

SUMMARY
PTEN is a frequently mutated tumor suppressor gene that opposes the PI3K-AKT pathway through dephosphorylation of phosphoinositide-3,4,5-triphosphate. Recently, nuclear compartmentalization of PTEN was found as a key component of its tumor suppressive activity, however its nuclear function remains poorly defined. Here we show that nuclear PTEN interacts with APC/C, promotes APC/C association with CDH1, and thereby enhances the tumor suppressive activity of the APC-CDH1 complex. We find that nuclear exclusion but not phosphatase inactivation of PTEN impairs APC-CDH1. This nuclear function of PTEN provides a straightforward mechanistic explanation for the fail-safe cellular senescence response elicited by acute PTEN loss and the tumor suppressive activity of catalytically-inactive PTEN. Importantly, we demonstrate that PTEN-mutant and PTEN-null states are not synonymous since they are differentially sensitive to pharmacological inhibition of APC-CDH1 targets such as PLK1 and Aurora Kinases. This finding identifies a strategy for cancer patient stratification and thus, optimization of targeted therapies.
INTRODUCTION
PTEN (phosphatase and tensin homolog) is amongst the most frequently lost or mutated tumor suppressors, with a frequency of monoallelic mutations estimated at 50–80% in endometrial carcinoma, glioblastoma and prostate cancer and at 30–50% in breast, colon and lung tumors (Cairns et al., 1997; Feilotter et al., 1998; Gray et al., 1998; Li and Sun, 1997; Steck et al., 1997). Complete loss of PTEN is observed at highest frequencies in endometrial cancer and glioblastoma and is generally associated with metastatic cancers (Ali et al., 1999; Salmena et al., 2008). Moreover, germline mutations of PTEN have been identified in cancer-susceptibility syndromes such as Cowden syndrome (Di Cristofano et al., 1998; Eng, 2003).
PTEN can dephosphorylate phosphoinositide-3,4,5-triphosphate (PIP3), a potent activator of AKT (Maehama and Dixon, 1998). Loss of PTEN function leads to derepression of the PI3K/AKT pathway, which stimulates cell growth and survival (Stambolic et al., 1998; Sun et al., 1999). However, emerging evidence suggests that PTEN also has PI3K/AKT-independent functions (Salmena et al., 2008). Furthermore, cells harboring phosphatase-inactive PTEN mutants retain residual tumor suppressive activity, leading to the hypothesis that PTEN exerts functions that are independent of its phosphatase activity (Blanco-Aparicio et al., 2007; Georgescu et al., 2000; Gildea et al., 2004; Koul et al., 2002; Maier et al., 1999).
Early studies proposed that PTEN was exclusively cytoplasmic. However, recent reports clearly demonstrate that nuclear PTEN has important tumor suppressive function (Fridberg et al., 2007; Perren et al., 2000; Whiteman et al., 2002; Zhou et al., 2002). Mechanistically, we have reported that ubiquitination of PTEN regulates its nuclear compartmentalization (Song et al., 2008; Trotman et al., 2007). However, the tumor suppressive functions of PTEN within the nucleus still remain poorly defined.
Cell cycle progression is controlled by ubiquitination-mediated proteolysis of cell-cycle machinery. The two major E3 ubiquitin ligases controlling this process are SCF (Skp1/Cullin/F-box protein complex) and APC/C (Anaphase Promoting Complex/Cyclosome). SCF mainly controls target protein levels during S-phase, whereas APC/C is thought to be active from mitosis to late G1 (Cardozo and Pagano, 2004; Peters, 2006). APC/C contains at least eleven different structural subunits and its activity is controlled through the binding of CDC20 and CDH1, which recognize and recruit specific substrates. CDC20 is active in early mitosis whereas CDH1 activity is restricted to late mitosis and G1 (Pines, 2006; Sullivan and Morgan, 2007). Specific APC/C substrates include mitotic cyclins (Cyclin A and B), mitotic kinases (Aurora kinases, PLK1, Nek2A), proteins involved in chromosome segregation (Securin, Sgo1), DNA replication proteins (Geminin, Cdc6), a F-box protein (SKP2) and transcription factors (Ets2, FoxM1) (Manchado et al., 2010; Wasch et al., 2010).
Of importance, APC-CDH1 substrates, such as Cyclin A, PLK1, Aurora A, CDC20 or SKP2, are overexpressed in human tumors and are associated with chromosomal instability and poor prognosis (Carter et al., 2006). In mice, Cdh1 heterozygosity results in the development of epithelial tumors, suggesting that CDH1 may be a haploinsufficient tumor suppressor (Garcia-Higuera et al., 2008). Down-regulation of CDH1 has been reported in many cancers, including those of prostate, ovary, liver and brain, and during the malignant progression of a B-cell lymphoma cell line (Bassermann et al., 2008; Wang et al., 2000). Therefore, inactivation of APC-CDH1 in cancer may lead to unchecked accumulation of its targets with profound consequences for cell cycle and genomic stability.
In this study, we demonstrate that nuclear PTEN directly enhances the activity of APC/C by promoting its association with CDH1. Conversely, PTEN loss impairs the activity of the APC-CDH1 tumor suppressive complex. Critically, PTEN activates APC-CDH1 in a phosphatase-independent manner, an observation that has important implications for cancer therapy.
RESULTS
Nuclear PTEN interacts with APC/C
In order to identify novel tumor suppressive pathways regulated by PTEN, we immunoprecipitated exogenous Myc-tagged PTEN from the nuclear extracts of PTEN-deficient PC3 cells and identified interacting proteins by mass spectrometry (Figure 1A and Suppl. Fig. S1A). Remarkably, four APC/C constituents including APC3/CDC27, APC4, APC5 and APC7 were identified as nuclear PTEN-associated proteins. We validated the binding between endogenous PTEN and the four APC/C components in DU145 cells by performing reciprocal immunoprecipitations (Figure 1B) and observed that the binding was restricted to the nucleus (Suppl. Figs. S1B and S1C). Notably, an in vitro binding assay revealed that the interaction of PTEN with APC3 is restricted to the COOH-terminus of PTEN, indicating that the N-terminal phophatase domain is dispensable for this interaction (Figure 1C). Critically, PTEN immunoprecipitation complexes displayed ubiquitinating ability towards the well-established APC/C target Cyclin B (Figure 1D).
Figure 1. Nuclear PTEN interacts with APC/C.
