

Abstract
The binding of EcoR1 to a 90-bp DNA duplex attached to colloidal microparticles and the subsequent cleavage by the enzyme was observed in real time and label-free with time-resolved second harmonic (SH) spectroscopy. This method provides a unique way to investigate biomolecular interactions based on its sensitivity to changes in structure and electrical charge on formation of a complex and subsequent dynamics. The binding of EcoR1 to the recognition sequence in DNA appears as a rapid increase in the SH signal, which is attributed to the enzyme-induced change in the DNA conformation, going from a rod-like to a bent shape. In the presence of the cofactor Mg2+, the subsequent decay in the SH signal was monitored in real time as the following processes occurred: cleavage of DNA, dissociation of the enzyme from the DNA, and diffusion of the 74-bp fragment into the bulk solution leaving the 16-bp fragment attached to the microparticle. The observed decay was dependent on the concentration of Mg2+, which functions as a cofactor and as an electrolyte. With SH spectroscopy the rehybridization dynamics between the rehybridized microparticle bound and free cleaved DNA fragments was observed in real time and label-free following the cleavage of DNA. Collectively, the experiments reported here establish SH spectroscopy as a powerful method to investigate equilibrium and time-dependent biological processes in a noninvasive and label-free way.
Keywords: cleavage kinetics, DNA-endonuclease, rehybridization kinetics, nonspecific binding
The highly selective binding of proteins to specific nucleotide sequences in DNA is an essential step in many biological processes, including gene regulation, gene expression, and DNA hybridization (1). The family of type II restriction enzymes, which includes EcoR1, have been, and continue to be, of major importance in serving as model systems of protein-DNA interactions. The endonucleases, because of their capability to bind and cleave specific DNA sequences, also play a key role in DNA recombinant and cloning methods (1, 2). The EcoR1-DNA equilibria and reaction dynamics have been probed extensively by a number of established methods, such as, but not limited to: FRET (3–5), gel electrophoresis (6–15), surface plasmon resonance (16), microfluidic trapping (17), two-color cross-correlation spectroscopy (18), and isothermal calorimetry (19).
The essential characteristic of the method used here is the application of second harmonic (SH) spectroscopy to selectively probe the interface of a polystyrene carboxylate (PSC) microparticle, to which target DNA molecules have been attached. Significant effort and progress has been made in hybridizing artificial colloidal nano- and micromaterials with naturally occurring biological reagents to build self-assembled superstructured materials (20–26), and to develop biologically coded probes for protein detection and disease treatment (27, 28). The present work builds on these pioneering studies, and on studies of DNA supported on flat surfaces using SH and sum-frequency generation spectroscopies (29–31). Using those spectroscopies, the electronic (29, 31) and vibrational (30, 31) spectra of DNA oligonucleotides at a solid/water interface were obtained. In addition, the kinetics of DNA hybridization and the equilibrium between ion-free and ion-bound DNA have been measured using the SH method (29, 31).
Surface-sensitive nonlinear spectroscopies allow one to probe equilibrium properties and time-dependent changes in both the electrical charge of molecules and changes in the structure of molecules that are located at interfaces (29–34). An attractive feature using DNA bound to the surface of a microparticle, which can react with free EcoR1 molecules in solution, is that the total surface area that can be irradiated and thereby interrogated is much larger than can be achieved using a planar surface. The larger surface area results in more surface-bound molecules, hence a stronger SH signal is generated. In addition, the total number of molecules that are bound to the microparticles can be readily changed without altering the surface density of the bound target molecules. A brief overview of second harmonic generation is given below.
Background: Second Harmonic Spectroscopy
The interaction of intense light at a frequency ω with interfacial chemical species can result in the generation of light at twice the frequency, 2ω, and is known as second harmonic generation. For reasons of symmetry, second-order spectroscopies, such as second harmonic and sum-frequency generation, only produce coherent signals in anisotropic environments such as those found at interfaces (32, 35). The generation of the SH light can be described by a model in which the incident light induces a second-order polarization, 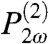 , which is proportional to the square of the incident electromagnetic field, Eω, and radiates an electromagnetic field, E2ω, at 2ω. It is expressed as,
, which is proportional to the square of the incident electromagnetic field, Eω, and radiates an electromagnetic field, E2ω, at 2ω. It is expressed as,
| [1] |
where χ(2) is the second-order susceptibility and contains all of the information regarding the interfacial species that are irradiated; it depends on the chemical identity, the electronic and vibrational states, the energies and transition dipole strengths, the number of each species irradiated, and molecular orientations at the interface, which can be used to probe equilibrium and time-dependent phenomena. The total SH intensity, I2ω(total) is the incoherent sum of the contributions made by the individual particles to the SH intensity,
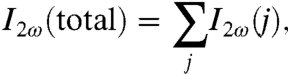 |
[2] |
where I2ω(j) is proportional to the absolute square of the second harmonic electric field generated by the microparticle j, i.e., I2ω(j) ∼ |E2ω(j)|2.
