

Abstract
EcoRII is a homodimer with two domains consisting of a DNA-binding N terminus and a catalytic C terminus and recognizes two specific sequences on DNA. It shows a relatively complicated cleavage reaction in bulk solution. After binding to either recognition site, EcoRII cleaves the other recognition site of the same DNA (cis-binding) strand and/or the recognition site of the other DNA (trans-binding) strand. Although it is difficult to separate these two reactions in bulk solution, we could simply obtain the binding and cleavage kinetics of only the cis-binding by following the frequency (mass) changes of a DNA-immobilized quartz-crystal microbalance (QCM) responding to the addition of EcoRII in aqueous solution. We obtained the maximum binding amounts (Δmmax), the dissociation constants (Kd), the binding and dissociation rate constants (kon and koff), and the catalytic cleavage reaction rate constants (kcat) for wild-type EcoRII, the N-terminal-truncated form (EcoRII N-domain), and the mutant derivatives in its C-terminal domain (K263A and R330A). It was determined from the kinetic analyses that the N-domain, which covers the catalytic C-domain in the absence of DNA, preferentially binds to the one DNA recognition site while transforming EcoRII into an active form allosterically, and then the secondary C-domain binds to and cleaves the other recognition site of the DNA strand.
DNA-binding proteins, which specifically recognize DNA sequences, play an important role in maintaining and managing genetic information in all living cells. To study the binding mechanisms of these proteins to DNA is important not only for a biological understanding of gene expression but also for the design of an artificial DNA-binding molecule. A restriction endonuclease is also a specific DNA-binding protein. Bacteria possess restriction endonucleases to protect them from infection by digesting the foreign DNA from viruses. In addition to being an essential tool for gene manipulation in the genetic engineering field, they have been widely used as tools to understand DNA recognition and cleavage mechanisms.
Thousands of restriction endonucleases have been discovered thus far, and classified into several types because of their cleavage reactions shown in Fig. 1A (1–3). Type IIP restriction endonucleases, which are commonly used in the genetic engineering field, recognize simply one palindromic sequence (4–8 base pairs), which is cleaved. On the other hand, Type IIE or IIF endonucleases need two recognition sites and cleave one or two sites, respectively (4). EcoRII, which is classified as a type IIE restriction endonuclease, is a homodimer (5, 6). It recognizes two units of recognition sequences (5′-CCWGG-3′) included in one DNA chain (cis-binding) or in two DNA chains one by one (trans-binding), and cleaves either site (see Fig. 1B) (7–10). Therefore, it is difficult to distinguish between cis and trans binding in the bulk solution by conventional analysis methods such as electrophoresis mobility shift assay and fluorescent anisotropy. Biochemical and structural analyses have revealed that EcoRII is a homodimer consisting of two domains: an N-terminal domain that only exhibits specific binding at the recognition site, and a C-terminal domain that recognizes and cleaves the second recognition site (10–12). In the absence of DNA, the N-domain covers the putative catalytic residues of Arg-330 and Lys-263 in the C-domain with hydrogen bonds and salt bridges (13). Therefore, an autoinhibition/activation model has been proposed such that the induction of a structural change in the N-domain with DNA causes the uncovering of the catalytic residue of the C-domain. However, this allosteric binding and cleavage reaction of EcoRII has not been confirmed kinetically.
FIGURE 1.

A, schematic illustrations of the recognition sites and cleavage styles of Type IIP, Type IIE, and Type IIF restriction endonucleases. B, proposed binding and cleavage mechanism for EcoRII, one of the Type IIE restriction enzymes. EcoRII is a homodimer with an N-domain showing specific binding and a C-domain with catalytic cleavage capacity that was originally covered by the N-domain.
To analyze these dynamic reactions of EcoRII on DNA and to understand the enzymatic reaction, it is advantageous to directly determine the formation and decomposition of an enzyme-DNA (ES) complex. The problem is that EcoRII may form a complicated ES complex including both cis- and trans-binding mechanisms in bulk solution. Furthermore, it is difficult to quantitatively analyze the formation and decomposition of the ES complex by means of a conventional gel-shift assay and fluorescent anisotropy in bulk solution.
In this study, we used a DNA-immobilized 27-MHz quartz-crystal microbalance (QCM)2 to monitor the allosteric DNA recognition and digestion catalyzed by a Type IIE restriction endonuclease of EcoRII. The QCM is a very sensitive massmeasuring device (14), and the resonance frequency has been shown to decrease linearly with increasing mass on the QCM electrode at the nanogram level in aqueous solutions (15–23, 38, 39). The sensitivity of the 27-MHz QCM in aqueous solutions has been calibrated to be 0.1 ng cm–2 per 1 Hz (21). When a DNA-immobilized QCM is employed, the ES complex formation can be followed as the frequency decreases (mass increases), and the decomposition of the ES complex into the product can be followed by frequency increases (mass decreases) on the same QCM plate (24, 25). Furthermore, it is advantageous to use the DNA-immobilized QCM system for EcoRII catalysis to allow only the cis-binding mechanism (see Fig. 2A). Several types of biotinylated DNAs can be immobilized: a 2-site DNA, a 1-site DNA, and a 0-site DNA with two recognition sites (5′-CCAGG-3′ and 5′-CCTGG-3′) with a 5-bp spacer, one recognition site (5′-CCAGG-3′), and no recognition sites in the 55-bp DNA, respectively (see Fig. 2B). We also prepared a biotinylated 2-site-digested DNA-α and a 2-site-digested DNA-β, responding residual DNAs after the cleavage at the inner and outer sites, respectively. We also prepared a truncated EcoRII N-domain and EcoRII mutations in the C-domain (K263A and R330A), which are expected to cause simple binding kinetics to the DNA. This is the first study to observe the formation and decomposition of the EcoRII/DNA (ES) complex with dynamic conformational changes, and to follow the cleavage process as mass changes, while obtaining kon, koff, and kcat values from the same device (see Fig. 2A). The kinetic analyses revealed a model of autoinhibition/activation and an autocatalysis mode of activated EcoRII.
FIGURE 2.

A, proposed reaction scheme for the DNA cleavage reaction of EcoRII on a DNA-immobilized 27-MHz QCM in aqueous solution. B, DNA structures immobilized on an avidin-covered QCM plate.
