

Abstract
Retinal membrane guanylyl cyclase (RetGC)1 in the outer segments of vertebrate photoreceptors is controlled by guanylyl cyclase activating proteins (GCAPs), responding to light-dependent changes of the intracellular Ca2+ concentrations. We present evidence that a different RetGC binding protein, retinal degeneration 3 protein (RD3), is a high-affinity allosteric modulator of the cyclase which inhibits RetGC activity at submicromolar concentrations. It suppresses the basal activity of RetGC in the absence of GCAPs in a non-competitive manner and it inhibits the GCAP-stimulated RetGC at low intracellular Ca2+ levels. RD3 opposes the allosteric activation of the cyclase by GCAP, but does not significantly change Ca2+ sensitivity of the GCAP-dependent regulation. We have tested a number of mutations in RD3 implicated in human retinal degenerative disorders and have found that several mutations prevent the stable expression of RD3 in HEK293 cells and decrease the affinity of RD3 for RetGC1. The RD3 mutant lacking the carboxy-terminal half of the protein and associated with Leber congenital amaurosis type 12 (LCA12) is unable to suppress the activity of the RetGC1/GCAP complex. Furthermore, the inhibitory activity of the G57V mutant implicated in cone-rod degeneration is strongly reduced. Our results suggest that inhibition of RetGC by RD3 may be utilized by photoreceptors to block RetGC activity during its maturation and/or incorporation into the photoreceptor outer segment rather than participate in dynamic regulation of the cyclase by Ca2+ and GCAPs.
Keywords: photoreceptors, guanylyl cyclase, guanylyl cyclase activating proteins, cGMP, retina, retinal degeneration, enzyme inhibition, Leber congenital amaurosis
INTRODUCTION
Cyclic GMP in vertebrate photoreceptors couples rhodopsin in disk membranes to cGMP-gated cation channels in the plasma membrane of rod outer segments (ROS) through a photoransduction cascade (reviewed in (1)). Following photoexcitation, cGMP in photoreceptors is first hydrolyzed by a light-activated phosphodiesterase (PDE6) and then replenished by a retinal membrane guanylyl cyclase (RetGC) (2–4). In addition to its role as a key enzyme in photoreceptor physiology, RetGC has been linked to multiple cases of congenital blinding disorders in human patients including Leber congenital amaurosis type 1 (LCA1) and dominant cone-rod dystrophy (5–7). RetGC activity in rods and cones is controlled by Ca2+-sensitive guanylyl cyclase activating proteins, GCAPs (8–11), via a Ca2+ feedback mechanism. When cGMP-gated channels close in response to light, the reduced influx of Ca2+ through the channels causes GCAP to switch from a Ca2+-bound (inhibitor) state to a Mg2+-bound (RetGC activator) state (reviewed in (12)). This accelerates the re-synthesis of cGMP for timely recovery of photoreceptors to their dark state or establishing light adaptation (13–14).
More recently, it has been shown that RD3, a 23-kDa protein unrelated to GCAPs and linked to Leber congenital amaurosis type12 (LCA12) and rapid retinal degeneration in the rd3 mouse (15), co-immunoprecipitates with RetGC and is essential for the normal expression of RetGC in rod and cone photoreceptor cells (16). The RD3 gene transcripts are highly abundant in the retina and concentrated in photoreceptors but undetectable in other mouse tissues (15). An anti-RD3 polyclonal antibody was observed to stain the inner and outer segments of mouse rod and cone photoreceptor cells by immunofluorescence microscopy (16). RD3 also co-localized with RetGC in intracellular vesicles when both proteins were co-expressed in HEK293 cells (16), thus leading to a hypothesis that at least one function of RD3 in photoreceptor cells is to participate in the intracellular trafficking of the cyclase.
In the present study, we have investigated whether the association of RetGC with RD3 can affect the catalytic function of RetGC. We find that RD3 acts as a high-affinity allosteric inhibitor of RetGC, capable of both effectively competing with GCAPs and suppressing the catalytic activity of the cyclase. Some mutations in the human RD3 gene found in patients with congenital retinal disorders strongly affect the inhibitory activity of the human recombinant RD3 protein in vitro.
EXPERIMENTAL PROCEDURES
Antibodies
Rabbit polyclonal antibody RD3 #497 was raised against the isolated full-length human recombinant RD3 expressed in E. coli as described below and purified from the serum by immunoaffinity chromatography on RD3 coupled to CNBr-activated Sepharose CL-4B (GE Heath Sciences). The Rho-1D4 mouse monoclonal antibody (17) was against the TETSQVAPA peptide used as a C-terminal tag in some RD3 constructs, and the anti-RD3 mouse monoclonal RD3-9D12 antibody was produced against the C-terminal peptide of RD3 (16). The rabbit polyclonal anti-RetGC1 antibody was produced against the catalytic domain of human RetGC1 (18) and the mouse monoclonal GC-8A5 antibody was raised against the C-terminus of mouse RetGC1.
Human recombinant RD3 expression in E. coli
Human RD3 cDNA was amplified from a pCMV-SPORT6/MHS1010-9206149 cDNA clone (Open Biosystems/Thermo Scientific) using high-fidelity Phusion Flash DNA polymerase (Finnzymes), subcloned into the NcoI/BamHI sites of pET11d vector (Novagen/Calbiochem), sequenced, and expressed in a BL21(DE3)CodonPlus (Agilent Technologies) E. coli strain in the presence of isopropyl β-D-1-thiogalactopyranoside for 2 hours. The protein which accumulated in inclusion bodies was purified by series of sonication and centrifugation cycles described for GCAP purification (19), solubilized in 10 mM Tris-HCl (pH 7.5) buffer containing 2 mM EDTA, 8 M urea, and 14 mM 2-mercaptoethanol, and dialyzed against 2 × 300 volumes of 10 mM Tris-HCl (pH 7.5) buffer containing 0.1 mM EDTA and 14 mM 2-mercaptoethanol at 4°C. Insoluble protein was removed by centrifugation at 10,000 × g for 10 min at 4°C, and the supernatant containing RD3 (typically 80 – 90% purity by SDS PAGE) was collected and used either immediately or after storage at −70°C with 50% v/v glycerol. RD3 has a tendency to precipitate under normal storage conditions. For expression of RD3 mutants (15) in E. coli, the corresponding mutations were introduced into the RD3 cDNA using Phusion Flash DNA polymerase (Finnzymes) by a conventional “splicing by overlap extension” method and the mutated cDNA was verified by sequencing the entire coding region of the resulting plasmid.