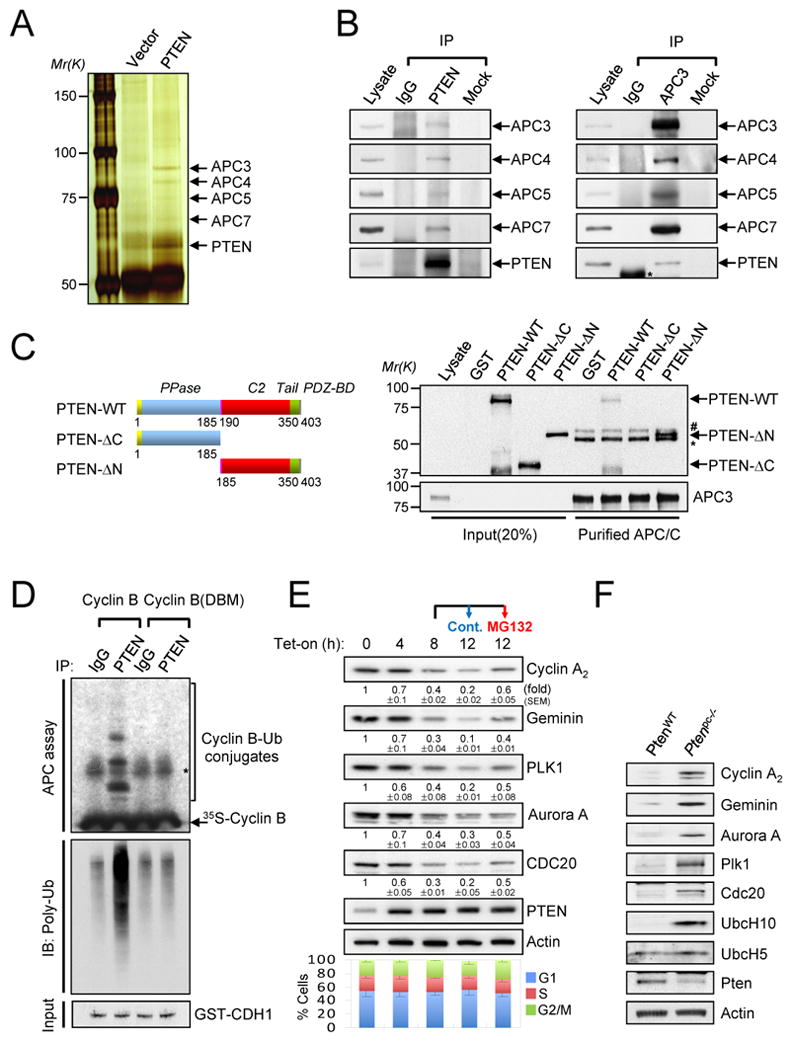
(A) Nuclear extracts from PTEN-deficient PC3 cells transfected with Myc-PTEN were immunoprecipitated with an anti-Myc antibody followed by mass spectrometric peptide sequencing. APC/C components including APC3, APC4, APC5 and APC7 were identified. (B) DU145 cell lysates were immunoprecipitated (IP) without (Mock) or with anti-PTEN (left) or anti-APC3 (right) antibody followed by immunoblotting. Asterisk indicates heavy chain of IgG. (C) Recombinant GST-PTEN (aa 1–403), GST-PTEN-ΔC (aa 1–185) or GST-PTEN-ΔN (aa 185–403) proteins, as shown in top diagram, were incubated with immunopurified APC/C from PC3 cells released 3 hr from nocodazole synchronization. * and # indicate heavy chain of IgG and non-specific band, respectively. (D). Lysates from 12 hr after tetracyclin induction as in (E) were immunoprecipitated (IP) with anti-PTEN antibody and subjected to in vitro ubiquitination assay using 35S-labeled Cyclin B (WT and destruction box mutant (DBM)) in the presence of GST-CDH1 protein (50 ng) and immunoblotting with anti-polyubiquitinated proteins antibody. (E) PC3 cells were co-transfected with pcDNA4/TO/Myc-His-PTEN and pcDNA6/TR to induce PTEN expression after the addition of tetracycline (Tet-on) for the indicated times or treated with 10 μM MG132 after 8hr-Tet induction for additional 4 hr, and then subjected to immunoblotting (top) and flow cytometric analysis (bottom). The quantification of the relative immunoreactivity of each protein normalized to Actin is represented as the mean and the standard error of the mean (SEM) from three different experiments. (F) Accumulation of APC-CDH1 targets in PtenloxP/loxP;Probasin- Cre4 (Ptenpc−/−) mouse prostate epithelium. Lysates from anterior prostates in wild-type (WT-Cre) and Ptenpc−/− mice at 11 weeks age were subjected to immunoblotting. “see also Figure S1A-F”.
We next assessed the functional relevance of this physical interaction. To test whether PTEN directly affects the level of APC/C targets, we developed a model system in which we could dissociate the consequences of acute PTEN induction from the ensuing cell cycle effects. For this we used a Tetracyclin inducible (Tet-on)-PTEN cellular system in asynchronously growing PC3 cells (Rubin et al., 1991). In this cellular system, acute PTEN induction precedes any sizable effects on cell cycle distribution for at least 12 hours (Figure 1E). By contrast, in this time frame PTEN induction led to a marked and rapid proteasome-dependent down-regulation of APC/C targets. This was reverted by MG132 treatment, at time points much earlier than any detectable effect on cell cycle distribution (Figure 1E). Moreover, we found that APC/CDH1 target levels where increased in vivo in Pten null prostates at time points where the incidence of prostate intraepithelial neoplasia (PIN) and the increase of cell proliferation are low (Figure 1F, Suppl. Fig. S1D and data not shown). Similarily, PTEN-silencing in DU145 cells led to the up-regulation of the APC/C targets: Cyclin A2, Geminin, PLK1, Aurora A and CDC20, whereas over-expression of PTEN in PTEN-deficient LNCaP and PC3 cells reduced of APC/C target levels concomitant with G1 cell cycle arrest (Suppl. Figs. S1E and S1F).
Nuclear PTEN enhances the activity of APC-CDH1
Our data show that PTEN interacts with components of the APC/C complex and PTEN status alters the level of APC/C complex target proteins. Thus, we hypothesized that PTEN can regulate the ubiquitin ligase activity of APC/C. Indeed, in vitro APC/C ubiquitin ligase assays revealed that Cyclin B ubiquitination by APC/C was increased in a dose-dependent manner by recombinant PTEN protein in the presence of CDH1 (Figure 2A). By contrast, the ubiquitin ligase activity of APC/C was reduced in Pten−/− mouse embryonic fibroblasts (MEFs) compared to wild-type cells (Figure 2B). Remarkably, nuclear but not cytoplasmic PTEN immunoprecipitates were capable of ubiquitinating Cyclin B (Figure 2C).
Figure 2. Nuclear PTEN regulates the activity of APC-CDH1.

(A) Immunopurified APC/C from PC3 cells, released 3hr post nocodazole synchronization, were subjected to in vitro ubiquitination assay using 35S-labeled in vitro-translated (IVT) wild-type or destruction box mutated (DBM) Cyclin B in the presence of IVT-CDH1 (1 μl) and different amounts of GST-PTEN proteins (0, 50, 100ng) and immunoblotting with anti-polyubiquitinated proteins antibody. Bottom panel illustrates the density unit (D.U.) of Cyclin B-Ub conjugates analyzed by the ImageJ 1.38× software (NIH). Asterisk indicates non-specific band. (B) Lysates from wild-type and Pten−/− MEFs subjected to 48hrs of serum depletion were immunoprecipitated (IP) with anti-APC3 or anti-Pten antibody and subjected to in vitro ubiquitination assay using 35S-labeled Cyclin B in the presence of GST-CDH1 protein (50 ng). Top panel shows the density unit (D.U.) of Cyclin B-Ub conjugates analyzed by the ImageJ 1.38x software. (C) Cytoplasmic (C) and nuclear (N) extracts from DU145 cells, released 3 hr from nocodazole synchronization, were IP with anti-APC3 or anti-PTEN antibody and subjected to in vitro ubiquitination assay using 35S-labeled Cyclin B. Top panel shows the density unit (D.U.) of Cyclin B-Ub conjugates analyzed by the ImageJ 1.38x software. (D) Lysates from PTEN-deficient PC3 cells complemented with empty vector or PTEN (top) or PTEN-proficient DU145 cells transfected with siRNAs for Renilla luciferase (siControl) or PTEN (siPTEN-1) (bottom) were IP with anti-CDH1 antibody and then subjected to immunoblotting. (E) Immunopurified APC/C from PC3 cells, released 3 hr from nocodazole synchronization, were incubated with GST-CDH1 (100 ng) and different amounts of His6-PTEN proteins (0, 50, 100, 200 ng) for 1 hr followed by immunoblotting. see also Figure S1G”.
Next, we sought to ascertain the mechanism by which PTEN regulates APC/C function. APC/C activation occurs upon the association with two different adaptor proteins, CDC20 and CDH1 (Cardozo and Pagano, 2004; Peters, 2006). While CDC20 activates APC/C in early mitosis, APC-CDH1 is active in late mitosis and during G1 (Pines, 2006; Sullivan and Morgan, 2007). In vitro Cyclin B ubiquitination assays revealed that the ubiquitin ligase activity of PTEN immunoprecipitates in interphase was higher than in mitosis (Suppl. Fig. S1G). We therefore examined whether PTEN regulates the formation of APC-CDH1 complex. In vivo, reintroduction of PTEN into PC3 cells increased the association between APC3 and CDH1 (Figure 2D, top panel), whereas PTEN knock-down in DU145 dramatically reduced the co-immunoprecipitation of APC3 and CDH1 cells (Figure 2D, bottom panel). In vitro binding assay revealed that PTEN favors the assembly between APC3 and CDH1 in a dose-dependent manner (Figure 2E). Taken together, these results suggest that PTEN promotes the association between APC and CDH1, thereby enhancing the activity of APC-CDH1.
