

Abstract
β-Hexosaminidases (β-hex) are a group of glycosyl hydrolase isozymes that break down neutral and sialylated glycosphingolipids in the lysosomes, thereby preventing their buildup in neuronal cells. Some mutants of β-hex have decreased folding stability that results in adult-onset forms of lysosomal storage diseases. However, prevention of the harmful accumulation of glycolipids only requires 10% of wild-type activity. Pyrimethamine (PYR) is a potential pharmacological chaperone that works by stabilizing these mutant enzymes sufficiently to allow more β-hex to arrive in the lysosome, where it can carry out its function. An X-ray structure of the complex between human β-hexosaminidase B (HexB) and PYR has been determined to 2.8 Å. PYR binds to the active site of HexB where several favorable van der Waals contacts and hydrogen bonds are introduced. Small adjustments of the enzyme structure are required to accommodate the ligand, and details of the inhibition and stabilization properties of PYR are discussed.
INTRODUCTION
Human β-hexosaminidase isozymes are glycosyl hydrolases that remove β-linked nonreducing-terminal GalNAc or GlcNAc residues from a number of macromolecules in the lysosome. There are two subunits of β-hexosaminidase, α (528 residues) and β (556 residues), that share roughly 60% sequence identity and are encoded by two unlinked but evolutionarily related structural genes. These two subunits each have their own active site; however, dimerization is necessary in order for them to become fully functional.1 Thus, these subunits can form three different hexosaminidase (Hex) isozymes, HexA (αβ), HexB (ββ), and HexS (αα). The HexS (αα) isozyme is unstable and only detectable unequivocally in samples from patients with Sandhoff disease, caused by a deficiency of β-subunits, and will not be considered further here. Notable differences between the HexA and HexB isozymes are that the latter is more thermostable and the active site binding pocket of the α subunit in the former can accommodate negatively charged substrates.
The unique natural substrate for HexA is GalNAc-1,4 (Neu-Ac-2,3) Gal-1,4 Glc-ceramide (GM2) ganglioside, a sialylated glycosphingolipid found in the plasma membrane and produced as a breakdown product of the “higher” gangliosides (e.g., GM1 ganglioside), predominately synthesized in neuronal tissue.2,3 The GM2 activator protein, a small substrate-specific cofactor for HexA, sequesters this lipid from the membrane, and the complex binds to HexA. The ceramide tail is enclosed in a unique β-cup fold, leaving the negatively charged neuraminic acid head-group accessible for binding by the HexA active site, ultimately resulting in the cleavage of the terminal β- GalNAc residue producing GM3 ganglioside.1 Substrates for HexB, as well as HexA, are neutral glycosphingolipids, protein oligosaccharides, and mucopolysaccharides with nonreducing terminal β-linked GalNAc or GlcNAc residues.
The Hex isozymes are members of the glycoside hydrolase family 20. All the members of this family use a substrate-assisted catalysis mechanism, which is achieved by positioning the C2-acetamido group of the terminal GalNAc into the appropriate position for nucleophilic attack of the carbonyl oxygen atom on C1.4,5 The substrate binding environment also ensures that a β-configuration is retained by directing the position of the incoming nucleophilic water molecule. A detailed description of the catalytic mechanism can be found in the paper by Lemieux et al.6
Genetic defects in either gene encoding the subunits of HexA can result in the accumulation of GM2 ganglioside in neural tissues and two of three lysosomal storage diseases collectively known as GM2 gangliosidosis. Tay–Sachs disease (defects in the α subunit) and Sandhoff disease (defects in the β subunit) are the most “common” and best studied of this family of diseases. The third form of the disease, the AB variant, is caused by a deficiency of the GM2 activator protein. Mutations that cause a complete enzyme deficiency are extremely severe, and children affected with these forms of the disease usually die before they reach four years of age, i.e., the classical infantile form of Tay–Sachs or Sandhoff disease. Mutations that affect folding or dimerization but are still compatible with the formation of some functional enzyme, resulting in residual HexA activity (1–5% of normal), are less severe and result in a “late-onset” clinical phenotype. Interestingly, it has been estimated that a residual HexA activity of only 5–10% of normal is required to prevent the accumulation of GM2.7,8
Lysosomal enzymes, like secretory and plasma membrane proteins, are synthesized in the endoplasmic reticulum (ER). The ER contains a highly conserved mechanism to protect the cell from misfolded and potentially toxic proteins, the endoplasmic reticulum associated degradation (ERAD) pathway. Thus, if the folding process of either of the subunits of HexA is disrupted, e.g., by a missense mutation, the improperly folded subunits are susceptible to ERAD, making these late-onset disease variants of GM2 gangliosidosis potential targets for enzyme enhancement therapy (EET).9 For this treatment strategy, small molecules that bind to the active site of the protein are introduced in order to stabilize the native enzyme fold, thereby allowing a higher proportion of newly synthesized subunits to successfully fold, dimerize, and escape becoming a substrate for ERAD. This would result in more of the functional enzyme passing through the ER and being transported to lysosomes. Once the enzyme reaches the lysosome, the low pH and mass action assist in the displacement of the small molecule chaperone by the abundant substrate, which can then be hydrolyzed. This has recently been demonstrated in late onset Tay–Sachs cells loaded with a fluorescent derivative of GM2 ganglioside.10
Several molecules that act as inhibitors of HexA and HexB have been identified as potential pharmacological chaperones (PC) for late-onset GM2 gangliosidosis, including known inhibitors of Hex such as N-acetylglucosamine thiazoline (NGT).11 Additionally, two libraries have been screened, the Maybridge library12 (50 000 compounds) and the NINDs library13 (1040 FDA approved compounds). These screens have led to the discovery that pyrimethamine (PYR, 5-(4-chlorophenyl)-6-ethyl-2,4-pyrimidinediamine) is also a good candidate for a PC.9 PYR is more widely recognized as an antimalarial drug that targets dihydrofolate reductase (DHFR). The structure of the DHFR:PYR complex revealed that the drug binds to the active site of DHFR and acts as a competitive inhibitor.14
We have determined the X-ray crystal structure of HexB bound with PYR to 2.8 Å resolution. PYR binds to the active site of HexB in a manner similar to the binding of NGT.1 A comparison of HexB:PYR to HexA:NGT6 suggests that PYR would also bind in the active site of the α-subunit of HexA in a similar manner. The enzyme–inhibitor complex structure presented here provides a framework for future modifications to PYR in order to improve its potency as a PC.