Expression of a full-length human RD3 in HEK293 cells used in RetGC activity assays
The RD3 cDNA was inserted into the HindIII/XbaI sites of a modified pRCCMV vector (Invitrogen), transfected into a 50 to 80%-confluent cell culture (ca. 20 μg of DNA per 100-mm dish) using the calcium phosphate DNA precipitation protocol and expressed for 24 – 36 hours. The soluble fraction containing RD3 was extracted from the harvested cells which were subjected to a hypotonic shock on ice in a buffer solution containing 10 mM Tris-HCl (pH 7.5), 0.1 mM EGTA, 14 mM 2-mercaptoethanol, 60 μg/ml aprotinin, and 30 μg/ml leupeptin and further disrupted by 10–15 strokes in a Dounce glass-to-glass homogenizer. The soluble fraction was clarified by centrifugation at 12,000 × g for 30 min, 4°C, and concentrated in a Millpore Amicon Ultra-4 MW10000 cartridge. A 10-μl sample was subjected to SDS PAGE on a 15% gel. Expression of RD3 was confirmed by immunoblotting probed with the rabbit polyclonal anti-RD3 antibody #497 and visualized using a SuperSignal chemiluminescence peroxidase substrate kit (Pierce/ThermoFisher) in a FotoDyne Luminous FX imaging system.
For comparative testing of the RD3 mutant expression in cultured HEK293T cells, wild type and the mutant RD3 cDNA constructs generated using the QuikChange Mutagenesis kit (Agilent Technologies) all contained an extension that coded for the C-terminal Rho-1D4 peptide, TETSQVAPA. HEK293T cells were transfected with 10 μg of the RD3 plasmid DNA per a 100-mm culture dish using the calcium phosphate precipitation protocol. Forty-eight hours after transfection, the cells from 2 plates were solubilized in 0.8 ml of 1% Triton X-100 in PBS containing Complete Protease Inhibitor cocktail (Roche) for 15 min at 4°C and the insoluble material was removed by centrifugation. Each sample was resolved on a 10% SDS polyacrylamide gel and analyzed on immunoblots sequentially probed with the Rho-1D4 mouse monoclonal antibody and secondary goat anti-mouse antibody conjugated to IR dye 680 for imaging on a LI-COR Odyssey infrared imaging system.
GCAP1 and GCAP2 expression
GCAP1 and GCAP2 were expressed from pET11d vector in a BLR(DE3) E. coli strain harboring yeast N-myristoyl transferase as previously described (19, 20).
RetGC activity
RetGC cyclase activity in ROS fraction isolated from mouse retinas using Optiprep density gradient centrifugation as described in (21) was assayed in the dark using infrared viewers as described previously (14, 22–23). The activity of the recombinant human RetGC1 expressed from pRCCMV vector in HEK293 cells transfected by calcium-phosphate precipitation method was assayed under normal illumination as described previously (22). The assay mixture (25 μL) contained (unless indicated otherwise) 30 mM MOPS – KOH (pH 7.2), 60 mM KCl, 4 mM NaCl, 1mM DTT, 2 mM Ca2+/EGTA buffer, 1 mM free Mg2+, 0.3 mM ATP, 4 mM cGMP, 1 mM GTP, 10 mM creatine phosphate, 0.5 unit of creatine phosphokinase, 1 μCi of [α-32P]GTP, 0.1 μCi of [8-3H]cGMP (Perkin Elmer), PDE6 inhibitors zaprinast and dipyridamole, and variable concentrations of GCAPs, RD3, and membranes containing RetGC1. The reaction continued at a linear time-course for 12 min at 30°C (40 min for the recombinant RetGC1) and subsequently heat-inactivated at 95°C for 2 min. The [32P]cGMP was quantified using a thin-layer chromatography on polyethylenimine cellulose plates. Statistical analysis of the data, where applicable, was performed using KaleidaGraph (Synergy Software).
Ca2+ buffers
Free Ca2+ and Mg2+ concentrations in the RetGC assay were maintained using a series of Ca2+/EGTA mixtures prepared using Tsien and Pozzan method (24), and their free Ca2+ and Mg2+ concentrations were calculated using the Marks and Maxfield algorithm in Bound and Determined and MaxChelator software which corrects for the effects of other components of the assay mixture. The calculated Ca2+/EGTA buffering was verified by fluorescent indicator dyes as previously described in detail (20, 25).
Co-immunoprecipitation (co-IP)
For the co-IP studies, the HEK293T cell cultures were transfected with 10 μg of the RD3 plasmid and 10 μg of human RetGC1 plasmid DNA per a 100-mm diameter culture dish. Twenty-four hours after the transfection, the cells were solubilized in 800 μl 1% Trition X-100 in PBS containing Complete Protease Inhibitor cocktail (Roche). After removal of the insoluble material by centrifugation, the supernatant was applied to an Rd3-9D12-Sepharose immunoaffinity column. After 1 hour the column was washed several times with 0.1% Triton X-100 in PBS and the fraction containing bound proteins was eluted with 80 μl of 3% SDS.
RESULTS
Inhibition of the basal and the GCAP-stimulated RetGC activity by RD3
Recombinant RD3 strongly inhibits the activity of the native RetGC1/GCAP1 complex in ROS membranes and the recombinant RetGC1 expressed in HEK293 membranes and reconstituted with purified GCAP1 (Fig. 1A–C). The suppression of the RetGC activity was observed by RD3 expressed and purified from E. coli (Fig. 1A, B) and RD3 present in a soluble extract from the HEK293 cells expressing recombinant protein (Fig. 1C). The experiment presented in Fig. 1D also demonstrates that RD3 suppresses the catalytic activity of RetGC in the absence of GCAPs. Two isoforms of GCAPs, GCAP1 and GCAP2, are the products of neighboring genes, which are both eliminated by a single knockout construct in GCAP1,2−/− mice (26). GCAP1 and GCAP 2 are the only two GCAP isoforms present in the mouse genome. Therefore, RetGC in the GCAP1,2−/− remains insensitive to Ca2+ (13, 26) and its basal cyclase activity is much lower than the GCAP-stimulated RetGC (21), even though the level of RetGC expression in the double knockout retinas remains normal (21, 26). Since RD3 inhibits RetGC activity in ROS membranes from the GCAP1,2−/− retinas (Fig. 1D), it must directly inhibit the catalytic activity of the cyclase per se.
Fig. 1.