If the interface is charged, as it is in the experiments described here, largely due to the attachment of DNA molecules to the microparticle surface, then there is an additional source of SH light due to the third-order polarization, 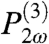 , of the interfacial species, which is a product of three electric fields given by,
, of the interfacial species, which is a product of three electric fields given by,
| [3] |
where Estatic(r) is the electric field, predominantly due to DNA, at a distance, r, from the microparticle that polarizes the surrounding molecules, and χ(3) is the third-order susceptibility. It is because Estatic(r) is a zero frequency electric field that the third-order polarization generates light at 2ω. It is primarily the water molecules that are polarized by the interfacial electric field established by the DNA that is attached to the microparticle surface and includes associated interfacial ions. To include the contributions to the third-order polarization, which include water molecules that are near and far from the microparticle, the static field is integrated over all space to give the interfacial electrostatic potential, Φ. The total SH field is thus given by
| [4] |
which simultaneously describes the interfacial chemistry and the electrostatic properties of the particle and surrounding molecules (33, 34). Because DNA is highly charged at physiological pHs, having a charge of -1 per nucleotide, the result of coupling DNA to a microparticle substantially increases the microparticle surface charge density, which increases the interface static electric field and thereby the contribution of the third-order polarization to the SH signal. Dissociation of cleaved DNA produces a fragment free to diffuse into the bulk solution. The detachment of a 74-bp fragment from the microparticle surface upon reaction with EcoR1 reduces both the number of nucleotides and the charge at the interface, thus resulting in the decay of the SH signal as the reaction proceeds. Changes in the SH signal due to the interactions of DNA with EcoR1 can be directly monitored in situ. It is to be noted that the binding and cleavage was observed without the need for labeled reporter molecules or invasive detection methods. The time-dependent SH field is linearly proportional to the number density of molecules at the particle surface at high electrolyte concentrations because the contribution to the third-order polarization at high electrolyte concentrations can be neglected. At low electrolyte concentration the time-dependent SH signal is more complex, owing to the third-order polarization.
Experimental Details
The experimental apparatus used to measure the time-resolved SH signal is similar to those described in previous reports (34, 36) with the addition of a Princeton Instruments CCD camera. The fundamental laser frequency was centered at 810 nm with approximately 400 mW of average power and a pulse width of approximately 75 fs. There was no evidence of photo damage to the sample as was established in control experiments where the SH signal was observed to be static in time (SI Text).
The PSC-DNA bioconjugate particles were prepared in an analogous way to previous synthetic reports (24–26); a sketch of the resulting particles is given in Fig. 1. Specifically, the biotin binding protein NeutrAvidin (NA) was covalently linked to the PSC surface (diameter = 1.0 μm, Invitrogen) according to the procedure supplied by the manufacturer. The resulting PSC-NA solution had a concentration of 5.6 × 109 particles/mL, which was suspended in water at pH = 7.5. To this solution ds biotinylated-DNA (90 bp, Integrated DNA Technologies) containing the EcoR1 recognition sequence, which is 15 bp from the attachment to the microparticle, was added to a DNA concentration of 0.69 nM and incubated at 55 °C for 1 h. As a control, an analogous DNA sample was prepared in an identical manner with the exception that the control DNA molecule had six base pairs that differed from the EcoR1 recognition sequence. The sequence of the recognition and control DNA is given in the legend of Fig. 1. The final particle concentration used in the reactions is 9.3 × 107 PSC-DNA particles/mL at pH = 7.5. There was no evidence of bead–bead interactions, i.e., no aggregation at this low bead density. The final DNA concentration was 170 pM, leading to a stoichiometric ratio of approximately 1.1 × 103 DNA/particle, which is a surface coverage of approximately 3.5 × 102 DNA/μm2. The Na+ concentration in the sample was kept at 2 mM for all experiments except where noted (i.e., those where additional NaCl is added).
Fig. 1.