RESULTS
Crystals of β-hexosaminidase B (HexB) soaked with PYR diffract to 2.8 Å and belong to space group P6122. Two monomers of HexB are present in the asymmetric unit. These two monomers do not form the biological dimer.1 Instead, the biological dimer is the result of applying the crystallographic 2-fold rotation axis to each monomer. Data collection and refinement statistics can be found in Table 1. The Rmerge appears high, but this indicator is dependent on redundancy, which is also high for this data set. Alternative indicators of the quality of a data set have been proposed by Weiss,15 and Rpim has been included in Table 1. Weiss has suggested that Rpim is a better indicator than is Rmerge of the performance of diffraction data with respect to refinement.
Table 1.
Data Collection and Refinement Statisticsa
| data collection | |
|---|---|
| beamline | SSRL BL 9-2 |
| wavelength | 0.97946 Å |
| space group | P6122 |
| cell dimensions | a = b = 113.78 Å, c = 397.34 Å, α = β = 90°, γ = 120° |
| resolution | 35–2.8 Å |
| mosaicity | 0.51° |
| total observations | 291 439 |
| unique reflections | 35 710 |
| completeness | 92.1% (87.5) |
| redundancy | 8.1 (5.4) |
| I/σI | 14.0 (2.1) |
| Rmergeb | 0.174 (0. 501) |
| Rpimc | 0.065 (0. 220) |
| Refinement | |
| resolution | 35–2.8 Å |
| no. of reflections used in reflnement | 33909 (Rfree = 1801) |
| Rwork,d Rfreee | 0.197, 0.251 |
| total number of atoms | 8006 |
| protein | 7750 |
| Water | 79 |
| carbohydrate atoms | 137 |
| ligands | 17 × 2PYR, 5 SO4 |
| average B | all atoms 43.0 Å2, main chain 41.7 Å2, side chain 42.6 Å2, carbohydrate 86.4 Å2, water 32.9 Å2, ligand 74.8 Å2 |
| Wilson B | 65.1 Å2 |
| B rmsd | bonded main chain 0.66 Å2, bonded side chain 2.41 Å2, nonbonded main chain 1.36 Å2, nonbonded side chain 4.14 Å2 |
| rmsd from ideal geometry | bond lengths 0.018 Å, bond angles 1.88° |
| Ramachandran Plot | |
| most favored region | 87.4% |
| allowed region | 12.5% |
| generously allowed region | 0.1% (D240β2) |
Values in parentheses refer to the high-resolution shell, 2.9–2.8 Å.
Rmerge = ΣhklΣ|Ij(hkl) − 〈I(hkl)〉|/ΣhklΣj jI(hkl) with Ij(hkl) representing the intensity of measurement j and 〈I(hkl)〉 is the mean of measurements for the reflection hkl.
Rpim = Σhkl[1/(N − 1)]1/2Σj|Ij(hkl) − 〈I(hkl)〉|/ΣhklΣj jI(hkl) with N representing the redundancy.15
Rwork = Σhkl||Fobs(hkl)| − |Fcalc(hkl)||/Σhkl|Fobs(hkl)|, where Fobs and Fcalc are the observed and calculated structure factors, respectively.
Rfree is calculated with the formula for Rwork with 5% of the structure factors that were not included in the model refinement.
