


Abstract
Boundary elements partition eukaryotic chromatin into active and repressive domains, and can also block regulatory interactions between domains. Boundary elements act via diverse mechanisms making accurate feature-based computational predictions difficult. Therefore, we developed an unbiased algorithm that predicts the locations of human boundary elements based on the genomic distributions of chromatin and transcriptional states, as opposed to any intrinsic characteristics that they may possess. Application of our algorithm to ChIP-seq data for histone modifications and RNA Pol II-binding data in human CD4+ T cells resulted in the prediction of 2542 putative chromatin boundary elements genome wide. Predicted boundary elements display two distinct features: first, position-specific open chromatin and histone acetylation that is coincident with the recruitment of sequence-specific DNA-binding factors such as CTCF, EVI1 and YYI, and second, a directional and gradual increase in histone lysine methylation across predicted boundaries coincident with a gain of expression of non-coding RNAs, including examples of boundaries encoded by tRNA and other non-coding RNA genes. Accordingly, a number of the predicted human boundaries may function via the synergistic action of sequence-specific recruitment of transcription factors leading to non-coding RNA transcriptional interference and the blocking of facultative heterochromatin propagation by transcription-associated chromatin remodeling complexes.
INTRODUCTION
Eukaryotic chromosomes are functionally organized into alternating active and repressive chromatin domains, referred to as euchromatin and heterochromatin respectively (1,2). Active chromatin domains are characterized by histone modifications that facilitate gene expression via the opening of chromatin, which provides transcription factors access to genomic DNA, whereas repressive domains are enriched with histone modifications that yield more tightly compact and less accessible chromatin leading to the repression of gene expression (3–9). Accordingly, the establishment and maintenance of distinct chromatin domains has important implications for gene regulation specific to cellular development and function (10,11).
The organization of eukaryotic chromatin into functionally distinct domains implies the existence of chromatin partitioning elements that can be used both to delineate active euchromatic and repressive heterochromatic domains, while preserving their structural integrity, and to prevent regulatory cross talk between different domains (12–15). Such chromatin partitioning elements do in fact exist and they are known as ‘boundary elements’ (16–18). Boundary element functionality is characterized by two fundamental properties: (i) the ability to protect from chromosomal position effects by acting as barriers against the self-propagation of repressive chromatin (16,19,20) and (ii) the ability to insulate or block regulatory interactions between distal enhancers and proximal gene promoters (15,21,22). Some boundary elements are able to act both as chromatin barriers and enhancer blocking insulators (18,23). Boundary elements that are cell type-specific help to establish alternating facultative, as opposed to constitutive, euchromatic and heterochromatic domains.
Known boundary elements are diverse, and several different mechanisms of boundary element activity have been uncovered. First, fixed boundary elements consist of specific DNA sequences and their associated proteins, which establish boundaries with well defined positions. Such precisely located boundaries are thought to form discrete physical barriers that partition distinct chromatin and/or regulatory domains. For example, the HS4 boundary element found upstream of the chicken β-globin locus is bound by the CCCTC-binding factor (CTCF), a well known vertebrate insulator associated protein with demonstrated enhancer blocking activity (24,25). The scs/scs′ elements in Drosophila provide fixed boundaries at the heat-shock domain locus (19,22,26), and the chromatin barrier activity of the scs/scs′ boundaries is dependent upon the binding of two protein factors Zw5 and BEAF (27).
Second, there are variable boundary elements that do not occupy specific DNA sequences or genomic locations. These variable boundaries are thought to be established and maintained through a dynamic balance of collisions between opposing chromatin modifying enzyme complexes responsible for the formation of euchromatin on one side of the boundary and heterochromatin on the other (28,29). For example, the phenomenon of position effect variegation (PEV) in Drosophila can be attributed to variable boundary elements (13,30). PEV refers to the variegated expression of genes located between adjacent euchromatic and heterochromatic domains. PEV occurs due to the changing locations of variable boundaries between cells, which result in genes being located in alternating euchromatic or heterochromatic environments in different cells.
Third, boundary element activity can depend upon transcriptional interference from small non-protein-coding transcriptional units, such as tRNA genes in yeast (20,31–34) or tRNA-derived SINE retrotransposons in mouse (18,35). Boundary elements that function via transcriptional interference contain specific sequence features needed to recruit transcription factors (e.g. the Pol II and Pol III machineries), and they may also provide a physical barrier to the propagation of heterochromatin via nucleosomal gaps close to transcription start sites. These nucleosomal gaps may also serve as entry sites for chromatin remodeling complexes that help to establish the boundaries (14,31).
Thus, many of the currently known boundary elements have been defined functionally, based on experimental confirmation of their activity, rather than categorically based on the presence of well defined features. Indeed, as detailed above, there are diverse mechanisms that underlie boundary element activity and no common sequence or protein features that unite all known boundaries. This lack of common boundary element features makes comprehensive prediction of boundaries difficult. To date, boundary element prediction methods have relied on specific features to identify mechanistically coherent subsets of boundaries. For example, genome-wide distributions of CTCF-binding sites considered together with chromatin domain borders have been used to infer the locations of putative fixed boundaries (36,37). This feature-based approach to boundary element prediction may overlook boundaries that function via diverse and possibly as yet unknown mechanisms.
Recently, a number of genome-wide maps of histone modifications have been computationally analyzed in order to describe chromatin architecture in terms of the distribution of distinct domains within and between cell types. For instance, studies in Drosophila melanogaster (38,39), Caenorhabditis elegans (40) and human (41,42) have characterized the genomic distributions of euchromatic and heterochromatic domains at high levels of resolution. The ability to characterize chromatin domain distributions in this way suggests that it should also be possible to more precisely define the locations of putative chromatin boundaries between domains along with their local properties. To address this issue here, we employed a computational analysis of histone modification maps in human CD4+ T cells. To date, CD4+ T cells represent the single best characterized system for studying chromatin architecture as there exist genome-wide maps for 38 histone modifications and one histone variant (36,43). The existence of multiple (five) repressive modifications, in particular, is a unique aspect of this data set that provides increased resolution for delineating active versus repressive domains. Furthermore, experimentally characterized genome-wide maps of chromatin accessibility (DNase I hypersensitive sites), binding sites for RNA Pol II and Pol III as well as several other protein factors exist for CD4+ T cells along with RNA-seq data for genome expression.
The goal of this study was to take advantage of the detailed genome-wide chromatin maps that exist for CD4+ T cells in order to predict and analyze a collection of putative human boundary elements that is unbiased with respect to the mechanisms of boundary activity. Such a set of predicted boundary elements could help to prioritize experimental interrogation of boundaries and further define the scope of possible boundary element mechanisms. To this end, we developed a boundary element prediction algorithm that does not rely on any previously characterized features of boundary element sequences, such as the binding of specific protein factors (e.g. CTCF), the presence of tRNA or tRNA-derived sequences or the expression of non-coding RNAs. Rather, our approach defines the genomic positions of putative boundaries in cell type-specific manner based solely on the locations of transition points between facultatively active (euchromatic) and repressive (heterochromatic) domains, along with the distributions of Pol II-binding sites. We chose this objective approach to avoid biasing our boundary element predictions with respect to a limited set of previously known features, and more importantly, to allow for the opportunity to discover boundary elements that may operate via novel, previously unreported mechanisms of action. Boundary element prediction proceeded in two steps. First, we defined euchromatic and heterochromatic domains based on the distributions of active versus repressive histone modifications, and the regions between adjacent domains were taken as possible locations for boundary elements. Second, the regions between chromatin domains were further analyzed with respect to the distributions of Pol II-binding sites to more precisely locate putative boundaries.
Application of this two-stage chromatin boundary element prediction algorithm to human CD4+ T-cell chromatin data resulted in the prediction of 2542 cell type-specific boundary elements genome wide. The functional relevance of the predicted boundaries, with respect to facultative chromatin and cell type-specific expression, was supported by the finding that pairs of genes immediately flanking the boundaries are more divergently expressed in CD4+ T cells than in other human cells. Feature analysis of the predicted human boundaries suggests the possibility of several novel and distinct modes of action: (i) predicted boundaries show a distinct local chromatin environment including peaks of open chromatin marked by enrichment for numerous histone acetylations. These results suggest that the establishment of boundaries involves the local action of specific chromatin remodeling proteins, (ii) while many of the predicted boundaries are shown to be bound by the well known insulator protein CTCF, there are a number of boundaries that may function in a CTCF-independent manner via the binding of protein factors that are known to function in chromatin remodeling but were not previously implicated in boundary activity, e.g. EVI1 and YY1, (iii) a number of predicted boundaries show evidence for the action of transcriptional interference including examples of putative tRNA derived boundaries. tRNA genes were previously shown to function as boundaries in yeast (20,31–33) but these are the first examples of putative tRNA derived boundaries in human.
MATERIALS AND METHODS
Datasets of histone modifications and Pol II binding in CD4+ T cells
We used publicly available genome-wide ChIP-seq data for 38 histone modifications and one histone variant (H2A.Z) defined in human CD4+ T cells (36,43). These 39 histone modifications are classified into active histone modifications and repressive histone modifications, based on previous results (43), for use in chromatin domain prediction. Active modifications are positively correlated with gene expression levels and are known to mark euchromatic genomic regions, whereas repressive modifications are negatively correlated with expression levels and mark heterochromatic domains. The 34 active modifications used here are: H2BK5ac, H2BK12ac, H2BK20ac, H2BK120ac, H2AK5ac, H2AK9ac, H2AZ, K3K4ac, H3K9ac, H3K14ac, H3K18ac, H3K23ac, H3K27ac, H3K36ac, H4K8ac, H3K12ac, H4K5ac, H4K16ac, H4K91ac, H2BK5me1, H3K4me1, H3K4me2, H3K4me3, H3K9me1, H3K27me1, H3K36me1, H3K36me3, H3K79me1, H3K79me2, H3K79me3, H3R2me1, H3R2me2, H4K20me1 and H4R3me2. The five repressive modifications are: H3K9me2, H3K9me3, H3K27me2, H3K27me3 and H4K20me3. Genome-wide ChIP-seq data for Pol II binding in CD4+ T cells was also obtained from Barski et al. 2007 (36).