A schematic of the experimental sample and reaction studied in the work is given here. The recognition sequence DNA sample is biotin-5′- AGACCGTAATTGCCTGAATTCCTGTTCCCAATCATCGTTGCTCACCAGAATCGCCCAGCAGTTAGGCCGTATGATATGTAAGGTATAACG (the complementary strand is implied). The underlined bold bases highlight the recognition sequence. The control DNA sequence replaces the recognition site with ACTCAT bases.
The restriction enzyme EcoR1 was purchased from New England Biolabs. The concentration of the enzyme used in the reaction was 17 pM. The concentration of MgSO4 was systematically varied. The reaction kinetics were obtained from measurements of the SH signal versus time after EcoR1 was added. Negative times correspond to the SH signal prior to injection of EcoR1; t = 0 is the time that the enzyme is added. The SH signals were integrated for 25 s. The kinetics traces shown are the result of averaging at least two individual experimental runs to improve signal-to-noise and to establish reproducibility.
Results and Discussion
EcoR1 cuts DNA at the recognition sequence between the G and A nucleotides (see legend for Fig. 1) to produce a 16-bp fragment that remains attached to the microparticle surface and a 74-bp fragment free to diffuse into the bulk solution. The reaction kinetics of the restriction enzyme with DNA are strongly dependent on the electrolyte concentration, the size and concentration of the DNA, the concentration of the enzyme, and the temperature at which the reaction is run (1, 4–18).
Representative kinetic traces of the reaction of EcoR1 with the attached DNA at various Mg2+ concentrations are shown in Fig. 2 for times spanning -750 s–4,500 s. A solid red line is plotted to guide the eye and represents the SH signal as a function of time; the average of the negative time SH signal (i.e., prereaction) is represented as a blue dashed line. The prereaction SH signal, was found to be strongly dependent on the Mg2+ concentration, decreasing as the Mg2+ concentration increased. This result is attributed to two factors, both of which reduce the SH signal. One is the binding of Mg2+ to the DNA molecules at the interface, which reduces the interfacial charge and thus the interfacial potential, which correspondingly decreases the SH signal. Second, the free Mg2+ in solution acts as an electrolyte that screens the electric field sensed by the bulk water molecules, which lowers the SH signal. Similar effects of counter ions on the SH signals from charged interfaces have been reported in previous experiments (29–34).
Fig. 2.

Time-resolved SH signals obtained from the reaction of the DNA-coated microparticles with the restriction enzyme EcoR1 at various Mg2+ concentrations indicated in each figure. The solid red line is intended to guide the eye as the reaction of DNA with EcoR1 proceeds. Dashed blue lines represent the prereaction SH signal level. The decay times, τ, are shown at the various Mg2+ concentrations.
Binding of EcoR1 to DNA—Initial Jump in the SH Intensity.
When EcoR1 was added to the reaction cell an abrupt jump in the SH signal was observed, followed by a slow decay over hundreds of seconds. To determine if the jump was due to the binding of EcoR1 to the DNA molecules and not to the surface of the microparticle, a control DNA was used. The control DNA is identical to the DNA that has the recognition sequence, except that the 6-bp EcoR1 recognition sequence has been altered. The control experiment did not show any jump in the SH signal when EcoR1 was added, nor did the amplitude of the SH signal change with time (SI Text). This observation indicates that EcoR1 does not induce a structural change to DNA, nor does it cleave DNA molecules that do not have the recognition sequence.
The jump in the SH signal is attributed to an EcoR1-induced conformational change in the DNA structure upon binding of the enzyme to the recognition sequence, which is known to induce a transition from a rod-like shape to a bent conformational state. The bending and unwinding of DNA, by approximately 25° (37, 38), results in changes of the orientations of the DNA bases relative to one another, which therefore changes the overall absorption strength of the interfacially bound DNA. It is the relative orientations of the bases that determine whether the transition-dipole–transition-dipole interbase interactions result in an increase or decrease in the absorption strength of DNA. It is known that dsDNA has a smaller absorption cross section, i.e., it is hypochromic, as compared to the absorption cross section of the individual nucleotides or ssDNA (39). The bending and unwinding of DNA can decrease the parallel stacking of the transition moments and lead to an increased collinear component of the transition-dipole moments that results in a stronger optical absorption (39, 40). In a similar way, the cancellation of the light induced nonlinear dipole moments of the individual bases can be reduced by conformational changes of DNA, which would increase the SH signal and is in accord with the SH measurements.