Nuclear exclusion of PTEN impairs activation of APC-CDH1
Despite the increasing number of studies emphasizing the importance of nuclear PTEN as a tumor suppressor (Planchon et al., 2008; Salmena et al., 2008), how nuclear PTEN exerts its tumor suppressive activity remains unclear. We have demonstrated that PTEN monoubiquitination at lysines 13 and 289 is essential for its nuclear localization and tumor suppressive function and, conversely, deubiquitination of PTEN by HAUSP renders PTEN predominantly cytoplasmic (Song et al., 2008; Trotman et al., 2007). Therefore, we tested whether a nuclear excluded PTENK13,289E mutant (Figure 3A) could still regulate APC-CDH1. In vivo ubiquitination assay showed that unlike wild-type PTEN, nuclear excluded PTENK13,289E was unable to promote the Cyclin B ubiquitination and to reduce the level of APC-CDH1 targets despite of antagonizing AKT activation (Figure 3B and Suppl. Fig. S2A). Importantly, co-immunoprecipitation analysis revealed that PTENK13,289E mutant failed to interact with APC3 (Figure 3C). Since both PTEN and APC-CDH1 are known negative regulators of cell proliferation (Salmena et al., 2008; Wasch et al., 2010), we next examined the effects of nuclear exclusion of PTEN on cell growth. BrdU incorporation analysis revealed that nuclear-excluded PTENK13,289E mutant was ineffective in suppressing S phase entry after release from nocodazole block, whereas PTENWT significantly delayed the G1-S transition accompanied with a reduction in the level of APC-CDH1 targets (Figure 3D and Suppl. Fig. S2B). This observation is of critical relevance as it allowed us to uncouple the effect of PTEN on Akt and APC-CDH1 towards cell cycle regulation. In line with this notion, PTENWT but not PTENK13,289E over-expression led to a down-regulation of CDK2 activity due to reduction in the level of Cyclin A2 rather than Cyclin E at the G1-S transition, suggesting that nuclear PTEN might regulate the G1-S transition through reduction in the level of Cyclin A2, an APC-CDH1 target (Figure 3E). We also ascertained whether the delay in S phase entry by PTEN could be due to failure to be released from the nocodazole block or to exit from mitosis. PTENK13,289E as well as PTENWT over-expression had little effects on mitotic exit, as measured by the positivity of phospho-histone H3 (Suppl. Figs. S2C and S2D). In addition, PTENWT or PTENK13,289E-expressing PC3 cells exhibited similar proportions of cells in mitosis by upon nocodazole block, suggesting that PTEN might not be involved in spindle checkpoint (SAC) as well. Taken together, these results suggest that nuclear localization of PTEN is necessary for activation of APC-CDH1, thereby regulating cell cycle progression.
Figure 3. Nuclear exclusion of PTEN impairs activation of APC-CDH1.

(A) Immunofluorescence analysis of GFP, GFP-tagged wild-type (PTENWT) and nuclear excluded PTEN mutant (PTENK13,289E). Arrows indicate the nucleus in PC3 cells. (B) PC3 cells were co-transfected with a combination of wild-type or destruction box mutated (DBM) Myc-Cyclin B, HA-ubiquitin (Ub), GFP, PTENWT and PTENK13,289E, and treated with the proteasome inhibitor MG132 (10 μM) for 4 hr before harvesting. Cell lysates were then immunoprecipitated (IP) with anti-Myc antibody and subjected to immunoblotting (IB). * and # indicate heavy chain of IgG and non-specific band, respectively. (C) Lysates from PC3 cells transfected with GFP, PTENWT or PTENK13,289E were IP with anti-GFP antibody and subjected to immunoblotting. (D) PC3 cells transfected with GFP, PTENWT or PTENK13,289E were synchronized by growth in nocodazole (400 nM) for 24 hr, released for the indicated times, pulsed with BrdU for 30 min and subjected to flow cytometric analysis. Data are the means from three different experiments and error bars represent the SEM. (E) Lysates from (D) were subjected to in vitro kinase assay with CDK2, Cyclin E1 or Cyclin A2. The relative kinase activities were normalized with histone H1 inputs. Asterisks indicate heavy chain of IgG. “see also Figure S2”. P value was determined by Student’s t test (*P<0.01).
Growth-suppressive activity of PTEN requires APC-CDH1
Next, we examined the requirement of APC-CDH1 function in PTEN-mediated cell growth suppression. The effects of PTEN on cell cycle distribution as well as APC-CDH1 target modulation were completely abrogated by depletion of APC3 or CDH1 (Figures 4A and 4B). We then evaluated the contribution of APC-CDH1 to the growth suppressive function of PTEN by using wild-type and Cdh1−/− MEFs (among which the phosphatase activity of endogenous Pten was indistinguishable, Figure 4C). Strikingly, while PTEN over-expression induced a growth inhibition in immortalized Cdh1+/+ (or in Cdh1−/− MEFs complemented with human CDH1), Cdh1−/− cells were refractory to this effect of PTEN over-expression (Figure 4D, top panel). Consistent with this data, over-expression of PTEN was accompanied by the down-regulation of Cyclin A2-associated CDK2 activity as well as other APC-CDH1 targets in Cdh1+/+ (or in Cdh1−/− MEFs complemented with CDH1), but not in Cdh1−/− cells (Figure 4D, bottom panel and Suppl. Fig. S3A). Conversely, the increase in proliferation and elevation of the level of APC-CDH1 targets induced by Pten-silencing was abrogated in the absence of Cdh1 (Figure 4E and Suppl. Fig. 3B). Taken together, these results suggest that the growth suppressive activity of PTEN is, at least in part, mediated by APC-CDH1.
Figure 4. Growth suppression by PTEN relies on APC-CDH1 function.

A–B) PC3 cells co-transfected with empty vector or HA-PTEN, and Renilla luciferase siRNA (siControl), two independent APC3 siRNAs (siAPC3-1 and -2) (A) or CDH1 siRNAs (siCDH1-1 and -2) (B) were subjected to immunoblotting (top) and flow cytometric analysis to measure cell cycle distribution (bottom). The relative immunoreactivity of each protein was quantified by normalizing with Hsp90. (C) diC8-PtdIns(3,4,5)P3 (40 μM) was incubated with and without (control) the immunopurified Pten (1 μg) at 37°C for 30 min. The amount of free phosphate was measured Absorbance 620 nm (A620nm) and calculated using the standard curve line-fit data in top panel. Error bars represent SEM from three different experiments. (D) Growth curves (top) and in vitro kinase assay (bottom) of SV40-LT-immortalized wild-type and Cdh1−/− MEFs, infected with a retroviral combination of human CDH1 and PTEN (with selection) as indicated, and followed over a 3-day period. Error bars represent the SEM from three different experiments. Asterisks indicate heavy chain of IgG. (E) Growth curves (top) and in vitro kinase assay (bottom) of immortalized wild-type and Cdh1−/− MEFs, infected with a retroviral combination of human CDH1 and Pten shRNA (with selection) as indicated, and followed over a 3-day period. Error bars represent SEM from three different experiments. The relative kinase activities were normalized with histone H1 inputs. Asterisks indicate heavy chain of IgG. “see also Figure S3”.
