

Abstract
Multi-component reactions have been extensively employed in many areas of organic chemistry. Despite significant progress, the discovery of such enabling transformations remains challenging. Here, we present the development of a parallel, label-free reaction-discovery platform, which can be used for identification of new multi-component transformations. Our approach is based on the parallel mass spectrometric screening of interfacial chemical reactions on arrays of self-assembled monolayers. This strategy enabled the identification of a simple organic phosphine that can catalyze a previously unknown condensation of siloxy alkynes, aldehydes and amines to produce 3-hydroxy amides with high efficiency and diastereoselectivity. The reaction was further optimized using solution phase methods.
Integration of innovative reaction screening formats with analytical techniques for rapid product identification can enable the discovery of an arsenal of new catalytic, synthetically useful processes. While significant progress in developing parallel reaction screening platforms has been made (1–19), rapid discovery of transformations that produce products with unanticipated structures remains highly challenging. To enable the identification of such reactions, analysis of the outcome of each individual experiment must be rapid, precise and structurally unbiased. Existing approaches that tackle this problem require either conjugation of reactants to information-coding DNA strands (20,21,22) or the use of LCMS or GCMS methods (23,24,25). Herein, we describe the development of an efficient reaction-discovery strategy, which entails the screening of interfacial chemical transformations on self-assembled monolayers (SAMs) of alkanethiolates on gold using matrix-assisted laser desorption/ionization and time-of flight mass spectrometry (MALDI-TOF-MS). This general approach, which we earlier termed SAMDI (26), enabled initial identification of a new three-component reaction of amines, aldehydes and siloxy alkynes, which was subsequently optimized using solution phase methods. This effort validated our structurally unbiased discovery strategy and uncovered a unique mode of catalytic activation of electron rich alkynes using simple organic phosphines.
Our general screening approach starts with a glass slide that is patterned with an array of gold islands, each of which is modified with a monolayer that presents the substrate of interest. Unique combinations of reagents are applied to each of the islands to create an array of reactions, which are allowed to proceed for a specified amount of time. The reactions are stopped by rinsing the array and are then rapidly analyzed by mass spectrometry to identify those combinations of reactants and reagents that give products in high yield. These ‘hits’ are easily identified and can then be investigated in more detail. This technique is highly sensitive—the amount of monolayer-linked substrate is on the order of picomoles per square millimeter—and because it uses mass spectrometry, it does not require work-up procedures that separate the products from the reaction mixture. We previously employed this method for a range of biochemical applications (27–30). Furthermore, we also found that the monolayers are compatible with many organic and organometallic reactions (31,32) and showed that the SAMDI method can rapidly detect all interfacial reactions that result in a mass change, providing a unique opportunity for label-free discovery of new catalytic processes.
Results and Discussion
SAMDI Reaction Screen
Multi-component condensations enable a rapid increase in structural complexity and molecular diversity by creating new products from more than two reactants in a single operation. Such reactions have been extensively employed for generating chemical libraries (33), assembling bioactive natural products (34) and inventing organocatalytic transformations (35). Despite significant progress, the discovery of new multi-component transformations remains challenging. As part of our ongoing investigation of the basic reactivity of siloxy alkynes, we decided to take advantage of the unique chemical profile of this class of organosilanes for developing new multi-component reactions. Siloxy alkynes were previously found to participate in a series of catalytic carbon-carbon and carbon-heteroatom bond-forming reactions (36–41). Depending on the mode of activation, such electron-rich alkynes underwent reactions with either nucleophiles (36–39) or electrophiles (40,41). Thus, we anticipated that a single catalyst could promote a three-component condensation of a siloxy alkyne with two other reaction partners with opposite electronic properties, for example a carbonyl-based electrophile and an amine-containing nucleophile. While various reaction products could be expected from such a three-component combination, the SAMDI method could efficiently identify formation of new products and enable substantial variation of possible reactants, catalysts, additives and solvents en route to such discovery.
We created screening arrays by first treating glass substrates with a dilute solution of (tridecafluoro-1,1,2,2-tetrahydrooctyl)-1-trichlorosilane followed by masking the substrate with a stencil to deposit titanium (10 nm), followed by gold (100 nm) in a pattern of islands. The fluorinated surface surrounding the islands serves to confine wetting by a wide range of organic solvents and therefore keeps the droplets on each monolayer in place and separate from each other. This platform is compatible with most non-fluorinated solvents. Monolayers were next prepared by treating the gold islands with an ethanolic solution of an amine-terminated alkanethiol containing multiple ethylene glycol spacers and a similar methyl ether-terminated alkanethiol with a tris(ethylene glycol) spacer (Fig. 1a). The latter serves as an inert background to control the density of reactive amine-containing functional groups on the monolayer surface. Figure 1b shows a mass spectrum of the initially formed monolayer and reveals a peak at m/z 838 that corresponds to the sodium adduct of a mixed disulfide derived from the amine-containing and methylether-terminated alkanethiolate.
Figure 1. Discovery of a new three-component reaction by SAMDI.