The third-order contribution to the SH must also be considered because upon bending, the electric field associated with the rod-like conformational state of DNA will change. The average static field from the DNA before EcoR1 is added is essentially symmetric about the DNA helical axis and thus the induced polarization of the unbound water molecules on opposite sides of the helical DNA axis would largely cancel. These molecules would therefore not contribute to the SH signal. However, at the tip of the DNA duplex there would be polarization of the water molecules extending into the bulk medium. Upon bending of DNA, the average static electric field is no longer radially symmetric with respect to the DNA helical axis; as a consequence there would be less cancellation of polarized water molecules, which would enhance the third-order polarization and thus result in an increase of the SH signal.
In addition to the effects of specific binding of EcoR1 on DNA conformational states it is noted that information on nonspecific binding is also provided by the SH control experiments. The SH measurements are consistent with the generally held view that the nonspecific binding of EcoR1 to DNA does not result in a distortion to the DNA conformation (1, 2). Combining these facts with the control experimental result supports our attribution that the jump in the SH signal is due to the bending and unwinding of DNA induced by specific binding of EcoR1. Thus, neither the nonspecific binding of EcoR1 to locations other than the recognition site, which is known to occur (1, 2, 9, 13, 15), nor the subsequent lateral diffusion of the enzyme along the DNA strands to reach the recognition sequence, induce a conformational change to DNA that was observed in the SH experiments.
It is seen in Fig. 2 that the amplitude of the jump in the SH signal increased as the concentration of Mg2+ increased. Because the SH jump occurs before a significant portion of DNA is cleaved, the Mg2+ acts at early times as an electrolyte that screens the repulsive interactions between the neighboring phosphate groups (3, 41–43). It thereby facilitates the binding of EcoR1 to DNA and allows for a more facile bending of DNA at the reaction site. Consequently, a better fit of the DNA into the active site of the enzyme, as compared to reactions where little or no electrolyte is present, is achieved.
To test the hypothesis as to whether Mg2+ acts as an electrolyte with respect to the appearance of the SH jump, a separate experiment was performed where NaCl was used in place of Mg2+. Because Mg2+ is needed as a cofactor for the cleavage of DNA by EcoR1 it is expected that replacing Mg2+ with Na+ should also result in a sharp jump that we associate with EcoR1 binding to DNA; however, the SH signal should not decay because cleavage cannot occur when Mg2+ is absent. The result of this experiment is shown in Fig. 3, which illustrates that an electrolyte is needed to produce an SH jump and that the amplitude of the jump depends strongly on the electrolyte concentration. Specifically, at an NaCl concentration of 8.50 mM the SH signal jumps by 30% when EcoR1 is added, as compared to a jump of 9.5%, at 2 mM NaCl. These results are consistent with recent experimental and theoretical investigations of the electrostatic interactions between phosphate groups on the DNA backbone and the effect of these interactions on the binding and DNA-bending dynamics of a different restriction endonuclease, namely EcoRV (3). Additionally, the present results and interpretation are consistent with experiments that measure DNA flexibility as a function of electrolyte concentration (41).
Fig. 3.

The signal measured when EcoR1 is added to a PSC-DNA solution containing 8.50 mM and 2.0 mM NaCl.
Time-Resolved Cleavage of DNA by EcoR1.
In the presence of Mg2+ the observed decay in the SH signal is attributed to the following processes: cleavage of the DNA, dissociation of EcoR1 from DNA, and the diffusion of the 74-bp fragment into the bulk (1, 2). In the absence of Mg2+ the SH signal does not decay over the span of several thousand seconds (Fig. 2 and Fig. 3). Control experiments successfully indicated that the recognition sequence is necessary for EcoR1 to cleave DNA. Inspection of Fig. 2 shows that the reaction rates are dependent on the concentration of Mg2+ cofactor, as expected, and range from several hundred to thousands of seconds, in agreement with a range of reported timescales for similar reaction conditions (6, 10, 12, 14, 17).
Fig. 2 shows that at low Mg2+ concentrations the SH signal measured at long times dip below the prereaction level. This result is due to the reduced size of the DNA fragment bound to the interface, which is 16 bp after the cleavage occurs, versus 90 bp before the reaction started. After cleavage, the 74-bp fragment is free in solution and is randomly oriented, and thus does not contribute to the SH signal originating from the microparticles. In contrast to the aforementioned experiments, it is seen in Fig. 2 that at higher Mg2+ concentration the SH level reaches the intensity that was observed before the reaction was initiated, within experimental uncertainty. In other words the SH signal is essentially the same after the cleavage reaction finishes as it was before any of the DNA was cut. One might not expect the SH signal to return to the same level after the reaction has been completed because a 74-bp fragment has been cleaved from the DNA attached to the microparticle. This surprising feature is addressed in the section below.