PTEN loss-induced cellular senescence involves APC-CDH1
Reduction of cellular levels of PTEN or CDH1 results in the decrease of their respective tumor suppressive activity (Di Cristofano et al., 1998; Garcia-Higuera et al., 2008). However, acute and complete loss of both Pten and Cdh1 leads to a failsafe cellular senescence response; this phenomenon has been shown to profoundly suppress tumor progression upon Pten loss (Chen et al., 2005; Li et al., 2008). We therefore hypothesized that Pten-loss induced senescence could be in part mediated by a reduction of Cdh1 activity towards mediators of cellular senescence. In order to test this hypothesis, we over-expressed CDH1 concomitantly with the acute inactivation of Pten in Ptenlox/lox MEFs. Strikingly, over-expression of CDH1 counteracted the senescence response (Figures 5A and 5B). Importantly, over-expression of CDH1 specifically prevented the increase of p16 mRNA and protein levels, but not those of Arf or p53 (Figures 5C and 5D, and Suppl. Fig. S4A). Conversely, complete concomitant inactivation of Cdh1 and Pten in MEFs dramatically increased the distinctive morphology of senescent cells (flattened large cells; data not shown) and the positivity for senescence-associated β-galactosidase (SA-β-Gal), a hallmark of senescent cells (Figure 5E and Suppl. Fig. S4B). Consistent with this data, complete loss of Cdh1 upon Pten loss led to reduced proliferation compared to Pten loss alone, as measured by growth curves and BrdU incorporation analysis (Figure 5F). Of note, the senescence response elicited upon acute loss of Pten was accompanied by elevation of p19ARF(Arf)/p53/p21 as well as p16INK4A(p16) (Figures 5C and 5G). We have previously reported that senescence elicited by acute loss of Pten is accompanied by the activation of the p53 pathway (Chen et al., 2005). Interestingly, complete loss of Cdh1 and Pten led to a dramatic accumulation of p16, whereas Arf or p53/p21 was not affected compared to Pten loss alone. Critically, silencing of p16 prevented Pten-loss-induced senescence without affecting the p53 axis (Suppl. Figs. S4C-E). In line with previous reports from our lab, p19 silencing also reduced cellular senescence in Pten null MEFs without affecting p53 (Suppl. Figs. S4C-E) (Chen et al., 2009). Since Ets2 has been shown to selectively regulate p16, but not Arf transcription (Huot et al., 2002; Ohtani et al., 2001), we analyzed Ets2 levels in Pten null cells to determine whether an increase in Ets2 could account for the enhanced expression of p16. Indeed, Ets2 protein but not mRNA level was markedly up-regulated by acute loss of Pten (Figures 5C and 5G, and Suppl. Fig. S4F). Of interest, consistent with the notion that APC-CDH1 regulates the ubiquitination-mediated degradation of Ets2 (Suppl. Fig. S4G) (Li et al., 2008), PTEN promoted the ubiquitination of wild-type but not destruction box mutated (DBM) Ets2 (Figure 5H). To determine the direct role of Ets2-p16 pathway on active senescence in Pten null cells, we over-expressed or silenced Ets2 upon acute Pten loss. Over-expression of destruction box mutated (DBM) Ets2 significantly promoted the senescence response by Pten loss even in the presence of CDH1 (Figure 5I), while silencing of Ets2 dramatically reduced senescence and the transcriptional induction of p16 (Suppl. Figs. S4H and S4I). Collectively, these data provide a compelling mechanism for the senescence response elicited by Pten-loss through parallel CDH1-Ets2-p16 and Arf or p53/p21 axis.
Figure 5. Pten loss-induced cellular senescence involves APC-CDH1.

Cellular senescence assay (A), growth curves and BrdU incorporation assay (B), and immunoblot (C) of primary Ptenlox/lox MEFs, infected with retroviral Cre and human CDH1 selected for 4 days. Error bars represent SEM from three different experiments. P value was determined by Student’s t test. (D) Real-time RT-PCR analysis of murine p16 expression was quantified. Error bars represent SEM. (EG) Cellular senescence assay (E), growth curves and BrdU incorporation assay (F) and immunoblotting (G) of primary conditional Ptenlox/lox;Cdh1+/+, Ptenlox/lox;Cdh1lox/+, Ptenlox/lox;Cdh1lox/lox MEFs, infected with retroviral Cre recombinase at 4 day after selection. Error bars represent the SEM from three different experiments. Asterisk indicates nonspecific band. (H) PC3 cells were co-transfected with wild-type or destruction box mutated (DBM) Flag-Ets2, His-ubiquitin (Ub) and HA-PTEN, and treated with the proteasome inhibitor MG132 (10 μM) for 4 hr before harvesting. His-Ub-conjugated Ets2 was purified from cell lysates using Ni2+-NTA spin column under denaturing conditions. (I) Cellular senescence assay of primary Ptenlox/lox MEFs, infected with a retroviral combination of wild-type or Ets2(DBM), CDH1 and Cre as indicated, at 4 day after selection. Error bars represent SEM from three different experiments. “see also Figure S4”.
PTEN regulates APC-CDH1 independently of its phosphatase activity
PTEN opposes the PI3K/AKT signaling pathway by catalyzing the dephosphorylation of PIP3 (Maehama and Dixon, 1998). Loss of PTEN leads to the activation of the PI3K/AKT cascade and stimulates cell growth and survival (Stambolic et al., 1998; Sun et al., 1999). Nevertheless, cells harboring phosphatase-inactive PTEN mutatnts are phenotypically distinct from cell lacking PTEN protein, suggesting that PTEN exerts functions that are independent of its phosphatase activity (Blanco-Aparicio et al., 2007; Georgescu et al., 2000; Gildea et al., 2004; Maier et al., 1999; Okumura et al., 2005). In order to examine the contribution of the phosphatase activity of PTEN in the regulation of APC-CDH1, we first complemented PTEN-null PC3 cells, with either wild-type or phosphatase-inactive PTEN (C124S or G129E) (Figure 6A). The level of several canonical APC-CDH1 targets was reduced upon expression of PTEN(C124S) or PTEN(G129E) phosphatase-inactive mutants in this system (Suppl. Fig. S5A). Consistently, expression of phosphatase-inactive PTEN (C124S) in PC3 cells resulted in a delayed entry in S phase phenotype similar to wild-type PTEN expressing-PC3 cells or PTEN-proficient DU145 cells, implying that PTEN regulates the G1-S transition independent of its phosphatase activity (Figure 6B and Suppl. Figs. S5B and S5C). Importantly, expression of wild-type or PTEN(C124S) led to reduced Cyclin A2-associated CDK2 activity in PC3 cells, suggesting that PTEN(C124S) as well as wild-type PTEN suppresses the G1-S transition, at least in part, through APC-CDH1. Consistent with this data, in vivo xenograft generation with these variants of PC3 cells revealed a significant growth suppressive activity of PTEN(C124S), in line with the modulation of APC-CDH1 targets (Figure 6C). Although expression of PTEN(C124S) led to a significant reduction in the level of late mitotic substrates of APC-CDH1, such as Aurora A, PLK1 and CDC20, the length of mitosis was not significantly shortened, compared to parental PC3 cells (Suppl. Figs. S5D and S5E). Importantly, phosphatase-inactive PTEN(C124S) mutant exhibited comparable activity to wild-type PTEN enhancing the ubiquitin ligase activity of APC-CDH1 in vitro (as measured by Cyclin B ubiquitination) and in vivo (as measured by ubiquitination of Cyclin B and Ets2) (Figure 6D and Suppl. Figs. S6A and S6B). Finally, we found that, like wild-type PTEN, over-expression of phosphatase inactive PTEN(C124S) resulted in a significant growth suppression accompanied with lower CDK2 activity in immortalized Cdh1+/+ (or Cdh1−/− MEFs complemented with human CDH1), but not Cdh1−/− cells (Figure 6E and Suppl. Fig. S6C).
Figure 6. PTEN regulates APC-CDH1 independently of its phosphatase activity.

(A) diC8-PtdIns(3,4,5)P3 (40 μM) was incubated without (control) and with PTEN (WT), PTEN(C124S) or PTEN(G129E) immunoprecipitates (1 μg) at 37°C for 30 min and then the free phosphate was measured using the Green Reagent. Error bars represent the SEM from three different experiments. (B) PC3 cells complemented with wild-type or phosphatase-inactive PTEN(C124S) were nocodazole synchronized, released for the indicated times and pulsed with BrdU 30 min before harvesting. The proportion of BrdU+ cells was measured (left) and cell lysates were subjected to western blot (right). Error bars represent SEM from 3 different experiments. (C) PC3 cells complemented with wild-type or phosphatase-inactive PTEN(C124S) were injected subcutaneously into nude mice. Tumor volume was monitored (top) and tissue lysates at 2 week after injection were subjected to immunoblotting (bottom). Error bars represent SEM (n=6 mice/group). (D) Immunopurified APC/C from PC3 cells, released 3hrs post nocodazole synchronization, were subjected to in vitro ubiquitination assay using 35S-labeled Cyclin B in the presence of in vitro-translated (IVT)-CDH1 (2 μl) and different amounts of wild-type or PTEN(C124S) proteins (0, 50, 100, 200 ng). Right panel illustrates the density unit (D.U.) of Cyclin B-Ub conjugates analyzed by the ImageJ 1.38× software. (E) Growth curves (left) and in vitro kinase assay (right) of immortalized wild-type and Cdh1−/− MEFs, infected with a retroviral combination of human CDH1 and PTEN(C124S) (with selection) as indicated, and followed over a 3-day period. Error bars represent SEM from three different experiments. The relative kinase activities were normalized with histone H1 inputs. Asterisk indicates heavy chain of IgG. “see also Figures S5 and S6”. P value was determined by Student’s t test (*P<0.01; #P<0.05).