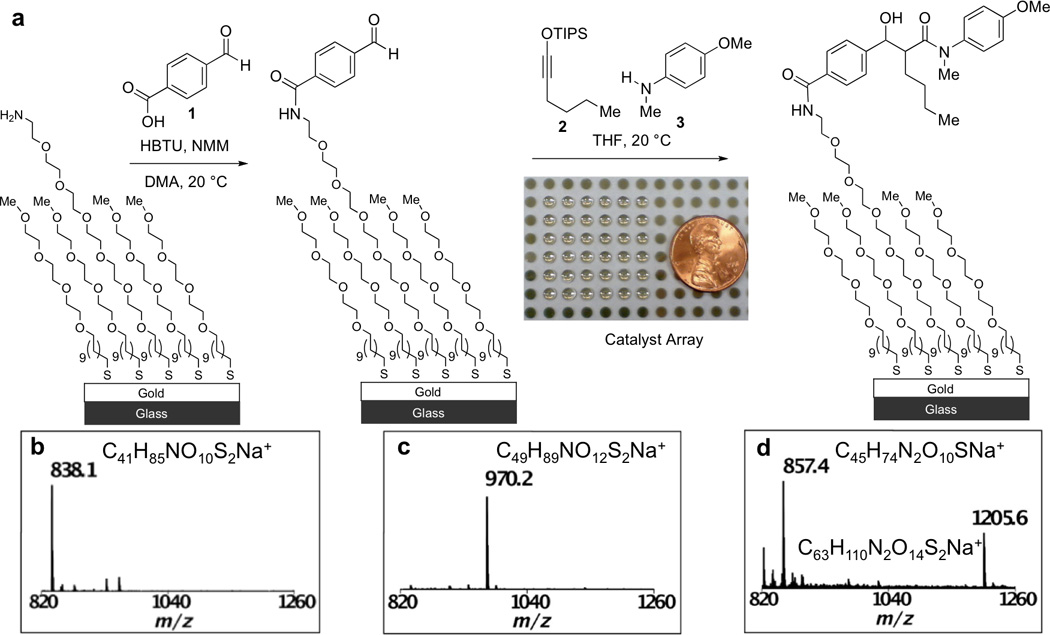
a, The reaction screen was set up by first immobilizing aldehyde 1 to the monolayer, followed by exposure of the resulting monolayer to 2 and 3 in the presence of various possible catalysts. c, SAMDI spectrum of the amine-terminated monolayer. d, SAMDI spectrum of an aldehyde-functionalized monolayer synthesized via amide coupling. e, SAMDI spectrum of a new three-component product formed in one of the interfacial reactions.
We introduced the aldehyde functionality by treating the amino-terminated monolayer with 4-formyl-benzoic acid (1), O-benzotriazole-N,N,N’,N’-tetramethyl-uronium-hexafluoro-phosphate (HBTU), and N-methylmorpholine. The monolayer was rinsed, dried, and analyzed by SAMDI to reveal a new peak at m/z 970 (Fig. 1c) corresponding to the sodium adduct of a mixed disulfide composed of a methyl ether-terminated alkanethiolate and another alkanethiolate containing the newly installed aldehyde. We note that this method of immobilizing substrates could be applied to introduce other functional groups of interest (e.g. alkynes, alkenes) onto monolayers and avoids the need to synthesize functionalized alkanethiols prior to assembly of the monolayer.
We performed reactions by applying a solution of 1-siloxy-1-hexyne (2) and 4-methoxy-N-methyl aniline (3) in tetrahydrofuran (THF, Figure 1B) to each aldehyde-terminated monolayer spot followed by the addition of a potential catalyst in THF with a 10:5:1 molar ratio of 2:3:catalyst. We evaluated several common Lewis acids, Brønsted acids and transition-metal complexes (Supplementary Fig S1). The screening array was assembled under a nitrogen atmosphere to prevent degradation of air- and moisture-sensitive reagents, and was next transferred to a glass chamber saturated with THF to prevent solvent evaporation after all reagents had been added. The reactions were allowed to proceed for 30 minutes at room temperature, after which the monolayer array was rinsed with acetone, THF, and ethanol. After drying, the array was treated with 2,5-dihydroxybenzoic acid and each spot was analyzed by MALDI-TOF mass spectrometry. The majority of conditions screened revealed that no reaction occurred with the immobilized aldehyde (Supplementary Fig. S1). For example, a mass spectrum of the reaction containing only siloxy alkyne 2 and amine 3 in THF showed a single peak at m/z 970 corresponding to the starting aldehyde. Although most of the conditions screened yielded no detectable or unidentified products, the mass spectrum from a reaction spot containing Pd(PPh3)4 revealed the formation of new peaks at m/z 857 and 1205 (Fig. 1d), which were consistent with the sodium adduct of an alkanethiolate terminated in a product derived from 1, 2 and 3, with loss of the triisopropylsilyl (TIPS) group as well as the sodium adduct of a mixed disulfide produced from this three-component product and the background methylether-terminated alkanethiolate. Since SAMDI simultaneously provides data on the presence of both the reactants and potential products, this analytical technique can be used to qualitatively access the conversion achieved in an interfacial reaction. In the mass spectrum from this reaction spot (Fig. 1d), no peaks derived from the unreacted aldehyde could be detected, indicating that this reaction proceeded efficiently on the monolayer.