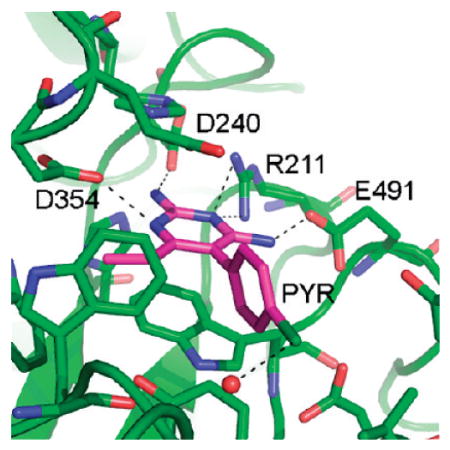
An electron density map in the vicinity of the active site calculated after the initial rigid body refinement and superimposed onto the final refined atomic coordinates is given in Figure 1. The PYR molecule represented in the figure had not yet been included at this stage of refinement. The positive difference density clearly indicated the position and orientation of the soaked inhibitor. Any attempt to rotate the PYR molecule by 180° resulted in a clash between C16 and the side chain atoms of Glu491. Any attempt to alleviate this clash resulted in additional bad contacts. The final electron density for the active site and the bound PYR after refinement is depicted in Figure 2. Oligosaccharides were observed covalently bonded to several Asn residues (84β2, 142β1β2, 190 β1β2, 327 β1β2), all in agreement with expected sites of glycosylation.16,17 The overall description of HexB remains the same as in the native structure1 (Figures 3 and 4). The N-terminal domain of the monomer consists of a six-stranded antiparallel β-sheet with two α-helices packing against it. The C-terminal TIM barrel belongs to family 20 of the glycoside hydrolases. This domain contains the active site and the dimerization site for the biological dimer. PYR was found bound in the active site in slightly different conformations in the β1 and β2 molecules of the asymmetric unit (Figure 5). The pyrimidine ring occupies a space similar to that of NGT6 and forms hydrogen bonding interactions with several residues of the pocket (Table 2, Figures 4 and 6). In order to accommodate the p-chlorophenyl ring, Tyr450 moves by approximately 10° around χ1 relative to its conformation observed in the native structure. The p-chlorophenyl ring adopts a torsion angle of −28.5° (C3–C4–C7-C8, β1) and −99.5° (β2) relative to the pyrimidine ring. The equivalent torsion angle for PYR bound to the active site of DHFR is −65.81°.14 In the β2 molecule, PYR C9 makes a close contact with Tyr450 Cε2 (2.72 Å) and the chlorine atom forms a hydrogen bond with HOH20. Root mean square deviations (rmsd values) for various superimpositions can be found in Table 3. All of the structures superpose well with the largest values calculated for superimpositions of HexA onto HexB (β onto β, 477 Cα atoms, 0.56 Å; α onto β, 472 Cα atoms, 0.77 Å).
Figure 1.

Initial electron density. (a) Electron density map, 2|Fo| − |Fc|, 1σ (purple), calculated after initial rigid body refinement but before the inclusion of PYR. Difference density (|Fo| − |Fc|) has been drawn in green (2.5σ) and orange (−2.5σ). (b) Structure of pyrimethamine. Atom numbering is the same as that found in PDB code 2BL9.14
Figure 2.
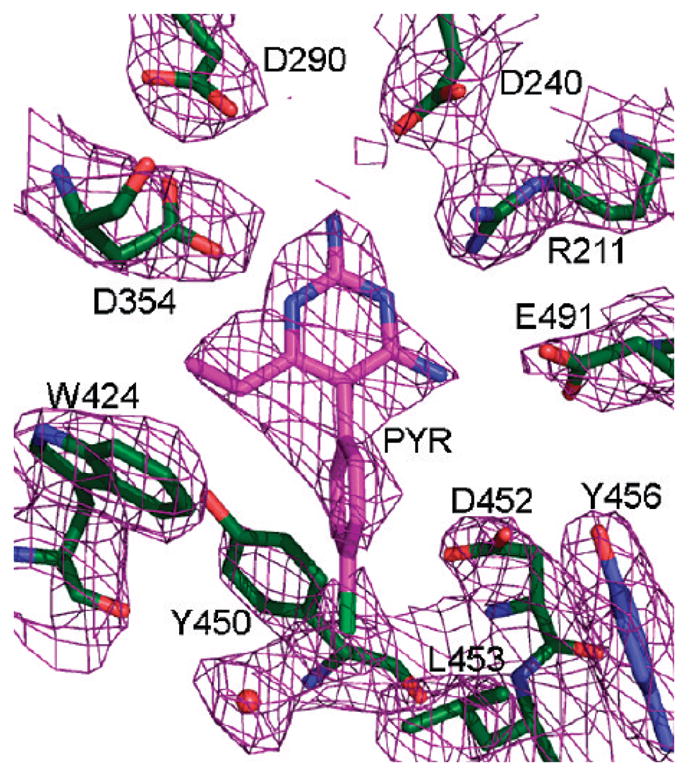
Final electron density map, 2|Fo| − |Fc|, 1σ (purple), drawn around the active site of HexB.
Figure 3.