We then investigated the effects of pharmacological inhibition of PTEN phosphatase activity on APC-CDH1 function. Di-potassium bisperoxo (picolinato) oxovanadate [bpV(Hopic)] inhibits PTEN phosphatase activity (Lai et al., 2007; Rosivatz et al., 2006). Co-immunoprecipitation analysis revealed that the treatment of DU145 cells with 1 μM bpV(Hopic) had no effects on the formation of APC-CDH1 despite of strong activation of AKT (Suppl. Fig. S6D). Additionally, the inhibition of PI3-Kinase by treatment with 1 μM LY294002 failed to alter the interaction between APC3 and CDH1 or the ubiquitin ligase activity of APC-CDH1 (Suppl. Fig. S6E). Taken together, these results demonstrate that PTEN regulates APC-CDH1 in a phosphatase-independent manner.
PTEN loss but not inactivation of its phosphatase activity results in hypersensitivity to pharmacological inhibition of APC-CDH1 targets
Inhibition of APC-CDH1 targets, such as PLK1 or Aurora kinases, has been pursued as a therapeutic modality to treat human cancers due to the fact that APC-CDH1 activity is reduced in tumors (Bassermann et al., 2008; Taylor and Peters, 2008). Indeed, PLK1 inhibitors as well as Aurora A inhibitors are currently being evaluated as anticancer agents (Harrington et al., 2004; Strebhardt and Ullrich, 2006). In our study we have demonstrated that levels of PLK1 and Aurora A are elevated upon PTEN-loss in both asynchronous and synchronized conditions, suggesting that PTEN-deficient tumors might exhibit addiction to these kinases and hence hypersensitivity to their pharmacological inhibition. On the basis of the dispensability of the phosphatase activity of PTEN, we hypothesized that PTEN-deficient tumor cells would be more sensitive to such therapeutic approaches compared to phosphatase inactive PTEN mutant cells. To test this hypothesis, we first pharmacologically inhibited PLK1 in wild type and PTEN-null cells. Both human and murine Pten null cells were highly sensitive to PLK1 pharmacological inhibition by BI 2536, a PLK1 inhibitor (Figures 7A and 7B, and Suppl. Figs. S7A and S7B; PLK1 inhibition was directly measured by spindle assembly (Suppl. Fig. S7C)). Importantly, the growth inhibitory effect of BI 2536 in PTEN-null cells was associated with an profound mitotic arrest followed by apoptosis, as measured by cleavage of Parp and Caspase-3 (Cpp32) (Figures 7A, right panel and 7C). Critically, reconstitution of PTEN-null cells with a phosphatase-inactive PTEN(C124S) significantly restored the resistance to pharmacological inhibition of PLK1 (Figures 7B and 7C, and Suppl. Fig. S7D). Similarly, PTEN-null cells were hypersensitive to Aurora A inhibition by VX680 (MK0457) (Figures 7D and 7E, and Suppl. Figs. S7E and S7F; Aurora A inhibition was directly measured by histone H3 phosphorylation on Ser10 (Suppl. Fig. S7G)). Of note, Aurora A inhibition by VX680 in PTEN-null cells induced a robust accumulation of >4 N DNA content - as measured by flow cytometry - and subsequent apoptosis (Figures 7D, right panel and 7F). Importantly, the sensitivity of a phosphatase-inactive PTEN(C124S)-expressing cells to VX680 was similar to that of wild-type PTEN-expressing cells (Figures 7E and 7F, and Suppl. Fig. S7H). Taken together, these data suggest that PTEN-loss but not phosphatase inactivation results in hypersensitivity to pharmacological inhibition of APC-CDH1 targets, PLK1 and Aurora A.
Figure 7. PTEN loss but not phosphatase inactivation results in hypersensitivity to pharmacological inhibition of APC-CDH1 targets.

(A) Growth inhibition of immortalized wild-type and Pten−/− MEFs treated with the PLK1 inhibitor BI 2536 (50 nM) for 48 hr was measured (left), western blots of cell lysates after BI2536 treatment for 48 hr (right). B) Growth inhibition of PC3 cells complemented with wild-type or PTEN(C124S) and treated with the PLK1 inhibitor BI 2536 (5 nM) was measured after the 48 hr-treatment. (C) The cell population in mitosis of PC3 cells complemented with wild-type or PTEN(C124S) and treated with 5 nM BI 2536 was analyzed by immunofluorescence with anti-phospo-histone H3 (P-H3(pS10)) (left) and quantified for a 48 h-period (right). Error bars represent SEM from three different experiments. (D) Pten-null cells are hypersensitive to Aurora A inhibition. Growth inhibition of immortalized wild-type and Pten−/− MEFs treated with the Aurora A inhibitor VX680 (500 nM) was measured after the 48 hr-treatment (left), western blots of cell lysates after the treatment with different concentrations of VX680 for 48 hr (right). Error bars represent SEM from three different experiments. (E) Growth inhibition of PC3 cells complemented with wild-type or PTEN(C124S) and treated with the Aurora A inhibitor VX680 (500 nM) was measured after the 48 hr-treatment. Error bars represent SEM from three different experiments. (F) The cell population harboring >4 N DNA content of PC3 cells complemented with wild-type or PTEN(C124S) and treated with VX680 (500 nM) was analyzed by flow cytometry (left) and quantified for a 48 h-period (right). Error bars represent SEM from three different experiments. (G) A proposed model for phosphatase-independent role of nuclear PTEN towards APC-CDH1 to regulate proliferation and cellular senescence. “see also Figure S7”. P values were determined by Student’s t test (*P<0.01; #P>0.05).
DISCUSSION
Our findings allow us to reach a number of relevant conclusions:
Firstly, we have identified a novel mechanism by which PTEN exerts its tumor-suppressive function within the nucleus by regulating the assembly and activity of APC-CDH1. While high concentrations of nuclear PTEN is associated with G0/G1 phase and differentiated cells, lower concentrations of nuclear PTEN level is observed during S phase and highly proliferating advanced tumors (Gimm et al., 2000; Ginn-Pease and Eng, 2003; Perren et al., 2000). Furthermore, Pten and Cdh1 are haploinsufficient tumor suppressors in the mouse and their heterozygous loss leads to diverse cancers, including cancers of the prostate and breast (Garcia-Higuera et al., 2008; Salmena et al., 2008). By contrast, complete acute loss of either tumor suppressor triggers a cellular senescence response (Chen et al., 2005; Li et al., 2008). In this study, we have demonstrated that Pten-loss induced senescence is dependent on the Cdh1-Ets2-p16 pathway. On the basis of these data, we propose that PTEN loss elicits a potent senescence response through both phosphatase-dependent (superactivation of a PI3K/mTOR/p53 pathway (Alimonti et al., 2010; Chen et al., 2005)) and phosphatase-independent (loss of APC-CDH1 function, unrelated to Arf or p53/p21 pathway) activities, in turn highlighting the complexity of the dose-dependent tumor promoting and fail safe cellular responses evoked by PTEN loss (Fig. 7G).
Secondly, p we find that the novel function of nuclear PTEN presented herein is independent of its phosphatase activity. Cytoplasmic but not nuclear pools of PIP3 are sensitive to catalysis by PTEN, implying a potential role for nuclear PTEN beyond its phosphatase activity (Lindsay et al., 2006). Indeed, growth inhibitory effects of nuclear PTEN are likely not mediated directly through PI3K/AKT pathway (Blanco-Aparicio et al., 2007; Liu et al., 2005). Since APC3 is highly phosphorylated on multiple residues in mitosis (Kraft et al., 2003), one could suspect that PTEN may be a potential phosphatase for APC3. However, in mitotic cells we did not observe dephosphorylation of APC3 by PTEN neither in vivo nor in vitro (M.S.S. and P.P.P, unpublished data), and in addition, a catalytically inactive form of PTEN is fully functional towards APC. Overall, the phosphatase-independent activity of nuclear PTEN towards APC-CDH1 provides a straightforward explanation for the remnant tumor suppressive activity associated with the phosphatase inactive PTEN (C124S) mutation.