EXPERIMENTAL PROCEDURES
Materials—1-Ethyl-3-(3-dimethylamino-propyl)carbodiimide (EDC) was purchased from DOJINDO, Co. (Kumamoto, Japan), and N-hydroxysuccinimide was from Wako Pure Chemical Industries, Ltd (Osaka, Japan). The oligonucleotides were purchased from Operon, Co. (Tokyo, Japan). All other materials were purchased from Nacalai Tesque, Co. (Kyoto, Japan), and were used without further purification.
Protein Preparation—The open reading frame of R.EcoRII was amplified from pNY30 (26) (a gift from Prof. Kobayashi, Tokyo University) using PCR with each primer connected to the BamHI site at the 5′-side, and the PstI site at the 3′-site, respectively. The plasmid pQE80RII for the overexpression of His-tagged EcoRII was constructed from pQE80L (Qiagen) as previously described (6). Escherichia coli strain JM109 was transformed with both pQE80RII and pNY31 (26) carrying the M.EcoRII gene for host genome protection. The transformant was cultured and induced for overexpression by isopropyl-1-thio-β-d-galactopyranoside. After cell disruption and centrifugation, the EcoRII was purified as previously described (6). It was then dialyzed against 10 mm NaH2PO4 (pH 7.0). The protein was loaded on a HiTrap Heparin HP 1-ml column (GE Healthcare) and eluted with a linear gradient of 0–1 m NaCl. The fractions containing EcoRII were collected and dialyzed against the stock buffer (10 mm Tris-HCl, 50 mm NaCl, 1 mm DTT, pH 7.5), and then changed to the assay buffer (stock buffer plus 5 mm CaCl2 for DNA binding or MgCl2 for DNA cleavage) by PD-10 (GE Healthcare) before the QCM measurements. The EcoRII N-domain was prepared as described in the literature (6). The expression plasmids for the K263A and R330A mutants were constructed using a QuikChange Mutagenesis kit (Stratagene), and the constructs were overexpressed and purified as described above.
Calibration of 27-MHz QCM in Aqueous Solution—AFFI-NIX Q4 was used as the QCM apparatus (Initium Co., Ltd, Tokyo) with four 500-μl cells equipped with a 27-MHz QCM plate (8.7-mm diameter quartz plate and 5.7-mm2 area Au electrode) on the bottom of the cell plus a stirring bar with a temperature control system (21–25). The relationship between mass and frequency changes in aqueous solutions when DNAs and/or proteins were immobilized onto the QCM were calibrated by comparing it against values in the air phase. 1 Hz of frequency represents a 0.10 ng cm–2 mass increase on the QCM plate. These detailed calibration experiments are described elsewhere (25). The noise level of the 27 MHz QCM was ±2 Hz in buffer solutions at 25 °C, and the stability of the frequency was ±2 Hz for 1 h in buffer at 25 °C. A sensitivity of 0.10 ng cm–2 per Hz is sufficiently large to detect the binding of enzyme and the DNA cleavage process.
Preparation of DNA-immobilized QCM Plates—The structures of the biotinylated oligonucleotides used in this study are summarized in Fig. 2B: 5′-biotinylated dsDNA (55 bp) containing two sites recognized by EcoRII (2-site DNA), a one site dsDNA (1-site DNA), and no-site dsDNA (0-site DNA). We also prepared 5′-biotinylated dsDNA containing cohesive ends (2-site-digested DNA-α and -β) representing residual DNAs after cleavage at the inner and outer recognition sites of the 2-site DNA, respectively. Oligonucleotide duplexes were formed by mixing a biotinylated strand and its complementary strand in a solution of 10 mm Tris-HCl, pH 7.8, 1 mm EDTA, and 200 mm NaCl, and then boiling for a few minutes, followed by cooling to room temperature over 3 h (24, 25). The amount of DNA immobilized was maintained at 19 ± 1 ng (0.55 ± 0.02 pmol) cm–2, which corresponds to a ∼1% coverage of the Au surface (5.7 mm2). This would allow for enough space, and thus accommodate the binding of a large enzyme molecule.
Enzyme Reactions in a DNA-immobilized QCM Cell—A DNA-immobilized QCM cell was filled with 500 μl of assay buffer (10 mm Tris-HCl, pH 7.5, 50 mm NaCl, 1 mm DTT in the presence of 5 mm CaCl2 or MgCl2). The frequency changes in response to the addition of enzymes were then followed over time. The solution was vigorously stirred to avoid any effect from the slow diffusion of the enzymes. The stirring did not affect the stability or magnitude of the frequency changes.
RESULTS AND DISCUSSION
Binding Behavior of EcoRII in the Presence of Ca2+ Ions—Restriction endonucleases are known to require Mg2+ ions as cofactors to digest dsDNA (1–3). However, in the presence of Ca2+ ions, EcoRII has been reported to bind to the target DNA but not cleave the DNA strand (10). In fact, transmission electron microscopy has confirmed the formation of the EcoRII/DNA complex in the presence of Ca2+ ions (10). We designed a 2-site dsDNA with two recognition sites with a 5-bp spacer, which has been reported to be the most appropriate length to cleave efficiently in cis-binding (6).
Fig. 3A shows typical frequency decreases (mass increases) as a function of time, in response to the addition of EcoRII in the presence of 5 mm Ca2+ ion. EcoRII mainly bound to the 2-site DNA and the 1-site DNA (curves a and b, respectively), and barely bound to the 0-site DNA (curve c). Fig. 3B shows that the amount bound (Δm) followed a simple saturation curve as a function of the EcoRII concentration. These binding behaviors are well-fitted by Equation 1 as a saturation curve against the concentration of EcoRII.
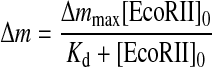 |
(Eq. 1) |
FIGURE 3.
A, typical frequency changes of: (curve a) 2-site DNA, (curve b) 1-site DNA, and (curve c) 0-site DNA immobilized onto a QCM, in response to the addition of EcoRII in the presence of Ca2+ ions. The arrow indicates the time of enzyme injection. ([EcoRII] = 3 nm, [DNA] = 19 ± 1 ng (0.55 ± 0.02 pmol) cm–2 on a QCM, in 5 mm CaCl2, 10 mm Tris-HCl, pH 7.5, 50 mm NaCl, 1 mm DTT, 25 °C). B, saturation binding of EcoRII to each DNA. Each curve was fitted with Equation 1 displayed in the text. C, linear reciprocal plots of the relaxation rate (τ–1) against the EcoRII concentration according to Equations 5 and 6 in the text.
