

Abstract
Telomere repeats in budding yeast are maintained at a constant average length and protected (‘capped’), in part, by mechanisms involving the TG1−3 repeat-binding protein Rap1. However, metazoan telomere repeats (T2AG3) can be maintained in yeast through a Rap1-independent mechanism. Here, we examine the dynamics of capping and telomere formation at an induced DNA double-strand break flanked by varying lengths of T2AG3 repeats. We show that a 60-bp T2AG3 repeat array induces a transient G2/M checkpoint arrest, but is rapidly elongated by telomerase to generate a stable T2AG3/TG1–3 hybrid telomere. In contrast, a 230-bp T2AG3 array induces neither G2/M arrest nor telomerase elongation. This capped state requires the T2AG3-binding protein Tbf1, but is independent of two Tbf1-interacting factors, Vid22 and Ygr071c. Arrays of binding sites for three other subtelomeric or Myb/SANT domain-containing proteins fail to display a similar end-protection effect, indicating that Tbf1 capping is an evolved function. Unexpectedly, we observed strong telomerase association with non-telomeric ends, whose elongation is blocked by a Mec1-dependent mechanism, apparently acting at the level of Cdc13 binding.
Keywords: DNA damage checkpoint, Tbf1, telomerase, telomere capping, yeast
Introduction
Telomeres, the protein–DNA complexes that constitute the ends of linear eukaryotic chromosomes, ensure the complete DNA replication of chromosome ends and protect these ends from recombination, degradation, and DNA damage checkpoint activation, the so-called ‘capping’ function (reviewed recently in Palm and de Lange, 2008; Lydall, 2009; de Lange, 2009; Wellinger, 2010). The molecular basis for these two essential telomere functions can be found in the telomere DNA sequences themselves, which are comprised of simple DNA repeats, T2AG3 in all vertebrates and most higher eukaryotes, and TG1−3 in the well-studied budding yeast Saccharomyces cerevisiae. These repeat sequences generate binding sites for factors (TRF1 and TRF2 in higher eukaryotes and Rap1 in the budding yeast) that act as platforms for the assembly of a complex set of proteins (referred to as the ‘shelterin’ complex in metazoans) that carry out both telomerase recruitment/activation and capping functions at chromosome ends. The extent to which the duplex DNA telomere-repeat binding proteins play a direct role in these processes is still poorly understood.
In Saccharomyces cerevisiae, at least two different protein complexes have been implicated in telomere protection. The first of these capping complexes to be characterized was the Cdc13, Stn1, Ten1 (CST) complex, a putative structural homologue of the more ubiquitous RPA hetero-trimer (Gao et al, 2007), which binds to the GT-rich single-stranded overhang at telomeres (Lin and Zakian, 1996; Nugent et al, 1996). Inactivation of any one of the three CST components causes extensive and specific telomere DNA degradation, primarily of the 5′-end strand, and a checkpoint-dependent G2/M cell-cycle arrest (Garvik et al, 1995; Grandin et al, 1997, 2001). Interestingly, loss of CST function only leads to telomere damage following DNA replication in cells with high CDK activity (Vodenicharov and Wellinger, 2006). The CST complex may act by specifically blocking telomere association of the Mec1 (homologue of mammalian ATR) kinase, a key transducer in the DNA damage checkpoint pathway (Hirano and Sugimoto, 2007). A second telomere-capping mechanism involves the Rap1 protein and two Rap1-interacting factors, Rif1 and Rif2 (Hardy et al, 1992; Wotton and Shore, 1997; Negrini et al, 2007; Marcand et al, 2008; Hirano et al, 2009), whose target may be the Tel1 (ATM) kinase, through direct binding of Rif2 to the Xrs2 component of the DNA end-binding MRX complex (Hirano et al, 2009). MRX, consisting of Mre11, Rad50, and Xrs2 (NBS1 in mammals), is a highly conserved complex that binds rapidly to accidental DNA double-strand breaks (DSBs) and plays key roles in both repair and checkpoint pathways. Experiments examining the effect of telomeric sequences adjacent to a DSB suggest in addition that Rap1 possesses capping functions independent of the two Rif proteins, as well as the CST complex (Negrini et al, 2007). Finally, the yeast Ku heterodimer, a conserved, ubiquitous DNA end-binding protein, plays an important role in telomere capping outside of S phase by blocking the initiation of DNA resection (Bonetti et al, 2010; Vodenicharov et al, 2010).
Both CST and Rap1–Rif protein complexes also regulate telomerase action at telomeres (reviewed in Bianchi and Shore, 2008 and Shore and Bianchi, 2009). Several lines of genetic and biochemical evidence point to a critical interaction between Cdc13 and the essential telomerase holoenzyme subunit Est1 in the recruitment and/or activation of telomerase at chromosome ends (Pennock et al, 2001; Taggart et al, 2002; Bianchi et al, 2004; Chan et al, 2008). Telomerase action at individual telomeres is regulated by a mechanism that senses the length of the TG-repeat tract such that short telomeres have a higher probability of being elongated by telomerase in a given cell cycle than do longer ones (Teixeira et al, 2004). Studies employing chromatin immunoprecipitation (ChIP) to examine protein association at individual telomeres in vivo as a function of TG-tract length suggest that the association of telomerase holoenzyme with ends is regulated by telomere length and that this effect is driven by increased binding of Tel1 kinase (Bianchi and Shore, 2007; Sabourin et al, 2007). A recent study (Gao et al, 2010) suggests, contrary to expectation, that Tel1 does not modulate the Cdc13–Est1 interaction, and its target or targets responsible for TG-tract length-dependent recruitment and/or activation of telomerase remain to be determined. Nevertheless, in vitro biochemical studies point to a role for the Cdc13–Est1 interaction in activation of a telomere-bound enzyme (DeZwaan and Freeman, 2009). Interestingly, the Rap1-bound Rif1 and Rif2 proteins are implicated in a TG-tract length-dependent mechanism that regulates MRX complex binding at DNA ends, and through this the recruitment of Tel1, which requires an interaction with the Xrs2 component of MRX (Negrini et al, 2007; Hirano et al, 2009; McGee et al, 2010). These findings suggest that the Rap1–Rif complex may employ related mechanisms to control both telomere end protection and telomerase recruitment or activation.
Previous studies have indicated that T2AG3 repeats can participate in telomerase regulation when present adjacent to native TG1−3 tracts in budding yeast. Such chimeric telomeres have been generated either by de novo telomere formation with seed sequences consisting of T2AG3 repeats (Alexander and Zakian, 2003; Brevet et al, 2003), or by replacement of the endogenous TLC1 telomerase template RNA with a mutated allele that directs the synthesis of T2AG3 repeats onto the native TG1−3 ends (Henning et al, 1998; Alexander and Zakian, 2003; Brevet et al, 2003). Strains with the altered telomerase RNA, so-called ‘humanized telomerase’ strains, have also been used to generate novel telomeres that contain only T2AG3 repeats (Alexander and Zakian, 2003). TG-tract length regulation by T2AG3-repeat sequences has been shown to involve an endogenous and essential yeast protein, Tbf1 (Brigati et al, 1993), which binds to T2AG3 repeats through an SANT/Myb type DNA-binding domain that is related to that of both TRF1 and TRF2 (Bilaud et al, 1996). Curiously, telomere length regulation by Tbf1 is partially inhibited by Tel1 (Berthiau et al, 2006) and independent of either Rap1 or the Rif proteins (Alexander and Zakian, 2003). The mechanism(s) by which Tbf1 regulates telomere length is unknown. Furthermore, it is unclear how, or even the extent to which Tbf1 can cap telomeres in yeast. Interestingly, strains carrying a humanized telomerase template RNA appear to be in a chronic state of checkpoint activation, raising the question of whether Tbf1 possesses a capping function (di Domenico et al, 2009).
Here, we exploit a simplified system, involving de novo telomere formation at an induced DNA DSB, to explore both the capping and telomerase regulatory functions of Tbf1 in yeast. Our data provide strong evidence that Tbf1 can efficiently block checkpoint activation at a DSB flanked by sufficiently long T2AG3-repeat arrays, through a mechanism strikingly similar to that of Rap1. In contrast, we show that shorter T2AG3-repeat arrays, though efficiently elongated by telomerase, more closely resemble an uncapped DSB. Our data also reveal a remarkably robust association of telomerase enzyme at non-telomeric DSBs and thus help to define situations in which telomerase activity is regulated at a step or steps following recruitment. Finally, this work strongly supports the idea (Berthiau et al, 2006; Arneric and Lingner, 2007) that Tbf1 may play a key ‘backup’ role in promoting the healing of telomeres that have experienced a catastrophic loss of terminal TG1−3 repeats.