RD3 inhibits RetGC activity at submicromolar concentrations. A. Wild type mouse ROS were titrated with human recombinant RD3 in the presence of 1 mM free Mg2+ and 2 mM EGTA. Inset, Coomassie Brilliant Blue R250 - stained gel of human RD3 isolated from E. coli (left lane) and molecular weight standards (right lane). B. Human recombinant RetGC1 activated by 1.5 μM GCAP1 in the presence of 1 mM free Mg2+ and 2 mM EGTA was assayed at different concentrations of the human recombinant RD3 expressed and purified from E. coli. C. Human recombinant RetGC1 was activated by 1.5 μM bovine GCAP1 in the presence of 1 mM free Mg2+ and 2 mM EGTA and titrated with protein extracts from HEK293 cells either expressing or not expressing RD3. To exclude the possibility of a non-specific effect of different total protein concentrations, the total amount of protein was equilibrated with a control protein extract in every assay mixture. Inset, immunoblotting of HEK293 cell extracts probed with anti-RD3 antibody 497, left lane - non-transfected cells; right lane – RD3 plasmid-transfected cells (notice that there is no endogenous RD3 expression in the non-transfected cells). The data in A–D are fitted by the equation, a = (amax − amin)/(1+[RD3]/IC50) + amin; where amax and amin are the maximal and minimal activity of guanylyl cyclase in the experiment, respectively, and the IC50 is the concentration of RD3 producing 50% inhibition. D–F. The effect of RD3 on guanylyl cyclase catalytic activity in ROS fractions measured in the absence of GCAPs. The RetGC activity in ROS fraction from GCAP1,2−/− mouse retinas (24) was titrated with the purified E. coli-expressed RD3 in the presence of saturating 10 mM Mg2+ and 2 mM EGTA; GTP concentration in D was 1 mM and in E–F varied as indicated. E. Michaelis plot of the non-stimulated RetGC catalytic activity in the absence (●) or in the presence of 30 nM (■) or 60 nM (▲) purified recombinant RD3, representative from three similar independent experiments. F. Lineweaver-Burke plot for data from panel E illustrates a respective ~2.5-fold and 5-fold suppression of the Vmax by 30 nM and 60 nM RD3 without a major effect on KmGTP. The Ki calculated using the equation for a noncompetitive inhibition, Vi=Vmax(1+[RD3]/Ki)−1, from three independent experiments was 19 nM ± 7 SD. The activity in assays containing retinal membranes is presented per rhodopsin content, in membranes expressing recombinant RetGC it is normalized by the maximal activity for each series of the membrane preparations.
Classical inhibitory analysis of Michaelis kinetics is presented in Fig. 1E–F for basal RetGC activity. There is no significant difference between the KmGTP values (mean ± SD from 3 independent measurements) in the absence (1.31 ± 0.17 mM) or in the presence of 30 nM (1.57 ± 0.25 mM) and 60 nM (0.94 ± 0.41 mM) RD3, whereas the Vmax was reduced nearly 2.5-fold and 5-fold – from 3.5 ± 0.7 to 1.4 ± 0.4 and 0.7 ± 0.4 nmol min−1 mg Rh−1, respectively (P<0.05). This argues that the inhibition of the basal catalytic activity of RetGC by RD3 in the absence of GCAPs is mostly non-competitive (Ki ~ 20 nM). Based on this, it is unlikely that RD3 directly binds to the catalytic pocket of the enzyme and affects substrate binding. Since the RetGC is active only as a homodimer (7, 27–29), one possible mechanism for the RD3 affecting RetGC catalytic activity is by altering the conformation of the cyclase dimer. This possible mechanism requires further study.
In addition to the basal activity, RD3 also inhibits the activity of RetGC reconstituted with GCAP as well as the RetGC activity in ROS fraction isolated from wild type mice (Fig. 1A). This suggests that the GCAP-stimulated activity of RetGC can also be affected by RD3, perhaps through its competition with GCAPs. Indeed, the data presented in Fig. 2 directly support the possibility that RD3 strongly competes with GCAP1 for the recombinant human RetGC1 when both regulator proteins are present in the assay. When RetGC1 is co-expressed with RD3, the concentration-dependence of its activation by GCAP measured in HEK293 cell homogenates shifts to a much higher range compared to the RetGC1 expressed in the absence of RD3 (Fig. 2A). Likewise, the competition between RD3 and GCAPs for the cyclase is also evident from the experiments when RetGC1 expressed in HEK293 cells is activated by purified GCAP1 (Fig. 2C) or GCAP2 (Fig. 2D) in the presence of different concentrations of purified recombinant RD3. While the maximal level of the cyclase activity stimulated by GCAPs at saturation is only slightly affected by RD3, the concentration-dependence of the activation by each GCAP shifts toward much higher EC50 values. The latter can only be interpreted as a competitive blocking of the GCAP-dependent stimulation of RetGC by RD3. We emphasize that in these experiments RD3 competes with the GCAPs regardless of what expression system is used to produce the RD3 protein, i.e. HEK 293 cells (Fig. 2A) or E. coli (Fig. 2C–D). Therefore, the effect we observe cannot be attributed to a non-specific artifact(s) of a particular RD3 expression system.
Fig. 2.

RD3 inhibits RetGC activation through competition with GCAPs. A. The guanylyl cyclase activity in HEK293 homogenates containing RetGC1 expressed alone (●) or co-expressed with human RD3 (▲) was assayed in the presence of added recombinant GCAP1. The data were fitted assuming Michaelis hyperbolic function, a=amax[GCAP]/(K1/2+(GCAP)); equalized by RetGC1 content in both samples. B. Immunoblotting. The cells expressing RetGC1, either alone or co-transfected with RD3, from panel A were probed with anti-RetGC1 (left) or anti-RD3 polyclonal antibody 497 (right). Non-transfected HEK293 cells (leftmost in each panel) were used as a specificity control. C–F. Competition of RD3 with GCAP1 and GCAP2 in RetGC assay. C, D. RetGC1 expressed in HEK293 cells was activated by purified GCAP1 (C) or GCAP2 (D) in the absence (●) or in the presence of 3 nM RD3 (▲) or 9 nM RD3 (■). The data were fitted by Michaelis hyperbolic function. Maximal RetGC1 activity (amax, mean ± SD) at 0 nm, 3 nM or 9 nM RD3 was 4.8 ± 0.13, 4.8 ± 0.11, and 4.6 ± 0.16 nmol/min/mg protein, respectively, when activated by GCAP1 and 2.1 ± 0.05, 2.6 ± 0.2 and 2.7 ± 0.1 nmol/min/mg when activated by GCAP2. The respective concentrations of GCAP producing half-maximal activation (K1/2) were 1.1 ± 0.12, 4.1 ± 0.6, and 7.5 ± 0.5 μM (GCAP1) and 5.9 ± 0.6, 19 ± 2.6, and 36 ± 1.5 μM (GCAP2). E, F. Double-reciprocal plots related to panels C and D, respectively.
RD3 affects the activation of RetGC rather than Ca2+ sensitivity of the GCAP-dependent regulation
The effect of RD3 on Ca2+ and Mg2+- dependence of RetGC regulation by GCAPs was tested using two different preparations – the native ROS fraction obtained from dark-adapted mouse retinas by density gradient centrifugation (Fig. 3A–C) and a membrane fraction from the HEK293 cells expressing RetGC1 and reconstituted with the recombinant GCAP1 (Fig. 3D–F). When RetGC activity partially suppressed by RD3 is measured as a function of Ca2+ concentration, its sensitivity to Ca2+ remains similar to that in the absence of RD3. The sensitivity of cyclase activity to Mg2+ is directly relevant to its sensitivity to Ca2+. This is because Mg2+ is required for maintaining the activator conformation of GCAP1 in the light (12, 20, 26, 30) and competes with Ca2+ binding in the EF hand domains of GCAP (20) thus shifting the Ca2+ sensitivity of the cyclase regulation by GCAPs (23). This shift appears in the presence of RD3 when the free Mg2+ level in the assay is increased from a near-physiological 1 mM to 6 mM and is similar to the control samples assayed in the absence of RD3 (Fig. 3C, F).