General scheme of chromatin boundary element prediction algorithm
In order to predict chromatin boundary elements in CD4+ T cells, we designed a two-stage algorithm (Figure 1A). First, we employed active versus repressive histone modification distribution information to define the locations of large-scale euchromatic and heterochromatic domains, respectively (Figure 1B). Regions in transitions (RIT) between adjacent euchromatic and heterochromatic domains are taken as possible locations containing chromatin boundary elements. Second, we predicted the specific locations of boundary elements using Pol II binding inside RITs. Boundary elements were taken as 8-kb windows flanking the precise transition points between high versus low Pol II-binding regions. Only RITs with one such Poll II transition point were considered to contain unambiguous boundary elements. Details for each stage of the algorithm are provided below.
Figure 1.

Boundary element prediction algorithm scheme. (A) Pipeline of the boundary element prediction algorithm. (B) Scheme of domain prediction: repressive modifications (R) and active modifications (A) at each genomic site are transformed to positive or negative scores. A maximal-segment algorithm is then applied on the score strings to locate contiguous regions with local maximal cumulative scores; such regions correspond to euchromatic or heterochromatic domains. (C) Scheme of the HMM for boundary element prediction. The two hidden states are heterochromatin and euchromatin. Each state is characterized by distinct emission probabilities of low, medium and high Pol II-binding levels.
Domain localization with a maximal-segment algorithm
Histone modifications were characterized as active versus repressive based on their correlation with gene expression levels as previously described (43). All active modifications were then considered together as a single set for subsequent analysis as were all repressive modifications. In order to infer heterochromatic domains, we set a positive score for each genomic location, which has repressive histone modification ChIP-seq tags and a negative score for each location with active modification tags. The tag counts of repressive and active modifications were further classified as small (≤8 tags), medium (>8 tags and ≤15 tags) and large (>15 tags). Based on Karlin's theorems (44), the scores for individual genomic sites are set as 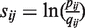 , where i={repressive, active} and j={small, medium, large}. pij represents the estimated frequency of the specific kind of sites in real heterochromatin domains, and qij represents the genomic background frequency of the specific kind of sites. Intuitively, in heterochromatic domains, the frequency of repressively modified sites is higher than the genomic frequency of repressively modified sites and the corresponding scores are positive and larger for sites with more tags. Likewise, the scores for actively modified sites are negative. We use the peri-centromeric regions to estimate pij, since peri-centromeric regions are believed to be heterochromatic regions. Peri-centromeric regions are defined as the regions on both sides of centromeres extending to the most proximal gene as previously described (45). After the scoring step, we applied the maximal-segment algorithm (46) to detect contiguous genomic regions with local maximal cumulative scores. Such contiguous regions represent domains that are enriched with repressive histone modifications, i.e. heterochromatic domains (Figure 1B). As previously suggested (1), we removed the candidate heterochromatic domains that are <10 kb. This cutoff was chosen to reflect the fact that domains, by definition, are thought to be broad and widely spread, and relatively short genomic regions <10 kb are more likely to represent discrete regulatory elements than bona fide domains. The remaining inferred heterochromatic domains were used in subsequent steps.
, where i={repressive, active} and j={small, medium, large}. pij represents the estimated frequency of the specific kind of sites in real heterochromatin domains, and qij represents the genomic background frequency of the specific kind of sites. Intuitively, in heterochromatic domains, the frequency of repressively modified sites is higher than the genomic frequency of repressively modified sites and the corresponding scores are positive and larger for sites with more tags. Likewise, the scores for actively modified sites are negative. We use the peri-centromeric regions to estimate pij, since peri-centromeric regions are believed to be heterochromatic regions. Peri-centromeric regions are defined as the regions on both sides of centromeres extending to the most proximal gene as previously described (45). After the scoring step, we applied the maximal-segment algorithm (46) to detect contiguous genomic regions with local maximal cumulative scores. Such contiguous regions represent domains that are enriched with repressive histone modifications, i.e. heterochromatic domains (Figure 1B). As previously suggested (1), we removed the candidate heterochromatic domains that are <10 kb. This cutoff was chosen to reflect the fact that domains, by definition, are thought to be broad and widely spread, and relatively short genomic regions <10 kb are more likely to represent discrete regulatory elements than bona fide domains. The remaining inferred heterochromatic domains were used in subsequent steps.
In order to infer euchromatic domains, we set positive scores for actively modified sites and negative scores for repressively modified sites, and the other steps were the same as described for inference of heterochromatic domains. As with heterochromatic domains, predicted euchromatic domains <10 kb were eliminated from further consideration. In order to estimate the frequency of actively modified sites in real euchromatic domains, we used the histone modification data for the top 5% of genes that are most highly expressed in CD4+ T cells (47) assuming those genes must be inside euchromatic regions.
After obtaining heterochromatic domains and euchromatic domains in this way, we define a list of RITs between adjacent heterochromatic and euchromatic domains. All possible boundary elements should reside within RITs, but it is not necessary that every RIT contains a boundary element. The next step in the algorithm narrows down these RITs to more precisely define the location of putative boundary elements.
Boundary element localization with a hidden Markov model
In order to more accurately predict specific chromatin boundary element locations within RITs, we took advantage of the fact that euchromatic regions have higher Pol II-binding signal levels than heterochromatic regions. We built a two-state hidden Markov model (HMM) on Pol II-binding data, and employed the Viterbi algorithm to find the most possible hidden state chain (Figure 1C). The two states in this chain are heterochromatin and euchromatin respectively. The emission probabilities of the Pol II signal in euchromatic regions are estimated based on Pol II data in genes which are the top 5% most highly expressed in CD4+ T cells, and the emission probabilities of Pol II signal in heterochromatic regions are estimated based on Pol II data in genes which are not expressed (the lowest 5%). The total size of heterochromatic domains is denoted as s1 and the total size of euchromatic domains as s2. The total size of RITs that go from heterochromatin to euchromatin is denoted as t12, and the total size of RITs that go from euchromatin to heterochromatin as t21. Then the transition probability from heterochromatin to euchromatin is estimated as 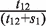 , and the transition probability from heterochromatin to heterochromatin is estimated as
, and the transition probability from heterochromatin to heterochromatin is estimated as 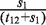 . The transition probability from euchromatin to heterochromatin is estimated as
. The transition probability from euchromatin to heterochromatin is estimated as 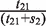 , and the transition probability from euchromatin to euchromatin is estimated as
, and the transition probability from euchromatin to euchromatin is estimated as 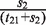 .
.
After running the Viterbi algorithm over all RITs, we recorded the most probable hidden state chains for each RIT. Transition points from one state to the other were taken as possible boundary element locations. To avoid bivalently modified regions and to eliminate small scale variations in Pol II binding, boundary elements were only predicted for RITs that show a single transition point in the hidden state chain. Since boundary elements may be expected to contain a combination of multiple regulatory elements around the precise transition points, putative boundary elements were taken as 8 kb regions around the exact transition points.
DNase I hypersensitivity analysis
Genome-wide DNase I hypersensitivity data in human CD4+ T cells were taken from (48). The genomic locations of DNase I hypersensitive sites are transformed to NCBI36/hg18 using the UCSC Genome Browser program Liftover (49,50). To check whether the predicted boundary elements are more DNase I hypersensitive than flanking regions on average, we extended the predicted boundary elements by 8-kb upstream and downstream and divided the extended regions into 1-kb non-overlapping bins. For each bin, we calculated the average DNase I hypersensitive scores and normalized them by the genomic average DNase I hypersensitive scores.
Histone modification signature analysis
Tag counts for each individual histone modification were computed for predicted boundary elements extended by 8-kb upstream and downstream. Extended regions were divided into 1-kb non-overlapping bins, and for each bin, the average tag counts are normalized by genomic averages.
Analysis of CTCF binding
Genome-wide ChIP-seq data for CTCF binding in human CD4+ T cells were taken from (36). We only considered locations with more than five tags as reliable CTCF-binding sites. To check whether predicted boundary elements have higher affinity to CTCF binding than flanking regions on average, we extended the predicted boundary elements by 8-kb upstream and downstream and divided the extended regions into 1-kb non-overlapping bins. For each bin, we calculated the average CTCF tag counts and normalized them by the genomic average CTCF tag count for 1-kb regions.
TFBS analysis
In order to look for putative protein factors associated with predicted chromatin boundary elements, we used the ‘TFBS Conserved’ track from the UCSC Genome Browser. We gathered those computationally predicted conserved TFBS (with Z-score >1.96) inside predicted boundary elements. For each transcription factor, we counted the number of its appearance within boundary elements and statistically tested whether the specific transcription factor is significantly associated with boundary elements using the hypergeometric test.