Results
Telomerase elongation of Tbf1 site arrays (T2AG3 repeats) is length dependent
In order to examine the dynamics of telomere formation at arrays of vertebrate-like (T2AG3) telomere repeats, we took advantage of a de novo telomere formation assay first described by Gottschling and colleagues that employs a galactose-inducible HO endonuclease gene (Diede and Gottschling, 1999). T2AG3 repeat telomere ‘seed’ sequences of either 60 or 230 bp in length were placed on the centromere-proximal side of an HO recognition site at cassettes placed near the left end of Chr. VII and the right end of Chr. V, respectively (Figure 1A). Galactose induction of HO in these strains triggers the production of a DSB with T2AG3 telomere repeat tracts at one end. We observed on a Southern blot that the short 60-bp T2AG3 tract is rapidly elongated following HO induction while the 230-bp tract, which is very similar in length to T2AG3-only telomeres generated in a humanized telomerase yeast strain (210–240 bp; Alexander and Zakian, 2003), is maintained at a constant length (Figure 1B). Because we did not modify the TLC1-encoded telomerase template RNA, telomerase adds TG1−3 repeats onto the T2AG3 ends, whose subsequent conversion to duplex DNA can be monitored by ChIP of Rap1 (Supplementary Figure S1), which has been shown previously to be a sensitive proxy for telomerase-mediated elongation at a DSB (Hirano et al, 2009; Zhou et al, 2011). Significantly, Tbf1–Myc binding remains remarkably constant following the induction of HO, even in cultures grown to saturation overnight, suggesting that there is little or no degradation of the T2AG3 tracts (Figure 1C; see below).
Figure 1.

Regulated elongation of short T2AG3 seed sequences by yeast telomerase is Tbf1 dependent. (A) Schematic representation of the modified subtelomeric regions of Chr. VII-L and Chr. V-R. (B) Southern blots monitoring HO cleavage and elongation of the indicated T2AG3 tract ends in a wild-type (TBF1) strain. An internal loading control (‘INT’), a fragment arising before HO cutting (‘U’), and a fragment derived from ‘U’ following HO digestion (‘C’) are marked. (C) Analysis by ChIP of the binding of Tbf1–myc in wild-type strains after HO induction. Results are reported as average fold enrichment (bar) and standard deviation (lines) relative to an internal control sequence within the PDI1 gene on Chr. III (see Materials and methods for details). (D) Southern blots monitoring HO cleavage and elongation of the indicated T2AG3 tract ends in a tbf1-Δi mutant strain.
To test the role of Tbf1 in telomere healing at the T2AG3 ends, we repeated the experiment described above in strains carrying the tbf1-Δi allele, which lacks an internal region (amino acids 327–403) immediately upstream of the C-terminal SANT/Myb-like DNA-binding domain (Berthiau et al, 2006). The tbf1-Δi protein binds DNA but is defective in length regulation of T2AG3-containing telomeres and apparently unable to support viability in humanized telomerase yeast strains (Berthiau et al, 2006). In tbf1-Δi cells, we observed elongation of both the short and the long T2AG3 tracts (Figure 1D), indicating that Tbf1 is indeed required for the regulation of TG1−3-repeat addition observed in wild-type cells. We confirmed by qPCR ChIP that the mutant tbf1-Δi protein is still able to bind T2AG3 tracts in vivo (Supplementary Figure S1). These results show that short T2AG3 arrays are recognized as telomeres and elongated by the yeast telomerase in a manner similar to that of short TG1−3 seeds. Likewise, a long T2AG3 tract, similar to a long TG1−3 array (Negrini et al, 2007), is maintained at a constant length, in this case due to a regulatory function provided, at least in part, by the binding of Tbf1.
Short T2AG3 arrays induce a transient G2/M arrest, but long arrays are capped
To determine if T2AG3 sequences are able to ‘cap’ the DSB, and thus prevent activation of a DNA damage checkpoint response, we used an assay developed by Weinert and colleagues (Michelson et al, 2005) to examine directly cell-cycle progression at the single-cell level. Since the stability and checkpoint status of the DNA end distal to the elongating telomeric end in the HO cut experiments is still controversial (Hirano and Sugimoto, 2007), and in any event would be expected to induce a checkpoint response immediately following induction of the break, we modified the set-up described in Figure 1 by placing identical T2AG3 tracts on the distal side of the HO site, oriented in the opposite direction (Figure 2A, right panel). The behaviour of these constructs (with either head-to-head 60 or 230 bp T2AG3 arrays) was compared with that of two controls strains, one with no HO cleavage site and the other with no telomere-like sequences flanking the HO site.
Figure 2.

Short T2AG3 tracts elicit a checkpoint delay and are actively resected, while long T2AG3 ends are capped. (A) Percent of large-budded cells (G2/M-arrested cells) after HO cleavage (left panel) for strains (wild-type or vid22 ygr071c double mutants) containing the wild-type or modified Chr. VII-L constructs indicated in the right panel. Single and triple black arrowheads indicate 60 and 230 bp T2AG3 tracts, respectively, flanking an HO site (orange bar). The open arrowheads indicate the native telomere, located ∼13 kb from the HO site cassette. (B) Percent of single-stranded DNA measured by QAOS at either short (subtelomeric region of Chr. VII-L) or long (subtelomeric region of Chr. V-R) T2AG3 telomeric ends, or ends containing no TG-repeat sequence (DSB; the distal, telomere-proximal end of the HO site on Chr. VII-L) after HO cleavage. (C) ChIP analysis of Mec1–Myc and Rfa1–Myc association at short or long T2AG3 ends, or at non-TG (DSB) ends in wild-type cells. Fold enrichment reported as described in Figure 1.
As expected (Michelson et al, 2005), the majority of cells from the strain containing no T2AG3 tracts flanking the HO site remained blocked in G2/M throughout the course of the experiment (7 h following galactose induction of HO), whereas most cells lacking the HO site had already traversed G2/M by 4.5 h. Remarkably, cells in which the HO site was flanked by 230 bp of T2AG3-repeat sequence passed through G2/M with kinetics similar to cells lacking the HO site (average restart time of 4.2 h versus 3.7 h), suggesting that the exposed 230 bp T2AG3 arrays were only very transiently recognized as DNA damage (Figure 2A). Furthermore, virtually all of these cells (82/84 or 97.6%) survived induction of the DSB (which was confirmed by Southern blotting; Supplementary Figure S2B), suggesting that stable telomere formation at the break was highly efficient. This behaviour is similar to that observed in cells carrying long arrays of native TG1−3 repeats (CR and DS, unpublished results). Interestingly, cells in which the HO site was flanked by a short (60 bp) T2AG3-repeat array displayed an intermediate phenotype, where passage through G2/M was clearly delayed, such that by 7.5 h only ∼50% of the cells examined had proceeded to the next cell cycle. Although these short T2AG3 tracts appeared to be efficiently elongated as judged by Southern analysis (Supplementary Figure S2B), reduced survival of these cells (83%; Supplementary Figure S2E) suggests that a small but significant fraction failed to be healed, and remained permanently arrested. Taken together, these data indicate that the short, elongating T2AG3 repeats were initially recognized as DNA damage that induced a G2/M checkpoint arrest. We presume that the elongation of these arrays in most cells (see Figure 1B) gradually converts them to a state that no longer promotes checkpoint activation.
Consistent with an initial capping defect at the 60-bp T2AG3-repeat ends, we detected a considerable amount of single-stranded DNA (ssDNA) upstream of these ends (Figure 2B). Since the probes used to monitor the ssDNA are internal to the T2AG3 sequences (>1.2 kb from HO site), the assay detects resection events that proceed well beyond the repeats themselves. Thus, although the population of short T2AG3 tracts is undergoing telomerase-mediated 3′-end elongation (Figure 1B), they are also being subjected to extensive 5′-nucleolytic attack. In contrast, and consistent with the cell-cycle arrest data, we measured little or no ssDNA at the long T2AG3 repeats following HO induction (Figure 2B). In line with these resection data, we detected by ChIP significant recruitment of Rfa1 protein, a subunit of the trimeric ssDNA-binding RPA complex, and Mec1, the yeast ATR checkpoint kinase, at both the short T2AG3 tract and non-TG ends, but little or no recruitment of either protein at the long T2AG3 array (Figure 2C). These results reinforce the conclusion that the long T2AG3-repeat tract is hidden from the DSB checkpoint machinery, whereas the short tract is not.