Fig. 3.

RetGC activation by GCAP suppressed by RD3 retains near normal sensitivity to Ca2+and Mg2+. A–C. ROS fraction isolated from C57B6 mouse retinas was assayed for RetGC activity (open symbols) at various free Ca2+ concentrations and either 1 mM (A) or 6 mM (B) free Mg2+ concentrations in the presence or in the absence of 0.1 μM or 0.5 μM recombinant RD3 as indicated; in panel C, the activities normalized to the maximal RetGC activity in each assay series are shown as a function of free Ca2+ concentrations. D–F. Membrane fraction from HEK 293 cells expressing human RetGC1 (filled symbols) was reconstituted with 5 μM recombinant GCAP1 in the absence or in the presence of 10 nM and 30 nM RD3 as indicated and assayed as described under Experimental Procedures.
Mutations in human RD3 affect its expression in HEK293 cells and RetGC activity in vitro
A nonsense mutation in rd3 mice that terminates RD3 synthesis after Glu106 and a mutation in LCA12 patients that alters a splicing signal and also generates a stop codon after Arg99 both delete the C-terminal half of the protein (15). In addition to the deletion mutation directly linked to LCA12, a number of missense mutations were detected in patients with other retinal disorders (15), although their roles in development of the disease remain to be established. We have tested the effect of several such mutations on human RD3 expression in HEK293 cells and their ability to to associate with RetGC in co-IP expertiments (Fig. 4). The F100ter mutation mimicking the LCA-linked truncation of RD3 as well as the G57V mutant failed to accumulate in transfected HEK293T cells at detectable levels (Fig. 4A, B). This is in agreement with the very low expression of the related mouse RD3 truncated protein observed in transfected COS-1 culture cells (15). Other tested RD3 mutants were expressed in HEK293 cells and co-immunoprecipitated with RetGC1, indicating that they retain their general ability to interact with the cyclase (Fig. 4C). We then evaluated the inhibitory activity of different mutants expressed and purified from E. coli (Fig. 5). Although the expression of the F100ter and G57V mutants was undetectable in HEK293 cells, their expression in E. coli is fairly robust. All RD3 mutants expressed in bacteria were purified in quantities sufficient for the in vitro RetGC assay (Fig. 5A). When reconstituted with the recombinant RetGC1 activated by GCAP1, the purified F100ter RD3 did not compete with the GCAP and was unable to suppress the RetGC activity (Fig. 5B). All other mutants are able to inhibit the activity of the GCAP1/RetGC1 complex, but to varying degrees. The W6R/E23D and the G35R mutants both inhibited RetGC1 activity in a manner nearly indistinguishable from the wild type protein, whereas the R68W, K130M, and G57V mutants were significantly less effective in suppressing cyclase activation by GCAP. This is particularly true for the G57V mutant, which has an IC50 value that is more than 10-fold higher than wild type RD3 (Fig. 5B).
Fig. 4.
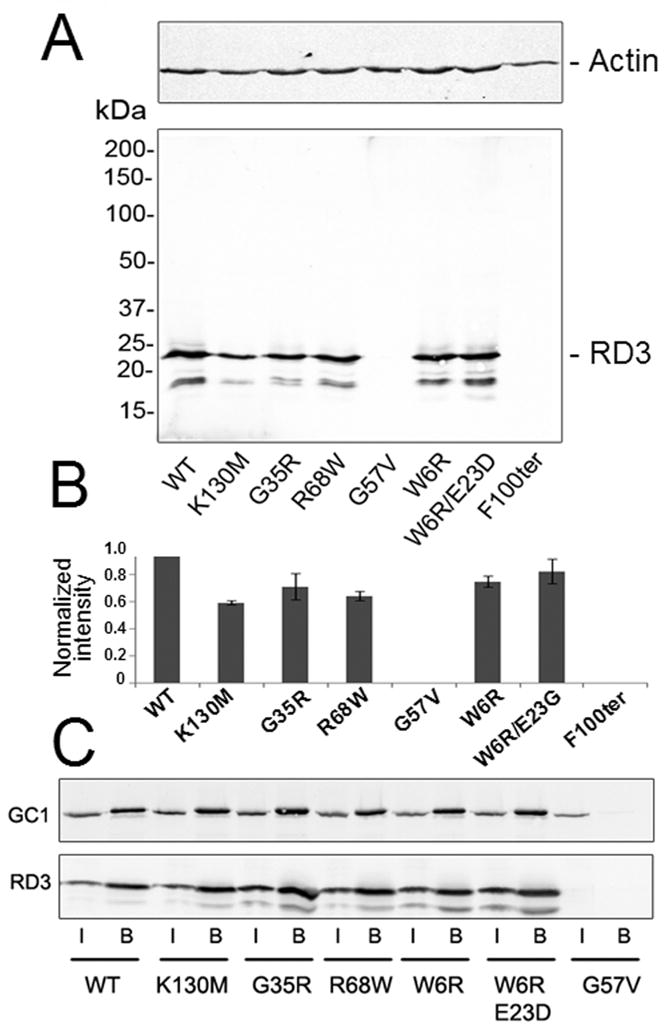
Mutations found in human patients with retinal diseases (15) affect RD3 expression in HEK293 cells. A. Expression of RD3 mutants in HEK293 cells. Human wild type and mutant RD3 tagged with the 1D4 peptide at the C-terminus were detected by immunoblotting as described in Experimental Procedures. Actin immunostaining (upper panel) was used as a protein load control. B. The average band intensities of the mutant RD3 proteins relative to WT RD3 band were determined from 3 independent experiments described in panel A (mean ± SD). C. Co-IP experiment. RetGC1 and RD3 were co-expressed as described in Experimental procedures and co-IP using Sepharose-coupled anti-RD3 9D12 antibody. The samples applied to the column (marked “I” for “input”) and the eluted fractions (marked “B” for “bound”) were analyzed by immunoblotting probed with anti-RetGC1 (GC-8A5; upper panel) and anti-RD3 (RD3-9D12; lower panel) antibodies. The G57V RD3 mutant that fails to express in HEK293 cells was used as a control to confirm that RetGC1 does not bind nonspecifically to the 9D12 antibody-coupled Sepharose. The lower band on the blot is specific for RD3-transfected cells and is most likely a partially proteolyzed RD3 polypeptide.
Fig. 5.