The dissociation constants (Kd) and maximum binding amounts (Δmmax) were calculated from Equation 1. The obtained Δmmax and Kd values are summarized in Table 1 (runs 3–5). The Δmmax for the 2-site DNA and the 1-site DNA were 51 ± 1 ng (0.53 ± 0.01 pmol) cm–2, and 51 ± 4 ng (0.53 ± 0.04 pmol) cm–2, respectively. Because the amount of DNA immobilized was 19 ± 1 ng (0.53 ± 0.02 pmol) cm–2, the EcoRII dimer could bind onto DNA with at a 1:1 molar ratio regardless of the number of sites. The Kd = 5.6 nm for the 2-site DNA was slightly lower than Kd = 11 nm for the 1-site DNA. Although the Kd = 71 nm for the 0-site DNA was 20-fold larger than those for the specific binding, EcoRII bound to the 0-site DNA (55-bp length) at a molar ratio of 1:1 (Δmmax = 51 ± 4 ng (0.53 ± 0.04 pmol) cm–2).
TABLE 1.
Summary of kinetic parameters
Maximum amount of binding (Δmmax), dissociation constant (Kd), binding (kon), dissociation rate constants (koff), and the cleavage rate constant (kcat) of EcoRII and its derivatives for different DNAs. [DNA] = 19 ± 1 ng (0.55 ± 0.02 pmol) cm–2 on a QCM, [EcoRII] = 1–30 nm, [EcoRII-N domain] = 1–500 nm, [K263A] = [R330A] = 1–5 nm, 10 mm Tris-HCl, pH 7.5, 50 mm NaCl, 1 mm DTT, [Mg2+ or Ca2+] = 5 mm, 25 °C. The obtained kinetic values contain ±5% experimental errors.
| Run | Enzymes | DNA | Divalent cations | Δmmaxa | Kd | kond | koffd | kcat |
|---|---|---|---|---|---|---|---|---|
| ng cm-2 | nm | 105m-1 s-1 | 10-3 s-1 | 10-3 s-1 | ||||
| 1 | EcoRII | 2-site DNA | Mg2+ | - | 3.3b | 3.0b | 1.0b | 18b |
| 2 | 1-site DNA | Mg2+ | 46 (0.9)c | 16a, 3.2d | 4.8 | 1.6 | 0 | |
| 3 | EcoRII | 2-site DNA | Ca2+ | 51 (1.0)c | 5.6a, 2.0d | 5.2 | 1.0 | 0 |
| 4 | 1-site DNA | Ca2+ | 51 (1.0)c | 11a, 3.4d | 5.0 | 1.7 | 0 | |
| 5 | 0-site DNA | Ca2+ | 51 (1.0)c | 71a, 77d | 0.10 | 0.69 | 0 | |
| 6 | 2-site digested DNA-α | Ca2+ | ∼15 (∼0.3)c | 150a | - | - | - | |
| 7 | 2-site digested DNA-β | Ca2+ | 37 (0.7)c | 13a | - | - | - | |
| 8 | EcoRII N-domain | 1-site DNA | Ca2+ | 14e (1.1)c, 32f (2.5)c | 10e, 530f | 1.4 | 2.2 | - |
| 9 | 0-site DNA | Ca2+ | 20 (1.6)c | 130a, 122d | 0.09 | 1.1 | - | |
| 10 | K263A | 2-site DNA | Ca2+ | - | 3.7d | 4.7 | 1.7 | 0 |
| 11 | 1-site DNA | Ca2+ | - | 3.4d | 5.1 | 1.7 | 0 | |
| 12 | R330A | 2-site DNA | Ca2+ | - | 3.3d | 4.6 | 1.6 | 0 |
| 13 | 1-site DNA | Ca2+ | - | 3.1d | 5.2 | 1.6 | 0 |
Obtained from the saturation binding experiments according to Eq. 1.
Binding ratio of EcoRII or the EcoRII N-domain to DNA.
[EcoRII N-domain] = 1-10 nm.
[EcoRII N-domain] = 100-500 nm.
The binding time course of EcoRII to the DNA shown in Fig. 3A can be described by Equations 2 and 3. The amount of EcoRII/DNA (ES) complex formed at time t after the injection is given by Equations 3 and 4. The relaxation rate (τ–1) of the EcoRII binding was calculated from each curve fitted to the frequency decreases at various enzyme concentrations. When the concentrations of EcoRII were increased from 3 to 30 nm, the amount of enzyme bound on the DNA increased (Fig. 3B). The τ–1 values obtained at each enzyme concentration were plotted against [EcoRII] in Fig. 3C according to Equation 5. The EcoRII binding and dissociation rate constants (kon and koff) could be obtained from the slope and the intercept, respectively.
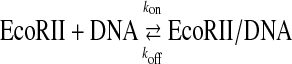 |
(Eq. 2) |
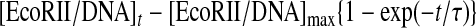 |
(Eq. 3) |
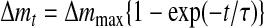 |
(Eq. 4) |
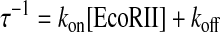 |
(Eq. 5) |
The aforementioned results for the 2-site, 1-site, and 0-site DNAs are summarized in Table 1 (runs 3–5). The Kd values obtained from the saturation binding method (Equation 1) consisted of Kd values obtained from the koff/kon values. When compared with the parameters for the 2-site DNA of kon = 5.2 × 105 m–1 s–1 and koff = 1.0 × 10–3 s–1, the binding rate constant for the 1-site DNA of kon = 5.0 × 105 m–1 s–1 was almost the same, but the dissociation rate constant of koff = 1.7 × 10–3 s–1 was about 2-fold larger. These parameters also indicated that the rate-determining step of EcoRII binding is the first 1-site binding step to DNA, and that the binding to the other site should occur simultaneously or sequentially, and thus the 2-site binding of EcoRII could decrease the koff value. These are unique characteristics of type IIE restriction endonucleases. Our data on the dissociation constant for the 1-site DNA (Kd = 3.4 nm) agreed well with a previous study (Kd = 1.0 nm) for a short DNA containing one cognate site determined by gel-shift assay (11). On the other hand, the Kd value of ∼70 nm for the 0-site DNA was only 10–40-fold less than binding to the specific sites. Our previous results revealed that EcoRV had a 300-fold greater specificity for site recognition, in which the kon (3.6 × 104 m–1 s–1) and koff (1.7 × 10–2 s–1) values for the nonspecific DNA were 20-fold smaller and 15-fold larger than the specific DNA, respectively (25). The relatively high affinity of EcoRII for the 0-site DNA was mostly due to the small dissociation rate constant (koff = 0.7 × 10–3 s–1). These results indicated that EcoRII would dissociate slowly from the DNA strand, which is a unique characteristic among restriction endonucleases, and could be attributed to linear diffusion on the DNA strand to search for a recognition site.