Tbf1 forms a stable complex with two BED domain-containing proteins, Vid22 and Ygr071c (Krogan et al, 2006), and these two proteins co-localize with Tbf1 at a large number (∼100) of promoter binding sites (Preti et al, 2010; CR and DS, unpublished data). Therefore, we asked if Vid22 and Ygr071c play a role in capping at T2AG3 array-containing ends by repeating the cell-cycle assay in strains where both VID22 and YGR071c genes were deleted. Interestingly, the capping function of the long T2AG3 array was unaffected by the double mutation (Figure 2A), despite the fact that Tbf1 binding at the end was considerably reduced, as measured by ChIP (Supplementary Figure S2A). Similarly, the weak capping function at the short T2AG3 array ends was not obviously affected by mutation of these two genes, though cell survival was reduced to <70% (Supplementary Figure S2E), probably reflecting a decrease in the efficiency of elongation, and thus stable telomere formation. Taken together, these data indicate that although Vid22 and Ygr071c promote more stable chromatin association of Tbf1, an effect observed also at promoter binding sites for Tbf1 (Preti et al, 2010), they are not required for Tbf1-mediated end protection. We also tested the effect of the tbf1-Δi mutation and of two mutations that affect the telomerase pathway: cdc13-2, and tlc1-Δ48. Only the tbf1-Δi mutation had a significant effect on capping at the long T2AG3 ends, leading to an ∼30-min delay in the cell-cycle arrest assay (Supplementary Figure S2D). The short array ends still displayed a prolonged arrest in these mutant backgrounds, as expected, and survival in these mutants was further reduced, compared with the vid22-Δ ygr071c-Δ double mutant (Supplementary Figure S2C and E). Interestingly, none of these mutations had as severe an effect on survival as did mre11-Δ (Supplementary Figure S2E).
Other subtelomeric or SANT/Myb domain DNA-binding proteins do not cap DNA ends
The ability of multiple Tbf1 molecules to cap DSBs, and to support their elongation by telomerase, prompted us to ask if other SANT/Myb domain-containing proteins, or factors known to bind at subtelomeric regions, would behave similarly. We, thus, constructed binding site arrays for Bas1 (TGACTCTG), an Myb-related transcription factor involved in purine and histidine biosynthesis, Reb1 (CCGGGTAAC), an SANT/Myb domain transcription factor that binds to many promoters, but also to subtelomeric sites, and Abf1 (GTCACTCTAGACG), another ubiquitous general regulatory factor (GRF) similar to Rap1 and Reb1, with both promoter and subtelomeric binding sites. Unlike the other factors tested (including Rap1 and Tbf1), Abf1 does not contain a SANT/Myb domain, but instead binds to DNA through an unusual bipartite Zn-finger domain. We generated binding site arrays of either 9 or 24 tandem copies for each of these three new factors, which were placed adjacent to the HO sites at the previously described cassettes near the telomeres of chromosome V-R and chromosome VII-L, respectively.
None of these new sequence arrays were maintained after induction of the HO endonuclease, despite strong association of their respective DNA-binding proteins, as measured by ChIP (Supplementary Figure S3; data not shown). Southern blots revealed the rapid disappearance of the restriction fragment containing the arrays following HO digestion, with no evidence for elongation of the ends seen, in contrast to what was observed for the Tbf1 site arrays, or previously with TG1−3 tracts (Diede and Gottschling, 1999; Negrini et al, 2007). These results suggest that DSB end capping and extension by telomerase are not general features conferred by extended arrays of DNA-binding proteins. The negative result obtained with Reb1 arrays is particularly significant since Reb1, like Tbf1, has been directly implicated in telomere length regulation both by insertion of its binding sites immediately upstream of TG1−3 ends or by protein-tethering experiments at individual telomeres (Berthiau et al, 2006).
Telomerase is excluded from long T2AG3 arrays but associates equally well with both short array or non-TG containing ends
To begin to characterize the capping effect of T2AG3 tracts at the molecular level, we first examined the repeat-length dependence of telomerase association with these ends by ChIP, monitoring Myc-tagged versions of both the catalytic subunit Est2 and the associated Est1 protein. Telomerase was shown previously to crosslink more strongly to short versus long TG1−3 seed sequences at a DSB (Negrini et al, 2007), and the same relationship holds for native telomeres of different lengths (Bianchi and Shore, 2007; Sabourin et al, 2007). As expected, both Est1 and Est2 were detected at the short T2AG3 tract, while the long tract displayed weak binding of both proteins (Figure 3, upper panels). These results are in accord with the Southern blot observations, which showed that only the short tract is elongated (Figure 1B). In contrast, both telomerase subunits associate at very similar levels to the short and long T2AG3 tracts in cells carrying the tbf1-Δi mutation (Figure 3, bottom panels). Again, this situation is consistent with the observed elongation of both ends (Figure 1D). These data demonstrate directly that Tbf1, like Rap1, can regulate telomerase recruitment at DNA ends in a manner dependent upon the number of molecules bound immediately adjacent to that end, as suggested by previous studies (Alexander and Zakian, 2003; Brevet et al, 2003; Berthiau et al, 2006).
Figure 3.

Yeast telomerase is not recruited at long T2AG3 ends in wild-type cells, but is bound to these ends in a tbf1-Δi mutant strains. Analysis by ChIP of Est1–Myc and Est2–Myc binding after HO induction at 60 bp T2AG3, 250 bp T2AG3, or non-TG ends (as marked), in wild-type cells (top panels) or in tbf1-Δi mutant strains (bottom panels). HO constructs, PCR probes, ChIP methods, and statistical analysis were as described in Figures 1 and 2, and in Materials and methods.
We also checked the binding of telomerase at the distal (telomere-proximal) side of the DSB at chromosome VII-L, even though this end does not contain TG-repeat sequences of any kind. Surprisingly, we detected strong binding of Est1 and Est2 at this end, similar to that observed at the elongating 60 bp T2AG3 end (Figure 3). However, this DSB end did not appear to be elongated, as expected, as judged by the absence of a slower migrating species on Southern blots and the failure to observe any Rap1 recruitment to this side of the DSB, which would have indicated the addition of TG1−3 repeats (data not shown). To validate these observations, we repeated the experiment using strains containing either Bas1 repeats or 300 bp of lambda DNA in place of the T2AG3 tracts, in this case upstream (on the centromere-proximal side) of the HO cut site. Again, we observed significant telomerase association following HO cutting at both types of ends, despite the fact that Southern blotting showed that they were not elongated (Supplementary Figure S3C and data not shown).
Long T2AG3 tracts inhibit Mre11 binding, which promotes telomerase association at short tracts
To investigate the underlying cause of reduced telomerase association at long T2AG3 tracts we turned to Mre11 protein. Mre11, a component of the conserved MRX (Mre11/Rad50/Xrs2) complex, is required for proper 3′ G-rich single-strand overhang production at telomeres (Diede and Gottschling, 2001; Larrivee et al, 2004), as well as normal recruitment of telomerase (Goudsouzian et al, 2006). MRX also binds rapidly to newly formed DSBs where it plays a key role in both non-homologous DNA-end joining and homologous recombination (reviewed in Rupnik et al, 2010). Given its role at both telomeres and accidental DSBs, we asked whether T2AG3 tracts influence in cis the association of Mre11 with a DSB. Notably, we observed severely reduced Mre11 binding at an end carrying the 230-bp T2AG3 array, compared with either a 60-bp T2AG3 end or a non-TG repeat containing end (DSB), where binding of Mre11 was equally strong (Figure 4A). We note that this reduction in Mre11 association at the long T2AG3 ends might also be sufficient to explain their relatively low levels of ssDNA and Rfa1 or Mec1 binding (Figure 2). These data indicate that long T2AG3 arrays block both the DNA-damage response and telomerase pathways at a very early step (MRX end recruitment), as was observed previously for long TG1−3 tracts (Negrini et al, 2007).
Figure 4.

Mre11 is required for efficient telomerase recruitment. (A) Analysis by ChIP of the binding of Mre11–Myc after HO induction in a wild-type strain. (B) Analysis by ChIP of the binding of Est1–Myc and Est2–Myc after HO induction in mre11-Δ. (C) Southern blot monitoring cleavage and elongation at both short (60 bp, left panel) and long (230 bp, right panel) T2AG3 tracts in mre11-Δ strains. HO constructs, PCR probes, ChIP methods, and statistical analysis were as described in Figures 1 and 2, and in Materials and methods.
Given these findings, we next asked whether Mre11 is required for telomerase recruitment at the short T2AG3 seed sequence or a DSB. Indeed, we found that association of both telomerase subunits is very strongly reduced at these ends in mre11-Δ cells compared with wild type, nearly to levels observed at the long T2AG3 arrays (Figure 4B). Consistent with these findings, Southern blot analysis and measurement of Rap1 binding by ChIP indicated that neither the short T2AG3 seed nor the DSB are elongated in the mre11-Δ mutant. Interestingly, we also observed a slight decrease in the length of the 230-bp T2AG3 tracts after 24 h of HO induction (Figure 4C and data not shown). We conclude that Mre11 is required for normal length regulation of telomeres consisting largely of Tbf1 binding sites, as is the case for native Rap1-binding TG1−3 ends.