DNA Rehybridization Equilibrium and Dynamics.
The behavior of the SH signal at long times can be understood by noting that EcoR1 does not cut DNA into fragments with blunt ends (i.e., no overhanging bases), but rather cuts the DNA into fragments that can rehybridize via H-bonding and stacking interactions (44–47); such DNA fragments are known as sticky-end pairs. The bonds in the phosphate backbone that are cleaved by EcoR1 are not reestablished, but rather base pair complementarity of the sticky ends can reestablish base pairing, which lowers the free energy of the system (44–47). To explain these seemingly anomalous results we propose that it is the equilibrium between the rehybridized DNA, 90∗, and the DNA fragments, namely the attached 16 bp and the free 74 bp,
The equilibrium concentrations of the rehybridized 90∗ bp, the free 74-bp fragment and the bound 16-bp fragment are strongly dependent on electrolyte concentration because the electrolyte screens the charged fragments, which reduces the repulsive electrostatic interactions between fragments. The increased screening enables the charged DNA strands to get close enough to form the H-bonding and stacking interactions that are necessary for rehybridization (44–47). The higher electrolyte concentrations favor rehybridization whereas the lower concentrations favor separation of the two cleaved fragments. Thus, the post reaction concentration of free (74 bp) versus reattached (90∗) fragments should not be thought of as a reaction end point, but rather as an equilibrium that is dependent on the electrolyte concentration.
If this description is correct then perturbing the equilibrium by adding electrolyte after the reaction has completed, should result in a time-dependent return to a new equilibrium. To test this idea, NaCl was injected into the reaction cell containing a low concentration of Mg2+, specifically 0.2 mM Mg2+, after the reaction was completed at approximately 9,000 s. The effect of adding NaCl to a final concentration of 9.3 mM resulted in an immediate decrease of the SH signal that was followed by a slow recovery, Fig. 4. The signal initially drops due to the increased electrolyte concentration, i.e., greater screening. The SH signal then recovers in time to a final SH level that is greater than the previously observed end point; specifically, it is larger by 3.4 ± 1.6%. The reason for this increase in the SH signal is that the addition of electrolytes results in a shift of the equilibrium between the separated and rehybridized fragments, which favors the rehybridized form. The time it takes to reestablish the new equilibrium condition is roughly 1,200 s. To the best of our knowledge, there have been no previous reports of real-time label-free observation of rehybridization dynamics following DNA cleavage.
Fig. 4.

The kinetic trace measured on addition of NaCl after the reaction was completed. The solid red line represents the SH jump and decay, the dashed blue line is the prereaction SH signal level, and the solid green trace tracks the drop and recovery of the SH signal.
In a second experiment performed in the presence of 6.1 mM Mg2+, the addition of Na+ did not produce an observable immediate drop nor a slow recovery in the SH signal (SI Text), because the third-order polarization contribution to the SH signal is negligible at high electrolyte concentrations. In addition, at this high electrolyte concentration the equilibrium strongly favors the rehybridized form.
Conclusions and Outlook
Time-resolved second harmonic generation spectroscopy was used to observe the binding of the restriction enzyme EcoR1 to its DNA recognition sequence, followed by cleaving the DNA into a large and small fragment, and also observed the subsequent rehybridization dynamics. A sharp increase in the SH intensity was observed upon addition of EcoR1 to the reaction vessel, which is attributed to conformational changes in the DNA that are induced by the binding of EcoR1. Specifically, the DNA is known to bend and unwind by approximately 25° in the EcoR1-DNA complex (37, 38) relative to its more rod-like conformation in the absence of EcoR1. It was found that the amplitude of the jump in the SH signal is dependent on the presence of electrolytes, which can facilitate the change in the DNA structure by shielding adjacent charges on the DNA backbone when the DNA is in the bent conformational state. The change in the structure of the DNA that contains the EcoR1 recognition sequence results in a change in the second-order polarization and simultaneously alters the static electric field, which consequently changes the SH signal. In contrast, the SH control experiments indicate the nonspecific binding of EcoR1 to DNA does not induce conformational changes to DNA. In experiments where Mg2+ was present with recognition DNA, it was observed that Mg2+ acts as both an electrolyte, facilitating the bending of DNA and associated binding of EcoR1, and also as a cofactor that effects the cleavage reaction rates. At lower electrolyte concentrations the cleavage of DNA with EcoR1 produced a free 74-bp DNA fragment and a 16-pb fragment that remains attached to the microparticle, which was seen in experiments where the post reaction SH signal was found to be smaller than its prereaction value. This result was expected because the quantity of DNA that remained bound had been reduced. However at higher electrolyte concentrations the SH signal at long times, within experimental uncertainty, was the same as before the binding and reaction. The explanation of this finding is that there is an equilibrium between the separated DNA fragments and the rehybridized DNA fragments. At higher electrolyte concentrations there is sufficient screening to reduce electrostatic repulsions between the fragments, which shifts the equilibrium population to the rehybridized form of DNA. To test this idea, the equilibrium was perturbed by adding NaCl after the reaction with EcoR1 was completed. A fast decrease followed by a gradual increase in the SH signal that reached an SH signal level that was larger than before adding the NaCl was observed. Following DNA cleavage, the DNA rehybridization was measured in real time and label-free.