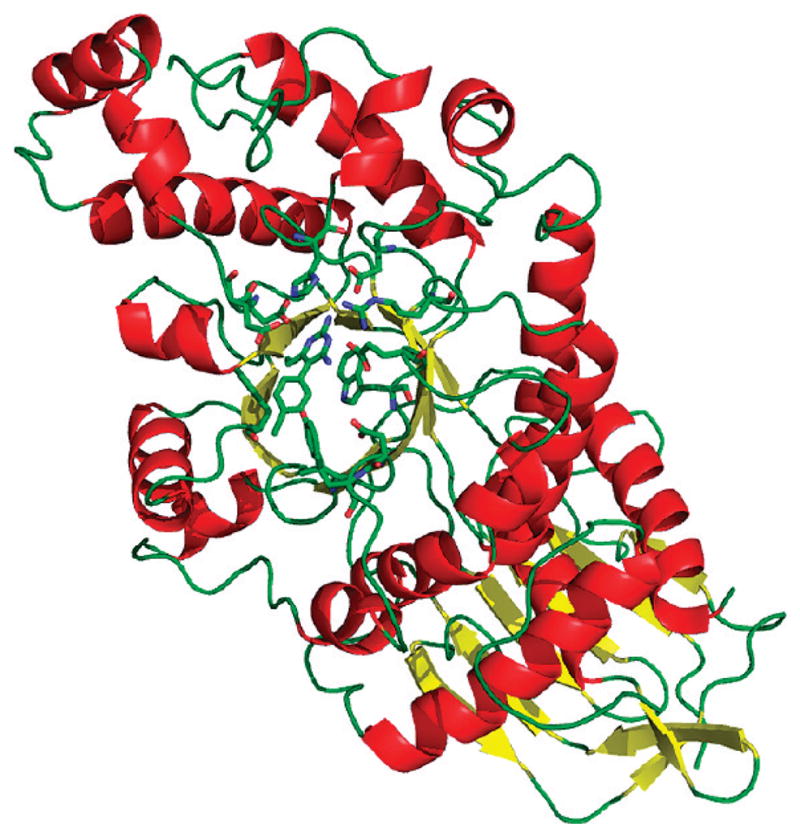
Overall structure of the HexB monomer. α-Helices are depicted in red, and β-strands are in yellow. Active site residues are drawn as ball and stick, as is the PYR molecule.
Figure 4.

Interface of the biological dimer. Molecule β1 has been drawn in green, and the second monomer has been drawn in blue. Residues from the dimer interface and active site have been included as ball and stick.
Figure 5.

Stereoview of the active site of HexB with both monomers in the asymmetric unit superposed. The darker shades of green (enzyme) and pink (PYR) belong to monomer β1 and the lighter shades to monomer β2.
Table 2.
Hydrogen Bonds Involving PYR
| donor | acceptor | distance (Å) |
|---|---|---|
| PYR 1P,a N6 | D354β1 Oδ2 | 2.69 |
| PYR 1P, N14 | D240β1 Oδ2 | 3.08 |
| Arg 211β1, NH1 | PYR1P, N1 | 3.23 |
| Arg 211β1, NH2 | PYR1P, N1 | 2.99 |
| PYR 1P, N13 | E491β1, Oε2 | 2.65 |
| PYR 2P,a N6 | D354β2 Oδ2 | 2.64 |
| PYR 2P, N14 | D240β2 Oδ2 | 3.03 |
| Arg 211β2, NH1 | PYR2P, N1 | 2.95 |
| Arg 211β2, NH2 | PYR2P, N1 | 3.15 |
| PYR 2P, N13 | E491β2, Oε2 | 2.73 |
| Wat 20 HOH | PYR 2P, Cl1 | 3.09 |
Figure 6.

HexB:NGT vs HexB:PYR. Superimposition of HexB:NGT (1NP0) and HexB:PYR. Carbon atoms from HexB:NGT are depicted in cyan, and carbon atoms from HexB:PYR are depicted in green. Hydrogen bonding interactions coordinating both ligands have been included as dashed lines.
Table 3.
Root Mean Square Deviation
| molecule A | molecule B | atoms chosen | number of atoms used | rmsd (Å) |
|---|---|---|---|---|
| HexB β1 | HexB β2 | N, Cα, C, O | 1912 atoms, 479 residues | 0.27 |
| HexB (1NOU) Aa | HexB:PYR β1 | Cα | 478 residues | 0.25 |
| HexB (1NOU) B | HexB:PYR β2 | Cα | 479 residues | 0.27 |
| HexB:NGT (1NP0) Aa | HexB:PYR β1 | Cα | 478 residues | 0.27 |
| HexB:NGT (1NP0) B | HexB:PYR β2 | Cα | 479 residues | 0.29 |
| HexA (2GK1) α | HexB:PYR β1 | Cα | 472 residues | 0.77 |
| HexA (2GK1) β | HexB:PYR β2 | Cα | 477 residues | 0.56 |
In the HexB structures from PDB codes 1NOU and 1NP0, the two β molecules from the asymmetric unit have been labeled as “A” and “B”, not to confuse the “A” for α.
DISCUSSION
HexB:PYR vs DHFR:PYR
PYR is an FDA approved antimalarial drug that targets folic acid synthesis. Specifically, it binds to the active site of DHFR, thereby preventing substrate access. In the structure of Plasmodium vivax (Pv) DHFR,14 PYR sits in a deep pocket containing the catalytic residues (Figures 7 and 8). The p-chlorophenyl ring makes van der Waals contact with the DHFR cofactor NADPH. In the structure of HexB:PYR, PYR also binds at the active site. Although the active site of HexB appears shallower than DHFR (Figure 8), there are many similarities to DHFR in binding the drug. The pyrimidine ring stacks against an aromatic residue (Trp489 in HexB and Phe57 in DHFR) and forms similar hydrogen bonding interactions with residues belonging to the active site (Figure 7).
Figure 7.

PYR binding site DHFR vs HexB. The overall secondary structures of the molecules have been drawn in blue (a, DHFR) and green (b, HexB). Residues and water molecules near the PYR binding site have been depicted as ball and stick. The NADPH molecule and the active site of DHFR have also been included as ball and stick. Hydrogen bonding interactions are indicated by dashed lines. Coordinates for DHFR are from PDB code 2BL9.14
Figure 8.