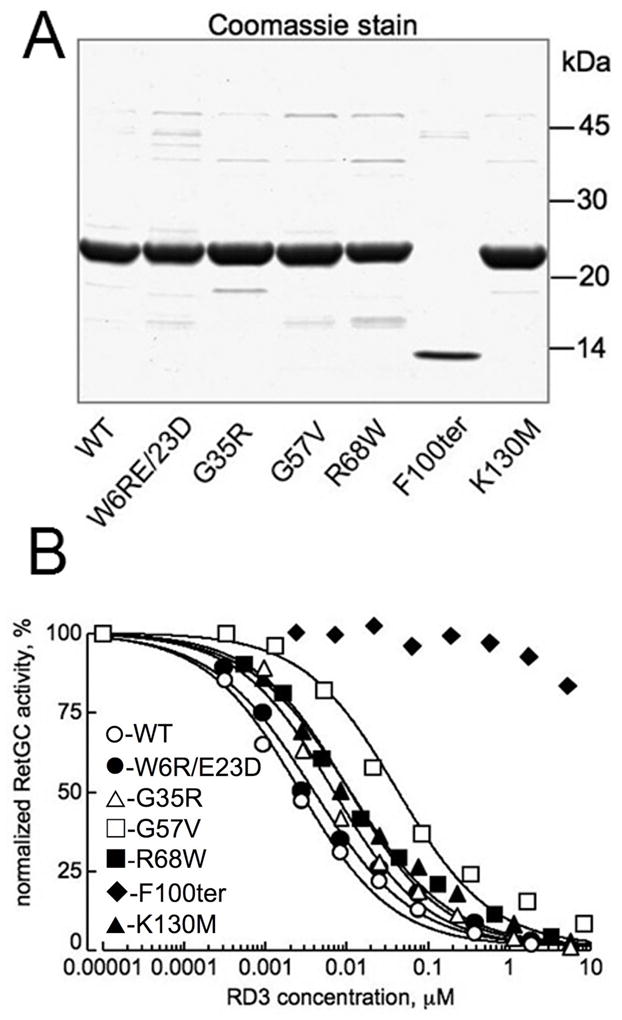
Effect of mutations in RD3 found in human patients with retinal diseases on RetGC activity in vitro. A. Wild type and mutant RD3 (15) expressed in E. coli were stained by Coomassie Brilliant Blue R-250 after SDS PAGE, 15% gel. For the use in subsequent RetGC assays, RD3 concentration in each case was that of the main band. B. Inhibition of RetGC1 activity by the E. coli - expressed RD3 mutants. RetGC activity in independent assays was normalized per activity measured in the absence of RD3. Recombinant RetGC1 expressed in HEK293 cells was reconstituted with 1.5 μM GCAP1 at indicated concentrations of RD3 (○) or its mutants: W6R/E23D (●), G35R (△), G57V (□), R68W (■), K130M (▲), and F100ter (◆). The IC50 values (mean ± SE, n) for the inhibition were 4.6 ± 1.3 nM, 5 (WT); 6.4 ± 2 nM, 3 (W6R/E23D); 7.9 ± 1 nM, 3 (G35R); 68 ± 13 nM, 3 (G57V); 17.3 ± 3.8 nM, 3 (R68W); and 14.3 ± 2.4 nM, 3 (K130M); the IC50 for the F100ter was ≫10 μM. The difference in the IC50 values from the wild type was significant (p in unpaired Student t-test assuming equal variance) for the G57V (<0.001), F100ter (<0.0001), R68W (0.012), and K130M (0.014) mutants.
DISCUSSION
It was recently found that a retina-specific protein RD3 linked to LCA12 blindness in humans and rapid retinal degeneration in rd3 mouse line (15) was expressed in rods and cones and was capable of associating with the photoreceptor-specific guanylyl cyclase RetGC (16). Our present data argue that this association has a strong functional consequence and that RD3 is a novel potent inhibitor of RetGC, capable of suppressing both the basal and GCAP-stimulated activity of the cyclase. A diagram summarizing the RD3 effects on the cyclase activity is presented in Fig. 6. In the absence of GCAPs RD3 suppresses the basal catalytic activity of the RetGC acting as a noncompetitive inhibitor (see also Fig. 1E–F). It also counteracts the GCAP-dependent activation of the cyclase acting as a negative allosteric modulator (see also Fig. 2). It should be emphasized that stimulation by GCAP increases the Vmax for RetGC in mouse ROS up to 20-fold (21) and up to 100-fold in HEK293 membranes (reference 31 and Fig. 2). Therefore, compared to the non-competitive inhibition of the basal RetGC activity in GCAPs−/− ROS (Fig. 1E–F), a direct competition with the stimulating effect of GCAPs clearly dominates the overall effect of RD3 on the cyclase regulation in a RetGC/GCAP complex (Fig. 2). Indeed, once RD3 partially or completely displaces GCAP from the complex with the cyclase, the activity of the recombinant RetGC would fall as much as 100-fold since the cyclase is no longer activated by GCAP. The additional suppression of the basal cyclase activity by RD3 would also contribute to the overall inhibition, but relatively less than the displacement of GCAP from the cyclase. As a result, the non-competitive inhibition of the catalytic activity evident in Fig. 1D–F becomes masked in experiments presented in Fig. 2 by the more robust allosteric modulation, i.e. RD3 competition with GCAPs. These results would be consistent with a model in which RD3 does not strongly inhibit the catalytic activity of the RetGC in complex with GCAP until after GCAP is displaced from the complex (Fig. 6).
Fig. 6.

A diagram depicting the inhibition of RetGC by RD3. A RetGC homodimer forms an active site by combining the Mg2+-coordinating center from one subunit with the GTP binding center from another subunit (27–29). There are two symmetrical active sites in the dimer, but only one such site is depicted here for simplicity. The low RetGC basal catalytic activity (left) becomes accelerated up to 20–100 fold (21, 31) when GCAPs bind the cytoplasmic portion of RetGC (middle). RD3 binding to the cyclase (right) inhibits the RetGC catalytic activity, but does not prevent binding of the Mg2+GTP substrate in the active site; at the same time, RD3 acts as a negative high-affinity modulator of the RetGC/GCAP complex by displacing GCAP – this prevents dynamic activation by GCAPs via Ca2+-feedback mechanism. The GCAP-stimulated activity is much higher than the basal RetGC activity (21, 31); therefore, the strongest effect of RD3 as a negative allosteric modulator of the cyclase results from its competition with GCAP. Other explanations are provided in the Discussion.
The exact binding sites for RD3 in RetGC1 have not yet been identified. Deletion of a 48-amino acid fragment from the C-terminus of RetGC hampered RD3 binding in co-immunoprecipitation experiments (16), but at this point it is unclear whether or not this is the only critically important region for RD3 binding and whether or not the binding sites for GCAP and RD3 in RetGC overlap. Multiple fragments of RetGC primary structure have been proposed by different groups to participate in binding of GCAP, including a portion of the cyclase catalytic domain (32–35). Yet, the fragments of the catalytic domain that may participate in GCAP binding (or at least be in a close proximity to the GCAP binding site, ref. 33–35) are thus far located upstream from the C-terminal fragment whose deletion affects co-immunoprecipitation with RD3. However, it is also important to note that although a competition for an overlapping epitope(s) in RetGC appears to be the simplest and the most likely possibility, RD3 and GCAP do not necessarily have to bind to exactly the same peptide epitopes – the effect of their mutual exclusion can even be potentially achieved through changing RetGC dimer conformation upon binding of the two allosteric protein regulators in different places.