Solution Phase Optimization
We began a detailed investigation of this novel transformation by performing the reaction in solution under the conditions initially found in the primary SAMDI reaction screen. An initial evaluation of several aldehydes revealed that 4-(trifluoromethyl)benzaldehyde (4) displayed the highest reactivity. We found that subjecting 4 to siloxy alkyne 2 and aniline 3 in the presence of Pd(PPh3)4 (20 mol%) and 5 equivalents of MeOH in THF at 20 °C produced an 85:15 mixture of two diastereomeric products 5 and 6 in 56% conversion as determined by monitoring this process by 1H NMR spectroscopy (Table 1, Entry 1). We determined the structure of the major anti product 5 using NMR, MS, and subsequent X-ray crystallographic analysis. We also found that the presence of methanol in the reaction mixture was critical to the reaction. While a proton source was not specifically introduced during the initial reaction screening on SAMs, we believe that adventitious moisture could have served this role in the interfacial reaction.
Table 1. Effect of catalysts, additives and solvents on the solution-phase reaction.
The experiments were performed by treating a 0.5 mL solution of 3 (0.25 mmol), 2 (0.37 mmol), and 4 (0.50 mmol) with the specified catalysts and additives listed above. After 24 h, an aliquot of the reaction was concentrated in vacuo, dissolved in 0.50 mL of CDCl3, and analyzed by 1H NMR. For each run, conversion and diastereoselectivity was calculated based on consumption of 4-methoxy-N-methylaniline (3) and formation of products 5 and 6. Diastereomeric ratios were determined by 500 MHz 1H NMR analysis of crude reaction mixtures prior to chromatographic purification.
 | |||||
|---|---|---|---|---|---|
| Entry | Catalyst (mol%) | Additive (Equiv.) | Solvent, Temp (°C) | Conversion | d.r. |
| 1 | Pd(PPh3)4(5) | MeOH(5) | THF(20) | 56% | 85:15 |
| 2 | Pd(OAc)2(5) | MeOH(5) | toluene(20) | <2 | |
| 3 | Pd(PCy3)2(5) | MeOH(5) | toluene(20) | 35% | 85:15 |
| 4 | Pd(PPh3)4(5) | MeOH(5) | toluene(60) | 65% | 85:15 |
| 5 | Ph3P(20) | MeOH(5) | toluene(60) | 50% | 88:12 |
| 6 | (4-MeC6H4)3P(10) | MeOH(2.5) | toluene(60) | 60% | 91:9 |
| 7 | (4-MeOC6H4)3P(10) | MeOH(2.5) | toluene(60) | 75% | 91:9 |
| 8 | (4-MeOC6H4)3P(20) | BnOH(2) | toluene(60) | 89% | 91:9 |
| 9 | (4-MeOC6H4)3P(20) | 4-FC6H4CH2OH(2) | toluene(60) | 96% | 91:9 |
Since Pd complexes had not been previously found to promote any reactions of siloxy alkynes, and the role of this metal in the three-component reaction was not evident, we examined other Pd-containing compounds. Interestingly, while Pd(OAc)2 did not give any previously observed products (Table 1, Entry 2), a Pd complex containing another phosphine ligand produced the same outcome albeit with slightly lower efficiency (Table 1, Entry 3). Raising the temperature of the reaction to 60 °C resulted in a further increase of reaction efficiency (Table 1, Entry 4). Since only phosphine-containing complexes proved effective thus far, we decided to perform the same reaction in the presence of only PPh3 and MeOH, with complete exclusion of Pd. Under such conditions, the same three-component product was obtained with similar efficiency and diastereoselectivity (Table 1, Entry 5), strongly suggesting that the presence of a transition metal was not required for this reaction. Since PPh3 was not included in the primary SAMDI reaction screen, the effect of this compound, which is typically used as a transition metal ligand, as the sole reaction promoter could not be evaluated at an earlier stage.
In order to increase the efficiency of the solution-phase three-component condensation of 2, 3 and 4, we next evaluated a variety of phosphines, other nucleophilic reagents and a range of proton sources. Selected results of our extensive studies are summarized in Table 1 (Entries 6–9). We found that electron-rich triaryl phosphines were the most reactive in promoting the desired transformation with tris(4-methoxyphenyl)phosphine being the best catalyst (Table 1, Entry 7). Throughout the optimization studies, the diastereoselectivity of this process was further improved to 91:9, while the reaction efficiency was also increased by replacing methanol with benzyl and 4-fluorobenzyl alcohols (Table 1, Entries 8–9). During these studies, we established that one equivalent of the alcohol is required for quantitative transfer of the TIPS group. Complete exclusion of a phosphine or its substitution by phosphine oxides resulted in no observed three-component reaction, indicating that a combination of a phosphine with an appropriate alcohol additive was uniquely responsible for successful product formation and catalytic turn-over. This finding is highly noteworthy as simple organic phosphines were not known to catalyze any reactions of siloxy alkynes prior to this study.