(a) Stereoview of the van der Waals surface (blue) of the PYR binding site in DHFR. Waters and NADPH molecule bound at the active site have been removed for clarity. Coordinates for DHFR are from PDB code 2BL9.14 (b) Stereoview of the van der Waals surface (green) of the PYR binding site of HexB.
It is important to note that one of the other reasons for the success of PYR as an antimalarial and potential PC drug candidate is that PYR does not seriously impair the function of human DHFR. Since the binding site of human DHFR, (hD-HFR) is slightly smaller than that of Pv DHFR, this might in part account for the reduced binding affinity of PYR to the human orthologue.18 More importantly, Zhang and Rathod have shown that the sensitivity of Pv versus hDHFR arises because different sites on the enzyme are involved in translational repression of its cognate mRNA. Whereas human DHFR protein represses translation of its own mRNA using the same site that binds substrate and PYR, Pv DHFR uses a site other than the substrate binding pocket.19 Thus, in the presence of high concentrations of substrate or PYR, competitive inhibition results in release of translation repression enabling increased synthesis of hDHFR protein that can compensate for the other molecules that are inhibited by PYR. In contrast neither PYR nor DHFR substrate are able to release translation repression of mRNA by Pv DHFR. Consequently, no additional Pv DHFR protein is synthesized to compensate for the PYR bound enzyme.
HexB:NGT vs HexB:PYR
Known inhibitors of HexA were initially screened as potential PCs. NGT, a sulfur analogue of the oxazolinium ion intermediate formed during substrate turnover, was found to be an excellent candidate with a Ki of 300 nM.11 Subsequent screens identified PYR as a potential PC having many favorable qualities such as its ability to cross the blood–brain barrier and the fact that it was already FDA approved, thereby eliminating the need for expensive preclinical trials.13 The encouraging results from a small phase I/II clinical trial of PYR for late onset GM2 gangliosidosis have recently been reported.20
An intriguing property of PYR is the pH profile of inhibition in comparison to that for NGT. NGT inhibits HexA activity optimally at pH 4.5, whereas PYR has an optimal inhibitory pH of 6.5.13 Thus, the pH profile for PYR suggests another advantage over NGT as a PC. That is, PYR inhibits best at the internal pH of the lumen of the ER (pH 7)21 where a chaperone is needed to assist mutant Hex subunits in their folding and dimerization, whereas NGT binds best at the internal pH of the lysosome (pH 5),22 where the chaperone has to be displaced in order to leave the enzyme free to hydrolyze its substrate. A superimposition of HexB:NGT and HexB:PYR is shown in Figure 6, and the rmsd values are given in Table 3. Most of the hydrogen-bonding partners for the two inhibitors are identical. The differing pKa values for NGT (4.5) and PYR (6.5)13 do not entirely explain the differences in inhibition pH profiles, since the positive charge that develops on each inhibitor with lowering pH probably locates to the same position in both complexes (N2 of NGT, N6 of PYR); both of these groups form a salt bridge with Asp354. However, while the other surrounding hydrogen bonding interactions involve the same residues, the “donor” and “acceptor” roles are not identical. The amino groups (N14 and N13) of PYR donate hydrogen bonds to Asp240 and Glu491 in HexB, respectively. As the pH drops, the carboxylic acid side chains can become protonated, and they become less ideal “acceptors”. In contrast, O4 of NGT can be both a hydrogen-bond donor (at neutral pH) and an acceptor (at low pH when Glu or Asp residues become protonated). Along with the flexibility of the water molecule at the equivalent position of N14 in PYR, NGT appears better equipped to accommodate a downward shift in pH.
Crystal structures have also been determined for HexB in complex with the mechanistic inhibitor GalNAc-isofagomine.1,5 The hydrogen bonding partners for this ligand overlap with those found for HexB:NGT, including the water molecule at position N14 in PYR. Therefore, this inhibitor would be expected to have a pH profile of inhibition similar to that of NGT.
Variation in PYR Binding: β1 vs β2
The p-chlorophenyl ring adopts a slightly different torsion angle in each monomer of the asymmetric unit of HexB (Figure 5). In molecule β1, the p-chlorophenyl ring lies approximately 30° out of the plane occupied by the pyrimidine ring. This torsion angle helps to minimize unfavorable contacts with Tyr450 of HexB. In molecule β2 (Figure 6) the p-chlorophenyl ring adopts a torsion angle that forms close contacts with the ring of Tyr450 but allows the Cl atom to form a stabilizing hydrogen bond with a water molecule, Wat20 (numbered Wat52 in β1, this water molecule is present in each active site). Neither conformation is an ideal fit, unlike the more complementary fit for PYR with DHFR (Figures 7 and 8). Whereas the electron density is clear for the pyrimidine ring (Figures 1 and 2), it is much weaker for the p-chlorophenyl ring in both monomers of the asymmetric unit, illustrating that there is some strain or conformational flexibilty in this region. However, it might be the less-than-ideal fit of PYR that contributes to its success as a PC. Once in the lysosome, the inhibitor needs to dissociate from the enzyme in order to allow the enzyme to access its physiological substrate. A more complementary fit might interfere with this particular property of the ideal chaperone.
