife RJ, Leach CA, Leake DS, Milliner KJ, Patterson RA, Suckling KE, Tew DG, Hickey DMB. Biochem. J. 1999;338:479. [PMC free article] [PubMed] [Google Scholar]
- 492.Benson GM, Grimsditch D, Milliner K, Moores K, Boyd H, Tew D, Hickey D, Ife R, Suckling K, Macphee C. Atherosclerosis. 2000;151:166. [Google Scholar]
- 493.Boyd HF, Flynn ST, Hickey DMB, Ife RJ, Jones M, Leach CA, Macphee CH, Milliner KJ, Rawlings DA, Slingsby BP, Smith SA, Stansfield IG, Tew DG, Theobald CJ. Bioorg. Med. Chem. Lett. 2000;10:395. doi: 10.1016/s0960-894x(00)00002-0. [DOI] [PubMed] [Google Scholar]
- 494.(a) Boyd HF, Fell SCM, Flynn ST, Hickey DMB, Ife RJ, Leach CA, Macphee CH, Milliner KJ, Moores KE, Pinto IL, Porter RA, Rawlings DA, Smith SA, Stansfield IG, Tew DG, Theobald CJ, Whittaker CM. Bioorg. Med. Chem. Lett. 2000;10:2557. doi: 10.1016/s0960-894x(00)00510-2. [DOI] [PubMed] [Google Scholar]; (b) Boyd HF, Hammond B, Hickey DMB, Ife RJ, Leach CA, Lewis VA, Macphee CH, Milliner KJ, Pinto IL, Smith SA, Stansfield IG, Theobald CJ, Whittaker CM. Bioorg. Med. Chem. Lett. 2001;11:701. doi: 10.1016/s0960-894x(01)00038-5. [DOI] [PubMed] [Google Scholar]; (c) Bloomer JC, Boyd HF, Hickey DMB, Ife RJ, Leach CA, Macphee CH, Milliner KJ, Pinto IL, Rawlings DA, Smith SA, Stansfield IG, Stanway SJ, Taylor MA, Theobald CJ, Whittaker CM. Bioorg. Med. Chem. Lett. 2001;11:1925. doi: 10.1016/s0960-894x(01)00346-8. [DOI] [PubMed] [Google Scholar]; (d) Boyd HF, Fell SCM, Hickey DMB, Ife RJ, Leach CA, Macphee CH, Milliner KJ, Pinto IL, Rawlings DA, Smith SA, Stansfield IG, Stanway SJ, Theobald CJ, Whittaker CM. Bioorg. Med. Chem. Lett. 2002;12:51. doi: 10.1016/s0960-894x(01)00678-3. [DOI] [PubMed] [Google Scholar]
- 495.Leach CA, Hickey DMB, Ife RJ, Macphee CH, Smith SA, Tew DG. Farmaco. 2001;56:45. doi: 10.1016/s0014-827x(01)01011-4. [DOI] [PubMed] [Google Scholar]
- 496.Blackie JA, Bloomer JC, Brown MJB, Cheng H-Y, Elliott RL, Hammond B, Hickey DMB, Ife RJ, Leach CA, Lewis VA, Macphee CH, Milliner KJ, Moores KE, Pinto IL, Smith SA, Stansfield IG, Stanway SJ, Taylor MA, Theobald CJ, Whittaker CM. Bioorg. Med. Chem. Lett. 2002;12:2603. doi: 10.1016/s0960-894x(02)00473-0. [DOI] [PubMed] [Google Scholar]
- 497.Blackie JA, Bloomer JC, Brown MJB, Cheng HY, Hammond B, Hickey DMB, Ife RJ, Leach CA, Lewis VA, Macphee CH, Milliner KJ, Moores KE, Pinto IL, Smith SA, Stansfield IG, Stanway SJ, Taylor MA, Theobald CJ. Bioorg. Med. Chem. Lett. 2003;13:1067. doi: 10.1016/s0960-894x(03)00058-1. [DOI] [PubMed] [Google Scholar]
- 498.Mohler ER, Iii, Ballantyne CM, Davidson MH, Hanefeld M, Ruilope LM, Johnson JL, Zalewski A. J. Am. Coll. Cardiol. 2008;51:1632. doi: 10.1016/j.jacc.2007.11.079. [DOI] [PubMed] [Google Scholar]
- 499.Serruys PW, García-García HM, Buszman P, Erne P, Verheye S, Aschermann M, Duckers H, Bleie O, Dudek D, Bøtker HE, Von Birgelen C, D'Amico D, Hutchinson T, Zambanini A, Mastik F, Van Es GA, Van Der Steen AFW, Vince DG, Ganz P, Hamm CW, Wijns W, Zalewski A. Circulation. 2008;118:1172. doi: 10.1161/CIRCULATIONAHA.108.771899. [DOI] [PubMed] [Google Scholar]