Binding Behavior of the EcoRII N-domain in the Presence of Ca2+ Ions—EcoRII is composed of two domains that are connected with a flexible polypeptide linker (12, 13). Before binding to DNA, it is believed that the catalytic active site of the EcoRII C-domain is covered by the N-domain (13). In the presence of the cognate DNA, it is suggested that the N-domain of EcoRII is removed from the C-domain and binds to the recognition site (see Figs. 1B and 2A). Therefore, we prepared a truncated protein of the EcoRII N-terminal domain (12) to study the binding ability of the EcoRII N-domain.
Fig. 4A shows the typical binding behaviors of the EcoRII N-domain to 1-site and 0-site DNAs in the presence of Ca2+ ions. In a low concentration of N-domain (<10 nm), EcoRII seems to bind selectively to the 1-site DNA, but not to the 0-site DNA. When the concentration of the EcoRII N-domain increased, a biphasic binding behavior was observed for the 1-site DNA, and the first binding was saturated near the binding amount of 14 ± 3 ng (0.60 ± 0.12 pmol) cm–2 of the EcoRII N-domain. Because the 1-site DNA was immobilized at 19 ± 1 ng (0.55 ± 0.02 pmol) cm–2 on the QCM, the first saturation of curve a in Fig. 4B shows a 1:1 binding of the N-domain to the one recognition site of the DNA. In contrast, a simple one-phase binding saturation was observed for the 0-site DNA, showing the low binding ability (curve b in Fig. 4B). From the Scatchard plot analyses, the dissociation constants (Kd) for the 1-site and the 0-site DNAs were 10 and 130 nm, respectively (see runs 8 and 9, Table 1). The Kd value for the first step of the EcoRII N-domain was close to the Kd = 3.4 nm obtained for the binding of wild-type EcoRII for the 1-site DNA. The Kd = 530 nm for the second step binding of the EcoRII N-domain to the 1-site DNA was larger than the Kd = 130 nm for the nonspecific binding of EcoRII to the 0-site DNA.
FIGURE 4.
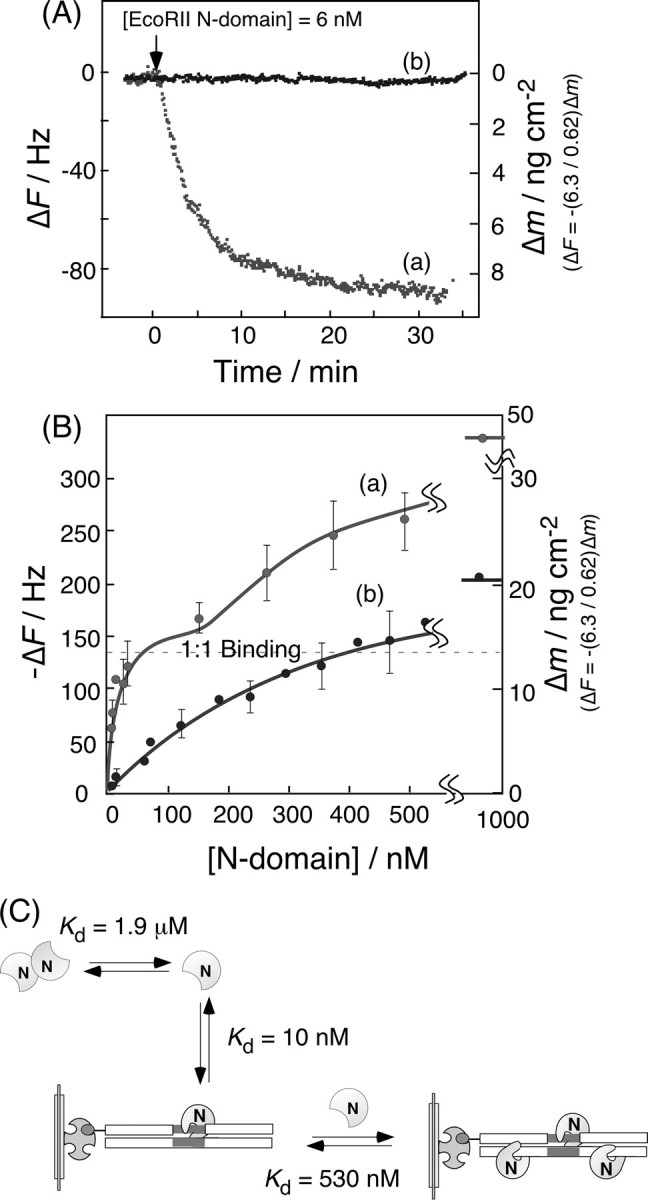
Binding behavior (A) and saturation binding (B) of the EcoRII N-domain to: (curve a) 1-site DNA and (curve b) 0-site DNA in the presence of Ca2+ ions. ([EcoRII N-domain] = 1–500 nm, [DNA] = 19 ± 1 ng (0.55 ± 0.02 pmol) cm–2 on a QCM, in 5 mm CaCl2, 10 mm Tris-HCl, pH 7.5, 50 mm NaCl, 1 mm DTT, 25 °C). C, schematic illustration of the binding behavior of the EcoRII N-domain to 1-site DNA.
The binding and dissociation rate constants (kon and koff) of the EcoRII N-domain to the 1-site DNA in the first saturation curve region (1–10 nm) were obtained by curve fitting of Fig. 4A, according to Equations 3, 4, 5. These results are summarized in Table 1 (runs 8 and 9). The kon = 1.4 × 105 m–1 s–1 and koff = 2.2 × 10–3 s–1 values for the EcoRII N-domain binding to the 1-site DNA were similar to those for wild-type EcoRII (kon = 5.0 × 105 m–1 s–1 and koff = 1.7 × 10–3 s–1 for the 1-site DNA, run 4 in Table 1).