Reaction Scope Study
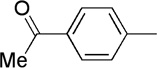
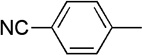
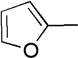
We next examined a series of aromatic aldehydes that could participate in a phosphine-catalyzed three-component condensation with alkyne 2 and amine 3. The major product 5 of the parent reaction with 4-(trifluoromethyl)benzaldehyde (4) was isolated in 87% yield (Table 2, Entry 1), which was fully consistent with the high degree of conversion observed in early NMR studies. Under the same reaction conditions, other electron deficient benzaldehydes afforded the expected products with good yields and high diastereoselectivity (Table 2, Entries 2–7). Both ortho and meta aromatic substitution of the aldehyde was well tolerated. It is also noteworthy that the reaction of a ketone-containing aldehyde (Table 2, Entry 2) proceeded with complete chemoselectivity with only the aldehyde being reactive under the conditions of the three-component reaction. Heteroaromatic and halogen-containing aldehydes also reacted efficiently (Table 2, Entries 8–9) but unsaturated and aliphatic aldehydes gave lower yields presumably due to other reactions that are promoted by secondary amines, including enolization of aliphatic aldehydes and conjugate additions to unsaturated aldehydes.
Table 2. Scope of the three-component condensation of aldehydes, siloxy alkynes and amines.
All reactions were carried out according to the general procedure described in the Methods section. The yields refer to those isolated for the major diastereomeric product of each three-component reaction following chromatographic purification. Diastereomeric ratios were determined by 500 MHz 1H NMR analysis of crude reaction mixtures prior to chromatographic purification.
 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | R1 | R2 | R3 | R4 | Major Product | Yield, % | d.r. |
| 1 | Me |  |
 |
5 | 87 | 91:9 | |
| 2 | Me |  |
 |
7 | 87 | 88:12 | |
| 3 | Me | 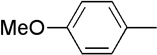 |
 |
8 | 73 | 88:12 | |
| 4 | Me | 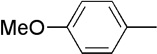 |
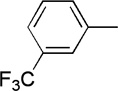 |
9 | 63 | 91:9 | |
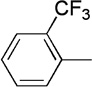
| 5 | Me | 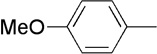 |
 |
10 | 74 | 93:7 | |
| 6 | Me | 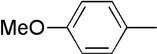 |
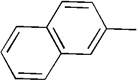 |
11 | 68 | 91:9 | |
| 7 | Me | 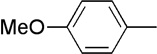 |
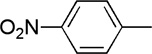 |
12 | 66 | 84:16 | |
| 8 | Me | 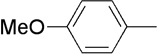 |
 |
13 | 59 | 88:12 | |
| 9 | Me | 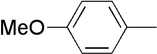 |
 |
14 | 63 | 90:10 | |
| 10 | Me | 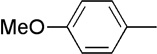 |
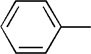 |
15 | 58 | 91:9 | |
| 11 | Me | 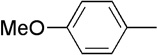 |
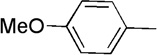 |
16 | 44 | 84:16 | |
| 12 | Me | 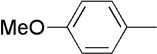 |
Me | 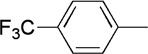 |
17 | 75 | 85:15 |
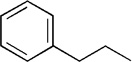
| 13 | Me | 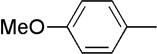 |
 |
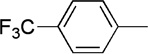 |
18 | 72 | 91:9 |
| 14 | Me | 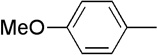 |
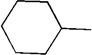 |
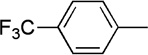 |
19 | 80 | 91:9 |
| 15 | Me | 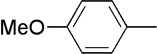 |
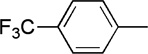 |
20 | 87 | 91:9 | |
| 16 | Me | 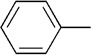 |
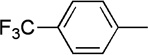 |
21 | 76 | 91:9 | |
| 17 | Me | 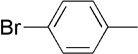 |
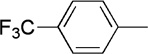 |
22 | 74 | 95:5 | |
| 18 | Me | 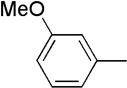 |
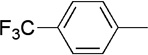 |
23 | 61 | 91:9 | |
| 19 | 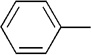 |
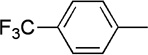 |
24 | 78 | 88:12 | ||
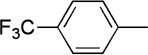
| 20 | 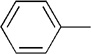 |
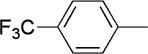 |
25 | 57 | 89:11 | ||
| 21 | 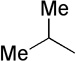 |
 |
 |
26 | 52 | 80:20 | |
| 22 | 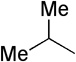 |
 |
 |
27 | 30 | 63:37 | |
We also examined the scope of the reaction with regard to siloxy alkyne substitution. The use of 1-siloxy-1-propyne resulted in formation of the desired product in high yield with slightly diminished diastereoselectivity (Table 2, Entry 12). The presence of aromatic groups in the siloxy alkyne structure was well tolerated (Table 2, Entry 13). Furthermore, introduction of bulky substituents, such as cyclohexyl or cyclopropyl groups, in direct proximity to the alkyne moiety did not seem to impact the efficiency or the diaseteroselectivity of the reaction (Table 2, Entries 14–15). Such results suggest that a wide range of siloxy alkyne substitution would be well tolerated in the three-component reaction.