- 500.Corson MA. Ther. Adv. Cardiovasc. Dis. 2010;4:241. doi: 10.1177/1753944710375820. [DOI] [PubMed] [Google Scholar]
- 501.Abe A, Shayman JA, Radin NS. J. Biol. Chem. 1996;271:14383. doi: 10.1074/jbc.271.24.14383. [DOI] [PubMed] [Google Scholar]
- 502.Abe A, Shayman JA. J. Biol. Chem. 1998;273:8467. doi: 10.1074/jbc.273.14.8467. [DOI] [PubMed] [Google Scholar]
- 503.Abe A, Hiraoka M, Wild S, Wilcoxen SE, Paine R, 3rd, Shayman JA. J. Biol. Chem. 2004;279:42605. doi: 10.1074/jbc.M407834200. [DOI] [PubMed] [Google Scholar]
- 504.Hiraoka M, Abe A, Shayman JA. J. Biol. Chem. 2002;277:10090. doi: 10.1074/jbc.M111977200. [DOI] [PubMed] [Google Scholar]
- 505.Abe A, Shayman JA. J. Lipid Res. 2009;50:2027. doi: 10.1194/jlr.M900008-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 506.Abe A, Hiraoka M, Shayman JA. J. Lipid Res. 2006;47:2268. doi: 10.1194/jlr.M600183-JLR200. [DOI] [PubMed] [Google Scholar]
- 507.Hiraoka M, Abe A, Shayman JA. J. Lipid Res. 2005;46:2441. doi: 10.1194/jlr.M500248-JLR200. [DOI] [PubMed] [Google Scholar]
- 508.Hiraoka M, Abe A, Lu Y, Yang K, Han X, Gross RW, Shayman JA. Mol. Cell Biol. 2006;26:6139. doi: 10.1128/MCB.00627-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 509.Abe A, Kelly R, Kollmeyer J, Hiraoka M, Lu Y, Shayman JA. J. Immunol. 2008;181:7873. doi: 10.4049/jimmunol.181.11.7873. [DOI] [PubMed] [Google Scholar]
- 510.Abe A, Hiraoka M, Shayman JA. Drug Metab. Lett. 2007;1:49. doi: 10.2174/187231207779814292. [DOI] [PubMed] [Google Scholar]
- 511.Taniyama Y, Fuse H, Satomi T, Tozawa R, Yasuhara Y, Shimakawa K, Shibata S, Hattori M, Nakata M, Taketomi S. Biochem. Biophys. Res. Commun. 2005;330:104. doi: 10.1016/j.bbrc.2005.02.126. [DOI] [PubMed] [Google Scholar]
- 512.(a) Hajnal A, Klemenz R, Schafer R. Oncogene. 1994;9:479. [PubMed] [Google Scholar]; (b) Sers C, Emmenegger U, Husmann K, Bucher K, Andres AC, Schafer R. J. Cell Biol. 1997;136:935. doi: 10.1083/jcb.136.4.935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 513.Jin XH, Okamoto Y, Morishita J, Tsuboi K, Tonai T, Ueda N. J. Biol. Chem. 2007;282:3614. doi: 10.1074/jbc.M606369200. [DOI] [PubMed] [Google Scholar]
- 514.Uyama T, Morishita J, Jin XH, Okamoto Y, Tsuboi K, Ueda N. J. Lipid Res. 2009;50:685. doi: 10.1194/jlr.M800453-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 515.Sers C, Husmann K, Nazarenko I, Reich S, Wiechen K, Zhumabayeva B, Adhikari P, Schroder K, Gontarewicz A, Schafer R. Oncogene. 2002;21:2829. doi: 10.1038/sj.onc.1205377. [DOI] [PubMed] [Google Scholar]
- 516.(a) Wang Y, Kowalski PE, Thalmann I, Ornitz DM, Mager DL, Thalmann R. Proc. Natl. Acad. Sci. U. S. A. 1998;95:15345. doi: 10.1073/pnas.95.26.15345. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Verpy E, Leibovici M, Petit C. Proc. Natl. Acad. Sci. U. S. A. 1999;96:529. doi: 10.1073/pnas.96.2.529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 517.(a) Zhao X, Yang H, Yamoah EN, Lundberg YW. Dev. Biol. 2007;304:508. doi: 10.1016/j.ydbio.2007.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Deans MR, Peterson JM, Wong GW. PLoS One. 2010;5 doi: 10.1371/journal.pone.0012765. e12765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 518.Kiss PJ, Knisz J, Zhang Y, Baltrusaitis J, Sigmund CD, Thalmann R, Smith RJ, Verpy E, Banfi B. Curr. Biol. 2006;16:208. doi: 10.1016/j.cub.2005.12.025. [DOI] [PubMed] [Google Scholar]