At the same time, we see no evidence that RD3 is able to critically affect Ca2+-sensitivity of RetGC regulation by GCAPs either in the native ROS fraction or as a recombinant cyclase (Fig. 3). This is generally consistent with the model presented in Fig. 6, where RD3 mainly competes with GCAPs rather than alters the activity of the GCAP/RetGC complex. Two different physiological forms of a metal-bound GCAP1 have been characterized – a Mg2+-bound activator form to which GCAP converts in the light and a Ca2+-bound inhibitor form which suppresses the activity of RetGC in the dark when the free Ca2+ rises (reviewed in 12). Competition between Ca2+ and Mg2+ for EF-hands in GCAP1 and GCAP2 therefore strongly affects Ca2+ sensitivity of RetGC regulation by GCAPs resulting in a prominent right-shift of the Ca2+ sensitivity curve as Mg2+ concentrations increase (23, 31). This same shift remains in the presence of RD3 (Fig. 3). These results argue that RD3 does not significantly affect metal binding to GCAP and is unlikely to alter the dynamic regulation of the cyclase in response to light induced changes in Ca2+ concentrations in photoreceptor cells (12,13).
What then is the possible role for the inhibitory activity of RD3 on RetGC activity in a photoreceptor cell? Evidently, the biochemical mechanism of RetGC inhibition by RD3 itself does not require the environment of the photoreceptor cell (Fig. 1, 2), yet it should be specific for the photoreceptors since both RD3 and RetGC (16) are both highly expressed in rod and cone cells and largely absent in other cells of the body (15). RD3 displays a much stronger apparent affinity than GCAP for RetGC since it inhibits the cyclase in a nanomolar range, whereas GCAPs activate RetGC at micromolar concentrations (Fig. 2). Furthermore, unlike GCAP/RetGC complex, the RD3/RetGC complex is sufficiently stable in detergent to sustain the co-IP procedures (Fig. 4 and reference 16). The precise binding stoichiometry of RD3 and RetGC1 remains unknown at this point, but the concentration of RD3 could hamper the GCAP-dependent stimulation based on the following estimate. Since nearly half of RetGC monomers may be co-immunoprecipitated with RD3 from the detergent-solubilized retinal membranes as an irreversible complex (16) and the concentration of RetGC dimer in mammalian ROS lies in a micromolar range (35, 36) reaching ~3.6 μM in mouse ROS (or 7.2 μM RetGC monomer)(21), the total concentration of RD3 could approach ~ 3.6 μM (assuming for simplicity that one RD3 binds per RetGC monomer). The total concentration of GCAPs can reach to as high as ~ 17 μM in the mouse ROS cytosol, nearly 5 times higher than RD3 (calculated based on ref. 21 and 35). However, RD3 exerts its allosteric modulation of the cyclase at concentrations nearly 100 times lower than GCAPs (Fig. 2, 3, 5). Therefore, even by a conservative estimate, RD3 has the potential for suppressing a large fraction of the cyclase activity. Even though we do not know at this time the fine mechanism of RD3/RetGC complex formation and quantification of RD3 in photoreceptors needs further investigation, we would not anticipate that RD3-dependent inhibition of the cyclase activity or its high-affinity competition with GCAP contributes to the dynamic regulation of RetGC by Ca2+ and GCAPs. Furthermore, based on our data it would also very likely require additional mechanisms to reduce or prevent the RD3 inhibitory effects on RetGC at the cellular level to allow the dynamic regulation of RetGC in the outer segment.
A possible functional role of RD3 in photoreceptor physiology is to suppress the basal and GCAP activated RetGC activity in the inner segment during its transport to the outer segment. Another possibility is that RD3 itself can actively participate in the process of the transport (16) and/or assembly of the cyclase complex into the proper structure of the outer segment, during which its ability to strongly compete with GCAPs can prevent GCAP from prematurely interfering with this photoreceptor-specific process.
An apparent involvement of mutated RD3 in congenital retinal diseases corroborates its possible physiological role in photoreceptors. The 195-amino acid product of Rd3 (C1of36) gene originally identified by Friedman and co-workers (15) has been linked to a recessive retinal degeneration in the rd3 mice (37, 38) and LCA12 patients (15). Although RD3 effectively competes with GCAPs for the target enzyme, it fails to do so when truncated by the F100ter mutation linked to LCA12 (Fig. 5). A similar shortening of RD3 in rd3 mice (15) also leads to the loss of interaction between RD3 and RetGC and dramatically suppresses the levels of RetGC1 and RetGC2 expression in mouse photoreceptors in vivo (16). A variety of other mutations in the RD3 gene were reported in patients with retinal disorders (15). While the importance of those mutations for the development of congenital diseases remains to be determined, we find that some of these mutations can affect RD3 expression in human cultured cells and reduce the inhibitor activity of RetGC (Fig. 4 and 5).
One could expect that a possible consequence of mutations hampering RD3 binding to the cyclase would be to prevent RetGC from reaching the outer segment resulting in its accumulation in the inner segment. Evidently, this is not the case in rd3 mice (16). When RD3 is truncated similar to the F100ter mutant found in LCA12 patients and is unable to bind RetGC, the photoreceptors suppress production of RetGC rather than allow it to accumulate in the inner segment. It is likely that the photoreceptors possess a safety check capability that prevents the accumulation of RetGC in the absence of RD3. It is therefore tempting to speculate that failure of the mutant RD3 to properly bind the cyclase could trigger the mislocalization and rapid destruction of the cyclase as a protection against unsuitable activation of cGMP synthesis. Depletion of RetGC in ROS would inevitably suppress cGMP production resulting in a drop in intracellular Ca2+ to a level which could cause photoreceptor degeneration (16). The consequence of the altered activity of several RD3 mutants shown in this study requires further in-depth analysis of the mutants in vivo.
Acknowledgments
Funding: This work was supported by NIH grants EY11522 (AMD) and EY02422 (RSM) and Formula Grant from Pennsylvania Department of Health (AMD)
AMD is a Martin and Florence Hafter Chair Professor of Pharmacology and RSM is a Canada Research Chair in Vision and Macular Degeneration.