Telomerase action at T2AG3 seeds, but not recruitment, requires Tel1, Cdc13, and Yku
Downstream of MRX association and 5′-end resection, telomerase recruitment has been shown to depend on a number of additional factors, including the Tel1 kinase, which localizes to telomeres and DSBs through an interaction with Xrs2 (Nakada et al, 2003). We, thus, asked whether Tel1 is recruited at T2AG3 seed sequences following their exposure by HO cutting. As expected, given our findings with Mre11, we noted that Tel1 is robustly recruited to the short T2AG3 ends, at levels comparable to those observed at a non-TG DSB, but is barely detectable at the long T2AG3 tract (Figure 5A).
Figure 5.

Tel1 is required for efficient elongation of T2AG3 tracts despite the binding of the telomerase. (A) Analysis by ChIP of the binding of Tel1–HA after HO induction, in wild-type cells. (B) Southern blots monitoring cleavage at the HO site in a tel1-Δ strain. The internal loading control (‘INT’), an uncut fragment (‘U’), and the fragment resulting from ‘U’ after induction of the HO cut (‘C’). (C) Analysis by ChIP of the binding of Est1–myc and Est2–myc after HO induction, in tel1-Δ cells. HO constructs, PCR probes, ChIP methods, and statistical analysis were as described in Figures 1 and 2, and in Materials and methods.
We then examined the effect of deleting the TEL1 gene on T2AG3 elongation and on telomerase association at these ends. We noticed that in tel1-Δ strains both the efficiency and extent of elongation at the 60-bp T2AG3 end are considerably reduced compared with wild type in the first 4 h following galactose addition (Figure 5B; compare with Figure 1B). Furthermore, the long T2AG3 end was reduced in length following overnight incubation in galactose, an effect not observed in wild-type cells (Figure 5B; compare with Figure 1B). Surprisingly, though, telomerase association at the 60-bp T2AG3 end was not significantly affected in the tel1-Δ strain (Figure 5C). These results suggest a much weaker requirement for Tel1 in telomerase recruitment at short T2AG3 ends compared with native (TG1−3) ends of roughly the same length (Goudsouzian et al, 2006). In addition, though, they reveal an apparent requirement for Tel1 in telomerase activation at the short T2AG3 ends, an effect that might be masked at TG1−3 ends due to a stronger requirement at these ends for Tel1 in telomerase recruitment.
Telomerase is thought to be recruited to telomeres through at least two independent pathways (Fisher et al, 2004; Chan et al, 2008), one involving an interaction between the G-rich overhang binding protein Cdc13 and Est1 (Evans and Lundblad, 1999; Pennock et al, 2001; Bianchi et al, 2004) and a second relying upon the Yku heterodimer (Yku70/Yku80), which interacts with a specific hairpin structure in the telomerase template RNA, TLC1 (Peterson et al, 2001; Stellwagen et al, 2003). Recruitment and/or activation of telomerase via the Cdc13 protein appears to be essential for telomere maintenance, based upon the senescence and telomerase recruitment phenotypes of a cdc13-2 mutant. We observed strong binding of Cdc13 to the short T2AG3 tract, moderate levels at the long T2AG3 tracts, and weaker, though clearly detectable binding, at a non-TG DSB (Supplementary Figure S4). Surprisingly, however, ChIP measurements in the cdc13-2 mutant strain show no decrease in Est2 association at the short T2AG3 tract compared with a CDC13 wild-type strain (Figure 6A), indicating that normal levels of telomerase recruitment at this type of end are not affected by the cdc13-2 mutation. We, thus, tested the role of Yku. Even though Yku70 is clearly detectable at both long and short T2AG3 tracts (Supplementary Figure S4), yku70-Δ mutant cells still displayed strong Est2 association at the short T2AG3 ends (Figure 6A). Similarly, deletion of a hairpin structure in TLC1 responsible for recruitment of telomerase by Yku also has no effect on Est2 binding. However, and as observed in the tel1-Δ mutant, elongation of the short T2AG3 tracts is significantly reduced in cdc13-2, tlc-Δ48 and yku70-Δ mutant cells, as judged by both Rap1 association (Figure 6B) and by Southern blot analysis (Supplementary Figure S5; compare with wild type, Figure 1B).
Figure 6.

Neither the Cdc13–Est1 interaction (affected in cdc13-2), nor the Yku70–TLC1 interaction, is required for Est2 association at short T2AG3 or non-TG ends, yet both contribute to elongation of the T2AG3 ends. (A) Analysis by ChIP of the binding of Est2-myc in wild-type or mutant strains (as indicated) at short or long T2AG3 ends or non-TG (DSB) ends. Only the increase in telomerase recruitment at both short tracts and the DSB in the yku70-Δ mutant, relative to wild type, is statistically significant (P<0.05 by Mann–Whitney test). (B) ChIP analysis of Rap1 binding (a read-out for telomerase elongation) at short T2AG3 ends following HO induction, in tel1-Δ, cdc13-2, yku70-Δ and tlc1-Δ48 strains. All of these mutants display a statistically significant (P<0.05) difference compared with wild type at t=4 h, with cdc13-2 already different at t=2 h. HO constructs, PCR probes, ChIP methods, and statistical analyses were as described in Figures 1 and 2, and in Materials and methods.
Together, these results suggest that robust telomerase recruitment at short T2AG3 tracts or non-TG ends requires the presence of the MRX complex, but is not strictly dependent upon Tel1, Yku70 or the pathway affected by the cdc13-2 mutation. Nevertheless, each of the latter three factors appears to be required for efficient telomerase-mediated elongation of the short T2AG3 ends.
Mec1 inhibits the binding of Cdc13, and prevents telomerase elongation of DSB
Because we detected high levels of Mec1 at short T2AG3 tracts (Figure 2C), we decided to ask if the protein might be involved in either telomerase recruitment or action at these ends. We found, though, that neither Est1 nor Est2 association at 60 bp T2AG3 ends are significantly altered in a mec1-Δ sml1-Δ strain compared with wild type (Figure 7A). Furthermore, the elongation of these ends, as measured by Southern blot, also appears to be normal (Supplementary Figure S6). To test if Mec1 function is redundant with Tel1 at short T2AG3 tracts, we measured telomerase recruitment in a strain deleted for both genes. Although the T2AG3 tracts were not maintained in the tel1-Δ mec1-Δ sml1-Δ strain, we detected no obvious effect on telomerase binding at these ends immediately following HO digestion (data not shown).
Figure 7.

Mec1 is not required for telomerase elongation at T2AG3 ends but reduces Cdc13 association and elongation by telomerase at non-TG (DSB) ends. Analysis by ChIP of the binding Est1–myc and Est2–myc (A), Rap1 (B), and Cdc13–myc (C) after HO induction, in sml1-Δ mec1-Δ strains. For Rap1 binding (B) only the differences between WT and mec1-Δ for the DSB at 3 and 4 h are significant (P=0.03 and P=0.028, respectively). For Cdc13 association (C), the statistically significant differences are indicated (*P=0.015 and **P=0.006 by Mann–Whitney test). HO constructs, PCR probes, ChIP methods, and statistical analysis were as described in Figures 1 and 2, and in Materials and methods.
Unexpectedly, however, we found that the mec1Δ mutation caused a significant increase in telomerase action at an end containing no TG-repeat sequence, as judged by a 3- to 4-fold increase in Rap1 binding (Figure 7B). Interestingly, we noticed a comparable (∼3-fold) increase in Cdc13 binding at the DSB end in mec1-Δ sml1-Δ compared with wild type (Figure 7C; P=0.015). In this regard, we also note that in wild-type cells Cdc13 binding is over four-fold greater at 60 bp T2AG3 ends compared with a DSB (P=0.006), whereas in mec1-Δ sml1-Δ cells this difference is <2-fold and not statistically significant (P=0.222). In contrast to the effects on TG1−3 tract (Rap1 binding site) addition and Cdc13 binding caused by mec1-Δ, we observed only a modest (two-fold) increase in Est2 association at the DSB end and no apparent difference in Est1 binding (Figure 7A). Finally, in mec1-Δ sml1-Δ rad52-Δ cells in which a non-TG DSB is exposed by HO cutting on the centromeric side of the break, survival through telomere formation is increased, as judged by the appearance of Ade+, Lys− colonies, compared with a sml1-Δ rad52-Δ control strain (Supplementary Figure S7). This genetic test strongly supports the conclusion from the ChIP data, namely that Mec1 acts to reduce Cdc13 binding at a DSB and subsequent telomerase action.