- 519.Zhao X, Jones SM, Yamoah EN, Lundberg YW. Neuroscience. 2008;153:289. doi: 10.1016/j.neuroscience.2008.01.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 520.Lu W, Zhou D, Freeman JJ, Thalmann I, Ornitz DM, Thalmann R. Hear Res. 2010;268:172. doi: 10.1016/j.heares.2010.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 521.Jones PM, Turner KM, Djordjevic JT, Sorrell TC, Wright LC, George AM. Biochemistry. 2007;46:10024. doi: 10.1021/bi7009508. [DOI] [PubMed] [Google Scholar]
- 522.(a) Fyrst H, Oskouian B, Kuypers FA, Saba JD. Biochemistry. 1999;38:5864. doi: 10.1021/bi9824590. [DOI] [PubMed] [Google Scholar]; (b) Merkel O, Fido M, Mayr JA, Pruger H, Raab F, Zandonella G, Kohlwein SD, Paltauf F. J. Biol. Chem. 1999;274:28121. doi: 10.1074/jbc.274.40.28121. [DOI] [PubMed] [Google Scholar]; (c) Lee KS, Patton JL, Fido M, Hines LK, Kohlwein SD, Paltauf F, Henry SA, Levin DE. J. Biol. Chem. 1994;269:19725. [PubMed] [Google Scholar]
- 523.(a) Masuda N, Kitamura N, Saito K. Eur J. Biochem. 1991;202:783. doi: 10.1111/j.1432-1033.1991.tb16433.x. [DOI] [PubMed] [Google Scholar]; (b) Fujii S, Unezaki S, Okumura T, Miura R, Saito K. J. Biochem. 1994;116:204. doi: 10.1093/oxfordjournals.jbchem.a124494. [DOI] [PubMed] [Google Scholar]
- 524.Maury E, Prevost MC, Nauze M, Redoules D, Tarroux R, Charveron M, Salles JP, Perret B, Chap H, Gassama-Diagne A. Biochem. Biophys. Res. Commun. 2002;295:362. doi: 10.1016/s0006-291x(02)00657-5. [DOI] [PubMed] [Google Scholar]
- 525.Senda K, Yoshioka H, Doke N, Kawakita K. Plant Cell Physiol. 1996;37:347. doi: 10.1093/oxfordjournals.pcp.a028952. [DOI] [PubMed] [Google Scholar]
- 526.Kienesberger PC, Lass A, Preiss-Landl K, Wolinski H, Kohlwein SD, Zimmermann R, Zechner R. J. Biol. Chem. 2008;283:5908. doi: 10.1074/jbc.M709598200. [DOI] [PubMed] [Google Scholar]
- 527.Rhee SG, Yang KS, Kang SW, Woo HA, Chang TS. Antioxid. Redox Signaling. 2005;7:619. doi: 10.1089/ars.2005.7.619. [DOI] [PubMed] [Google Scholar]
- 528.Kang SW, Baines IC, Rhee SG. J. Biol. Chem. 1998;273:6303. doi: 10.1074/jbc.273.11.6303. [DOI] [PubMed] [Google Scholar]
- 529.Kim TS, Sundaresh CS, Feinstein SI, Dodia C, Skach WR, Jain MK, Nagase T, Seki N, Ishikawa K, Nomura N, Fisher AB. J. Biol. Chem. 1997;272:2542. doi: 10.1074/jbc.272.4.2542. [DOI] [PubMed] [Google Scholar]
- 530.Kim TS, Dodia C, Chen X, Hennigan BB, Jain M, Feinstein SI, Fisher AB. Am. J. Physiol. 1998;274:L750. doi: 10.1152/ajplung.1998.274.5.L750. [DOI] [PubMed] [Google Scholar]
- 531.Fisher AB, Dodia C, Manevich Y, Chen JW, Feinstein SI. J. Biol. Chem. 1999;274:21326. doi: 10.1074/jbc.274.30.21326. [DOI] [PubMed] [Google Scholar]
- 532.Nevalainen TJ. Biochimie. 2010;92:638. doi: 10.1016/j.biochi.2010.01.019. [DOI] [PubMed] [Google Scholar]