In summary, the work reported here demonstrates the utility of time-resolved SH spectroscopy, together with biomolecule functionalized microparticles, to probe time-dependent and equilibrium biological processes noninvasively, without labels, and with high sensitivity.
Supplementary Material
Acknowledgments.
The authors acknowledge many useful and stimulating conversations with Professor Ruben R. Gonzalez, Jr. Also, discussions with Dr. Louis H. Haber, Dr. Yi Rao, Dr. Jerry Dadap, and Sheldon J. J. Kwok are appreciated. Support was generously provided by the National Science Foundation Eager Award CHE-1041980.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1115498108/-/DCSupplemental.
References
- 1.Pingoud A, Jeltsch A. Structure and function of type II restriction endonucleases. Nucleic Acids Res. 2001;29:3705–3727. doi: 10.1093/nar/29.18.3705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jen-Jacobson L. Protein-DNA recognition complexes: Conservation of structure and binding energy in the transition state. Biopolymers. 1997;44:153–180. doi: 10.1002/(SICI)1097-0282(1997)44:2<153::AID-BIP4>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 3.Hancock SP, Hiller DA, Perona JJ, Jen-Jacobson L. The energetic contribution of induced electrostatic asymmetry to DNA bending by a site-specific protein. J Mol Biol. 2011;406:285–312. doi: 10.1016/j.jmb.2010.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li XM, Song C, Zhao MP, Li YZ. Continuous monitoring of restriction endonuclease cleavage activity by universal molecular beacon light quenching coupled with real-time polymerase chain reaction. Anal Biochem. 2008;381:1–7. doi: 10.1016/j.ab.2008.06.027. [DOI] [PubMed] [Google Scholar]
- 5.Urata H, Tamaki C, Matsuno M, Wada S, Akagi M. FRET-based kinetic analysis of highly reactive heterochiral DNA toward EcoRI endonuclease. Biochem Biophys Res Commun. 2009;390:192–195. doi: 10.1016/j.bbrc.2009.08.164. [DOI] [PubMed] [Google Scholar]
- 6.Halford SE, Johnson NP. The EcoRI restriction endonuclease with bacteriophage lambda-DNA—equilibrium-studies. Biochem J. 1980;191:593–604. doi: 10.1042/bj1910593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Halford SE, Johnson NP, Grinsted J. Reactions of the EcoRI and other restriction endonucleases. Biochem J. 1979;179:353–365. doi: 10.1042/bj1790353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Halford SE, Johnson NP, Grinsted J. The EcoRI restriction endonuclease with bacteriophage lambda-DNA—kinetic-studies. Biochem J. 1980;191:581–592. doi: 10.1042/bj1910581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jack WE, Terry BJ, Modrich P. Involvement of outside DNA-sequences in the major kinetic path by which EcoR1 endonuclease located and leaves its recognition sequence. Proc Natl Acad Sci USA. 1982;79:4010–4014. doi: 10.1073/pnas.79.13.4010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jeltsch A, Alves J, Wolfes H, Maass G, Pingoud A. Substrate-assisted catalysis in the cleavage of DNA by the EcoRI and EcoRV restriction enzymes. Proc Natl Acad Sci USA. 1993;90:8499–8503. doi: 10.1073/pnas.90.18.8499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lesser DR, Kurpiewski MR, Jen-Jacobson L. The energetic basis of specificity in the EcoRI endonuclease DNA interaction. Science. 1990;250:776–786. doi: 10.1126/science.2237428. [DOI] [PubMed] [Google Scholar]
- 12.Pingoud V, et al. On the divalent metal ion dependence of DNA cleavage by restriction endonucleases of the EcoRI family. J Mol Biol. 2009;393:140–160. doi: 10.1016/j.jmb.2009.08.011. [DOI] [PubMed] [Google Scholar]