Although the dimerization of EcoRII occurs easily in low concentrations (Kd = 2.9 nm) (10), the dimerization of the N-domain has not been reported below 1 μm (Kd = 1.9 μm) (12, 13). Therefore, as schematically shown in Fig. 4C, the EcoRII N-domain can bind specifically to the recognition site at a 1:1 ratio as a monomer at low concentrations of 1–10 nm, and the nonspecific binding to DNA occurs in the high concentration region (100–500 nm). Based on the result that the EcoRII N-domain can bind to the specific sequence at a 1:1 molar ratio, but not the 2:1 molar ratio of the N-domain to the 1-site DNA, only one N-domain from the EcoRII dimer can bind to the recognition site, and the other N-domain may exist as a free, unbound site, as illustrated in Fig. 2A.
Binding Behavior of EcoRII Mutants in the Presence of Ca2+ Ions—It has been reported that the Lys-263 and Arg-330 residues of the catalytic C-domain are covered by the binding N-domain of EcoRII before binding to DNA, and that those residues in the C-domain act as recognition residues for DNA sequences after the N-domain is removed (13). The positively charged residues neighboring Lys-263 are quite well-conserved residues, which recognize DNA as well as other restriction endonucleases such as PspGI and SsoII (13, 27). Arg-330 is also a quite well-conserved residue which recognizes this sequence as well as NgoMIV (28) and Cfr101 (29, 30).
We prepared EcoRII mutants of K263A and R330A, which were expected to show the specific binding activity of the N-domain but not the C-domain. Binding experiments were performed on K263A and R330A for 2-site DNA and 1-site DNA similar to the wild-type EcoRII over the range of [enzyme] = 1.3–5.2 nm under the same conditions (5 mm Ca2+ ions). The binding behavior of these mutants were similar to the wild-type EcoRII as shown in Fig. 3, and the kon, koff, and Kd values were obtained according to Equations 3, 4, 5. These results are summarized in Table 1 (runs 10–13). The kon and koff values of K263A and R330A for the 2-site and 1-site DNAs (kon = (4.6–5.2) × 105 m–1 s–1 and koff = (1.6–1.7) × 10–3 s–1) were very close to those of the wild-type EcoRII for the 1-site DNA (kon = 5.0 × 105 m–1 s–1 and kon = 1.7 × 10–3 s–1, run 4). These results indicated that the N-domain of EcoRII would be the main binding domain and binds to the first cognate site, and then the C-domain would bind to an adjacent second site for the catalytic cleavage.
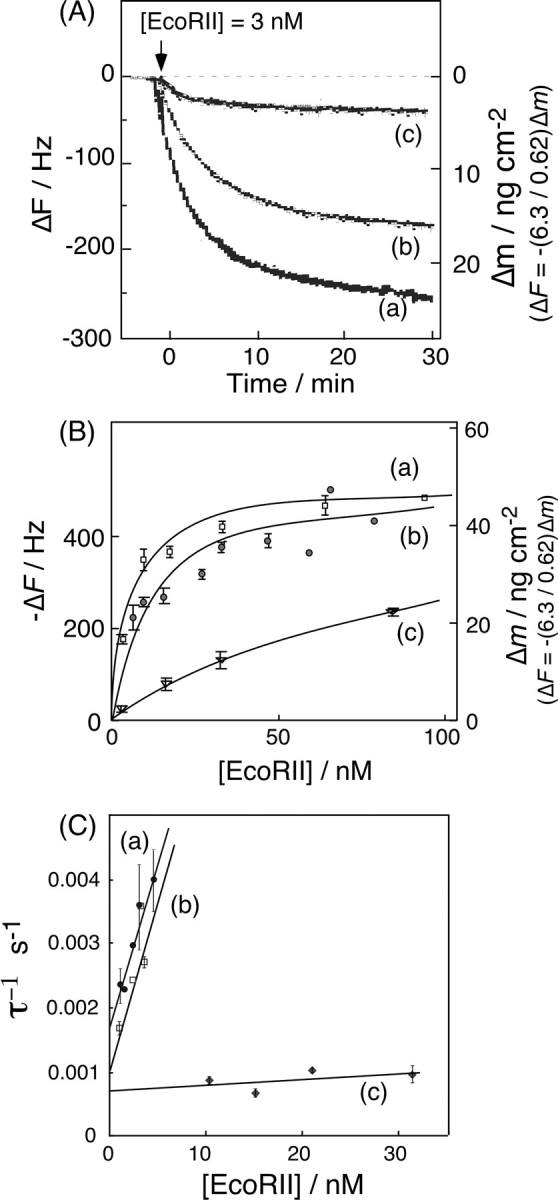
Binding and Cleavage Behaviors of EcoRII in the Presence of Mg2+ Ions—Although EcoRII shows only binding behaviors to the cognate site of DNA in the presence of Ca2+ ions, it was expected to also bind and cleave the cognate site in the presence of Mg2+ ions (1–3). Fig. 5A shows typical frequency decreases of the DNA-immobilized QCM, in response to the addition of EcoRII in the presence of Mg2+ or Ca2+ ions. For the 2-site DNA-immobilized QCM with 5 mm Mg2+ ions (curve a), the frequency decrease (mass increase) was about one-half of that in the presence of Ca2+ ions, in which only 1:1 binding was observed without any cleavage (curve b). Because the 2-site DNA was immobilized at 19 ± 1 ng (0.55 ± 0.02 pmol) cm–2 (–200 ± 10 Hz cm–2) on the QCM, and the cleavage site existed near the middle of the length of the DNA, a frequency increase of about 100 Hz (9 ng (0.55 pmol) cm–2) from the starting point was expected when the DNA was cleaved, and the enzyme was released from the residual DNA on the QCM. In contrast, for the 1-site DNA-immobilized QCM in the presence of Mg2+ ions, only the binding behavior was observed (curve c), which clearly indicated that two recognition sites are required to cleave the DNA. The N-domain of EcoRII can bind either the inner or outer recognition site of the 2-site DNA on the QCM (see Fig. 2A). When the N-domain binds to the outer cognate and the C-domain cleaves the inner cognate (α-type digestion, Fig. 2A), EcoRII will be released from the QCM plate with the digested DNA. On the other hand, when the N-domain binds to the inner cognate and cleaves the outer site (β-type digestion), then EcoRII will remain on the residual DNA because of the strong affinity of the N-domain. Thus, the frequency change of curve a in Fig. 5A would indicate that both α- and β-type digestions occurred simultaneously on the QCM. Actually, we prepared a 2-site-digested DNA-α that mimicked the residual DNA after the α-type digestion (see Fig. 2B), and a 2-site-digested DNA-β that mimicked the residual DNA after the β-type digestion. The Kd values of EcoRII for the 2-site-digested DNA-α and -β were 150 nm and 13 nm, respectively, with similar saturation binding in the presence of Ca2+ ions (see runs 6 and 7 in Table 1). Thus, EcoRII remains with the β-type-digested DNA on the QCM (Kd = 13 nm), but releases from the α-type-digested DNA (Kd = 150 nm). The results were consistent with our proposed cleavage mechanism shown in Fig. 2A. The binding and cleavage reactions are expressed by Equation 6,
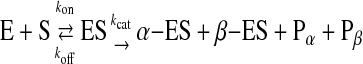 |
(Eq. 6) |
where, E, S, ES, α-ES, β-ES, Pα, and Pβ are EcoRII, the 2-site DNA, the EcoRII/DNA complex, the α-type-digested EcoRII/DNA complex, the β-type-digested EcoRII/DNA complex, the α-type-cleaved DNA, and the β-type-cleaved DNA, respectively. The formation of the ES complex at time t is shown by Equation 7, which was revised from Equation 3 by the addition of the product formation step to the [α-ES]max and [β-ES]max formation step, because the [ES]max (= [α-ES]max + [β-ES]max) decrease corresponded to each cleaved DNA ([Pα] and [Pβ], respectively).