Discussion
Here, we have exploited the inducible HO endonuclease system to investigate the DNA-end capping and telomere maintenance properties of Tbf1, a TRF1/2-like protein in budding yeast that binds to the common eukaryotic telomere repeat sequence T2AG3. We show that Tbf1 binding at long (230 bp) arrays of T2AG3 repeats plays a direct role in blocking 5′-end resection and checkpoint activation at adjacent DNA ends, indicating that Tbf1 possesses a robust capping function, similar to that of Rap1. The end-capping function that we observe for Tbf1 is likely to be an evolved property, since several other related DNA-binding proteins (Reb1, Bas1, and Abf1), two of which are also Myb/SANT domain factors, fail to protect DNA ends flanked by long arrays of their binding sites.
Evidence for a common capping mechanism for Tbf1 and Rap1
Significantly, Tbf1 binding site arrays, as noted previously for the case of Rap1 (Negrini et al, 2007), block checkpoint activation at a very early step, namely the binding and/or accumulation of Mre11, and presumably the whole MRX complex. This strong inhibition of Mre11 association may be sufficient to explain the reduced binding of other factors that we observed (e.g., Rfa1, Mec1, Tel1, Cdc13, and telomerase subunits), though we cannot exclude the possibility that Tbf1 arrays also directly affect the association of these and/or other factors with DNA ends. Despite, or perhaps because of, this general inhibition of protein binding and resection, these ends are still bound strongly by the Yku70/80 heterodimer (Supplementary Figure S4), as observed previously for long Rap1 array ends (Negrini et al, 2007). Nevertheless, deletion of YKU70 does not affect the ability of long Tbf1 or Rap1 arrays to limit Mre11 binding, or that of other downstream factors (data not shown). Although the mechanism(s) underlying Tbf1- (or Rap1-) dependent capping are still unclear, previous studies have proposed that Rap1 can promote the formation of a number-dependent ‘closed’ state at telomeres through protein–protein interactions together with the interacting Rif1/2 proteins (Wotton and Shore, 1997; Levy and Blackburn, 2004; Negrini et al, 2007; Marcand et al, 2008; Hirano et al, 2009). However, we show here that the two known Tbf1-interacting proteins Vid22 and Ygr071c are not required for capping at long T2AG3-flanked ends. Similarly, a significant degree of Rap1-mediated capping is observed at long TG1−3 ends in the absence of both Rif1 and Rif2 (CR and DS, unpublished results). We, thus, suggest that Tbf1 (and Rap1), when present in sufficient amounts at a DNA end, either possess autonomous capping activity or act together with as yet unidentified capping factors. We suggest furthermore that the capping function of Tbf1 (like that of Rap1) acts in parallel to that of Cdc13.
In contrast to long (230 bp) T2AG3 array ends, the short (60 bp) T2AG3 ends displayed a more severe DSB-like phenotype than that observed at similar length TG1−3 tracts, characterized by extensive resection at sequences internal to the repeats themselves, high levels of Cdc13 and Rfa1/Mec1 binding, and prolonged though still transient G2/M cell-cycle arrest. Taken together, these data suggest that arrays of Tbf1 are somewhat less effective in capping than similar Rap1 arrays, though we cannot rule out that this is simply due to a quantitative difference in protein binding at the respective site arrays, perhaps leading to a slightly higher density of Rap1 at TG1−3 arrays than that of Tbf1 at T2AG3 arrays of comparable length. A weaker intrinsic capping function of Tbf1 might explain why yeast cells carrying a ‘humanized’ telomerase template RNA, where Tbf1 is the predominant telomere duplex binding protein, are in a chronic state of checkpoint activation (di Domenico et al, 2009).
Regulation of telomerase recruitment and activity at DNA ends
The similarity between short T2AG3 repeat and non-telomeric (DSB) ends with respect to Rfa1, Mec1, Mre11, and Tel1 binding prompted us to ask whether telomerase might also be recruited to the non-telomeric ends. Indeed, we found comparable levels of Est2 and its associated co-factor Est1 at all five non-telomeric ends that we examined (one containing 300 bp of phage lambda DNA, one distal to the HO site at the ADH4 locus on Chr. VII-L, the others flanked by arrays of Bas1, Abf1, or Reb1 binding sites; Figures 4, 5 and 6 and data not shown). Our results are consistent with a recent finding that revealed strong Est2 binding at the MAT locus (the natural site of action of HO endonuclease) in cells lacking a ‘donor’ sequence for repair of the DSB (Oza et al, 2009). Therefore, it appears that telomerase recruitment, at least as measured by a ChIP assay, is a common feature of DSBs in yeast that is actively suppressed at ends flanked by long arrays of either Tbf1 or Rap1 binding sites.
Telomerase (both Est1 and Est2) recruitment at short T2AG3 repeat and non-telomeric (DSB) ends is strikingly insensitive to loss of Tel1 or mutation of either the Cdc13–Est1 (cdc13-2) or the Yku–TLC1 telomerase recruitment pathways, suggesting that either pathway alone might be sufficient for robust telomerase binding. Alternatively, the high levels of Rfa1 (and presumably the whole RPA hetero-trimeric complex) might be sufficient to promote robust telomerase recruitment at these ends (Schramke et al, 2004). These data stand in contrast to findings at native telomeres (Fisher et al, 2004; Goudsouzian et al, 2006; Chan et al, 2008) or at TG1−3-containing DSBs (Bianchi et al, 2004), where these mutations can significantly reduce telomerase recruitment. It should be noted, however, that these studies mostly examined synchronized cells and found defects specific to either late S phase, when telomerase normally acts, or G1 (in the case of Yku). In the experiments described here, telomerase recruitment was assayed in cells undergoing checkpoint arrest in G2/M.
Our finding of similar levels of telomerase binding at short T2AG3 tract ends and ends lacking TG repeats begs the question of why the former ends are efficiently elongated by telomerase while that latter are not. One factor is likely to be the strong preference for telomere repeat addition at short runs of G or G-T sequences, first documented by Kramer and Haber (1993), and correctly predicted by these authors to reflect base pairing with a specific sequence in the telomerase template RNA. Consistent with these early observations, we found that TG1−3-repeat addition occurs immediately following the terminal T2AG3 in our constructs (data not shown). The surprising observation to emerge from our study is that the telomerase ‘dwell time’ at non-TG ends, which we assume is what the ChIP assay measures, is very similar to that at the efficiently elongated T2AG3 ends. Perhaps the action of Pif1 helicase, which is known to suppress telomerase action at accidental DSB ends (Schulz and Zakian, 1994; Myung et al, 2001), strongly reduces telomerase association in a productive template base-pairing conformation at the non-TG ends without having a significant effect on its ChIP-detectable association with these ends. Finally, it is important to note that telomerase association with short T2AG3 or non-TG ends is remarkably insensitive to mutations in the two telomerase recruitment pathways (Cdc13–Est and Yku–TLC1) known to play an important role at native telomeres or TG1−3-containing ends. If anything, cdc13-2 and yku70-Δ mutations lead to increased Est2 association at these ends. Nevertheless, each of the mutants tested caused a quantitative decrease in telomerase action at the short T2AG3 end, as measured by Rap1 binding. These data suggest that the tel1-Δ, cdc13-2, yku70-Δ and tlc1-Δ48 mutations, in addition to affecting telomerase recruitment, at least in some circumstances, are also defective in one (or more) steps in telomerase activation that occur following its recruitment to ends. We would also point out that Tbf1 itself might directly participate in telomerase activation, as suggested by an elegant protein-tethering experiment described by Lingner and colleagues (Arneric and Lingner, 2007).
One significant additional difference we observed between short T2AG3 tract ends and non-TG ends is in their amount of Cdc13 binding. Several recent studies have documented Cdc13 binding at DSBs (Oza et al, 2009; Zhang and Durocher, 2010), and we show here that this binding is ∼3- to 6-fold weaker than that which occurs at 60 bp T2AG3 ends. As pointed out previously, this relatively small difference may reflect an unexpectedly weak in vivo binding preference of Cdc13 for TG-rich ssDNA. Significantly, we find that this binding difference is considerably reduced by mutation of MEC1. This result is consistent with the model proposed by Zhang and Durocher (2010), who provided evidence that Mec1 and Pph3 act in a kinase-phosphatase regulatory loop that directly controls Cdc13 S316 phosphorylation, and through this its ability to bind and promote telomere formation, particularly at ends containing little or no TG-repeat sequence. Alternatively, or in addition, our findings may be explained by the recently observed Mec1-dependent phosphorylation of the Pif1 helicase, which was shown to prevent inappropriate telomerase action at DSBs (Makovets and Blackburn, 2009). The increased Est2 association we observed at a DSB in mec1-Δ cells may reflect a reduced ability of Pif1 helicase to remove telomerase from these ends (Boulé et al, 2005). The resulting increased action of telomerase, which would in principle generate Cdc13 binding sites, might in part explain the concomitant increase in Cdc13 binding. In any event, our data, taken together with previous findings, reveal the permissiveness of DSBs to telomerase binding and highlight the importance of downstream regulatory events in preventing inappropriate telomerase action at these ends.