- 13.Rau DC, Sidorova NY. Diffusion of the restriction nuclease EcoRI along DNA. J Mol Biol. 2010;395:408–416. doi: 10.1016/j.jmb.2009.10.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vipond IB, Baldwin GS, Halford SE. Divalent metal-ions at the active-sites of the EcoRV and EcoRI restriction endonucleases. Biochemistry. 1995;34:697–704. doi: 10.1021/bi00002a037. [DOI] [PubMed] [Google Scholar]
- 15.Wright DJ, Jack WE, Modrich P. The kinetic mechanism of EcoRI endonuclease. J Biol Chem. 1999;274:31896–31902. doi: 10.1074/jbc.274.45.31896. [DOI] [PubMed] [Google Scholar]
- 16.Bier FF, Kleinjung F, Schmidt PM, Scheller FW. Determination of the turnover number of the restriction endonuclease EcoRI using evanescent wave technology. Anal Bioanal Chem. 2002;372:308–313. doi: 10.1007/s00216-001-1216-4. [DOI] [PubMed] [Google Scholar]
- 17.Xu WL, Muller SJ. Exploring both sequence detection and restriction endonuclease cleavage kinetics by recognition site via single-molecule microfluidic trapping. Lab Chip. 2011;11:435–442. doi: 10.1039/c0lc00176g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kettling U, Koltermann A, Schwille P, Eigen M. Real-time enzyme kinetics monitored by dual-color fluorescence cross-correlation spectroscopy. Proc Natl Acad Sci USA. 1998;95:1416–1420. doi: 10.1073/pnas.95.4.1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jen-Jacobson L, Engler LE, Ames JT, Kurpiewski MR, Grigorescu A. Thermodynamic parameters of specific and nonspecific protein-DNA binding. Supramol Chem. 2000;12:143–160. [Google Scholar]
- 20.Alivisatos AP, et al. Organization of ‘nanocrystal molecules’ using DNA. Nature. 1996;382:609–611. doi: 10.1038/382609a0. [DOI] [PubMed] [Google Scholar]
- 21.Dillenback LM, Goodrich GP, Keating CD. Temperature-programmed assembly of DNA: Au nanoparticle bioconjugates. Nano Lett. 2006;6:16–23. doi: 10.1021/nl0508873. [DOI] [PubMed] [Google Scholar]
- 22.Soto CM, Srinivasan A, Ratna BR. Controlled assembly of mesoscale structures using DNA as molecular bridges. J Am Chem Soc. 2002;124:8508–8509. doi: 10.1021/ja017653f. [DOI] [PubMed] [Google Scholar]
- 23.Dreyfus R, et al. Simple quantitative model for the reversible association of DNA coated colloids. Phys Rev Lett. 2009;102:048301. doi: 10.1103/PhysRevLett.102.048301. [DOI] [PubMed] [Google Scholar]
- 24.Leunissen ME, et al. Switchable self-protected attractions in DNA-functionalized colloids. Nat Mater. 2009;8:590–595. doi: 10.1038/nmat2471. [DOI] [PubMed] [Google Scholar]
- 25.Leunissen ME, Dreyfus R, Sha R, Seeman NC, Chaikin PM. Quantitative study of the association thermodynamics and kinetics of DNA-coated particles for different functionalization schemes. J Am Chem Soc. 2010;132:1903–1913. doi: 10.1021/ja907919j. [DOI] [PubMed] [Google Scholar]
- 26.Valignat MP, Theodoly O, Crocker JC, Russel WB, Chaikin PM. Reversible self-assembly and directed assembly of DNA-linked micrometer-sized colloids. Proc Natl Acad Sci USA. 2005;102:4225–4229. doi: 10.1073/pnas.0500507102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hong R, et al. Glutathione-mediated delivery and release using monolayer protected nanoparticle carriers. J Am Chem Soc. 2006;128:1078–1079. doi: 10.1021/ja056726i. [DOI] [PubMed] [Google Scholar]
- 28.Zheng D, Seferos DS, Giljohann DA, Patel PC, Mirkin CA. Aptamer nano-flares for molecular detection in living cells. Nano Lett. 2009;9:3258–3261. doi: 10.1021/nl901517b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Boman FC, et al. DNA at aqueous/solid interfaces: Chirality-based detection via second harmonic generation activity. J Am Chem Soc. 2009;131:844–848. doi: 10.1021/ja808007b. [DOI] [PubMed] [Google Scholar]