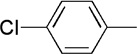
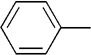
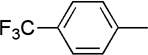
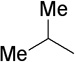
The final aspect of the scope study entailed evaluation of a series of amines. Secondary anilines proved to be the most suitable substrates for this reaction. This observation is not surprising since most of the initial optimization work was performed employing aromatic amines, which displayed the most promising reactivity profile. For instance, N-methyl aniline, N-methyl-4-bromoaniline and N-methyl-3-methoxyaniline produced the corresponding three-component reaction products 21–23 in 61–74% isolated yields (Table 2, Entries 16–18). Other N-substituted anilines readily participated in this reaction (Table 2, Entries 19–21). Not surprisingly, increasing the steric bulk of the N-alkyl substituent resulted in lower yields of the desired product, as well as diminished diastereoselectivity. While primary anilines did not afford any three-component products under the same reaction conditions, N,N-dialkyl amines displayed a variable pattern of reactivity and diastereoselectivity. For instance, the three-component reaction using diisopropylamine afforded the desired major anti-diastereomeric product in a decreased isolated yield as a result of a diminished level of diastereoselection (Table 2, entry 22).
Having established the reaction scope, we next demonstrated the utility of this three-component transformation for rapid synthesis of a representative small-molecule library of hydroxy amides, which was generated from 3 siloxy alkynes, 3 aldehydes and 4 amines (Supplementary Fig. S2). The library was produced and purified by mass-triggered preparative LCMS to successfully deliver 36 requisite compounds on 20–66 mg scale in high chemical purity (see Supplementary Information).
Formation of the observed three-component products can be tentatively rationalized based on the mechanism depicted in Figure 2. Reaction of a siloxy alkyne with phosphine and alcohol is expected to produce a ketene. This step is supported by our deuterium incorporation studies, which demonstrated exclusive incorporation of deuterium at the α-position of the carbonyl in the final three-component product using a deuterated alcohol (R2-OD). The silyl group is exclusively transferred during the course of the reaction to the corresponding siloxy ether (R2-OSiR3) as shown in Figure 2. Subsequent addition of the secondary amine to a ketene, followed by an aldol reaction with an aldehyde would generate the observed hydroxy amide product. The closed transition state shown in Figure 2 could explain the predominant formation of the major anti-diastereomer. The proposed mechanism is further supported by a range of other experiments, including the preliminary kinetic analysis of this process.
Figure 2. A proposed mechanism of the phosphine-catalyzed three-component reaction of siloxy alkynes with amines and aldehydes.

The mechanism involves initial desilyation of the siloxy alkyne, which is promoted by phosphine and alcohol, followed by the reaction of the resulting ketene with amine and aldehyde to generate the observed three-component product. The stereochemistry of the final product can be rationalized based on the shown chair-like transition state.
Other phosphine-catalyzed reactions of siloxy alkynes
We also identified several other phosphine-catalyzed transformation of siloxy alkynes, which are fully consistent with possible generation of ketenes under the reaction conditions. For example, treatment of siloxy alkyne 28 with amine 30 in the presence of tris(4-methoxyphenyl)phosphine (20 mol%) and 2 equivalents of 4-fluorobenzylalcohol at 20 °C afforded the corresponding amide 31 in 95% yield (Fig. 3A). Primary aniline 32 was also employed under the same conditions to afford amide 33 (Fig. 3B). Furthermore, treatment of siloxy alkyne 28 with hindered phenol 34 yielded the corresponding ester 35 in excellent yield (Fig. 3C). The use of 2 equivalents of the alcohol was required in this experiment, as one equivalent was converted to the corresponding TIPS ether. Finally, we examined if phosphine catalysis would enable C-acylation of malonates with siloxy alkynes. Treatment of malonate 36 with alkyne 29 in the in the presence of tris(4-methoxyphenyl)phospine (20 mol%), followed by subsequent desilylation with HF afforded the C-acylated product 37 (Fig. 3D). The above phosphine-catalyzed reactions of siloxy alkynes with several classes of commonly used nucleophiles offer unique and exceedingly mild reaction conditions for such transformations and further expand the newly identified concept of nucleophilic activation of this class of organosilicon compounds.
Figure 3. Other representative phosphine-catalyzed reactions of siloxy alkynes.

Such reactions include amidations of siloxy alkynes with aliphatic and aromatic amines (a and b), esterification of a siloxy alkyne with a highly hindered phenols (c) and condensation of a siloxy alkyne with diethyl malonate (d).
Conclusions
The work described herein establishes that SAMDI-based screening can be used to identify novel multi-component reactions. This method does not require labels to identify specific products, is well suited for the discovery of transformations that give products with structures not easily predicted based on the prior knowledge of substrate reactivity, and is adaptable to multi-parameter reaction screening. This effective reaction screening strategy enabled the discovery of the first three-component reaction of siloxy alkynes with aldehydes and amines and uncovered a conceptually novel mode of catalytic activation of electron rich alkynes using simple organic phosphines.