Boundary element transcription analysis
RNA-seq data of transcription in human CD4+ T cells were taken from (51). We extended the putative chromatin boundary elements by 8-kb upstream and downstream and divided them into 1-kb non-overlapping bins. We calculated the average non-protein-coding RNA-seq tag counts for each bin and normalized them by the genomic average tag counts. The data was then log2 transformed. Predicted boundary elements were classified into two groups: boundaries containing RNA genes and boundaries without RNA genes, and the above calculations were done on the two groups of boundaries separately. The annotations of RNA gene locations are from the ‘RNA gene’ track (52,53) on UCSC Genome Browser.
Gene expression analysis
Gene expression profiles were taken from (47). For genes located within predicted euchromatic domains and heterochromatic domains, we calculated their average expression levels in human CD4+ T cells. For each predicted boundary element, we took the two genes most proximal to it on the two opposite sides (the euchromatic side and the heterochromatic side) and calculated the expression differences between these pairs for CD4+ T cells and for all other tissues together.
Gene function annotations
Gene Ontology (GO) analysis and KEGG pathway analysis were performed using MSigDB (54,55) for predicted euchromatic domains with high gene densities (>1 gene/20 kb).
RESULTS
Datasets and chromatin boundary element prediction algorithm
In recent years, a substantial body of data detailing the chromatin structure of eukaryotic genomes has been accumulated. For the human genome in particular, there are now genomic maps with experimentally characterized locations of numerous histone modifications as well as binding sites for a variety of proteins. Such data provide opportunities for the discovery of novel chromatin related regulatory elements across the genome.
Human CD4+ T cells represent one of the best characterized systems for the genome-scale analysis of chromatin. Keji Zhao and colleagues have used chromatin immunoprecipitation followed by high-throughput sequencing experiments (ChIP-seq) to generate genome-wide maps for 38 histone modifications and one histone variant (H2A.Z), CTCF binding, Pol II binding and Pol III binding (36,43,51). Chromatin accessibility in CD4+ T cells has been evaluated genome wide using DNase I hypersensitivity assays coupled to high-throughput sequencing (48), and genome-wide CD4+ T cell expression levels have been determined using microarray and RNA-seq technologies (47,51).
We took advantage of the existence of these genome-scale chromatin datasets to facilitate the discovery of boundary elements in the human genome. The goal of this work was to provide a comprehensive list of likely boundary element candidates, and then to evaluate the features of these putative boundaries with respect to possible mechanisms of action. We designed a two-stage algorithm to predict the locations of putative boundary elements (Figure 1A). In the first stage, we defined the locations of large-scale active (euchromatic) and repressive (heterochromatic) chromatin domains based on the genomic distributions of active and repressive histone modifications. The histone modifications analyzed here were characterized active or repressive as previously described (see ‘Materials and Methods’ section) (43). For each genomic position, a specific score (negative or positive) was assigned according to the relative abundance of active or repressive modifications. A maximal-segment algorithm was then applied to the resulting string of scores to locate contiguous genomic regions with maximal local cumulative scores (Figure 1B). The maximal-segment algorithm was chosen because it can detect such contiguous regions over variant lengths, and it is robust to small-scale stochastic noise in the ChIP-seq data. The maximal-segment algorithm also worked well here because the parameters that define the relative negative or positive scores can be directly estimated from the ChIP-seq data. Further details on our maximal-segment algorithm for domain detection can be found in the ‘Materials and Methods’ section (see Domain localization with a maximal-segment algorithm).
We searched for chromatin boundary elements that reside within regions between adjacent euchromatic and heterochromatic domains—hereafter referred to as RITs. However, it should be noted that not all RITs will necessarily contain discretely located boundary elements. For instance, some RITs may contain regions with fuzzy patterns of active and repressive modification distributions that would not allow for precise delineation of boundary element locations. Such fuzzy patterns may represent boundaries that act via PEV-related mechanisms, owing to different boundary locations among heterogeneous cell populations, and these imprecisely located boundaries will not be detected by our method. Furthermore, because the sizes of RITs can be relatively large (>50 kb) in some cases, a method is needed to narrow down the genomic regions where predicted boundary elements can be located. In light of both of these issues, we developed a second stage of the algorithm that uses a HMM of Pol II-binding distributions along RITs in order to more precisely locate boundary elements (Figure 1C). This approach is based on the rationale that euchromatin is transcriptionally active, whereas heterochromatin is largely transcriptionally silent. Accordingly, euchromatin is expected to have higher levels of Pol II binding, and heterochromatin is expected to have lower levels of Pol II binding. Furthermore, Pol II protein complexes are known to associate with proteins that have acetyltransferase and/or chromatin remodeling functions (56). Thus, boundary elements are expected to be located in genomic regions with particularly sharp transitions between low and high Pol II binding; HMMs are ideal for delineating such abrupt transitions.
HMMs were used to model RITs by predicting the facultative chromatin state—euchromatin or heterochromatin—for each genomic site that best explains the Pol II-binding distribution along each RIT. To do this, the Viterbi algorithm was used to infer the most probable chromatin state chain along the RITs based on Pol II-binding emission probabilities and chromatin state transition probabilities (Figure 1C). Details on the HMM we used for boundary element localization can be found in the ‘Materials and Methods’ section (see Boundary element localization with a HMM). After obtaining the most probable hidden state chains of euchromatin and heterochromatin, we removed RITs that contain more than one transition point between the two chromatin states, since these represent ambiguously located boundaries. Sequence features of the remaining RITs are summarized in Supplementary Table S1. For RITs with single chromatin state transition points, we take 8-kb regions centered on those transition points as putative boundary element regions (Supplementary Files S1 and S2). The 8-kb window size was chosen to strike a balance between the utility of precisely locating predicted boundary elements and the biological reality that boundary element activity may be spread over multiple adjacently located regulatory elements.
Chromatin domain localization
In the first stage of the algorithm (Figure 1B), we predicted the locations of large-scale active and repressive chromatin domains, i.e. facultative euchromatic and heterochromatic regions. An example of several adjacent euchromatic and heterochromatic domains on chromosome 2 can be seen in Figure 2. The predicted euchromatic domains are enriched with the active histone modification H3K79me1, and the predicted heterochromatic domains are enriched with the repressive modification H3K27me2. The same pattern can be seen when all 34 active and all 5 repressive modifications are considered together (Supplementary Figure S1). In this example, we also observe higher Pol II binding and RNA-seq expression levels in the predicted euchromatic domains than seen for the predicted heterochromatic domains (Figure 2), consistent with the expectation that euchromatin is more actively transcribed than heterochromatin. Furthermore, predicted euchromatic domains genome wide have significantly higher average CD4+ T cell expression levels than the predicted heterochromatic domains (Figure 3; Mann–Whitney U test P < 1E–10). The observations on expression levels serve to validate the maximum segment algorithm we use to delineate active (euchromatic) and repressive (heterochromatic) domains based on the analysis of histone modification data alone.
Figure 2.

Example of predicted chromatin domains. An ideogram of chromosome 2 shows the cytogenetic banding pattern along with the location of this specific example. The distributions of ChIP-seq tag mapping peaks for the active histone modification H3K79me1 (red bars), the repressive histone modification H3K27me2 (blue bars), Pol II binding (black bars) and RNA-seq tags (purple bars) are shown in separate tracks. The predicted euchromatic domains (red bands) and heterochromatic domains (blue bands) are shown in the tracks denoted as ‘Euchromatin’ and ‘Heterochromatin’. The locations of RefSeq Genes are shown below the chromatin domains.
Figure 3.

Validation and analysis of predicted chromatin domains. (A) Average human CD4+ T cell expression levels for genes located in predicted euchromatic domains (gray bar) and heterochromatic domains (black bar). (B) Average RNA-seq tags per site in CD4+ T cell for predicted euchromatic domains (gray bar) and heterochromatic domains (black bar).
We also used GO and KEGG pathway analyses to interrogate the functional relevance of the euchromatic and heterochromatic domains predicted with our algorithm. Genes found in predicted euchromatic domains are enriched with functional terms and pathways related to CD4+ T cell functions, such as defense response (GO), systemic lupus erythematosus (KEGG) and antigen processing and presentation (KEGG) (Supplementary Table S2).
Boundary element prediction
Application of the two-stage maximal segment algorithm and HMM approach (Figure 1) to the CD4+ T cell ChIP-seq data resulted in the identification of 2542 putative chromatin boundary elements (Supplementary File S2). Sequence features of these boundary elements are summarized in Supplementary Table S1. It should be noted that our prediction method is not mechanistically biased in the sense that it does not rely on any previously known features of boundary element sequences, e.g. CTCF protein binding (37,57), the presence of tRNA genes (31) or the expression of non-coding RNAs originating from SINE repeats (18,35). By predicting boundaries in this way, without regard to previously known features, we can evaluate the associations of putative boundaries with such features a posteriori and, more importantly, look for novel boundary element related features, which may be indicative of as yet unknown boundary element mechanisms.
Examples of three predicted chromatin boundaries are shown in Figure 4; the locations of the boundaries are compared to the locations of the chromatin domains defined by active and repressive histone modification distributions along with the locations of CTCF binding, Pol II binding and RNA-seq expression levels. All of these boundaries are located close to the edges of borders between adjacent chromatin domains and at sharp transition points of Pol II binding and RNA-seq levels. The two boundaries shown in Figure 4A are co-located with CTCF-binding sites. The boundary shown in Figure 4B shows a similar chromatin profile to those in Figure 4A but is not related to CTCF binding. More detailed illustrations of these boundaries showing all of the individual histone modifications can be found in Supplementary Figures S2 and S3.
Figure 4.