ABBREVIATIONS
- BSA
bovine serum albumin
- EGTA
ethylene glycol-bis(2-aminoethylether)-N,N,N′,N′-tetraacetic acid
- DTT
dithiothreitol
- GCAP
guanylyl cyclase activating protein
- co-IP
co-immunoprecipitation
- IPTG
isopropyl-β-D-thio-galactoside
- KHD
kinase homology domain
- LCA
Leber congenital amaurosis
- LDAO
lauryldimethylamine-oxide
- MOPS
3-(N-morpholino) propanesulfonic acid
- RD3
retinal degeneration 3 protein
- RetGC
retinal guanylyl cyclase
- ROS
rod outer segment
- SDS
sodium dodecyl sulfate
References
- 1.Fu Y, Yau KW. Phototransduction in mouse rods and cones. Pflugers Arch. 2007;454:805–819. doi: 10.1007/s00424-006-0194-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lowe DG, Dizhoor AM, Liu K, Gu Q, Spencer M, Laura R, Lu L, Hurley JB. Cloning and expression of a second photoreceptor-specific membrane retina guanylyl cyclase (RetGC), RetGC-2. Proc Natl Acad Sci U S A. 1995;92:5535–5539. doi: 10.1073/pnas.92.12.5535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yang RB, Foster DC, Garbers DL, Fulle HJ. Two membrane forms of guanylyl cyclase found in the eye. Proc Natl Acad Sci USA. 1995;92:602–606. doi: 10.1073/pnas.92.2.602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pugh EN, Jr, Duda T, Sitaramayya A, Sharma RK. Photoreceptor guanylate cyclases: a review. Biosci Rep. 1997;17:429–473. doi: 10.1023/a:1027365520442. [DOI] [PubMed] [Google Scholar]
- 5.Perrault I, Rozet JM, Gerber S, Ghazi I, Ducroq D, Souied E, Leowski C, Bonnemaison M, Dufier JL, Munnich A, Kaplan J. Spectrum of retGC1 mutations in Leber’s congenital amaurosis. Eur J Hum Genet. 2000;8:578–582. doi: 10.1038/sj.ejhg.5200503. [DOI] [PubMed] [Google Scholar]
- 6.Perrault I, Rozet JM, Gerber S, Kelsell RE, Souied E, Cabot A, Hunt DM, Munnich A, Kaplan J. A retGC-1 mutation in autosomal dominant cone-rod dystrophy. Am J Hum Genet. 1998;63:651–654. doi: 10.1086/301985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ramamurthy V, Tucker C, Wilkie SE, Daggett V, Hunt DM, Hurley JB. Interactions within the coiled-coil domain of RetGC-1 guanylyl cyclase are optimized for regulation rather than for high affinity. J Biol Chem. 2001;276:26218–26229. doi: 10.1074/jbc.M010495200. [DOI] [PubMed] [Google Scholar]
- 8.Gorczyca WA, Gray-Keller MP, Detwiler PB, Palczewski K. Purification and physiological evaluation of a guanylate cyclase activating protein from retinal rods. Proc Natl Acad Sci U S A. 1994;91:4014–4018. doi: 10.1073/pnas.91.9.4014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Palczewski K, Subbaraya I, Gorczyca WA, Helekar BS, Ruiz CC, Ohguro H, Huang J, Zhao X, Crabb JW, Johnson RS, Walsh KA, Gray-Keller MP, Detwiller PB, Baehr W. Molecular cloning and characterization of retinal photoreceptor guanylyl cyclase-activating protein. Neuron. 1994;13:395–404. doi: 10.1016/0896-6273(94)90355-7. [DOI] [PubMed] [Google Scholar]
- 10.Dizhoor AM, Lowe DG, Olshevskaya EV, Laura RP, Hurley JB. The human photoreceptor membrane guanylyl cyclase, RetGC, is present in outer segments and is regulated by calcium and a soluble activator. Neuron. 1994;12:1345–1352. doi: 10.1016/0896-6273(94)90449-9. [DOI] [PubMed] [Google Scholar]
- 11.Dizhoor AM, Olshevskaya EV, Henzel WJ, Wong SC, Stults JT, Ankoudinova I, Hurley JB. Cloning, sequencing, and expression of a 24-kDa Ca(2+)-binding protein activating photoreceptor guanylyl cyclase. J Biol Chem. 1995;270:25200–25206. doi: 10.1074/jbc.270.42.25200. [DOI] [PubMed] [Google Scholar]
- 12.Dizhoor AM, Olshevskaya EV, Peshenko IV. Mg2+/Ca2+ cation binding cycle of guanylyl cyclase activating proteins (GCAPs): role in regulation of photoreceptor guanylyl cyclase. Mol Cell Biochem. 2010;334:117–124. doi: 10.1007/s11010-009-0328-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Burns ME, Mendez A, Chen J, Baylor DA. Dynamics of cyclic GMP synthesis in retinal rods. Neuron. 2002;36:81–91. doi: 10.1016/s0896-6273(02)00911-x. [DOI] [PubMed] [Google Scholar]
- 14.Makino CL, Peshenko IV, Wen XH, Olshevskaya EV, Barrett R, Dizhoor AM. A role for GCAP2 in regulating the photoresponse. Guanylyl cyclase activation and rod electrophysiology in GUCA1B knock-out mice. J Biol Chem. 2008;283:29135–43. doi: 10.1074/jbc.M804445200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Friedman JS, Chang B, Kannabiran C, Chakarova C, Singh HP, Jalali S, Hawes NL, Branham K, Othman M, Filippova E, Thompson DA, Webster AR, Andreasson S, Jacobson SG, Bhattacharya SS, Heckenlively JR, Swaroop A. Premature truncation of a novel protein, RD3, exhibiting subnuclear localization is associated with retinal degeneration. Am J Hum Genet. 2006;79:1059–70. doi: 10.1086/510021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Azadi S, Molday LL, Molday RS. RD3, the protein associated with Leber congenital amaurosis type 12, is required for guanylate cyclase trafficking in photoreceptor cells. Proc Natl Acad Sci U S A. 2011;107:21158–21163. doi: 10.1073/pnas.1010460107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hodges RS, Heaton RJ, Parker JM, Molday L, Molday RS. Antigen-antibody interaction. Synthetic peptides define linear antigenic determinants recognized by monoclonal antibodies directed to the cytoplasmic carboxyl terminus of rhodopsin. J Biol Chem. 1988;263:11768–11775. [PubMed] [Google Scholar]
- 18.Laura RP, Dizhoor AM, Hurley JB. The membrane guanylyl cyclase, retinal guanylyl cyclase-1, is activated through its intracellular domain. J Biol Chem. 1996;271:11646–11651. doi: 10.1074/jbc.271.20.11646. [DOI] [PubMed] [Google Scholar]