Methods
Preparation of monolayers and SAMDI reaction screen
Glass slides (50 mm×35 mm) were placed in a dilute solution of (tridecafluoro-1,1,2,2-tetrahydrooctyl)-1-trichlorosilane (1 vol% in anhydrous toluene) for 10 min to create a fluorinated glass surface. The resulting slides were rinsed with acetone, sonicated in ethanol twice for 20 min and heated to 80 °C for 2 h. The glass platform was masked with a stencil (made from aluminum) containing evenly spaced holes having a diameter of 2.5 mm and center-to-center distance of 4 mm. Titanium (10 nm), followed by gold (100 nm) were evaporated onto the masked glass substrate using an electron beam evaporator (Thermionics VE-100) at a rate of 0.2 Å/s for Ti and 0.4 Å/s for Au, and at a pressure of less than 5×10−7 Torr) to generate the array of gold-coated islands. The modified glass plate was then immersed in 1.5 mL of an ethanolic solution containing the background methyl ether-terminated alkanethiol and amino-functionalized alkanethiol (for structures, see Figure 1B) in a 3:2 ratio (total concentration of 0.2 mmol/L) for 12 h. The monolayer was then treated with 2,5-dihydroxybenzoic acid (1 µL of an 10 mg/mL solution in acetonitrile), allowed to air dry, and was analyzed on a 4800 MALDI-TOF/TOF Biospectrometry mass spectrometer (Applied Biosystems, Framingham, MA) equipped with a 355 nm Nd:YAG laser, using positive ion reflector mode. Aldehyde 1 was immobilized by immersing the amine-functionalized monolayer in a solution containing 4-formylbenzoic acid (100 mmol/L), HBTU (100 mmol/L) and N-methylmorpholine (200 mmol/L) in N,N,-dimethylacetamide. The reaction was allowed to proceed for 30 min at 20 °C. This process was repeated, and the slides were rinsed with acetone, dried under a stream of nitrogen and analyzed by MALDI-TOF-MS as described above. The array was next placed in a glove bag filled with nitrogen, and each reaction spot was treated with 3 µL of a solution of alkyne 2 (0.67 mol/L) and amine 3 (0.33 mol/L). Following addition of all reagents, 1 µL of a solution of a potential catalyst (0.2 mol/L) in THF was applied to each spot. The array was transferred to a glass chamber saturated with THF and reactions were allowed to proceed for 30 min at 20 °C. Monolayers were then rinsed with acetone, THF and ethanol and dried under a stream of nitrogen. Each reaction spot was treated with 2,5-dihydroxybenzoic acid (1 µL per spot of 10 mg/mL solution in acetonitrile) and analyzed by mass spectrometry as described above.
General procedure for three-component condensation of siloxy alkynes, aldehydes and amines
All solution-phase reactions were performed under an atmosphere of argon in flame-dried (10×75 mm) test tubes equipped with stir bars and sealed with rubber septa. A solution of tris(4-methoxyphenyl)phosphine (35.2 mg, 0.100 mmol, 20 mol%), amine (0.500 mmol), and siloxy alkyne (0.750 mmol) in 1.00 mL of toluene was treated with aldehyde (1.00 mmol) and 4-fluorobenzyl alcohol (0.109 mL, 2.00 mmol). The resulting solution was heated to 60 °C for 48 h. The reaction mixture was cooled to room temperature, diluted with 1.00 mL of CH2Cl2, concentrated by rotary evaporation, and subjected to purification by flash chromatography to afford the resulting three-component products shown in Table 2.
Supplementary Material
Acknowledgements
This work was funded by the National Institutes of Health (P50 GM086145) and the Chicago Biomedical Consortium with support from the Searle Funds at the Chicago Community Trust. We thank Dr. Ian Steele for X-ray crystallographic analysis of 5.
Footnotes
Author Contribution
T.J.M. and J.L. contributed equally to the work. J.L. performed and analyzed all interfacial reactions on monolayers. T.J.M. carried out most of the solution-based studies. J.R.C.-P. assisted with scope studies. M.M. and S.A.K. equally provided project management. The manuscript was written by T.J.M., J.L., S.A.K. and M.M.