Examples of predicted chromatin boundary elements. (A) Examples of predicted boundary elements with CTCF binding. (B) Example of a predicted boundary element without CTCF binding. The predicted boundary elements are shown as green bands. ChIP-seq peaks for active and repressive histone modifications, CTCF binding, Pol II binding and RNA-seq tags along with the locations of euchromatic domains, heterochromatic domains and RefSeq genes are illustrated as separate tracks (as in Figure 2).
In order to test the relevance of the predicted chromatin boundaries to facultative chromatin and cell type-specific gene regulation, we compared the expression level differences for pairs of genes located on immediately opposing sides of the boundaries for CD4+ T cells to their expression level differences among a set of 78 different human tissues and cell types (47). If the predicted boundary elements do in fact represent CD4+ T cell specific regulatory elements that help to establish facultative chromatin domains, then the expression level differences of gene pairs that flank the boundaries should be greater for CD4+ T cells than for other tissue-types. Consistent with this expectation, gene pairs that flank the predicted boundaries have significantly greater expression level differences in CD4+ T cells than in other tissues and cell-types (Figure 5; Mann–Whitney U test P < 1E–10).
Figure 5.

Expression differences between gene pairs that flank boundary elements. Expression differences of gene pairs located on immediately opposing sides of predicted boundary elements are shown for CD4+ T cells (gray bar) and 78 other human tissues together (black bar).
In an attempt to further evaluate the potential functional significance of the boundaries predicted here, we searched for overlaps between the predictions and previously experimentally characterized boundaries. Among the few known boundaries that have been functionally verified, only one boundary element, the BEAD-1 element, was identified in human T cells. BEAD-1 is a ∼2-kb region located between the divergently transcribed Vδ3 and TEA gene segments within the T cell receptor α/δ locus, and it has been shown to have enhancer-blocking activity (58). BEAD-1 is located within a RIT defined by our algorithm and overlaps one of the predicted boundary elements (Figure 6 and Supplementary Figure S4). Previously, the BEAD-1 sequence was shown to have a CTCF-binding site and its enhancer blocking activity was found to be CTCF dependent in an erythroleukemia cell line (59). However, there is no evidence for CTCF binding of BEAD-1 from the genome-wide ChIP-seq analysis of CD4+ T cells (36) suggesting that boundary element activity at this locus may be CTCF independent in some conditions.
Figure 6.

Co-location of a predicted boundary element with BEAD-1. A boundary element predicted by our method (green band) is shown to overlap with the experimentally characterized BEAD-1 boundary element (purple band). The BEAD-1 element is located between the Vδ3 and TEA gene segments (black boxes) of the T cell receptor α/δ locus. ChIP-seq peaks for active and repressive histone modifications, CTCF binding, Pol II binding and RNA-seq tags along with the locations of euchromatic domains, heterochromatic domains are illustrated as separate tracks (as in Figure 4). The inset shows greater detail at the BEAD-1 locus.
Chromatin features of predicted boundaries
The boundary element predictions reported here are based solely on chromatin states inferred from histone modifications and Pol II binding and do not rely on any previously characterized features of boundary element sequences. Since boundary elements are known to have diverse mechanisms of action (13,14,60,61), we analyzed our predicted boundaries for enrichment with a number of previously characterized boundary features and also with respect to as yet unknown features that may suggest novel mechanisms of boundary element activity.
We evaluated the chromatin environment of predicted boundaries using enrichment analysis of a number of genome-scale chromatin data sets. To do this, the 2542 predicted boundary element regions were co-oriented and center aligned in such a way as to observe 8-kb boundary element regions flanked by 8-kb heterochromatic and euchromatic regions respectively. Predicted boundary elements show marked enrichment for DNase I hypersensitivity consistent with an open chromatin environment (Figure 7A). Twelve histone acetylation marks all show similar peaked patterns of enrichment over predicted boundaries compared to flanking heterochromatic and euchromatic regions, suggesting that the predicted boundary elements are specifically acetylated to facilitate opening of the chromatin and recruitment of sequence-specific DNA-binding factors (Figure 7B).
Figure 7.

Chromatin signatures of predicted boundary elements. (A–F) The 8-kb boundary regions are shown together with 8-kb flanking heterochromatic and euchromatic regions. Normalized levels of DNase I hypersensitivity (A), fold enrichment profiles of 12 histone acetylations (B), normalized levels of CTCF binding (C), normalized levels of YY1 binding (D), fold enrichment profiles of H3K27 mono-, di- and tri-methylations (E) and fold enrichment profiles of H3K9 mono-, di- and tri-methylations (F) are compared for flanking regions and boundaries.
Levels of binding for the CTCF insulator protein are also elevated in predicted boundary element regions compared to adjacent heterochromatic and euchromatic regions (Figure 7C). Thus, the apparent acetylation activity at predicted boundary elements may be recruited by specific protein factors such as CTCF. The importance of CTCF in establishing chromatin regulatory domains recently was underscored by results indicating that numerous functional CTCF-binding sites are constitutively occupied among different cell types, and more remarkably, conserved among syntenic regions in the human, mouse and chicken genomes (62). However, it should be noted that only a minority of predicted boundary elements (777 or 30.6%) contain CTCF-binding sites (Supplementary File S3), suggesting that at some of the predicted boundaries acetylation events occur in a CTCF independent manner or may be indicative of the recruitment of different DNA-binding factors.
We used the conserved TFBS data from the UCSC Genome Browser (49,50) to search for protein-binding sites that are significantly enriched among the set of predicted chromatin boundaries (Supplementary File S4). There are a number of significantly enriched TFBS that interact with proteins directly or indirectly involved in chromatin remodeling events (Table 1). For example, EVI1, CEBP, CREBP1, USF and YY1 are all involved in chromatin remodeling via their interactions with chromatin modifying enzymes such as HAT, HDAC and HMT (63–69). In addition, the transcription factor USF has previously been implicated as mediating chromatin boundary element activity (70,71). The presence of distinct TFBS often overlap at individual boundaries indicating that a number of predicted boundaries have common binding sites (Supplementary Figure S5).
Table 1.
Protein factors enriched in predicted boundary elements
| Protein | No.a | P-valueb | Annotationsc |
|---|---|---|---|
| EVI1 | 382 | 0.022 | Interacts with histone deacetylase, histone methyltransferases and CBP and P/CAF |
| CEBP | 249 | 2.27E-17 | Interacts with CBP and p300 and promotes histone acetylation |
| YY1 | 157 | 1.44E-17 | Directs histone deacetylases and HATs to promoter |
| CREBP1 | 150 | 5.87E-24 | Essential in H2B and H4 acetylation, can interact with CBP HAT domain |
| USF | 140 | 2.50E-28 | Recruits histone modifications at vertebrate boundary elements |
aThe number of boundary elements containing the corresponding protein factor-binding sites.
bThe statistical significance of the enrichment of the protein factor in predicted boundary elements assessed by hypergeometric test.
cFunctional annotations for the proteins based on the relevant literature (cited in the text).
Inferences on protein binding based on the presence of TFBS are prone to false positives (although the use of conserved sites greatly mitigates this possibility) and also do not yield information on cell type-specific binding. For these reasons, we searched for ChIP-seq data sets from CD4+ T cells to validate the TFBS observed to be enriched at our predicted boundaries with experimentally characterized cell type-specific binding events. There are CD4+ T cell ChIP-seq data for YY1 (72), and analysis of these data reveal that the predicted boundaries are significantly overrepresented for YY1 binding (n = 918; P ≤ 10−16 hypergeometric test), and YY1-binding peaks at boundaries relative to adjacent chromatin (Figure 7D and Supplementary File S5). Interestingly, there are far more boundaries bound by YY1 (n = 918) than boundaries with conserved YY1 TFBS (n = 157). This may be due to the presence of lineage-specific or non-canonical YY1-binding site motifs among the predicted boundaries. Consistent with observations that YY1 is a cofactor of CTCF for X-chromosome inactivation (73), there is a highly significant overlap between boundaries bound by CTCF and YY1 (n = 534; P ≤ 10−113 hypergeometric test) suggesting the possibility of synergistic action between these two factors. Nevertheless, there remain 384 boundaries with YY1 binding only suggesting CTCF-independent mechanisms of action. For example, evidence showing that YY1 can interact with both HDAC and HAT (74–80) led to a potential model proposing that YY1 can activate or repress transcription via changing the local chromatin environment (78). YY1 was also shown to be able to interact with components of nuclear matrix (81,82), which may also facilitate partitioning of active and repressive chromatin domains.
The specific methylation status, mono- di- or tri-methylation, of the H3K27 and H3K9 histone marks show divergent trends across predicted boundary elements containing regions and adjacent heterochromatic and euchromatic regions (Figure 7E and F). H3K27 and H3K9 mono-methylation (H3K27me1 and H3K9me1) levels increase steadily from facultative heterochromatic domains across boundary element containing regions and into euchromatic domains. On the other hand, di- and tri-methylation of the same residues (H3K27me2, H3K27me3, H3K9me2 and H3K9me3) gradually decrease from heterochromatin through the boundary element regions to euchromatin.
A number of other histone methylation marks, along with non-protein-coding RNA-seq accumulation, also show steadily increasing levels across boundary element regions from facultative heterochromatin to euchromatin (Figure 8A and B), consistent with a gradual opening of the chromatin. However, all of the modifications of histone H3K4 analyzed here (H3K4me1, H3K4me2, H3K4me3 and H3K4ac) show distinct peaks over the predicted boundaries relative to flanking heterochromatic and euchromatic regions (Figure 8C). These particular histone modifications have been associated with promoter and/or enhancer activity, suggesting that boundary element mechanisms may be related to initiation of transcription (14), in the case of promoters, and/or perturbation of the local chromatin environment, as has been suggested for enhancers (13). The enrichment profiles of all histone modifications could be found in Supplementary Figures S6, S7 and S8.