Finally, we have demonstrated that PTEN-deficient and PTEN-mutant cancer cells are differentially sensitive to pharmacological inhibition of PLK1 and Aurora A. Thus, our study indicates that patients with cancers harboring complete PTEN-loss may benefit from pharmacological targeting of APC-CDH1 pathway, while we predict mutant PTEN tumors to be much less sensitive. The APC-CDH1 targets, PLK1 and Aurora kinases are over-expressed in human tumors and this has prognostic and therapeutic potential in cancers (Meraldi et al., 2004; Strebhardt and Ullrich, 2006). Clinical trials in cancer patients are currently underway to test the effects of various PLK1 and Aurora A inhibitors as monotherapy or in combination with conventional chemotherapy. For example, phase II trials of BI 2536 in advanced SCLC, NSCLC and AML, and phase II trials of VX680 (MK0457) in CML are currently ongoing (weblink:http://www.cancer.gov). It has been recently reported that oncogenic Ras activation engages the cells to be hypersensitive (synthetic lethal) to various mitotic inhibitors including BI 2536 (Luo et al., 2009). By contrast, and in full agreement with our findings, oncogenic PI3K activation does not respond to PLK1 inhibition, suggesting that PTEN-loss driven hypersensitivity to inhibition of PLK1 and Aurora A is unlikely due to PI3K/AKT pathway (Luo et al., 2009). Hence, our findings provide a rationale for cancer patient stratification based on PTEN-loss vs. PTEN mutation towards the optimization of targeted therapies. This principle is of great importance given the widespread incidence of these two states of PTEN gene disruption in human cancer. Furthermore, since the functions of PTEN towards PI3K/AKT signaling and APC-CDH1 pathway are uncoupled, it is tempting to speculate that combinatorial therapy with PLK1/Aurora kinases and PI3K/mTOR inhibitors may be an effective approach in PTEN-null cancers.
EXPERIMENTAL PROCEDURES
Reagents and Antibodies
Chemical reagents are from Sigma unless otherwise described. G418, puromycin, and hygromycin, which are used for establishment of stable cell lines, are purchased from Invitrogen. BrdU and anti-BrdU antibody were from BD Biosciences. The PLK1 inhibitor BI 2536 and the Aurora A inhibitor VX680 were from Chemie Tek. All the sources of antibodies used are listed in the Extended Experimental Procedures.
Mass Spectrometry
PTEN-deficient PC3 cells transfected with pRK5-Myc-PTEN were subjected to cellular fractionation. The details of nuclear fractionation method are described in the Extended Experimental Procedures. The nuclear extracts were immunoprecipitated with anti-Myc antibody and the PTEN-associated proteins were eluted with Myc peptide. The eluates were resolved by SDS-PAGE on 4–12% gradient gel (Invitrogen) for silver staining (Pierce). Specific bands were cut out from the gel and subjected to mass-spectrometric peptide sequencing.
Immunoprecipitation and in vitro Binding Assay
The details of immunoprecipitation and in vitro binding assay are described in the Extended Experimental Procedures.
In vitro Ubiquitination Assay
The APC/C or PTEN was immunopurified with anti-APC3 (Sigma) or anti-PTEN (Cell Signaling) antibody, respectively, from PC3, DU145, wild-type and Pten−/− MEFs subjected to nocodazole synchronization and release for 2 or 3 hr. The ubiquitin ligase activity of APC-CDH1 complex was performed as described in detail in the Extended Experimental Procedures.
In vivo Ubiquitination Assay
PC3 cells were co-transfected with pCS2-6Myc-Cyclin B (wild-type or destruction box mutant (DBM)), provided by H. Yu, GFP-PTENWT or PTENK13,289E, and HA-ubiquitin (Ub). Alternatively, cells were transfected with a combination of His-Ub and wild-type or destruction box mutated (DBM) Ets2, provided by P. Zhang (Li et al., 2008). The details of in vivo ubiquitination assay are described in the Extended Experimental Procedures.
Cell Culture and Flow Cytometry
Primary MEFs and PC3 cells complemented with wild-type or phosphatase-inactive PTEN(C124S) were established and maintained as described in the Extended Experimental Procedures. The details of cell cycle analysis by flow cytometry are also described in the Extended Experimental Procedures.
siRNA and shRNA
All the sources of siRNA duplexes and shRNA constructs used are described in the Extended Experimental Procedures.
PtdIns(3,4,5)P3 Phophatase Assay
We purchased diC8-PtdIns(3,4,5)P3 and the Green Reagent from Echelon and Biomol, respectively. Measurement of phosphate released from the substrate was performed according to the manufacturer’s instructions and described in detail in the Extended Experimental Procedures.
In vitro Kinase Assay
The immunoprecipitates with Cyclin A2, Cyclin E1 or CDK2 prepared from cell lysates were incubated with histone H1 (Roche) and [γ-32P]ATP (PerkinElmer) for 30 min at room temperature. Reaction mixture was resolved by SDS-PAGE and then phosphorylated histone H1 was analyzed by SDS-PAGE and autoradiography.
Cell Proliferation and Senescence Assay
Typically, cell proliferation and senescence assays were performed as described (Chen et al., 2005) and the details of methods are described in the Extended Experimental Procedures. For cell proliferation assays with the PLK1 inhibitor BI 2536 (Lenart et al., 2007; Steegmaier et al., 2007) or the Aurora A inhibitor VX680 (Harrington et al., 2004), SV40-LT-immortalized wild-type and Pten−/− MEFs or PC3 cells complemented with wild type or PTEN(C124S) were incubated in the presence of the indicated concentrations of BI 2536 or VX680 (Chemie Tek) at a 48 hr-period, and then cell growth was measured.
Mouse Xenograft Tumor Model
PC3 cells complemented with wild type or PTEN(C124S) (1 × 107 cells per site) in suspension were mixed with equal volumes of matrigel (BD Bioscience) and injected subcutaneously into 6-week-old male nude mice (Charles River laboratory). Measurement of tumor size was performed twice a week, and tumor volume was estimated using the formula: volume = Length × 010215 Width2 × 0.526.
RNA Isolation and Quantitative Real-Time RT-PCR
Total RNA was isolated with Trizol reagent (Invitrogen) and reverse-transcribed with the Superscript III reverse transcriptase (Invitrogen). Expression of specific mRNAs was determined with the LightCycler (Roche) using the SYBR Green PCR Master Mix (Qiagen). All the sources of quantitative real-time RT-PCR primers used are listed in the Extended Experimental Procedures.