References
- 1.Menger FM, Eliseev AV, Migulin VA. Phosphatase catalysis via combinatorial organic chemistry. J. Org. Chem. 1995;60:6666–6661. [Google Scholar]
- 2.Liu G, Ellman JA. A general solid-phase synthesis strategy for the preparation of 2-pyrrolidinemethanol ligands. J. Org. Chem. 1995;60:7712–7713. [Google Scholar]
- 3.Burgess K, Lim H-J, Porte AM, Sulikowski GA. New catalysts and conditions for a C-Hinsertion reaction identified by high-throughput catalyst screening. Angew. Chem. Int. Ed. Engl. 1996;35:220–222. [Google Scholar]
- 4.Cole BM, Shimizu KD, Krueger CA, Harrity JPA, Snapper MC, Hoveyda AH. Discovery of chiral catalysts through ligand diversity: Ti-Catalyzed enantioselective addition of TMSCN to mesoepoxides. Angew. Chem. Int. Ed. Engl. 1996;35:1668–1671. [Google Scholar]
- 5.Francis MB, Finney NS, Jacobsen EN. Combinatorial approach to the discovery of novel coordination complexes. J. Am. Chem. Soc. 1996;118:8983–8984. [Google Scholar]
- 6.Taylor SJ, Morken JP. Thermographic selection of effective catalysts from an encoded polymer-bound library. Science. 1998;280:267–270. doi: 10.1126/science.280.5361.267. [DOI] [PubMed] [Google Scholar]
- 7.Reetz MT, Becker MH, Kühling KM, Holzwarth A. Time-resolved IR-thermographic detection and screening of enantioselectivity in catalytic reactions. Angew. Chem. Int. Ed. Engl. 1998;37:2647–2650. doi: 10.1002/(SICI)1521-3773(19981016)37:19<2647::AID-ANIE2647>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 8.Senkan SM. High-throughput screening of solid-state catalyst libraries. Nature. 1998;394:350–353. [Google Scholar]
- 9.Reddington E, Sapienza A, Gurau B, Viswanathan R, Sarangapani S, Smotkin ES, Mallouk TE. Combinatorial electrochemistry: A highly parallel, optical screening method for discovery of better electrocatalysts. Science. 1998;280:1735–1737. doi: 10.1126/science.280.5370.1735. [DOI] [PubMed] [Google Scholar]
- 10.Sigman MS, Jacobsen EN. Schiff base catalysts for the asymmetric Strecker reaction identified and optimized from parallel synthetic libraries. J. Am. Chem. Soc. 1998;120:4901–4902. [Google Scholar]
- 11.Cooper AC, McAlexander LH, Lee D-H, Torres MT, Crabtree RH. Reactive dyes as a method for rapid screening of homogeneous catalysts. J. Am. Chem. Soc. 1998;120:9971–9972. [Google Scholar]
- 12.Shaughnessy KH, Kim P, Hartwig JF. A fluorescence-based assay for high-throughput screening of coupling reactions. Application to Heck chemistry. J. Am. Chem. Soc. 1999;121:2123–2132. [Google Scholar]
- 13.Copeland GT, Miller SJ. A Chemosensor-based approach to catalyst discovery in solution and on solid support. J. Am. Chem. Soc. 1999;121:4306–4307. [Google Scholar]
- 14.Berkowitz DB, Bose M, Choi S. In situ enzymatic screening (ISES): a tool for catalyst discovery and reaction development. Angew. Chem., Int. Ed. 2002;41:1603–1607. doi: 10.1002/1521-3773(20020503)41:9<1603::aid-anie1603>3.0.co;2-d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stauffer SR, Hartwig JF. Fluorescence resonance energy transfer (FRET) as a high-throughput assay for coupling reactions. Arylation of amines as a case study. J. Am. Chem. Soc. 2003;125:6977–6985. doi: 10.1021/ja034161p. [DOI] [PubMed] [Google Scholar]
- 16.Yao S, Meng J-C, Siuzdak G, Finn MG. New catalysts for the asymmetric hydrosilylation of ketones discovered by mass spectrometry screening. J. Org. Chem. 2003;68:2540–2546. doi: 10.1021/jo026365e. [DOI] [PubMed] [Google Scholar]
- 17.Chen P. Electrospray ionization tandem mass spectrometry in high-throughput screening of homogeneous catalysts. Angew. Chem., Int. Ed. 2003;42:2832–2847. doi: 10.1002/anie.200200560. [DOI] [PubMed] [Google Scholar]
- 18.Teichert T, Pfaltz A. Mass Spectrometric Screening of Enantioselective Diels-Alder Reactions. Angew. Chem. Int. Ed. 2008;47:3360–3362. doi: 10.1002/anie.200705082. [DOI] [PubMed] [Google Scholar]
- 19.Wassenaar J, Jansen E, van Zeist W-J, Bickelhaupt FM, Siegler MA, Spek AL, Reek JNH. Catalyst selection based on intermediate stability measured by mass spectrometry. Nat. Chem. 2010;2:417–421. doi: 10.1038/nchem.614. [DOI] [PubMed] [Google Scholar]