Figure 8.
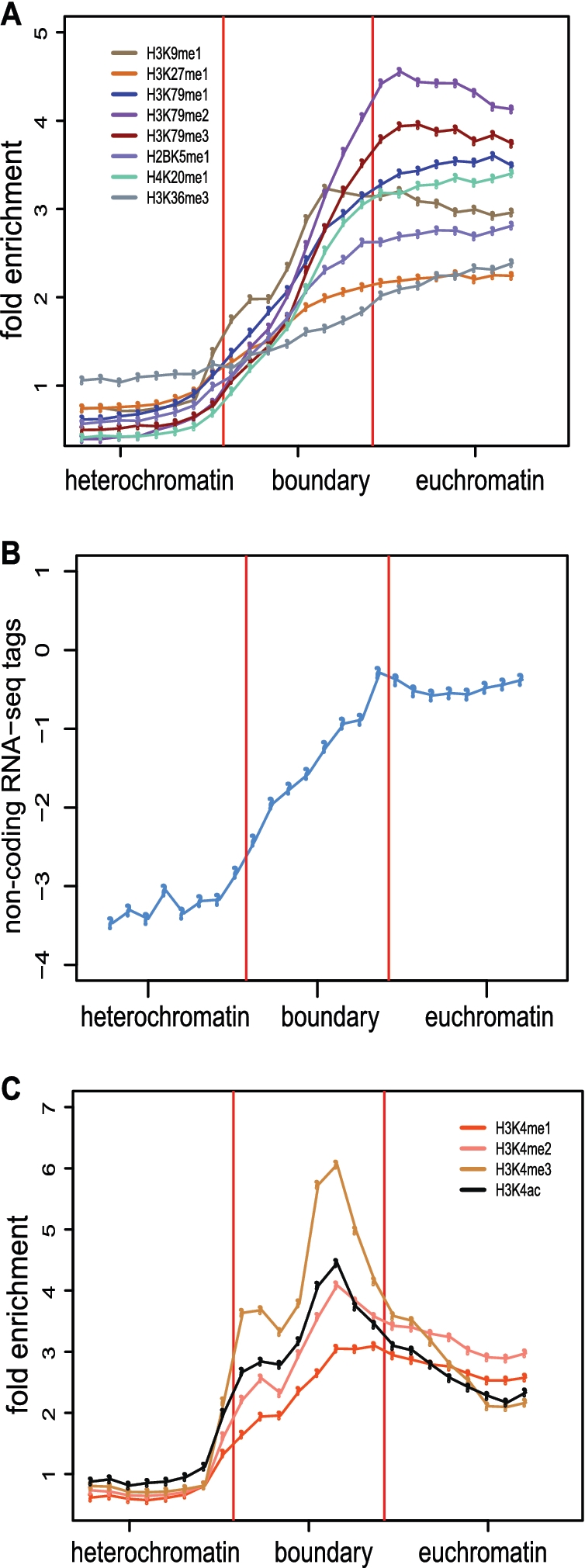
Chromatin and transcriptional transitions across predicted boundary elements. (A–C) The 8-kb boundary regions are shown together with 8-kb flanking heterochromatic and euchromatic regions. Fold enrichment profiles of eight histone methylations (A), log2 transformed normalized non-protein-coding RNA-seq tags (B) and fold enrichment profiles of H3K4 histone modifications (C) are compared for flanking regions and boundaries.
Transcriptional interference at predicted boundaries
Transcription of non-coding RNA has been shown to be important for boundary element function from yeast to higher eukaryotes (18,31,32,35). Therefore, we analyzed RNA-seq data from CD4+ T cells in order to evaluate whether our predicted boundaries are transcriptionally active (51). Across the predicted boundary elements, RNA-seq levels increase steadily with the transition from heterochromatin (low levels) to euchromatin (high levels) (Figure 8B). Interestingly, a subset of 77 predicted boundary elements contain annotated non-coding RNA genes (52,53) and show distinct peaks of RNA accumulation relative to the adjacent chromatin domains (Figure 9A), which coincide with Pol III binding (Figure 9B). The RNA-seq peaks indicate that these particular boundary locations are transcribed at markedly higher levels than genomic background consistent with a role for transcriptional interference.
Figure 9.

Features of boundary elements containing RNA genes. (A and B) The 8-kb boundary regions are shown together with 8-kb flanking heterochromatic and euchromatic regions. Log2 transformed normalized RNA-seq tags (A) and normalized Pol III-binding levels (B) of boundary elements containing RNA genes are compared for flanking regions and boundary regions. (C) Example of boundary element containing tRNA genes. The predicted boundary element is shown as the green band. ChIP-seq peaks for active and repressive histone modifications, CTCF binding, Pol II binding and RNA-seq tags along with the locations of euchromatic domains, heterochromatic domains and RefSeq genes are illustrated as separate tracks (as in Figure 2). Pol III binding (yellow bars) and RNA genes are also shown separately.
Figure 9C shows an example of a predicted boundary element that contains a cluster of four tRNA genes along with peaks of RNA-seq expression and Pol III and CTCF binding, suggesting a possible relationship between CTCF binding and tRNA gene transcription. The example shown in Figure 9C suggests that, similar to yeast, tRNA genes in the human genome may operate as genomic boundaries, although definitive assessment of their functional significance awaits further experimental analysis. Consistent with this prediction, clusters of mouse tRNA genes have been shown to encode chromatin barrier activity (83).
DISCUSSION
A chromatin based approach to unbiased boundary element prediction
Boundary elements are known to organize chromatin into functionally distinct domains and to prevent regulatory crosstalk between domains. Distinct boundary elements may act through a variety of mechanisms, and accordingly boundaries have been characterized phenotypically based on their activity rather than the presence of characteristic features. Thus, boundary element prediction algorithms that use pattern detection methods to search for known boundary element characteristic features will result in biased sets of predictions that only reflect one or another of the known mechanisms of action. This fundamental challenge to the computational prediction of boundary elements motivated our development and application of an unbiased algorithm that predicts the locations of putative boundary elements genome wide based on their functional consequences, with respect to both chromatin and transcription states, as opposed to any intrinsic characteristics that they may possess.
Our approach to boundary element prediction relies on the delineation of adjacent active (euchromatic) and repressive (heterochromatic) domains based on the genomic distributions of active versus repressive histone modifications. RITs between adjacent chromatin domains are further interrogated for the presence and location of putative boundaries using distributions of Pol II-binding sites that serve as marks of active cell type-specific transcription. Application of this two-stage chromatin boundary element prediction algorithm (Figure 1) to CD4+ T cell data resulted in the prediction of 2542 boundary elements across the human genome. The role of these predicted boundary elements in cell type-specific chromosomal domain organization was confirmed by the finding that genes immediately flanking boundaries are more highly differentially expressed in CD4+ T cells than seen for other human cells/tissues (Figure 5). Having predicted boundary elements in this way, we then analyzed the putative boundaries for the presence of a variety of features that may yield specific clues as to their potential mechanisms of action.
Models for human boundary element activity
Previous studies on boundary elements have suggested competing models that explain the mechanisms underlying boundary element activity. The fixed model for boundary element activity implicates specific DNA sequences and their associated proteins, whereas the transcriptional interference model emphasizes the role of transcription from non-protein-coding transcriptional units. We have previously noted that these two models are not necessarily mutually exclusive (14). Under the fixed model, boundaries are precisely located and contain specific sequences that form discrete physical barriers between domains. Specific sequence features are also needed to recruit Pol II and Pol III machineries for the transcriptional interference model, and transcriptional units that act as boundaries may also form physical barriers that block the propagation of repressive chromatin. The features uncovered for our predicted boundary elements can similarly be taken to suggest that the mechanisms of human boundary activity include aspects of both the fixed and transcriptional interference models.
Analysis of the predicted boundary elements and surrounding RITs revealed two main features: (i) position-specific acetylation and open chromatin coincident with the recruitment of transcription factors such as EVI1, YY1 and USF (Figure 7A, B, D and Table 1), and (ii) a gradual transition across RITs, from heterochromatin to euchromatin, of increasing histone lysine methylation and non-protein-coding RNA levels (Figures 7E, F, 8A and B). Considered together, these two observations lead us to propose a possible model for human boundary element activity (Figure 10). Under this model, the specific positions of boundaries are established via the local recruitment of histone acetyltransferase (HAT) activity and transcription factors leading to the expression of non-protein-coding RNAs (Figure 10A). Boundary element function is maintained more broadly across RITs by the superposition of distinct and opposing chromatin modifying activities leading to the observed gradual transitions between heterochromatic and euchromatic histone lysine methylation and mediated by transcriptional interference (Figure 10B).
Figure 10.

Model for human chromatin boundary element activity. Broader RITs from heterochromatin to euchromatin are shown along with more precisely located boundary elements. (A) Boundary element locations are characterized by position-specific open chromatin environment (DNase I Hypersensitivity Site) and hyperacetylation (Ac) that are coincident with the recruitment of transcription factors (TF) and non-protein-coding RNA transcription. (B) RITs are characterized by gradual changes in the levels of histone methylation (me) from heterochromatin to euchromatin.