Supplementary Material
Acknowledgments
We are grateful to former and present members of the Pandolfi lab for experimental support, advice or discussion. We are thankful to Drs. Manuel Serrano, Pumin Zhang, Wenyi Wei and Hongtao Yu for providing reagents. We thank Dr. John Asara for mass spectrometric analysis. This work was supported by NIH grants to P.P.P. A.C. is supported by a Long-Term Fellowship Award from the European Molecular Biology Organization and L.S. is supported by a Fellowship from the Canadian Institutes of Health Research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ali IU, Schriml LM, Dean M. Mutational spectra of PTEN/MMAC1 gene: a tumor suppressor with lipid phosphatase activity. J Natl Cancer Inst. 1999;91:1922–1932. doi: 10.1093/jnci/91.22.1922. [DOI] [PubMed] [Google Scholar]
- Alimonti A, Nardella C, Chen Z, Clohessy JG, Carracedo A, Trotman LC, Cheng K, Varmeh S, Kozma SC, Thomas G, et al. A novel type of cellular senescence that can be enhanced in mouse models and human tumor xenografts to suppress prostate tumorigenesis. J Clin Invest. 2010;120:681–693. doi: 10.1172/JCI40535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassermann F, Frescas D, Guardavaccaro D, Busino L, Peschiaroli A, Pagano M. The Cdc14B-Cdh1-Plk1 axis controls the G2 DNA-damage-response checkpoint. Cell. 2008;134:256–267. doi: 10.1016/j.cell.2008.05.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco-Aparicio C, Renner O, Leal JF, Carnero A. PTEN, more than the AKT pathway. Carcinogenesis. 2007;28:1379–1386. doi: 10.1093/carcin/bgm052. [DOI] [PubMed] [Google Scholar]
- Cairns P, Okami K, Halachmi S, Halachmi N, Esteller M, Herman JG, Jen J, Isaacs WB, Bova GS, Sidransky D. Frequent inactivation of PTEN/MMAC1 in primary prostate cancer. Cancer Res. 1997;57:4997–5000. [PubMed] [Google Scholar]
- Cardozo T, Pagano M. The SCF ubiquitin ligase: insights into a molecular machine. Nat Rev Mol Cell Biol. 2004;5:739–751. doi: 10.1038/nrm1471. [DOI] [PubMed] [Google Scholar]
- Carter SL, Eklund AC, Kohane IS, Harris LN, Szallasi Z. A signature of chromosomal instability inferred from gene expression profiles predicts clinical outcome in multiple human cancers. Nat Genet. 2006;38:1043–1048. doi: 10.1038/ng1861. [DOI] [PubMed] [Google Scholar]
- Chen Z, Carracedo A, Lin HK, Koutcher JA, Behrendt N, Egia A, Alimonti A, Carver BS, Gerald W, Teruya-Feldstein J, et al. Differential p53-independent outcomes of p19(Arf) loss in oncogenesis. Sci Signal. 2009;2:ra44. doi: 10.1126/scisignal.2000053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Trotman LC, Shaffer D, Lin HK, Dotan ZA, Niki M, Koutcher JA, Scher HI, Ludwig T, Gerald W, et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature. 2005;436:725–730. doi: 10.1038/nature03918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Cristofano A, Pesce B, Cordon-Cardo C, Pandolfi PP. Pten is essential for embryonic development and tumour suppression. Nat Genet. 1998;19:348–355. doi: 10.1038/1235. [DOI] [PubMed] [Google Scholar]
- Eng C. PTEN: one gene, many syndromes. Hum Mutat. 2003;22:183–198. doi: 10.1002/humu.10257. [DOI] [PubMed] [Google Scholar]
- Feilotter HE, Nagai MA, Boag AH, Eng C, Mulligan LM. Analysis of PTEN and the 10q23 region in primary prostate carcinomas. Oncogene. 1998;16:1743–1748. doi: 10.1038/sj.onc.1200205. [DOI] [PubMed] [Google Scholar]
- Fridberg M, Servin A, Anagnostaki L, Linderoth J, Berglund M, Soderberg O, Enblad G, Rosen A, Mustelin T, Jerkeman M, et al. Protein expression and cellular localization in two prognostic subgroups of diffuse large B-cell lymphoma: higher expression of ZAP70 and PKC-beta II in the non-germinal center group and poor survival in patients deficient in nuclear PTEN. Leuk Lymphoma. 2007;48:2221–2232. doi: 10.1080/10428190701636443. [DOI] [PubMed] [Google Scholar]
- Garcia-Higuera I, Manchado E, Dubus P, Canamero M, Mendez J, Moreno S, Malumbres M. Genomic stability and tumour suppression by the APC/C cofactor Cdh1. Nat Cell Biol. 2008;10:802–811. doi: 10.1038/ncb1742. [DOI] [PubMed] [Google Scholar]
- Georgescu MM, Kirsch KH, Kaloudis P, Yang H, Pavletich NP, Hanafusa H. Stabilization and productive positioning roles of the C2 domain of PTEN tumor suppressor. Cancer Res. 2000;60:7033–7038. [PubMed] [Google Scholar]
- Gildea JJ, Herlevsen M, Harding MA, Gulding KM, Moskaluk CA, Frierson HF, Theodorescu D. PTEN can inhibit in vitro organotypic and in vivo orthotopic invasion of human bladder cancer cells even in the absence of its lipid phosphatase activity. Oncogene. 2004;23:6788–6797. doi: 10.1038/sj.onc.1207599. [DOI] [PubMed] [Google Scholar]
- Gimm O, Perren A, Weng LP, Marsh DJ, Yeh JJ, Ziebold U, Gil E, Hinze R, Delbridge L, Lees JA, et al. Differential nuclear and cytoplasmic expression of PTEN in normal thyroid tissue, and benign and malignant epithelial thyroid tumors. Am J Pathol. 2000;156:169–700. doi: 10.1016/s0002-9440(10)65040-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginn-Pease ME, Eng C. Increased nuclear phosphatase and tensin homologue deleted on chromosome 10 is associated with G0-G1 in MCF-7 cells. Cancer Res. 2003;63:282–286. [PubMed] [Google Scholar]
- Gray IC, Stewart LM, Phillips SM, Hamilton JA, Gray NE, Watson GJ, Spurr NK, Snary D. Mutation and expression analysis of the putative prostate tumour-suppressor gene PTEN. Br J Cancer. 1998;78:1296–1300. doi: 10.1038/bjc.1998.674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrington EA, Bebbington D, Moore J, Rasmussen RK, Ajose-Adeogun AO, Nakayama T, Graham JA, Demur C, Hercend T, Diu-Hercend A, et al. VX-680, a potent and selective small-molecule inhibitor of the Aurora kinases, suppresses tumor growth in vivo. Nat Med. 2004;10:262–267. doi: 10.1038/nm1003. [DOI] [PubMed] [Google Scholar]
- Huot TJ, Rowe J, Harland M, Drayton S, Brookes S, Gooptu C, Purkis P, Fried M, Bataille V, Hara E, et al. Biallelic mutations in p16(INK4a) confer resistance to Ras- and Ets-induced senescence in human diploid fibroblasts. Mol Cell Biol. 2002;22:8135–8143. doi: 10.1128/MCB.22.23.8135-8143.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koul D, Jasser SA, Lu Y, Davies MA, Shen R, Shi Y, Mills GB, Yung WK. Motif analysis of the tumor suppressor gene MMAC/PTEN identifies tyrosines critical for tumor suppression and lipid phosphatase activity. Oncogene. 2002;21:2357–2364. doi: 10.1038/sj.onc.1205296. [DOI] [PubMed] [Google Scholar]
- Kraft C, Herzog F, Gieffers C, Mechtler K, Hagting A, Pines J, Peters JM. Mitotic regulation of the human anaphase-promoting complex by phosphorylation. EMBO J. 2003;22:6598–6609. doi: 10.1093/emboj/cdg627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai JP, Dalton JT, Knoell DL. Phosphatase and tensin homologue deleted on chromosome ten (PTEN) as a molecular target in lung epithelial wound repair. Br J Pharmacol. 2007;152:1172–1184. doi: 10.1038/sj.bjp.0707501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenart P, Petronczki M, Steegmaier M, Di Fiore B, Lipp JJ, Hoffmann M, Rettig WJ, Kraut N, Peters JM. The small-molecule inhibitor BI 2536 reveals novel insights into mitotic roles of polo-like kinase 1. Curr Biol. 2007;17:304–315. doi: 10.1016/j.cub.2006.12.046. [DOI] [PubMed] [Google Scholar]
- Li DM, Sun H. TEP1, encoded by a candidate tumor suppressor locus, is a novel protein tyrosine phosphatase regulated by transforming growth factor beta. Cancer Res. 1997;57:2124–2129. [PubMed] [Google Scholar]