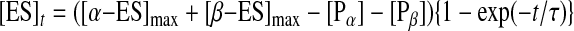 |
(Eq. 7) |
FIGURE 5.
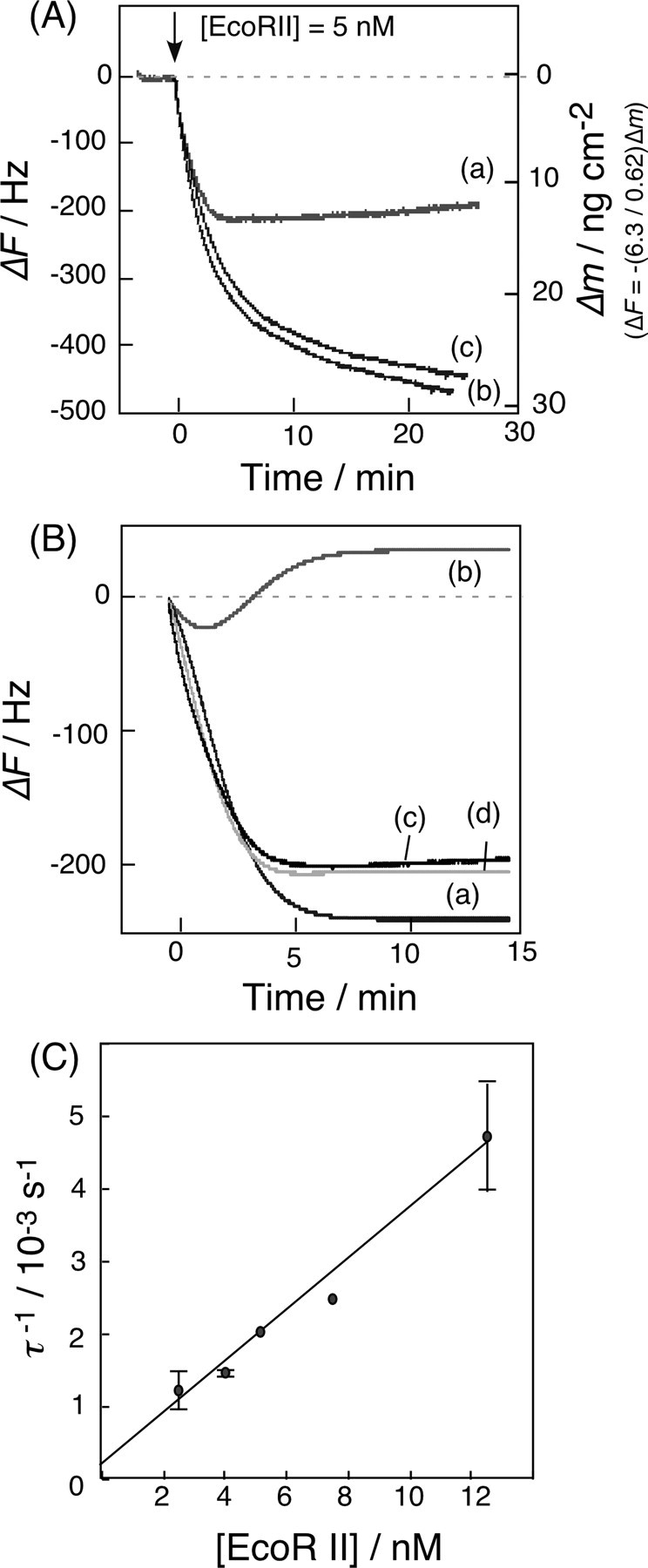
A, typical frequency changes of the DNA-immobilized QCM, in response to the addition of EcoRII. Curve a, 2-site DNA with Mg2+ ions; curve b, 2-site DNA with Ca2+ ions; and curve c, 1-site DNA with Mg2+ ions. ([EcoRII] = 5 nm, [DNA] = 19 ± 1 ng (0.55 ± 0.02 pmol) cm–2 on a QCM, [MgCl2 or CaCl2] = 5 mm, 10 mm Tris-HCl, pH 7.5, 50 mm NaCl, 1 mm DTT, 25 °C). B, curve a, calculated time dependence of the [β-ES] complex as shown in Equation 7 in the text; curve b, calculated time dependence of the [α-ES] as shown in Equations 8 and 9 in the text; curve c, fitted curve obtained from the simultaneous Equations 7, 8, 9; and curve d, experimental curve at [EcoRII] = 5 nm and [2-site DNA] = 19 ng (0.55 pmol) cm–2 on a QCM. C, linear reciprocal plots of the relaxation rate (τ–1) obtained from Equation 7 against the EcoRII concentration. The kon and koff values can be obtained from the slope and the intercept, respectively, according to Equation 5 in the text.
The amount of DNA cleaved ([P]α and [P]β) at time t is shown by Equations 8 and 9.