Predicted boundary elements reside in regions of distinctly open chromatin and also show position-specific accumulations of 12 different histone acetylation marks (Figure 7A and B). Previous studies have suggested boundary element activity is dependent upon the local recruitment of HAT activities to counteract the spread of repressive chromatin (71,84,85). The patterns of histone lysine acetylation enrichment observed at position-specific location within predicted boundaries are in agreement with already reported prominent role for histone acetylation at boundary elements and further corroborate the boundary prediction method used here.
Along with the position-specific chromatin features and recruitment seen at predicted boundaries, we also observe distinct chromatin dynamics spread across the RITs that lie between adjacent facultative heterochromatic and euchromatic domains. For instance, H3K27 and H3K9 mono-methylation levels increase steadily from heterochromatic domains across boundary element containing regions and into euchromatic domains, whereas H3K27 and H3K9 di- and tri-methylation levels gradually decrease across the same intervals (Figure 7E and F). This pattern can be taken to indicate a unidirectional activity of histone demethylation across RITs from heterochromatin to euchromatin. At the same time, a number of other mono- di- and tri-methylation histone marks show steady accumulations across RITs from heterochromatin to euchromatin (Figure 8A) and are indicative of increased transcriptional activity (Figure 8B) and/or the action of chromatin modifying enzymatic complexes associated with transcriptional elongation.
H3K79 mono-, di- and tri-methylation all show progressively increasing levels across RITs from facultative heterochromatin to euchromatin (Figure 8A). While the exact function of H3K79 methylation is currently unknown, accumulation of these marks, catalyzed by the lysine methyltransferase (KMT) DOT1 (86), is correlated with actively transcribed protein-coding genes (87). Accordingly, it is possible that H3K79 methylation also marks active transcription of non-protein-coding RNAs across RITs as observed here (Figure 8A and B). In fact, H3K79 methylation has previously been implicated in the stable maintenance of distinct chromatin states in yeast and mammalian cells (88), and our data also suggests a possible, and previously unexplored, role for DOT1 in the establishment and maintenance of chromatin boundaries.
Transcriptional regulators at predicted boundary elements
The observations that predicted boundary elements contain binding site motifs for a number of proteins implicated in both the regulation of transcription and chromatin remodeling (Table 1), along with experimentally characterized YY1 binding (Figure 7D), are consistent with a role for transcriptional interference in human boundary element activity. Involvement of transcription factors capable of maintaining a local active chromatin environment at boundaries has previously been reported by the Felsenfeld group in the context of the USF1 factor (70). USF transcription factors can regulate Pol II transcription via direct interaction with components of the basic transcription machinery, such as TFIID and TBP associated factors (89), or through the recruitment of co-factors such as the HAT PCAF or the H3K4 histone methyltransferase SET7/92 (71). Here, we observe a significant enrichment of the USF-binding site motif (E-box element) among predicted boundaries. Thus, we speculate that USF participates in the establishment and/or maintenance of human boundary element activity by triggering transcriptional interference, which may be mediated, at least in part, by the action of the aforementioned co-factors.
EVI1 is another sequence-specific transcription regulator with binding sites that are over-represented among the boundary elements predicted here (Table 1). EVI1 has been shown to interact with the HAT PCAF, the histone deacetylase HDAC1 and the histone methyltransferases SUV39H1 and G9A (63,64). Thus, we speculate that EVI1 may function in boundary element activity by serving as a switch between distinct chromatin remodeling activities thereby mediating the transition from heterochromatin to euchromatin in a cell type-dependent manner.
CONCLUSIONS AND PROSPECTS
Chromatin boundary elements are major players in genome organization and regulation, but at this time there are relatively few examples of known boundary elements. Here, we report a large collection of putative boundary elements for CD4+ T cells that span the entire human genome. The boundaries reported here are computational predictions and thus must be treated with all due caution; nevertheless, analysis of the features of these boundaries yields results that are consistent with their roles as chromatin related regulatory elements. We hope that the boundaries predicted here can serve as a prioritized list of targets for further experimental validation. If validated experimentally, the predictions reported here could help to substantially enlarge the catalog of known chromatin boundary elements. Our feature analysis of the predicted boundaries also raises the possibility of a mechanism of chromatin boundary activity in the human genome related to transcriptional interference. This possibility awaits further detailed investigations.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Alfred P. Sloan Research Fellowship in Computational and Evolutionary Molecular Biology (BR-4839 to J.W. and I.K.J.); Georgia Tech Integrative BioSystems Institute pilot program grant (to J.W. and I.K.J.); the Buck Institute Trust Fund (to V.V.L.). Funding for open access charge: Alfred P. Sloan Research Fellowship; Georgia Institute of Technology; and Buck Institute for Age Research.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to acknowledge members of the Jordan and Lunyak labs along with Lluis Montoliu for useful discussions.
REFERENCES
- 1.Thurman RE, Day N, Noble WS, Stamatoyannopoulos JA. Identification of higher-order functional domains in the human ENCODE regions. Genome Res. 2007;17:917–927. doi: 10.1101/gr.6081407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pauler FM, Sloane MA, Huang R, Regha K, Koerner MV, Tamir I, Sommer A, Aszodi A, Jenuwein T, Barlow DP. H3K27me3 forms BLOCs over silent genes and intergenic regions and specifies a histone banding pattern on a mouse autosomal chromosome. Genome Res. 2009;19:221–233. doi: 10.1101/gr.080861.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 4.Bernstein BE, Humphrey EL, Erlich RL, Schneider R, Bouman P, Liu JS, Kouzarides T, Schreiber SL. Methylation of histone H3 Lys 4 in coding regions of active genes. Proc. Natl Acad. Sci. USA. 2002;99:8695–8700. doi: 10.1073/pnas.082249499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bernstein BE, Kamal M, Lindblad-Toh K, Bekiranov S, Bailey DK, Huebert DJ, McMahon S, Karlsson EK, Kulbokas EJ, 3rd, Gingeras TR, et al. Genomic maps and comparative analysis of histone modifications in human and mouse. Cell. 2005;120:169–181. doi: 10.1016/j.cell.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 6.Bernstein BE, Meissner A, Lander ES. The mammalian epigenome. Cell. 2007;128:669–681. doi: 10.1016/j.cell.2007.01.033. [DOI] [PubMed] [Google Scholar]
- 7.Boyer LA, Plath K, Zeitlinger J, Brambrink T, Medeiros LA, Lee TI, Levine SS, Wernig M, Tajonar A, Ray MK, et al. Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature. 2006;441:349–353. doi: 10.1038/nature04733. [DOI] [PubMed] [Google Scholar]
- 8.Roh TY, Cuddapah S, Cui K, Zhao K. The genomic landscape of histone modifications in human T cells. Proc. Natl Acad. Sci. USA. 2006;103:15782–15787. doi: 10.1073/pnas.0607617103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roh TY, Cuddapah S, Zhao K. Active chromatin domains are defined by acetylation islands revealed by genome-wide mapping. Genes Dev. 2005;19:542–552. doi: 10.1101/gad.1272505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dillon N, Sabbattini P. Functional gene expression domains: defining the functional unit of eukaryotic gene regulation. Bioessays. 2000;22:657–665. doi: 10.1002/1521-1878(200007)22:7<657::AID-BIES8>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 11.Kamakaka RT, Thomas JO. Chromatin structure of transcriptionally competent and repressed genes. EMBO J. 1990;9:3997–4006. doi: 10.1002/j.1460-2075.1990.tb07621.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Capelson M, Corces VG. Boundary elements and nuclear organization. Biol Cell. 2004;96:617–629. doi: 10.1016/j.biolcel.2004.06.004. [DOI] [PubMed] [Google Scholar]
- 13.Gaszner M, Felsenfeld G. Insulators: exploiting transcriptional and epigenetic mechanisms. Nat. Rev. Genet. 2006;7:703–713. doi: 10.1038/nrg1925. [DOI] [PubMed] [Google Scholar]
- 14.Lunyak VV. Boundaries. Boundaries … boundaries??? Curr. Opin. Cell Biol. 2008;20:281–287. doi: 10.1016/j.ceb.2008.03.018. [DOI] [PubMed] [Google Scholar]