HexB:PYR vs HexA
Crystals of HexA6 (αβ dimer) were difficult to obtain in comparison to those of HexB (ββ). We were therefore only able to obtain crystals of the complex between HexB and PYR. A superimposition of the active site residues in the structures of HexB:PYR and HexA(NGT) (Figure 9, Table 3) suggests that a HexA:PYR interaction would closely resemble that of HexB:PYR. The only notable differences at the active site/binding pocket are residues Asp452β:Asn423α and Leu453β:Arg424α. The observed conformations of these side chains in the HexA:NGT structure do not present any steric hindrance to PYR binding. Tyr421β1 would have to rotate slightly (as does Tyr450β2 in HexB) in order to accommodate the p-chlorophenyl ring. The pyrimidine ring superimposes onto the NGT molecule and forms hydrogen bonds with some of the same residues from the active site, as well as stacking against Trp489β2 (PYR) and Trp460β1(NGT).6
Figure 9.

Dimer interface of HexA vs HexB. Superimposition of HexA:NGT6 and HexB:PYR is shown. Molecules from HexA have been drawn in pink (α) and light green (β). Both molecules from HexB have been drawn in dark green. Molecules of PYR bound to HexB are drawn in orange. Carbon atoms of NGT bound to HexA are drawn in cyan.
PYR and Late-Onset Mutations for Tay–Sachs and Sandh-off Disease
The binding of PYR to the active site of HexB introduces five hydrogen bonds per active site and several favorable van der Waals contacts, thereby potentially increasing the stability of the protein fold and developing the correct conformation for the active site. Common mutations observed in late-onset Tay–Sachs or Sandhoff patients disrupt the folding stability of the enzyme, and many of these mutations have previously been mapped onto the structure of HexA.6,9,13 Examples include Gly269Ser from the α subunit and the neighboring residues from the β subunit, Pro504Ser and Arg-505Gln. Gly269Ser is located at the C-terminus of an α-helix that begins near the active site. The Cαatom of residue Gly269 is 3.19 Å from the backbone O of Glu220. Introduction of a side chain at this position will cause a serious clash and would likely disrupt the α-helix.6 Substitutions at Pro504 and Arg505 can also destabilize the protein by interfering with the backbone conformation at Pro504 and by a loss of hydrogen-bond and salt-bridge interactions at Arg505. Since most of the destabilizing mutations are not in the immediate vicinity of the active site, they will not interfere with PYR binding, making the small molecule suitable for EET. The introduction of hydrogen bonds and hydrophobic interactions that are associated with PYR binding will compensate for these destabilizing interactions that were introduced by the mutations. The HexA:PYR complex will then be able to bypass the ERAD pathway and successfully relocate to the lysosome where substrate molecules will displace PYR and HexA activity will be restored to adequate levels.
Future Directions
Compared to DHFR, the active-site binding pockets of HexB and HexA are not perfectly matched to bind pyrimethamine. While it is possible that the fit of the p-chlorophenyl ring provides an advantage for dissociation in the lysosome, decreasing the size of the PYR molecule by elimination of the Cl atom might be beneficial for binding in the endoplasmic reticulum. Another potential site of alteration is C16 from the ethyl group of PYR. This atom is 3.45 Å from Asp 354 Oδ2 in β1 and 3.31 Å in β2 (Figure 1). Replacing the ethyl group by an aminomethyl group could introduce a stabilizing salt bridge. The ultimate goal is to create a PC that will alleviate symptoms of late-onset Tay–Sachs and Sandhoff disease, and toward this goal, many chemical modifications associated with experimental analysis to the drug are currently underway (J. Zhang and M. Ciufolini, personal communication).
EXPERIMENTAL PROCEDURES
Crystallization
β-Hexosaminidase B was purified from human placenta as described previously.23 Hexagonal crystals of β-hexosaminidase B were grown from seeds as reported in Mark et al.1 PYR was dissolved in ethanol at 1 mM and was soaked into the crystals 24 h before they were transferred to a glycerol solution and flash-cooled in liquid nitrogen.
Data Collection and Refinement
X-ray diffraction data were collected on beamline 9-2 at Stanford Synchrotron Radiation Laboratory (SSRL)24–26 and processed with HKL2000.27 In order to locate the PYR ligand, a difference Fourier map was calculated with the Collaborative Computational Project 4 (CCP4)28 suite of programs (SFall,29 Sigma-A,30 and Refmac531 for rigid body refinement) using the PDB coordinates (1NOU) of the native β-hexosaminidase B.1 PYR, sugars, SO4 ions, and water molecules were not included in the initial rigid body refinement and map calculations but were added in subsequent rounds. Further rounds of refinement were also carried out with Refmac5.28,31 Between refinement rounds, coordinates and electron density maps were studied and adjusted where needed with Xfit.32 Noncrystallographic symmetry (NCS) restraints were applied to the molecules of the asymmetric unit during refinement and removed for the final round. Superimpositions were calculated with Coot33 and O34, and figures were made with the program Pymol.35 Coordinates have been deposited in the RCSB PDB36 with accession code 3LMY.