- Li M, Shin YH, Hou L, Huang X, Wei Z, Klann E, Zhang P. The adaptor protein of the anaphase promoting complex Cdh1 is essential in maintaining replicative lifespan and in learning and memory. Nat Cell Biol. 2008;10:1083–1089. doi: 10.1038/ncb1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsay Y, McCoull D, Davidson L, Leslie NR, Fairservice A, Gray A, Lucocq J, Downes CP. Localization of agonist-sensitive PtdIns(3,4,5)P3 reveals a nuclear pool that is insensitive to PTEN expression. J Cell Sci. 2006;119:5160–5168. doi: 10.1242/jcs.000133. [DOI] [PubMed] [Google Scholar]
- Liu JL, Sheng X, Hortobagyi ZK, Mao Z, Gallick GE, Yung WK. Nuclear PTEN-mediated growth suppression is independent of Akt down-regulation. Mol Cell Biol. 2005;25:6211–6224. doi: 10.1128/MCB.25.14.6211-6224.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J, Emanuele MJ, Li D, Creighton CJ, Schlabach MR, Westbrook TF, Wong KK, Elledge SJ. A genome-wide RNAi screen identifies multiple synthetic lethal interactions with the Ras oncogene. Cell. 2009;137:835–848. doi: 10.1016/j.cell.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maehama T, Dixon JE. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem. 1998;273:13375–13378. doi: 10.1074/jbc.273.22.13375. [DOI] [PubMed] [Google Scholar]
- Maier D, Jones G, Li X, Schonthal AH, Gratzl O, Van Meir EG, Merlo A. The PTEN lipid phosphatase domain is not required to inhibit invasion of glioma cells. Cancer Res. 1999;59:5479–5482. [PubMed] [Google Scholar]
- Manchado E, Eguren M, Malumbres M. The anaphase-promoting complex/cyclosome (APC/C): cell-cycle-dependent and -independent functions. Biochem. 2010;38:65–71. doi: 10.1042/BST0380065. [DOI] [PubMed] [Google Scholar]
- Meraldi P, Honda R, Nigg EA. Aurora kinases link chromosome segregation and cell division to cancer susceptibility. Curr Opin Genet Dev. 2004;14:29–36. doi: 10.1016/j.gde.2003.11.006. [DOI] [PubMed] [Google Scholar]
- Ohtani N, Zebedee Z, Huot TJ, Stinson JA, Sugimoto M, Ohashi Y, Sharrocks AD, Peters G, Hara E. Opposing effects of Ets and Id proteins on p16INK4a expression during cellular senescence. Nature. 2001;409:1067–1070. doi: 10.1038/35059131. [DOI] [PubMed] [Google Scholar]
- Okumura K, Zhao M, DePinho RA, Furnari FB, Cavenee WK. PTEN: a novel anti-oncogenic function independent of phosphatase activity. Cell Cycle. 2005;4:540–542. doi: 10.4161/cc.4.4.1614. [DOI] [PubMed] [Google Scholar]
- Perren A, Komminoth P, Saremaslani P, Matter C, Feurer S, Lees JA, Heitz PU, Eng C. Mutation and expression analyses reveal differential subcellular compartmentalization of PTEN in endocrine pancreatic tumors compared to normal islet cells. Am J Pathol. 2000;157:1097–1103. doi: 10.1016/S0002-9440(10)64624-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters JM. The anaphase promoting complex/cyclosome: a machine designed to destroy. Nat Rev Mol Cell Biol. 2006;7:644–656. doi: 10.1038/nrm1988. [DOI] [PubMed] [Google Scholar]
- Pines J. Mitosis: a matter of getting rid of the right protein at the right time. Trends Cell Biol. 2006;16:55–63. doi: 10.1016/j.tcb.2005.11.006. [DOI] [PubMed] [Google Scholar]
- Planchon SM, Waite KA, Eng C. The nuclear affairs of PTEN. J Cell Sci. 2008;121:249–253. doi: 10.1242/jcs.022459. [DOI] [PubMed] [Google Scholar]
- Rosivatz E, Matthews JG, McDonald NQ, Mulet X, Ho KK, Lossi N, Schmid AC, Mirabelli M, Pomeranz KM, Erneux C, et al. A small molecule inhibitor for phosphatase and tensin homologue deleted on chromosome 10 (PTEN) ACS Chem Biol. 2006;1:780–790. doi: 10.1021/cb600352f. [DOI] [PubMed] [Google Scholar]
- Rubin SJ, Hallahan DE, Ashman CR, Brachman DG, Beckett MA, Virudachalam S, Yandell DW, Weichselbaum RR. Two prostate carcinoma cell lines demonstrate abnormalities in tumor suppressor genes. J Surg Oncol. 1991;46:31–36. doi: 10.1002/jso.2930460108. [DOI] [PubMed] [Google Scholar]
- Salmena L, Carracedo A, Pandolfi PP. Tenets of PTEN tumor suppression. Cell. 2008;133:403–414. doi: 10.1016/j.cell.2008.04.013. [DOI] [PubMed] [Google Scholar]
- Song MS, Salmena L, Carracedo A, Egia A, Lo-Coco F, Teruya-Feldstein J, Pandolfi PP. The deubiquitinylation and localization of PTEN are regulated by a HAUSP-PML network. Nature. 2008;455:813–817. doi: 10.1038/nature07290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stambolic V, Suzuki A, de la Pompa JL, Brothers GM, Mirtsos C, Sasaki T, Ruland J, Penninger JM, Siderovski DP, Mak TW. Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell. 1998;95:29–39. doi: 10.1016/s0092-8674(00)81780-8. [DOI] [PubMed] [Google Scholar]
- Steck PA, Pershouse MA, Jasser SA, Yung WK, Lin H, Ligon AH, Langford LA, Baumgard ML, Hattier T, Davis T, et al. Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat Genet. 1997;15:356–362. doi: 10.1038/ng0497-356. [DOI] [PubMed] [Google Scholar]
- Steegmaier M, Hoffmann M, Baum A, Lenart P, Petronczki M, Krssak M, Gurtler U, Garin-Chesa P, Lieb S, Quant J, et al. BI 2536, a potent and selective inhibitor of polo-like kinase 1, inhibits tumor growth in vivo. Curr Biol. 2007;17:316–322. doi: 10.1016/j.cub.2006.12.037. [DOI] [PubMed] [Google Scholar]
- Strebhardt K, Ullrich A. Targeting polo-like kinase 1 for cancer therapy. Nat Rev Cancer. 2006;6:321–330. doi: 10.1038/nrc1841. [DOI] [PubMed] [Google Scholar]
- Sullivan M, Morgan DO. Finishing mitosis, one step at a time. Nat Rev Mol Cell Biol. 2007;8:894–903. doi: 10.1038/nrm2276. [DOI] [PubMed] [Google Scholar]
- Sun H, Lesche R, Li DM, Liliental J, Zhang H, Gao J, Gavrilova N, Mueller B, Liu X, Wu H. PTEN modulates cell cycle progression and cell survival by regulating phosphatidylinositol 3,4,5,-trisphosphate and Akt/protein kinase B signaling pathway. Proc Natl Acad Sci U S A. 1999;96:6199–6204. doi: 10.1073/pnas.96.11.6199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor S, Peters JM. Polo and Aurora kinases: lessons derived from chemical biology. Curr Opin Cell Biol. 2008;20:77–84. doi: 10.1016/j.ceb.2007.11.008. [DOI] [PubMed] [Google Scholar]
- Trotman LC, Wang X, Alimonti A, Chen Z, Teruya-Feldstein J, Yang H, Pavletich NP, Carver BS, Cordon-Cardo C, Erdjument-Bromage H, et al. Ubiquitination regulates PTEN nuclear import and tumor suppression. Cell. 2007;128:141–156. doi: 10.1016/j.cell.2006.11.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang CX, Fisk BC, Wadehra M, Su H, Braun J. Overexpression of murine fizzy-related (fzr) increases natural killer cell-mediated cell death and suppresses tumor growth. Blood. 2000;96:259–263. [PubMed] [Google Scholar]
- Wasch R, Robbins JA, Cross FR. The emerging role of APC/CCdh1 in controlling differentiation, genomic stability and tumor suppression. Oncogene. 2010;29:1–10. doi: 10.1038/onc.2009.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiteman DC, Zhou XP, Cummings MC, Pavey S, Hayward NK, Eng C. Nuclear PTEN expression and clinicopathologic features in a population-based series of primary cutaneous melanoma. Int J Cancer. 2002;99:63–67. doi: 10.1002/ijc.10294. [DOI] [PubMed] [Google Scholar]
- Zhou XP, Loukola A, Salovaara R, Nystrom-Lahti M, Peltomaki P, de la Chapelle A, Aaltonen LA, Eng C. PTEN mutational spectra, expression levels, and subcellular localization in microsatellite stable and unstable colorectal cancers. Am J Pathol. 2002;161:439–447. doi: 10.1016/S0002-9440(10)64200-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.