Conclusions and implications for telomere evolution
In summary, we demonstrate here that long Tbf1 arrays can protect a DNA end from being recognized as DNA damage, and we provide mechanistic insight into this activity. Notably, we show that Tbf1, like Rap1, can block the checkpoint response at the early step of Mre11 binding (or accumulation) when present in sufficient numbers at a DNA end. The strongly reduced Mre11 binding at long T2AG3 ends may explain the similar effect on binding of Rfa1/Mec1, as well as Tel1, and the near absence of detectable 5′-end resection. The finding that Tbf1 can block checkpoint activation at long T2AG3 ends even in the absence of two interacting proteins (both of which enhance Tbf1 DNA binding) suggests either that Tbf1 possesses an autonomous, direct capping function, or that it acts with the help of other factors yet to be identified. We note that Rap1 also appears to act autonomously from its interacting proteins, Rif1 and Rif2, at sufficiently long TG1−3 tracts (Negrini et al, 2007; CR and DS, unpublished data).
In wild-type cells, Tbf1 does not bind at telomere termini in S. cerevisiae, but instead associates strongly with somewhat dispersed T2AG3 sequences found just internal to the native TG1−3 telomere repeats (Preti et al, 2010). This raises the question of what, if any, telomere function the protein might have. Tbf1 can participate in length regulation of both all-T2AG3 ends and mixed sequence ends (Brevet et al, 2003; Cagney et al, 2006), and may directly affect nature telomeres, since a hypomorphic TBF1 allele displays a short telomere phenotype (Ungar et al, 2009). Consistent with this, and further indicative of a complex role for Tbf1 in native telomere length regulation, we found that the tbf1-Δi mutation actually causes a mild telomere elongation phenotype (Supplementary Figure S8). As pointed out above, another study also points to a direct role for subtelomere-bound Tbf1 in telomerase regulation (Arneric and Lingner, 2007). Findings reported here strongly support an additional telomeric role for Tbf1 in the rescue of native telomeres that have suffered a critical loss of terminal TG1−3 repeats, as a consequence, for example, of replication fork collapse and subsequent telomere repeat breakage (Miller et al, 2006). Consistent with this notion, we found that a short, internal T2AG3 array can very efficiently rescue (through telomere healing) a DNA end carrying only 11 bp of TG1−3 repeat, which by itself is incapable of promoting telomere function (Supplementary Figure S9).
Results reported here provide additional support for the proposal that Tbf1 is indeed a descendent of an ancestral telomere-binding protein in yeasts (Teixeira and Gilson, 2005; Berthiau et al, 2006). In this scenario, the presence of subtelomeric binding sites for Tbf1 (Preti et al, 2010) in present-day budding yeast may be viewed as a remnant from a period of transition between a Tbf1- and Rap1-based telomere regulatory system, which presumably involved both the emergence of a Rap1 protein with a duplicated Myb/SANT domain capable of strong binding to TG1−3-repeat sequences and mutation of TLC1 to generate these novel repeats (Lue, 2010). The evolutionary origins of Tbf1, which presently appears to be largely fungal specific, are still unclear. Tbf1 DNA-binding domains are highly homologous to those of TRF proteins, but these proteins lack a recognizable TRFH domain found in the N-terminal part of TRF proteins and their fission yeast orthologue Taz1 (Li et al, 2000). Nevertheless, Tbf1 N-terminal domains may be structurally related to the TRFH domain (Pitt et al, 2008), suggesting the possibility of a common ancestor. Further characterization of Tbf1 function at DNA ends and telomeres may help to resolve these and other issues raised by this work.
Materials and methods
Strains and plasmids
All strains used in this study (listed in Supplementary Table S1) are derived from a W303 strain carrying a deletion of the HO site at the MAT locus (mat::loxP), a galactose-inducible HO endonuclease gene integrated at the LEU2 locus and a copy of the LYS2 gene at the MNT2 locus (Bianchi et al, 2004). For ChIP experiments, this strain was further modified by the insertion of qPCR amplicons: a sequence from the mouse Dbp gene (amplicon 7) between the ADH4 and MNT2 genes in a cassette containing the ADE2 gene (Negrini et al, 2007) and either a 60-bp T2AG3-repeat sequence with a 200 bp of phage DNA, or a 300 bp of phage DNA, followed by the HO cut site. In the same strain, we modified the subtelomeric YER188W locus of Chr. V-R with a cassette containing the TRP1 gene, a different sequence from the mouse Dbp gene (amplicon 9), a 230-bp T2AG3-repeat sequence, and the HO cut site. For checkpoint assays, the subtelomeric region of Chr. VII-L was modified with the same cassette described above containing either T2AG3-230 or T2AG3-60 tracts, with each sequence present on both sides of the HO site in a head-to-head orientation. The Bas1 binding sequences (TGACTCTG) were inserted in the same constructions described above. The binding sequences repeated 9 and 24 times were introduced into the Chr. V-R and Chr. VII-L subtelomeric regions, respectively.
The MRE11 and YKU70 deletion mutants (mre11::kanMX and yku70::kanMX) were generated as described (Negrini et al, 2007). The TEL1 deletion (tel1::kanMX) was constructed by transformation of a PCR product obtained by amplification of pFA6a-kanMX6 (Wach et al, 1994). The SML1 and MEC1 deletions were generated by transformation of the PCR products obtained from pAG32 and pAG25, respectively (Goldstein and McCusker, 1999). For the triple mutants sml1 mec1 tel1, the plasmid pCdc13Est2-U was introduced to prevent senescence. These strains were placed under 5-FOA selection prior to the experiment to select for loss of this plasmid. The tbf1-Δi mutant allele was constructed from the successive pop-in and pop-out of pVR7, a derivative of pRS314-tbf1-Δi (a gift of Eric Gilson; Berthiau et al, 2006). The cdc13-2 strains were constructed as described previously (Bianchi et al, 2004). The tlc1-Δ48 mutant was generated by the integration of the ptlc1-Δ48int plasmid at TLC1 and subsequent screening for loss of the wild-type allele following 5-FOA selection. The Myc epitope-tagged (13 × Myc) versions of TBF1, tbf1-Δi, EST1, CDC13, MRE11, RPA1, and YKU70 (all C-terminal tags) were generated from pFA6a–13Myc–HIS3MX6 (Longtine et al, 1998) PCR products, as described previously (Negrini et al, 2007). Construction of N-terminal Myc-tagged EST2 and MEC1 strains, as well as the TEL1–5HA allele have been described previously (Bianchi et al, 2004).
Chromatin immunoprecipitation
ChIP assays were performed as described previously (Negrini et al, 2007), with some modifications. Briefly, cells are grown in YPLG medium and HO endonuclease was induced by addition of galactose to 2%. After crosslinking in 1% formaldehyde, cells were lysed and sonicated using a Bioruptor device (Diagenode) (one round of 15min: 30s sonication interspersed with 60s pause). Immunoprecipitations were carried out with culture supernatant as a source of anti-Myc 9E10 antibody or anti-HA HA.11 (Covance) antibodies, and Dynabeads M280 coupled to sheep anti-mouse IgG (Dynal). Quantification of immunoprecipitated DNA was achieved by real-time qPCR on a Bio-Rad iCycler or a Roche LightCycler 480. Amplicon 7 is located next to either T2AG3-60 or 300 bp of phage DNA on Chr. VII-L; amplicon 9 is followed by T2AG3-230 on Chr. V-R; amplicon 14 is located distal to the HO site on Chr. VII-L; and the internal control is located within the PDI1 gene (50 kb from left telomere of Chr. III). Fold enrichment (FE) of amplicons 7, 9, and 14 (sample), over an internal control (ref, PDI1 gene) was determined after normalization with values obtained for input samples and correction for the efficiency of HO cutting (%HO cut) at Chr. VII-L and Chr. V-R using the following equation (where E refers to measured efficiency of PCR amplicons):
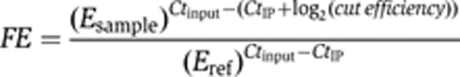 |
Results were obtained from at least three independent experiments for each strain tested. Data are reported as averages (bars) and standard deviations are indicated by lines above and below.
Southern blotting and quantification of HO cutting efficiency
Southern blots were carried out as described previously (Negrini et al, 2007). HO cleavage efficient was determined by measuring the amount of the uncut band relative to the internal loading control, normalized to the uninduced (t=0) sample, using a Bio-Rad PersonalFX Imager and Quantity-One software.