- 20.Kanan MW, Rozenman MM, Sakurai K, Snyder TM, Liu DR. Reaction discovery enabled by DNA-templated synthesis and in vitro selection. Nature. 2004;431:545–549. doi: 10.1038/nature02920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rozenman MM, Kanan MW, Liu DR. Development and initial application of a hybridization-independent, DNA-encoded reaction discovery system compatible with organic solvents. J. Am. Chem. Soc. 2007;129:14933–14938. doi: 10.1021/ja074155j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen Y, Kamlet AS, Steinman JB, Liu DR. A Biomolecule-compatible visible light-induced azide reduction from a DNA-encoded reaction discovery system. Nature Chem. 2011;3:146–153. doi: 10.1038/nchem.932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Beeler AB, Su S, Singleton CA, Porco JA., Jr. Discovery of chemical reactions through multidimensional screening. J. Am. Chem. Soc. 2007;129:1413–1419. doi: 10.1021/ja0674744. [DOI] [PubMed] [Google Scholar]
- 24.Goodell JR, McMullen JP, Zaborenko N, Maloney JR, Ho C-X, Jensen KF, Porco JA, Jr., Beeler AB. Development of an automated microfluidic reaction platform for multidimensional screening: reaction discovery employing bicyclo[3.2.1]octanoid scaffolds. J. Org. Chem. 2009;74:6169–6180. doi: 10.1021/jo901073v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Robbins DW, Hartwig JF. A simple, multidimensional approach to high-throughput discovery of catalytic reactions. Science. 2011;333:1423–1427. doi: 10.1126/science.1207922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Min D-H, Yeo W-S, Mrksich M. A method for connecting solution phase enzyme activity assays with immobilized format analysis by mass spectrometry. Anal. Chem. 2004;76:3923–3929. doi: 10.1021/ac049816z. [DOI] [PubMed] [Google Scholar]
- 27.Min D-H, Tang W-J, Mrksich M. Chemical screening by mass spectrometry to identify inhibitors of anthrax lethal factor. Nature Biotechnol. 2004;22:717–720. doi: 10.1038/nbt973. [DOI] [PubMed] [Google Scholar]
- 28.Su J, Bringer MR, Ismagilov RF, Mrksich M. Combining microfluidic networks and peptide arrays for multi-enzyme assays. J. Am. Chem. Soc. 2005;127:7280–7281. doi: 10.1021/ja051371o. [DOI] [PubMed] [Google Scholar]
- 29.Ban L, Mrksich M. On-chip synthesis and label-free assays of oligosaccharide arrays. Angew. Chem. Int. Ed. 2008;47:3396–3399. doi: 10.1002/anie.200704998. [DOI] [PubMed] [Google Scholar]
- 30.Gurard-Levin ZA, Kim J, Mrksich M. Combining mass spectrometry and peptide arrays to profile the specificities of histone deacetylases. ChemBioChem. 2009;10:2159–2161. doi: 10.1002/cbic.200900417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Su J, Mrksich M. Using MALDI-TOF Mass spectrometry to characterize interfacial reactions on self-assembled monolayers. Langmuir. 2003;19:4867–4870. [Google Scholar]
- 32.Li J, Thiara PS, Mrksich M. Rapid evaluation and screening of interfacial reactions on self-assembled monolayers. Langmuir. 2007;23:11826–11835. doi: 10.1021/la701638d. [DOI] [PubMed] [Google Scholar]
- 33.Dömling A, Ugi I. Multicomponent reactions with isocyanides. Angew. Chem. Int. Ed. 2000;39:3168–3210. doi: 10.1002/1521-3773(20000915)39:18<3168::aid-anie3168>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 34.Touré BB, Hall DG. Natural product synthesis using multicomponent reaction strategies. Chem. Rev. 2009;109:4439–4486. doi: 10.1021/cr800296p. [DOI] [PubMed] [Google Scholar]
- 35.Guillena G, Ramon DJ, Yus M. Organocatalytic enantioselective multicomponent reactions (OEMCRs) Tetrahedron: Asymmetry. 2007;18:693–700. [Google Scholar]
- 36.Zhang L, Kozmin SA. Brønsted acid-promoted cyclizations of siloxyalkynes with arenes and alkenes. J. Am. Chem. Soc. 2004;126:10204–10205. doi: 10.1021/ja046586x. [DOI] [PubMed] [Google Scholar]
- 37.Zhang L, Kozmin SA. Gold-catalyzed cycloisomerizations of siloxy enynes to cyclohexadienes. J. Am. Chem. Soc. 2004;126:11806–11807. doi: 10.1021/ja046112y. [DOI] [PubMed] [Google Scholar]
- 38.Sun J, Kozmin SA. Brønsted Acid-Promoted Cyclizations of 1-Siloxy-1,5-diynes. J. Am. Chem. Soc. 2005;127:13512–13513. doi: 10.1021/ja055054t. [DOI] [PubMed] [Google Scholar]
- 39.Sun J, Kozmin SA. Silver-catalyzed hydroamination of siloxy alkynes. Angew. Chem. Int. Ed. 2006;45:4991–4993. doi: 10.1002/anie.200601276. [DOI] [PubMed] [Google Scholar]
- 40.Sweis R, Schramm MP, Kozmin SA. Silver-catalyzed [2+2] cycloadditions of siloxyalkynes. J. Am. Chem. Soc. 2004;126:7442–7443. doi: 10.1021/ja048251l. [DOI] [PubMed] [Google Scholar]
- 41.Sun J, Keller VA, Meyer ST, Kozmin SA. Silver-catalyzed aldehyde olefination using siloxy alkynes. Adv. Synth. Catal. 2010;352:839–842. doi: 10.1002/adsc.200900835. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.