- 15.Raab JR, Kamakaka RT. Insulators and promoters: closer than we think. Nat. Rev. Genet. 2010;11:439–446. doi: 10.1038/nrg2765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gdula DA, Gerasimova TI, Corces VG. Genetic and molecular analysis of the gypsy chromatin insulator of Drosophila. Proc. Natl Acad. Sci. USA. 1996;93:9378–9383. doi: 10.1073/pnas.93.18.9378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Labrador M, Corces VG. Setting the boundaries of chromatin domains and nuclear organization. Cell. 2002;111:151–154. doi: 10.1016/s0092-8674(02)01004-8. [DOI] [PubMed] [Google Scholar]
- 18.Lunyak VV, Prefontaine GG, Nunez E, Cramer T, Ju BG, Ohgi KA, Hutt K, Roy R, Garcia-Diaz A, Zhu X, et al. Developmentally regulated activation of a SINE B2 repeat as a domain boundary in organogenesis. Science. 2007;317:248–251. doi: 10.1126/science.1140871. [DOI] [PubMed] [Google Scholar]
- 19.Kellum R, Schedl P. A position-effect assay for boundaries of higher order chromosomal domains. Cell. 1991;64:941–950. doi: 10.1016/0092-8674(91)90318-s. [DOI] [PubMed] [Google Scholar]
- 20.Oki M, Kamakaka RT. Barrier function at HMR. Mol. Cell. 2005;19:707–716. doi: 10.1016/j.molcel.2005.07.022. [DOI] [PubMed] [Google Scholar]
- 21.Recillas-Targa F, Pikaart MJ, Burgess-Beusse B, Bell AC, Litt MD, West AG, Gaszner M, Felsenfeld G. Position-effect protection and enhancer blocking by the chicken beta-globin insulator are separable activities. Proc. Natl Acad. Sci. USA. 2002;99:6883–6888. doi: 10.1073/pnas.102179399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Udvardy A, Maine E, Schedl P. The 87A7 chromomere. Identification of novel chromatin structures flanking the heat shock locus that may define the boundaries of higher order domains. J. Mol. Biol. 1985;185:341–358. doi: 10.1016/0022-2836(85)90408-5. [DOI] [PubMed] [Google Scholar]
- 23.Noma K, Allis CD, Grewal SI. Transitions in distinct histone H3 methylation patterns at the heterochromatin domain boundaries. Science. 2001;293:1150–1155. doi: 10.1126/science.1064150. [DOI] [PubMed] [Google Scholar]
- 24.Pikaart MJ, Recillas-Targa F, Felsenfeld G. Loss of transcriptional activity of a transgene is accompanied by DNA methylation and histone deacetylation and is prevented by insulators. Genes Dev. 1998;12:2852–2862. doi: 10.1101/gad.12.18.2852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chung JH, Whiteley M, Felsenfeld G. A 5′ element of the chicken beta-globin domain serves as an insulator in human erythroid cells and protects against position effect in Drosophila. Cell. 1993;74:505–514. doi: 10.1016/0092-8674(93)80052-g. [DOI] [PubMed] [Google Scholar]
- 26.Kellum R, Schedl P. A group of scs elements function as domain boundaries in an enhancer-blocking assay. Mol. Cell. Biol. 1992;12:2424–2431. doi: 10.1128/mcb.12.5.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhao K, Hart CM, Laemmli UK. Visualization of chromosomal domains with boundary element-associated factor BEAF-32. Cell. 1995;81:879–889. doi: 10.1016/0092-8674(95)90008-x. [DOI] [PubMed] [Google Scholar]
- 28.Fourel G, Magdinier F, Gilson E. Insulator dynamics and the setting of chromatin domains. Bioessays. 2004;26:523–532. doi: 10.1002/bies.20028. [DOI] [PubMed] [Google Scholar]
- 29.Kimura A, Horikoshi M. Partition of distinct chromosomal regions: negotiable border and fixed border. Genes Cell. 2004;9:499–508. doi: 10.1111/j.1356-9597.2004.00740.x. [DOI] [PubMed] [Google Scholar]
- 30.Henikoff S. Position-effect variegation after 60 years. Trends Genet. 1990;6:422–426. doi: 10.1016/0168-9525(90)90304-o. [DOI] [PubMed] [Google Scholar]
- 31.Donze D, Kamakaka RT. RNA polymerase III and RNA polymerase II promoter complexes are heterochromatin barriers in Saccharomyces cerevisiae. EMBO J. 2001;20:520–531. doi: 10.1093/emboj/20.3.520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Noma K, Cam HP, Maraia RJ, Grewal SI. A role for TFIIIC transcription factor complex in genome organization. Cell. 2006;125:859–872. doi: 10.1016/j.cell.2006.04.028. [DOI] [PubMed] [Google Scholar]
- 33.Scott KC, Merrett SL, Willard HF. A heterochromatin barrier partitions the fission yeast centromere into discrete chromatin domains. Curr. Biol. 2006;16:119–129. doi: 10.1016/j.cub.2005.11.065. [DOI] [PubMed] [Google Scholar]
- 34.Valenzuela L, Kamakaka RT. Chromatin insulators. Annu. Rev. Genet. 2006;40:107–138. doi: 10.1146/annurev.genet.39.073003.113546. [DOI] [PubMed] [Google Scholar]
- 35.Roman AC, Gonzalez-Rico FJ, Molto E, Hernando H, Neto A, Vicente-Garcia C, Ballestar E, Gomez-Skarmeta JL, Vavrova-Anderson J, White RJ, et al. Dioxin receptor and SLUG transcription factors regulate the insulator activity of B1 SINE retrotransposons via an RNA polymerase switch. Genome Res. 2011;21:422–432. doi: 10.1101/gr.111203.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 37.Cuddapah S, Jothi R, Schones DE, Roh TY, Cui K, Zhao K. Global analysis of the insulator binding protein CTCF in chromatin barrier regions reveals demarcation of active and repressive domains. Genome Res. 2009;19:24–32. doi: 10.1101/gr.082800.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kharchenko PV, Alekseyenko AA, Schwartz YB, Minoda A, Riddle NC, Ernst J, Sabo PJ, Larschan E, Gorchakov AA, Gu T, et al. Comprehensive analysis of the chromatin landscape in Drosophila melanogaster. Nature. 2011;471:480–485. doi: 10.1038/nature09725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Riddle NC, Minoda A, Kharchenko PV, Alekseyenko AA, Schwartz YB, Tolstorukov MY, Gorchakov AA, Jaffe JD, Kennedy C, Linder-Basso D, et al. Plasticity in patterns of histone modifications and chromosomal proteins in Drosophila heterochromatin. Genome Res. 2011;21:147–163. doi: 10.1101/gr.110098.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu T, Rechtsteiner A, Egelhofer TA, Vielle A, Latorre I, Cheung MS, Ercan S, Ikegami K, Jensen M, Kolasinska-Zwierz P, et al. Broad chromosomal domains of histone modification patterns in C. elegans. Genome Res. 2011;21:227–236. doi: 10.1101/gr.115519.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ernst J, Kellis M. Discovery and characterization of chromatin states for systematic annotation of the human genome. Nat. Biotechnol. 2010;28:817–825. doi: 10.1038/nbt.1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ernst J, Kheradpour P, Mikkelsen TS, Shoresh N, Ward LD, Epstein CB, Zhang X, Wang L, Issner R, Coyne M, et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature. 2011;473:43–49. doi: 10.1038/nature09906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang Z, Zang C, Rosenfeld JA, Schones DE, Barski A, Cuddapah S, Cui K, Roh TY, Peng W, Zhang MQ, et al. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat. Genet. 2008;40:897–903. doi: 10.1038/ng.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Karlin S, Altschul SF. Methods for assessing the statistical significance of molecular sequence features by using general scoring schemes. Proc. Natl Acad. Sci. USA. 1990;87:2264–2268. doi: 10.1073/pnas.87.6.2264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rosenfeld JA, Wang Z, Schones DE, Zhao K, DeSalle R, Zhang MQ. Determination of enriched histone modifications in non-genic portions of the human genome. BMC Genomics. 2009;10:143. doi: 10.1186/1471-2164-10-143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ruzzo WL, Tompa M. A linear time algorithm for finding all maximal scoring subsequences. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1999:234–241. [PubMed] [Google Scholar]
- 47.Su AI, Wiltshire T, Batalov S, Lapp H, Ching KA, Block D, Zhang J, Soden R, Hayakawa M, Kreiman G, et al. A gene atlas of the mouse and human protein-encoding transcriptomes. Proc. Natl Acad. Sci. USA. 2004;101:6062–6067. doi: 10.1073/pnas.0400782101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Boyle AP, Davis S, Shulha HP, Meltzer P, Margulies EH, Weng Z, Furey TS, Crawford GE. High-resolution mapping and characterization of open chromatin across the genome. Cell. 2008;132:311–322. doi: 10.1016/j.cell.2007.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Karolchik D, Baertsch R, Diekhans M, Furey TS, Hinrichs A, Lu YT, Roskin KM, Schwartz M, Sugnet CW, Thomas DJ, et al. The UCSC genome browser database. Nucleic Acids Res. 2003;31:51–54. doi: 10.1093/nar/gkg129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Karolchik D, Hinrichs AS, Furey TS, Roskin KM, Sugnet CW, Haussler D, Kent WJ. The UCSC Table Browser data retrieval tool. Nucleic Acids Res. 2004;32:D493–D496. doi: 10.1093/nar/gkh103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Barski A, Chepelev I, Liko D, Cuddapah S, Fleming AB, Birch J, Cui K, White RJ, Zhao K. Pol II and its associated epigenetic marks are present at Pol III-transcribed noncoding RNA genes. Nat. Struct. Mol. Biol. 2010;17:629–634. doi: 10.1038/nsmb.1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pasquinelli AE, Reinhart BJ, Slack F, Martindale MQ, Kuroda MI, Maller B, Hayward DC, Ball EE, Degnan B, Muller P, et al. Conservation of the sequence and temporal expression of let-7 heterochronic regulatory RNA. Nature. 2000;408:86–89. doi: 10.1038/35040556. [DOI] [PubMed] [Google Scholar]