Acknowledgments
This research was supported in part by the Canadian Institutes of Health Research (CIHR) and by the Alberta Heritage Foundation for Medical Research. K.S.B. and M.N.G.J. are grateful for the support by the CIHR Team Grant CTP-82944. Portions of this research were carried out at the Stanford Synchrotron Radiation Laboratory, a national user facility operated by Stanford University on behalf of the U.S. Department of Energy, Office of Basic Energy Sciences. The SSRL Structural Molecular Biology Program is supported by the Department of Energy, Office of Biological and Environmental Research, and by the National Institutes of Health, National Center for Research Resources, Biomedical Technology Program, and the National Institute of General Medical Sciences.
ABBREVIATIONS USED
- CCP4
Collaborative Computational Project 4
- DHFR
dihydrofolate reductase
- ERAD
endoplasmic reticulum associated degradation
- GM2
GalNAc-1,4 (NeuAc-2,3) Gal-1,4 Glc-ceramide
- HexA
β-hexosaminidase A
- HexB
β-hexosaminidase B
- NAG
N-acetylglucosamine
- NCS
noncrystallographic symmetry
- NGT
N-acetylglucosamine thiazoline
- PC
pharmacological chaperone
- Pv
Plasmodium vivax
- PYR
5-(4-chlorophenyl)-6-ethyl-2,4-pyrimidinediamine (pyrimethamine)
- rmsd
root mean square deviation
- SSRL
Stanford Synchrotron Radiation Laboratory
Footnotes
Accession Codes
PDB code: 3LMY.
References
- 1.Mark BL, Mahuran DJ, Cherney MM, Zhao D, Knapp S, James MN. Crystal structure of human beta-hexosaminidase B: understanding the molecular basis of Sandhoff and Tay–Sachs disease. J Mol Biol. 2003;327:1093–1109. doi: 10.1016/s0022-2836(03)00216-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hepbildikler ST, Sandhoff R, Kolzer M, Proia RL, Sandhoff K. Physiological substrates for human lysosomal beta-hexosaminidase S. J Biol Chem. 2002;277:2562–2572. doi: 10.1074/jbc.M105457200. [DOI] [PubMed] [Google Scholar]
- 3.Kresse H, Fuchs W, Glossl J, Holtfrerich D, Gilberg W. Liberation of N-acetylglucosamine-6-sulfate by human beta-N-acetyl-hexosaminidase A. J Biol Chem. 1981;256:12926–12932. [PubMed] [Google Scholar]
- 4.Mark BL, Vocadlo DJ, Knapp S, Triggs-Raine BL, Withers SG, James MN. Crystallographic evidence for substrate-assisted catalysis in a bacterial beta-hexosaminidase. J Biol Chem. 2001;276:10330–10337. doi: 10.1074/jbc.M011067200. [DOI] [PubMed] [Google Scholar]
- 5.Maier T, Strater N, Schuette CG, Klingenstein R, Sandhoff K, Saenger W. The X-ray crystal structure of human beta-hexosaminidase B provides new insights into Sandhoff disease. J Mol Biol. 2003;328:669–681. doi: 10.1016/s0022-2836(03)00311-5. [DOI] [PubMed] [Google Scholar]
- 6.Lemieux MJ, Mark BL, Cherney MM, Withers SG, Mahuran DJ, James MN. Crystallographic structure of human beta-hexosaminidase A: interpretation of Tay–Sachs mutations and loss of GM2 ganglioside hydrolysis. J Mol Biol. 2006;359:913–929. doi: 10.1016/j.jmb.2006.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Leinekugel P, Michel S, Conzelmann E, Sandhoff K. Quantitative correlation between the residual activity of beta-hexosaminidase A and arylsulfatase A and the severity of the resulting lysosomal storage disease. Hum Genet. 1992;88:513–523. doi: 10.1007/BF00219337. [DOI] [PubMed] [Google Scholar]
- 8.Mahuran DJ. Biochemical consequences of mutations causing the GM2 gangliosidoses. Biochim Biophys Acta. 1999;1455:105–138. doi: 10.1016/s0925-4439(99)00074-5. [DOI] [PubMed] [Google Scholar]
- 9.Tropak MB, Mahuran D. Lending a helping hand, screening chemical libraries for compounds that enhance beta-hexosaminidase A activity in GM2 gangliosidosis cells. FEBS J. 2007;274:4951–4961. doi: 10.1111/j.1742-4658.2007.06040.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tropak MB, Bukovac SW, Rigat BA, Yonekawa S, Wakarchuk W, Mahuran DJ. A sensitive fluorescence-based assay for monitoring GM2 ganglioside hydrolysis in live patient cells and their lysates. Glycobiology. 2010;20:356–365. doi: 10.1093/glycob/cwp183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tropak M, Reid SP, Guiral M, Withers SG, Mahuran D. Pharmacological enhancement of beta-hexosaminidase activity in fibroblasts from adult Tay–Sachs and Sandhoff patients. J Biol Chem. 2004;279:13478–13487. doi: 10.1074/jbc.M308523200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tropak MB, Blanchard JE, Withers SG, Brown ED, Mahuran DJ. High-throughput screening for human lysosomal beta-N-acetyl hexosaminidase inhibitors acting as pharmacological chaperones. Chem Biol. 2007;14:153–164. doi: 10.1016/j.chembiol.2006.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maegawa GH, Tropak M, Buttner J, Stockley T, Kok F, Clarke JT, Mahuran DJ. Pyrimethamine as a potential pharmacological chaperone for late-onset forms of GM2 gangliosidosis. J Biol Chem. 2007;282:9150–9161. doi: 10.1074/jbc.M609304200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kongsaeree P, Khongsuk P, Leartsakulpanich U, Chitnumsub P, Tarnchompoo B, Walkinshaw MD, Yuthavong Y. Crystal structure of dihydrofolate reductase from Plasmodium vivax: pyrimethamine displacement linked with mutation-induced resistance. Proc Natl Acad Sci US A. 2005;102:13046–13051. doi: 10.1073/pnas.0501747102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Weiss MS. Global indicators of X-ray data quality. J Appl Crystallogr. 2000;34:130–135. [Google Scholar]