Single-cell checkpoint arrest analysis
Strains were grown overnight in YPA with 2% raffinose then diluted in the same medium the following morning. After 2 h of growth, cells were washed with water and then resuspended in YPLG (lactic acid/glycerol) medium containing 2% galactose to induce the HO endonuclease gene. After a further 2 h of growth, the cells were washed and spread on YPAD plates. For each strain, at least 70 small budded cells were dissected into a grid for analysis. Every 30 min, cells were checked for cell-cycle restart (second round of budding). After growth of cells into colonies, only Lys− colonies are scored as having been subjected to a DNA break (the LYS2 gene is located telomere proximal to the HO site; see Figure 1A). Lys+ colonies are thus excluded from the data set. The average restart time has been estimated by Kaplan–Meier analysis using Sigmaplot 11 software. Statistical differences between the curves have been calculated using a Log-Rank test.
QAOS assay
QAOS analysis (Booth et al, 2001) was performed as described previously (Negrini et al, 2007).
Supplementary Material
Acknowledgments
We are grateful to Eric Gilson, Victoria Lundblad, Daniel Durocher, P Paul Liu and Fiorentina Ascenzioni for gifts of plasmids and strains, and to Alessandro Bianchi for advice and reagents during early phases of this work, as well as for his comments on the manuscript. We thank N Roggli for expert graphics work, and members of the Shore laboratory for helpful advice and comments on the manuscript. This work was supported by grants from the Swiss National Science Foundation, by the National Center for Competence in Research (NCCR) program ‘Frontiers in Genetics’ (sponsored by the Swiss National Science Foundation), and by the Canton of Geneva. VR was supported by a NCCR ‘Frontiers in Genetics’ doctoral student fellowship.
Author contributions: VR, CR and PD performed the experiments. VR, CR and DS analysed the data. DS conceived the study. VR and DS planned the experiments and wrote the manuscript.
Footnotes
The authors declare that they have no conflict of interest.
References
- Alexander MK, Zakian VA (2003) Rap1p telomere association is not required for mitotic stability of a C(3)TA(2) telomere in yeast. EMBO J 22: 1688–1696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arneric M, Lingner J (2007) Tel1 kinase and subtelomere-bound Tbf1 mediate preferential elongation of short telomeres by telomerase in yeast. EMBO Rep 8: 1080–1085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berthiau AS, Yankulov K, Bah A, Revardel E, Luciano P, Wellinger RJ, Geli V, Gilson E (2006) Subtelomeric proteins negatively regulate telomere elongation in budding yeast. EMBO J 25: 846–856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi A, Negrini S, Shore D (2004) Delivery of yeast telomerase to a DNA break depends on the recruitment functions of Cdc13 and Est1. Mol Cell 16: 139–146 [DOI] [PubMed] [Google Scholar]
- Bianchi A, Shore D (2007) Increased association of telomerase with short telomeres in yeast. Genes Dev 21: 1726–1730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi A, Shore D (2008) How telomerase reaches its end: mechanism of telomerase regulation by the telomeric complex. Mol Cell 31: 153–165 [DOI] [PubMed] [Google Scholar]
- Bilaud T, Koering CE, Binet-Brasselet E, Ancelin K, Pollice A, Gasser SM, Gilson E (1996) The telobox, a Myb-related telomeric DNA binding motif found in proteins from yeast, plants and human. Nucleic Acids Res 24: 1294–1303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonetti D, Clerici M, Anbalagan S, Martina M, Lucchini G, Longhese MP (2010) Shelterin-like proteins and Yku inhibit nucleolytic processing of Saccharomyces cerevisiae telomeres. PLoS Genet 6: e1000966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booth C, Griffith E, Brady G, Lydall D (2001) Quantitative amplification of single-stranded DNA (QAOS) demonstrates that cdc13-1 mutants generate ssDNA in a telomere to centromere direction. Nucleic Acids Res 29: 4414–4422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulé JB, Vega LR, Zakian VA (2005) The yeast Pif1p helicase removes telomerase from telomeric DNA. Nature 438: 57–61 [DOI] [PubMed] [Google Scholar]
- Brevet V, Berthiau AS, Civitelli L, Donini P, Schramke V, Geli V, Ascenzioni F, Gilson E (2003) The number of vertebrate repeats can be regulated at yeast telomeres by Rap1-independent mechanisms. EMBO J 22: 1697–1706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brigati C, Kurtz S, Balderes D, Vidali G, Shore D (1993) An essential yeast gene encoding a TTAGGG repeat-binding protein. Mol Cell Biol 13: 1306–1314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cagney G, Alvaro D, Reid RJ, Thorpe PH, Rothstein R, Krogan NJ (2006) Functional genomics of the yeast DNA-damage response. Genome Biol 7: 233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan A, Boule JB, Zakian VA (2008) Two pathways recruit telomerase to Saccharomyces cerevisiae telomeres. PLoS Genet 4: e1000236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lange T (2009) How telomeres solve the end-protection problem. Science 326: 948–952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeZwaan DC, Freeman BC (2009) The conserved Est1 protein stimulates telomerase DNA extension activity. Proc Natl Acad Sci USA 106: 17337–17342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- di Domenico EG, Auriche C, Viscardi V, Longhese MP, Gilson E, Ascenzioni F (2009) The Mec1p and Tel1p checkpoint kinases allow humanized yeast to tolerate chronic telomere dysfunctions by suppressing telomere fusions. DNA Repair (Amst) 8: 209–218 [DOI] [PubMed] [Google Scholar]
- Diede SJ, Gottschling DE (1999) Telomerase-mediated telomere addition in vivo requires DNA primase and DNA polymerases alpha and delta. Cell 99: 723–733 [DOI] [PubMed] [Google Scholar]
- Diede SJ, Gottschling DE (2001) Exonuclease activity is required for sequence addition and Cdc13p loading at a de novo telomere. Curr Biol 11: 1336–1340 [DOI] [PubMed] [Google Scholar]
- Evans SK, Lundblad V (1999) Est1 and Cdc13 as comediators of telomerase access. Science 286: 117–120 [DOI] [PubMed] [Google Scholar]
- Fisher TS, Taggart AK, Zakian VA (2004) Cell cycle-dependent regulation of yeast telomerase by Ku. Nat Struct Mol Biol 11: 1198–1205 [DOI] [PubMed] [Google Scholar]
- Gao H, Cervantes RB, Mandell EK, Otero JH, Lundblad V (2007) RPA-like proteins mediate yeast telomere function. Nat Struct Mol Biol 14: 208–214 [DOI] [PubMed] [Google Scholar]
- Gao H, Toro TB, Paschini M, Braunstein-Ballew B, Cervantes RB, Lundblad V (2010) Telomerase recruitment in Saccharomyces cerevisiae is not dependent on Tel1-mediated phosphorylation of Cdc13. Genetics 186: 1147–1159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garvik B, Carson M, Hartwell L (1995) Single-stranded DNA arising at telomeres in cdc13 mutants may constitute a specific signal for the RAD9 checkpoint. Mol Cell Biol 15: 6128–6138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein AL, McCusker JH (1999) Three new dominant drug resistance cassettes for gene disruption in Saccharomyces cerevisiae. Yeast 15: 1541–1553 [DOI] [PubMed] [Google Scholar]
- Goudsouzian LK, Tuzon CT, Zakian VA (2006) S cerevisiae Tel1p and Mre11p are required for normal levels of Est1p and Est2p telomere association. Mol Cell 24: 603–610 [DOI] [PubMed] [Google Scholar]
- Grandin N, Damon C, Charbonneau M (2001) Ten1 functions in telomere end protection and length regulation in association with Stn1 and Cdc13. EMBO J 20: 1173–1183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grandin N, Reed SI, Charbonneau M (1997) Stn1, a new Saccharomyces cerevisiae protein, is implicated in telomere size regulation in association with Cdc13. Genes Dev 11: 512–527 [DOI] [PubMed] [Google Scholar]
- Hardy CFJ, Sussel L, Shore D (1992) A RAP1-interacting protein involved in silencing and telomere length regulation. Genes Dev 6: 801–814 [DOI] [PubMed] [Google Scholar]
- Henning KA, Moskowitz N, Ashlock MA, Liu PP (1998) Humanizing the yeast telomerase template. Proc Natl Acad Sci USA 95: 5667–5671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano Y, Fukunaga K, Sugimoto K (2009) Rif1 and rif2 inhibit localization of tel1 to DNA ends. Mol Cell 33: 312–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano Y, Sugimoto K (2007) Cdc13 telomere capping decreases Mec1 association but does not affect Tel1 association with DNA ends. Mol Biol Cell 18: 2026–2036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer KM, Haber JE (1993) New telomeres in yeast are initiated with a highly selected subset of TG1-3 repeats. Genes Dev 7: 2345–2356 [DOI] [PubMed] [Google Scholar]