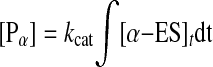 |
(Eq. 8) |
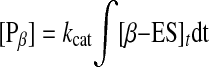 |
(Eq. 9) |
Curves a and b in Fig. 5B were obtained from calculations based on Equations 7, 8, and 9, respectively. Curve c is the fitted curve from the simultaneous Equation 7 and Equations 8 and 9, and curve d is the experimental curve at [EcoRII] = 5 nm and [2-site DNA] = 19 ng (0.55 pmol) cm–2 on the QCM. When the relaxation rates (τ–1) were plotted against [EcoRII] (2–12 nm), kon and koff values were obtained from the linear plot of Equation 5. The dissociation constant (Kd) of the ES complex was obtained from the ratio koff/kon. The cleavage rate constant (kcat) was obtained from the fitting of Equation 8. The obtained kinetic parameters are summarized in Table 1 (run 1). The kcat values can be obtained separately from each enzyme concentration experiment at 1–12 nm from Equation 8, and the kcat value in Table 1 is the average from each experiment (±10% error). The ratio of the α-type and β-type digestions was ∼0.4 from the fitting equation. Thus, there are no options for EcoRII binding to select the inner versus outer cognates of the DNA strand. The kon = 3.0 × 105 m–1 s–1 and koff = 1.0 × 10–3 s–1 values for the 2-site DNA obtained from Equations 6, 7, 8, 9 (binding and cleavage reactions, run 1) were consistent with those obtained from the binding kinetics of the only binding reaction according to Equations 3, 4, 5 (kon = 5.2 × 105 m–1 s–1 and koff = 1.0 × 10–3 s–1, run 3). We noted no cleavage reaction for EcoRII (kcat = 0 s–1) for the 1-site DNA in the presence of Mg2+ (curve c of Fig. 5A and run 2 of Table 1) despite the binding to the DNA. This result indicates that even if the C-domain binds to the 1-site DNA, it has no cleavage activity. Alternatively, the activated C-domain would not exist in bulk solution before binding to the DNA, which is direct evidence of an allosteric activation that EcoRII requires for the binding of the N-domain to the recognition site to enable the cleavage reaction.
EcoRII Reaction in Solution—Using a DNA-immobilized QCM system, we determined the binding (kon) and dissociation rate constants (koff) of the EcoRII/DNA complex in the presence of Ca2+, and both the binding process (kon and koff) and the cleavage rate constant (kcat) of the EcoRII/DNA complex in the presence of Mg2+. From the obtained kinetic parameters (kon, koff, and kcat) of the EcoRII reaction, we suggest a new aspect to the mechanism of the EcoRII reaction: first EcoRII binds onto one (inner or outer) of two cognate sites of the DNA strand without any selectivity with its N-domain. Next, the secondary active site of the C-domain covered by the N-domain is uncovered and can approach the DNA after the allosteric conformation change. Afterward, the C-domain can bind to the other cognate site and cleaves within this site, and EcoRII can efficiently seek by anchoring the N-domain on the initial cognate site. Thus, EcoRII usually inactivates itself and is activated via the DNA binding of the N-domain, and then repeats the cleavage reaction with the activated complex. We have reported the binding and cleavage reactions of EcoRV as typical of a Type IIP restriction endonuclease, in which EcoRV simply recognizes (Kd = 1.6 nm) and cleaves one site on the DNA (kcat = 0.6 s–1), quickly releases from the digested DNA, and then repeats the reaction on the other site (24). In this reaction, the recognition sites of the Type IIP enzyme should be cleaved completely in the presence of the enzyme at a concentration near the Kd value. In contrast, because cleavage by EcoRII requires DNA-binding by the N-domain (Kd = 2.2 nm), the recognition sites of EcoRII would be cleaved until a concentration near the Kd value of the N-domain, and then remains at a constant concentration. Thus, the catalytic cycle of EcoRII is controlled by a positive feedback mechanism (31) to maintain its activated condition by anchoring its N-domain to the first cognate site. The dissociation rate of koff = 1.7 × 10–3 s–1 of EcoRII for the 1-site DNA was 10-fold smaller than the catalytic cleavage rate of kcat = 1.8 × 10–2 s–1 of EcoRII. Therefore, the several chances at site digestion could occur before the dissociation of the N-domain from the first bound site, which would support an autocatalysis mode of the activated EcoRII.
Many DNA-related enzymes have several domains in the enzyme or form a complex with other proteins, such as transcriptional regulators (32, 33) and recombinases (34–36). They would work cooperatively or allosterically to represent their functional expression effectively depending on their concentration in the biological network. It has been suggested that EcoRII evolved by acquiring the N-domain, which functions as an effector such that the C-domain cannot easily access the DNA (12). This evolution can be regarded as a regulation of the cleavage activity by blocking the N-domain. However, considering that the effect of post-segregational cell killing was stronger than other restriction endonucleases, EcoRII still has rather high restriction activity in vivo (26, 37).
In summary, the kinetic parameters obtained by the QCM method can explain the mechanism of action of EcoRII following an analysis of the consecutive reactions on immobilized DNA. This QCM technology gave information on the stepwise binding of EcoRII, and the anchoring effect of the N-domain after the cleavage reaction as a positive feedback control.
Acknowledgments
We thank Prof. Kobayashi, Tokyo University, for providing the plasmids encoding EcoRII and for valuable discussions.
This work was supported in part by a Grant-in aid for Scientific Research from the Ministry of Education, Culture, Sports, Science, and Technology of Japan. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement”in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: QCM, quartz-crystal microbalance; EDC, 1-ethyl-3-(3-dimethylamino-propyl)carbodiimide; DTT, dithiothreitol; ds, double-stranded.