- 16.O’Dowd BF, Cumming DA, Gravel RA, Mahuran D. Oligosaccharide structure and amino acid sequence of the major glycopeptides of mature human beta-hexosaminidase. Biochemistry. 1988;27:5216–5226. doi: 10.1021/bi00414a041. [DOI] [PubMed] [Google Scholar]
- 17.Schuette CG, Weisgerber J, Sandhoff K. Complete analysis of the glycosylation and disulfide bond pattern of human beta-hexosaminidase B by MALDI-MS. Glycobiology. 2001;11:549–556. doi: 10.1093/glycob/11.7.549. [DOI] [PubMed] [Google Scholar]
- 18.Bowman AL, Lerner MG, Carlson HA. Protein flexibility and species specificity in structure-based drug discovery: dihydrofolate reductase as a test system. J Am Chem Soc. 2007;129:3634–3640. doi: 10.1021/ja068256d. [DOI] [PubMed] [Google Scholar]
- 19.Zhang K, Rathod PK. Divergent regulation of dihydrofolate reductase between malaria parasite and human host. Science. 2002;296:545–547. doi: 10.1126/science.1068274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Clarke JT, Mahuran DJ, Sathe S, Kolodny EH, Rigat BA, Raiman JA, Tropak MB. An open-label phase I/II clinical trial of pyrimethamine for the treatment of patients affected with chronic GM2 gangliosidosis (Tay–Sachs or Sandhoff variants) Mol Genet Metab. doi: 10.1016/j.ymgme.2010.09.004. [Online early access] Published Online: Sep 17, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim JH, Johannes L, Goud B, Antony C, Lingwood CA, Daneman R, Grinstein S. Noninvasive measurement of the pH of the endoplasmic reticulum at rest and during calcium release. Proc Natl Acad Sci US A. 1998;95:2997–3002. doi: 10.1073/pnas.95.6.2997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ohkuma S, Poole B. Fluorescence probe measurement of the intralysosomal pH in living cells and the perturbation of pH by various agents. Proc Natl Acad Sci US A. 1978;75:3327–3331. doi: 10.1073/pnas.75.7.3327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mahuran D, Lowden JA. The subunit and polypeptide structure of hexosaminidases from human placenta. Can J Biochem. 1980;58:287–294. doi: 10.1139/o80-038. [DOI] [PubMed] [Google Scholar]
- 24.Cohen AE, Ellis PJ, Miller MD, Deacon AM, Phizackerley RP. An automated system to mount cryo-cooled protein crystals on a synchrotron beamline, using compact sample cassettes and a small-scale robot. J Appl Crystallogr. 2002;35:82–86. doi: 10.1107/s0021889802016709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gonzalez A, Moorhead P, McPhillips SE, Song J, Sharp K, Taylor JR, Adams PD, Sauter NK, Soltis SM. Web-ice: integrated data collection and analysis for macromolecular crystallography. J Appl Crystallogr. 2008;41:176–184. [Google Scholar]
- 26.McPhillips TM, McPhillips SE, Chiu HJ, Cohen AE, Deacon AM, Ellis PJ, Garman E, Gonzalez A, Sauter NK, Phizackerley RP, Soltis SM, Kuhn P. Blu-ice and the distributed control system: software for data acquisition and instrument control at macromolecular crystallography beamlines. J Synchrotron Radiat. 2002;9:401–406. doi: 10.1107/s0909049502015170. [DOI] [PubMed] [Google Scholar]
- 27.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 28.CCP4. The CCP4 suite: programs for protein crystallography. Acta Crystallogr. 1994;D50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 29.Brunger AT. Free R value: a novel statistical quantity for assessing the accuracy of crystal structures. Nature. 1992;355:472–474. doi: 10.1038/355472a0. [DOI] [PubMed] [Google Scholar]
- 30.Read RJ. Acta Crystallogr. 1990;A46:900–912. [Google Scholar]
- 31.Pannu NS, Murshudov GN, Dodson EJ, Read RJ. Incorporation of prior phase information strengthen maximum-likelihood structure refinement. Acta Crystallogr. 1998;D54:1285–1294. doi: 10.1107/s0907444998004119. [DOI] [PubMed] [Google Scholar]
- 32.McRee DE. XtalView/Xfit, a versatile program for manipulating atomic coordinates and electron density. J Struct Biol. 1999;125:156–165. doi: 10.1006/jsbi.1999.4094. [DOI] [PubMed] [Google Scholar]
- 33.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jones TA, Zou JY, Cowan SW, Kjeldgaard D. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. 1991;A47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 35.DeLano WL. The PyMOL Molecular Graphics System. DeLano Scientific; Palo Alto, CA: 2002. [Google Scholar]
- 36.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]