- 30.Holland JG, Malin JN, Jordan DS, Geiger FM. Specific and nonspecific metal ion-nucleotide interactions at aqueous/solid interfaces functionalized with adenine, thymine, guanine, and cytosine oligomers. J Am Chem Soc. 2011;133:2567–2570. doi: 10.1021/ja107883x. [DOI] [PubMed] [Google Scholar]
- 31.Walter SR, Geiger FM. DNA on stage: Showcasing oligonucleotides at surfaces and interfaces with second harmonic and vibrational sum frequency generation. J Phys Chem Lett. 2010;1:9–15. [Google Scholar]
- 32.Eisenthal KB. Second harmonic spectroscopy of aqueous nano- and microparticle interfaces. Chem Rev. 2006;106:1462–1477. doi: 10.1021/cr0403685. [DOI] [PubMed] [Google Scholar]
- 33.Ong SW, Zhao XL, Eisenthal KB. Polarization of water molecules at a charged interface—2nd harmonic studies of the silica water interface. Chem Phys Lett. 1992;191:327–335. [Google Scholar]
- 34.Yan ECY, Liu Y, Eisenthal KB. New method for determination of surface potential of microscopic particles by second harmonic generation. J Phys Chem B. 1998;102:6331–6336. [Google Scholar]
- 35.Shen YR. The Principles of Nonlinear Optics. New York: Wiley Interscience; 2003. [Google Scholar]
- 36.Haber LH, Kwok SJJ, Semeraro M, Eisenthal KB. Probing the colloidal gold nanoparticle/aqueous interface with second harmonic generation. Chem Phys Lett. 2011;507:11–14. [Google Scholar]
- 37.Kim YC, Grable JC, Love R, Greene PJ, Rosenberg JM. Refinement of Eco RI endonuclease crystal-structure—a revised protein chain tracing. Science. 1990;249:1307–1309. doi: 10.1126/science.2399465. [DOI] [PubMed] [Google Scholar]
- 38.Frederick CA, et al. Kinked DNA in crystalline complex with EcoRI endonuclease. Nature. 1984;309:327–331. doi: 10.1038/309327a0. [DOI] [PubMed] [Google Scholar]
- 39.Tinoco I. Hypochromism in polynucleotides. J Am Chem Soc. 1960;82:4785–4790. [Google Scholar]
- 40.Waters TR, Connolly BA. Continuous spectrophotometric assay for restriction endonucleases using synthetic oligodeoxynucleotides and based on the hyperchromic effect. Anal Biochem. 1992;204:204–209. doi: 10.1016/0003-2697(92)90162-z. [DOI] [PubMed] [Google Scholar]
- 41.Baumann CG, Smith SB, Bloomfield VA, Bustamante C. Ionic effects on the elasticity of single DNA molecules. Proc Natl Acad Sci USA. 1997;94:6185–6190. doi: 10.1073/pnas.94.12.6185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mirzabekov AD, Rich A. Asymmetric lateral distribution of unshielded phosphate groups in nucleosomal DNA and its role in DNA bending. Proc Natl Acad Sci USA. 1979;76:1118–1121. doi: 10.1073/pnas.76.3.1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Range K, Mayaan E, Maher LJ, York DM. The contribution of phosphate-phosphate repulsions to the free energy of DNA bending. Nucleic Acids Res. 2005;33:1257–1268. doi: 10.1093/nar/gki272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gidwani V, Riahi R, Zhang DD, Wong PK. Hybridization kinetics of double-stranded DNA probes for rapid molecular analysis. Analyst. 2009;134:1675–1681. doi: 10.1039/b906077d. [DOI] [PubMed] [Google Scholar]
- 45.Li JWJ, Tan WH. A real-time assay for DNA sticky-end pairing using molecular beacons. Anal Biochem. 2003;312:251–254. doi: 10.1016/s0003-2697(02)00375-5. [DOI] [PubMed] [Google Scholar]
- 46.Protozanova E, Yakovchuk P, Frank-Kamenetskii MD. Stacked-unstacked equilibrium at the nick site of DNA. J Mol Biol. 2004;342:775–785. doi: 10.1016/j.jmb.2004.07.075. [DOI] [PubMed] [Google Scholar]
- 47.Sponer J, Riley KE, Hobza P. Nature and magnitude of aromatic stacking of nucleic acid bases. Phys Chem Chem Phys. 2008;10:2595–2610. doi: 10.1039/b719370j. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.