References
- 1.Pingoud, A., and Jektsch, A. (1997) Eur. J. Biochem. 246 1–22 [DOI] [PubMed] [Google Scholar]
- 2.Pingoud, A., and Jektsch, A. (2001) Nucleic Acids Res. 29 3705–3727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pingoud, A. Fuxreiter, M., Pingoud, V., and Wende, W. (2005) Cell. Mol. Life. Sci. 62 685–707 [DOI] [PubMed] [Google Scholar]
- 4.Mücke, M., Krüger, D. H., and Reuter, M. (2003) Nucleic Acids Res. 31 6079–6084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kosykh, V. G., Puntezhis, S. A., Buryanov, Y. I., and Bayev, A. A. (1982) Biokhimiya 47 619–625 [PubMed] [Google Scholar]
- 6.Reuter, M., Kupper, D., Meisel, A., Schroeder, C., and Krüger, D. H. (1998) J. Biol. Chem. 273 8294–8300 [DOI] [PubMed] [Google Scholar]
- 7.Krüger, D. H., Barcak, G. J., Reuter, M., and Smith, H. O. (1988) Nucleic Acids Res. 16 3997–4008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Krüger, D. H., Kupper, D., Meisel, A., Reuter, M., and Schroeder, C. (1995) FEMS Microbiol. Rev. 17 177–184 [DOI] [PubMed] [Google Scholar]
- 9.Petraouskene, O. V., Babkina, O. V., Tashlitsky, V. N., Kazankov, G. M., and Gromova, E. S. (1998) FEBS Lett. 425 29–34 [DOI] [PubMed] [Google Scholar]
- 10.Mücke, M., Lurz, R., Mackeldanz, P., Behlke, J., Krüger, D. H., and Reuter, M. (2000) J. Biol. Chem. 275 30631–30637 [DOI] [PubMed] [Google Scholar]
- 11.Reuter, M., Schneider, M. J., Kupper, F., Meisel, A., Mackeldanz, P., Krüger, D. H., and Schroeder, C. (1999) J. Biol. Chem. 274 5213–5221 [DOI] [PubMed] [Google Scholar]
- 12.Mücke, M., Grelle, G., Behlke, J., Kraft, R., Krüger, D. H., and Reuter, M. (2002) EMBO J. 21 5262–5268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhou, X. E., Wang, Y., Reuter, M., Mücke, M., Krüger, D. H., Meehan, E. J., and Chen, L. (2004) J. Mol. Biol. 335 307–319 [DOI] [PubMed] [Google Scholar]
- 14.Sauerbrey, G. Z. (1959) Z. Phyzik. 155 206–222 [Google Scholar]
- 15.Rickert, J., Brecht, A., and Göpel, W. (1997) Anal. Chem. 69 1441–1448 [DOI] [PubMed] [Google Scholar]
- 16.Höök, F., Voros, J., Rodahl, M., Kurrat, R., Böni, P., Ramsden, J. J., Textor, M., Spencer, N. D., Tengvall, P., Gold, J., and Kasemo, B. (2002) Coll. Surf. B. 24 155–170 [Google Scholar]
- 17.Munro, J. C., and Frank, C. W. (2004) Macromolecules 37 925–938 [Google Scholar]
- 18.Larsson, C., Rodahl, M., and Höök, F. (2003) Anal. Chem. 75 5080–5087 [DOI] [PubMed] [Google Scholar]
- 19.Graneli, A., Rydstrom, J., Kasemo, B., and Höök, F. (2003) Langmuir 19 842–850 [Google Scholar]
- 20.Otzen, D. E., Oliveberg, M., and Höök, F. (2003) Coll. Surf. B. 29 67–73 [Google Scholar]
- 21.Ozeki, T., Morita, M., Yoshimine, H., Furusawa, H., and Okahata, Y. (2007) Anal. Chem. 79 79–88 [DOI] [PubMed] [Google Scholar]
- 22.Nishino, H., Nihira, T., Mori, T., and Okahata, Y. (2004) J. Am. Chem. Soc. 126 2264–2265 [DOI] [PubMed] [Google Scholar]
- 23.Nishino, H., Murakawa, A., Mori, T., and Okahata, Y. (2004) J. Am. Chem. Soc. 126 14752–14757 [DOI] [PubMed] [Google Scholar]
- 24.Matsuno, H., Niikura, K., and Okahata, Y. (2001) Biochemistry 40 3615–3622 [DOI] [PubMed] [Google Scholar]
- 25.Takahashi, S., Matsuno, H., Furusawa, H., and Okahata, Y. (2007) Anal. Biochem. 361 210–217 [DOI] [PubMed] [Google Scholar]
- 26.Chinen, A., Naito, Y., Handa, N., and Kobayashi, I. (2000) Mol. Biol. Evol. 17 1610–1619 [DOI] [PubMed] [Google Scholar]
- 27.Pingoud, V., Conzelmann, C., Kinzebach, S., Sudina, A., Metelev, V., Kubareva, E., Bujnicki, J. M., Lurz, R., Lüder, G., Xu, S. Y., and Pingoud, A. (2003) J. Mol. Biol. 329 913–929 [DOI] [PubMed] [Google Scholar]
- 28.Deibert, M., Grazulis, S., Sasnauskas, G., Siksnys, V., and Huber, R. (2000) Nat. Struct. Biol. 7 792–799 [DOI] [PubMed] [Google Scholar]
- 29.Bozic, D., Grazulis, S., Siksnys, V., and Huber, R. (1996) J. Mol. Biol. 255 176–186 [DOI] [PubMed] [Google Scholar]
- 30.Siksnys, V., Skirgaila, R., Sasnauskas, G., Urbanke, C., Cherny, D., Grazulis, S., and Huber, R. (1999) J. Mol. Biol. 291 1105–1118 [DOI] [PubMed] [Google Scholar]
- 31.Ferrell, J. E., Jr., and Machleder, R. M. (1998) Science 280 895–898 [DOI] [PubMed] [Google Scholar]
- 32.Schleif, R. (1992) Annu. Rev. Biochem. 61 199–223 [DOI] [PubMed] [Google Scholar]
- 33.Lewis, M., Chang, G., Horton, N. C., Kercher, M. A., Pace, H. C., Schumacher, M. A., Brennan, R. G., and Lu, P. (1996) Science 71 1247–1254 [DOI] [PubMed] [Google Scholar]
- 34.Guo, F., Gopaul, D. N., and van Duyne, G. D. (1997) Nature 389 40–46 [DOI] [PubMed] [Google Scholar]
- 35.Gopaul, D. N., Guo, F., and van Duyne, G. D. (1998) EMBO J. 17 4175–4187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Guo, F., Gopaul, D. N., and van Duyne, G. D. (1999) Proc. Natl. Acad. Sci. U. S. A. 96 7143–7146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Takahashi, N., Naito, Y., Handa, N., and Kobayashi, I. (2002) J. Bacteriol. 184 6100–6108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fant, C., Elwing, H., and Höök, F. (2002) Biomacromolecules 3 732–741 [DOI] [PubMed] [Google Scholar]
- 39.Höök, F., Rodahl, M., Kasemo, B., and Brezezinski, P. (1998) Proc. Natl. Acad. Sci. U. S. A. 95 12271–12276 [DOI] [PMC free article] [PubMed] [Google Scholar]