- 19.Olshevskaya EV, Hughes RE, Hurley JB, Dizhoor AM. Calcium binding, but not a calcium-myristoyl switch, controls the ability of guanylyl cyclase-activating protein GCAP-2 to regulate photoreceptor guanylyl cyclase. J Biol Chem. 1997;272:14327–14333. doi: 10.1074/jbc.272.22.14327. [DOI] [PubMed] [Google Scholar]
- 20.Peshenko IV, Dizhoor AM. Ca2+ and Mg2+ binding properties of GCAP-1. Evidence that Mg2+-bound form is the physiological activator of photoreceptor guanylyl cyclase. J Biol Chem 281, 23830–23841. J Biol Chem. 2006;281:23830–23841. doi: 10.1074/jbc.M600257200. [DOI] [PubMed] [Google Scholar]
- 21.Peshenko IV, Olshevskaya EV, Savchenko AB, Karan S, Palczewski K, Baehr W, Dizhoor AM. Enzymatic properties and regulation of the native isozymes of retinal membrane guanylyl cyclase (RetGC) from mouse photoreceptors. Biochemistry. 2011;50:5590–600. doi: 10.1021/bi200491b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Peshenko IV, Olshevskaya EV, Dizhoor AM. Binding of guanylyl cyclase activating protein 1 (GCAP1) to retinal guanylyl cyclase (RetGC1). The role of individual EF-hands. J Biol Chem. 2008;283:21747–21757. doi: 10.1074/jbc.M801899200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Peshenko IV, Dizhoor AM. Guanylyl cyclase-activating proteins (GCAPs) are Ca2+/Mg2+ sensors: implications for photoreceptor guanylyl cyclase (RetGC) regulation in mammalian photoreceptors. J Biol Chem. 2004;279:16903–16906. doi: 10.1074/jbc.C400065200. [DOI] [PubMed] [Google Scholar]
- 24.Tsien R, Pozzan T. Measurement of cytosolic free Ca2+ with quin2. Methods Enzymol. 1989;172:230–62. doi: 10.1016/s0076-6879(89)72017-6. [DOI] [PubMed] [Google Scholar]
- 25.Peshenko IV, Dizhoor AM. Activation and inhibition of photoreceptor guanylyl cyclase by guanylyl cyclase activating protein 1 (GCAP-1): the functional role of Mg2+/Ca2+ exchange in EF-hand domains. J Biol Chem. 2007;282:21645–21652. doi: 10.1074/jbc.M702368200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mendez A, Burns ME, Sokal I, Dizhoor AM, Baehr W, Palczewski K, Baylor DA, Chen J. Role of guanylate cyclase-activating proteins (GCAPs) in setting the flash sensitivity of rod photoreceptors. Proc Natl Acad Sci U S A. 2001;98:9948–9953. doi: 10.1073/pnas.171308998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu Y, Ruoho AE, Rao VD, Hurley JH. Catalytic mechanism of the adenylyl and guanylyl cyclases: modeling and mutational analysis. Proc Natl Acad Sci U S A. 1997;94:13414–13419. doi: 10.1073/pnas.94.25.13414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tucker CL, Hurley JH, Miller TR, Hurley JB. Two amino acid substitutions convert a guanylyl cyclase, RetGC-1, into an adenylyl cyclase. Proc Natl Acad Sci U S A. 1998;95:5993–5997. doi: 10.1073/pnas.95.11.5993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ramamurthy V, Tucker C, Wilkie SE, Daggett V, Hunt DM, Hurley JB. Interactions within the coiled-coil domain of RetGC-1 guanylyl cyclase are optimized for regulation rather than for high affinity. J Biol Chem. 2001;276:26218–26229. doi: 10.1074/jbc.M010495200. [DOI] [PubMed] [Google Scholar]
- 30.Lim S, Peshenko I, Dizhoor A, Ames JB. Effects of Ca2+, Mg2+, and myristoylation on guanylyl cyclase activating protein 1 structure and stability. Biochemistry 48, 850–862. Biochemistry. 2009;48:850–862. doi: 10.1021/bi801897p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Peshenko IV, Moiseyev GP, Olshevskaya EV, Dizhoor AM. Factors that determine Ca2+ sensitivity of photoreceptor guanylyl cyclase. Kinetic analysis of the interaction between the Ca2+-bound and the Ca2+-free guanylyl cyclase activating proteins (GCAPs) and recombinant photoreceptor guanylyl cyclase 1 (RetGC-1) Biochemistry. 2004;43:13796–13804. doi: 10.1021/bi048943m. [DOI] [PubMed] [Google Scholar]
- 32.Lange C, Duda T, Beyermann M, Sharma RK, Koch KW. Regions in vertebrate photoreceptor guanylyl cyclase ROS-GC1 involved in Ca(2+)-dependent regulation by guanylyl cyclase-activating protein GCAP-1. FEBS Lett. 1999;460:27–31. doi: 10.1016/s0014-5793(99)01312-5. [DOI] [PubMed] [Google Scholar]
- 33.Sokal I, Haeseleer F, Arendt A, Adman ET, Hargrave PA, Palczewski K. Identification of a guanylyl cyclase-activating protein-binding site within the catalytic domain of retinal guanylyl cyclase 1. Biochemistry. 1999;38:1387–93. doi: 10.1021/bi982512k. [DOI] [PubMed] [Google Scholar]; Krylov DM, Hurley JB. J Biol Chem. 2001;276:30648–30654. doi: 10.1074/jbc.M104121200. [DOI] [PubMed] [Google Scholar]
- 34.Duda T, Fik-Rymarkiewicz E, Venkataraman V, Krishnan R, Koch KW, Sharma RK. The calcium-sensor guanylate cyclase activating protein type 2 specific site in rod outer segment membrane guanylate cyclase type 1. Biochemistry. 2005;44:7336–7345. doi: 10.1021/bi050068x. [DOI] [PubMed] [Google Scholar]
- 35.Hwang JY, Lange C, Helten A, Hoppner-Heitmann D, Duda T, Sharma RK, Koch KW. Regulatory modes of rod outer segment membrane guanylate cyclase differ in catalytic efficiency and Ca2+-sensitivity. Eur J Biochem. 2003;270:3814–3821. doi: 10.1046/j.1432-1033.2003.03770.x. [DOI] [PubMed] [Google Scholar]
- 36.Helten A, Saftel W, Koch KW. Expression level and activity profile of membrane bound guanylate cyclase type 2 in rod outer segments. J Neurochem. 2007;103:1439–46. doi: 10.1111/j.1471-4159.2007.04923.x. [DOI] [PubMed] [Google Scholar]
- 37.Chang B, Heckenlively JR, Hawes NL, Roderick TH. New mouse primary retinal degeneration (rd-3) Genomics. 1993;16:45–49. doi: 10.1006/geno.1993.1138. [DOI] [PubMed] [Google Scholar]
- 38.Danciger M, Ogando D, Yang H, Matthes MT, Yu N, Ahern K, Yasumura D, Williams RW, Lavail MM. Genetic modifiers of retinal degeneration in the rd3 mouse. Invest Ophthalmol Vis Sci. 2008;49:2863–2869. doi: 10.1167/iovs.08-1715. [DOI] [PMC free article] [PubMed] [Google Scholar]