- Krogan NJ, Cagney G, Yu H, Zhong G, Guo X, Ignatchenko A, Li J, Pu S, Datta N, Tikuisis AP, Punna T, Peregrin-Alvarez JM, Shales M, Zhang X, Davey M, Robinson MD, Paccanaro A, Bray JE, Sheung A, Beattie B et al. (2006) Global landscape of protein complexes in the yeast Saccharomyces cerevisiae. Nature 440: 637–643 [DOI] [PubMed] [Google Scholar]
- Larrivee M, LeBel C, Wellinger RJ (2004) The generation of proper constitutive G-tails on yeast telomeres is dependent on the MRX complex. Genes Dev 18: 1391–1396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy DL, Blackburn EH (2004) Counting of Rif1p and Rif2p on Saccharomyces cerevisiae telomeres regulates telomere length. Mol Cell Biol 24: 10857–10867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Oestreich S, de Lange T (2000) Identification of human Rap1: implications for telomere evolution. Cell 101: 471–483 [DOI] [PubMed] [Google Scholar]
- Lin JJ, Zakian VA (1996) The Saccharomyces CDC13 protein is a single-strand TG1-3 telomeric DNA-binding protein in vitro that affects telomere behavior in vivo. Proc Natl Acad Sci USA 93: 13760–13765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longtine MS, McKenzie A III, Demarini DJ, Shah NG, Wach A, Brachat A, Philippsen P, Pringle JR (1998) Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 14: 953–961 [DOI] [PubMed] [Google Scholar]
- Lue NF (2010) Plasticity of telomere maintenance mechanisms in yeast. Trends Biochem Sci 35: 8–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lydall D (2009) Taming the tiger by the tail: modulation of DNA damage responses by telomeres. EMBO J 28: 2174–2187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makovets S, Blackburn EH (2009) DNA damage signalling prevents deleterious telomere addition at DNA breaks. Nat Cell Biol 11: 1383–1386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcand S, Pardo B, Gratias A, Cahun S, Callebaut I (2008) Multiple pathways inhibit NHEJ at telomeres. Genes Dev 22: 1153–1158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGee JS, Phillips JA, Chan A, Sabourin M, Paeschke K, Zakian VA (2010) Reduced Rif2 and lack of Mec1 target short telomeres for elongation rather than double-strand break repair. Nat Struct Mol Biol 17: 1438–1445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michelson RJ, Rosenstein S, Weinert T (2005) A telomeric repeat sequence adjacent to a DNA double-stranded break produces an anticheckpoint. Genes Dev 19: 2546–2559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller KM, Rog O, Cooper JP (2006) Semi-conservative DNA replication through telomeres requires Taz1. Nature 440: 824–828 [DOI] [PubMed] [Google Scholar]
- Myung K, Chen C, Kolodner RD (2001) Multiple pathways cooperate in the suppression of genome instability in Saccharomyces cerevisiae. Nature 411: 1073–1076 [DOI] [PubMed] [Google Scholar]
- Nakada D, Matsumoto K, Sugimoto K (2003) ATM-related Tel1 associates with double-strand breaks through an Xrs2-dependent mechanism. Genes Dev 17: 1957–1962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negrini S, Ribaud V, Bianchi A, Shore D (2007) DNA breaks are masked by multiple Rap1 binding in yeast: implications for telomere capping and telomerase regulation. Genes Dev 21: 292–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nugent CI, Hughes TR, Lue NF, Lundblad V (1996) Cdc13p: a single-strand telomeric DNA-binding protein with a dual role in yeast telomere maintenance. Science 274: 249–252 [DOI] [PubMed] [Google Scholar]
- Oza P, Jaspersen SL, Miele A, Dekker J, Peterson CL (2009) Mechanisms that regulate localization of a DNA double-strand break to the nuclear periphery. Genes Dev 23: 912–927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palm W, de Lange T (2008) How shelterin protects mammalian telomeres. Annu Rev Genet 42: 301–334 [DOI] [PubMed] [Google Scholar]
- Pennock E, Buckley K, Lundblad V (2001) Cdc13 delivers separate complexes to the telomere for end protection and replication. Cell 104: 387–396 [DOI] [PubMed] [Google Scholar]
- Peterson SE, Stellwagen AE, Diede SJ, Singer MS, Haimberger ZW, Johnson CO, Tzoneva M, Gottschling DE (2001) The function of a stem-loop in telomerase RNA is linked to the DNA repair protein Ku. Nat Genet 27: 64–67 [DOI] [PubMed] [Google Scholar]
- Pitt CW, Valente LP, Rhodes D, Simonsson T (2008) Identification and characterization of an essential telomeric repeat binding factor in fission yeast. J Biol Chem 283: 2693–2701 [DOI] [PubMed] [Google Scholar]
- Preti M, Ribeyre C, Pascali C, Bosio MC, Cortelazzi B, Rougemont J, Guarnera E, Naef F, Shore D, Dieci G (2010) The telomere-binding protein Tbf1 demarcates snoRNA gene promoters in Saccharomyces cerevisiae. Mol Cell 38: 614–620 [DOI] [PubMed] [Google Scholar]
- Rupnik A, Lowndes NF, Grenon M (2010) MRN and the race to the break. Chromosoma 119: 115–135 [DOI] [PubMed] [Google Scholar]
- Sabourin M, Tuzon CT, Zakian VA (2007) Telomerase and Tel1p preferentially associate with short telomeres in S. cerevisiae. Mol Cell 27: 550–561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schramke V, Luciano P, Brevet V, Guillot S, Corda Y, Longhese MP, Gilson E, Geli V (2004) RPA regulates telomerase action by providing Est1p access to chromosome ends. Nat Genet 36: 46–54 [DOI] [PubMed] [Google Scholar]
- Schulz VP, Zakian VA (1994) The Saccharomyces PIF1 DNA helicase inhibits telomere elongation and de novo telomere formation. Cell 76: 145–155 [DOI] [PubMed] [Google Scholar]
- Shore D, Bianchi A (2009) Telomere length regulation: coupling DNA end processing to feedback regulation of telomerase. EMBO J 28: 2309–2322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stellwagen AE, Haimberger ZW, Veatch JR, Gottschling DE (2003) Ku interacts with telomerase RNA to promote telomere addition at native and broken chromosome ends. Genes Dev 17: 2384–2395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taggart AK, Teng SC, Zakian VA (2002) Est1p as a cell cycle-regulated activator of telomere-bound telomerase. Science 297: 1023–1026 [DOI] [PubMed] [Google Scholar]
- Teixeira MT, Arneric M, Sperisen P, Lingner J (2004) Telomere length homeostasis is achieved via a switch between telomerase-extendible and -nonextendible states. Cell 117: 323–335 [DOI] [PubMed] [Google Scholar]
- Teixeira MT, Gilson E (2005) Telomere maintenance, function and evolution: the yeast paradigm. Chromosome Res 13: 535–548 [DOI] [PubMed] [Google Scholar]
- Ungar L, Yosef N, Sela Y, Sharan R, Ruppin E, Kupiec M (2009) A genome-wide screen for essential yeast genes that affect telomere length maintenance. Nucleic Acids Res 37: 3840–3849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vodenicharov MD, Laterreur N, Wellinger RJ (2010) Telomere capping in non-dividing yeast cells requires Yku and Rap1. EMBO J 29: 3007–3019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vodenicharov MD, Wellinger RJ (2006) DNA degradation at unprotected telomeres in yeast is regulated by the CDK1 (Cdc28/Clb) cell-cycle kinase. Mol Cell 24: 127–137 [DOI] [PubMed] [Google Scholar]
- Wach A, Brachat A, Pohlmann R, Philippsen P (1994) New heterologous modules for classical or PCR-based gene disruptions in Saccharomyces cerevisiae. Yeast 10: 1793–1808 [DOI] [PubMed] [Google Scholar]
- Wellinger RJ (2010) When the caps fall off: responses to telomere uncapping in yeast. FEBS Lett 584: 3734–3740 [DOI] [PubMed] [Google Scholar]
- Wotton D, Shore D (1997) A novel Rap1p-interacting factor, Rif2p, cooperates with Rif1p to regulate telomere length in Saccharomyces cerevisiae. Genes Dev 11: 748–760 [DOI] [PubMed] [Google Scholar]
- Zhang W, Durocher D (2010) De novo telomere formation is suppressed by the Mec1-dependent inhibition of Cdc13 accumulation at DNA breaks. Genes Dev 24: 502–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou BO, Wang SS, Zhang Y, Fu XH, Dang W, Lenzmeier BA, Zhou JQ (2011) Histone H4 lysine 12 acetylation regulates telomeric heterochromatin plasticity in Saccharomyces cerevisiae. PLoS Genet 7: e1001272. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.