- 53.Mourelatos Z, Dostie J, Paushkin S, Sharma A, Charroux B, Abel L, Rappsilber J, Mann M, Dreyfuss G. miRNPs: a novel class of ribonucleoproteins containing numerous microRNAs. Genes Dev. 2002;16:720–728. doi: 10.1101/gad.974702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstrale M, Laurila E, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003;34:267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- 56.Cho H, Orphanides G, Sun X, Yang XJ, Ogryzko V, Lees E, Nakatani Y, Reinberg D. A human RNA polymerase II complex containing factors that modify chromatin structure. Mol. Cell Biol. 1998;18:5355–5363. doi: 10.1128/mcb.18.9.5355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Phillips JE, Corces VG. CTCF: master weaver of the genome. Cell. 2009;137:1194–1211. doi: 10.1016/j.cell.2009.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhong XP, Krangel MS. An enhancer-blocking element between alpha and delta gene segments within the human T cell receptor alpha/delta locus. Proc. Natl Acad. Sci. USA. 1997;94:5219–5224. doi: 10.1073/pnas.94.10.5219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bell AC, West AG, Felsenfeld G. The protein CTCF is required for the enhancer blocking activity of vertebrate insulators. Cell. 1999;98:387–396. doi: 10.1016/s0092-8674(00)81967-4. [DOI] [PubMed] [Google Scholar]
- 60.West AG, Gaszner M, Felsenfeld G. Insulators: many functions, many mechanisms. Genes Dev. 2002;16:271–288. doi: 10.1101/gad.954702. [DOI] [PubMed] [Google Scholar]
- 61.Bushey AM, Dorman ER, Corces VG. Chromatin insulators: regulatory mechanisms and epigenetic inheritance. Mol. Cell. 2008;32:1–9. doi: 10.1016/j.molcel.2008.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Martin D, Pantoja C, Fernandez Minan A, Valdes-Quezada C, Molto E, Matesanz F, Bogdanovic O, de la Calle-Mustienes E, Dominguez O, Taher L, et al. Genome-wide CTCF distribution in vertebrates defines equivalent sites that aid the identification of disease-associated genes. Nat. Struct. Mol. Biol. 2011;18:708–714. doi: 10.1038/nsmb.2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Spensberger D, Delwel R. A novel interaction between the proto-oncogene Evi1 and histone methyltransferases, SUV39H1 and G9a. FEBS Lett. 2008;582:2761–2767. doi: 10.1016/j.febslet.2008.06.056. [DOI] [PubMed] [Google Scholar]
- 64.Chakraborty S, Senyuk V, Sitailo S, Chi Y, Nucifora G. Interaction of EVI1 with cAMP-responsive element-binding protein-binding protein (CBP) and p300/CBP-associated factor (P/CAF) results in reversible acetylation of EVI1 and in co-localization in nuclear speckles. J. Biol. Chem. 2001;276:44936–44943. doi: 10.1074/jbc.M106733200. [DOI] [PubMed] [Google Scholar]
- 65.Bruhat A, Cherasse Y, Maurin AC, Breitwieser W, Parry L, Deval C, Jones N, Jousse C, Fafournoux P. ATF2 is required for amino acid-regulated transcription by orchestrating specific histone acetylation. Nucleic Acids Res. 2007;35:1312–1321. doi: 10.1093/nar/gkm038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Karanam B, Wang L, Wang D, Liu X, Marmorstein R, Cotter R, Cole PA. Multiple roles for acetylation in the interaction of p300 HAT with ATF-2. Biochemistry. 2007;46:8207–8216. doi: 10.1021/bi7000054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sano Y, Tokitou F, Dai P, Maekawa T, Yamamoto T, Ishii S. CBP alleviates the intramolecular inhibition of ATF-2 function. J. Biol. Chem. 1998;273:29098–29105. doi: 10.1074/jbc.273.44.29098. [DOI] [PubMed] [Google Scholar]
- 68.Kovacs KA, Steinmann M, Magistretti PJ, Halfon O, Cardinaux JR. CCAAT/enhancer-binding protein family members recruit the coactivator CREB-binding protein and trigger its phosphorylation. J. Biol. Chem. 2003;278:36959–36965. doi: 10.1074/jbc.M303147200. [DOI] [PubMed] [Google Scholar]
- 69.Yao YL, Yang WM, Seto E. Regulation of transcription factor YY1 by acetylation and deacetylation. Mol. Cell Biol. 2001;21:5979–5991. doi: 10.1128/MCB.21.17.5979-5991.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Huang S, Li X, Yusufzai TM, Qiu Y, Felsenfeld G. USF1 recruits histone modification complexes and is critical for maintenance of a chromatin barrier. Mol. Cell Biol. 2007;27:7991–8002. doi: 10.1128/MCB.01326-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.West AG, Huang S, Gaszner M, Litt MD, Felsenfeld G. Recruitment of histone modifications by USF proteins at a vertebrate barrier element. Mol. Cell. 2004;16:453–463. doi: 10.1016/j.molcel.2004.10.005. [DOI] [PubMed] [Google Scholar]
- 72.Cuddapah S, Schones DE, Cui K, Roh TY, Barski A, Wei G, Rochman M, Bustin M, Zhao K. Genomic profiling of HMGN1 reveals an association with chromatin at regulatory regions. Mol. Cell. Biol. 2011;31:700–709. doi: 10.1128/MCB.00740-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Donohoe ME, Zhang LF, Xu N, Shi Y, Lee JT. Identification of a Ctcf cofactor, Yy1, for the X chromosome binary switch. Mol. Cell. 2007;25:43–56. doi: 10.1016/j.molcel.2006.11.017. [DOI] [PubMed] [Google Scholar]
- 74.Austen M, Luscher B, Luscher-Firzlaff JM. Characterization of the transcriptional regulator YY1. The bipartite transactivation domain is independent of interaction with the TATA box-binding protein, transcription factor IIB, TAFII55, or cAMP-responsive element-binding protein (CPB)-binding protein. J. Biol. Chem. 1997;272:1709–1717. doi: 10.1074/jbc.272.3.1709. [DOI] [PubMed] [Google Scholar]
- 75.Galvin KM, Shi Y. Multiple mechanisms of transcriptional repression by YY1. Mol. Cell. Biol. 1997;17:3723–3732. doi: 10.1128/mcb.17.7.3723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lee JS, Galvin KM, See RH, Eckner R, Livingston D, Moran E, Shi Y. Relief of YY1 transcriptional repression by adenovirus E1A is mediated by E1A-associated protein p300. Genes Dev. 1995;9:1188–1198. doi: 10.1101/gad.9.10.1188. [DOI] [PubMed] [Google Scholar]
- 77.Shi Y, Lee JS, Galvin KM. Everything you have ever wanted to know about Yin Yang 1. Biochim. Biophys. Acta. 1997;1332:F49–F66. doi: 10.1016/s0304-419x(96)00044-3. [DOI] [PubMed] [Google Scholar]
- 78.Thomas MJ, Seto E. Unlocking the mechanisms of transcription factor YY1: are chromatin modifying enzymes the key? Gene. 1999;236:197–208. doi: 10.1016/s0378-1119(99)00261-9. [DOI] [PubMed] [Google Scholar]
- 79.Yang WM, Inouye C, Zeng Y, Bearss D, Seto E. Transcriptional repression by YY1 is mediated by interaction with a mammalian homolog of the yeast global regulator RPD3. Proc. Natl Acad. Sci. USA. 1996;93:12845–12850. doi: 10.1073/pnas.93.23.12845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yang WM, Yao YL, Sun JM, Davie JR, Seto E. Isolation and characterization of cDNAs corresponding to an additional member of the human histone deacetylase gene family. J. Biol. Chem. 1997;272:28001–28007. doi: 10.1074/jbc.272.44.28001. [DOI] [PubMed] [Google Scholar]
- 81.Bushmeyer SM, Atchison ML. Identification of YY1 sequences necessary for association with the nuclear matrix and for transcriptional repression functions. J. Cell. Biochem. 1998;68:484–499. [PubMed] [Google Scholar]
- 82.Guo B, Odgren PR, van Wijnen AJ, Last TJ, Nickerson J, Penman S, Lian JB, Stein JL, Stein GS. The nuclear matrix protein NMP-1 is the transcription factor YY1. Proc. Natl Acad. Sci. USA. 1995;92:10526–10530. doi: 10.1073/pnas.92.23.10526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ebersole T, Kim JH, Samoshkin A, Kouprina N, Pavlicek A, White RJ, Larionov V. tRNA genes protect a reporter gene from epigenetic silencing in mouse cells. Cell Cycle. 2011;10 doi: 10.4161/cc.10.16.17092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chiu YH, Yu Q, Sandmeier JJ, Bi X. A targeted histone acetyltransferase can create a sizable region of hyperacetylated chromatin and counteract the propagation of transcriptionally silent chromatin. Genetics. 2003;165:115–125. doi: 10.1093/genetics/165.1.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Donze D, Kamakaka RT. Braking the silence: how heterochromatic gene repression is stopped in its tracks. Bioessays. 2002;24:344–349. doi: 10.1002/bies.10072. [DOI] [PubMed] [Google Scholar]
- 86.Feng Q, Wang H, Ng HH, Erdjument-Bromage H, Tempst P, Struhl K, Zhang Y. Methylation of H3-lysine 79 is mediated by a new family of HMTases without a SET domain. Curr. Biol. 2002;12:1052–1058. doi: 10.1016/s0960-9822(02)00901-6. [DOI] [PubMed] [Google Scholar]
- 87.Steger DJ, Lefterova MI, Ying L, Stonestrom AJ, Schupp M, Zhuo D, Vakoc AL, Kim JE, Chen J, Lazar MA, et al. DOT1L/KMT4 recruitment and H3K79 methylation are ubiquitously coupled with gene transcription in mammalian cells. Mol. Cell. Biol. 2008;28:2825–2839. doi: 10.1128/MCB.02076-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ng HH, Ciccone DN, Morshead KB, Oettinger MA, Struhl K. Lysine-79 of histone H3 is hypomethylated at silenced loci in yeast and mammalian cells: a potential mechanism for position-effect variegation. Proc. Natl Acad. Sci. USA. 2003;100:1820–1825. doi: 10.1073/pnas.0437846100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Meisterernst M, Roeder RG. Family of proteins that interact with TFIID and regulate promoter activity. Cell. 1991;67:557–567. doi: 10.1016/0092-8674(91)90530-c. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.